

ABSTRACT
Protein acetylation plays an important role during virus infection. Thus, it is not surprising that viruses always evolve elaborate mechanisms to regulate the functions of histone deacetylases (HDACs), the essential transcriptional and epigenetic regulators for deacetylation. Porcine deltacoronavirus (PDCoV), an emerging enteropathogenic coronavirus, causes severe diarrhea in suckling piglets and has the potential to infect humans. In this study, we found that PDCoV infection inhibited cellular HDAC activity. By screening the expressions of different HDAC subfamilies after PDCoV infection, we unexpectedly found that HDAC2 was cleaved. Ectopic expression of HDAC2 significantly inhibited PDCoV replication, while the reverse effects could be observed after treatment with an HDAC2 inhibitor (CAY10683) or the knockdown of HDAC2 expression by specific siRNA. Furthermore, we demonstrated that PDCoV-encoded nonstructural protein 5 (nsp5), a 3C-like protease, was responsible for HDAC2 cleavage through its protease activity. Detailed analyses showed that PDCoV nsp5 cleaved HDAC2 at glutamine 261 (Q261), and the cleaved fragments (amino acids 1 to 261 and 262 to 488) lost the ability to inhibit PDCoV replication. Interestingly, the Q261 cleavage site is highly conserved in HDAC2 homologs from other mammalian species, and the nsp5s encoded by seven tested mammalian coronaviruses also cleaved HDAC2, suggesting that cleaving HDAC2 may be a common strategy used by different mammalian coronaviruses to antagonize the antiviral role of HDAC2.
IMPORTANCE As an emerging porcine enteropathogenic coronavirus that possesses the potential to infect humans, porcine deltacoronavirus (PDCoV) is receiving increasing attention. In this work, we found that PDCoV infection downregulated cellular histone deacetylase (HDAC) activity. Of particular interest, the viral 3C-like protease, encoded by the PDCoV nonstructural protein 5 (nsp5), cleaved HDAC2, and this cleavage could be observed in the context of PDCoV infection. Furthermore, the cleavage of HDAC2 appears to be a common strategy among mammalian coronaviruses, including the emerging severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), to antagonize the antiviral role of HDAC2. To our knowledge, PDCoV nsp5 is the first identified viral protein that can cleave cellular HDAC2. Results from our study provide new targets to develop drugs combating coronavirus infection.
KEYWORDS: 3C-like protease, antiviral activity, cleavage, porcine deltacoronavirus (PDCoV), histone deacetylase 2 (HDAC2)
INTRODUCTION
Porcine deltacoronavirus (PDCoV), which is an emerging porcine enteropathogenic coronavirus (CoV) belonging to the genus Deltacoronavirus of the subfamily Orthocoronavirinae within the family Coronaviridae, was first detected in pig rectal swab samples for virological surveillance in wet markets in Hong Kong in 2012 (1). However, its pathogenicity was not defined until 2014. The first outbreak of PDCoV was reported in several pig farms with animals demonstrating acute diarrhea in Ohio in early 2014, with the virus then rapidly spreading throughout the United States and globally (2, 3). To date, PDCoV has been reported in many countries and regions, including China, South Korea, Thailand, Japan, Lao People’s Democratic Republic, Vietnam, and Mexico (4–9). Although PDCoV mainly causes acute diarrhea, vomiting, and death in suckling piglets, it also possesses cross-species transmission and zoonotic potential (10). Accumulating evidence has demonstrated that PDCoV can infect chickens, mice, turkeys, and cattle (11–14). More recently, Lednicky et al. reported for the first time that PDCoV was detected in plasma samples of three Haitian children with acute undifferentiated febrile illness (15), suggesting that PDCoV may have jumped from pigs to humans, highlighting the significant threat to human health posed by this emerging CoV and attracting tremendous attention to the topic.
Acetylation is one of the most common posttranslational modifications of proteins. It occurs in various nuclear and cytoplasmic proteins and plays a vital role in regulating gene expression, the cell cycle, signal transduction, and innate immune responses (16–19). Acetylation and deacetylation are reversible processes, controlled by histone acetyltransferases (HATs) and histone deacetylases (HDACs), respectively. Histone acetylation by HATs relaxes the chromatin structure to enhance gene expression, while HDAC-mediated deacetylation results in chromatin condensation to suppress the interactions of transcription factors with their target promoters (20, 21). Furthermore, HDACs are also responsible for modifying the activity of diverse types of nonhistone proteins, including transcription factors and signal transduction mediators (21–26). At least 18 HDACs have been identified in mammals, and they can be divided into four subfamilies: class I (HDAC 1, 2, 3, and 8), class II (HDAC 4, 5, 6, 7, 9, and 10), class III (sirtuins 1 to 7), and class IV (HDAC11) (27, 28). Accumulating evidence has demonstrated that HDACs are frequently dysregulated during viral infection and can even be hijacked by some viruses, which modulates viral replication (29–31). For example, HDAC1 binds to the corepressor element 1 silencing transcription factor/repressor element 1 silencing transcription factor (CoREST/REST) complex to inhibit viral gene expression during herpes simplex virus 1 (HSV-1) infection (32). HDAC inhibitors block hepatitis C virus (HCV) replication by modulating the expressions of liver-specific antimicrobial peptide 1 (LEAP-1), osteopontin (OPN), and apoliprotein-A1 (Apo-A1) (33). HDACs play an important role in the maintenance of human immunodeficiency virus (HIV) latency, while HIV-encoded proteins also downregulate class I HDACs by proteasomal degradation, thereby promoting viral promoter reactivation (34, 35). Transgenic pigs that constitutively overexpress porcine HDAC6 demonstrate an enhanced resistance to porcine reproductive and respiratory syndrome virus (PRRSV) infection in vivo (36). HDAC6 inhibits influenza A virus (IAV) replication by deacetylating the viral RNA polymerase PA subunit, thereby restricting IAV RNA transcription (37). HDAC2 has also been demonstrated to be a component of the host’s innate antiviral response induced by IAV; HDAC2 can reduce the IAV-induced phosphorylation of the signal transducer and activator of transcription 1 (STAT1) and interferon (IFN)-stimulated gene (ISG) expression to exert antiviral effects (38). As for CoVs and HDACs, Xu et al. have reported that porcine epidemic diarrhea virus (PEDV) inhibits HDAC1 expression to facilitate its replication via the binding of its nucleocapsid protein to host transcription factor Sp1 (39). Pitt et al. recently proposed that valproic acid, an HDAC inhibitor, may have therapeutic potential to prevent severe coronavirus disease 2019 (COVID-19) caused by the emerging severe acute respiratory syndrome CoV 2 (SARS-CoV-2), and this is because valproic acid can reduce the expression of angiotensin-converting enzyme 2 (ACE2) and transmembrane serine protease 2 (TMPRSS2) as well as modulate the immune cellular and cytokine responses to SARS-CoV-2 infection, thereby reducing organ damage (40). However, the relationship between PDCoV and HDACs has not yet been reported.
In this study, we initially analyzed the activity of cellular deacetylases and the expression profiles of different HDAC subfamily members after PDCoV infection. We found that PDCoV infection downregulated cellular deacetylase activity, and some HDACs, particularly HDAC2, were downregulated. Although HDAC2 exhibited significant anti-PDCoV effects, PDCoV nonstructural protein 5 (nsp5), a 3C-like protease, cleaved HDAC2 to antagonize its antiviral role. Furthermore, nsp5s of other mammalian CoVs also cleaved HDAC2.
RESULTS
PDCoV infection inhibits cellular deacetylase activity and downregulates expression of some HDACs.
Previous studies have suggested that cellular deacetylases are involved in the replication of some viruses, while concurrently, viruses regulate cellular deacetylase activity to benefit viral replication (30, 38, 41, 42). To investigate whether PDCoV infection changes cellular deacetylase activity, LLC-PK1 (a porcine kidney cell line) and IPI-FX (a porcine intestinal epithelial cell line) cells were infected with PDCoV strain CHN-HN-2014 at various multiplicities of infection (MOI). At 24 h postinfection (hpi), cellular deacetylase activity was detected. Compared with mock-infected controls, PDCoV infection significantly decreased cellular deacetylase activity in both cell lines in a dose-dependent manner (Fig. 1). To rule out the possibility that the decrease of cellular deacetylase activity in infected cells is a side effect of virus-induced cytotoxicity, we detected the cell viability after PDCoV infection. The results showed that the cell viability was above 95% in PDCoV-infected LLC-PK1 and IPI-FX cells at all tested infection doses (MOI = 0.5, 1.0, 2.0) (data not shown).
FIG 1.
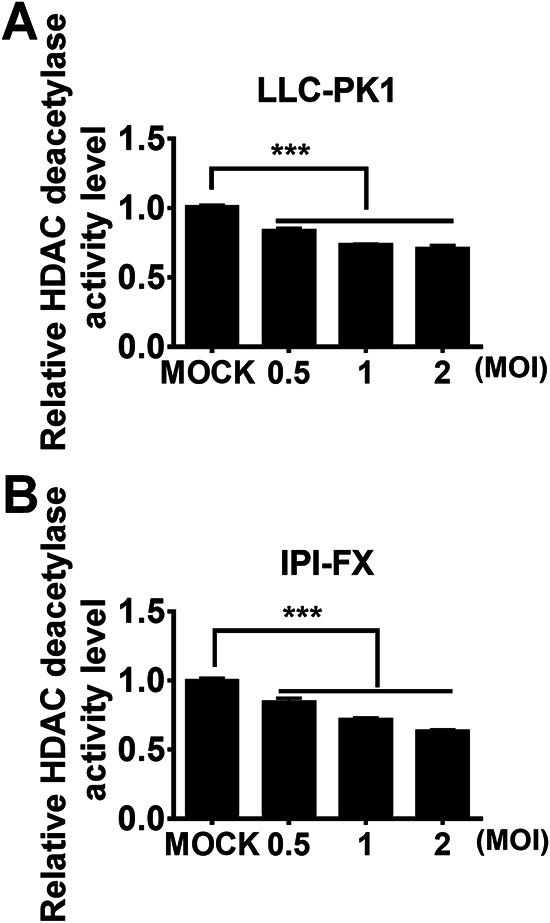
PDCoV infection inhibits cellular deacetylase activity. LLC-PK1 (A) and IPI-FX (B) cells were mock-infected or infected with PDCoV at different infectious doses (MOI 0.5, 1, or 2). At 24 h after infection (hpi), the cells were collected for HDAC activity assay. The relative HDAC activity in PDCoV-infected cells was normalized to that of mock-infected cells. All experiments were performed in triplicate, and the data are presented as means ± SD of three independent experiments. ***, P < 0.001.
