

Abstract
Background -
Truncating variants in the desmosomal gene PKP2 (PKP2tv) cause arrhythmogenic right ventricular cardiomyopathy (ARVC) yet display varied penetrance and expressivity.
Methods -
We identified individuals with PKP2tv from the UK Biobank (UKB) and determined the prevalence of an ARVC phenotype and other cardiovascular traits based on clinical and procedural data. The PKP2tv minor allelic frequency (MAF) in the UKB was compared with a second cohort of probands with a clinical diagnosis of ARVC (ARVC cohort), with a figure of 1:5000 assumed for disease prevalence. In silico predictors of variant pathogenicity (CADD and Splice AI) were assessed.
Results -
PKP2tv were identified in 193/200,643 (0.10%) UKB participants, with 47 unique PKP2tv. Features consistent with ARVC were present in 3 (1.6%), leaving 190 with PKP2tv without manifest disease (UKB cohort; MAF 4.73x10−4). The ARVC cohort included 487 ARVC probands with 144 distinct PKP2tv, with 25 PKP2tv common to both cohorts. The odds ratio (OR) for ARVC for the 25 common PKP2tv was 0.047 (95% CI 0.001-0.268; p 2.43x10−6), and only favored ARVC (OR>1) for a single variant, p.Arg79*. In silico variant analysis did not differentiate PKP2tv between the 2 cohorts. Atrial fibrillation was over-represented in the UKB cohort in those with PKP2tv (7.9% vs. 4.3%; OR 2.11; p=0.005).
Conclusions -
PKP2tv are prevalent in the population and associated with ARVC in only a small minority, necessitating a more detailed understanding of how PKP2tv cause ARVC in combination with associated genetic and environmental risk factors.
Keywords: Arrhythmogenic right ventricular cardiomyopathy, plakophilin-2, truncating variants, atrial fibrillation
Introduction
Arrhythmogenic right ventricular cardiomyopathy (ARVC), the right dominant form of arrhythmogenic cardiomyopathy, is an inherited cardiac disorder characterized by ventricular arrhythmia, heart failure and sudden cardiac death. 1,2 The diagnosis is based on varying electrocardiographic, structural, and histological features as defined by the 2010 International Task Force criteria (TFC). 3 In its early course ARVC is characterized by ventricular arrhythmias, with the later development of ventricular dysfunction leading to heart failure 4 and atrial remodeling associated with atrial fibrillation. 5
In two thirds of patients meeting TFC, a causal genetic variant can be identified, most commonly in the gene PKP2, encoding the desmosomal armadillo repeat protein, plakophilin-2. Credible disease variants are almost exclusively truncating variants (PKP2tv) with the mutant transcript exposed to nonsense medicated mRNA decay (NMD) leading to haploinsufficiency as the inferred mechanism, suggesting the vast majority, if not all, of PKP2tv share a common mechanism.
Cascade screening within families affected by ARVC has demonstrated reduced penetrance and variable expressivity in family members compared to probands. 6 Additionally, multiple PKP2tv associated with ARVC have been identified in large-scale population sequencing projects and as secondary findings, supporting the concept of genetic complexity beyond a simple monogenic disorder.
Prior studies have assessed sequence variation in cardiomyopathy genes identified within control populations compared to patients with a clinical diagnosis of cardiomyopathy,7 or the presence of a cardiomyopathy phenotype in those with desmosomal variants. 8,9 In this analysis we focused specifically on PKP2; firstly, to identify PKP2tv in UK Biobank participants and to assess for the presence of a phenotype consistent with ARVC and other cardiovascular traits such as atrial fibrillation and heart failure and identify specific PKP2tv present in those without manifest cardiomyopathy. Secondly, we identified probands with ARVC and a PKP2tv reported in the medical literature and compared the frequency of PKP2tv amongst the 2 cohorts.
Methods
Detailed methods are available in the Supplementary Material. All data used in this analysis is open source and publicly available. The ARVC cohort was derived from published cases in the medical literature (https://pubmed.ncbi.nlm.nih.gov) using the search terms as defined in the Supplementary Material. Genetic and clinical data in the UK Biobank cohort was identified in the UK Biobank (https://www.ukbiobank.ac.uk) as described. No human subjects were used for this study. The authors declare that all supporting data are available within the article (and the online associated supplementary files).
Results
UK Biobank participants with PKP2tv
PKP2tv were identified in 193 UKB participants, who were 58.2 ± 8.4 years old at recruitment, of whom 94 (48.7%) were male. Clinical findings consistent with ARVC were reported in 3 (1.6%) individuals. A 58-year-old male with ventricular tachycardia (VT) and implantable cardioverter-defibrillator (ICD) and family history of cardiomyopathy, and a 44-year-old male with VT and an ICD. Both had the Arg735X variant. The third, a 51-year-old male with the splice variant c.2146-1G>C had a history of premature ventricular beats, VT and prior electrophysiology study and ablation, left bundle branch block and AF. None of the 3 had hypertension or coronary artery disease. Overall, the identifiable prevalence of ARVC in the UK Biobank cohort was 3/193 (1.6%).
UKB Cohort
The remaining 190 UKB participants with PKP2tv (MAF 4.73x10−4) were considered to have insufficient symptoms or evidence of ARVC to result in clinical investigations or management and constituted the UKB cohort. In this cohort there were 45 distinct PKP2tv of which 20 were reported more than once, and 25 were singletons. Three variants were present in more than 10 of the UKB cohort (c.2146-1G>C, p.Arg735* and Thr50Serfs*61) all of which have been associated with ARVC in numerous individuals (Figure 1; Supplementary table I). Based on location no variants were predicted to escape NMD.
Figure 1.
