

Abstract
Breast cancer (BC) is a highly heterogeneous neoplasm of the mammary tissue, causing the deaths of a large number of women worldwide. Nearly 70% and 20% of BC cases are estrogen receptor alpha positive (ERα+) and human epidermal growth factor receptor 2-positive (HER2+), respectively; therefore, ER and HER2 targeted therapies have been employed in BC treatment. However, resistance to these therapies has been reported, indicating a need for developing novel therapeutic strategies. Proteolysis-targeting chimeras (PROTACs) are new, promising therapeutic tools designed with a bimodular structure: one module allows specific binding to target proteins, and the other module allows efficient degradation of these target proteins. In this paper, PROTACs and their potential in controlling the progression of ERα and HER2+ BC are discussed.
Keywords: Estrogen receptor alpha, breast cancer, degradation via the ubiquitin-proteasome system, proteolysis-targeting chimeras
Introduction
The ubiquitin-proteasome system (UPS) refers to the ubiquitination of proteins followed by their degradation via the 26S proteasome. Ubiquitin (Ub) is an 8-kDa and 76-amino acid protein that is conserved from yeast to humans. Because of a conjugation motif on its C-terminus, Ub can form an isopeptide bond to specific lysine residues of several proteins. This process called ubiquitination occurs by three sequential reactions, requiring E1-Ub activating enzyme, E2-Ub conjugating enzyme, and E3-Ub ligase enzyme. The E3-Ub enzymes allow for a high degree of substrate specificity in ubiquitination [1–3].
Proteasome 26S is composed of catalytic core 20S and regulatory unit 19S. The 19S unit allows polyubiquitinated proteins to generate unfolded proteins that are then translocated to the 20S catalytic unit [4]. In summation, a polyubiquitinated protein is recognized by proteasome 26S, transported to the 20S core particle, and converted into oligopeptides by a variety of enzymes that promote oligopeptide release from the proteasome. Ub is recycled at the end of this process [5].
There are approximately 600 members of the E3-Ub ligase family, with really interesting new gene (RING) E3 ligases being the most common [1, 6–8] (Table 1). The bonds of Ub can form monoubiquitination of different residues on one target protein and polyubiquitination. Polyubiquitination with Ub chains occurs because of Ub lysine residues 6, 11, 27, 29, 33, 48, and 63, allowing for the possibility of branched Ub structures [9–12].
Table 1.
E3-Ub ligases types
| Main types of E3 ligases | Characteristics | Action mode |
|---|---|---|
| RING E3 ligases | Contains a RING domain or U-box domain | Ub is transferred in one step from the E2-conjugating enzyme to the substrate |
| Homologous to the HECT E3 ligases | Subfamilies: 1. RCC1-like domains; 2. tryptophan-tryptophan domains; 3. ligases lacking these domains | Ub is transferred in two steps: these ligases first transfer Ub from the E2-conjugating enzyme to themselves and subsequently to the substrate |
| RBR E3 ligases | Contain an IBR which separates two predicted RING domains (RING1 and RING2) |
RCC1: regulator of chromosome condensation 1; RBR: RING-in-between-RING; HECT: E6AP carboxyl terminus; IBR: in between ring
The ubiquitination process is modulated through deubiquitinases (DUBs) that reverse ubiquitination, and almost 100 DUBs are known in humans [9–11]. Furthermore, Ub receptors are Ub-binding domains that recognize the Ub mark from proteins modified by ubiquitination, and this interaction can impact the function or signaling of these proteins [5, 13–16]. Although polyubiquitination is classically associated with proteolytic activities, it is important to note that non-proteolytic functions can also be performed as a result of polyubiquitination in specific cases [17–21]. Additionally, monoubiquitination has been described as a modification implemented in the regulation of protein stability, transcription, and DNA repair.
In recent years, there has been an increasing number of studies on the deregulation of Ub-associated pathways and the functional implications of ubiquitination in many diseases, including cancer. Much of the knowledge about ubiquitination and the UPS is being utilized to generate new technologies that are useful as therapeutic strategies, such as proteolysis-targeting chimeras (PROTACs). We discuss PROTACs in the context of breast cancer (BC) in this review.
PROTACs
PROTACs have a bifunctional structure designed to initiate the degradation of specific proteins through UPS. The bifunctional structure of PROTACs contains a module with the ability to bind to the target protein (core region) and another module responsible for recruiting an E3-Ub ligase (degron), both of which are linked to the target protein by a spacer [22]. The E3-Ub ligase marks the target protein for degradation, reducing the abundance and functionality of the target protein (Figure 1) [23, 24].
Figure 1.
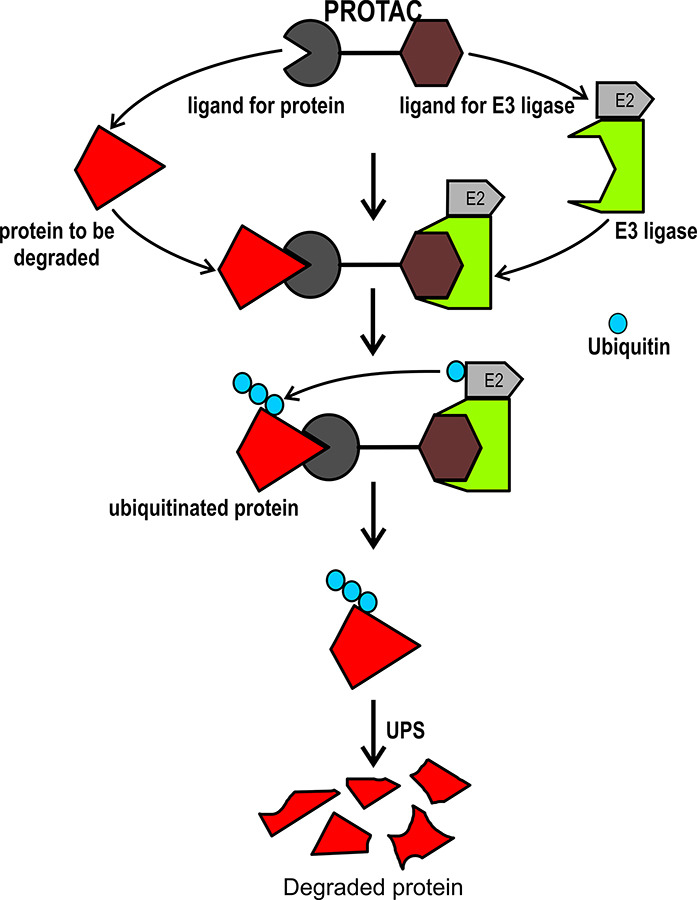
PROTACs. Representation of molecular mechanism that is used by PROTACs
Domains and motifs required for interaction with E3-Ub ligases need to be considered in the construction of PROTACs. The large peptides which were initially used in PROTACS were not completely functional due to their limited penetration into the cell [23]. In one of the first studies, PROTACs containing phosphopeptide IkappaB (IκB) have been shown to recruit the SCFβ-TrCP Ub ligase complex, subsequently promoting the degradation of methionine aminopeptidase 2 (MetAP-2), which has been linked to cancer [23, 24]. To date, several E3-Ub ligases have been considered in the design of PROTACs, some of which are shown in Table 2 [23, 25–27].
Table 2.