To further investigate the expression profiles of different HDAC subfamily members, we analyzed the mRNA expression levels of different HDACs (HDAC1, HDAC2, HDAC3, and HDAC8 of class I; HDAC4 of class II; Sirt1 and Sirt2 of class III; and HDAC11 of class IV) in PDCoV-infected LLC-PK1 and IPI-FX cells. The results showed that PDCoV infection significantly inhibited the mRNA expression of HDAC2, HDAC8, SIRT2, and HDAC11 in both cell lines (Fig. 2A and B). Histone acetylation is reversible, and it is determined by competing activities of HDACs and HATs (43, 44). Thus, we also analyzed the mRNA expression of some representative HATs, including HAT1, EP300, CREBBP, and MYST2, in PDCoV-infected LLC-PK1 and IPI-FX cells. As expected, PDCoV infection significantly upregulated mRNA expression of these tested HATs (Fig. 2C and D).
FIG 2.

PDCoV infection downregulates expression of some HDACs and upregulates expression of tested HATs. (A–D) LLC-PK1cells (A, C) and IPI-FX cells (B, D) were mock-infected or infected with PDCoV (MOI 1). At 24 hpi, the cells were collected for qRT-PCR to detect HDAC mRNA expression (A, B) and HATs (C, D). Relative mRNA expressions in PDCoV-infected cells were normalized to those of mock-infected cells. All experiments were performed in triplicate, and the data are presented as means ± SD of three independent experiments (*, P < 0.05; **, P < 0.01; ***, P < 0.001). (E) LLC-PK1 cells were infected with PDCoV at increasing infectious doses (MOI 0.25, 0.5, or 1). At 24 hpi, cells were collected for Western blotting with antibodies against HDAC1, HDAC2, HDAC3, HDAC8, SIRT2, and HDAC11. Anti-N protein antibody was used to confirm PDCoV infection. (F) Density analysis of panel E represents the relative HDACs expression levels compared to the control group. The analysis was performed with the ImageJ software package.
We further detected the protein levels of some HDACs (HDAC2, HDAC8, SIRT2, and HDAC11) in which mRNA expression was significantly decreased. HDAC1 and HDAC3, in which mRNA expression was not significantly changed after PDCoV infection, were used as controls. The results showed that the protein levels of HDAC2, HDAC8, SIRT2, and HDAC11 were also decreased to various degrees (Fig. 2E and F), while no significant change could be observed for HDAC1 and HDAC3. Interestingly, we found that for HDAC2 in PDCoV-infected cells, there were two specific bands (Fig. 2E) which were not present in mock-infected cells, indicating that HDAC2 may be cleaved during PDCoV infection. Considering that early studies have demonstrated that HDAC2 plays a crucial role in the modulation of cell signaling, innate immune responses, and expression of genes involved in antiviral innate immunity (45–48), as well as the possible cleavage observed in this study, we chose to focus our subsequent experiments on HDAC2.
HDAC2 negatively regulates PDCoV replication.
HDAC2 is an important member of the class I HDAC family, and previous studies have suggested that HDAC2 is involved in antiviral innate immunity (15, 18, 28, 37). We initially investigated whether HDAC2 affects PDCoV replication through three strategies: ectopic expression, chemical inhibition, and small interfering RNA (siRNA). To this end, increasing concentrations of Flag-HDAC2 were overexpressed in LLC-PK1 cells, which were then infected with PDCoV for 12 h. The results showed that the overexpression of HDAC2 inhibited the replication of PDCoV, as evidenced by decreased viral mRNA expression (Fig. 3A), viral titers (Fig. 3B), and viral protein expression (nucleocapsid protein, N) (Fig. 3C and D). Notably, we also used the Flag antibody to detect the expression of Flag-HDAC2 and found a specific protein band that migrated faster (Fig. 3C). Similar results could be observed in IPI-FX cells (Fig. 3E and F), consistent with previous results in detecting endogenous HDAC2 after PDCoV infection (Fig. 2E) and further demonstrating that HDAC2 was cleaved during PDCoV infection.
FIG 3.

HDAC2 negatively regulates PDCoV replication. (A–C) LLC-PK1 cells were transfected with increasing doses of pCAGGS-Flag-HDAC2 or empty vector. At 24 h after transfection, the cells were infected with PDCoV (MOI 0.5) for 12 h. Then, the cells were collected for qRT-PCR (A), TCID50 assay (B), and Western blotting (C). (D) Density analysis of panel C represents the relative expression levels of PDCoV N protein compared to the control group. The analysis was performed with the ImageJ software package. (E) IPI-FX cells were treated as described above, and Western blotting was performed with antibodies against Flag or PDCoV N protein. (F) Density analysis of panel E represents the relative expression levels of PDCoV N protein compared to the control group. The analysis was performed with the ImageJ software package. (G) The cytotoxic effects of CAY on LLC-PK1 cells. LLC-PK1 cells were treated with increasing concentrations of CAY. At 24 h after treatment, cell viability was measured by using a CCK-8 detection kit. (H–K) LLC-PK1 cells were pretreated with CAY (75 μM) for 2 h. Then, the cells were infected with PDCoV (MOI 0.5). At 12 hpi, cells were collected for qRT-PCR (H), Western blotting (I), density analysis of panel I in the ImageJ software package (J), and TCID50 assay (K). (L–O) The effects of HDAC2 knockdown on PDCoV replication. LLC-PK1 cells were transfected with siRNA against HDAC2 or with control siRNA for 24 h. Then, the cells were infected with PDCoV (MOI 0.5) for 12 h. Cell samples were harvested and subjected to qRT-PCR (L, N), Western blotting (M), and TCID50 assay (O) to determine relative HDAC2 mRNA, viral mRNA, and viral titers. The presented results represent the means and standard deviations of data from three independent experiments. **, P < 0.01; ***, P < 0.001; ns, nonsignificant difference.
Santacruzamate A (CAY10683), hereinafter referred to as CAY, is a potent and selective inhibitor of HDAC2. We first tested the inhibitory effect of CAY on HDAC2 in LLC-PK1 cells, and the results showed that CAY treatment had little effect on the total protein levels of HDAC2 and histone H3. However, it significantly promoted the acetylation levels of histone H3 in a dose-dependent manner (data not shown), indicating that CAY inhibits the activity of HDAC2 rather than its protein expression. To investigate whether CAY affects PDCoV replication, LLC-PK1 cells were pretreated with different doses of CAY for 2 h with no detectable cytotoxicity (Fig. 3G) and then infected with PDCoV (MOI = 0.5). The results showed that the inhibition of HDAC2 activity by CAY treatment resulted in increased copies of viral genomic RNA (Fig. 3H), N protein expression (Fig. 3I and J), and viral titers (Fig. 3K). To further confirm the anti-PDCoV function of HDAC2, specific siRNA was used to knock down HDAC2 expression in LLC-PK1 cells, and then the cells were infected with PDCoV. Compared with the control siRNA, HDAC2-specific siRNA notably knocked down HDAC2 expression and promoted PDCoV replication, as demonstrated by the results from quantitative reverse transcription polymerase chain reaction (RT-qPCR), Western blotting, and TCID50 assay (Fig. 3L to O). Collectively, these findings indicated that HDAC2 has anti-PDCoV effects.
PDCoV nsp5 cleaves HDAC2.
The above results showed that both endogenous (Fig. 2E) and overexpressed (Fig. 3C and E) HDAC2 may be cleaved, which prompted us to investigate the mechanisms of HDAC2 cleavage after PDCoV infection. Because CoV nsp5, also known as the main protease or 3C-like protease, possesses the ability to cleave viral polyproteins and host proteins (49–51), we first investigated whether PDCoV nsp5 cleaves HDAC2. To this end, HEK-293T cells were cotransfected with pCAGGS-Flag-HDAC2 and increasing concentrations of pCAGGS-nsp5-HA, followed by Western blot analyses with anti-Flag and anti-HA antibodies, respectively. As shown in Fig. 4A, Flag-HDAC2 was cleaved by nsp5 in a dose-dependent manner. In addition, we found that the overexpression of PDCoV nsp5 had no effect on the mRNA expression of endogenous HDAC2 (data not shown). Furthermore, nsp5-mediated HDAC2 cleavage depended on its protease activity because no cleavage band could be observed in cells coexpressing Flag-HDAC2 and nsp5 mutants (H41A or C144A), in both of which the nsp5 protease activity was inactivated (Fig. 4B). To further confirm the cleavage of HDAC2 by PDCoV nsp5, a recombinant expression plasmid, pCAGGS-Flag-HDAC2-Myc with N-terminal Flag and C-terminal Myc tags, was constructed and cotransfected into HEK-293T cells with pCAGGS-nsp5-HA for 28 h, followed by a Western blotting assay. As shown in Fig. 4C, the molecular weights of the 5′-products and the 3′-products generated by the PDCoV nsp5 cleavage of HDAC2 are similar and are approximately 34 kDa, suggesting that the cleavage site is located in the middle of HDAC2.
