

Abstract
Cobalt(II) halides in combination with phenoxy-imine (FI) ligands generated efficient precatalysts in situ for the C(sp2)–C(sp3) Suzuki–Miyaura cross coupling between alkyl bromides and neopentylglycol (hetero)arylboronic esters. The protocol enabled efficient C–C bond formation with a host of nucleophiles and electrophiles (36 examples, 34–95%) with precatalyst loadings of 5 mol%. Studies with alkyl halide electrophiles that function as radical clocks support the intermediacy of alkyl radicals during the course of the catalytic reaction. The improved performance of the FI-cobalt catalyst was correlated with decreased lifetimes of cage-escaped radicals as compared to diamine-type ligands. Studies of the phenoxy(imine)-cobalt coordination chemistry validate the L,X interaction leading to the discovery of an optimal, well defined, air-stable mono-FI cobalt(II) precatalyst structure.
Keywords: catalysis, cobalt, cross-coupling, Suzuki–Miyaura, phenoxy(imine)
Graphical Abstract
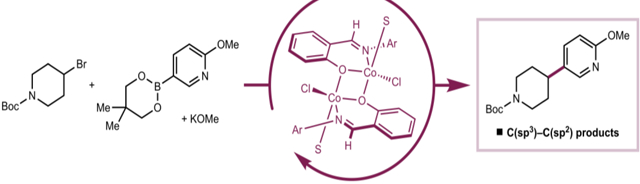
INTRODUCTION
Transition metal–catalyzed cross coupling is an enabling and widely applied technology for the synthesis of pharmaceuticals, agrochemicals and fine chemicals.1 Among the many variants used for C(sp2)–C(sp2) bond formation, the Suzuki–Miyaura reaction is the most widely used in medicinal chemistry,2,3 and is by far the most common type of cross-coupling employed on scale for the synthesis of drug candidates.1 The bench-stability and availability of the coupling partners, particularly neutral organoboron nucleophiles; the reliability and scalability with advanced intermediates; and the mild reaction conditions, distinguishes the Suzuki–Miyaura reaction from other catalytic cross-coupling methods.1,4
Evolution of the metal-catalyzed Suzuki-Miyaura cross coupling to include C(sp3)-based partners is of interest in drug discovery because increasing three-dimensionality that has been linked to the performance and success of drug candidates.5,6 First-row transition metals offer considerable promise for C(sp2)–C(sp3) cross coupling owing to a lower propensity for competitive β-hydrogen elimination when compared to palladium catalysts.7,8 Examples with copper have been demonstrated but are principally limited to primary alkyl electrophiles.9,10 Nickel catalysts have been widely explored for this transformation,11,12,13,14 however widespread adoption has thus far been limited.15
Cobalt is appealing as a catalyst for this transformation because it is Earth-abundant, relatively inexpensive, and available in quantities that are amenable to large-scale pharmaceutical applications.16,17,18 Cobalt catalysis would also provide an alternative in the synthetic toolbox for the installation of C(sp2)–C(sp3) bonds. Cobalt catalysts have demonstrated promosing reactivity with typically “unactivated” alkyl (pseudo)halide electrophiles in related couplings employing more reactive organomagnesium and organozinc nucleophiles.19,20,21,22 However, unlike other first-row metals like nickel, cobalt-catalyzed Suzuki–Miyaura coupling remains in its infancy.
Examples of cobalt-catalyzed C(sp2)–C(sp2) cross-coupling with organoboron nucleophiles have been reported using select neutral ligands, including bis(phosphino)pyridine pincers,23 terpyridine chelates,24 and aryl-substituted N-heterocyclic carbenes (Scheme 1A).25,26 The understanding of ligand-to-metal complexation has significant implications on the reproducibility of a cross-coupling reaction, and demonstrates the inherent difficulty in cross-coupling reaction development. More broadly, understanding catalytically relevant oxidation states, the nature of the active species, and redox cycles in cobalt-catalyzed cross-coupling lags behind iron- and nickel-catalyzed variants.27,28
Scheme 1.

Cobalt-catalyzed C(sp2)–C(sp3) Suzuki–Miyaura cross-coupling
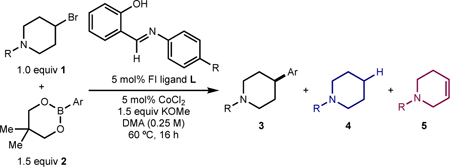
Our laboratory recently reported that the combination of cobalt(II) salts with trans-N,N’-dimethylcyclohexane-1,2-diamine in the presence of KOMe was catalytically active for the C(sp2)–C(sp3) Suzuki–Miyaura cross-coupling between neopentylglycol (hetero)arylboronic esters and alkyl bromides (Scheme 1B).29 While the scope of the transformation included sterically and electronically diverse aryl boronate nucleophiles, the catalyst loadings were high (15 mol%), and little is known about metal-ligand interactions, the origin of the effects of the specific diamine substituents, or the mechanism of the C–C bond-forming event.4 For the development of new catalytic reactions with improved performance, an understanding of metal–ligand interactions and reactivity is a key aspect of successful reaction design.30,31,32
Here we describe the application of readily prepared and modular phenoxyimine (FI) ligands to next-generation cobalt catalysts for C(sp2)–C(sp3) Suzuki–Miyaura cross-coupling (Scheme 1C). These compounds operate with lower cobalt loadings (5 mol%) and enable broader substrate scope, with lower amounts of side-products arising from alkyl bromide reduction. Substrates with known radical clocks support the intermediacy of radicals derived from the alkyl electrophile, which are stabilized by the [(FI)Co] complexes to a higher degree compared to complexes with diamine ligands. X-ray crystallographic and EPR spectroscopic studies provided insight into the metal–ligand interaction in cobalt precatalysts as well as to identify deactivation pathways of cobalt(II) sources.
RESULTS AND DISCUSSION
Part I. Discovery and Evaluation of the Methodology.
The discovery of next generation cobalt catalysts for C(sp2)–C(sp3) Suzuki–Miyaura reaction began with high-throughput experimentation (HTE)33 to evaluate classes of ligands (Table 1). A representative catalytic reaction was selected using N-Cbz-4-bromopiperidine 1a (1 equiv) as a prototypical electrophile with pharmaceutical relevance,34 neopentylglycol phenylboronic ester 2a (PhB(neo)) (1.5 equiv) as the nucleophilic partner, and potassium methoxide (1.25 equiv, according to the previously method) as the base.29 Various solvent classes including amide, nitrile, ester, aromatic, ether, and protic/aprotic, cobalt(II) sources such as CoCl2•6H2O or CoBr2, and ligand classes were evaluated. Reaction success was measured according to relative area percent of C(sp2)–C(sp3) cross-coupled product 3a as determined by ultra-performance liquid chromatography–mass spectrometry (UPLC-MS).35 A key design feature in the screening procedure was the use of 5 mol% of the cobalt(II) source, significantly lower than the 15 mol% loading required for the previously reported cobalt–diamine catalyst.29 Among the evaluated ligand classes, ligands previously reported for cobalt-catalyzed cross-coupling such as bipyridine-/terpyridine-type ligands24,26a,36 or phosphines (see SI) were generally inadequate (Table 1).16 Consistent with previous studies,29 the use of diamine-type ligands generally resulted in modest to good yield of 3a (up to 48% UPLCMS area) (Table 1).20,23,37 Another class of ligand that exhibited promising performance was 8-hydroxyquinolines (QNOL), which gave the desired product ( 3a ) in up to 42% UPLCMS area (Table 1).38 Notably, this class of ligands likely forms “L,X-type” interactions where deprotonation of the phenol generates an anionic donor (vide infra). Related N,O ligands such as amino alcohols have also been successful in nickel-catalyzed Suzuki–Miyaura arylation reactions of unactivated alkyl electrophiles.39
Table 1.
Evaluation of ligands by high-throughput experimentation (HTE)a
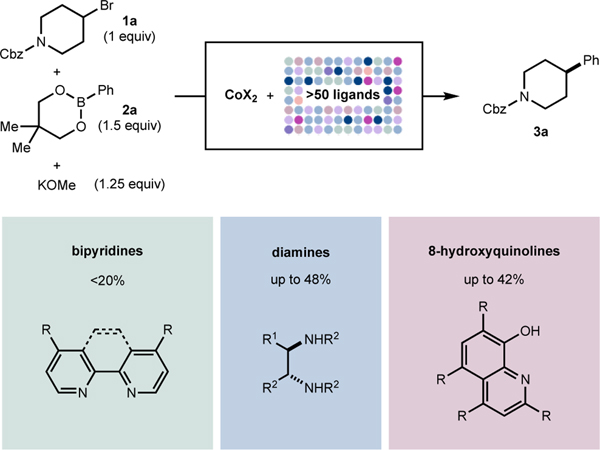
|
Reactions performed on 0.010-mmol scale using CoX2 (5 mol %) and various polar aprotic solvents at 60 °C. Yields represent relative UPLC-MS area percent. See SI for details.