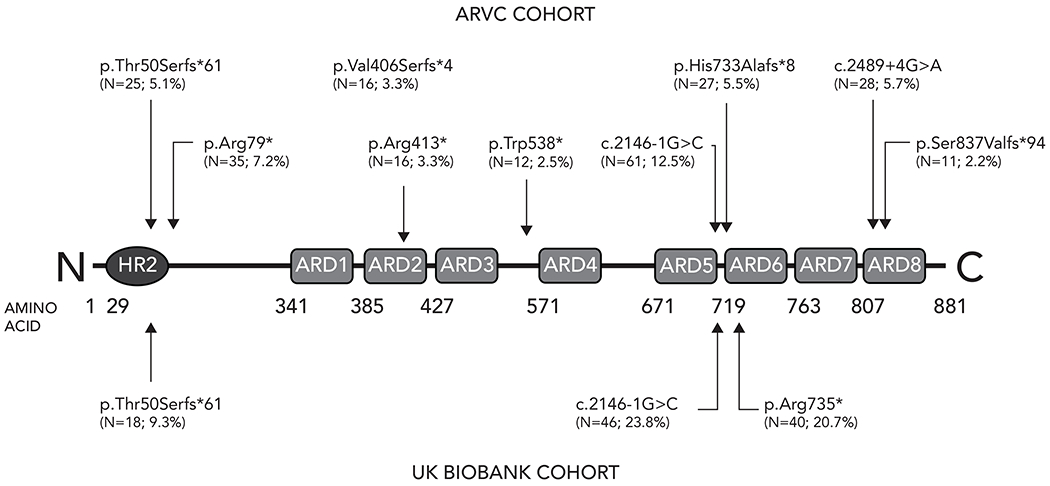
PKP2 truncating variants in ARVC cohort and UK Biobank cohort
Topological location of PKP2 protein truncating variants identified in more than 10 individuals in the ARVC and UK Biobank cohorts. The number of times the variant was identified, and percentage of overall burden is displayed with each variant.
CMR data was reported for 17/190 individuals: LV ejection fraction was 58.2 ± 5.7%; LVEDVI 69.8 ± 14.3mL/m2; and cardiac index 2.6 ± 0.4mL/min/m2. ECG data was also available in 17/190 individuals with QRS duration measured at 91 ± 18ms and corrected QT interval 425 ± 35ms.
Atrial fibrillation was present in 15/190 (7.9%) in the UKB cohort with PKP2tv compared to 8699 (4.3%) of those without PKP2tv and was therefore over-represented in those with PKP2tv (OR 2.11; p=0.005). The prevalence of cardiac failure was 4/190 (2.1%) and VT 3/190 (1.6%), which were not different from the overall cohort. All 3 individuals with VT had coronary artery disease.
Previously reported PKP2tv associated with ARVC
We identified 487 ARVC probands previously reported in the medical literature and Leiden University Medical Center database with 144 distinct PKP2tv. Ten variants in 22 probands were copy number variants (4.5% of the overall number) varying from deletion of the entire PKP2 coding sequence (9/22; 40.9%) to deletion of single or multiple exons, which were excluded from further analysis. Of the remaining 134 PKP2tv, 53 were reported more than once and 81 were singletons. Nine different PKP2tv were identified in >10 individuals in the ARVC cohort, representing 47% of the total. (Figure 1; Supplementary table II) Four variants were predicted to escape NMD.
PKP2tv seen in both UKB cohort and ARVC cohort
Twenty-five (25) PKP2tv were common to both the ARVC cohort and UKB cohort, accounting for 220/487 (45.0%) of individuals in the ARVC cohort and 157/190 (82.6%) in UKB cohort respectively. The OR for all 25 variants was 0.047 (95% CI 0.001-0.268; p = 2.43x10−6) and was <1 for all individual variants except for p.Arg79*, suggesting that the majority with variants associated ARVC in other individuals will not develop clinical features of ARVC. (Table 1; Figure 2)
TABLE 1:
PKP2 truncating variants identified in both ARVC cohort and UKB cohort
| Nucleotide position | Nucleotide substitution | Consequence | UKB cohort (n) | UKB cohort (%) | UKB cohort (MAF) | ARVC cohort (n) | ARVC cohort (%) | ARVC cohort (MAF) Prevalence 1:5000 | Odds ratio (OR) | 95% CI (Lower) | 95% CI (Upper) | Fisher’s exact test (p) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ALL | 157 | 48.61% | 3.91E-04 | 220 | 45.0% | 1.89E-05 | 0.0473 | 0.0012 | 0.2675 | 2.43E-06 | ||
| 14 | c.14delG | p.Gly5Alafs*34 | 1 | 0.31% | 2.49E-06 | 1 | 0.20% | 8.61E-08 | 0.0345 | 0.0004 | 2.7095 | - |
| 148 | c.148_151delACAG | p.Thr50Serfs*61 | 18 | 5.52% | 4.49E-05 | 25 | 5.11% | 2.15E-06 | 0.0479 | 0.0012 | 0.3036 | 4.58E-04 |