Some E3-ligases used for PROTACs
| Abbreviation | Name |
|---|---|
| MDM2 | Murine double minute 2 |
| CRBN | Cereblon |
| VHL | Von Hippel-Lindau tumor suppressor |
| IAPs | Inhibitor of apoptosis proteins |
PROTAC technology has advantages over other treatments. PROTACs directly degrades the target protein, even undruggable targets, since PROTACs design allows the binding to any site of the target protein without requiring a high affinity. Moreover, resistance to conventional treatments for cancer is common, whereas the development of resistance to PROTACs is not feasible. Thus, PROTACs degrade overexpressed or mutated proteins associated with resistance to drugs. In addition, PROTACs have high potency, efficacy, and less toxicity [28].
Although PROTACs are a promising means of inducing the degradation of specific target proteins, they also have disadvantages. Future PROTAC studies could investigate improvements regarding molecule size, pharmacokinetics, and administration. Since PROTACs are designed to bind to specific targets, sometimes the large size of molecules can affect their bioavailability and solubility. PROTAC bioavailability is also affected by intraperitoneal and subcutaneous administration which are the two current standards of administration; accordingly, new intravenous and oral methods of administration are being investigated [22, 29–31].
BC: luminal and human epidermal growth factor receptor 2-overexpressing
BC is one of the main causes of death among women worldwide. There is high molecular heterogeneity in mammary tumors, indicating the molecular complexity that influences BC’s classification, biomarker determination, and pharmacological treatments (Table 3) [32–34].
Table 3.
BC types and therapies
| BC type | Biomarkers | Therapy |
|---|---|---|
| Luminal A-like | ERα+, PR ≥ 20%, HER2-, Ki67 < 20% | Endocrine therapy |
| Luminal B-like | ERα+, PR < 20% and/or HER2+ and/or Ki67 ≥ 20% | Endocrine therapy |
| HER2-overexpression | ERα-, PR-, HER2+ | HER2-targeted therapy |
| Basal-like | ERα-, PR-, HER2- (triple-negative) | Not responsive to endocrine or anti-HER2 therapies |
ERα+: estrogen receptor alpha positive; PR: progesterone receptor; HER2-: human epidermal growth factor receptor 2-negative; HER2+: HER2-positive; Ki67: marker of proliferation Ki-67
ERα+ BC
Most BC cases (~70%) are luminal type and thus are ERα+. ERα is a nuclear receptor activated by estrogens. It is also activated via phosphorylation induced by growth factors such as insulin-like growth factor (IGF) and epidermal growth factor (EGF) in BC cells [32, 33, 35, 36]. Although ERα is a nuclear receptor, it is also associated with cytoplasmic proteins and transmembrane receptors. ERα can also be membrane-associated via its palmitoylation [37, 38]. Extranuclear ERα is part of the signaling route that may lead to gene regulation [39–41]. Nuclear ERα can act as a transcription factor or a coregulator that modulates gene expression [42, 43]. Estradiol, the most abundant estrogen, induces the expression of its target genes through ERα activation. Many of those target genes are associated with pro-tumor activity in BC cells (Figure 2) [44, 45].
Figure 2.

ER+ and HER2+ BC and PROTACs. A) Signaling pathways triggered by ER; B) representation of PROTACs designed for ER; C) signaling pathways of HER2+; D) design of antibody (Ab)-PROTAC conjugated using an Ab for HER2. ERE: estrogen response element; IGFR: IGF receptor; PI3K: phosphatidylinositol-4,5-bisphosphate 3-kinase; EGFR: EGF receptor; MAPK: mitogen-activated protein kinase; TF: transcription factor; TFRE: TF response elements; ER+: ER-positive
The structure of ERα, from the N-terminus and C-terminus, consists of the activation function domain-1 (AF-1), the DNA-binding domain (DBD), a hinge region, the ligand-binding domain (LBD), and the activation function domain-2 (AF-2) [46, 47]. AF-2 recruits coregulators in an estradiol-dependent manner. AF-1 does the same in an estradiol-independent manner. AF-1 (in residues that include S118 and S167) is a target of phosphorylation by EGF and IGF signaling, activating the action of the receptor [44, 48–51]. The expression and recruitment of coactivators for ERα are usually increased in BC, enhancing the expression of their target genes [30, 45, 52–54]. Through DBD, ERα binds to EREs contained in the enhancers and promoters of their target genes to modulate its expression. Moreover, forkhead box A1 (FOXA1) and GATA binding protein 3 (GATA3) are considered pioneer factors that facilitate the recruitment of ERα to ERE in DNA regulatory sequences [55, 56].
HER2-overexpression in BC cells
Approximately 20% of all BCs are HER2+ [57]. HER2 is a member of the EGF receptor family and is also known as the proto-oncogene neu and ErbB2. HER2+ BC cases are characterized as being highly aggressive, recurrent, and having a poor prognosis. Nearly 50% of these cancer cases also display co-expression with hormone receptors [58, 59]. The gene that encodes HER2 is on chromosome 17q12 and is known as ErbB2 receptor tyrosine kinase 2 (ERBB2) [60, 61]. HER2 is a transmembrane receptor with tyrosine-protein kinase activity, but it does not have the ability to recognize growth factors nor can it be activated by growth factor ligands directly, as it lacks the ligand-binding extracellular domain [60, 62, 63]. HER2 associates with other members of the EGF receptor family that do possess the ligand-binding ability, forming heterodimers that trigger downstream signaling, such as phosphatidylinositol-3 kinase and MAPK activation, promoting BC progression [60, 62].
ERα and HER2-targeted therapies for BC
Endocrine therapies
Endocrine therapies (ET) are used for ERα+ BC cases because they enclose aromatase inhibitors (AIs) and selective estrogen receptor modulators (SERMs). AIs block estradiol production, whereas SERMs are a type of ET that acts by competing with estradiol by binding to ERα and inhibiting the recruitment of coactivators. SERMs promote the interaction with corepressors to avoid the transcription of estradiol-target genes in BC cells [64–66]. A common problem of these types of ET is de novo resistance and acquired resistance [65–69]. The mechanisms underlying the resistance to ET are not completely understood, but they have been shown to be related to signaling pathways activated by ERα [69, 70].
Interestingly, mutations in estrogen receptor 1 (ESR1), the gene that encodes ERα, specifically the mutations Y537S, Y537N, Y537C, D538G, and E380Q, have been found in circulating tumor DNA and biopsies of metastatic BC with resistance to ET [71–73]. These mutations encompass the LBD and allow ERα to act in an estradiol-independent manner, modifying the profile of ERα target genes, some of them associated with endocrine resistance to ET [72, 74–77].
Selective estrogen receptor degraders (SERDs) are another ET that are considered as antiestrogens and antagonize estradiol [66–68]. However, the binding of SERDs to ERα promotes the polyubiquitination and degradation of this receptor through the UPS [64, 78, 79]. To date, fulvestrant is the most important SERD described since even the commonly reported ESR1 mutations seem to be sensitive to this SERD [72, 75, 76]. Although several SERDs are being evaluated, fulvestrant is the first SERD approved as a first-line ET to date [80]. It is hoped that the new generation of SERDs will have improved bio-dispensability and administration routes in comparison with fulvestrant, which is administrated intramuscularly and has a limited bio-dispensability [81–86].