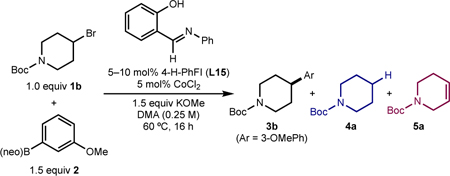
With this preliminary ligand data in hand, a more comprehensive ligand comparison was performed in batch on 0.25-mmol scale (Table 2A). Reactions were conducted using N-Boc-4-bromopiperidine (1b), 3-methoxyphenylboronic acid neopentylglycol ester (2b), CoCl2 (5 mol %), and KOMe (1.5 equiv) in DMA at 60 °C, and analyzed by GC to determine the yield of desired product (3b) as well as the yields of alkyl bromide-derived side-products, namely reduction (4a) and elimination (5a). The head-to-head comparison of the preferred diamine ligand from the first-generation catalyst, DMCyDA (L1) and the potential “L,X-type” 2,5,7-tri-Me-QNOL (L2) demonstrated that both ligands delivered the desired product (3b) in comparable yield, within GC-FID error (57% and 61%, respectively) (Table 2B). Notably, the distribution of side products arising from catalysts derived from L1 or L2 differed, with L1 forming 4a and 5a in 18% and 13% yield, respectively, and L2 forming these side-products in 4% and 29%, respectively. These data suggest that the QNOL-based ligand is more effective in suppressing formation of the reduced side-product (4a) and that formation of 4a is likely catalyst-dependent (i.e., dependent on (i) the rate of cobalt-catalyzed product formation and (ii) the ability of the cobalt catalyst to “stabilize” cage-escaped radicals; vide infra). By contrast, formation of 5a is likely a background reaction arising from interaction of the base with the electrophile that is independent of the cobalt catalyst (vide infra). These results suggested that L,X-type ligands may be able to maintain high selectivity for 3b versus 4a, and that in an optimized case, could accelerate the reaction rate to outpace background elimination (5a).
Table 2.
Optimization of ligand structurea
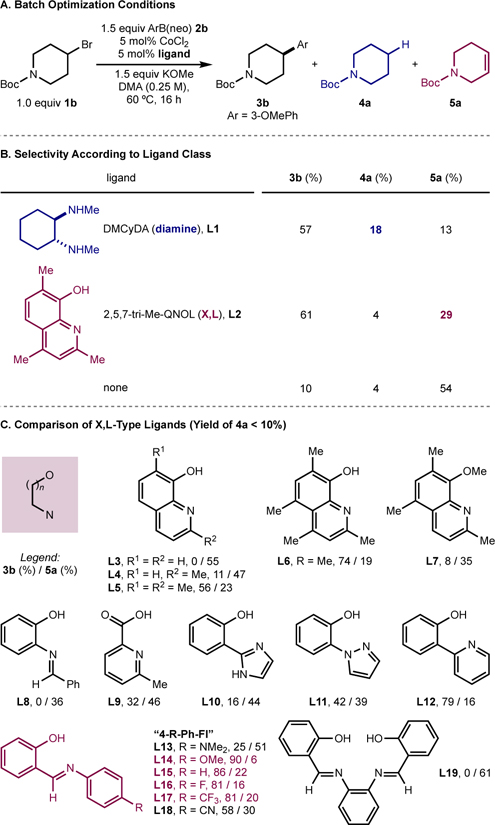
|
Reactions performed on 0.10-mmol scale using 5 mol% CoCl2 and 5 mol% ligand; yields determined by GC-FID using n-dodecane as an internal standard. Mass balances >100% reflect error of GC yields. DMA = N,N-dimethylacetamide; neo = neo-pentylglycol.
Based on these findings, other structurally similar and potential L,X-type ligands were explored and evaluated for the yield of desired product (3b) relative to elimination byproduct (5a) (Table 2C). Within the series of QNOL ligands, minimal formation of the reduced side product (4a) was consistently observed (typically <5%; in all cases <10%, see SI) with the remaining mass balance usually being unreacted starting material. QNOL ligands without substituents at the 2- and 7-positions performed poorly (L3, L4) while 2,7-diMe-QNOL (L5) displayed good reactivity (56%). 2,4,5,7-TetraMe-QNOL (L6) performed better than 2,5,7-triMe-QNOL (L2) (74% vs. 61%). Methylation of 5,7-diMe-QNOL (L7) resulted in comparable yield (8%) and selectivity to the background reaction in the absence of ligand (Table 2B), supporting the role of phenol deprotonation for improved catalytic performance.
Evaluation of other potential L,X-type ligands was conducted to determine the general role of this structural type on catalytic performance and to explore if ligand more readily synthesized and modular than QNOL were effective. A benzylidene-ortho-aminophenol (L8) produced no cross coupled product (0%) and a pyridine-2-carboxylate (L9) furnished the desired product in modest yield (32%). While ortho-2-imidazolylphenol (L10) and ortho-pyrazolylphenol (L11) did not outperform QNOL ligands, ortho-2-pyridinylphenol (L12) gave the desired product in an improved 79% yield by GC-FID. Inspired by reports in the polymerization literature that phenoxy-imine-based (FI) ligands performed similarly to QNOLs,40 the former class of ligands was explored for cobalt-catalyzed C(sp2)–C(sp3) Suzuki-Miyuara cross coupling. This ligand class has been previously studied in the context of cobalt-catalyzed C(sp2)–C(sp2) Suzuki–Miyaura cross-coupling,41 and modest reactivity with yields up to 47% as judged by GC-MS using activated lithium borate nucleophiles were reported.42
FI ligands derived from strongly electron-donating (L13) or electron-withdrawing (L18) anilines performed poorly for C(sp2)–C(sp3) coupling; however a range of FI ligands with electron-neutral or moderately electron-withdrawing N-aryl substituents (L14–L17)43 yielded product 3b with high yield (81–90%) and selectivity. Under these same conditions, a control reaction in the absence of CoCl2 resulted in full conversion to side-product 5a, indicating that formation of 5a arises from background elimination rather than β-hydride elimination from a cobalt complex with an FI ligand (see SI). Notably, extension of the FI-type ligand to the related tetradentate salphen44 ligand class (L19) resulted in no formation of cross coupled product. Variations to the FI were also explored on the phenol ring; however no change to steric or electronic structure of the phenol resulted in a discernable improvement in performance (see SI).
To gain insight into the reactivity of diamine and FI ligands under optimized conditions, experiments were performed to probe alkyl radical behavior (Scheme 2). Radical cyclization was examined (Scheme 2A). Namely, for a cage-escaped radical derived from the activation of 6-bromo-1-hexene (1c), the proportion of linear (L) (6) to cyclized (C) (7) cross-coupled product will reflect the lifetime of the free radical in solution, because the radical can undergo 5-exo-trig cyclization (kcyclization ≃ 1 × 106 s−1)45,46 to yield the cyclized intermediate. In related experiments, Biswas and Weix47 and Shenvi and coworkers48 demonstrated that, in the presence of a nickel or cobalt catalyst that can recombine with an alkyl radical, the proportion of radical cyclization will change as a function of catalyst loading; higher concentrations of catalyst tend to result in more recombination and less 5-exo-trig radical cyclization, according to the recombination-propensity of the catalyst. Employing either 4-CF3-Ph-FI (L17) or DMCyDA (L1), L/C ratios were measured with varying catalyst concentrations (2–20 mol%) and a 1:1 CoCl2:ligand ratio (Scheme 2A). In both cases, a positive slope was obtained, indicating that both L17- and L1-supported cobalt catalysts undergo recombination with cage-escaped alkyl radicals as a function of catalyst loading. However, the L17 data set ratios were shifted upwards compared to those for L1 across the same catalyst concentrations (b = 0.112 and 0.049, respectively), indicating that radical capture is more efficient with L17 than with L1 under otherwise identical conditions. It should be noted that a coordination–insertion mechanism cannot be excluded based on these observations.
Scheme 2. Experiments to probe the intermediacy of alkyl radicals.

aSee SI for details; bDetermined by GC-FID; cDetermined by 1H NMR spectroscopy.
These data suggest that an important catalytic feature of L,X-type FI ligands versus diamine ligands is the ability to productively capture alkyl radicals, an advantageous property for reactions involving C(sp3) coupling partners.49 In other chemistry, FI ligands are common for reactions involving alkyl partners, e.g. nickel-catalyzed ethylene polymerization.50 Related chelating ligands with X and L donors for cobalt, such as salen or cobaloxime, are known to participate in recombination with alkyl radicals to act as stabilizing “reservoirs” of alkyl fragments in catalytic reactions.48,51 The reduced side product (4) likely forms from a cage-escaped radical (R•) that undergoes HAT from solvent (SH) to yield RH and S•. Yield of 4a (Table 2) is expected to be dependent on radical lifetime and likely the origin as to why FI ligands (L14–L17) tend to produce lower quantities of the reduced side-product than DMCyDA.
The formation of alkyl radicals was corroborated with a cyclopropylcarbinyl radical probe (Scheme 2B). When bromide 1d was used as the substrate, the corresponding arylated product (8) was formed in moderate yield with exclusive selectivity for ring-opening.
With an optimized metal-ligand combination and reaction conditions, the scope of the cobalt-catalyzed C(sp2)–C(sp3) Suzuki-Miyaura cross coupling was explored (Table 3). Standard conditions employed 5 mol% of each of the FI ligand and CoCl2 along with 1.5 equivalents of KOMe in 0.25 M DMA at 60 °C. Notably, FI ligands of interest were routinely prepared from commodity chemicals on decagram scale by condensation and collection by filtration, leading to large quantities of bench-stable materials (Table 3, photos). For alkyl bromide coupling partners, a variety of secondary substrates were tolerated, including hydrocarbon (3e–3f, 3h), ether-containing (3d, 3j, 3l, 3n), and carbamate-containing (3c, 3g, 3i, 3m) substrates. A TBS-protected alcohol was compatible (3k). Primary electrophiles were also compatible (3o–3q), including an acyclic acetal (3p), albeit with higher catalyst loadings (10 mol% each of CoCl2 and L14), which resulted in less formation of the elimination side-product compared to the standard 5 mol% loading. Substrates where elimination was facile (3r) yielded lower amounts of product. Among compatible alkyl substituents, a few are also of note due to their beneficial physiochemical properties,52 namely oxetane (3l),53 azetadine (3m),54 and an oxaspirocycle (3n).55
Table 3.