| 235 | c.235C>T | p.Arg79* | 1 | 0.31% | 2.49E-06 | 35 | 7.16% | 3.01E-06 | 1.2081 | 0.0154 | 94.7665 | - |
| 253 | c.253_256delGAGT | p.Glu85Metfs*26 | 5 | 1.53% | 1.25E-05 | 4 | 0.82% | 3.44E-07 | 0.0276 | 0.0006 | 0.2468 | - |
| 275 | c.275T>A | p.Leu92* | 6 | 1.84% | 1.50E-05 | 3 | 0.61% | 2.58E-07 | 0.0173 | 0.0004 | 0.1423 | 1.14E-04 |
| 337 | c.337-2A>T | 8 | 2.45% | 1.99E-05 | 1 | 0.20% | 8.61E-08 | 0.0043 | 0.0001 | 0.0322 | 3.49E-10 | |
| 368 | c.368G>A | p.Trp123* | 1 | 0.31% | 2.49E-06 | 1 | 0.20% | 8.61E-08 | 0.0345 | 0.0004 | 2.7095 | - |
| 658 | c.658C>T | p.Gln220* | 1 | 0.31% | 2.49E-06 | 1 | 0.20% | 8.61E-08 | 0.0345 | 0.0004 | 2.7095 | - |
| 663 | c.663C>A | p.Tyr221* | 4 | 1.23% | 9.97E-06 | 2 | 0.41% | 1.72E-07 | 0.0172 | 0.0004 | 0.1744 | 2.14E-03 |
| 1063 | c.1063>T | p.Arg355* | 1 | 0.31% | 2.49E-06 | 2 | 0.41% | 1.72E-07 | 0.0691 | 0.0009 | 5.4190 | - |
| 1132 | c.1132C>T | p.Gln378* | 2 | 0.61% | 4.98E-06 | 8 | 1.64% | 6.89E-07 | 0.1381 | 0.0023 | 2.6522 | - |
| 1177 | c.1177C>T | p.Gln393* | 1 | 0.31% | 2.49E-06 | 1 | 0.20% | 8.61E-08 | 0.0345 | 0.0004 | 2.7095 | - |
| 1237 | c.1237C>T | p.Arg413* | 3 | 0.92% | 7.48E-06 | 16 | 3.27% | 1.38E-06 | 0.1841 | 0.0035 | 2.2928 | - |
| 1369 | c.1369_1372delCAAA | p.Gln457* | 1 | 0.31% | 2.49E-06 | 5 | 1.02% | 4.30E-07 | 0.1726 | 0.0022 | 13.5475 | - |
| 1643 | c.1643delG | p.Gly548Valfs*15 | 1 | 0.31% | 2.49E-06 | 9 | 1.84% | 7.75E-07 | 0.3107 | 0.0040 | 24.3856 | - |
| 1689 | c.1689-1G>C | 1 | 0.31% | 2.49E-06 | 1 | 0.20% | 8.61E-08 | 0.0345 | 0.0004 | 2.7095 | - | |
| 1709 | c.1709delC | p.Ala570Valfs*7 | 2 | 0.61% | 4.98E-06 | 1 | 0.20% | 8.61E-08 | 0.0173 | 0.0003 | 0.3315 | - |
| 1821 | c.1821dupC | p.Val608Cysfs*6 | 6 | 1.84% | 1.50E-05 | 3 | 0.61% | 2.58E-07 | 0.0173 | 0.0004 | 0.1423 | 1.14E-04 |
| 2013 | c.2013delC | p.Lys672Argfs*12 | 1 | 0.31% | 2.49E-06 | 3 | 0.61% | 2.58E-07 | 0.1036 | 0.0013 | 8.1285 | - |
| 2146 | c.2146-1G>C | 45 | 13.80% | 1.12E-04 | 61 | 12.47% | 5.25E-06 | 0.0468 | 0.0012 | 0.2743 | 1.62E-05 | |
| 2198 | c.2918_2202delACACC | p.His733Alafs*8 | 1 | 0.31% | 2.49E-06 | 1 | 0.20% | 8.61E-08 | 0.0345 | 0.0004 | 2.7095 | - |
| 2203 | c.2203C>T | p.Arg735* | 38 | 11.66% | 9.47E-05 | 6 | 1.23% | 5.17E-07 | 0.0055 | 0.0001 | 0.0323 | 6.83E-27 |
| 2299 | c.2299+1G>A | 2 | 0.61% | 4.98E-06 | 1 | 0.20% | 8.61E-08 | 0.0173 | 0.0003 | 0.3315 | - | |
| 2421 | c.2421C>A | p.Tyr807* | 1 | 0.31% | 2.49E-06 | 2 | 0.41% | 1.72E-07 | 0.0691 | 0.0009 | 5.4190 | - |
| 2489 | c.2489+1G>A | 6 | 1.84% | 1.50E-05 | 27 | 5.52% | 2.32E-06 | 0.1553 | 0.0034 | 1.2803 | - |
Figure 2.
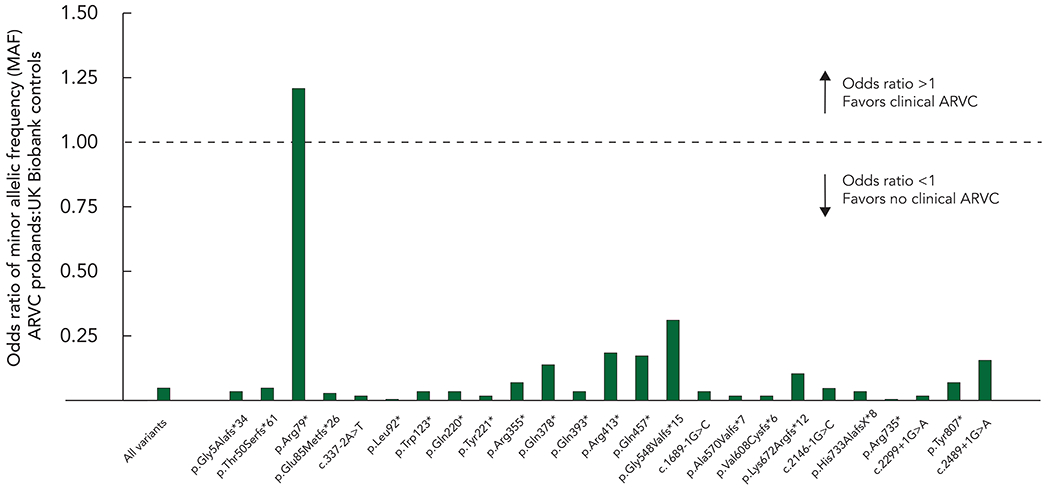
Odds ratio of PKP2 truncating variants identified in both ARVC cohort and UK Biobank cohort.
The odds ratio (OR) of minor allelic frequency for PKP2 truncating variants identified in both ARVC and UK Biobank cohorts. The dotted line denotes an OR of 1, and variants above that line are more commonly associated with ARVC than not.
