

SUMMARY
Epidemiological studies reveal that marijuana increases the risk of cardiovascular disease (CVD); however, little is known about the mechanism. Δ9-tetrahydrocannabinol (Δ9-THC), the psychoactive component of marijuana, binds to cannabinoid receptor 1 (CB1/CNR1) in the vasculature and is implicated in CVD. A UK Biobank analysis found that cannabis was an risk factor for CVD. We found that marijuana smoking activated inflammatory cytokines implicated in CVD. In silico virtual screening identified genistein, a soybean isoflavone, as a putative CB1 antagonist. Human-induced pluripotent stem cell-derived endothelial cells were used to model Δ9-THC-induced inflammation and oxidative stress via NF-κB signaling. Knockdown of the CB1 receptor with siRNA, CRISPR interference, and genistein attenuated the effects of Δ9-THC. In mice, genistein blocked Δ9-THC-induced endothelial dysfunction in wire myograph, reduced atherosclerotic plaque, and had minimal penetration of the central nervous system. Genistein is a CB1 antagonist that attenuates Δ9-THC-induced atherosclerosis.
In brief
Marijuana use is on the rise and is associated with cardiovascular disease. Δ9-tetrahydrocannabinol (Δ9-THC), the psychedelic component of marijuana, causes vascular inflammation, oxidative stress, and atherosclerosis via cannabinoid receptor 1. Genistein, a soybean isoflavone, blocks harmful cardiovascular effects of Δ9-THC while preserving clinically useful effects such as sedation and analgesia.
Graphical Abstract
INTRODUCTION
Marijuana is one of the most widely used illicit drugs worldwide (Thomas et al., 2014). The growing legalization of marijuana is expected to increase the use of marijuana, highlighting the need to learn about its adverse effects, particularly those affecting the cardiovascular systems. Retrospective studies indicate that marijuana increases the risk of cardiovascular disease (CVD), including myocardial infarction (MI), angina, and arrhythmias (DeFilippis et al., 2018; Thomas et al., 2014). Marijuana exposure is known to induce endothelial dysfunction, atherosclerosis, cardiomyopathy, and metabolic dysfunction in animal models (Wang et al., 2016; Pacher et al., 2018). Nevertheless, the US Food and Drug Administration (FDA) has approved two synthetic cannabinoids for treating chemotherapy-induced nausea and vomiting and HIV-associated anorexia: dronabinol and nabilone (Whiting et al., 2015). Synthetic cannabinoids are also associated with adverse cardiovascular effects (Pacher et al., 2018). Thus, there is a need to understand the link between marijuana and CVD.
Two cannabinoid receptors mediate the effects of Δ9-tetrahydrocannabinol (Δ9-THC), cannabinoid receptor 1 (CB1/CNR1) and cannabinoid receptor 2 (CB2/CNR2), which belong to the G-protein-coupled receptor (GPCR) superfamily (Atakan, 2012; Hoffman and Lupica, 2013). The most abundant GPCR in the mammalian brain, CB1, regulates the psychoactive effects of marijuana. The CB1 receptor is also expressed in other peripheral tissues such as the heart, vasculature, and smooth muscle. The CB2 receptor is not only expressed in immune cells, particularly in macrophages, but also in endothelial cells (Pacher and Mechoulam, 2011). Cannabinoid receptors have endogenous ligands known as endocannabinoids. Endocannabinoid signaling is important for metabolic regulation, memory, mood, pain, and immune function (Mackie, 2006). The endocannabinoid anandamide is made on-demand, unlike classical neurotransmitters (Di Marzo et al., 1994), and causes vasodilation, bradycardia, and hypotension (Movahed et al., 2005). Previous studies found that CB1 activation is proatherogenic by promoting inflammation and oxidative stress that cause endothelial dysfunction and atherosclerosis, whereas CB2 activation is anti-atherogenic (Pacher et al., 2018; Ibsen et al., 2017). CB2 agonists are in development for vascular disorder (Pacher and Mechoulam, 2011), and CB1 antagonists are anti-atherogenic (Pacher et al., 2018).
CB1 signaling is also involved in various pathophysiological processes, including obesity, smoking cessation, diabetes, liver cirrhosis, and cancer (Sugamura et al., 2009). In 2006, rimonabant became the first CB1 antagonist approved for treating obesity (Després et al., 2005). However, rimonabant was withdrawn in 2008 due to psychiatric side effects (Onakpoya et al., 2016; Moreira et al., 2009). In an effort to reduce the psychiatric side effects, pharmaceutical companies developed peripherally restricted CB1 antagonists, which may soon enter clinical development (Cinar et al., 2020). The development of CB1 antagonists that lack psychiatric side effects would be clinically significant for treating obesity, diabetes, metabolic syndrome, and CVD.
The discovery of induced pluripotent stem cells (iPSCs) based on somatic cell reprogramming has transformed the field of stem cell biology (Shi et al., 2017). Human iPSCs (hiPSCs) avoid the ethical dilemmas surrounding the use of human embryonic stem cells (hESCs). Patient-specific hiPSC-derived cell lineages (i.e., neurons, cardiomyocytes, hepatocytes, and smooth muscle cells) hold great promise in regenerative medicine and drug discovery for their ability to generate a limitless supply of differentiated cell lineages for disease modeling. hiPSC-derived endothelial cells (hiPSC-ECs) contain any given individual’s genetic information and permit the investigation of patient-specific causes of vascular disease (Lau et al., 2019), making them an ideal platform for vascular disease modeling and drug discovery.
In this study, we found that cannabis users from the UK Biobank database analysis showed a higher risk of MI than non-cannabis users. We recruited volunteer subjects and found marijuana smoking elevated plasma markers of inflammation associated with atherosclerosis. Using in silico modeling, we discovered that genistein, an isoflavone abundantly present in soybeans, could bind to the CB1 receptor and inhibit its activity. Complementary in vitro and in vivo studies uncovered that genistein is a neutral antagonist of the CB1 receptor that blocks the pathophysiological effects of the CB1 receptor in the vasculature with minimal central nervous system (CNS) penetration. Thus, our study reveals that genistein attenuates Δ9-THC-induced endothelial dysfunction via CB1 inhibition and can serve as a CB1 antagonist.
RESULTS
UK Biobank analysis reveals an association between cannabis use and myocardial infarction
The UK Biobank is the largest prospective cohort study to date, containing genetic and phenotypic data on 500,000 individuals aged 40–69 (Bycroft et al., 2018). Participants were recruited between 2006 and 2010 and serially followed over time. The UK Biobank provides a unique opportunity to characterize relationships across a wide assortment of phenotypes. Here, we utilized the UK Biobank to determine the relationship between cannabis use, MI, and other cardiovascular events (Figure 1A). Prior studies demonstrating an association between cannabis use and MI were conducted in individuals who developed MI before the age of 50 (DeFilippis et al., 2018). When controlling for age, body mass index (BMI), and sex, we demonstrated that cannabis use was a statistically significant positive predictor for MI, using a logistic regression model.
Figure 1. Analysis of the UK Biobank dataset demonstrates the relationship between cannabis and myocardial infarction.