FIG 4.

PDCoV nsp5 cleaves HDAC2. (A) HEK-293T cells were cotransfected with pCAGGS-Flag-HDAC2 and empty vector or increasing doses of pCAGGS-nsp5-HA. After 28 h, the cells were lysed for Western blotting with antibodies against Flag and HA. (B) HEK-293T cells were cotransfected with pCAGGS-Flag-HDAC2 and expression constructs encoding wild-type PDCoV nsp5 (nsp5-WT) or its protease-defective mutants (H41A or C144A). At 28 h after cotransfection, cells were lysed for Western blotting. (C) HEK-293T cells were cotransfected with pCAGGS-Flag-HDAC2-Myc and pCAGGS-HA-nsp5 for 28 h, followed by Western blotting for detecting the 5′-products or the 3′-products of HDAC2 cleavage using antibodies against Flag, Myc and HA, respectively. (D) Schematic diagram of a single point mutant of HDAC2. (E) HEK-293T cells were cotransfected with pCAGGS-nsp5-HA and expression constructs encoding HDAC2 WT or the indicated HDAC2 mutants. At 28 h after cotransfection, cells were collected for Western blotting. (F) LLC-PK1 cells were transfected with pCAGGS-Flag-HDAC2, pCAGGS-Flag-HDAC2-Q261A, pCAGGS-Flag-HDAC2-Q254A, or empty vector, followed by PDCoV infection (MOI 0.5). Cells were collected for Western blotting with antibodies against human HDAC2 or PDCoV N protein. (G) LLC-PK1 cells were mock-infected or infected with PDCoV (MOI 0.5). At 24 hpi, cells were collected, and the nuclear and cytoplasmic fractions were extracted and subjected to Western blotting with anti-HDAC2 antibody. The antibodies against HSP90 and LaminA+C were used to confirm the isolation of cytoplasmic and nuclear proteins, respectively. (H) Density analysis of panel G represents the relative protein levels of total HDAC2 (nucleus and cytoplasm) that was normalized to the total protein levels of LaminA+C and HSP90. The analysis was performed with the ImageJ software package, and the value of mock-infected control group was set to 1. The presented results represent the means and standard deviations of data from three independent experiments. **, P < 0.01.
Previous studies have shown that CoV nsp5 recognizes the glutamine (Q) residue at the P1 position for substrate cleavage (52, 53). After analyzing the possible Q residues in the middle of HDAC2, we selected five Q residues (Q240, Q254, Q261, Q354, and Q365) and mutated these to alanine (A) (Fig. 4D). HEK-293T cells were cotransfected with pCAGGS-nsp5-HA and the different point mutants: HDAC2-Q240A, -Q254A, -Q261A, -Q354A, or -Q365A. Western blotting showed that HDAC2-Q261A was resistant to PDCoV nsp5-mediated cleavage, while the cleavage of the other mutants did not change (Fig. 4E). These results suggested that PDCoV nsp5 targets HDAC2 at Q261. To further strengthen this conclusion, LLC-PK1 cells were transfected with pCAGGS-Flag-HDAC2, pCAGGS-Flag-HDAC2-Q261A, or pCAGGS-Flag-HDAC2-Q254A followed by PDCoV infection to detect whether HDAC2-Q261A is also resistant to cleavage in the context of a viral infection. As expected, HDAC2-Q261A was not cleaved, while the HDAC2-WT (wild-type) and mutant HDAC2-Q254A were cleaved successfully (Fig. 4F), consistent with the results observed in nsp5-overexpressing cells and further demonstrating that Q261 is the cleavage site.
Previous studies have shown that class I HDACs, including HDAC2, are mainly located in the nucleus (54). However, some studies have also suggested that HDAC2 is localized to both the nucleus and the cytoplasm, particularly under pathological conditions (55–57). Thus, we further analyzed the subcellular localization and cleavage of HDAC2 after PDCoV infection. Because no commercial antibody against porcine HDAC2 is available and because the antibody against human HADC2 does not work well for immunofluorescence assays in LLC-PK1 and IPI-FX cells, we chose to perform nucleocytoplasmic separation. As shown in Fig. 4G, HDAC2 was mainly expressed in the nucleus, but it was also expressed in the cytoplasm of LLC-PK1 cells. After PDCoV infection, nuclear HDAC2 was decreased, while cytoplasmic HDAC2 was increased, suggesting that PDCoV infection promoted nuclear to cytoplasmic translocation of HDAC2. Of particular interest, only cytoplasmic HDAC2 was cleaved by PDCoV, demonstrating that the cleavage of HDAC2 occurred in the cytoplasm after PDCoV infection (Fig. 4G and H).
Cleaved HDAC2 loses the ability to induce ISGs.
Previous studies have demonstrated that HDAC2 is required for type I IFN signaling (46, 58) because HDAC2 acts as a critical positive coactivator for the transcriptional responses of the ISG factor 3 (ISGF3) complex (STAT1/STAT2/IRF9) (46). The activated ISGF3 complex translocates into the nucleus, where it binds to the IFN-sensitive response element (ISRE) sequence of ISG promoters, leading to the expression of ISGs (59). Considering that HDAC2 was cleaved into two fragments (amino acids 1 to 261 and 262 to 488) by PDCoV, we wanted to know whether cleaved HDAC2 loses the ability to induce ISRE promoter activation and subsequent ISG expression. To this end, LLC-PK1 cells were cotransfected with an ISRE-luciferase (ISRE-Luc) reporter plasmid, pRL-TK (an internal control for the normalization of transfection efficiency), and the expression plasmids for HDAC2-wild-type (WT), HDAC2-Δ1-261, or HDAC2-Δ262-488. At 24 h after cotransfection, cells were stimulated with IFN-α (1,000 U/mL) for 12 h, followed by luciferase activity assays. As shown in Fig. 5A, ISRE promoter activities were induced after IFN-α stimulation in cells transfected with empty vector, and overexpression of the full-length HDAC2 further enhanced ISRE promoter activity. However, the induction was significantly reduced in cells expressing the two cleaved HDAC2 products, HDAC2-Δ1-261 and HDAC2-Δ262-488, compared with HDAC2-WT. Similar results were obtained in HEK-293T cells (Fig. 5B).
FIG 5.
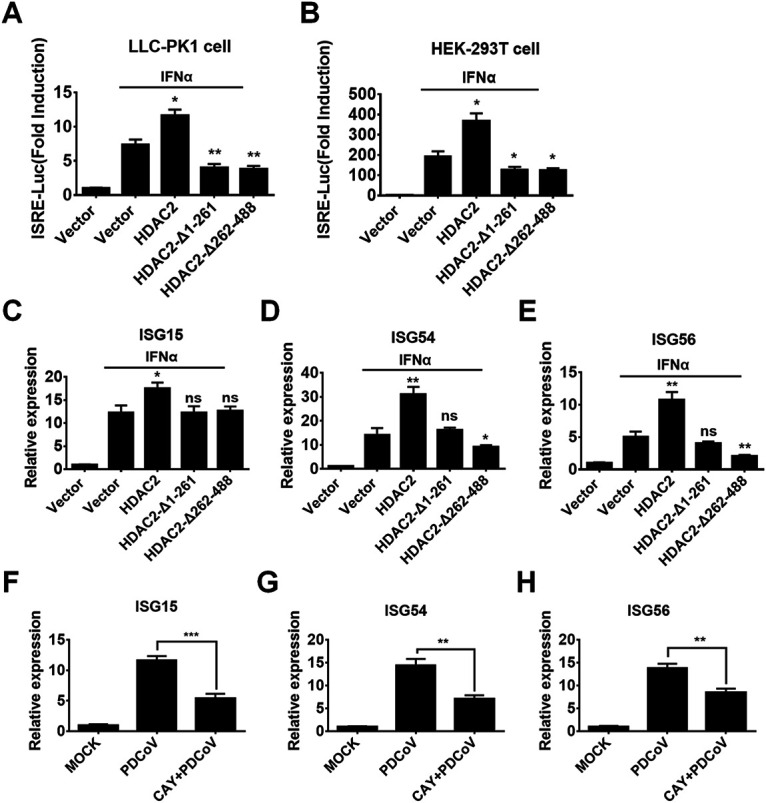
Cleaved HDAC2 loses the ability to induce ISGs. LLC-PK1 cells (A) and HEK-293T cells (B) were transfected with expression plasmids for WT HDAC2, HDAC2-Δ1-261, HDAC2-Δ262-488, or empty vector along with ISRE-Luc plasmid and pRL-TK plasmid. After 24 h, cells were treated with 1,000 U/mL of IFN-α for 12 h, followed by dual-luciferase assays. (C–E) LLC-PK1 cells were treated as described above. At 12 h IFN-α stimulation, the cells were collected for qRT-PCR for the purpose of detecting mRNA expression of ISG15 (C), ISG54 (D), and ISG56 (E). (F–H) LLC-PK1 cells were mock-treated or pretreated with CAY (75 μM) for 2 h, followed by PDCoV infection (MOI 0.5). At 24 h after infection, cells were collected for qRT-PCR for the purpose of detecting mRNA expression of ISG15 (F), ISG54 (G), and ISG56 (H). All experiments were performed in triplicate, and the data are presented as means ± SD of three independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ns, not significant.