The mechanism of SERDs is based on ERα degradation via the UPS, which has significant advantages compared to the mechanisms of SERMs. New therapeutic strategies that are also based on ERα polyubiquitination as a marker for its degradation via the UPS are PROTACs (Figure 2C), as will be discussed in the latter half of this review.
HER2-targeted therapies
An improvement in the prognosis of HER2+ BC has been observed as a result of different therapeutic strategies. The first approved therapy for HER2+ BC was adjuvant chemotherapy called trastuzumab, which consists of a humanized monoclonal Ab that specifically recognizes HER2, inhibiting HER2 dimerization [87]. Other HER2-directed monoclonal antibodies, such as pertuzumab, have now been generated. Additionally, novel strategies have emerged, including Ab-drug conjugates (ADCs) and tyrosine kinase inhibitors (TKIs) [88].
ADCs are recombinant monoclonal antibodies covalently bonded to a cytotoxin, generating a chemotherapeutic drug specifically targeted against HER2. This allows for intracellular delivery of the cytotoxic drug [89]. The bonds between the monoclonal Ab and cytotoxic drug are cleaved once the Ab binds to its target antigen, allowing the cytotoxic drug for use on other cells [89, 90]. Trastuzumab emtansine (T-DM1) is a non-cleavable ADC, showing antitumor activity in HER2+ BC [91]. Despite its therapeutic effects, since HER2 can be expressed in other cells and since cytotoxic drugs can be internalized in cells with low HER2 expression, gastrointestinal, hematologic, and hepatic toxicity has been reported. Therefore, further investigations are required to ensure the safe use of the ADCs in BC [92, 93].
TKIs, such as lapatinib, neratinib, tucatinib, and pyrotinib, are also used to treat HER2+ BC. TKIs can be used in combination with chemotherapy [88]. TKIs are of interest for the treatment of HER2+ cells that are resistant to the other therapies. The resistance to therapies has been associated with HER2 reactivation, HER2-dependent downstream signaling, upregulation of the HER receptor family, and HER2 mutations [94–97]. For example, L755S is an acquired activating mutation of HER2 identified in HER2+ metastatic BC, which may prevent the binding of the monoclonal antibodies and lapatinib activity [94, 95, 98]. With the use of TKIs as a BC treatment even with HER2 mutations, there is an increasing interest in developing novel TKIs for HER2+ BC [94, 96].
PROTAC for BC
PROTACs for ERα
PROTAC-2, PROTAC-B, compound 24, compound 11, and estrogen receptor degrader (ERD)-148, among other PROTACs, have been designed to induce the reduction of ERα levels in BC cells (Figure 3) [99–101]. PROTAC-2 was designed with an estradiol molecule to bind to ERα and the phosphopeptide of IκBα that recruits SCFβ-trcp Ub ligase complex, leading to ERα degradation [102]. PROTAC-B displays a peptide from hypoxia-inducible factor-1α (HIF-1α) that allows for the recruitment of the E3-Ub ligase VHL [103, 104]. E2-octa and E2-penta contain synthetic peptides derived from HIF-1α, resulting in an efficient reduction of ERα levels [101]. Additionally, specific and non-genetic IAP-dependent protein eraser (SNIPER) is a PROTAC formed by a derivative of estradiol along with a bestatin amide that binds the cellular inhibitor of apoptosis protein 1 (cIAP1) ligand which induces ERα degradation [23, 105, 106].
Figure 3.

Representative PROTACs for ER
Another example is compound 1–6, which was designed as a peptide-based PROTAC with better penetration [26]. Similarly, TD-PROTAC is another peptide-based PROTAC constructed from TD-PERM (a stabilized peptide that binds to ERα), the linker, and the hydroxyproline-containing pentapeptide from HIF-1α to recruit the VHL E3 Ub ligase complex. TD-PERM promotes 75% reduction in tumor volume in michigan cancer foundation-7 (MCF-7) mouse xenograft model [99].
Furthermore, ERD-308 PROTAC induces ERα degradation by reducing cell proliferation to a greater extent than fulvestrant (> 95% of ERa degradation at concentrations as low as 5 nmol/L in MCF-7 and T47D BC cells) [25]. ARV-471 PROTAC reduces ERα levels and its Y537S and D538G variants. Furthermore, ARV-471 significantly reduces estradiol-dependent MCF-7 xenograft tumors and tumor ER protein levels (> 90%) [27, 107]. Other PROTACs have also been evaluated in vitro as well as using in vivo models, and indicated a reduction in cell proliferation, inhibition of estradiol target genes, and induction of apoptosis in both models. This suggests that PROTACs have a strong antitumor therapeutic effect [99]. However, ARV-471 has been proposed as the best-in-class oral ERa PROTAC due to its results in preclinical trials. Currently, ARV-471 is in phase 2 studies to treat advanced or metastatic ER+/HER2+ BC [107, 108].
PROTACs in HER2+ BC
New PROTAC designs consider the use of monoclonal antibodies to target specific proteins. For example, a study generated a new PROTAC with Ab linker named Ab-PROTAC3, a trastuzumab-PROTAC conjugate. This conjugate has the ability of PROTACs to recruit E3-Ub ligase specifically to HER2+ cells through trastuzumab. Then, trastuzumab-PROTAC can be internalized into the cell, and active PROTAC is released to promote the degradation of bromodomain-containing protein 4 (BRD4) [109].
PROTACs for other proteins in BC
PROTACs can be applied to other proteins implicated in the progression of BC. For example, B-cell lymphoma-extra large (Bcl-xL) is overexpressed in several cancer types, including BC. The PROTAC named 8a can promote the Bcl-xL degradation in BC cells [110]. Additionally, the bromo and extra terminal domain proteins (BET) such as BRD2, BRD3, and BRD4 are epigenetic regulators considered potential targets for BC treatment. It has been generated PROTACs for BRD4 using VHL E3 ligase and for BRD2, BRD3, and BRD4, using E3 Ub ligase cereblon (CRBN), showing results in triple-negative BC [111, 112]. Thus, several proteins involved in BC may be targeted by PROTACs.
Conclusions
Most of the BC cases are ERα+ and use endocrine therapy, but a common problem is resistance to these therapies. SERDs exhibit better therapeutic results with few instances of resistance compared to SERMs and AI. However, to date, fulvestrant is the only approved SERD. New SERDs are being evaluated, focusing specifically on improving bioavailability and administration routes [66]. PROTACs have emerged as promising therapies. Because their mechanisms are also linked to the UPS, it is hypothesized that PROTACs will be specific and efficient, without drug-resistance problems.
PROTACs require the definition of new “degron” motifs to have more recognition regions for their use [100]. This is important since PROTACs have demonstrated effective inhibition of ERα target genes and proliferation. However, further investigations are necessary to improve their cell penetration ability [99, 113–115]. PROTAC technology is associated with other therapeutic strategies, such as SERD-like PROTACs or Ab-PROTACs. It is expected that these molecules will show a higher specificity and adequate bioavailability and administration route than other therapies. Complete inhibition of tumor progression by new PROTACs is also expected. Moreover, it is relevant to consider that several mechanisms inhibit ERα degradation in BC cells, many of which are based on monoubiquitination by E3-Ub ligases and other cases by the actions of the DUBs [116]. Therefore, ERα stability in BC must be considered in future applications and designs of therapies based on the UPS.