Scope of the cobalt-catalyzed C(sp2)–C(sp3) Suzuki-Miyaura cross coupling reactiona
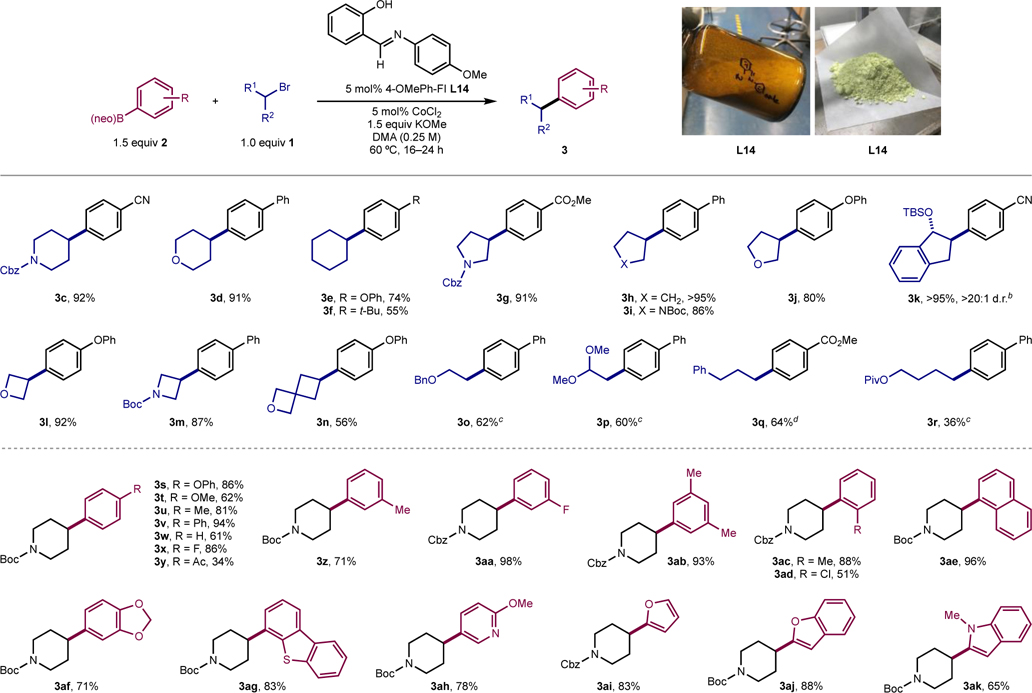
|
With respect to arylboronic esters, the reaction was compatible with electron-donating (3s, 3t), electron-neutral (3u–x, 3z, 3ab) and electron-withdrawing aryl substituents (3c, 3g, 3k, 3q, 3aa). A ketone-containing substrate furnished the coupled product in 34% yield (3y). Substrates with one ortho substituent (3ac–3ad) were compatible, including an ortho-chloro-substituted arylboronic acid (3ad). A cyclic-acetal-substituted aryl nucleophile gave the desired product in good yield (3af). Extended aromatic substrates (3ae, 3ag) also worked. Some heteroarylboronic esters were also viable, including 3-pyridyl (3ah), 2-furanyl (2ai), 2-benzofuranyl (2aj), and N-Me 2-indolyl (3ak). Substrates incompatible with the current method and gave low yields (<10%) of product and include those bearing functional groups such as benzaldehyde, primary alcohol, and phthalimide, among others (see SI for specific examples).
The method was also viable for gram-scale synthesis (Scheme 3). With 37.5 mmol of arylboronic ester 2b and 25 mmol of bromocyclohexane 1d, standard reaction conditions were employed with the modifications that instead of CoCl2, 5 mol% of less hygroscopic CoBr2 was used to allow weighing on benchtop, and that the reaction was stirred in an 80 °C oil bath to ensure adequate heat-transfer to the round-bottom flask (cf. Table S9). These conditions yielded 3.37 g of the desired cross-coupled product 3al, representing 71% yield.
Scheme 3.

Application of the method on 25-mmol scale
Part II. Synthesis of Phenoxyimine Cobalt Complexes and Relevance to Catalysis.
The improved catalytic performance of the phenoxy-imine based cobalt catalysts prompted more detailed studies into the coordination chemistry of this ligand class, elucidation of the importance of the “L,X” interaction, and understanding the oxidation state of the cobalt precursor. These studies were of importance as results initially obtained when translating reactions with FI ligands from HTE to batch were often variable. Specifically, the productivity and yield of the reactions depended on the order of addition of the reagents (Table 4). Optimized performance was observed when the aryl boronate (2) was premixed with the FI ligand (L) and KOMe, followed by mixing with CoCl2 for 1–2 minutes, then addition of alkyl bromide (1) and immediate heating of the reaction mixture. Using this procedure, the cross-coupled product (3) was obtained in 83% yield as judged by GC after 16 hours (Entry 1).
Table 4.
Impact of the order of addition of reagents on yield and selectivity for cobalt-catalyzed cross coupling in batch and HTEa
| ||||||
|---|---|---|---|---|---|---|
| Entry | Setup | Order of addition to DMA solution | 1 (%)b | 3 (%)b | 4 (%)b | 5 (%)b |
|
| ||||||
| 1 | batch | (i) 2 + L + KOMe; (ii) Cocl2; (iii) 1 | 0 | 83 | 2 | 20 |
| 2 | (i) L + CoCl2; (ii) 2 + KOMe; (iii) 1 | 8 | 2 | 0 | 87 | |
| 3 | (i) 2 + L + KOMe + CoCl2; (ii) 1 | 27 | 41 | 2 | 44 | |
| 4 | (i) KOMe + L;c (ii) 2; (iii) CoCl2; (iv) 1 | 8 | 83 | 2 | 11 | |
|
| ||||||
| 5d | HTE | (i) L + CoCl2; (ii) 1 + 2; (iii) KOMee | 4 | 43 | 1 | 23 |
See SI for details
Determined by GC-FID
Using preformed FI-phenoxide
Performed by HTE, yields represent relative area percent as determined by UPLC-MS
Using KOMe (1.25 equiv) prepared from MeOH (2 equiv) and KHMDS (1.25 equiv).
This order of addition was optimal as premixing 2 and KOMe establishes an equilibrium with the borate species (eq. 1, above), minimizing competing elimination upon addition of the electrophile. If L and CoCl2 were premixed prior to the addition of 2 and KOMe, minimal yield (~2%) of 3 was observed with the elimination product 5 accounting for most of the mass balance (entry 2). Combination of all of the reagents prior to addition of 1 also resulted in a reduced yield of 41% (Entry 3). Stirring KOMe and L to promote deprotonation also resulted in an 83% yield, identical to the optimized conditions (Entries 4 and 1). It should be noted that in HTE procedures, the KOMe base was added last and a lower yield of 43% was obtained (Entry 5). Given the acidity of the FI ligand (pKa of MeOH = 29.0 in DMSO56; pKa of ArOH ≃ 13–19 in DMSO57), it was expected that KOMe would promote rapid deprotonation. Experimentally, mixing 4-F-Ph-FI (L16) with KOMe DMSO-d6 resulted in >95% conversion to the anion as judged by 19F NMR spectroscopy.58
With a reproducible set-up for the reaction, the impact of the L:CoCl2 ratio on the performance and reproducibility of the catalytic cross coupling reaction was studied (Table 5). Namely, reactions with FI ligands (e.g., 4-H-PhFI, L15) were sensitive to higher ligand loadings. For example, an optimal reaction using L15 (5 mol %, 1:1 L:CoCl2) gave the desired product in 83% yield (Entry 1), whereas a reaction using double the amount of L15 (10 mol %, 2:1 L:CoCl2) resulted in an inefficient reaction (10% yield), with the major product being elimination side-product 5a (70%) (Entry 2).
Table 5.
Conversion and selectivity according to ligand loadinga
Reactions performed on 0.10-mmol scale.
Determined by GC-FID.
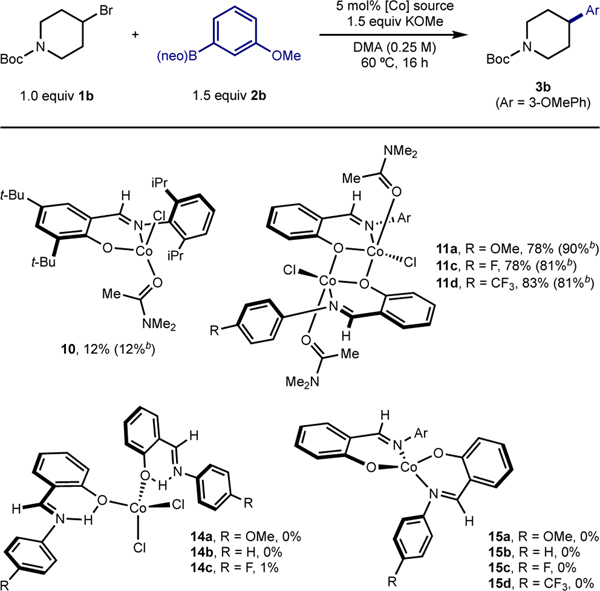
To better understand the effects of the L:CoCl2 ratio, the interactions of FI ligands with cobalt precursors were investigated. With the sterically hindered 2,6-di-i-PrPhdi-t-BuFI (9) ligand, deprotonation with one equivalent of NaH in DMA followed by addition to a DMA slurry containing one equivalent of CoCl2 produced a green solution (Scheme 4A). Stirring for 16 hours followed by treatment with Et2O and recrystallization from a DMA-Et2O mixture at −30 °C produced red crystals suitable for X-ray diffraction. A representation of the solid-state structure is shown in Scheme 4B, which identified the product of the reaction as the four-coordinate, idealized tetrahedral cobalt(II) complex (τ4 = 0.91), (2,6-di-i-PrPhdi-t-BuFI)CoCl(DMA) 10, containing a deprotonated FI ligand coordinated in an “L,X” fashion along with a molecule of DMA to complete the coordination sphere. The 1H NMR spectrum in benzene-d6 of paramagnetic 10 exhibits broad signals with chemical shifts ranging from 451 to −34 ppm.42 This is a rare example of a monomeric cobalt complex supported by an FI ligand.59,60 The structure of 10 provides insight to at least one role of the DMA solvent; namely, to serve as a ligand to stabilize the resulting [(FI)Co] precatalyst.