To further define the effect of varied ARVC prevalence in the population, we recalculated the OR assuming a maximum disease prevalence of 1 in 2000 and minimum of 1 in 8000, for all PKP2tv common to the 2 cohorts, and for the variant most favoring (p.Arg79*; OR = 1.2) and least favoring (p.Arg735*; OR = 0.0034) clinical ARVC in the initial analysis. At 1 in 2000 the OR were 3.02 for p.Arg79* and 0.01 for p.Arg735*, and at 1:8000 was 0.76 for p.Arg79* and 0.003 for p.Arg735* (Table 2). Irrespective of the disease prevalence the OR was >1 for only a single variant (p.Arg79*).
TABLE 2:
Odds ratio for all PKP2 truncating variants, p.Arg79* and p.Arg735* based on ARVC population prevalence
| Nucleotide position | Nucleotide substitution | Consequence | UKB cohort (n) | UKB cohort (%) | UKB cohort (MAF) | ARVC cohort (n) | ARVC cohort (%) | ARVC cohort (MAF) Prevalence 1:2000 | Odds ratio (OR) | 95% CI (Lower) | 95% CI (Upper) | Fisher’s exact test (p) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ALL | 157 | 48.61% | 4.16E-04 | 220 | 45.0% | 4.74E-05 | 0.113571 | 0.002863 | 0.641817 | - | ||
| 235 | c.235C>T | p.Arg79* | 1 | 0.31% | 2.49E-06 | 35 | 7.16% | 7.53E-06 | 3.020255 | 0.038476 | 236.6325 | - |
| 2203 | c.2203C>T | p.Arg735* | 38 | 11.66% | 1.20E-04 | 6 | 1.23% | 1.29E-06 | 0.000001 | 0.013639 | 0.000341 | 1.20E-15 |
| ARVC cohort (MAF) Prevalence 1:3500 | ||||||||||||
| ALL | 157 | 48.61% | 4.16E-04 | 220 | 45.0% | 2.71E-05 | 0.064881 | 0.001638 | 0.366663 | 2.66E-04 | ||
| 235 | c.235C>T | p.Arg79* | 1 | 0.31% | 2.49E-06 | 35 | 7.16% | 4.30E-06 | 1.725788 | 0.021986 | 135.3357 | - |
| 2203 | c.2203C>T | p.Arg735* | 38 | 11.66% | 1.20E-04 | 6 | 1.23% | 7.38E-07 | 0.007801 | 0.000198 | 0.046071 | 3.31E-22 |
| ARVC cohort (MAF) Prevalence 1:6500 | ||||||||||||
| ALL | 157 | 48.61% | 4.16E-04 | 220 | 45.0% | 1.46E-05 | 0.034924 | 0.000884 | 0.197432 | 4.86E-09 | ||
| 235 | c.235C>T | p.Arg79* | 1 | 0.31% | 2.49E-06 | 35 | 7.16% | 2.32E-06 | 0.929335 | 0.011846 | 72.909721 | - |
| 2203 | c.2203C>T | p.Arg735* | 38 | 11.66% | 1.20E-04 | 6 | 1.23% | 3.97E-07 | 0.004208 | 0.000110 | 0.024819 | 1.54E-30 |
| ARVC cohort (MAF) Prevalence 1:8000 | ||||||||||||
| ALL | 157 | 48.61% | 4.16E-04 | 220 | 45.0% | 1.18E-05 | 0.028371 | 0.000719 | 0.160413 | 2.43E-11 | ||
| 235 | c.235C>T | p.Arg79* | 1 | 0.31% | 2.49E-06 | 35 | 7.16% | 1.88E-06 | 0.755075 | 0.009627 | 59.245524 | - |
| 2203 | c.2203C>T | p.Arg735* | 38 | 11.66% | 1.20E-04 | 6 | 1.23% | 3.23E-07 | 0.003422 | 0.000091 | 0.020161 | 1.60E-33 |
ARVC = arrhythmogenic right ventricular cardiomyopathy; UKB = UK Biobank; MAF = minor allelic frequency
In silico analysis
CADD and Splice AI scores were available for 74/109 (68%) and 26/26 (100%) of the PKP2tv in ARVC cohort. CADD (38) and Splice AI (7) scores were available for all variants in the UKB cohort. There was no difference between the two cohorts for either CADD score (33.7 ± 6.5 vs. 32.2 ± 6.9; p=0.26) or Splice AI (0.91 ± 0.22 vs. 0.98 ± 0.02; p=0.39). Similarly, there was no difference in CADD score between the 20 variants in UKB cohort associated with ARVC compared to those not (33.7 ± 6.5 vs. 31.7 ± 7.0; p=0.15).
Discussion
Since the seminal description by Gerull in 2004, the relationship correlation between PKP2tv and ARVC has become ever more recognized. Yet despite this the evidence for low penetrance and expressivity continues to evolve, initially from evaluation of large families and more recently on a genotype first approach. In this analysis we identified that PKP2tv, including those with an established disease association, are prevalent in the population yet only a very small minority of individuals harboring these variants appear to develop clinically manifest ARVC.
This poses a number of important questions in regard to the relationship between PKP2tv and ARVC. PKP2tv have historically been considered rare disease-causing variants with large effect, yet as in several genetic disorders the advent of large scale databases incorporating comprehensive sequencing has identified many such variants in seemingly unaffected individuals.10 Over 80% of individuals in the UK Biobank cohort had a variant previously associated with ARVC, making this a highly relevant and comparative cohort. This evolving picture has important implications for our understanding of the relationship between purportedly monogenic cardiovascular diseases and associated genetic variants, with increasing evidence suggesting many variants are of limited effect, and require genetic or environmental modifiers to reach disease threshold. 11 Previously reported factors promoting penetrance include second desmosomal genetic variants,12 and most recently endurance exercise which may place excessive strain on susceptible right ventricular myocardium.13 As experience of the natural history of PKP2tv (and those in other genes) grows through resources such as UK Biobank, the reporting and management of secondary findings on non-phenotype testing will become better understood.