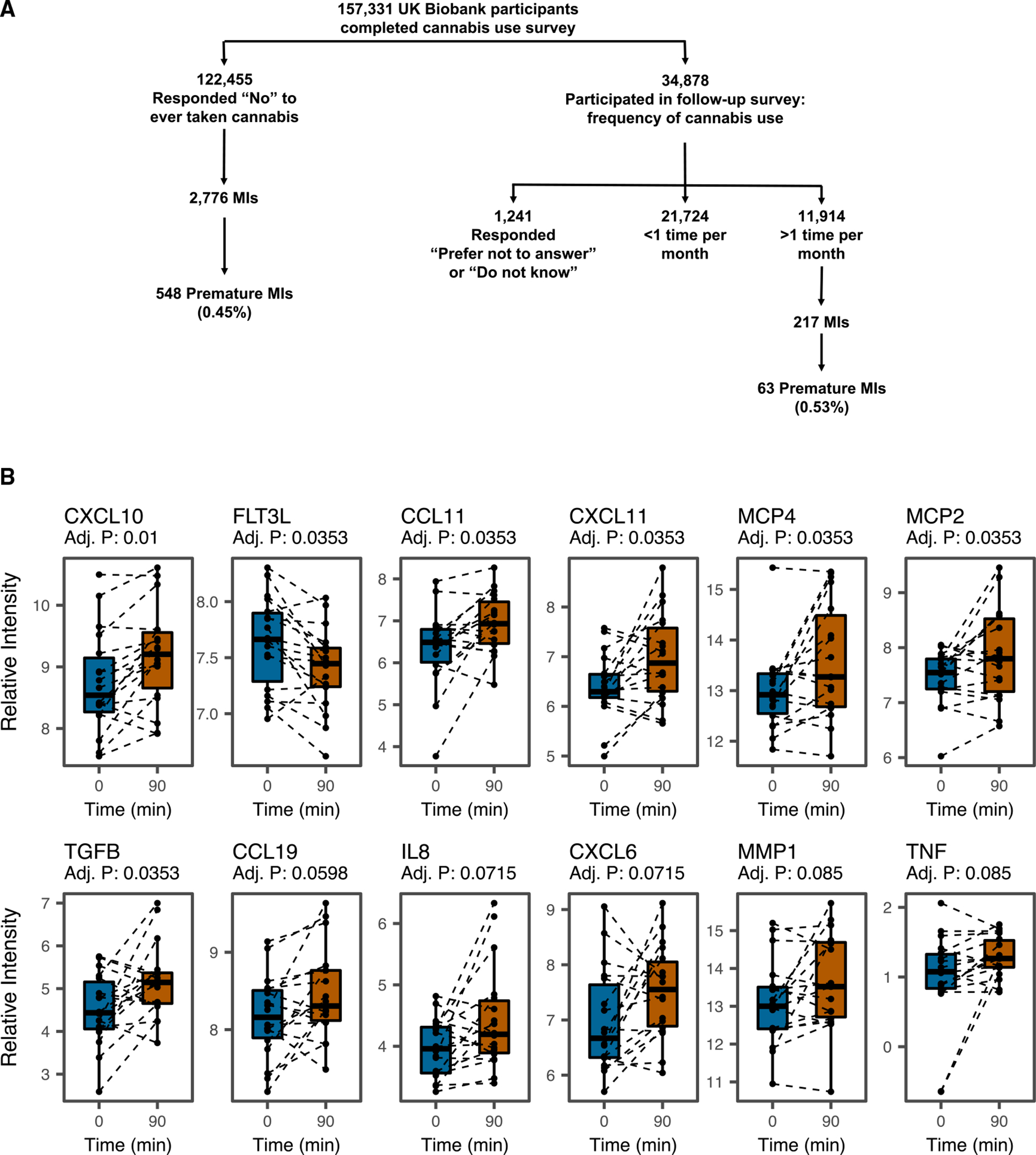
(A) Biobank Engine, a tool to search case-control association results from the UK Biobank hospital in-patient health-related outcomes summary information data, was utilized to obtain insight into the genetic mechanism of cannabis-associated cardiovascular disease. Schematic of the stratification of the UK Biobank dataset phenotypes for analysis. To determine the population-level/clinical impact of cannabis on cardiovascular events, patient data acquired from the UK Biobank were analyzed. To identify the relevant study population, self-reported patient surveys collected as part of the UK Biobank dataset were utilized to stratify the UK Biobank population. A total of 157,331UK Biobank participants were surveyed on whether they have ever used cannabis. A subset of 34,878 individuals who responded “yes” were subsequently surveyed for the frequency of cannabis use, and 11,914 responded that they utilized cannabis more than 1 time per month. On the other hand, 122,445 individuals responded “no” to ever taking cannabis. Among these individuals, cannabis use was associated with an increased incidence of MI in a logistic regression model with normalized age, sex, and body mass index (odds ratio 1.16 (95% confidence interval 1.00–1.34); p < 0.05). Cannabis users also showed a higher incidence of premature MI (under the age of 50) than non-users (0.53% versus 0.45%).
(B) Investigating inflammatory cytokine release in response to smoking marijuana. Twenty recreational marijuana smokers were recruited to smoke a single marijuana cigarette, and serial blood draws were performed to assess the Δ9-THC levels in blood. Inflammatory cytokine production was assayed using the Olink proteomic platform. Participants were asked to abstain from using marijuana for 24 h before testing. Two participants did not consent to subsequent testing and were excluded from the Olink analysis. Plasma was isolated from n = 18 individuals (13 males/5 females) using sodium citrate tubes at 15 min intervals from 0 min to 180 min after smoking. An Olink inflammation panel with 92 cytokines implicated in inflammation and oxidative stress was used to analyze the blood samples at 0, 90, and 180 min. The Olink panel revealed that several circulating cytokines were upregulated in marijuana smokers as implicated in atherosclerosis.
Inflammation is central in the pathogenesis of atherosclerosis, so we next investigated for markers of inflammation using an Olink proteomic analysis of plasma from recreational smokers (Figure 1B). After smoking a marijuana cigarette, plasma samples were serially drawn over 180 min. The Olink inflammation panel analysis revealed that several inflammatory cytokines were upregulated at 90 min, including TGFB, CCL4, CCL19, CXCL6, CXCL10, CXCL11, IL-8, MCP2, MCP4, TNF, and CCL11. These upregulated cytokines are all implicated in atherosclerosis (Chen et al., 2019; Chang et al., 2020; Damås et al., 2007; Paramel et al., 2020; Heller et al., 2006; Szentes et al., 2018; Cavusoglu et al., 2015; Ardigo et al., 2007; McKellar et al., 2009; Zernecke and Weber, 2010). FTL3 was downregulated after marijuana smoking, which is also associated with accelerated atherosclerosis in Flt3(−/−)Ldlr(−/−) mice (Choi et al., 2011).
Genistein binds to the CB1 receptor and inhibits CB1 activity
The SWEETLEAD chemical database (Moshiri et al., 2020) was screened against 4 selective CB1 antagonists using the ROCS software suite (Figure 2A). Ligand-based high-throughput virtual screening with a chemical database was used to discover new selective CB1 antagonists (Figure 2B). We found 62 chemical compounds that were structurally homologous with the 4 selective CB1 antagonists. To refine our search, a molecular docking analysis was performed to probe the interactions between the chemical compounds and the CB1 receptor with the selective CB1 antagonist AM6538 as a positive control. The Schrödinger (Portland, OR, United States of America) suite with glide protein-ligand docking functionality was used to validate that genistein was a ligand for the CB1 receptor (Table S1). We discovered that genistein, a natural soybean flavonoid, binds to the CB1 receptor (Figures 2C and 2D). Genistein adopted a similar shape to the selective CB1 antagonist query structures (Figure 2E). An in vitro GTPase assay found that genistein functions as a neutral antagonist (Figure 2F). Radioligand binding assays for predicted targets of genistein binding (Tables S2 and S3) revealed that genistein binds human CB1 with an IC50 of 150 nM (Table S4).
Figure 2. Identification of cannabinoid receptor 1 (CB1) inhibitor by high-throughput virtual screening and molecular docking.