Scheme 4. Synthesis of (FI)Co(Cl)(DMA)a.

aSee SI for details; bRepresentation of the molecular structure of (2,6-di-i-PrPhdi-t-BuFI)CoCl(DMA) at 30% probability ellipsoids; hydrogen atoms omitted for clarity. Selected bond distances (Å) and angles (°): Co1–O1 1.9025(12), Co1–O2 1.9882(13), Co1–N1 1.9864(14), Co1–Cl1 2.2258(5); O1–Co1–N1 96.12(5), Cl1–Co1–O2 118.34(4).
Compound 10 was stable in DMA solution for months as determined by 1H NMR spectroscopy but decomposed upon attempted isolation. Removal of the solvent by high-vacuum resulted in decomposition of 10 after 6 hours. Dilution of the DMA stock solution with MeCN, MeOH, CDCl3 or benzene-d6 also produced decomposition of 10 within 1 hour as determined by 1H NMR spectroscopy, likely a result of DMA dissociation to form an unstable three-coordinate cobalt compound. Attempted synthesis of 10 using stoichiometric DMA in THF as the solvent resulted in isolation of the corresponding bis(chelate) compound, (2,6-di-i-Pr-Phdi-t-BuFI)2Co, which was characterized by independent synthesis (see SI). This bis(chelate) species was also identified as a side-product formed during the synthesis of 10 that was obtained in variable amounts (0–20%).
The synthesis of related compounds with catalytically optimal ligands L14–L17 was targeted (Scheme 5). Deprotonation of ligands L14–L17 was accomplished by addition of NaH, followed by treatment with CoCl2 and a stoichiometric quantity of DMA.61 The resulting reaction mixtures were stirred for 16 hours, and green solids identified as 11a–d were isolated. Single crystals were obtained by vapor diffusion recrystallization of 11c (R = F) from DMF–Et2O and established the formation of a dimeric cobalt complex, [(4-F-Ph-FI)CoCl(DMF)]2 (12c) (Figure 1). Each subunit contained an idealized trigonal bipyramidal cobalt (τ5 = 0.55) ligated by an L,X FI ligand, a chloride and a molecule of DMF. A bridging oxygen from an adjacent FI ligand and the imine nitrogen of the FI define the axial positions.62 The presence of DMF in the crystal structure also established the lability of the coordinating solvent.
Scheme 5. Preparation of [(FI)CoCl(DMA)]2.

aInseparable from bisligand chelate (4-F-PhFI)2Co (15c). Sample represents an 80:20 molar ratio of 11c:15c; yield is with respect to desired compound.
Figure 1.

Representation of the molecular structure of [(4-F-Ph-FI)CoCl(DMF)]2 12 with 30% probability ellipsoids; hydrogen atoms omitted for clarity. Selected bond distances (Å) and angles (°): Co1–O1 1.9609(12), Co1–O2 2.0473(11), Co1–O1’ 2.1537(12), Co1–N1 2.0651(13), Co1–Cl1 2.3069(9); O1–Co1–N1 92.31(4), Cl1–Co1–O2 113.77(3).
The corresponding dimers with different aniline para-substituents (OMe, H, F, CF3, 11a–d respectively) were obtained in comparable yield (34–38%) (Scheme 5). These compounds were stored on benchtop and were stable to air and moisture for months. The remaining mass balance for these reactions was attributed to formation of bis-FI chelates, namely (FI)2Co complexes (vide infra), which were removed from the desired dimer by washing with Et2O or PhMe. The formation of (FI)2Co under these conditions may be partially attributed to the poor solubility of CoCl2 in THF. A coordinating solvent (e.g., DMSO-d6) could be used instead of THF to improve the yield of dimeric mono-FI compounds 11a–11d as determined by 1H and 19F NMR (vide infra), highlighting the importance of coordinating solvent for the synthesis and stability of mono-FI cobalt(II) compounds.
The X-band EPR spectra of the dimeric cobalt compounds 11a–d were recorded in DMA glass at 8 K and exhibited rhombic signals (Figure 2). The simulation parameters were indistinguishable for each entry in the series (Table 6) with notably large g1 values (5.07–5.29).63 In the solid state, magnetic moments of 6.6–7.1 μB were obtained for the dimers (Table 6).64 Together, the X-ray, EPR and magnetic moment data all support high-spin (S = 3/2) cobalt(II) for 11a–d.
Figure 2.

X-band EPR spectra of compounds 11a, 11b, and 11d recorded in DMA glass at 8 K. Black = experimental; red = simulated. Collection parameters for 11a: microwave frequency = 9.363 GHz, power = 2.0 mW, modulation amplitude = 4 G. Collection parameters for 11b: microwave frequency = 9.364 GHz, power = 0.06 mW, modulation amplitude = 4 G. Collection parameters for 11d: microwave frequency = 9.367 GHz, power = 0.6 mW, modulation amplitude = 4 G.
Table 6.
EPR simulation parameters and solid-state magnetic moment data for compounds 11a–d
| EPR Simulation Parameters |
|||||||
|---|---|---|---|---|---|---|---|
| Compound | FI 4-Ph-N Substituent | S | g 1 | g 2 | g 3 | g strain | μeff (μB) |
| 11a | OMe | 3/2 | 5.07 | 1.71 | 3.39 | (1.50, 0.71,2.58) | 7.02 |
| 11b | H | 3/2 | 5.13 | 1.63 | 3.29 | (1.38, 0.58, 2.60) | 7.13 |
| 11d | CF3 | 3/2 | 5.29 | 1.76 | 3.06 | (1.45, 0.79, 2.22) | 6.59 |
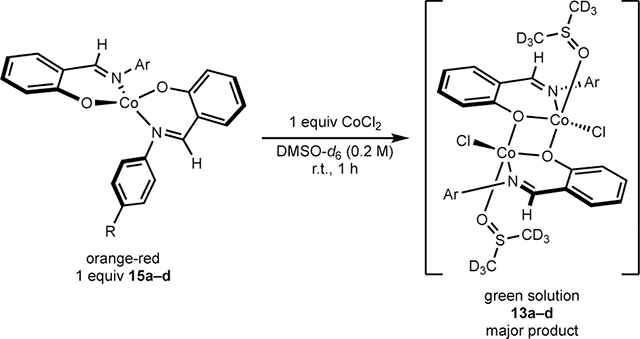
1H and 19F NMR spectroscopy were used to correlate isolated dimer 11d (R = CF3) with the cobalt compounds generated in situ during precatalyst formation (Scheme 6). Deprotonation of L17 (R = CF3) with one equivalent of KOMe in DMSO-d6 followed by addition of one equivalent of CoCl2 and monitoring the reaction by 1H and 19F NMR spectroscopies established dimeric cobalt compound, 13d as the major product (Scheme 6). Repeating this experiment with L16 (R = F) using C6H5F as internal standard, also resulted in formation of the corresponding dimer in 80% 19F NMR yield (see SI). Together, these data suggest [(FI)CoCl(DMA)]2 is likely the relevant precatalyst for the cobalt-catalyzed C(sp2)–C(sp3) Suzuki–Miyaura cross-coupling procedure.
Scheme 6.

In situ formation of FI–cobalt(II) compound
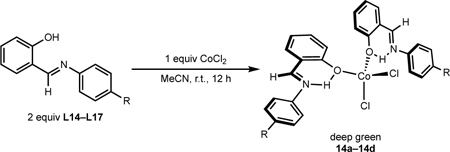
The synthesis of bis(FI) cobalt(II) complexes was also explored. Stirring two equivalents of neutral ligands L14–L16 with CoCl2 yielded deep green solids assigned as neutral FI coordination complexes, namely (H-FI)2CoCl2 compounds (14a–14c) (Table 7). For compounds 14a–14c (R = OMe, H, F, respectively), moderate to excellent yields of the desired deep green compounds were obtained (50–95%). For 14d (R = CF3), free ligand (L17) was obtained as the major product, potentially due to the weaker donor ability of the electron-deficient salicylaldimine. Employing L14 and CoCl2•6H2O and stirring in refluxing EtOH yielded the same product (4-OMe-H-FI)2CoCl2, 14a (80%) (see SI).41,42 The solid-state structure of 14a was confirmed by X-ray crystallography (Figure 3), establishing an idealized tetrahedral geometry at the metal (τ4 = 0.89). Solid-state magnetic data were collected for the isolated compounds and values of μeff between 4.1 and 4.3 μB were obtained (Table 7), consistent with high spin (S = 3/2) cobalt(II) center and the observed solid-state structure of 14a. Attempts to prepare similar neutral bis(chelate) cobalt compounds using FI ligands bearing substituents ortho to the phenol resulted in recovery of free ligand, likely due to steric interference preventing O–Co coordination.
Table 7.