PKP2tv associated with ARVC
These findings provide a detailed overview of the genetic architecture of PKP2tv associated with ARVC, an increasingly recognized correlation within desmosomal arrhythmogenic cardiomyopathies.14–17 Although this analysis includes only cases published in the medical literature, the cohort was large enough that it is likely an accurate representation of the spectrum and prevalence of different PKP2tv associated with ARVC. Whilst many variants are encountered only once, a number were reported in several probands in the ARVC cohort consistent with founder effects as previously described using haplotype analysis in multiple kindreds.15
PKP2tv in UK Biobank cohort
To provide a meaningful comparison with probands with manifest disease in the ARVC cohort we identified UKB participants with PKP2tv. Based on the assumption that all PKP2tv have the same impact on functional protein levels within the desmosome, the 190 individuals in the UKB cohort should theoretically have the same genetic predisposition as those with established disease. The UK Biobank provides comprehensive phenotypic data together with whole exome sequencing, allowing determination of the prevalence and disease association of specific variants in an adult population, thereby reducing the potential confounder of age-related penetrance. The average age of the UK Biobank cohort in this study was 57 years, compared to an average age of symptomatic disease onset of 33 years in individuals presenting with ARVC.18 As the onset of ARVC continues into later life but at lower incidence, the possibility of subsequent disease onset cannot be excluded.6
A phenotype consistent with ARVC was only seen in a very small minority (1.6%), with an OR of 0.047 for the development of ARVC assessing all PKP2tv identified in both cohorts. This finding is concordant with, and significantly scales the findings of, prior studies where a genotype first approach identified 19 individuals with PKP2tv, and although 5 exhibited features of the condition none had a ‘demonstrable ARVC phenotype’. 9 To accurately define penetrance of any PKP2tv in the UKB cohort (i.e., a binary phenomenon where features consistent with ARVC are present or not) would require a level of clinical evaluation beyond that provided in the data available. That said if these variants are indeed penetrant, the expressivity appears insufficient to generate symptoms requiring evaluation or documented clinical findings and intervention. The finding that AF was more common in the UKB cohort with PKP2tv requires further clinical validation and mechanistic characterization but may suggest an increased risk of common cardiovascular traits in those with variants in known disease genes typically associated with monogenic disorders.
Comparison of PKP2tv between ARVC and UKB cohorts
There was significant commonality in PKP2tv identified in ARVC cohort and UKB cohort, with many of the same founder variants seen in both groups and many well recognized ARVC-associated variants seen in apparently unaffected individuals in the UK Biobank. For example, the splice acceptor variant c.2146-1G>C, the most common variant associated with ARVC, was only associated with clinically manifest ARVC in 1 individual of 46 UK Biobank participants. On direct comparison between the two groups the OR was greater than 1 for only a single variant, p.Arg79*. This statistic was not affected by reducing the estimated disease prevalence to 1 in 2000. (Table 2)
In silico predictors of variant pathogenicity may be an important additional mechanism to help identify variants more likely to be disease associated. Using both CADD and SpliceAI we were unable to identify any statistically significant difference in scores between the variants in the ARVC and UKB cohorts. This may not be surprising given the significant commonality between PKP2tv in the 2 cohorts and predicted common mechanism for all variants.
Haploinsufficiency as primary mechanism of disease
The consequence of all PKP2tv involved in this study makes haploinsufficiency a credible disease mechanism, where reduced levels of plakophilin-2 protein may fundamentally affect desmosomal integrity and binding with associated proteins. This is further supported by a classical ARVC phenotype in patients with whole gene deletions and heterozygous knock-out murine models. 19,20 Plakophilin-2 plays a key role in linking the desmosomal cadherins within the intercellular junction to intermediate filaments and the sarcomere affecting force transmission, regulate beta-catenin pathways, and control the expression of multiple genes critical to cellular calcium cycling. 21,22
Western blot analysis of myocardial samples from multiple patients with differing PKP2tv demonstrates a reduction of plakophilin-2 to ~50% of the levels seen in control samples. 23 Protein and transcript levels were similarly reduced in cultured keratinocytes mirroring that seen in myocardial samples, and notably were also reduced in keratinocytes from unaffected family members with PKP2tv suggesting haploinsufficiency alone is not a prerequisite for disease expression. 23 Analysis of induced pluripotent stem cells (iPSC) has provided further insights into cellular derangements associated with PKP2tv. iPSC derived cardiomyocytes from a proband with ARVC (p.Ala324Glyfs*11) exhibited significant reduction in plakophilin-2 and connexin-43 immunostaining, with marked distortion of desmosomal architecture.24 iPSC modeling in cells derived from unaffected individuals such as those in the UKB cohort may provide a valuable insight into cellular and molecular differences between those with and without ARVC.
A major question arising from this study is an explanation for why so many individuals with proven or seemingly disease-causing variants apparently have non-penetrant or sub-clinical disease, and well-known disease-associated variants such as c.2146-1G>C and p.Arg735* appear to be associated with ARVC in only a minority of genetically susceptible individuals. Possible explanations for tolerance of PKP2tv include upregulation of the wild type allele to create sufficient protein to maintain normal function, or that plakophilin-2 haploinsufficiency requires an additional modifier to cause manifest disease. Allelic imbalance via cis-promoter or enhancer elements may increase production of the wild-type protein as opposed to the mutant and has been demonstrated in other cardiac genetic disorders. Levels of wild type myosin binding protein C in patients with hypertrophic cardiomyopathy resulting from MYBPC3 truncating variants may be 70% that of unaffected individuals. 25 However, upregulation of the wild type allele to fully compensate for the any truncating variants transcribed by the mutant allele is rare in humans, 26 although what threshold exists for plakophilin-2 expression required to prevent ARVC is unknown.