We also analyzed the mRNA expression of some ISGs, including ISG15 (Fig. 5C), ISG54 (Fig. 5D), and ISG56 (Fig. 5E). The results showed that full-length HDAC2, but not the cleaved products (HDAC2-Δ1-261 and HDAC2-Δ262-488), enhanced IFN-α-induced ISG expression. To further confirm the regulatory role of HDAC2 for PDCoV-induced ISG expression, we analyzed the mRNA expression of ISGs in cells treated with CAY and then infected with PDCoV. As shown in Fig. 5F–H, the PDCoV-induced expression of ISG15 (Fig. 5F), ISG54 (Fig. 5G), and ISG56 (Fig. 5H) was significantly decreased after CAY treatment. Furthermore, we found that CAY treatment had no influence on Sendai virus (SeV)-mediated IFN-β promoter activation, while it had a significant inhibitory effect on IFN-α-induced ISRE promoter activities (data not shown), suggesting that CAY does not affect IFN production but does inhibit IFN-α-induced ISG expressions. Taken together, these results confirmed that HDAC2 can positively regulate type I IFN signaling and that PDCoV-mediated cleavage impairs the ability of HDAC2 to induce ISG production.
PDCoV-mediated cleavage damages HDAC2 antiviral activity.
Because HDAC2 negatively regulated PDCoV replication and because the cleaved HDAC2 lost the ability to induce ISGs, it is reasonable to speculate that PDCoV-mediated HDAC2 cleavage damages its antiviral activity. To confirm this speculation, LLC-PK1 cells were transfected with expression plasmids encoding HDAC2-WT, HDAC2-Q261A, HDAC2-Δ1-261, HDAC2-Δ262-488, or empty vector for 24 h, followed by PDCoV infection. At 12 h after infection, cells were collected to determine viral titers. As shown in Fig. 6, compared with HDAC2-WT, HDAC2-Δ1-261 and HDAC2-Δ262-488 did not inhibit PDCoV replication. However, a stronger inhibitory effect could be observed in cells transfected with mutant HDAC2-Q261A compared with HDAC2-WT. These results suggested that the cleavage of HDAC2 by PDCoV completely abrogates the ability to inhibit PDCoV replication and that the mutation of the cleavage site enhances the antiviral activity of HDAC2.
FIG 6.
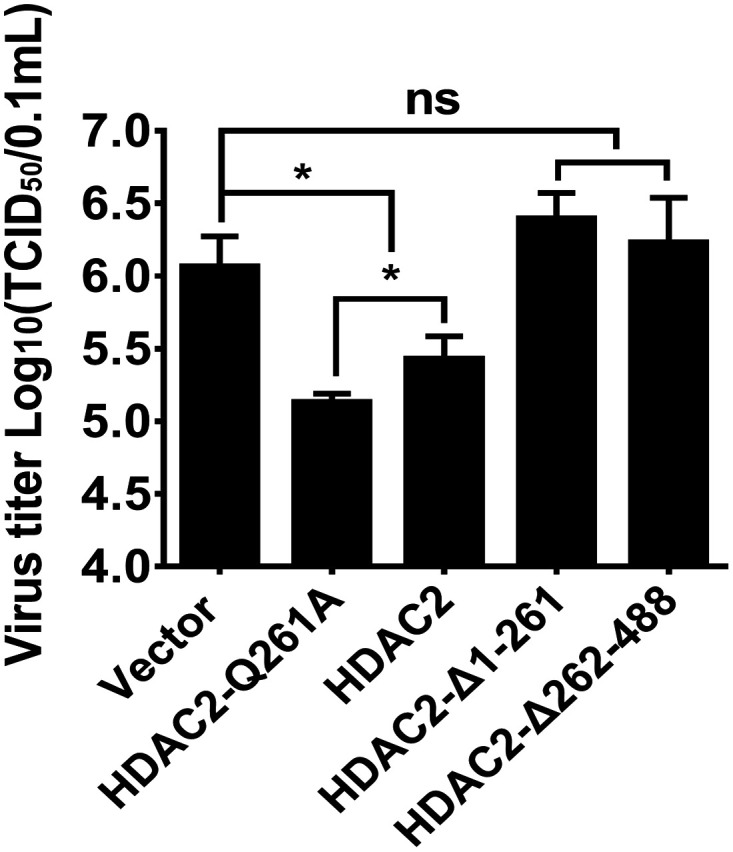
PDCoV-mediated cleavage abrogates HDAC2 antiviral activity. LLC-PK1 cells were transfected with expression plasmid encoding WT HDAC2, HDAC2-Q261A, HDAC2-Δ1-261, HDAC2-Δ262-488, or empty vector. At 24 h after transfection, cells were infected with PDCoV (MOI 0.5). Viral titer was detected by TCID50 assay. The experiments were performed in triplicate, and the data are presented as means ± SD of three independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ns, not significant.
HDAC2 is a common target of nsp5 in various coronaviruses.
To further investigate whether HDAC2 cleavage is species-restricted, we analyzed the diversity of HDAC2 homologs among different mammalian species. Multiple sequence alignment showed that the amino acid sequences of HDAC2 from different mammalian species were highly conserved. Particularly, the Q261 residue recognized by PDCoV nsp5 is identical among HDAC2 of different mammalian species (Fig. 7A), suggesting that HDAC2 cleavage may not be species-dependent. To support this view, we selected two alpha-CoVs, PEDV and human coronavirus 229E (HCoV-229E), and four beta-CoVs, HCoV-HKU1, SARS-CoV, Middle Eastern respiratory syndrome (MERS)-CoV, and SARS-CoV-2, to investigate whether the nsp5 from these CoVs cleaves HDAC2. HEK-293T cells were cotransfected with expression plasmids encoding Flag-tagged human HDAC2 (hHDAC2) or porcine HDAC2 (pHDAC2) and the respective expression plasmids encoding HA-tagged nsp5 from PEDV, HCoV-229E, HCoV-HKU1, SARS-CoV, MERS-CoV, or SARS-CoV2. PDCoV nsp5 was used as a positive control. As shown in Fig. 7B and C, all nsp5s from the tested CoVs successfully cleaved hHDAC2 and pHDAC2, suggesting that HDAC2 cleavage is a conserved mechanism across CoVs to antagonize the antiviral activity of HDAC2.
FIG 7.

HDAC2 is a common target of nsp5 of different CoVs. (A) Alignment of the amino acid sequences of porcine HDAC2 (GenBank accession number XP_001925353.2), human HDAC2 (GenBank accession number NP_001518.3), rat HDAC2 (GenBank accession number NP_445899.1), bovine HDAC2 (GenBank accession number NP_001068614.1), horse HDAC2 (GenBank accession number XP_023506728.1), and mouse HDAC2 (GenBank accession number NP_032255.2). The cleaved Q261 site is indicated with an pentagram. (B, C) HEK-293T cells were cotransfected with Flag-tagged human HDAC2 (hHDAC2) or porcine HDAC2 (pHDAC2) and expression plasmids encoding nsp5 of PEDV, HCoV-229E, HCoV-HKU1, MERS-CoV, SARS-CoV, SARS-CoV-2, or PDCoV. At 28 h after cotransfection, cells were collected for Western blotting with anti-Flag and anti-HA antibodies.
DISCUSSION
Owing to the significance of HDACs during viral infection and their requirement for essential host functions, it is not surprising that viruses always evolve finely tuned mechanisms for targeting HDACs to either appropriate or inhibit their enzymatic activities. In this study, we found that PDCoV infection downregulated cellular deacetylase activity and the expression of some HDACs. Of particular interest, PDCoV infection cleaved HDAC2, and PDCoV-encoded 3C-like protease was associated with this process. Although HDAC2 could function as a host restriction factor to inhibit viral replication, PDCoV-mediated cleavage almost completely abolished the antiviral activity of HDAC2 by impairing, at least, its ability to induce ISGs. Thus, cleavage of HDAC2 is an immune evasion strategy used by PDCoV.
In this study, our initial experiments showed that PDCoV infection downregulated cellular HDAC activity, and the expression levels of HDAC2, HDAC8, HDAC11, and SIRT2 were significantly downregulated among the tested HDACs. Apart from HDAC2, which was studied in detail in our present study, HDAC8, HDAC11, and SIRT2 have also been reported to be involved in the replication of some viruses and to be associated with innate immune regulation. For example, several studies have demonstrated that HDAC8 regulates the replication of IAV and hepatitis C virus (60, 61). Overexpression of HDAC11 exhibits stronger inhibitory effects on IAV and hepatitis B virus (HBV) by inducing ISG expression; however, IAV antagonizes the antiviral activity of HDAC11 by downregulating its expression in host cells (62, 63). AGK2, a SIRT2 inhibitor, significantly inhibits HBV replication in vitro and in vivo (64). The roles and mechanisms of HADC8, HDAC11, and SIRT2 in PDCoV infection need further investigation. In addition, a recent study reported changes in the expression of some HDACs in cells infected with PEDV, another alpha-CoV, and found that PEDV infection led to significant inhibition of HDAC1, HDAC3, HDAC4, HDAC11, and Sirt2 expression (39). However, no significant changes in HADC1, HDAC3, or HDAC4 were observed in cells after PDCoV infection, suggesting that different CoVs use different mechanisms to regulate cellular HDAC activities. In addition, we found that the expressions of all selected HATs were significantly upregulated after PDCoV infection. Previous studies reported that lysine acetyltransferase 8 (KAT8) selectively inhibits antiviral immunity by acetylating IRF3 (65) and that PDCoV infection inhibits host antiviral innate immune responses (50, 51). Whether the upregulated HATs are associated with the immunosuppression induced by PDCoV requires further study.