Bis(chelate) Cobalt Compounds, (H-FI)2CoCl2 Compounds 14a–d with Protonated Ligands.
| |||
|---|---|---|---|
| Compound | H-FI 4-Ph-N Substituent | Yield (%)a | μeff (μB)b |
| 15a | OMe | 95 | 4.33 |
| 15b | H | 95 | 4.32 |
| 15c | F | 50 | 4.14 |
| 15d | CF3 | <20 | n.d. |
Isolated yields.
Measured in the solid state.
Figure 3.

Representation of the molecular structure of (4-OMe-Ph-H-FI)2CoCl2 14a at 30% probability ellipsoids; hydrogen atoms omitted for clarity. [H1] and [H2] have been located. Selected bond distances (Å) and angles (°): Co1–O1 1.9596(12), Co1–O2 1.9432(12), Co1–Cl1 2.273(6); O1–Co1–O2 117.79(5), Cl1–Co1–Cl2 112.43(14).
The 1H NMR spectra of (H-FI)2CoCl2 compounds 14a–c in DMSO-d6 did not exhibit signals in the paramagnetic region of the spectrum. Instead, slightly broadened signals nearly identical to the free ligand were observed (see SI). These observations support that in the presence of coordinating solvent, (H-FI)2CoCl2 compounds 14a–c are in equilibrium with free ligand and solvated [CoCl2]x(S)y. These bis(ligated) cobalt compounds (14a–14c) were insoluble in noncoordinating solvents, prohibiting further characterization in solution.
The synthesis of bis(chelate) cobalt(II) derivatives containing anionic FI ligands, (FI)2Co (15a–d), was accomplished by deprotonating ligands L14–L17 with NaH, followed by addition of CoCl2 (Scheme 7A). (FI)2Co compounds 15a–d were paramagnetic orange-red solids, consistent with known (FI)2Co compounds.62,63,65 A bulkier derivative derived from ligand 9 was also synthesized using the same procedure (see SI).
Scheme 7. Synthesis of (FI)2Co Compounds (15a-d).

aSolid-state structure of (4-OMePhFI)2Co 15a at 30% probability ellipsoids; hydrogen atoms omitted for clarity. Selected bond distances (Å) and angles (°): Co1–O2 1.8998(11), Co1–N1 2.0039(12); O1–Co1–N2 96.96(5). bX-Band EPR spectrum of compound 15a at 10 K in DMA glass. Collection parameters: microwave frequency = 9.363 GHz, power = 0.02 mW, modulation amplitude = 4 G. Simulation parameters: S = 3/2, g1 = 4.21, g2 = 1.99, g3 = 4.75, gstrain = (1.20, 0.42, 1.58).
A representation of the molecular structure of 15a (R = OMe) determined by X-ray diffraction is presented in Scheme 7B. As expected from other known (FI)2Co complexes,66 an idealized tetrahedral geometry was observed (τ4 = 0.82). The series of (FI)2Co compounds (15a–d) were further characterized by low-temperature X-band EPR spectroscopy (e.g., 15a, Scheme 7C; see SI). In each case, rhombic spectra were obtained, consistent with metal-centered radical character. Together, these data support these complexes being high-spin (S = 3/2) cobalt(II).63
The role of base was evaluated with respect to the protonation state of the FI chelate as premixing the ligand and CoCl2 before addition of KOMe was detrimental for catalysis (cf. Table 4, Entry 2). The 4-F-Ph-FI ligand (L16) was added to CoCl2•6H2O and stirred in refluxing EtOH for 12 h (Scheme 8). This produced a heterogeneous mixture composed of a green EtOH solution and green precipitate, indicating formation of 14c, consistent with observations made during the independent synthesis of this class of compounds (eq. 2). Subsequently, two equivalents of KOH were added, and the reaction was stirred at 80 °C for 6 hours. This resulted in the formation of 15c, obtained in 47% isolated yield, confirming that 14c was converted to 15c in the presence of base.
Scheme 8.

Conversion of (H-FI)2CoCl2 to (FI)2Co
The impact of FI:CoCl2 stoichiometry was evaluated with respect to precatalyst formation and thermodynamics in solution, as 2:1 FI:CoCl2 stoichiometry was detrimental to catalysis compared to 1:1 stoichiometry (cf. Table 5, entries 3–4). Namely, the series of (FI)2Co compounds 15a–d was separately added to one equivalent of CoCl2 in DMSO-d6 and stirred at room temperature for one hour (eq. 1). Analysis of the reaction mixtures by 1H NMR spectroscopy revealed the mono-FI dimers 13a–d to be the major components in all cases, and (FI)2Co chelates 15a–d to be minor components. Analysis by 19F NMR spectroscopy for the reactions employing 15c (R = F) and 15d (R = CF3) revealed 63% and 88% conversion to the mono-FI compounds 13c and 13d, respectively. These data indicate that FI:CoCl2 stoichiometry in a coordinating solvent like DMSO-d6 affects the formation of mono- versus bis-FI cobalt(II) compounds. Empirically, the results in DMSO-d6 contrast with the protocol used to synthesize [(FI)CoCl(DMA)]2 compounds 11a–d that utilizes THF as solvent, for which a significant product is undesired (FI)2Co (Scheme 6). These data imply that coordinating solvent like DMA may help favor mono-FI cobalt intermediates, especially in catalysis.
 |
(1) |
The competence of the isolated [(FI)Co] coordination complexes in the C(sp2)–C(sp3) Suzuki–Miyaura cross-coupling reaction of 1b and 2b was evaluated using 5 mol% of each isolated cobalt compound (Table 8). These reactions were set up and performed on the benchtop using standard Schlenk techniques; besides 10, all complexes were air-stable over a period of months. Modest cross coupling reactivity was observed for reactions conducted with a stock solution of model complex 10 in DMA under otherwise standard catalytic conditions. The 12% yield of 3b using 10 was the same as when the in situ catalyst generation method was used, highlighting the detrimental effect of bulky substituents ortho to the FI phenol moiety, with respect to catalytic performance; the remaining mass balance in this reaction was predominantly elimination (5b). Dimers [(FI)CoCl(DMA)]2, 11a–d, were all competent in catalysis (78–95% yield), consistent with the yields for in situ precatalyst generation. These data support that the isolated cobalt compounds lead to on-cycle species upon exposure to the components of the cross-coupling reaction. In contrast, the bisligated (H-FI)CoCl2 compounds with neutral H-FI ligands, 14a–c, performed poorly in catalysis. Finally, with chelate complexes bearing two deprotonated FI ligands (15a–d), no turnover was observed.
Table 8.
Performance of isolated phenoxyimine cobalt complexes in catalytic C(sp2)–C(sp3) cross couplinga
|
Yields determined by GC-FID using n-dodecane as internal standard. The designation of “5 mol%” is with respect to equiv of [Co] (5 mol% compound loading for monomers, 2.5 mol% compound loading for dimers 11a, 11c, and 11d)
Using 5 mol% of the corresponding FI ligand and 5 mol% CoCl2 instead of the isolated cobalt complex.
From the experimental observations and synthesis and evaluation of isolated compounds, a unified picture for pathways leading to active and inactive precatalysts is presented in Scheme 9. Neutral FI ligands (L14–L17) undergo deprotonation with KOMe to yield potassium phenoxides 16a–d. Treatment of phenoxides 16a–d with one equivalent of CoCl2 generates [(FI)CoCl(DMA)]2 11a–d, which are active precatalysts for C(sp2)–C(sp3) Suzuki–Miyaura cross-coupling. Treatment of ligands L14–L17 with half an equivalent of CoCl2 prior to deprotonation results in an equilibrium with neutral (H-FI)2CoCl2 compounds 14a–d in coordinating solvent such as DMSO-d6. Compounds 14a–d do not produce active catalysts as treatment with KOMe resulted in rapid formation of (FI)2Co complexes 15a–d that are not catalytically competent. The bis(chelate) cobalt complexes 15a–d were also formed if excess phenoxide (16a–d) is added to CoCl2 (middle arrow) or to dimers [(FI)CoCl(DMA)]2 11a–d. However, the treatment of (FI)2Co chelates 15a–d with stoichiometric CoCl2 in a coordinating solvent yields the dimers [(FI)CoCl(DMA)]2 11a–d, albeit with lower conversion than the direct treatment of CoCl2 to phenoxides 16a–d. Together, this picture highlights the importance of coordinating solvent and of the FI:Co stoichiometry to access precatalysts for cobalt-catalyzed C(sp2)–C(sp3) Suzuki–Miyaura cross-coupling.
Scheme 9.

Formation of catalytically active and inactive phenoxy(imine) cobalt complexes.
CONCLUDING REMARKS
A readily synthesized and modular class of “L,X”-type ligands for cobalt-catalyzed C(sp2)–C(sp3) Suzuki–Miyaura cross-coupling has been found to enable efficient C–C bond formation and a host of products were isolated in synthetically useful yields. A Studies into the coordination chemistry established a well-defined mono-FI cobalt(II) precatalyst and demonstrate the importance of FI:CoCl2 stoichiometry, order of addition, and coordinating solvent (DMA), which solvent acts as L-type ligand to stabilize catalytically relevant cobalt compounds.
Supplementary Material
ACKNOWLEDGMENTS
Financial support from the the National Institutes of Health (5R01GM121441) and Bristol Myers Squibb through the Princeton Catalysis Initiative is gratefully acknowledged. L.R.M. thanks the Natural Sciences and Engineering Research Council of Canada for a Banting Postdoctoral Fellowship. Amy Sarjeant and Bhupinder Sandhu are thanked for performing X-ray crystallography. Paul O. Peterson and Lauren N. Mendelsohn (both Princeton University) are thanked for assistance with low-temperature EPR studies.