All probands in the ARVC cohort included in this analysis had a single PKP2tv in the heterozygous state effectively eliminating a gene dosage effect from a second desmosomal variant promoting ARVC, a mechanism previously reported as an important disease modifier. 12 Endurance exercise has attracted much recent attention as an important disease environmental risk factor, promoting early-onset, severe disease in PKP2-mediated ARVC in several patient cohorts and experimental models, 13,27,28 a factor likely related to the disproportionate hemodynamic stress to which the right ventricle is exposed during exertion. 29 This has led to recommendations of exercise restriction in individuals with a PKP2tv to limit disease expression. 1 However, the true role of exercise as a disease modifier needs to be assessed in the wider at-risk population, to determine to what degree this may be broadly applicable or applies only to a subset of individuals with PKP2tv. Such information is not part of the UK Biobank. Given the long-term medical, psychological and social benefits of exercise, a more nuanced understanding of this genotype-environmental relationship has important implications for future clinical management of this population as recently described,30 especially those increasingly ascertained through genetic testing in the absence of cardiac indications. 31
Assumptions and limitations
This study relied on several assumptions and has inherent limitations. Firstly, in calculating the MAF for the ARVC cohort, and hence odds ratio, we assumed the disease prevalence to be 1:5000. This figure is frequently quoted, although accurate determination of the true population prevalence of any rare disease is challenging. We assessed the impact of deviations from this estimated prevalence rate using a sensitivity analysis. Increasing the prevalence in ARVC probands to 1 in 2000 did not alter the main finding that PKP2tv are more prevalent in those without overt disease. Secondly, we assumed all PKP2tv identified in the ARVC cohort were the main driver of disease, which is difficult to prove in the absence of detailed clinical and genetic evaluation of the wider family to demonstrate segregation.
Third, in an attempt to create two groups that were genetically comparable we assumed all PKP2tv lead to haploinsufficiency, which cannot be proven without detailed analysis of mRNA and protein levels for each variant, and to date such studies have only been performed for a minority. Mutant transcripts have been detected resulting from splice variants c.2146-1G>C, c.2489+1G>A, c.2489+4A>C and c.2490-1G>C, thereby seemingly escaping NMD, but to what degree these translate to functional protein incorporated into the desmosome is unknown. 16,32 In other genes implicated in cardiomyopathies (e.g., MYBPC3) mutant transcripts similarly escape NMD, although truncated products which could exert a dominant negative effect on the wild type protein within the sarcomere have not been identified.25,33 Finally, considerable ascertainment bias could be present in the cases reported in the literature such that the true prevalence and contribution to disease of different variants (and hence MAF) could be inaccurate. We have attempted to mitigate this potential bias by including all cases reported.
Conclusions:
Although PKP2tv appear unequivocally associated with ARVC, they are also prevalent in the general population and are associated with clinically overt disease in only a small minority of genetically at-risk individuals. Many variants are recurrent and common to both those with ARVC and seemingly unaffected individuals yet seem more common in the latter despite an apparently common pathway leading to haploinsufficiency, suggesting a more complex etiology beyond simple Mendelian inheritance. A better understanding of the complex genetic and environmental interactions with PKP2tv leading to ARVC will allow for more personalized patient management.
Supplementary Material
Sources of Funding:
The Inherited Cardiac Arrhythmia Program is supported by the Mannion and Roberts Families. Dr. Roston is funded by the University of British Columbia Clinician Investigator Program and is a Friedman Scholar in Health and George Mines Traveling Fellow in Cardiac Electrophysiology. Drs. Christine and John Seidman are supported by National Institute of Health grants (NIH 2R01HL080494, NIH 2R01HL084553) and the Fondation Leducq (16 CVD 03).
Nonstandard abbreviations and acronyms
- ARVC
Arrhythmogenic right ventricular cardiomyopathy
- TFC
Task Force Criteria
- PKP2tv
Plakophilin-2 truncating variants
- VT
Ventricular tachycardia
- ICD
Implantable cardioverter defibrillator
- NMD
Nonsense mediated mRNA decay
- CADD
Combined annotation-dependent depletion
- UKB
UK Biobank
- OR
Odds ratio
- LVEDVI
Left ventricular end diastolic volume indexed
Footnotes
Disclosures: Dr. Abrams is a consultant for Dinaqor. The other authors declare no relevant conflicts of interest.