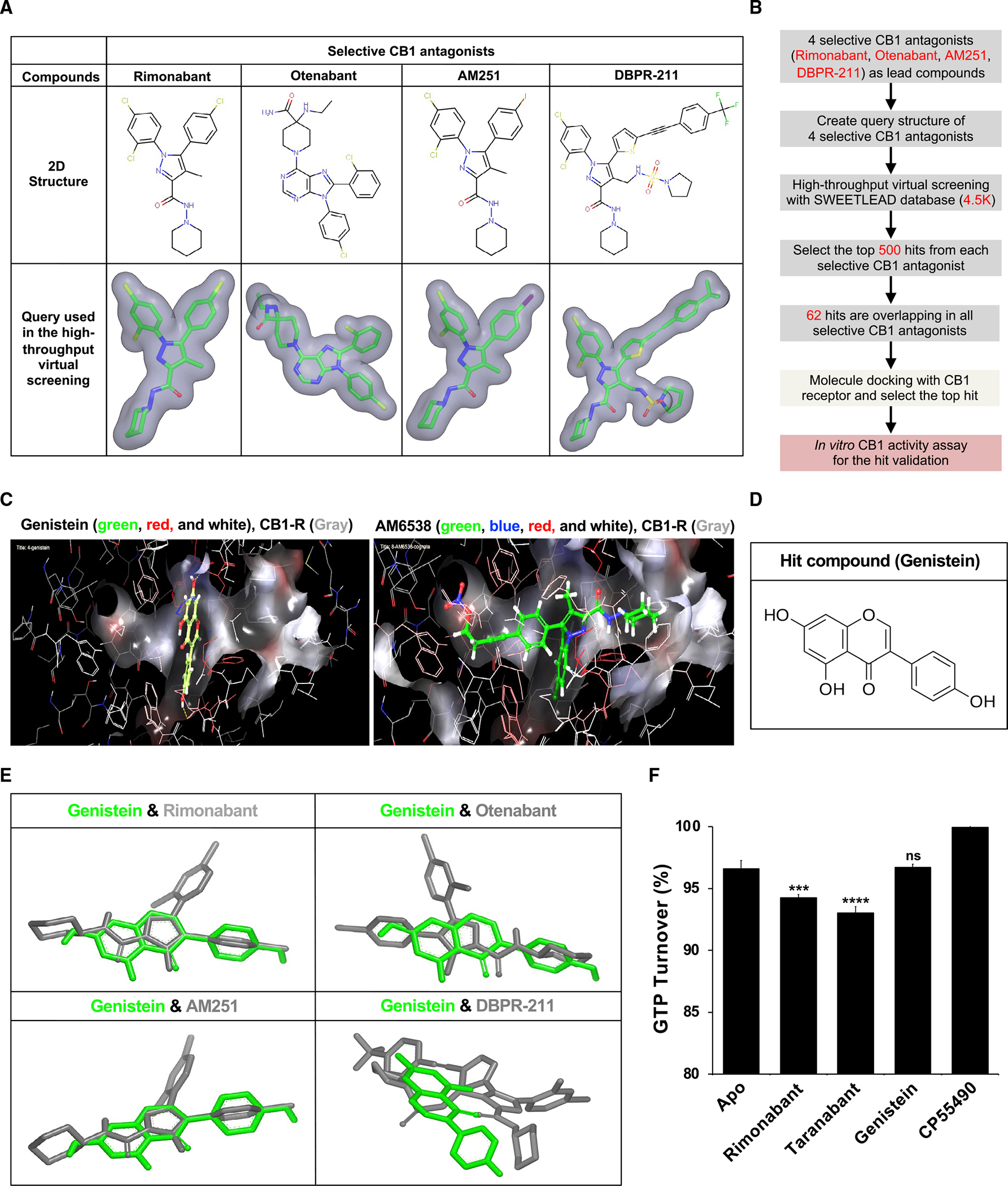
(A) Query structure of 4 selective CB1 antagonists (rimonabant, otenabant, AM251, and DBPR-211) was used in the ligand-based high-throughput virtual screening.
(B) High-throughput virtual screening workflow for lead compound identification.
(C) Docking of the selective CB1 antagonist AM6538 and genistein into the CB1 receptor. The best fit of AM6538 and genistein into CB1 was shown using Schrödinger molecule docking software.
(D) Molecular structure of hit compound genistein.
(E) Genistein shared structural homology with selective CB1 antagonists in ligand-based virtual screening.
(F) Genistein is a neutral antagonist of the CB1 receptor. GTPase-Glo assay reveals rimonabant and taranabant to decrease GTP turnover compared with Apo and GTPase without ligand, indicating that they are inverse agonists. Genistein elicits the same GTP turnover as Apo, suggesting that genistein is a neutral antagonist. CP55490 causes increased GTP turnover consistent with its function as a CB1 agonist. ***p < 0.001 versus Apo; ****p < 0.001 versus Apo; ns, not significant versus Apo.
Δ9-THC induces cytotoxicity in human endothelial cells
We next evaluated Δ9-THC cytotoxicity in three different cell types: (1) human endothelial cells (human umbilical vein endothelial cells [HUVECs] and human coronary artery endothelial cells [HCAECs]), (2) human embryonic stem cell-derived cardiomyocytes (H7 hESC-CMs), and (3) normal human cardiac fibroblasts-ventricular (NHCF-V) cells. We observed that Δ9-THC induced cytotoxicity in human endothelial cells without any adverse effect on cardiomyocytes or cardiac fibroblasts (Figure 3A). Inflammation and oxidative stress cause endothelial dysfunction (Heitzer et al., 2001), so we investigated the expression of these genes in HUVECs exposed to Δ9-THC to uncover the mechanism of such endothelial cytotoxicity. We observed that Δ9-THC induced the expression of inflammation-related genes and reduced the antioxidant-related gene expression (Figures 3B and 3C). HUVECs were derived from large pools of female patients with environmental exposures that might affect the phenotype with Δ9-THC. However, hiPSC-ECs are free of previous environmental exposures. Therefore, hiPSC-EC lines were generated from 4 healthy individuals through a chemically defined differentiation protocol (Figure S1A). hiPSC-ECs formed capillary-like tubes mimicking angiogenesis in vitro (Figure S1B), and Δ9-THC also induced cytotoxicity (Figure S1C; key resource table).
Figure 3. Δ9-THC-induced cytotoxicity in endothelial cells is associated with inflammation and oxidative stress.
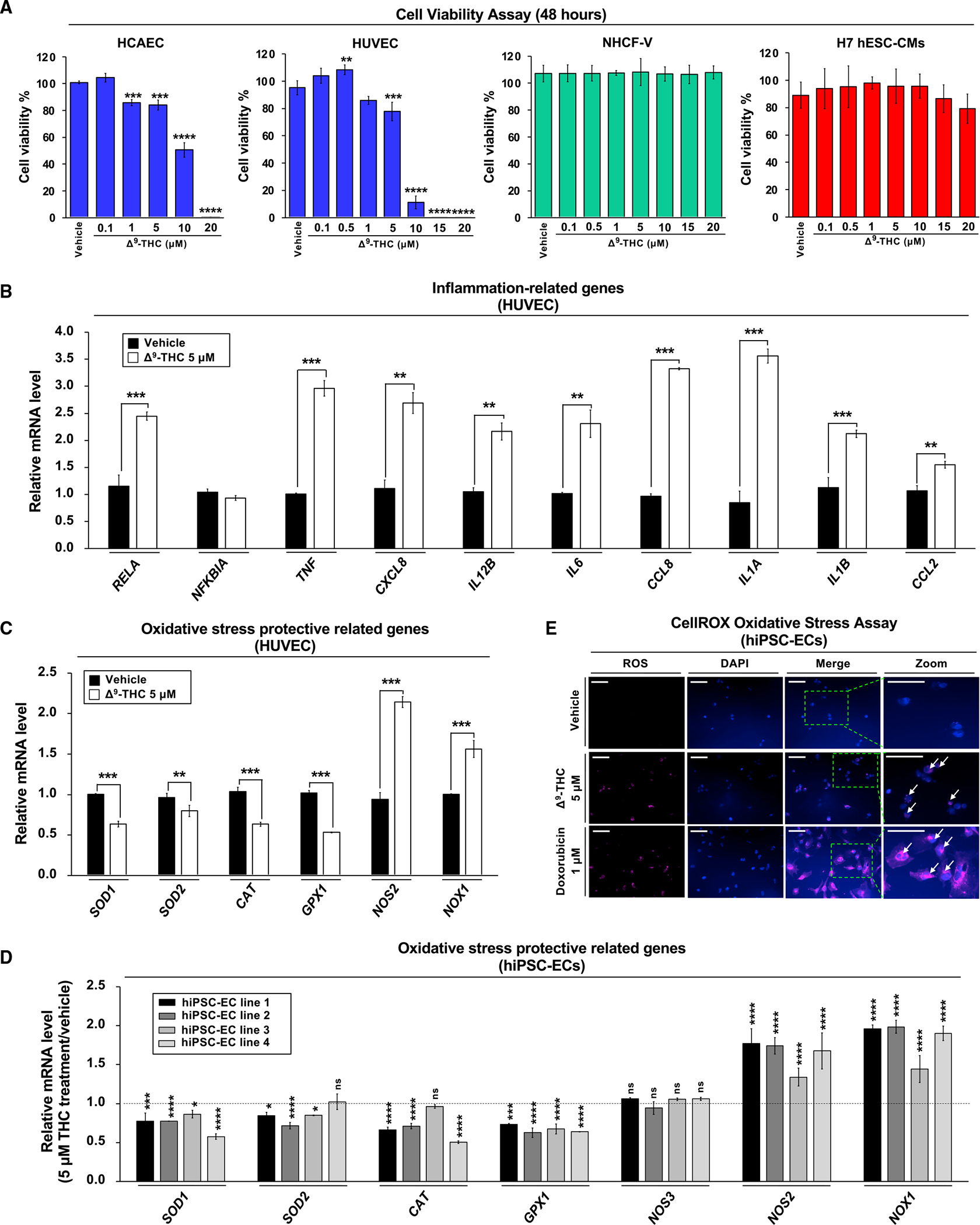
(A) The effects of Δ9-THC on cell viability of human coronary artery endothelial cells (HCAECs), human umbilical vein endothelial cells (HUVECs), normal human cardiac fibroblasts-ventricular (NHCF-V), and human embryonic stem cell-derived cardiomyocytes (hESC-CMs). Cells were treated with increasing concentrations of Δ9-THC for 48 h, and cell viability was measured by the CellTiter-Glo luminescent cell viability assay.