HDAC2 is a well-known member of the class I HDAC subfamily, and a plethora of cellular proteins and physiological functions have been demonstrated to be influenced by HDAC2. In addition to histones, nonhistone proteins, such as the IFN-inducible MHC class II transactivator CIITA (66), tumor suppressor p53 (67), nuclear factor κB (NF-κB) (68), steroid receptors, and STATs (58), are targets or substrates for HDAC2. Among these nonhistone protein targets, the regulatory roles of HDACs for STAT-associated signaling have been extensively investigated. Genin et al. reported that the treatment of murine cells by trichostatin A (TSA), a deacetylase inhibitor, inhibits Newcastle disease virus (NDV)-induced ISG expression by impairing the nuclear accumulation of STAT2 and ISGF3 complex formation (69). Chang and colleagues reported that TSA treatment also inhibits IFN-α-induced ISG expressions (41); however, their results suggested that TSA does not affect the phosphorylation of STAT1/STAT2 or the nuclear accumulation of ISGF3. Klampfer et al. demonstrated that HDAC inhibitors impede IFN-γ-induced phosphorylation of STAT1 to negatively regulate the expression of a subset of IFN-responsive genes, and HDAC1, HDAC2, and HDAC3 are required for IFN-γ-induced STAT1-dependent transcription (58). Although these studies reported different targets or action mechanisms of HDAC inhibitors, which might be due to different stimuli, different cell lines, or different treatments (simultaneous treatment with HDAC inhibitors and IFN or preincubation of cells with HDAC inhibitors prior to treatment with IFN), all of these studies demonstrated that HDAC activity is required for STAT signaling and for subsequent ISG expression. In this study, we found that the overexpression of HDAC2 significantly enhanced IFN-α-induced ISRE promoter activity and ISG expression. Inhibiting the activity of HDAC2 with CAY increased the replication of PDCoV, likely via the reduced expression of antiviral ISGs. We also found that PDCoV infection decreased HDAC2 expression. Thus, it is reasonable to assume that downregulation of HDAC2 expression is an immune evasion mechanism used by PDCoV. Certainly, PDCoV infection also downregulated the expression of HDAC8, HDAC11, and SIRT2, which may be associated with PDCoV replication.
An interesting finding of this study is that PDCoV nsp5 cleaves HDAC2, and this cleavage could be detected in the context of PDCoV infection. Previous studies showed that the expression and activity of HDAC2 are regulated at transcriptional, posttranscriptional, and posttranslational levels under stimulus or virus infection (70–73). Furthermore, HDAC2 can act as either a restriction or dependency factor for various viruses. For example, Liao et al. reported that the Marek’s disease virus Meq oncoprotein interacts with chicken HDAC1 and HDAC2 and mediates their degradation via the proteasome-dependent pathway (42). IAV infection downregulates host HDAC2 expression, which potentially occurs mainly at the protein level via proteasomal degradation (38). In this study, we found that the HDAC2 transcription level was significantly downregulated by PDCoV infection in both LLC-PK1 and IPI-FX cells, indicating that PDCoV restrains HDAC2 activity directly by regulating HDAC2 transcription. At the same time, we also found that PDCoV infection cleaves HDAC2 and demonstrated that PDCoV nsp5 is associated with this cleavage, although nsp5 does not affect the mRNA expression of endogenous HDAC2. To our knowledge, PDCoV nsp5 is the first identified viral protein that can cleave HDAC2. Considering that nsp5 is the main protease of CoVs and is responsible for cleaving the viral polyprotein and host proteins, we focused on the cleavage mechanism and its biological significance in this study. We and other groups have shown that CoV nsp5 can cleave many host proteins; however, the proteins cleaved by CoV nsp5 are mainly located in the cytoplasm. Although early studies suggested that HDAC2 is predominantly located in the nucleus of the resting cell, some studies reported that HDAC2 can be detected in both the nucleus and the cytoplasm (55–57, 74). Furthermore, HDAC2 can be translocated from the nucleus to the cytoplasm under stimulus or pathological conditions. The translocation of HDAC2 is speculated to be required for interaction with some necessary regulators (74). In this study, we found that PDCoV infection resulted in translocation of HDAC2 from the nucleus to the cytoplasm, and nsp5 cleaved cytoplasmic HDAC2 rather than nuclear HDAC2, which will decrease HDAC2 expression and impair its activity and ISGF3 assembly, thereby inhibiting the transcription of downstream ISGs. However, the mechanism by which PDCoV infection promotes the translocation of HDAC2 from the nucleus to the cytoplasm needs further investigation. Taken together, at least two kinds of mechanisms are utilized by PDCoV to inhibit HDAC2-mediated ISGs expression: decreasing HDAC2 expression and cleaving HDAC2.
HDAC2 is highly conserved in different species. Our present study provided evidence that nsp5s from different mammalian CoVs cleave HDAC2 at Q261 and that the cleaved products lose the ability to induce ISG expression. Thus, cleaving HDAC2 may be a common strategy used by different mammalian CoVs to antagonize the antiviral role of HDAC2. In addition, HDAC2 has been reported to negatively regulate inflammatory responses, and decreased expression of HDAC2 is considered to contribute to disease severity in chronic obstructive pulmonary disease (COPD) (75). Intestinal inflammation and lung inflammation are typically clinicopathological features for enteropathogenic CoVs, such as PDCoV, and respiratory CoVs, such as SARS-CoV-2, respectively. Thus, downregulating or cleaving HDAC2 may be associated with the inflammatory responses to CoVs. Targeting HDAC2 may be a novel target for the development of broad anti-CoV drugs, and this is worthy of further study.
MATERIALS AND METHODS
Cells and virus.
HEK-293T cells were obtained from the China Center for Type Culture Collection (Wuhan, China). IPI-FX cells were derived from IPI-2I cells (porcine ileum epithelial cells) by subcloning through limited serial dilution (76). LLC-PK1 cells were purchased from the American Type Culture Collection (Manassas, VA; ATCC CL-101). All cells were cultured in Dulbecco’s modified Eagle’s medium (Invitrogen, Madison, WI) supplemented with 10% fetal bovine serum at 37°C in 5% CO2. The PDCoV strain CHN-HN-2014 (GenBank accession number KT336560) used in this study was isolated from a piglet with severe diarrhea in China in 2014 (77).
Plasmid construction.
The full-length cDNA of porcine HDAC2 (GenBank accession number XM_001925318.6) was amplified from LLC-PK1 cells and then cloned into a pCAGGS-Flag vector containing an N-terminal Flag tag or a pCAGGS-Flag-Myc vector with N-terminal Flag and C-terminal Myc tags, generating the expression plasmids pCAGGS-Flag-HDAC2 and pCAGGS-Flag-HDAC2-Myc, respectively. The human HDAC2 gene (GenBank accession number NM_001527.4) was amplified from the cDNA of HEK-293T cells and cloned into the pCAGGS-Flag vector. The porcine HDAC2 mutants, including the substitution mutants HDAC2-Q240A, -Q254A, -Q261A, -Q354A, and -Q365A, as well as the truncated mutants HDAC2-Δ1-261 and HDAC2-Δ262-488 were also cloned into a pCAGGS-Flag vector. The nsp5-coding sequences of PDCoV strain CHN-HN-2014 and mutants were both amplified and cloned into pCAGGS-HA-C with an HA tag in the C terminus.
RNA extraction and quantitative real-time RT-PCR.
Cells in six-well plates were treated under various experimental conditions, followed by PDCoV infection. At the indicated time points, total RNA was extracted from the cells using TRIzol reagent (Invitrogen) and then reverse transcribed into cDNA by avian myeloblastosis virus reverse transcriptase (TaKaRa, Kusatsu, Japan) with the indicated primers (Table 1). Quantitative real-time PCR (qRT-PCR) experiments were performed in triplicate. mRNA expression levels were normalized to the level of glyceraldehyde-3-phosphate dehydrogenase (GAPDH).
TABLE 1.
Primers used in this study
| Primers | Sequences(5′-3′) |
|---|---|
| HDAC2-F | TTTGGTACC ATGGCGTACAGTCAGGGAGGCGG |
| HDAC2-R | TTTCTCGAGTCAAGGGTTGCTGAGCTGTTCTGA |
| HDAC1-F | ATGAGGAGGGAGAAGGTG |
| HDAC1-R | GGTTGTGGGATAAAGACG |
| HDAC2-F | CCCCATAAAGCCACTGCTGA |
| HDAC2-R | AGCCACCAGTTGAAAGCTGA |
| HDAC3-F | CGCAGACCTCCTGACCTAC |
| HDAC3-R | TTCCCTCCCACCCAACTA |
| HDAC4-F | GAAGTGCGAGAACGAGGAA |
| HDAC4-R | GACGGAGACAAACAGACAAGAG |
| HDAC8-F | TCCAGAAGGTCAGCCAAGA |
| HDAC8-R | TTTCCGTCGCAA TCGTAG |
| HDAC11-F | CGGCTTCCACCATTGCTC |
| HDAC11-R | GTCCCGCTCGTGTCCATT |
| Sirt1-F | AGAAGGAAACAATGGGCCG |
| Sirt1-R | CCAAACAGAAGGTTATCTCGGTA |
| Sirt2-F | CCCTTCGCA TCCCTCA T |
| Sirt2-R | A TCCCGACTGGGCA TCT |
| PDCoV-N-F | AGCTGCTACCTCTCCGATTC |
| PDCoV-N-R | ACATTGGCACCAGTACGAGA |
| PDCoV-nsp16-F | GCCCTCGGTGGTTCTATCTT |
| PDCoV-nsp16-R | TCCTTAGCTTGCCCCAAATA |
| GAPDH-F | ACATGGCCTCCAAGGAGTAAGA |
| GAPDH-R | GATCGAGTTGGGGCTGTGACT |
| CREBBP-F | CCTGTTTGCCTCCCTTTG |
| CREBBP-R | GGCTGTGCTGGTTGCTG |
| EP300-F | GTTCATTTCTTCCGTCCTA |
| EP300-R | GCTTGGGTATCTTCTGGTC |
| HAT1-F | TTACTCCATACATACTCGGTTCT |
| HAT1-R | AGCCTACGGTCGCAAAG |
| MYST2-F | TAGCGAGTATGACTTGGAT |
| MYST2-R | CAGGATAGGGAGAATGGTA |
| pISG15-F | CCTGTTGATGGTGCAAAGCT |
| pISG15-R | TGCACATAGGCTTGAGGTCA |
| pISG54-F | CTGGCAAAGAGCCCTAAGGA |
| pISG54-R | CTCAGAGGGTCAATGGAATTCC |
| pISG56-F | AAATGAATGAAGCCCTGGAGTATT |
| pISG56-R | AGGGATCAAGTCCCACAGATTTT |
Western blotting.