References:
- 1.Towbin JA, McKenna WJ, Abrams DJ, Ackerman MJ, Calkins H, Darrieux FCC, Daubert JP, de Chillou C, DePasquale EC, Desai MY, et al. 2019 HRS Expert Consensus Statement on Evaluation, Risk Stratification, and Management of Arrhythmogenic Cardiomyopathy. Hear Rhythm. 2019; [DOI] [PubMed] [Google Scholar]
- 2.Austin KM, Trembley MA, Chandler SF, Sanders SP, Saffitz JE, Abrams DJ, Pu WT. Molecular mechanisms of arrhythmogenic cardiomyopathy. Nat Rev Cardiol. 2019;16:519–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Marcus FI, McKenna WJ, Sherrill D, Basso C, Bauce B, Bluemke DA, Calkins H, Corrado D, Cox MGPJ, Daubert JP, et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/Dysplasia: Proposed modification of the task force criteria. Circulation. 2010;121:1533–1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gilotra NA, Bhonsale A, James CA, Te Riele ASJ, Murray B, Tichnell C, Sawant A, Ong CS, Judge DP, Russell SD, et al. Heart failure is common and under-recognized in patients with arrhythmogenic right ventricular cardiomyopathy/dysplasia. Circ Hear Fail. 2017;10:1–11. [DOI] [PubMed] [Google Scholar]
- 5.Zghaib T, Bourfiss M, van der Heijden JF, Loh P, Hauer RN, Tandri H, Calkins H, Nazarian S, Te Riele ASJM, Zimmerman SL, et al. Atrial Dysfunction in Arrhythmogenic Right Ventricular Cardiomyopathy. Circ Cardiovasc Imaging. 2018;11:e007344. [DOI] [PubMed] [Google Scholar]
- 6.Te Riele ASJM, James CA, Groeneweg JA, Sawant AC, Kammers K, Murray B, Tichnell C, Van Der Heijden JF, Judge DP, Dooijes D, et al. Approach to family screening in arrhythmogenic right ventricular dysplasia/cardiomyopathy. Eur Heart J. 2016;37:755–763. [DOI] [PubMed] [Google Scholar]
- 7.Walsh R, Thomson KL, Ware JS, Funke BH, Woodley J, McGuire KJ, Mazzarotto F, Blair E, Seller A, Taylor JC, et al. Reassessment of Mendelian gene pathogenicity using 7,855 cardiomyopathy cases and 60,706 reference samples. Genet Med. 2017;19:192–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carruth ED, Young W, Beer D, James CA, Calkins H, Jing L, Raghunath S, Hartzel DN, Leader JB, Kirchner HL, et al. Prevalence and Electronic Health Record-Based Phenotype of Loss-of-Function Genetic Variants in Arrhythmogenic Right Ventricular Cardiomyopathy-Associated Genes. Circ Genomic Precis Med. 2019;12:487–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carruth ED, Beer D, Alsaid A, Schwartz MLB, McMinn M, Kelly MA, Buchanan AH, Nevius CD, Calkins H, James CA, et al. Clinical Findings and Diagnostic Yield of Arrhythmogenic Cardiomyopathy through Genomic Screening of Pathogenic or Likely Pathogenic Desmosome Gene Variants. Circ Genomic Precis Med. 2021; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O’Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cerrone M, Remme CA, Tadros R, Bezzina CR, Delmar M. Beyond the one gene-one disease paradigm complex genetics and pleiotropy in inheritable cardiac disorders. Circulation. 2019;140:595–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu T, Yang Z, Vatta M, Rampazzo A, Beffagna G, Pillichou K, Scherer SE, Saffitz J, Kravitz J, Zareba W, et al. Compound and Digenic Heterozygosity Contributes to Arrhythmogenic Right Ventricular Cardiomyopathy. J Am Coll Cardiol [Internet], 2010;55:587–597. Available from: 10.1016/j.jacc.2009.11.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.James CA, Bhonsale A, Tichnell C, Murray B, Russell SD, Tandri H, Tedford RJ, Judge DP, Calkins H. Exercise increases age-related penetrance and arrhythmic risk in arrhythmogenic right ventricular dysplasia/cardiomyopathy-associated desmosomal mutation carriers. J Am Coll Cardiol. 2013;62:1290–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Groeneweg JA, Bhonsale A, James CA, Te Riele AS, Dooijes D, Tichnell C, Murray B, Wiesfeld ACP, Sawant AC, Kassamali B, et al. Clinical Presentation, Long-Term Follow-Up, and Outcomes of 1001 Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy Patients and Family Members. Circ Cardiovasc Genet. 2015;8:437–446. [DOI] [PubMed] [Google Scholar]
- 15.van Lint FHM, Murray B, Tichnell C, Zwart R, Amat N, Lekanne Deprez RH, Dittmann S, Stallmeyer B, Calkins H, van der Smagt JJ, et al. Arrhythmogenic Right Ventricular Cardiomyopathy-Associated Desmosomal Variants Are Rarely De Novo. Circ Genomic Precis Med. 2019;12:e002467. [DOI] [PubMed] [Google Scholar]
- 16.Gerull B, Heuser A, Wichter T, Paul M, Basson CT, McDermott DA, Lerman BB, Markowitz SM, Ellinor PT, MacRae CA, et al. Mutations in the desmosomal protein plakophilin-2 are common in arrhythmogenic right ventricular cardiomyopathy. Nat Genet. 2004;36:1162–1164. [DOI] [PubMed] [Google Scholar]
- 17.Dries AM, Kirillova A, Reuter CM, Garcia J, Zouk H, Hawley M, Murray B, Tichnell C, Pilichou K, Protonotarios A, et al. The genetic architecture of Plakophilin 2 cardiomyopathy. Genet Med [Internet], 2021;1–8. Available from: 10.1038/s41436-021-01233-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bhonsale A, Groeneweg JA, James CA, Dooijes D, Tichnell C, Jongbloed JDH, Murray B, Te Riele ASJM, Van Den Berg MP, Bikker H, et al. Impact of genotype on clinical course in arrhythmogenic right ventricular dysplasia/cardiomyopathy-associated mutation carriers. Eur Heart J. 2015;36:847–855. [DOI] [PubMed] [Google Scholar]
- 19.Pilichou K, Lazzarini E, Rigato I, Celeghin R, De Bortoli M, Perazzolo Marra M, Cason M, Jongbloed J, Calore M, Rizzo S, et al. Large genomic rearrangements of desmosomal genes in Italian arrhythmogenic cardiomyopathy patients. Circ Arrhythmia Electrophysiol. 2017;10:1–14. [DOI] [PubMed] [Google Scholar]