Moreover, antibodies cannot be used for intracellular targets, requiring new PROTACs with good ability to penetrate cell membranes and excellent stability. Interestingly, an Ab-PROTAC has been designed in which trastuzumab Ab is used to recognize HER2. Through this specific system, the PROTACs are directed to the site where it has to induce degradation. These models indicate the versatility of PROTACs and the many ways that they can be studied to generate better degradation strategies for several targets. The current PROTACs could be improved with the fusion of peptides to penetrate cell membranes. More PROTACs targeting intracellular components related to BC with improved biochemical characteristics will need to be discovered and fully characterized. Thus, there is a clear need to find novel safe, and efficient drugs in treating BC. A central challenge going forward in this field will be to improve the pharmacokinetic properties of PROTACs.
In conclusion, PROTAC technology is being evaluated for its application in ERα+ and HER2+ BC treatments with promising results since PROTACs reduce the pro-tumor effects. Further investigation and improvements of these drugs are still required before PROTACs can be used as part of a new generation of drugs for the treatment of BC.
Abbreviations
- Ab:
antibody
- ADCs:
antibody-drug conjugates
- AF-1:
activation function domain-1
- BC:
breast cancer
- BRD4:
bromodomain-containing protein 4
- DUBs:
deubiquitinases
- EGF:
epidermal growth factor
- ER+:
estrogen receptor-positive
- ERE:
estrogen response element
- ERα+:
estrogen receptor alpha positive
- ET:
endocrine therapies
- HER2-:
human epidermal growth factor receptor 2-negative
- HER2+:
human epidermal growth factor receptor 2-positive
- HIF-1α:
hypoxia-inducible factor-1α
- IGF:
insulin-like growth factor
- MCF-7:
michigan cancer foundation-7
- PR:
progesterone receptor
- PROTACs:
proteolysis-targeting chimeras
- RING:
really interesting new gene
- SERDs:
selective estrogen receptor degraders
- SERMs:
selective estrogen receptor modulators
- TKIs:
tyrosine kinase inhibitors
- Ub:
ubiquitin
- UPS:
ubiquitin-proteasome system
- VHL:
von Hippel-Lindau tumor suppressor
Declarations
Acknowledgments
We thank Alfonso Ramos Flores for technical help with this manuscript.
Author contributions
ACTC designed the research, integrated the information, and wrote the manuscript. JZC helped in the research and the integration of information and improving the manuscript. JORJ helped in the manuscript and prepared the figures. ARO helped in the research.
Conflicts of interest
The authors declare no conflict of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent to publication
Not applicable.
Availability of data and materials
Not applicable.
Funding
Not applicable.
Copyright
© The Author(s) 2021.
References
- 1.Bernassola F, Karin M, Ciechanover A, Melino G. The HECT family of E3 ubiquitin ligases: multiple players in cancer development. Cancer Cell. 2008;14:10–21. 10.1016/j.ccr.2008.06.001 [DOI] [PubMed] [Google Scholar]
- 2.Deshaies RJ, Joazeiro CA. RING domain E3 ubiquitin ligases. Annu Rev Biochem. 2009;78:399–434. 10.1146/annurev.biochem.78.101807.093809 [DOI] [PubMed] [Google Scholar]
- 3.Metzger MB, Pruneda JN, Klevit RE, Weissman AM. RING-type E3 ligases: master manipulators of E2 ubiquitin-conjugating enzymes and ubiquitination. Biochim Biophys Acta. 2014;1843:47–60. 10.1016/j.bbamcr.2013.05.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sahu I, Glickman MH. Proteasome in action: substrate degradation by the 26S proteasome. Biochem Soc Trans. 2021;49:629–44. 10.1042/BST20200382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Woelk T, Sigismund S, Penengo L, Polo S. The ubiquitination code: a signalling problem. Cell Div. 2007;2:11. 10.1186/1747-1028-2-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Morreale FE, Walden H. Types of ubiquitin ligases. Cell. 2016;165:248–e1. 10.1016/j.cell.2016.03.003 [DOI] [PubMed] [Google Scholar]
- 7.Scheffner M, Kumar S. Mammalian HECT ubiquitin-protein ligases: biological and pathophysiological aspects. Biochim Biophys Acta. 2014;1843:61–74. 10.1016/j.bbamcr.2013.03.024 [DOI] [PubMed] [Google Scholar]
- 8.Zheng N, Shabek N. Ubiquitin ligases: structure, function, and regulation. Annu Rev Biochem. 2017;86:129–57. 10.1146/annurev-biochem-060815-014922 [DOI] [PubMed] [Google Scholar]
- 9.D’Arcy P, Wang X, Linder S. Deubiquitinase inhibition as a cancer therapeutic strategy. Pharmacol Ther. 2015;147:32–54. 10.1016/j.pharmthera.2014.11.002 [DOI] [PubMed] [Google Scholar]
- 10.Helzer KT, Hooper C, Miyamoto S, Alarid ET. Ubiquitylation of nuclear receptors: new linkages and therapeutic implications. J Mol Endocrinol. 2015;54:R151–67. 10.1530/JME-14-0308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mevissen TET, Komander D. Mechanisms of deubiquitinase specificity and regulation. Annu Rev Biochem. 2017;86:159–92. 10.1146/annurev-biochem-061516-044916 [DOI] [PubMed] [Google Scholar]
- 12.Sadowski M, Suryadinata R, Tan AR, Roesley SN, Sarcevic B. Protein monoubiquitination and polyubiquitination generate structural diversity to control distinct biological processes. IUBMB Life. 2012;64:136–42. 10.1002/iub.589 [DOI] [PubMed] [Google Scholar]
- 13.Di Fiore PP, Polo S, Hofmann K. When ubiquitin meets ubiquitin receptors: a signalling connection. Nat Rev Mol Cell Biol. 2003;4:491–7. 10.1038/nrm1124 [DOI] [PubMed] [Google Scholar]
- 14.Wang X, Terpstra EJ. Ubiquitin receptors and protein quality control. J Mol Cell Cardiol. 2013;55:73–84. 10.1016/j.yjmcc.2012.09.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dikic I, Wakatsuki S, Walters KJ. Ubiquitin-binding domains-from structures to functions. Nat Rev Mol Cell Biol. 2009;10:659–71. 10.1038/nrm2767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Husnjak K, Dikic I. Ubiquitin-binding proteins: decoders of ubiquitin-mediated cellular functions. Annu Rev Biochem. 2012;81:291–322. 10.1146/annurev-biochem-051810-094654 [DOI] [PubMed] [Google Scholar]
- 17.Bonacci T, Audebert S, Camoin L, Baudelet E, Iovanna JL, Soubeyran P. Regulation of NUB1 activity through non-proteolytic Mdm2-mediated ubiquitination. PLoS One. 2017;12:e0169988. 10.1371/journal.pone.0169988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fei C, Li Z, Li C, Chen Y, Chen Z, He X, et al. Smurf1-mediated Lys29-linked nonproteolytic polyubiquitination of axin negatively regulates Wnt/β-catenin signaling. Mol Cell Biol. 2013;33:4095–105. 10.1128/MCB.00418-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hurley JH, Lee S, Prag G. Ubiquitin-binding domains. Biochem J. 2006;399:361–72. 10.1042/BJ20061138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kuslansky Y, Sominsky S, Jackman A, Gamell C, Monahan BJ, Haupt Y, et al. Ubiquitin ligase E6AP mediates nonproteolytic polyubiquitylation of β-catenin independent of the E6 oncoprotein. J Gen Virol. 2016;97:3313–30. 10.1099/jgv.0.000624 [DOI] [PubMed] [Google Scholar]