(B and C) (B) The RNA expression of inflammation-related and (C) oxidative stress-related genes in HUVECs. Cells were treated with 5 μM Δ9-THC for 48 h, and gene expression was measured by qPCR and normalized to GAPDH.
(D) Oxidative stress-related gene expression in hiPSC-ECs after Δ9-THC treatment. Cells were treated with 5 μM Δ9-THC for 48 h, and mRNA expression of various genes was measured by qPCR analysis and normalized to GAPDH.
(E) hiPSC-ECs were treated with 5 μM Δ9-THC or 1 μM doxorubicin (positive control) for 48 h and oxidative stress was measured by CellROX oxidative stress assay. Images were obtained by fluorescence microscopy. The white arrowhead indicates cells producing reactive oxygen species (scale bar, 50 μm). Error bars represent mean ± SEM. *p < 0.05 versus vehicle; **p < 0.01 versus vehicle; ***p < 0.001 versus vehicle; ****p < 0.0001 versus vehicle; ns, not significant versus vehicle.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE Antibodies | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| CD144 (VE-Cadherin) MicroBeads | MiltenyiBiotec | 130-097-857 |
| anti-phospho-NF-kB p65 antibody | Cell Signaling | #3033; AB_2341216 |
| anti-NF-κB p65 antibody | Cell Signaling | #8242; AB_10859369 |
| anti-IKB alpha antibody | Abcam | ab32518; AB_733068 |
| anti-phospho-IκBα antibody | Cell Signaling | #9246; RRID: AB_2267145 |
| anti-TLR4 antibody | Abcam | ab89455; RRID: AB_2043061 |
| anti-MyD88 antibody | Cell Signaling | #3699; RRID: AB_2282236 |
| anti-p38 MAPK antibody | Cell Signaling | #8690; RRID: AB_10999090 |
| anti-phospho-p38 MAPK antibody | Cell Signaling | #9211; RRID: AB_331641 |
| anti-F4/80 antibody | Abcam | ab111101; RRID:AB_10859466 |
| anti-CD68 antibody | Bio-Rad | MCA1957; RRID:AB_322219 |
| anti-beta actin antibody | GeneTex | GTX109639; RRID:AB_1949572 |
|
| ||
| Bacterial and virus strains | ||
|
| ||
| Escherichia coli DH5a competent cells | Zymo Research | T3007 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Δ9-Tetrahydrocannabinol (THC) | Sigma-Aldrich | T-005 |
| Genistein | Cayman Chemicals | 10005167 |
| Matrigel® Matrix Basement Membrane Growth Factor Reduced | Corning | 356231 |
| Matrigel® Basement Membrane Matrix | Corning | 354234 |
| Y-27632 2HCl (ROCK Inhibitor) | Selleck Chemicals | S1049 |
| CHIR-99021 (CT99021) HCl 5mg | Selleck Chemicals | S2924 |
| IWR-1 | Selleck Chemicals | S7086 |
| B-27 Supplement, minus insulin | Thermo Fisher Scientific | A1895601 |
| B-27 Supplement (50x), serum free | Thermo Fisher Scientific | 17504044 |
| Recombinant Human FGF-2 | PeproTech | 100-18B |
| Recombinant Human VEGF | R&D Systems | 293-VE-010/CF |
| EGM-2 Endothelial Cell Growth Medium-2 Bullet Kit | Lonza | CC-3162 |
| Doxycycline | Sigma-Aldrich | D3072 |
| Polyethylenimine HCl MAX | Polysciences | 24765-1 |
| Polybrene | Santa Cruz Biotechnology | sc-134220 |
| Puromycin | Cayman Chemical Company | 13884 |
| Oil Red O | Sigma-Aldrich | O0625 |
| BstXI | Thermo Fisher Scientific | FD1024 |
| BlpI | NEB | R0585S |
| DNA ligation kit | Takara Bio | 6023 |
| Lipofectamine 2000 | Thermo Fisher Scientific | 11668019 |
| CellTiter-Glo 2.0 Assay | Promega | G9241 |
| ROS-Glo™ H2O2 Assay | Promega | G8820 |
| GTPase-Glo™ Assay | Promega | V7681 |
| Cell ROX™ Deep Red Reagent, for oxidative stress detection | Thermo Fisher Scientific | C10422 |
| Calcein-AM | R&D Systems | 4892-010-01 |
| TRIzol™ Reagent | Thermo Fisher Scientific | 15596026 |
| RIPA buffer | Thermo Fisher Scientific | 89901 |
| NuPAGE 4-12% Mini protein gels | Thermo Fisher Scientific | NP0321BOX |
| TaqMan™ Universal PCR Master Mix | Thermo Fisher Scientific | 4305719 |
| TaqMan™ Fast Advanced Master Mix | Thermo Fisher Scientific | 4444557 |
|
| ||
| Deposited data | ||
|
| ||
| Olink and Luminex (https://data.mendeley.com/datasets/s6j8h9yy57/1) | Mendeley Data | (https://doi.org/10.17632/s6j8h9yy57.1) |
| Bulk RNA sequencing of EHTs | NCBI | GSE198578 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| Human iPSC Line 1 | Stanford Cardiovascular Institute (SCVI) Biobank | SCVI15 |
| Human iPSC Line 2 | SCVI Biobank | SCVI113 |
| Human iPSC Line 3 | SCVI Biobank | SCVI273 |
| Human iPSC Line 4 | SCVI Biobank | SCVI274 |
| CRISPRi iPSC line | Mandegar et al. 2016 | Gen1C and Gen2C |
| Human coronary artery endothelial cells (HCAEC) | The American Type Culture Collection (ATCC) | PCS-100-020 |
| Human umbilical vein endothelial cells (HUVEC) | ATCC | CRL-1730 |
| Human cardiac fibroblasts-ventricular (NHCF-V) | Lonza | CC-2904 |
| Human erythroleukemia cells (HEL92.1.7) | ATCC | TIB-180 |
| Human neuroblastoma cells (SK-N-FI) | ATCC | CRL-2142 |
| U-937 monocytes | ATCC | CRL-1593.2 |
| H7 Human embryonic stem cells | Wicell | WA07 (H7) |
| HEK293T packaging cells | Takara Bio | 632180 Lenti-X™ 293T Cell Line |
| CB1-expressing Chem-1 cells | EMD Millipore | HTS019RTA |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| Mouse: wild type | The Jackson Laboratory | C57BL/6J |
| Mouse: LDLR knockout | The Jackson Laboratory | B6.129S7-Ldlr<tm1Her>/J |
| Mouse: C57BL/6-Apoe em1Narl/Narl (Apoe knockout mice) | National Laboratory Animal Center (Taiwan) | RMRC13302 |
| Mouse: BALB/cAnN.Cg-Foxn1nu/CrlNarl (Nude mice) | National Laboratory Animal Center (Taiwan) | RMRC12005 |
|
| ||
| Oligonucleotides | ||
|
| ||
| Protospacer sequences of sgRNAs: GGACTAAGCGCAAGCACCTA (control sgRNA); GACAGCGCGAGCGGAGGGAG (CB1 sgRNA#1); GCGCGGGAGACAAGAAGAGG (CB1 sgRNA#2) | Addgene #83969 | N/A |
| ON-TARGET plus Human CNR1 (1268) siRNA-SmartPool: GCGAGAAACUGCAAUCUGU (CB1 siRNA#1); GACCAUAGCCAUUGUGAUC (CB1 siRNA#2); GGACAUAGAGUGUGUUUCAUG (CB1 siRNA#3); CAAGAGCACGGUCAAGAUU (CB1 siRNA#4) | Dharmacon | L-004711-00-0005 |
| ON-TARGETplus Non-targeting Control Pool | Dharmacon | D-001810-10-20 |
|
| ||
| Recombinant DNA | ||
|
| ||
| sgRNA backbone plasmid | Addgene | #60955 |
| psPAX2 (Lentiviral packaging plasmid) | Addgene | #12260 |
| pMD2.G (VSV-G envelope expressing plasmid) | Addgene | #12259 |
|
| ||
| Software and algorithms | ||
|
| ||
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| R version 3.5.3 | The R Project | https://www.r-project.org/ |
| BROOD version 3.0 | Open Eye Scientific Software | N/A |
| ROCS version 2.3 | Open Eye Scientific Software | N/A |
|
| ||
| Other | ||
|
| ||
| High fat diet (40% fat, 1.25% cholesterol) | Research Diets, Inc | D12079B, modified |
| Osmotic pumps | Alzet | Model 2006 |
| Feeding needle | Fine Science Tools | 18065-20 |
| Non-invasive blood pressure system | Kent Scientific Corporation | CODA-HT4 |
| Silicone rack | EHT Technologies | C0001 |
| Teflon spacer | EHT Technologies | C0002 |
| Measurement of lipid profiles in mice | Stanford Animal Diagnostic Lab | N/A |
| SWEETLEAD | SWEETLEAD Database (Stanford University) | N/A |
| UK Biobank | UK Biobank | N/A |
Cannabinoids exert their effects through the CB1 and CB2 receptors. We, therefore, evaluated the expression of cannabinoid receptors in hiPSC-ECs using hiPSC-derived neurons and human erythroblast cells (HEL92.1.7) as positive controls. The CB1 receptor was expressed in hiPSC-ECs, but CB2 receptor expression was not detected in our assays (Figure S1D). Thus, hiPSC-ECs can model the effects of Δ9-THC on the vasculature via the CB1 receptor. hiPSC-ECs can also reflect the paucity of CB2 receptor expression found in adult vascular endothelial cells.