- 20.van Opbergen CJM, Noorman M, Pfenniger A, Copier JS, Vermij SH, Li Z, van der Nagel R, Zhang M, de Bakker JMT, Glass AM, et al. Plakophilin-2 haploinsuffciency causes calcium handling deficits and modulates the cardiac response towards stress. Int J Mol Sci. 2019;20:1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cerrone M, Montnach J, Lin X, Zhao YT, Zhang M, Agullo-Pascual E, Leo-Macias A, Alvarado FJ, Dolgalev I, Karathanos TV, et al. Plakophilin-2 is required for transcription of genes that control calcium cycling and cardiac rhythm. Nat Commun. 2017;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen X, Bonné S, Hatzfeld M, Van Roy F, Green KJ. Protein binding and functional characterization of plakophilin 2. Evidence for its diverse roles in desmosomes and β-catenin signaling. J Biol Chem. 2002;277:10512–10522. [DOI] [PubMed] [Google Scholar]
- 23.Rasmussen TB, Nissen PH, Palmfeldt J, Gehmlich K, Dalager S, Jensen UB, Kim WY, Heickendorff L, Mlgaard H, Jensen HK, et al. Truncating plakophilin-2 mutations in arrhythmogenic cardiomyopathy are associated with protein haploinsufficiency in both myocardium and epidermis. Circ Cardiovasc Genet. 2014;7:230–240. [DOI] [PubMed] [Google Scholar]
- 24.Caspi O, Huber I, Gepstein A, Arbel G, Maizels L, Boulos M, Gepstein L. Modeling of arrhythmogenic right ventricular cardiomyopathy with human induced pluripotent stem cells. Circ Cardiovasc Genet. 2013;6:557–568. [DOI] [PubMed] [Google Scholar]
- 25.Glazier AA, Thompson A, Day SM. Allelic imbalance and haploinsufficiency in MYBPC3-linked hypertrophic cardiomyopathy. Pflugers Arch Eur J Physiol. 2019;471:781–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rivas MA, Pirinen M, Conrad DF, Lek M, Tsang EK, Karczewski KJ, Maller JB, Kukurba KR, DeLuca DS, Fromer M, et al. Human genomics. Effect of predicted protein-truncating genetic variants on the human transcriptome. Science. 2015;348:666–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mazzanti A, Ng K, Faragli A, Maragna R, Chiodaroli E, Orphanou N, Monteforte N, Memmi M, Gambelli P, Novelli V, et al. Arrhythmogenic Right Ventricular Cardiomyopathy. J Am Coll Cardiol [Internet], 2016;68:2540–2550. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0735109716365809 [DOI] [PubMed] [Google Scholar]
- 28.Cruz FM, Sanz-Rosa D, Roche-Molina M, García-Prieto J, García-Ruiz JM, Pizarro G, Jiménez-Borreguero LJ, Torres M, Bernad A, Ruíz-Cabello J, et al. Exercise triggers ARVC phenotype in mice expressing a disease-causing mutated version of human plakophilin-2. J Am Coll Cardiol. 2015;65:1438–1450. [DOI] [PubMed] [Google Scholar]
- 29.La Gerche A, Rakhit DJ, Claessen G. Exercise and the right ventricle: a potential Achilles’ heel. Cardiovasc Res. 2017;113:1499–1508. [DOI] [PubMed] [Google Scholar]
- 30.Wang W, Tichnell C, Murray BA, Agafonova J, Cadrin-Tourigny J, Chelko S, Tandri H, Calkins H, James CA. Exercise restriction is protective for genotype-positive family members of arrhythmogenic right ventricular cardiomyopathy patients. Europace. 2020;22:1270–1278. [DOI] [PubMed] [Google Scholar]
- 31.Haggerty CM, Murray B, Tichnell C, Judge DP, Tandri H, Schwartz M, Sturm AC, Matsumura ME, Murray MF, Calkins H, et al. Managing Secondary Genomic Findings Associated With Arrhythmogenic Right Ventricular Cardiomyopathy: Case Studies and Proposal for Clinical Surveillance. Circ Genomic Precis Med. 2018;11:e002237. [DOI] [PubMed] [Google Scholar]
- 32.Van Der Smagt JJ, Van Der Zwaag PA, Peter Van Tintelen J, Cox MGPJ, Wilde AAM, Van Langen IM, Ummels A, Hennekam FAM, Dooijes D, Gerbens F, et al. Clinical and genetic characterization of patients with arrhythmogenic right ventricular dysplasia/cardiomyopathy caused by a plakophilin-2 splice mutation. Cardiol. 2012;123:181–189. [DOI] [PubMed] [Google Scholar]
- 33.Helms AS, Thompson AD, Glazier AA, Hafeez N, Kabani S, Rodriguez J, Yob JM, Woolcock H, Mazzarotto F, Lakdawala NK, et al. Spatial and functional distribution of MYBPC3 pathogenic variants and clinical outcomes in patients with hypertrophic cardiomyopathy. Circ Genomic Precis Med. 2020;396–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gandjbakhch E, Charron P, Fressart V, Lorin de la Grandmaison G, Simon F, Gary F, Vite A, Hainque B, Hidden-Lucet F, Komajda M et al. Plakophilin 2A is the dominant isoform in human heart tissue: Consequences for the genetic screening of arrhythmogenic right ventricular cardiomyopathy. Heart. 2011;97(10):844–849. doi: 10.1136/hrt.2010.205880 [DOI] [PubMed] [Google Scholar]
- 35.Dries AM, Kirillova A, Reuter CM, Garcia J, Zouk H, Hawley M, Murray B, Tichnell C, Pilichou K, Protonotarios A, et al. The genetic architecture of Plakophilin 2 cardiomyopathy. Genet Med. 2021;10:1961–1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nagy E, Maquat LE. A rule for termination-codon position within intron-containing genes: When nonsense affects RNA abundance. Trends Biochem Sci. 1998;23(6):198–199. doi: 10.1016/S0968-0004(98)01208-0 [DOI] [PubMed] [Google Scholar]
- 37.Corrado D, Thiene G. Arrhythmogenic Right Ventricular Cardiomyopathy/Dysplasia. Circulation. 2006;113(13):1634–1637. doi: 10.1161/CIRCULATIONAHA.105.616490 [DOI] [PubMed] [Google Scholar]
- 38.Rentzsch P, Witten D, Cooper GM, Shendure J, Kircher M. CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019;47(D1):D886–D894. doi: 10.1093/nar/gky1016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jaganathan K, Panagiotopoulou SK, Mcrae JF, Darbandi SF, Knwoles D, Li Y, Kosmicki JA, Arbelaez J, Ciu W, Schwartz DB et al. Article Predicting Splicing from Primary Sequence with Deep Learning. Cell. 2019;176(3):535–548.e24. doi: 10.1016/j.cell.2018.12.015 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.