- 21.Scott D, Oldham NJ, Strachan J, Searle MS, Layfield R. Ubiquitin-binding domains: mechanisms of ubiquitin recognition and use as tools to investigate ubiquitin-modified proteomes. Proteomics. 2015;15:844–61. 10.1002/pmic.201400341 [DOI] [PubMed] [Google Scholar]
- 22.Dragovich PS, Adhikari P, Blake RA, Blaquiere N, Chen J, Cheng YX, et al. Antibody-mediated delivery of chimeric protein degraders which target estrogen receptor alpha (ERα). Bioorg Med Chem Lett. 2020;30:126907. 10.1016/j.bmcl.2019.126907 [DOI] [PubMed] [Google Scholar]
- 23.Lin X, Xiang H, Luo G. Targeting estrogen receptor α for degradation with PROTACs: a promising approach to overcome endocrine resistance. Eur J Med Chem. 2020;206:112689. 10.1016/j.ejmech.2020.112689 [DOI] [PubMed] [Google Scholar]
- 24.Sakamoto KM, Kim KB, Kumagai A, Mercurio F, Crews CM, Deshaies RJ. Protacs: chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc Natl Acad Sci U S A. 2001;98:8554–9. 10.1073/pnas.141230798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hu J, Hu B, Wang M, Xu F, Miao B, Yang CY, et al. Discovery of ERD-308 as a highly potent proteolysis targeting chimera (PROTAC) degrader of estrogen receptor (ER). J Med Chem. 2019;62:1420–42. 10.1021/acs.jmedchem.8b01572 [DOI] [PubMed] [Google Scholar]
- 26.Dai Y, Yue N, Gong J, Liu C, Li Q, Zhou J, et al. Development of cell-permeable peptide-based PROTACs targeting estrogen receptor α. Eur J Med Chem. 2020;187:111967. 10.1016/j.ejmech.2019.111967 [DOI] [PubMed] [Google Scholar]
- 27.Qi SM, Dong J, Xu ZY, Cheng XD, Zhang WD, Qin JJ. PROTAC: an effective targeted protein degradation strategy for cancer therapy. Front Pharmacol. 2021;12:692574. 10.3389/fphar.2021.692574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pei H, Peng Y, Zhao Q, Chen Y. Small molecule PROTACs: an emerging technology for targeted therapy in drug discovery. RSC Adv. 2019;9:16967–76. 10.1039/C9RA03423D [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Edmondson SD, Yang B, Fallan C. Proteolysis targeting chimeras (PROTACs) in ‘beyond rule-of-five’ chemical space: recent progress and future challenges. Bioorg Med Chem Lett. 2019;29:1555–64. 10.1016/j.bmcl.2019.04.030 [DOI] [PubMed] [Google Scholar]
- 30.Manavathi B, Samanthapudi VS, Gajulapalli VN. Estrogen receptor coregulators and pioneer factors: the orchestrators of mammary gland cell fate and development. Front Cell Dev Biol. 2014;2:34. 10.3389/fcell.2014.00034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pesiri V, Di Muzio E, Polticelli F, Acconcia F. Selective binding of estrogen receptor α to ubiquitin chains. IUBMB Life. 2016;68:569–77. 10.1002/iub.1514 [DOI] [PubMed] [Google Scholar]
- 32.Curigliano G, Burstein HJ, Winer EP, Gnant M, Dubsky P, Loibl S, et al. De-escalating and escalating treatments for early-stage breast cancer: the st. gallen international expert consensus conference on the primary therapy of early breast cancer 2017. Ann Oncol. 2017;28:1700–12. 10.1093/annonc/mdx308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hammond ME, Hayes DF, Dowsett M, Allred DC, Hagerty KL, Badve S, et al. American Society of Clinical Oncology/College of American Pathologists guideline recommendations for immunohistochemical testing of estrogen and progesterone receptors in breast cancer. J Clin Oncol. 2010;28:2784–95. 10.1200/JCO.2009.25.6529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yi M, Huo L, Koenig KB, Mittendorf EA, Meric-Bernstam F, Kuerer HM, et al. Which threshold for ER positivity? a retrospective study based on 9639 patients. Ann Oncol. 2014;25:1004–11. 10.1093/annonc/mdu053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen T, Zhang N, Moran MS, Su P, Haffty BG, Yang Q. Borderline ER-positive primary breast cancer gains no significant survival benefit from endocrine therapy: a systematic review and meta-analysis. Clin Breast Cancer. 2018;18:1–8. 10.1016/j.clbc.2017.06.005 [DOI] [PubMed] [Google Scholar]
- 36.Collins LC, Botero ML, Schnitt SJ. Bimodal frequency distribution of estrogen receptor immunohistochemical staining results in breast cancer: an analysis of 825 cases. Am J Clin Pathol. 2005;123:16–20. 10.1309/hcf035n9wk40etj0 [DOI] [PubMed] [Google Scholar]
- 37.Acconcia F, Ascenzi P, Fabozzi G, Visca P, Marino M. S-palmitoylation modulates human estrogen receptor-alpha functions. Biochem Biophys Res Commun. 2004;316:878–83. 10.1016/j.bbrc.2004.02.129 [DOI] [PubMed] [Google Scholar]
- 38.Pedram A, Razandi M, Deschenes RJ, Levin ER. DHHC-7 and -21 are palmitoylacyltransferases for sex steroid receptors. Mol Biol Cell. 2012;23:188–99. 10.1091/mbc.E11-07-0638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tecalco-Cruz AC, Pérez-Alvarado IA, Ramírez-Jarquín JO, Rocha-Zavaleta L. Nucleo-cytoplasmic transport of estrogen receptor alpha in breast cancer cells. Cell Signal. 2017;34:121–32. 10.1016/j.cellsig.2017.03.011 [DOI] [PubMed] [Google Scholar]
- 40.Caligiuri I, Toffoli G, Giordano A, Rizzolio F. pRb controls estrogen receptor alpha protein stability and activity. Oncotarget. 2013;4:875–83. 10.18632/oncotarget.1036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kumar R, Wang RA, Mazumdar A, Talukder AH, Mandal M, Yang Z, et al. A naturally occurring MTA1 variant sequesters oestrogen receptor-alpha in the cytoplasm. Nature. 2002;418:654–7. 10.1038/nature00889 [DOI] [PubMed] [Google Scholar]
- 42.Björnström L, Sjöberg M. Estrogen receptor-dependent activation of AP-1 via non-genomic signalling. Nucl Recept. 2004;2:3. 10.1186/1478-1336-2-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Frasor J, Weaver A, Pradhan M, Dai Y, Miller LD, Lin CY, et al. Positive cross-talk between estrogen receptor and NF-kappaB in breast cancer. Cancer Res. 2009;69:8918–25. 10.1158/0008-5472.CAN-09-2608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vrtačnik P, Ostanek B, Mencej-Bedrač S, Marc J. The many faces of estrogen signaling. Biochem Med (Zagreb). 2014;24:329–42. 10.11613/BM.2014.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hah N, Murakami S, Nagari A, Danko CG, Kraus WL. Enhancer transcripts mark active estrogen receptor binding sites. Genome Res. 2013;23:1210–23. 10.1101/gr.152306.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kumar R, Zakharov MN, Khan SH, Miki R, Jang H, Toraldo G, et al. The dynamic structure of the estrogen receptor. J Amino Acids. 2011;2011:812540. 10.4061/2011/812540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nardulli AM, Greene GL, Shapiro DJ. Human estrogen receptor bound to an estrogen response element bends DNA. Mol Endocrinol. 1993;7:331–40. 10.1210/mend.7.3.8483477 [DOI] [PubMed] [Google Scholar]