Because antioxidant-related genes were reduced by Δ9-THC treatment in HUVECs, we evaluated those genes in hiPSC-ECs and found that Δ9-THC also decreased mRNA expression of antioxidant-related genes, including superoxide dismutase 1 (SOD1), superoxide dismutase 2 (SOD2), catalase (CAT), and glutathione peroxidase 1 (GPX1) (Figure 3C). In addition, ROS-related genes implicated in endothelial dysfunction (Förstermann and Sessa, 2012), such as NADPH oxidase 1 (NOX1) and inducible nitric oxide synthase (NOS2), were upregulated in hiPSC-ECs after Δ9-THC treatment (Figure 3D). Immunofluorescence staining showed that Δ9-THC treatment of hiPSC-ECs also induced cellular oxidative stress with increased hydrogen peroxide (H2O2) levels (Figures 3E and S2A).
Δ9-THC induces inflammation and NF-κB activation in hiPSC-ECs
Media reports have touted the anti-inflammatory effects of cannabis and Δ9-THC despite a paucity of data (Nagarkatti et al., 2009; Klein and Newton, 2007). We, therefore, sought to add to the literature by investigating the expression of inflammation-related genes in hiPSC-ECs after treatment with Δ9-THC. We observed that proinflammatory cytokines and chemokines were increased in response to Δ9-THC (Figure 4A), while I kappa B (NFKBIA), a specific inhibitor of the NF-κB transcription factor, was inhibited by Δ9-THC (Figure 3A). TNF-α contributes to vascular dysfunction (Boesten et al., 2005). After Δ9-THC treatment, TNF-α concentration was increased in the cell culture medium of hiPSC-ECs (Figure 4B). Monocyte adhesion to endothelial cells is associated with the development of atherosclerosis (Mestas and Ley, 2008). We examined the adhesion of monocytes to hiPSC-ECs after Δ9-THC treatment and found that monocytes had increased adhesion to hiPSC-ECs (Figures 4C and 4D).
Figure 4. Δ9-THC causes inflammation in hiPSC-ECs, TNF-α release, and monocyte adhesion.
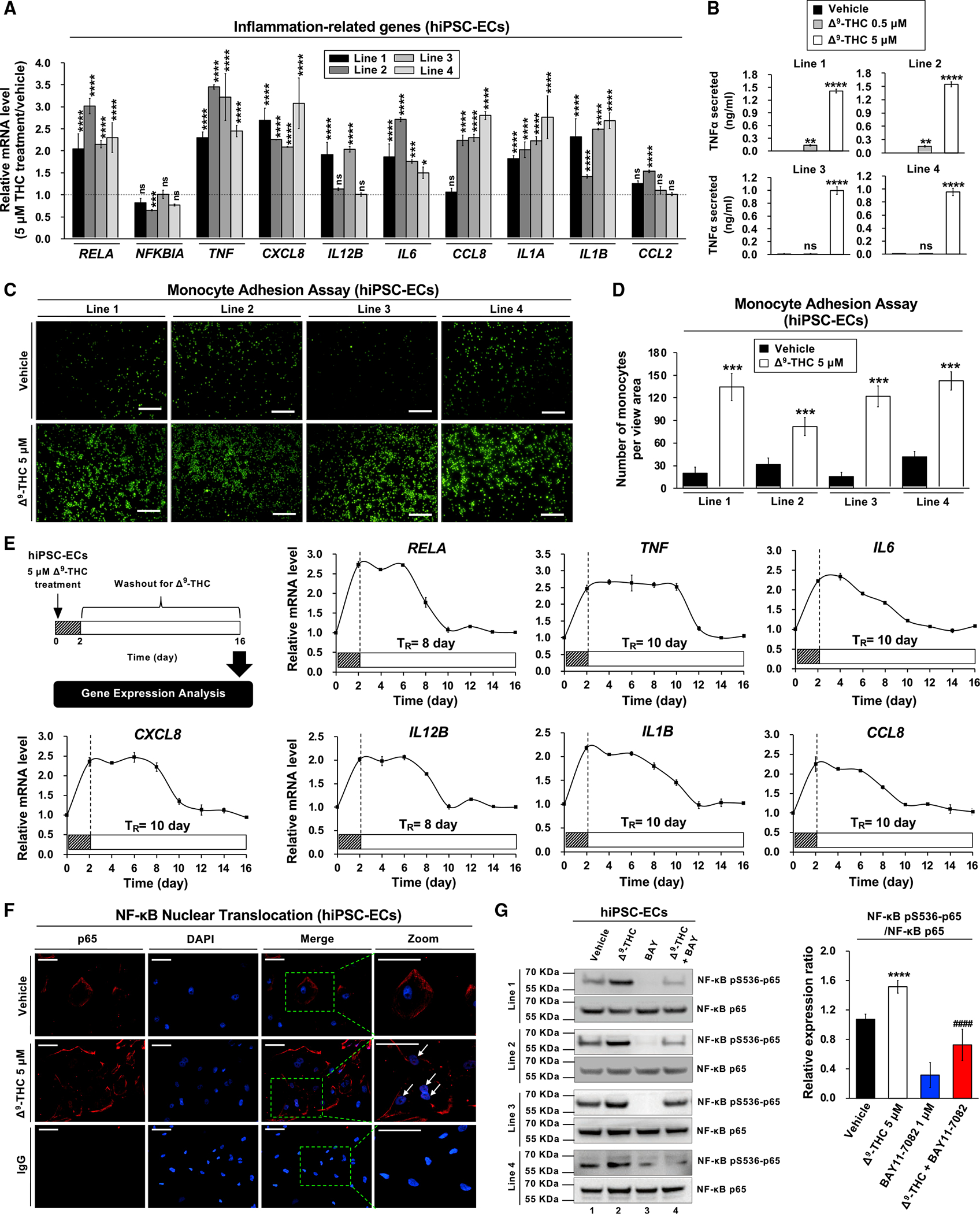
(A) RNA expression of inflammation-related genes in hiPSC-ECs after Δ9-THC treatment for 48 h, as determined by qPCR normalized to GAPDH.