Footnotes
ASSOCIATED CONTENT
Supporting Information
Reaction optimization, experimental and characterization data, supplementary figures, and NMR spectra (PDF)
X-ray crystallographic data for compound 10 (cif)
X-ray crystallographic data for compound 12 (cif)
X-ray crystallographic data for compound 14a (cif)
X-ray crystallographic data for compound 15a (cif)
X-ray crystallographic data for compound S21 (cif)
Accession Codes
CCDC deposition numbers 2110299–2110303 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
REFERENCES
- 1.Magano J; Dunetz JR Large-Scale Applications of Transition Metal-Catalyzed Couplings for the Synthesis of Pharmaceuticals. Chem. Rev. 2011, 111, 2177–2250. [DOI] [PubMed] [Google Scholar]
- 2.Miyaura N; Suzuki A Palladium-Catalyzed Cross-Coupling Reactions of Organoboron Compounds. Chem. Rev. 1995, 95, 2457–2483. [Google Scholar]
- 3.Brown DG; Boström J Analysis of Past and Present Synthetic Methodologies on Medicinal Chemistry: Where Have All the New Reactions Gone? J. Med. Chem. 2016, 59, 4443–4458. [DOI] [PubMed] [Google Scholar]
- 4.Martin R; Buchwald SL Palladium-Catalyzed Suzuki-Miyaura Cross- Coupling Reactions Employing Dialkylbiaryl Phosphine Ligands. Acc. Chem. Res. 2008, 41, 1461–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Lovering F; Bikker J; Humblet C Escape from Flatland: Increasing Saturation as an Approach to Improving Clinical Success. J. Med. Chem. 2009, 52, 6752–6756; [DOI] [PubMed] [Google Scholar]; (b) Clemons PA; Bodycombe NE; Carrinski HA; Wilson JA; Shamji AF; Wagner BK; Koehler AN; Schreiber SL Small molecules of different origins have distinct distributions of structural complexity that correlate with protein-binding profiles. PNAS 2010, 107, 18787–18792; (c) Lovering, F. Escape from Flatland 2: complexity and promiscuity. Med. Chem. Commun. 2013, 4, 515–519; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Dombrowski AW; Gesmundo NJ; Aguirre AL; Sarris KA; Young JM; Bogdan AR; Martin MC; Gedeon S; Wang Y Expanding the Medicinal Chemist Toolbox: Comparing Seven C(sp2)−C(sp3) Cross-Coupling Methods by Library Synthesis. ACS Med. Chem. Lett. 2020, 11, 597–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mussari CP; Dodd DS; Sreekantha RK; Pasunoori L; Wan H; Posy SL; Critton D; Ruepp S; Subramanian M; Watson A; Davies P; Schieven GL; Salter-Cid LM; Srivastava R; Tagore DM; Dudhgaonkar S; Poss MA; Carter PH; Dyckman AJ Discovery of Potent and Orally Bioavailable Small Molecule Antagonists of Toll-like Receptors 7/8/9 (TLR7/8/9). ACS Med. Chem. Lett. 2020, 11, 1751–1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kambe N; Iwasaki T; Terao J Pd-catalyzed cross-coupling reactions of alkyl halides. Chem. Soc. Rev. 2011, 40, 4937–4947. [DOI] [PubMed] [Google Scholar]
- 8.(a) Ishiyama T; Abe S; Miyaura N; Suzuki A Palladium-Catalyzed Alkyl-Alkyl Cross-Coupling Reaction of 9-Alkyl-9-BBN Derivatives with Iodoalkanes Possessing β-Hydrogens. Chem. Lett. 1992, 21, 691–694; [Google Scholar]; (b) Kirchoff JH; Dai C; Fu GC A Method for Palladium-Catalyzed Cross-Couplings of Simple Alkyl Chlorides: Suzuki Reactions Catalyzed by [Pd2(dba)3]/PCy3. Angew. Chem. Int. Ed. 2002, 41, 1945–1947; [PubMed] [Google Scholar]; (c) Netherton MR; Fu GC Suzuki Cross-Couplings of Alkyl Tosylates that Possess β Hydrogen Atoms: Synthetic and Mechanistic Studies. Angew. Chem. Int. Ed. 2002, 41, 3910–3912. [DOI] [PubMed] [Google Scholar]
- 9.Thapa S; Shrestha B; Gurung SK; Giri R Copper-catalysed cross-coupling: an untapped potential. Org. Biomol. Chem. 2015, 13, 4816–4827. [DOI] [PubMed] [Google Scholar]
- 10.Evano G; Blanchard N, Eds. Copper-Mediated Cross-Coupling Reactions. John Wiley & Sons: Hoboken, NJ, 2014; pp. 1–798. [Google Scholar]
- 11.Zhou J; Fu GC Suzuki Cross-Couplings of Unactivated Secondary Alkyl Bromides and Iodides. J. Am. Chem. Soc. 2004, 126, 1340–1341. [DOI] [PubMed] [Google Scholar]
- 12.(a) Saito B; Fu GC Enantioselective Alkyl−Alkyl Suzuki Cross-Couplings of Unactivated Homobenzylic Halides. J. Am. Chem. Soc. 2008, 130, 6694–6695; [DOI] [PubMed] [Google Scholar]; (b) Lundin PM; Fu GC Asymmetric Suzuki Cross-Couplings of Activated Secondary Alkyl Electrophiles: Arylations of Racemic α-Chloroamides. J. Am. Chem. Soc. 2010, 132, 11027–11029; (a) Zultanski, S. L.; Fu, G. C. Nickel-Catalyzed Carbon−Carbon Bond-Forming Reactions of Unactivated Tertiary Alkyl Halides: Suzuki Arylations. J. Am. Chem. Soc. 2013, 135, 624–627; (b) Wang, J.; Qin, T.; Chen, T.-G.; Wimmer, L.; Edwards, J. T.; Cornella, J.; Vokits, B.; Shaw, S. A.; Baran, P. S. Nickel-Catalyzed Cross-Coupling of Redox-Active Esters with Boronic Acids. Angew. Chem. Int. Ed. 2016, 55, 9676–9679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.(a) Hu X Nickel-catalyzed cross coupling of non-activated alkyl halides: a mechanistic perspective. Chem. Sci. 2011, 2, 1867–1886; [Google Scholar]; (b) Tasker SZ; Standley EA; Jamison TF Recent advances in homogeneous nickel catalysis. Nature 2014, 509, 299–309; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Fu GC Transition-Metal Catalysis of Nucleophilic Substitution Reactions: A Radical Alternative to SN1 and SN2 Processes. ACS Cent. Sci. 2017, 3, 692–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.For pioneering iron-catalyzed cross-coupling reactions involving activated borate nucleophiles and unactivated alkyl electrophiles: Hatakeyama T; Hashimoto T; Kondo Y; Fujiwara Y; Seike H; Takaya H; Tamada Y; Oono T; Nakamura M Iron-Catalyzed Suzuki-Miyaura Coupling of Alkyl Halides. J. Am. Chem. Soc. 2010, 132, 10674–10676; Hatakeyama T; Hashimoto T; Kathriarachchi KKADS; Zenmyo T; Seike H; Nakamura M Iron-Catalyzed Alkyl–Alkyl Suzuki–Miyaura Coupling. Angew. Chem. Int. Ed. 2012, 51, 8834–8837; Hashimoto T; Hatakeyama T; Nakamura M Stereospecific Cross-Coupling between Alkenylboronates and Alkyl Halides Catalyzed by Iron–Bisphosphine Complexes. J. Org. Chem. 2012, 77, 1168–1173; Bedford RB; Brenner PB; Carter E; Carvell TW; Cogswell PM; Gallagher T; Harvey JN; Murphy DM; Neeve EC; Nunn J; Pye DR Expedient Iron-Catalyzed Coupling of Alkyl, Benzyl and Allyl Halides with Arylboronic Esters. Chem. – Eur. J. 2014, 20, 7935–7938; Daifuku SL; Kneebone JL; Snyder BER; Neidig ML Iron(II) Active Species in Iron−Bisphosphine Catalyzed Kumada and Suzuki−Miyaura Cross-Couplings of Phenyl Nucleophiles and Secondary Alkyl Halides. J. Am. Chem. Soc. 2015, 137, 11432–11444.