Experiments to determine HDAC2 cleavage were conducted in six-well plates. We cotransfected 2.5 μg of HDAC2 or mutants with 0.5 μg of nsp5 or empty vector. Cells were collected in lysis buffer (Beyotime, Shanghai, China), added to sample loading buffer (Beyotime), and boiled for 10 min. Subsequently, the same samples were separated in parallel on the different gels and transferred to polyvinylidene difluoride membranes (Millipore, Burlington, MA). The anti-Flag antibody (Macgene, Beijing, China) and anti-HA antibody (MBL, Nagoya, Japan) were used to detect the respective proteins. The rabbit anti-HDAC11 and anti-SIRT2 monoclonal antibodies were purchased from Cell Signaling Technology (Danvers, MA). The mouse anti-HDAC1, anti-HDAC2, anti-HDAC3, and anti-HDAC8 monoclonal antibodies were purchased from Santa Cruz (Dallas, TX). The rabbit anti-β-actin antibody was purchased from Abclonal (Wuhan, China). The mouse anti-PDCoV N protein monoclonal antibody used herein was described previously (78).
Nuclear cytosol fractionation assay.
To prepare the nuclear and cytoplasmic fractions, cells were lysed using a nuclear and cytoplasmic protein extraction kit (Beyotime), following the manufacturer’s instructions. The cytoplasmic and nuclear fractions were subjected to Western blotting. Successful isolation was assessed using rabbit polyclonal antibodies against heat shock protein 90 and Lamin A+C (Abclonal) as cytoplasmic and nuclear protein markers, respectively.
Cell infection and drug treatment.
To determine the effects of the HDAC2 inhibitor on PDCoV infection, we used the inhibitor when cells were grown to 90% confluence. LLC-PK1 cells were pretreated for 2h with CAY10683 (Selleck, Radnor, PA) and then infected with PDCoV (MOI = 0.5) for 12 h. Cellular total RNA was extracted using TRIzol reagent, followed by the measurement of viral genomic RNA by qRT-PCR.
Luciferase reporter assay.
For luciferase reporter assays, cells were transfected with the reporter plasmids ISRE-Luc and pRL-TK for 24 h. The cells were stimulated with IFN-α (catalog no. 11101-2; PBL Assay Science) for 12 h at a final concentration of 1,000 U/mL. The lysed cells were prepared and subjected to double luciferase analysis using a luciferase reporter assay system (Promega, Madison, WI). Representative data from three independently conducted experiments are shown as the relative firefly luciferase activities with normalization to the Renilla luciferase activities.
Cell viability analysis.
The cytotoxic effects of the drugs on cells were measured using a CCK-8-based cell viability assay (Beyotime).
Antiviral analysis of HDAC2.
LLC-PK1 cells cultured in six-well plates were transfected with HDAC2 or empty vector (4 μg for each plasmid). At 24 h after transfection, the cells were infected with PDCoV (MOI = 0.5) and then collected for qRT-PCR to detect viral genomic RNA copies, Western blotting was used to detect viral protein expression, and a 50% tissue culture infectious dose (TCID50) assay was used to determine viral titers. The TCID50 assay was performed as described previously (77).
HDAC activity assay.
Cellular HDAC activity was detected using a fluorogenic HDAC activity assay kit (AAT Bioquest, Sunnyvale, CA) as described previously (79). Briefly, extracts were transferred to a black 96-well plate, and the fluorescence intensity at 490 ex/525 em was monitored using a multifunction microplate reader.
Sequence alignment.
The amino acid sequences of HDAC2 from different species, including porcine HDAC2 (GenBank accession number XP_001925353.2), human HDAC2 (GenBank accession number NP_001518.3), rat HDAC2 (GenBank accession number NP_445899.1), mouse HDAC2 (GenBank accession number NP_032255.2), horse HDAC2 (GenBank accession number XP_023506728.1), and bovine HDAC2 (GenBank accession number NP_001068614.1), were collected. Multiple-sequence alignment was conducted using Clustal Omega (https://www.ebi.ac.uk/Tools/msa/clustalo/).
Statistical analysis.
All experiments were performed in triplicate. Significant differences were determined using Student's t test. P values of <0.05 were considered to be indicative of statistical significance.
ACKNOWLEDGMENTS
This work is supported by the National Research and Development Program of China (2021YFD1801104) and the National Natural Science Foundation of China (31730095, 32072846, 31902247).
Contributor Information
Liurong Fang, Email: fanglr@mail.hzau.edu.cn.
Shaobo Xiao, Email: vet@mail.hzau.edu.cn.
Tom Gallagher, Loyola University Chicago.
REFERENCES
- 1.Woo PC, Lau SK, Lam CS, Lau CC, Tsang AK, Lau JH, Bai R, Teng JL, Tsang CC, Wang M, Zheng BJ, Chan KH, Yuen KY. 2012. Discovery of seven novel mammalian and avian coronaviruses in the genus deltacoronavirus supports bat coronaviruses as the gene source of alphacoronavirus and betacoronavirus and avian coronaviruses as the gene source of gammacoronavirus and deltacoronavirus. J Virol 86:3995–4008. 10.1128/JVI.06540-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li G, Chen Q, Harmon KM, Yoon KJ, Schwartz KJ, Hoogland MJ, Gauger PC, Main RG, Zhang J. 2014. Full-length genome sequence of porcine deltacoronavirus strain USA/IA/2014/8734. Genome Announc 2. 10.1128/genomeA.00278-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang L, Byrum B, Zhang Y. 2014. Detection and genetic characterization of deltacoronavirus in pigs, Ohio, USA, 2014. Emerg Infect Dis 20:1227–1230. 10.3201/eid2007.140296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lorsirigool A, Saeng-Chuto K, Madapong A, Temeeyasen G, Tripipat T, Kaewprommal P, Tantituvanont A, Piriyapongsa J, Nilubol D. 2017. The genetic diversity and complete genome analysis of two novel porcine deltacoronavirus isolates in Thailand in 2015. Virus Genes 53:240–248. 10.1007/s11262-016-1413-z. [DOI] [PubMed] [Google Scholar]
- 5.Dong N, Fang L, Zeng S, Sun Q, Chen H, Xiao S. 2015. Porcine deltacoronavirus in mainland China. Emerg Infect Dis 21:2254–2255. 10.3201/eid2112.150283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jang G, Lee KK, Kim SH, Lee C. 2017. Prevalence, complete genome sequencing and phylogenetic analysis of porcine deltacoronavirus in South Korea, 2014–2016. Transbound Emerg Dis 64:1364–1370. 10.1111/tbed.12690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Saeng-Chuto K, Lorsirigool A, Temeeyasen G, Vui DT, Stott CJ, Madapong A, Tripipat T, Wegner M, Intrakamhaeng M, Chongcharoen W, Tantituvanont A, Kaewprommal P, Piriyapongsa J, Nilubol D. 2017. Different lineage of porcine deltacoronavirus in Thailand, Vietnam and Lao PDR in 2015. Transbound Emerg Dis 64:3–10. 10.1111/tbed.12585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xu Z, Zhong H, Zhou Q, Du Y, Chen L, Zhang Y, Xue C, Cao Y. 2018. A highly pathogenic strain of porcine deltacoronavirus caused watery diarrhea in newborn piglets. Virol Sin 33:131–141. 10.1007/s12250-018-0003-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Suzuki T, Shibahara T, Imai N, Yamamoto T, Ohashi S. 2018. Genetic characterization and pathogenicity of Japanese porcine deltacoronavirus. Infect Genet Evol 61:176–182. 10.1016/j.meegid.2018.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li W, Hulswit RJG, Kenney SP, Widjaja I, Jung K, Alhamo MA, van Dieren B, van Kuppeveld FJM, Saif LJ, Bosch BJ. 2018. Broad receptor engagement of an emerging global coronavirus may potentiate its diverse cross-species transmissibility. Proc Natl Acad Sci USA 115:E5135–E5143. 10.1073/pnas.1802879115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liang Q, Zhang H, Li B, Ding Q, Wang Y, Gao W, Guo D, Wei Z, Hu H. 2019. Susceptibility of chickens to porcine deltacoronavirus infection. Viruses 11:573. 10.3390/v11060573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boley PA, Alhamo MA, Lossie G, Yadav KK, Vasquez-Lee M, Saif LJ, Kenney SP. 2020. Porcine deltacoronavirus infection and transmission in poultry, United States(1). Emerg Infect Dis 26:255–265. 10.3201/eid2602.190346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jung K, Hu H, Saif LJ. 2017. Calves are susceptible to infection with the newly emerged porcine deltacoronavirus, but not with the swine enteric alphacoronavirus, porcine epidemic diarrhea virus. Arch Virol 162:2357–2362. 10.1007/s00705-017-3351-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu Y, Wang B, Liang QZ, Shi FS, Ji CM, Yang XL, Yang YL, Qin P, Chen R, Huang YW. 2021. Roles of two major domains of the porcine deltacoronavirus S1 subunit in receptor binding and neutralization. J Virol 95:e0111821. 10.1128/JVI.01118-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lednicky JA, Tagliamonte MS, White SK, Elbadry MA, Alam MM, Stephenson CJ, Bonny TS, Loeb JC, Telisma T, Chavannes S, Ostrov DA, Mavian C, Beau De Rochars VM, Salemi M, Morris JG. Jr., 2021. Independent infections of porcine deltacoronavirus among Haitian children. Nature 600:133–137. 10.1038/s41586-021-04111-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Benedetti R, Conte M, Altucci L. 2015. Targeting histone deacetylases in diseases: where are we? Antioxid Redox Signal 23:99–126. 10.1089/ars.2013.5776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, Olsen JV, Mann M. 2009. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 325:834–840. 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- 18.Haery L, Thompson RC, Gilmore TD. 2015. Histone acetyltransferases and histone deacetylases in B- and T-cell development, physiology and malignancy. Genes Cancer 6:184–213. 10.18632/genesandcancer.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shakespear MR, Halili MA, Irvine KM, Fairlie DP, Sweet MJ. 2011. Histone deacetylases as regulators of inflammation and immunity. Trends Immunol 32:335–343. 10.1016/j.it.2011.04.001. [DOI] [PubMed] [Google Scholar]