(B) Δ9-THC promoted the release of TNF-α from hiPSC-ECs. Cells were treated with 0.5 or 5 μM Δ9-THC for 48 h, and TNF-α concentration in the cell culture medium was measured by ELISA.
(C) Monocyte adhesion assays of hiPSC-ECs treated with 5 μM Δ9-THC for 48 h. U937 adherence was observed by a fluorescence microscope.
(D) The intensity of fluorescence-labeled-adherent U937 monocytes was measured.
(E) The effect of Δ9-THC on the inflammation-related gene expression in hiPSC-ECs. Cells were treated with 5 μM Δ9-THC for 48 h. The medium was replaced with fresh cell culture medium, and total RNA was isolated from cells every other day for 14 days. Expression of inflammation-related genes was quantified by qPCR analysis and normalized to GAPDH. The retention time (TR) means the time required for the recovery of mRNA basal level.
(F) The effects of Δ9-THC on p65 nuclear translocation in hiPSC-ECs were determined in co-localization studies. hiPSC-ECs were treated with 5 μM Δ9-THC for 48 h and then assayed with anti-p65 antibody for localization of p65 (red fluorescence). The nuclei were counterstained with DAPI (blue fluorescence), and cells were visualized using immunofluorescence microscopy; merged images are shown. The white arrowhead indicates co-localization (purple) of p65 (red) and DAPI (blue) (scale bar, 50 μm).
(G) Δ9-THC caused p65 phosphorylation in hiPSC-ECs. Cells were treated with 5 μM Δ9-THC, 1 μM BAY11–7082, or their combination for 48 h. Western blot analysis was performed on total cell lysates using indicated antibodies (left panel). Images were quantified by ImageJ software (right panel). Error bars represent mean ± SEM. *p < 0.05 versus vehicle; **p < 0.01 versus vehicle; ***p < 0.001 versus vehicle; ****p < 0.0001 versus vehicle; ns, not significant versus vehicle; ####p < 0.0001 versus Δ9-THC.
The longitudinal effects of Δ9-THC-induced inflammation were assessed in hiPSC-ECs using wash-out experiments. After treatment with Δ9-THC for 48 h, the supernatant in hiPSC-ECs was replaced with fresh medium. The expression of inflammation-related genes in hiPSC-ECs was evaluated every other day for 2 weeks. The Δ9-THC-induced expressions of proinflammatory cytokines and chemokines were sustained for 8 to 10 days after the initial exposure (Figure 4E). Thus, Δ9-THC-induced inflammation persisted four to five times longer than the initial exposure.
We next sought to elucidate the mechanisms of how Δ9-THC causes oxidative stress in the vasculature via CB1. The elevated levels of TNF-α suggested that NF-κB could cause activation of inflammatory genes and promote oxidative stress via the nuclear translocation of the p65 component of the complex (Mattioli et al., 2004). The Δ9-THC-binding activates the mitogen-activated protein (MAP) kinase pathway, which is known to regulate NF-kB by increasing transcription of NF-kB and facilitating NF-kB translocation to the nucleus (Saha et al., 2007). In hiPSC-ECs, Δ9-THC also causes increased phosphorylation of p38 (Figure S2H). The cellular distribution of NF-κB was subsequently investigated in hiPSC-ECs after Δ9-THC treatment. Immunofluorescent staining revealed that Δ9-THC induced NF-κβ’s nuclear translocation of NF-κB (Figure 4F) and phosphorylation of NF-κB in hiPSC-ECs. The Δ9-THC-mediated phosphorylation of NF-κB was abrogated by the selective NF-κB inhibitor BAY11–7082 (Figures 4G and S2I).
Genistein reverses Δ9-THC-induced oxidative stress and inflammation in hiPSC-ECs by inhibiting CB1
The CB1 receptor is implicated in the pathological effects of Δ9-THC on the vasculature. We next employed pharmacologic and genetic inactivation of the CB1 receptor to determine whether the CB1 receptor plays a role in Δ9-THC-induced effects in hiPSC-ECs. The selective CB1 antagonists AM6545, AM251, and NESS-0327 reversed Δ9-THC-induced mRNA expression of inflammation-related genes and oxidative stress protective-related genes (Figure S3A). The Δ9-THC-induced effects were also reversed by the knockdown of CB1 by siRNA (Figure S3B). Similarly, CRISPR interference (CRISPRi) of CB1 expression reversed Δ9-THC-induced inflammation and oxidative stress in hiPSC-ECs (Figures 5A and 5B). Genistein is known to ameliorate TNF-α-mediated inflammation and oxidative stress in endothelial cells (Jia et al., 2013). We, therefore, tested if the CB1 receptor was required for genistein to block the effects of TNF-α in hiPSC-ECs using CRISPRi to assess CB1 expression. We discovered that the CB1 receptor was required for genistein to attenuate the inflammatory effects of TNF-α (Figures S3C–S3F).
Figure 5. Inhibition of CB1 or genistein treatment mitigates the effects of Δ9-THC on hiPSC-ECs.
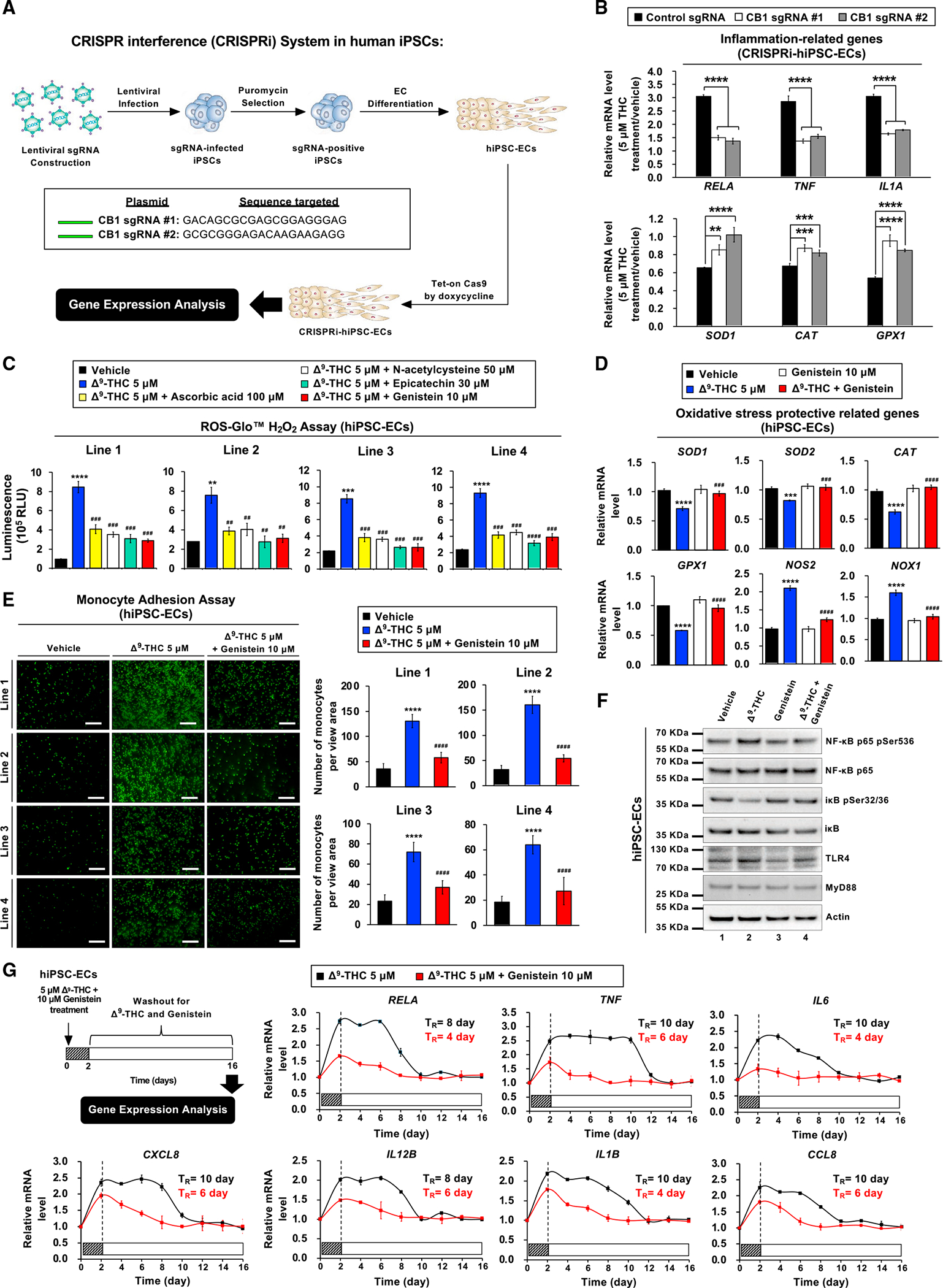
(A) Schematic overview of the CRISPR interference (CRISPRi) in hiPSC-ECs.