- 15.(a) Beutner GL; Simmons EM; Ayers S; Bemis CY; Goldfogel MJ; Joe CL; Marshall J; Wisniewski SR A Process Chemistry Benchmark for sp2–sp3 Cross Couplings. J. Org. Chem. 2021, 86, 10380–10396; [DOI] [PubMed] [Google Scholar]; (b) Goldfogel MJ; Guo X; Meléndez Matos JL; Gurak JA; Joannou MV; Moffat WB; Simmons EM; Wisniewski SR Advancing Base-Metal Catalysis: Development of a Screening Method for Nickel-Catalyzed Suzuki−Miyaura Reactions of Pharmaceutically Relevant Heterocycles. Org. Process Res. Dev. 2021, Articles ASAP. DOI: 10.1021/acs.oprd.1c00210. [DOI] [Google Scholar]
- 16.Cahiez G; Moyeux A Cobalt-Catalyzed Cross-Coupling Reactions. Chem. Rev. 2010, 110, 1435–1462. [DOI] [PubMed] [Google Scholar]
- 17.Garrett CE; Prasad K The Art of Meeting Palladium Specifications in Active Pharmaceutical Ingredients Produced by Pd-Catalyzed Reactions. Adv. Synth. Catal. 2004, 346, 889–900. [Google Scholar]
- 18.For a discussion of comparative transition metal toxicities: Egorova KS; Ananikov VP Toxicity of Metal Compounds: Knowledge and Myths. Organometallics 2017, 36, 4071–4090. [Google Scholar]
- 19.Hartwig J Organotransition Metal Chemistry: From Bonding to Catalysis. University Science Books: Sausalito, 2010; pp. 1–1100. [Google Scholar]
- 20.Guérinot A; Cossy J Cobalt-Catalyzed Cross-Couplings between Alkyl Halides and Grignard Reagents. Acc. Chem. Res. 2020, 53, 1351–1363. [DOI] [PubMed] [Google Scholar]
- 21.(a) Takai K; Nitta K; Fujimura O; Utimoto K Preparation of Alkylchromium Reagents by Reduction of Alkyl Halides with Chromium(II) Chloride under Cobalt Catalysis. J. Org. Chem. 1989, 54, 4732–4734; [Google Scholar]; (b) Ackerman LKG; Anka-Lufford LL; Naodovic M; Weix DJ Cobalt co-catalysis for cross-electrophile coupling: diarylmethanes from benzyl mesylates and aryl halides. Chem. Sci. 2015, 6, 1115–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Buskes MJ; Blanco M-J Impact of Cross-Coupling Reactions in Drug Discovery and Development. Molecules 2020, 25, 3493, DOI: 10.3390/molecules25153493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Neely JM; Bezdek MJ; Chirik PJ Insight into Transmetalation Enables Cobalt-Catalyzed Suzuki−Miyaura Cross Coupling. ACS Cent. Sci. 2016, 2, 935–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Duong HA; Wu W; Teo Y-Y Cobalt-Catalyzed Cross-Coupling Reactions of Arylboronic Esters and Aryl Halides. Organometallics 2017, 36, 4363–4366. [Google Scholar]
- 25.Tailor SB; Manzotti M; Smith GJ; David SA; Bedford RB Cobalt-Catalyzed Coupling of Aryl Chlorides with Aryl Boron Esters Activated by Alkoxides. ACS Catal. 2021, 11, 3856–3866. [Google Scholar]
- 26.Reports from Bedford and from Duong have shown that activated, anionic borates can participate in C(sp2)–C(sp2) Suzuki–Miyaura coupling catalyzed by cobalt: Asghar S; Tailor SB; Elorriaga D; Bedford RB Cobalt-Catalyzed Suzuki Biaryl Coupling of Aryl Halides. Angew. Chem. Int. Ed. 2017, 56, 16367–16370; Duong HA; Yeow Z-H; Tiong Y-L; Kamal NHBM; Wu W Cobalt-Catalyzed Cross-Coupling Reactions of Aryl Triflates and Lithium Arylborates. J. Org. Chem. 2019, 84, 12686–12691.
- 27.(a) Bedford RB How Low Does Iron Go? Chasing the Active Species in Fe-Catalyzed Cross-Coupling Reactions. Acc. Chem. Res. 2015, 48, 1485–1493; [DOI] [PubMed] [Google Scholar]; (b) Cassani C; Bergonzini G; Wallentin C-J Active Species and Mechanistic Pathways in Iron-Catalyzed C–C Bond-Forming Cross-Coupling Reactions. ACS Catal. 2016, 6, 1640–1648. [Google Scholar]
- 28.Diccianni JB; Diao T Mechanisms of Nickel-Catalyzed Cross-Coupling Reactions. Trends Chem. 2019, 1, 830–844. [Google Scholar]
- 29.Ludwig JR; Simmons EM; Wisniewski SR; Chirik PJ Cobalt-Catalyzed C(sp2)−C(sp3) Suzuki−Miyaura Cross Coupling. Org. Lett. 2021, 23, 625–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.(a) Fu GC The Development of Versatile Methods for Palladium-Catalyzed Coupling Reactions of Aryl Electrophiles through the Use of P(t-Bu)3 and PCy3 as Ligands. Acc. Chem. Res. 2008, 41, 1555–1564; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Maron N; Nolan SP Well-Defined N-Heterocyclic Carbenes−Palladium(II) Precatalysts for Cross-Coupling Reactions. Acc. Chem. Res. 2008, 41, 1440–1449. [DOI] [PubMed] [Google Scholar]
- 31.(a) Lavoie CM; MacQueen PM; Rotta-Loria NL; Sawatzky RS; Borzenko A; Chisolm AJ; Hargreaves BKV; McDonald R; Ferguson MJ; Stradiotto M Challenging nickel-catalysed amine arylations enabled by tailored ancillary ligand design. Nat. Commun. 2016, 7, 11073, DOI: 10.1038/ncomms11073; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wu K; Doyle AG Parameterization of phosphine ligands demonstrates enhancement of nickel catalysis via remote steric effects. Nat. Chem. 2017, 9, 779–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hazari N; Melvin PR; Mohadjer Beromi M Well-defined nickel and palladium precatalysts for cross-coupling. Nat. Rev. Chem. 2017, 1, 0025, DOI: 10.1038/s41570-017-0025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mennen SM; Alhambra C; Allen CL; Barberis M; Berritt S; Brandt TA; Campbell AD; Castañón J; Cherney AH; Christensen M; Damon DB; do Diego JE; García-Cerrada S; García-Losada P; Haro R; Janey J; Leitch DC; Li L; Liu F; Lobben PC; MacMillan DWC; Magano J; McInturff E; Monfette S; Post RJ; Schultz D; Sitter BJ; Stevens JM; Strambeanu II; Twilton J; Wang K; Zajac MA The Evolution of High-Throughput Experimentation in Pharmaceutical Development and Perspectives on the Future. Org. Process Res. Dev. 2019, 23, 1213–1242. [Google Scholar]
- 34.(a) Taylor RD; MacCoss M; Lawson ADG Rings in Drugs. J. Med. Chem. 2014, 57, 5845–5859; [DOI] [PubMed] [Google Scholar]; (b) Vitaku E; Smith DT; Njardarson JT Analysis of the Structural Diversity, Substitution Patterns, and Frequency of Nitrogen Heterocycles among U.S. FDA Approved Pharmaceuticals. J. Med. Chem. 2014, 57, 10257–10274. [DOI] [PubMed] [Google Scholar]
- 35.In keeping with standard industrial practice for optimization, in-process yields are assessed using “area percentage” (AP) yields. AP values are uncorrected for the difference in LC response factor for the starting alkyl halide versus its boronate ester derivative. These data, while approximate, give insight into general trends and aid in determining optimal conditions for lead reactions.
- 36.Sandford C; Fried LR; Ball TE; Minteer SD; Sigman MS Mechanistic Studies into the Oxidative Addition of Co(I) Complexes: Combining Electroanalytical Techniques with Parameterization. J. Am. Chem. Soc. 2019, 141, 18877–18889. [DOI] [PubMed] [Google Scholar]
- 37.Saito B; Fu GC Alkyl-Alkyl Suzuki Cross-Couplings of Unactivated Secondary Alkyl Halides at Room Temperature. J. Am. Chem. Soc. 2007, 129, 9602–9603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.In a recent report, 8-hydroxyquinoline ligands were used to support a nickel-catalyzed conjunctive coupling employing neopentylglyocol arylboronic esters as nucleophilic reagents: Dhungana RK; Aryal V; Niroula D; Sapkota RR; Lakomy MG; Giri R Ni-Catalyzed Regioselective Alkenylarylation of γ,δ-Alkenyl Ketones via Carbonyl Coordination. Angew. Chem. Int. Ed. 2021, DOI: 10.1002/anie.202104871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.González-Bobes F; Fu GC Amino Alcohols as Ligands for Nickel-Catalyzed Suzuki Reactions of Unactivated Alkyl Halides, Including Secondary Alkyl Chlorides, with Arylboronic Acids. J. Am. Chem. Soc. 2006, 128, 5360–5361. [DOI] [PubMed] [Google Scholar]
- 40.(a) Bakewell C; Platel RH; Cary SK; Hubbard SM; Roaf JM; Levine AC; White AJP; Long N; Naaf M; Williams CK Bis(8-quinolinolato)aluminum ethyl complexes: Iso-Selective Initiators for rac-Lactide Polymerization. Organometallics 2012, 31, 4729–4736; [Google Scholar]; (b) Siega P; Dreos R; Brancatelli G; Demitri N; Geremia S Formation and Structure of a Cobalt(III) Complex Containing a Nonstabilized Pyridinium Ylide Ligand. Organometallics 2014, 33, 6076–6080; [Google Scholar]; (c) Gao Y; Christianson MD; Wang Y; Chen J; Marshall S; Klosin J; Lohr TL; Marks TJ Unexpected Precatalyst σ-Ligand Effects in Phenoxyimine Zr-Catalyzed Ethylene/1-Octene Copolymerizations. J. Am. Chem. Soc. 2019, 141, 7822–7830; [DOI] [PubMed] [Google Scholar]; (d) Li M; Behzadi S; Chen M; Pang W; Wang F; Tan C Phenoxyimine Ligands Bearing Nitrogen-Containing Second Coordination Spheres for Zinc Catalyzed Stereoselective Ring- Opening Polymerization of rac-Lactide. Organometallics 2019, 38, 461–468. [Google Scholar]
- 41.In a series of reports from Bhat and coworkers, FI ligands were proposed to promote cobalt-catalyzed C(sp2)–C(sp2) Suzuki–Miyaura cross-coupling using arylboronic acids; however, as described by Bedford and coworkers,42 these reports from Bhat were found to be false; Ansari RM; Bhat BR Schiff base transition metal complexes for Suzuki–Miyaura cross-coupling reaction. J. Chem. Sci. 2017, 129, 1483–1490; Ansari RM; Kumar LM; Bhat BR Air-Stable Cobalt(II) and Nickel(II) Complexes with Schiff Base Ligand for Catalyzing Suzuki–Miyaura Cross-Coupling Reaction. Russ. J. Coord. Chem. 2018, 44, 1–8; Ansari RM; Mahesh LK; Bhat BR RETRACTED: Cobalt Schiff base complexes: Synthesis characterization and catalytic application in Suzuki–Miyaura reaction. Chin. J. Chem. Eng. 2019, 27, 556–563.