- 20.Hildmann C, Riester D, Schwienhorst A. 2007. Histone deacetylases–an important class of cellular regulators with a variety of functions. Appl Microbiol Biotechnol 75:487–497. 10.1007/s00253-007-0911-2. [DOI] [PubMed] [Google Scholar]
- 21.Song W, Tai YT, Tian Z, Hideshima T, Chauhan D, Nanjappa P, Exley MA, Anderson KC, Munshi NC. 2011. HDAC inhibition by LBH589 affects the phenotype and function of human myeloid dendritic cells. Leukemia 25:161–168. 10.1038/leu.2010.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Drummond DC, Noble CO, Kirpotin DB, Guo Z, Scott GK, Benz CC. 2005. Clinical development of histone deacetylase inhibitors as anticancer agents. Annu Rev Pharmacol Toxicol 45:495–528. 10.1146/annurev.pharmtox.45.120403.095825. [DOI] [PubMed] [Google Scholar]
- 23.Minucci S, Pelicci PG. 2006. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat Rev Cancer 6:38–51. 10.1038/nrc1779. [DOI] [PubMed] [Google Scholar]
- 24.Buchwald M, Kramer OH, Heinzel T. 2009. HDACi–targets beyond chromatin. Cancer Lett 280:160–167. 10.1016/j.canlet.2009.02.028. [DOI] [PubMed] [Google Scholar]
- 25.Spange S, Wagner T, Heinzel T, Kramer OH. 2009. Acetylation of non-histone proteins modulates cellular signalling at multiple levels. Int J Biochem Cell Biol 41:185–198. 10.1016/j.biocel.2008.08.027. [DOI] [PubMed] [Google Scholar]
- 26.Yang XJ, Seto E. 2008. The Rpd3/Hda1 family of lysine deacetylases: from bacteria and yeast to mice and men. Nat Rev Mol Cell Biol 9:206–218. 10.1038/nrm2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Falkenberg KJ, Johnstone RW. 2014. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat Rev Drug Discov 13:673–691. 10.1038/nrd4360. [DOI] [PubMed] [Google Scholar]
- 28.Haberland M, Montgomery RL, Olson EN. 2009. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet 10:32–42. 10.1038/nrg2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nusinzon I, Horvath CM. 2003. Interferon-stimulated transcription and innate antiviral immunity require deacetylase activity and histone deacetylase 1. Proc Natl Acad Sci USA 100:14742–14747. 10.1073/pnas.2433987100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lu Y, Stuart JH, Talbot-Cooper C, Agrawal-Singh S, Huntly B, Smid AI, Snowden JS, Dupont L, Smith GL. 2019. Histone deacetylase 4 promotes type I interferon signaling, restricts DNA viruses, and is degraded via vaccinia virus protein C6. Proc Natl Acad Sci USA 116:11997–12006. 10.1073/pnas.1816399116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nagesh PT, Husain M. 2016. Influenza A virus dysregulates host histone deacetylase 1 that inhibits viral infection in lung epithelial cells. J Virol 90:4614–4625. 10.1128/JVI.00126-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gu H, Liang Y, Mandel G, Roizman B. 2005. Components of the REST/CoREST/histone deacetylase repressor complex are disrupted, modified, and translocated in HSV-1-infected cells. Proc Natl Acad Sci USA 102:7571–7576. 10.1073/pnas.0502658102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhou Y, Wang Q, Yang Q, Tang J, Xu C, Gai D, Chen X, Chen J. 2018. Histone deacetylase 3 inhibitor suppresses hepatitis C virus replication by regulating Apo-A1 and LEAP-1 expression. Virol Sin 33:418–428. 10.1007/s12250-018-0057-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zaikos TD, Painter MM, Sebastian Kettinger NT, Terry VH, Collins KL. 2018. Class 1-selective histone deacetylase (HDAC) inhibitors enhance HIV latency reversal while preserving the activity of HDAC isoforms necessary for maximal HIV gene expression. J Virol 92 10.1128/JVI.02110-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Romani B, Baygloo NS, Hamidi-Fard M, Aghasadeghi MR, Allahbakhshi E. 2016. HIV-1 Vpr protein induces proteasomal degradation of chromatin-associated class I HDACs to overcome latent infection of macrophages. J Biol Chem 291:2696–2711. 10.1074/jbc.M115.689018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lu T, Song Z, Li Q, Li Z, Wang M, Liu L, Tian K, Li N. 2017. Overexpression of histone deacetylase 6 enhances resistance to porcine reproductive and respiratory syndrome virus in pigs. PLoS One 12:e0169317. 10.1371/journal.pone.0169317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen H, Qian Y, Chen X, Ruan Z, Ye Y, Chen H, Babiuk LA, Jung YS, Dai J. 2019. HDAC6 restricts influenza A virus by deacetylation of the RNA polymerase PA subunit. J Virol 93 10.1128/JVI.01896-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nagesh PT, Hussain M, Galvin HD, Husain M. 2017. Histone deacetylase 2 is a component of influenza A virus-induced host antiviral response. Front Microbiol 8:1315. 10.3389/fmicb.2017.01315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xu J, Mao J, Han X, Shi F, Gao Q, Wang T, Zhang Z, Shan Y, Fang W, Li X. 2021. Porcine epidemic diarrhea virus inhibits HDAC1 expression to facilitate its replication via binding of its nucleocapsid protein to host transcription factor Sp1. J Virol 95:e0085321. 10.1128/JVI.00853-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pitt B, Sutton NR, Wang Z, Goonewardena SN, Holinstat M. 2021. Potential repurposing of the HDAC inhibitor valproic acid for patients with COVID-19. Eur J Pharmacol 898:173988. 10.1016/j.ejphar.2021.173988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chang HM, Paulson M, Holko M, Rice CM, Williams BR, Marie I, Levy DE. 2004. Induction of interferon-stimulated gene expression and antiviral responses require protein deacetylase activity. Proc Natl Acad Sci USA 101:9578–9583. 10.1073/pnas.0400567101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liao Y, Lupiani B, Izumiya Y, Reddy SM. 2021. Marek's disease virus Meq oncoprotein interacts with chicken HDAC 1 and 2 and mediates their degradation via proteasome dependent pathway. Sci Rep 11:637. 10.1038/s41598-020-80792-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Saw G, Tang FR. 2020. Epigenetic regulation of the hippocampus, with special reference to radiation exposure. Int J Mol Sci 21. 10.3390/ijms21249514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang L, Sheng C, Zhou F, Zhu K, Wang S, Liu Q, Yuan M, Xu Z, Liu Y, Lu J, Liu J, Zhou L, Wang X. 2021. CBP/p300 HAT maintains the gene network critical for beta cell identity and functional maturity. Cell Death Dis 12:476. 10.1038/s41419-021-03761-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wu C, Li A, Hu J, Kang J. 2019. Histone deacetylase 2 is essential for LPS-induced inflammatory responses in macrophages. Immunol Cell Biol 97:72–84. 10.1111/imcb.12203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xu P, Ye S, Li K, Huang M, Wang Q, Zeng S, Chen X, Gao W, Chen J, Zhang Q, Zhong Z, Lin Y, Rong Z, Xu Y, Hao B, Peng A, Ouyang M, Liu Q. 2019. NOS1 inhibits the interferon response of cancer cells by S-nitrosylation of HDAC2. J Exp Clin Cancer Res 38:483. 10.1186/s13046-019-1448-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ren WB, Xia XJ, Huang J, Guo WF, Che YY, Huang TH, Lei LC. 2019. Interferon-gamma regulates cell malignant growth via the c-Abl/HDAC2 signaling pathway in mammary epithelial cells. J Zhejiang Univ Sci B 20:39–48. 10.1631/jzus.B1800211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Marie IJ, Chang HM, Levy DE. 2018. HDAC stimulates gene expression through BRD4 availability in response to IFN and in interferonopathies. J Exp Med 215:3194–3212. 10.1084/jem.20180520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang D, Fang L, Shi Y, Zhang H, Gao L, Peng G, Chen H, Li K, Xiao S. 2016. Porcine epidemic diarrhea virus 3C-like protease regulates its interferon antagonism by cleaving NEMO. J Virol 90:2090–2101. 10.1128/JVI.02514-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhu X, Fang L, Wang D, Yang Y, Chen J, Ye X, Foda MF, Xiao S. 2017. Porcine deltacoronavirus nsp5 inhibits interferon-beta production through the cleavage of NEMO. Virology 502:33–38. 10.1016/j.virol.2016.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhu X, Wang D, Zhou J, Pan T, Chen J, Yang Y, Lv M, Ye X, Peng G, Fang L, Xiao S. 2017. Porcine deltacoronavirus nsp5 antagonizes type I interferon signaling by cleaving STAT2. J Virol 91. 10.1128/JVI.00003-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhou J, Fang L, Yang Z, Xu S, Lv M, Sun Z, Chen J, Wang D, Gao J, Xiao S. 2019. Identification of novel proteolytically inactive mutations in coronavirus 3C-like protease using a combined approach. FASEB J 33:14575–14587. 10.1096/fj.201901624RR. [DOI] [PubMed] [Google Scholar]