(B) hiPSC-ECs were treated with 5 μM Δ9-THC for 48 h. The mRNA expression of inflammation-related genes and oxidative stress protective-related genes in hiPSC-ECs treated with sgRNA versus control was quantified by qPCR analysis and normalized to GAPDH.
(C) Compounds that mitigate reactive oxygen species production caused by Δ9-THC in hiPSC-ECs were screened. The cells were treated with 5 μM Δ9-THC plus antioxidant reagent or vehicle control for 48 h, and the level of hydrogen peroxide was measured by ROS-Glo™ H2O2 assay.
(D) Expression of oxidative stress protective-related genes in hiPSC-ECs as determined by qPCR analysis. The hiPSC-ECs were treated with 5 μM Δ9-THC, 10-μM genistein, or their combination for 48 h, and normalized to GAPDH.
(E) Genistein blocked monocyte adhesion in hiPSC-ECs treated with Δ9-THC. hiPSC-ECs were treated with 5 μM Δ9-THC and 10 μM genistein or vehicle (control) for 48 h. U937 cells adherence to hiPSC-ECs was visualized by fluorescence microscope (left panel) and the intensity was quantified (right panel).
(F) Genistein prevents Δ9-THC-induced NF-κB phosphorylation in hiPSC-ECs. Cells were treated with 5 μM Δ9-THC,10 μM genistein, or their combination for 48 h. Total cell lysates were subjected to western blot analysis.
(G) Genistein attenuates Δ9 -THC-induced inflammation in hiPSC-ECs. The cells were treated with 5μM Δ9-THC and 10 μM genistein for 48 h and then replaced with fresh cell culture medium. Total RNA was isolated from hiPSC-ECs every other day for 2 weeks. The mRNA expression of inflammation-related genes was quantified by qPCR and normalized to GAPDH. The retention time (TR) is the time required for the recovery of gene expression to basal level. Error bars represent mean ± SEM. *p < 0.05 versus vehicle; **p < 0.01 versus vehicle; ***p < 0.001 versus vehicle; ****p < 0.0001 versus vehicle; ns, not significant versus vehicle; ##p < 0.01 versus Δ9-THC; ###p < 0.001 versus Δ9-THC; ####p < 0.0001 versus Δ9-THC.
We next experimentally validated that the putative CB1 antagonist genistein prevents vascular dysfunction caused by Δ9-THC. We screened a series of antioxidant reagents and found that genistein was the best at preventing Δ9-THC-induced oxidative stress in hiPSC-ECs (Figures 5C and S4A). In hiPSC-ECs, genistein ameliorated the expression profile of antioxidant genes suppressed by Δ9-THC while also having a salutatory effect on NOS2 and NOX1 expression (Figure 5D).
With an attenuated inflammatory profile, we postulated that hiPSC-ECs treated with genistein and Δ9-THC were more likely to remain quiescent without contributing to the pathogenesis of atherosclerosis. We tested this hypothesis using monocyte adhesion assays. Genistein not only reversed Δ9-THC-induced monocyte adhesion to hiPSC-ECs but also attenuated NF-κB phosphorylation (Figures 5E and 5F).
Toll-like receptors (TLRs) are components of the innate immune system that recognize molecular patterns of microbial components, and TLR4/NF-κB signaling pathways can contribute to vascular inflammation in endothelial cells (Akira et al., 2006). We, therefore, investigated the effects of Δ9-THC and the anti-inflammatory properties of genistein on the TLR4/NF-κB signaling pathway. We observed that Δ9-THC induced TLR4 expression in hiPSC-ECs, whereas genistein disrupted its expression (Figures 5F and S4B). Moreover, genistein reversed the Δ9-THC-induced mRNA expression of proinflammatory cytokines and chemokines (Figure S4C). Cotreatment with genistein also shortened the recovery time of Δ9-THC-induced inflammation from 8–10 days to 4–6 days (Figure 5G), and treatment with genistein after Δ9-THC exposure also reduced the recovery time to 6–8 days (Figure S4D). These experimental results indicated that genistein attenuated Δ9-THC-induced oxidative stress and inflammation in hiPSC-ECs.
Genistein reverses Δ9-THC-induced endothelial dysfunction, oxidative stress, and inflammation in a mouse model
To evaluate the effects of genistein in vivo, male C57BL/6J mice were treated with vehicle, Δ9-THC, or Δ9-THC plus genistein (Figure 6A). While oral ingestion or smoking are more common routes of ingestion in humans, our mouse model produced a plasma concentration of Δ9-THC of ~100 ng/mL as revealed by LC-MS analysis, which is comparable to the concentration from smoking a single marijuana cigarette (Huestis and Cone, 2004) (Figures S5A and S5B). Wire myograph revealed that Δ9-THC induced endothelial dysfunction in mice, whereas genistein mitigated the effect (Figure 6B).
Figure 6. Genistein blocks Δ9-THC-induced endothelial dysfunction.
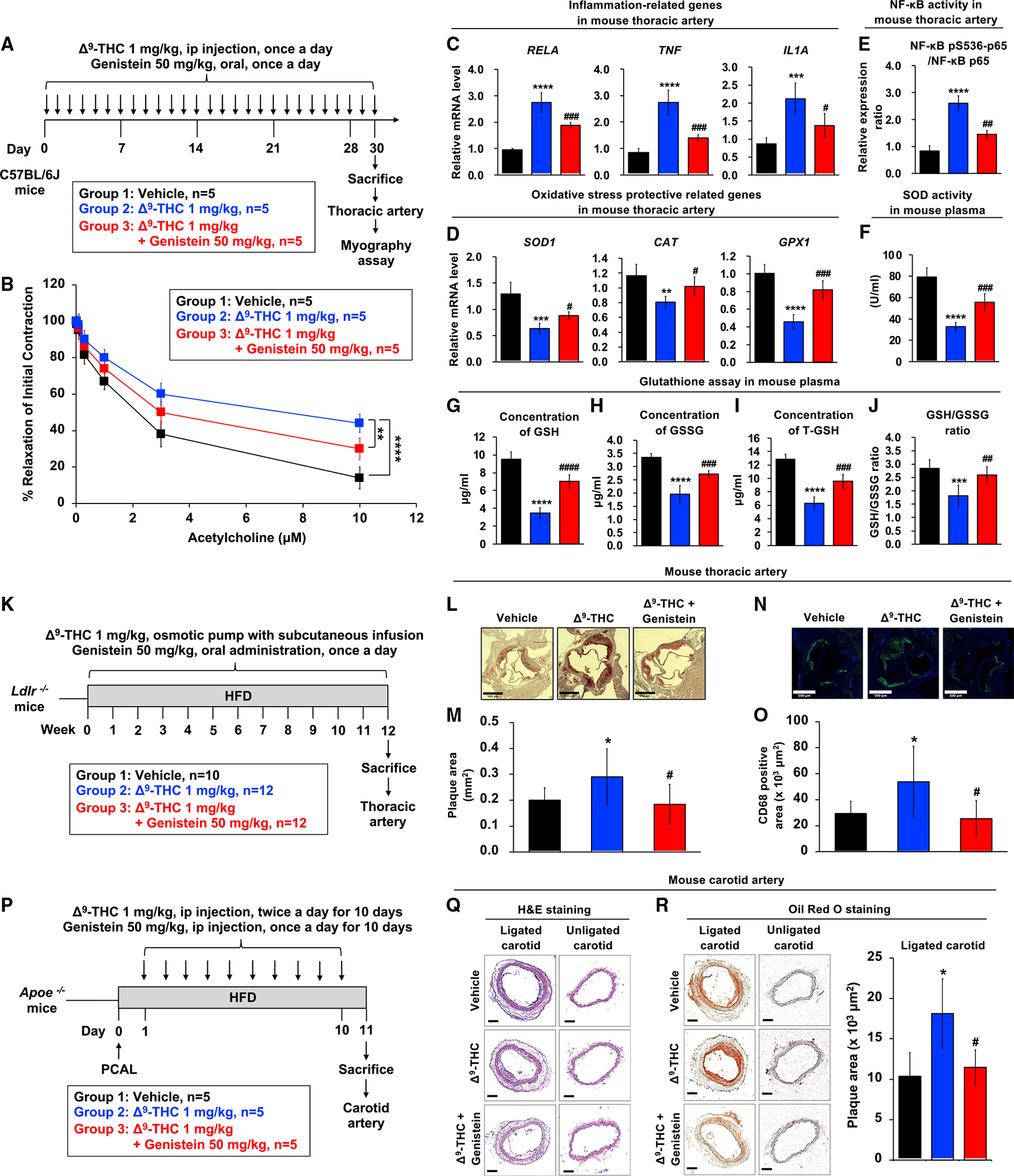
(A) Schematic overview of the wire myograph experimental design in the mouse model.