- 48.Billon-Galés A, Krust A, Fontaine C, Abot A, Flouriot G, Toutain C, et al. Activation function 2 (AF2) of estrogen receptor-alpha is required for the atheroprotective action of estradiol but not to accelerate endothelial healing. Proc Natl Acad Sci U S A. 2011;108:13311–6. 10.1073/pnas.1105632108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Métivier R, Penot G, Flouriot G, Pakdel F. Synergism between ERalpha transactivation function 1 (AF-1) and AF-2 mediated by steroid receptor coactivator protein-1: requirement for the AF-1 alpha-helical core and for a direct interaction between the N- and C-terminal domains. Mol Endocrinol. 2001;15:1953–70. 10.1210/mend.15.11.0727 [DOI] [PubMed] [Google Scholar]
- 50.Smirnova NF, Fontaine C, Buscato M, Lupieri A, Vinel A, Valera MC, et al. The activation function-1 of estrogen receptor alpha prevents arterial neointima development through a direct effect on smooth muscle cells. Circ Res. 2015;117:770–8. 10.1161/CIRCRESAHA.115.306416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zwart W, de Leeuw R, Rondaij M, Neefjes J, Mancini MA, Michalides R. The hinge region of the human estrogen receptor determines functional synergy between AF-1 and AF-2 in the quantitative response to estradiol and tamoxifen. J Cell Sci. 2010;123(Pt 8):1253–61. 10.1242/jcs.061135 [DOI] [PubMed] [Google Scholar]
- 52.Fleming FJ, Hill AD, McDermott EW, O’Higgins NJ, Young LS. Differential recruitment of coregulator proteins steroid receptor coactivator-1 and silencing mediator for retinoid and thyroid receptors to the estrogen receptor-estrogen response element by beta-estradiol and 4-hydroxytamoxifen in human breast cancer. J Clin Endocrinol Metab. 2004;89:375–83. 10.1210/jc.2003-031048 [DOI] [PubMed] [Google Scholar]
- 53.Hah N, Kraus WL. Hormone-regulated transcriptomes: lessons learned from estrogen signaling pathways in breast cancer cells. Mol Cell Endocrinol. 2014;382:652–64. 10.1016/j.mce.2013.06.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hervouet E, Cartron PF, Jouvenot M, Delage-Mourroux R. Epigenetic regulation of estrogen signaling in breast cancer. Epigenetics. 2013;8:237–45. 10.4161/epi.23790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Carroll JS, Liu XS, Brodsky AS, Li W, Meyer CA, Szary AJ, et al. Chromosome-wide mapping of estrogen receptor binding reveals long-range regulation requiring the forkhead protein FoxA1. Cell. 2005;122:33–43. 10.1016/j.cell.2005.05.008 [DOI] [PubMed] [Google Scholar]
- 56.Theodorou V, Stark R, Menon S, Carroll JS. GATA3 acts upstream of FOXA1 in mediating ESR1 binding by shaping enhancer accessibility. Genome Res. 2013;23:12–22. 10.1101/gr.139469.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Siegel RL, Miller KD, Jemal A. Cancer statistics. CA Cancer J Clin. 2020;70:7–30. 10.3322/caac.21590 [DOI] [PubMed] [Google Scholar]
- 58.Wolff AC, Hammond ME, Hicks DG, Dowsett M, McShane LM, Allison KH, et al. American Society of Clinical Oncology; College of American Pathologists. Recommendations for human epidermal growth factor receptor 2 testing in breast cancer: American Society of Clinical Oncology/College of American Pathologists clinical practice guideline update. J Clin Oncol. 2013;31:3997–4013. 10.1200/JCO.2013.50.9984 [DOI] [PubMed] [Google Scholar]
- 59.Wolff AC, Hammond MEH, Allison KH, Harvey BE, Mangu PB, Bartlett JMS, et al. Human epidermal growth factor receptor 2 testing in breast cancer: American Society of Clinical Oncology/College of American Pathologists clinical practice guideline focused update. J Clin Oncol. 2018;36:2105–22. 10.1200/JCO.2018.77.8738 [DOI] [PubMed] [Google Scholar]
- 60.Moasser MM. The oncogene HER2: its signaling and transforming functions and its role in human cancer pathogenesis. Oncogene. 2007;26:6469–87. 10.1038/sj.onc.1210477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Perez EA, Reinholz MM, Hillman DW, Tenner KS, Schroeder MJ, Davidson NE, et al. HER2 and chromosome 17 effect on patient outcome in the N9831 adjuvant trastuzumab trial. J Clin Oncol. 2010;28:4307–15. 10.1200/JCO.2009.26.2154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Iqbal N, Iqbal N. Human epidermal growth factor receptor 2 (HER2) in cancers: overexpression and therapeutic implications. Mol Biol Int. 2014;2014:852748. 10.1155/2014/852748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yan M, Schwaederle M, Arguello D, Millis SZ, Gatalica Z, Kurzrock R. HER2 expression status in diverse cancers: review of results from 37,992 patients. Cancer Metastasis Rev. 2015;34:157–64. 10.1007/s10555-015-9552-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Howell SJ, Johnston SR, Howell A. The use ofselectiveestrogenreceptor modulators and selectiveestrogen receptor down-regulators in breast cancer. Best Pract Res Clin Endocrinol Metab. 2004;18:47–66. 10.1016/j.beem.2003.08.002 [DOI] [PubMed] [Google Scholar]
- 65.Osborne CK, Schiff R. Mechanisms of endocrine resistance in breast cancer. Annu Rev Med. 2011;62:233–47. 10.1146/annurev-med-070909-182917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Patel HK, Bihani T. Selective estrogen receptor modulators (SERMs) and selective estrogen receptor degraders (SERDs) in cancer treatment. Pharmacol Ther. 2018;186:1–24. 10.1016/j.pharmthera.2017.12.012 [DOI] [PubMed] [Google Scholar]
- 67.Osborne CK, Wakeling A, Nicholson RI. Fulvestrant: an oestrogen receptor antagonist with a novel mechanism of action. Br J Cancer. 2004;90 Suppl 1:S2–6. 10.1038/sj.bjc.6601629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wardell SE, Marks JR, McDonnell DP. The turnover of estrogen receptor α by the selective estrogen receptor degrader (SERD) fulvestrant is a saturable process that is not required for antagonist efficacy. Biochem Pharmacol. 2011;82:122–30. 10.1016/j.bcp.2011.03.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.AlFakeeh A, Brezden-Masley C. Overcoming endocrine resistance in hormone receptor-positive breast cancer. Curr Oncol. 2018;25 Suppl 1:S18–27. 10.3747/co.25.3752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Acconcia F, Kumar R. Signaling regulation of genomic and nongenomic functions of estrogen receptors. Cancer Lett. 2006;238:1–14. 10.1016/j.canlet.2005.06.018 [DOI] [PubMed] [Google Scholar]