- 53.Ziebuhr J, Siddell SG. 1999. Processing of the human coronavirus 229E replicase polyproteins by the virus-encoded 3C-like proteinase: identification of proteolytic products and cleavage sites common to pp1a and pp1ab. J Virol 73:177–185. 10.1128/JVI.73.1.177-185.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gregoretti IV, Lee YM, Goodson HV. 2004. Molecular evolution of the histone deacetylase family: functional implications of phylogenetic analysis. J Mol Biol 338:17–31. 10.1016/j.jmb.2004.02.006. [DOI] [PubMed] [Google Scholar]
- 55.Liu J, Xu D, Wang H, Zhang Y, Chang Y, Zhang J, Wang J, Li C, Liu H, Zhao M, Lin C, Zhan Q, Huang C, Qian H. 2014. The subcellular distribution and function of MTA1 in cancer differentiation. Oncotarget 5:5153–5164. 10.18632/oncotarget.2095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zou C, Synan MJ, Li J, Xiong S, Manni ML, Liu Y, Chen BB, Zhao Y, Shiva S, Tyurina YY, Jiang J, Lee JS, Das S, Ray A, Ray P, Kagan VE, Mallampalli RK. 2016. LPS impairs oxygen utilization in epithelia by triggering degradation of the mitochondrial enzyme Alcat1. J Cell Sci 129:51–64. 10.1242/jcs.176701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.La Noce M, Mele L, Laino L, Iolascon G, Pieretti G, Papaccio G, Desiderio V, Tirino V, Paino F. 2019. Cytoplasmic interactions between the glucocorticoid receptor and HDAC2 regulate osteocalcin expression in VPA-treated MSCs. Cells 8:217. 10.3390/cells8030217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Klampfer L, Huang J, Swaby LA, Augenlicht L. 2004. Requirement of histone deacetylase activity for signaling by STAT1. J Biol Chem 279:30358–30368. 10.1074/jbc.M401359200. [DOI] [PubMed] [Google Scholar]
- 59.Nusinzon I, Horvath CM. 2005. Histone deacetylases as transcriptional activators? Role reversal in inducible gene regulation. Sci STKE 2005:re11. 10.1126/stke.2962005re11. [DOI] [PubMed] [Google Scholar]
- 60.Kozlov MV, Konduktorov KA, Malikova AZ, Kamarova KA, Shcherbakova AS, Solyev PN, Kochetkov SN. 2019. Structural isomers of cinnamic hydroxamic acids block HCV replication via different mechanisms. Eur J Med Chem 183:111723. 10.1016/j.ejmech.2019.111723. [DOI] [PubMed] [Google Scholar]
- 61.Xia B, Lu J, Wang R, Yang Z, Zhou X, Huang P. 2018. miR-21-3p regulates influenza A virus replication by targeting histone deacetylase-8. Front Cell Infect Microbiol 8:175. 10.3389/fcimb.2018.00175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nutsford AN, Galvin HD, Ahmed F, Husain M. 2019. The class IV human deacetylase, HDAC11, exhibits anti-influenza A virus properties via its involvement in host innate antiviral response. Cell Microbiol 21:e12989. 10.1111/cmi.12989. [DOI] [PubMed] [Google Scholar]
- 63.Yuan Y, Zhao K, Yao Y, Liu C, Chen Y, Li J, Wang Y, Pei R, Chen J, Hu X, Zhou Y, Wu C, Chen X. 2019. HDAC11 restricts HBV replication through epigenetic repression of cccDNA transcription. Antiviral Res 172:104619. 10.1016/j.antiviral.2019.104619. [DOI] [PubMed] [Google Scholar]
- 64.Yu HB, Jiang H, Cheng ST, Hu ZW, Ren JH, Chen J. 2018. AGK2, A SIRT2 inhibitor, inhibits hepatitis B virus replication in vitro and in vivo. Int J Med Sci 15:1356–1364. 10.7150/ijms.26125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Huai W, Liu X, Wang C, Zhang Y, Chen X, Chen X, Xu S, Thomas T, Li N, Cao X. 2019. KAT8 selectively inhibits antiviral immunity by acetylating IRF3. J Exp Med 216:772–785. 10.1084/jem.20181773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kong X, Fang M, Li P, Fang F, Xu Y. 2009. HDAC2 deacetylates class II transactivator and suppresses its activity in macrophages and smooth muscle cells. J Mol Cell Cardiol 46:292–299. 10.1016/j.yjmcc.2008.10.023. [DOI] [PubMed] [Google Scholar]
- 67.Stojanovic N, Hassan Z, Wirth M, Wenzel P, Beyer M, Schafer C, Brand P, Kroemer A, Stauber RH, Schmid RM, Arlt A, Sellmer A, Mahboobi S, Rad R, Reichert M, Saur D, Kramer OH, Schneider G. 2017. HDAC1 and HDAC2 integrate the expression of p53 mutants in pancreatic cancer. Oncogene 36:1804–1815. 10.1038/onc.2016.344. [DOI] [PubMed] [Google Scholar]
- 68.Wagner T, Kiweler N, Wolff K, Knauer SK, Brandl A, Hemmerich P, Dannenberg JH, Heinzel T, Schneider G, Kramer OH. 2015. Sumoylation of HDAC2 promotes NF-kappaB-dependent gene expression. Oncotarget 6:7123–7135. 10.18632/oncotarget.3344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Genin P, Morin P, Civas A. 2003. Impairment of interferon-induced IRF-7 gene expression due to inhibition of ISGF3 formation by trichostatin A. J Virol 77:7113–7119. 10.1128/jvi.77.12.7113-7119.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Malhotra D, Thimmulappa RK, Mercado N, Ito K, Kombairaju P, Kumar S, Ma J, Feller-Kopman D, Wise R, Barnes P, Biswal S. 2014. Retraction: denitrosylation of HDAC2 by targeting Nrf2 restores glucocorticosteroid sensitivity in macrophages from COPD patients. J Clin Invest 124:5521–5521. 10.1172/JCI79606. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 71.Heideman MR, Wilting RH, Yanover E, Velds A, de Jong J, Kerkhoven RM, Jacobs H, Wessels LF, Dannenberg JH. 2013. Dosage-dependent tumor suppression by histone deacetylases 1 and 2 through regulation of c-Myc collaborating genes and p53 function. Blood 121:2038–2050. 10.1182/blood-2012-08-450916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Liu J, Qian C, Cao X. 2016. Post-translational modification control of innate immunity. Immunity 45:15–30. 10.1016/j.immuni.2016.06.020. [DOI] [PubMed] [Google Scholar]
- 73.Nott A, Nitarska J, Veenvliet JV, Schacke S, Derijck AA, Sirko P, Muchardt C, Pasterkamp RJ, Smidt MP, Riccio A. 2013. S-nitrosylation of HDAC2 regulates the expression of the chromatin-remodeling factor Brm during radial neuron migration. Proc Natl Acad Sci USA 110:3113–3118. 10.1073/pnas.1218126110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hou N, Gong M, Bi Y, Zhang Y, Tan B, Liu Y, Wei X, Chen J, Li T. 2014. Spatiotemporal expression of HDAC2 during the postnatal development of the rat hippocampus. Int J Med Sci 11:788–795. 10.7150/ijms.8417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tan C, Xuan L, Cao S, Yu G, Hou Q, Wang H. 2016. Decreased histone deacetylase 2 (HDAC2) in peripheral blood monocytes (PBMCs) of COPD patients. PLoS One 11:e0147380. 10.1371/journal.pone.0147380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wang X, Fang L, Liu S, Ke W, Wang D, Peng G, Xiao S. 2019. Susceptibility of porcine IPI-2I intestinal epithelial cells to infection with swine enteric coronaviruses. Vet Microbiol 233:21–27. 10.1016/j.vetmic.2019.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Dong N, Fang L, Yang H, Liu H, Du T, Fang P, Wang D, Chen H, Xiao S. 2016. Isolation, genomic characterization, and pathogenicity of a Chinese porcine deltacoronavirus strain CHN-HN-2014. Vet Microbiol 196:98–106. 10.1016/j.vetmic.2016.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Luo J, Fang L, Dong N, Fang P, Ding Z, Wang D, Chen H, Xiao S. 2016. Porcine deltacoronavirus (PDCoV) infection suppresses RIG-I-mediated interferon-beta production. Virology 495:10–17. 10.1016/j.virol.2016.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Nott A, Watson PM, Robinson JD, Crepaldi L, Riccio A. 2008. S-Nitrosylation of histone deacetylase 2 induces chromatin remodelling in neurons. Nature 455:411–415. 10.1038/nature07238. [DOI] [PubMed] [Google Scholar]