- 42.Tailor SB; Manzotti M; Asghar S; Rowsell BJS; Luckham SLJ; Sparkes HA; Bedford RB Revisiting Claims of the Iron-, Cobalt-, Nickel-, and Copper- Catalyzed Suzuki Biaryl Cross-Coupling of Aryl Halides with Aryl Boronic Acids. Organometallics 2019, 38, 1770–1777. [Google Scholar]
- 43.Vatsadze SZ; Loginova YD; dos Passos Gomes G; Alabugin IV Stereoelectronic Chameleons: The Donor–Acceptor Dichotomy of Functional Groups. Chem. – Eur. J. 2017, 23, 3225–3245. [DOI] [PubMed] [Google Scholar]
- 44.Bedford RB; Bruce DW; Frost RM; Goodby JW; Hird M Iron(III) salen-type catalysts for the cross-coupling of aryl Grignards with alkyl halides bearing β-hydrogens. Chem. Commun. 2004, 2822–2823. [DOI] [PubMed] [Google Scholar]
- 45.Griller D; Ingold KU Free-radical clocks. Acc. Chem. Res. 1980, 13, 317–323. [Google Scholar]
- 46.Samsel EG; Kochi JK Radical chain mechanisms for alkyl rearrangement in organocobalt complexes. J. Am. Chem. Soc. 1986, 108, 4790–4804. [Google Scholar]
- 47.Biswas S; Weix DJ Mechanism and Selectivity in Nickel-Catalyzed Cross-Electrophile Coupling of Aryl Halides with Alkyl Halides. J. Am. Chem. Soc. 2013, 135, 16192–16197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shevick SL; Obradors C; Shenvi RA Mechanistic Interrogation of Co/Ni-Dual Catalyzed Hydroarylation. J. Am. Chem. Soc. 2018, 140, 12056–12068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.It should be noted that the slopes obtained (Scheme 2A) are comparatively modest.48 However, the data obtained do not necessarily reflect the rate of recombination alone, but also reflect (i) the rate of product formation compared to recombination and (ii) the rate of Co–alkyl homolysis, which homolysis is possible with Co(salen) and related compounds.46 See: Kyne SH; Lefèvre G; Ollivier C; Petit M; Ramis Cladera V-A; Fensterbank L Iron and cobalt catalysis: new perspectives in synthetic radical chemistry. Chem. Soc. Rev. 2020, 49, 8501–8542 and references cited therein.
- 50.(a) Wang C; Friedrich S; Younkin TR; Li RT; Grubbs RH; Bansleben DA; Day MW Neutral Nickel(II)-Based Catalysts for Ethylene Polymerization. Organometallics 1998, 17, 3149–3151; [Google Scholar]; (b) Zuideveld MA; Wehrmann P; Röhr C; Mecking S Remote Substituents Controlling Catalytic Polymerization by Very Active and Robust Neutral Nickel(II) Complexes. Angew. Chem. Int. Ed. 2004, 43, 869–873 and references cited therein. [DOI] [PubMed] [Google Scholar]
- 51.(a) Demarteau J; Debuigne A; Detrembleur C Organocobalt Complexes as Sources of Carbon-Centered Radicals for Organic and Polymer Chemistries. Chem. Rev. 2019, 119, 6906–6955. [DOI] [PubMed] [Google Scholar]
- 52.Subbaiah MAM; Meanwell NA Bioisosteres of the Phenyl Ring: Recent Strategic Applications in Lead Optimization and Drug Design. J. Med. Chem. 2021, 64, 14046–14128. [DOI] [PubMed] [Google Scholar]
- 53.Bull JA; Croft RA; Davis OA; Doran R; Morgan KF Oxetanes: Recent Advances in Synthesis, Reactivity, and Medicinal Chemistry. Chem. Rev. 2016, 116, 12150–12233. [DOI] [PubMed] [Google Scholar]
- 54.Bauer MR; Di Fruscia P; Lucas SCC; Michaelides IN; Nelson JE; Storer RI; Whitehurst BC Put a ring on it: application of small aliphatic rings in medicinal chemistry. RSC Med. Chem. 2021, 12, 448–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hiesinger K; Dar’in D; Proschak E; Krasavin M Spirocyclic Scaffolds in Medicinal Chemistry. J. Med. Chem. 2021, 64, 150–183. [DOI] [PubMed] [Google Scholar]
- 56.Olmstead WN; Margolin Z; Bordwell FG Acidities of water and simple alcohols in dimethyl sulfoxide solution. J. Org. Chem. 1980, 45, 3295–3299. [Google Scholar]
- 57.Bordwell FG; McCallum RJ; Olmstead WN Acidities and hydrogen bonding of phenols in dimethyl sulfoxide. J. Org. Chem. 1984, 49, 1424–1427. [Google Scholar]
- 58.Deprotonation could be visualized by 1H NMR via a disappearance of the phenol resonance at 12.94 ppm and general upfield shifts of the aromatic and imine resonances, and by 19F NMR via an upfield shift of the 4-F signal (see SI).
- 59.(a) Wang L; Sun W-H; Han L; Li Z; Hu Y; He C; Yan C Cobalt and nickel complexes bearing 2,6-bis (imino) phenoxyligands: syntheses, structures and oligomerization studies. J. Organomet. Chem. 2002, 650, 59–64; [Google Scholar]; (b) Du J; Han L; Cui Y; Li J; Li Y; Sun W-H Synthesis, Characterization, and Ethylene Oligomerization of 2,6-Bis(imino)phenoxy Cobalt Complexes. Aust. J. Chem. 2003, 56, 703–706, DOI: 10.1071/CH02193. [DOI] [Google Scholar]
- 60.For a review on the synthesis of transition metal–FI complexes: Garnovskii AD; Vasilchenko IS; Garnovskii DA; Kharisov BI Molecular design of mononuclear complexes of acyclic Schiff-base ligands. J. Coordination Chem. 2009, 62, 151–204, DOI: 10.1080/00958970802398178. [DOI] [Google Scholar]
- 61.KHMDS can be used on large scale.
- 62.Salehi M; Hasanzadeh M Characterization, crystal structures, electrochemical and antibacterial studies of four new binuclear cobalt(III) complexes derived from o-aminobenzyl alcohol. Inorg. Chim. Acta 2015, 426, 6–14; [Google Scholar]; (b) Ngcobo M; Nyamato GS; Ojwach SO Structural elucidation of NÔ (ethylimino-methyl)phenol Fe(II) and Co(II) complexes and their applications in ethylene oligomerization catalysis. Molecular Catal. 2019, 478, 110590, DOI: 10.1016/j.mcat.2019.110590 [DOI] [Google Scholar]
- 63.(a) Gaber M; El-Baradie KY; Amer SA; Issa RM Spectroscopic Investigation of Divalent Mn, Co, Ni, Cu and Trivalent Fe Complexes With Indazole Schiff Bases. Synthesis and Reactivity in Inorganic, Metal-Organic, and Nano-Metal Chemistry 1991, 21, 349–360, DOI: 10.1080/15533179108018344; [DOI] [Google Scholar]; (b) Scarpellini M; Wu AJ; Kampf JW; Pecoraro VL Corroborative Models of the Cobalt(II) Inhibited Fe/Mn Superoxide Dismutases. Inorg. Chem. 2005, 44, 5001–5010; [DOI] [PubMed] [Google Scholar]; (c) Gätjens J; Mullins CS; Kampf JW; Thuéry P; Pecoraro VL Corroborative cobalt and zinc model compounds of α-amino-β-carboxymuconic-ε-semialdehyde decarboxylase (ACMSD). Dalton Trans. 2009, 51–62; [DOI] [PubMed] [Google Scholar]; (d) Pavlov AA; Nehrkorn J; Zubkevich SV; Fedin MV; Holldack K; Schnegg A; Novikov VV A Synergy and Struggle of EPR, Magnetometry and NMR: A Case Study of Magnetic Interaction Parameters in a Six-Coordinate Cobalt(II) Complex. Inorg. Chem. 2020, 59, 10746–10755. [DOI] [PubMed] [Google Scholar]
- 64.Teotia MP; Gurtu JN; Rana VB Dimeric 5-and 6-coordinate complexes of tri and tetradentate ligands. J. Inorg. Nuc. Chem. 1980, 42, 821–831. [Google Scholar]
- 65.Bertini I; Lanini G; Luchinat C 1H NMR Relaxation Rate and Coordination Number in High Spin Cobalt(II) Complexes. Inorganica Chimica Acta 1983, 80, 123–126. [Google Scholar]
- 66.(a) Elmali A; Elerman Y; Svoboda I; Fuess H trans-Bis[N-(4-bromophenyl)salicylaldiminato]cobalt(II). Acta Cryst. 1996, C52, 553–554. [Google Scholar]; (b) Ziegenbalg S; Hornig D; Görls H; Plass W Cobalt(II)-Based Single-Ion Magnets with Distorted Pseudotetrahedral [N2O2] Coordination: Experimental and Theoretical Investigations. Inorg. Chem. 2016, 55, 4047–4058. [DOI] [PubMed] [Google Scholar]; (c) Belhadj N; Ourari A; Keraghel S; Schöllhorn B; Matt D Crystal Structure and Corrosion Inhibition Properties of Ferrocenyl- and Phenylendiamine-Iminomethylphenoxy Cobalt Complexes. J. Chem. Crystallogr. 2017, 47, 40–46. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.