- 71.Angus L, Beije N, Jager A, Martens JW, Sleijfer S. ESR1 mutations: moving towards guiding treatment decision-making in metastatic breast cancer patients. Cancer Treat Rev. 2017;52:33–40. 10.1016/j.ctrv.2016.11.001 [DOI] [PubMed] [Google Scholar]
- 72.Toy W, Weir H, Razavi P, Lawson M, Goeppert AU, Mazzola AM, et al. Activating ESR1 mutations differentially affect the efficacy of ER antagonists. Cancer Discov. 2017;7:277–87. 10.1158/2159-8290.CD-15-1523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Reinert T, Gonçalves R, Bines J. Implications of ESR1 mutations in hormone receptor-positive breast cancer. Curr Treat Options Oncol. 2018;19:24. 10.1007/s11864-018-0542-0 [DOI] [PubMed] [Google Scholar]
- 74.Jeselsohn R, Bergholz JS, Pun M, Cornwell M, Liu W, Nardone A, et al. Allele-specific chromatin recruitment and therapeutic vulnerabilities of ESR1 activating mutations. Cancer Cell. 2018;33:173–86.e5. 10.1016/j.ccell.2018.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bahreini A, Li Z, Wang P, Levine KM, Tasdemir N, Cao L, et al. Mutation site and context dependent effects of ESR1 mutation in genome-edited breast cancer cell models. Breast Cancer Res. 2017;19:60. 10.1186/s13058-017-0851-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Harrod A, Fulton J, Nguyen VTM, Periyasamy M, Ramos-Garcia L, Lai CF, et al. Genomic modelling of the ESR1 Y537S mutation for evaluating function and new therapeutic approaches for metastatic breast cancer. Oncogene. 2017;36:2286–96. 10.1038/onc.2016.382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Martin LA, Ribas R, Simigdala N, Schuster E, Pancholi S, Tenev T, et al. Discovery of naturally occurring ESR1 mutations in breast cancer cell lines modelling endocrine resistance. Nat Commun. 2017;8:1865. 10.1038/s41467-017-01864-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Long X, Nephew KP. Fulvestrant (ICI 182,780)-dependent interacting proteins mediate immobilization and degradation of estrogen receptor-alpha. J Biol Chem. 2006;281:9607–15. 10.1074/jbc.M510809200 [DOI] [PubMed] [Google Scholar]
- 79.Yeh WL, Shioda K, Coser KR, Rivizzigno D, McSweeney KR, Shioda T. Fulvestrant-induced cell death and proteasomal degradation of estrogen receptor α protein in MCF-7 cells require the CSK c-Src tyrosine kinase. PLoS One. 2013;8:e60889. 10.1371/journal.pone.0060889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Deeks ED. Fulvestrant: a review in advanced breast cancer not previously treated with endocrine therapy. Drugs. 2018;78:131–7. 10.1007/s40265-017-0855-5 [DOI] [PubMed] [Google Scholar]
- 81.De Savi C, Bradbury RH, Rabow AA, Norman RA, de Almeida C, Andrews DM, et al. Optimization of a novel binding motif to (E)-3-(3,5-Difluoro-4-((1R,3R)-2-(2-fluoro-2-methylpropyl)-3-methyl-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indol-1-yl)phenyl)acrylic acid (AZD9496), a potent and orally bioavailable selective estrogen receptor downregulator and antagonist. J Med Chem. 2015;58:8128–40. 10.1021/acs.jmedchem.5b00984 [DOI] [PubMed] [Google Scholar]
- 82.Wardell SE, Nelson ER, Chao CA, McDonnell DP. Bazedoxifene exhibits antiestrogenic activity in animal models of tamoxifen-resistant breast cancer: implications for treatment of advanced disease. Clin Cancer Res. 2013;19:2420–31. 10.1158/1078-0432.CCR-12-3771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Garner F, Shomali M, Paquin D, Lyttle CR, Hattersley G. RAD1901: a novel, orally bioavailable selective estrogen receptor degrader that demonstrates antitumor activity in breast cancer xenograft models. Anticancer Drugs. 2015;26:948–56. 10.1097/CAD.0000000000000271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lai A, Kahraman M, Govek S, Nagasawa J, Bonnefous C, Julien J, et al. Identification of GDC-0810 (ARN-810), an orally bioavailable selective estrogen receptor degrader (SERD) that demonstrates robust activity in tamoxifen-resistant breast cancer xenografts. J Med Chem. 2015;58:4888–904. 10.1021/acs.jmedchem.5b00054 [DOI] [PubMed] [Google Scholar]
- 85.Guo S, Zhang C, Bratton M, Mottamal M, Liu J, Ma P, et al. ZB716, a steroidal selective estrogen receptor degrader (SERD), is orally efficacious in blocking tumor growth in mouse xenograft models. Oncotarget. 2018;9:6924–37. 10.18632/oncotarget.24023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tria GS, Abrams T, Baird J, Burks HE, Firestone B, Gaither LA, et al. Discovery of LSZ102, a potent, orally bioavailable selective estrogen receptor degrader (SERD) for the treatment of estrogen receptor positive breast cancer. J Med Chem. 2018;61:2837–64. 10.1021/acs.jmedchem.7b01682 [DOI] [PubMed] [Google Scholar]
- 87.Moja L, Tagliabue L, Balduzzi S, Parmelli E, Pistotti V, Guarneri V, et al. Trastuzumab containing regimens for early breast cancer. Cochrane Database Syst Rev. 2012;2012:CD006243. 10.1002/14651858.CD006243.pub2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Pondé N, Brandão M, El-Hachem G, Werbrouck E, Piccart M. Treatment of advanced HER2-positive breast cancer: 2018 and beyond. Cancer Treat Rev. 2018. Jun;67:10–20. 10.1016/j.ctrv.2018.04.016 [DOI] [PubMed] [Google Scholar]
- 89.Beck A, Goetsch L, Dumontet C, Corvaía N. Strategies and challenges for the next generation of antibody-drug conjugates. Nat Rev Drug Discov. 2017;16:315–37. 10.1038/nrd.2016.268 [DOI] [PubMed] [Google Scholar]
- 90.Staudacher AH, Brown MP. Antibody drug conjugates and bystander killing: is antigen-dependent internalisation required? Br J Cancer. 2017;117:1736–42. 10.1038/bjc.2017.367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Verma S, Miles D, Gianni L, Krop IE, Welslau M, Baselga J, et al. EMILIA Study Group. Trastuzumab emtansine for HER2-positive advanced breast cancer. N Engl J Med. 2012;367:1783–91. 10.1056/NEJMoa1209124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A, et al. Tissue-based map of the human proteome. Science. 2015;347:1260419. 10.1126/science.1260419 [DOI] [PubMed] [Google Scholar]
- 93.Press MF, Cordon-Cardo C, Slamon DJ. Expression of the HER-2/neu proto-oncogene in normal human adult and fetal tissues. Oncogene. 1990;5:953–62. [PubMed] [Google Scholar]
- 94.Xu X, De Angelis C, Burke KA, Nardone A, Hu H, Qin L, et al. HER2 reactivation through acquisition of the HER2 L755S mutation as a mechanism of acquired resistance to HER2-targeted therapy in HER2+ breast cancer. Clin Cancer Res. 2017;23:5123–34. 10.1158/1078-0432.CCR-16-2191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Scaltriti M, Rojo F, Ocaña A, Anido J, Guzman M, Cortes J, et al. Expression of p95HER2, a truncated form of the HER2 receptor, and response to anti-HER2 therapies in breast cancer. J Natl Cancer Inst. 2007;99:628–38. 10.1093/jnci/djk134 [DOI] [PubMed] [Google Scholar]
- 96.Veeraraghavan J, Mistry R, Nanda S, Sethunath V, Shea M, Mitchell T, et al. Abstract 1911: HER2 L755S mutation is associated with acquired resistance to lapatinib and neratinib, and confers cross-resistance to tucatinib in HER2-positive breast cancer models. Am Assoc Cancer Res. 2020;80:1911. 10.1158/1538-7445.AM2020-1911 [DOI] [Google Scholar]
- 97.Ritter CA, Perez-Torres M, Rinehart C, Guix M, Dugger T, Engelman JA, et al. Human breast cancer cells selected for resistance to trastuzumab in vivo overexpress epidermal growth factor receptor and ErbB ligands and remain dependent on the ErbB receptor network. Clin Cancer Res. 2007;13:4909–19. 10.1158/1078-0432.CCR-07-0701 [DOI] [PubMed] [Google Scholar]
- 98.Goutsouliak K, Veeraraghavan J, Sethunath V, De Angelis C, Osborne CK, Rimawi MF, et al. Towards personalized treatment for early stage HER2-positive breast cancer. Nat Rev Clin Oncol. 2020;17:233–50. 10.1038/s41571-019-0299-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Jiang Y, Deng Q, Zhao H, Xie M, Chen L, Yin F, et al. Development of stabilized peptide-based PROTACs against estrogen receptor α. ACS Chem Biol. 2018;13:628–35. 10.1021/acschembio.7b00985 [DOI] [PubMed] [Google Scholar]
- 100.Li Y, Zhang S, Zhang J, Hu Z, Xiao Y, Huang J, et al. Exploring the PROTAC degron candidates: OBHSA with different side chains as novel selective estrogen receptor degraders (SERDs). Eur J Med Chem. 2019;172:48–61. 10.1016/j.ejmech.2019.03.058 [DOI] [PubMed] [Google Scholar]
- 101.Bafna D, Ban F, Rennie PS, Singh K, Cherkasov A. Computer-aided ligand discovery for estrogen receptor alpha. Int J Mol Sci. 2020;21:4193. 10.3390/ijms21124193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Sakamoto KM, Kim KB, Verma R, Ransick A, Stein B, Crews CM, et al. Development of protacs to target cancer-promoting proteins for ubiquitination and degradation. Mol Cell Proteomics. 2003;2:1350–8. 10.1074/mcp.T300009-MCP200 [DOI] [PubMed] [Google Scholar]
- 103.Rodriguez-Gonzalez A, Cyrus K, Salcius M, Kim K, Crews CM, Deshaies RJ, et al. Targeting steroid hormone receptors for ubiquitination and degradation in breast and prostate cancer. Oncogene. 2008;27:7201–11. 10.1038/onc.2008.320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kargbo RB. PROTAC-mediated degradation of estrogen receptor in the treatment of cancer. ACS Med Chem Lett. 2019;10:1367–9. 10.1021/acsmedchemlett.9b00397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Okuhira K, Demizu Y, Hattori T, Ohoka N, Shibata N, Nishimaki-Mogami T, et al. Development of hybrid small molecules that induce degradation of estrogen receptor-alpha and necrotic cell death in breast cancer cells. Cancer Sci. 2013;104:1492–8. 10.1111/cas.12272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ohoka N. Development of protein knockdown technology as emerging drug discovery strategy. Yakugaku Zasshi. 2018;138:1135–43. Japanese. 10.1248/yakushi.18-00113 [DOI] [PubMed] [Google Scholar]
- 107.Snyder LB, Flanagan JJ, Qian Y, Gough SM, Andreoli M, Bookbinder M, et al. Abstract 44: the discovery of ARV-471, an orally bioavailable estrogen receptor degrading PROTAC for the treatment of patients with breast cancer, in: experimental and molecular therapeutics. Am Assoc Cancer Res. 2021;81:44. 10.1158/1538-7445.AM2021-44 [DOI] [Google Scholar]
- 108.Arvinas and pfizer announce global collaboration to develop and commercialize PROTAC® protein degrader ARV-471 [Internet]. New York: Pfizer Inc; c2002-2021 [cited 2021 Jul 22]. Available from: https://www.pfizer.com/news/press-release/press-release-detail/arvinas-and-pfizer-announce-global-collaboration-develop?utm_source=researcher_app&utm_medium=referral&utm_campaign=RESR_MRKT_Researcher_inbound [Google Scholar]
- 109.Maneiro MA, Forte N, Shchepinova MM, Kounde CS, Chudasama V, Baker JR, et al. Antibody-PROTAC conjugates enable HER2-dependent targeted protein degradation of BRD4. ACS Chem Biol. 2020;15:1306–12. 10.1021/acschembio.0c00285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Zhang X, He Y, Zhang P, Budamagunta V, Lv D, Thummuri D, et al. Discovery of IAP-recruiting BCL-XL PROTACs as potent degraders across multiple cancer cell lines. Eur J Med Chem. 2020;199:112397. 10.1016/j.ejmech.2020.112397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Noblejas-López MDM, Nieto-Jimenez C, Burgos M, Gómez-Juárez M, Montero JC, Esparís-Ogando A, et al. Activity of BET-proteolysis targeting chimeric (PROTAC) compounds in triple negative breast cancer. J Exp Clin Cancer Res. 2019;38:383. 10.1186/s13046-019-1387-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Bai L, Zhou B, Yang CY, Ji J, McEachern D, Przybranowski S, et al. Targeted degradation of BET proteins in triple-negative breast cancer. Cancer Res. 2017;77:2476–87. 10.1158/0008-5472.CAN-16-2622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Cyrus K, Wehenkel M, Choi EY, Lee H, Swanson H, Kim KB. Jostling for position: optimizing linker location in the design of estrogen receptor-targeting PROTACs. ChemMedChem. 2010;5:979–85. 10.1002/cmdc.201000146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Demizu Y, Okuhira K, Motoi H, Ohno A, Shoda T, Fukuhara K, et al. Design and synthesisofestrogenreceptor degradation inducer based on a protein knockdown strategy. Bioorg Med Chem Lett. 2012;22:1793–6. 10.1016/j.bmcl.2011.11.086 [DOI] [PubMed] [Google Scholar]
- 115.Wang L, Guillen VS, Sharma N, Flessa K, Min J, Carlson KE, et al. New class of selective estrogen receptor degraders (SERDs): expanding the toolbox of PROTAC degrons. ACS Med Chem Lett. 2018;9:803–8. 10.1021/acsmedchemlett.8b00106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Tecalco-Cruz AC, Ramírez-Jarquín JO, Cruz-Ramos E. Estrogen receptor alpha and its ubiquitination in breast cancer cells. Curr Drug Targets. 2019;20:690–704. 10.2174/1389450119666181015114041 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.