(B) Isometric tension recordings of isolated mice thoracic aortas were performed using a wire myograph. Vascular concentration-dependent relaxation was induced by acetylcholine (ACh) in pre-constricted mouse thoracic arteries.
(C and D) (C) The mRNA expression of inflammation-related genes and (D) oxidative stress protective-related genes in thoracic artery tissues from mice is shown after normalizing to GAPDH.
(E) Effect of Δ9-THC and genistein on NF-κB phosphorylation in mouse thoracic artery. Total cell lysates were prepared, and the expressions of phosphor-NF-κB were analyzed by ELISA.
(F) Superoxide dismutase (SOD) activity of serum from mouse.
(G) Reduced glutathione (GSH) levels in the serum samples of mice were detected. Plasma isolated from C57BL/6J mice (n = 5) treated with vehicle control, Δ9-THC, genistein, or their combination every day for 30 days was analyzed by the glutathione colorimetric assay kit (BioVision, K261).
(H) Oxidized glutathione (GSSG) levels in the serum samples of mice.
(I) Total glutathione (T-GSH) levels in the serum samples of mice.
(J) The GSH/GSSG ratio in the serum samples of mice.
(K) Schematic overview of the chronic atherosclerosis model. Ldlr−/− mice (9–12 weeks old) were divided into three groups: (1) vehicle control (n = 10), (2) Δ9-THC (n = 12), and (3) Δ9-THC plus genistein (n = 12). Δ9-THC (1 mg/kg/day) or vehicle (90% saline, 5% ethanol, 5% cremophor) was administered subcutaneously using osmotic pumps, and genistein (50 mg/kg/day) or vehicle (corn oil 100 μL/d) was orally administered daily. All experimental animals were fed with a high-fat diet (HFD) for the duration of the treatment protocol. At the end of 12 weeks, the mice were euthanized to determine the extent of atherosclerotic plaque formation.
(L) Oil red O staining of atherosclerotic plaques in cross-sections at the aortic root level with scale bars at 500 μm.
(M) Quantitation of atherosclerotic plaques: (1) vehicle control (n = 10), (2) Δ9-THC (n = 10), and (3) Δ9-THC plus genistein (n = 11).
(N) Immunostaining of CD68 in cross-sections at the aortic root level with scale bars at 500 μm.
(O) Quantitation of CD68-positive area from (1) vehicle control (n = 10), (2) Δ9-THC (n = 9), and (3) Δ9-THC plus genistein (n = 10).
(P) Schematic overview of the experimental design in the Apoe−/− mouse model. Partial carotid artery ligation (PCAL) was performed in Apoe−/− mice (10–16 weeks old). One day after PCAL, Apoe−/− mice were divided into three groups (n = 5/group): (1) vehicle control, (2) Δ9-THC (1 mg/kg intraperitoneally, twice daily for a total of 10 days), and (3) Δ9-THC (1 mg/kg intraperitoneally, twice daily for a total of 10 days) plus genistein (50 mg/kg intraperitoneally, once daily for a total of 10 days). All experimental animals were fed with a high-fat diet (HFD) following PCAL. After 10 days of HFD and treatment exposure, carotid atherosclerosis plaque burden was assayed in all three groups.
(Q) Carotid artery sections were counterstained with hematoxylin and eosin (H&E), and a representative slide was presented with scale bars at 100 μm.
(R) Oil red O staining of atherosclerotic plaques in cross-section of mouse carotid artery (lower panel) with scale bar at 100 μm. The atherosclerotic plaques were quantified. Error bars represent mean ± SEM. *p < 0.05 versus vehicle; **p < 0.01 versus vehicle; ***p < 0.001 versus vehicle, ****p < 0.0001 versus vehicle; ns, not significant versus vehicle; #p < 0.05 versus Δ9-THC; ##p < 0.01 versus Δ9-THC; ###p < 0.001 versus Δ9-THC; ####p < 0.0001 versus Δ9-THC.
Genistein and the known CB1 antagonist rimonabant ameliorated mRNA expressions of inflammation-related genes induced by Δ9-THC in mouse thoracic artery tissues (Figures 6C and S5C). Analysis of C57BL/6J plasma revealed that IL-6, IL-3, and IL-10, which are associated with an increased risk of atherosclerosis (Ridker et al., 2018; Welsh et al., 2011; Brizzi et al., 2001), were elevated after treatment with Δ9-THC, and genistein cotreatment significantly reduced the expression of these inflammatory cytokines (Figure S5D). Genistein also rescued the oxidative stress protective-related gene expression (Figure 6D). Expression of inflammation and oxidative stress protective-related genes was correlated with vascular relaxation (Figure S5E). NF-κB phosphorylation and SOD expression in mouse serum were also attenuated with genistein (Figures 6E, 6F, and S5F). Δ9-THC administration decreased circulating levels of the antioxidant glutathione, which was ameliorated with genistein cotreatment (Figures 6G–6J). Collectively, these results indicated that genistein could reverse Δ9-THC-induced effects in vivo.
While genistein had a protective effect on the vasculature, it did not exhibit any toxicity based on body weight and hematoxylin and eosin (H&E) staining of various organs (Figure S5G). We used engineered heart tissues (EHTs) composed of hiPSC-ECs and hiPSC-derived cardiomyocytes (hiPSC-CMs) to interrogate the effects of genistein and Δ9-THC on contractility in vitro (Videos S1, S2, and S3). We found Δ9-THC and genistein cotreatment had no significant effect on contraction, relaxation, and peak force (Figures S2B, S2C, and S2F). Δ9-THC decreased beat rate and increased the contraction-relaxation interval, and genistein cotreatment did not attenuate these effects (Figures S2D and S2E). Bulk RNA sequencing analysis of EHT did not reveal any significant transcriptomic signature implicated in cardiac function, but some pathways were downregulated in response to Δ9-THC treatment compared with vehicle control (Figure S2G). Reflecting the EHT studies, echocardiographic analysis of C57BL/6J revealed that Δ9-THC, genistein, and cotreatment with genistein did not affect cardiac structure or function (Figures S6M–S6P), with no significant difference in ejection fraction (EF), heart rate (HR), left ventricular end-diastolic volume (LVEDV), and left ventricular end-diastolic mass (LVEDM). The CB1 antagonist rimonabant, however, reduced HR when used alone or in cotreatment with Δ9-THC but did not affect other cardiac parameters.
Effect of THC and genistein on atherosclerosis
To investigate the effect of Δ9-THC and genistein in chronic atherosclerosis formation, LDL receptor knockout mice (B6.129S7-Ldlrtm1Her/J) were fed a high-fat diet (HFD) for 12 weeks and treated with (1) vehicle control, (2) Δ9-THC, or (3) Δ9-THC and genistein (n = 10–12/group) (Figure 6K). All groups showed an increase in body weight during the HFD period (Figure S7A). The successful administration of Δ9-THC was confirmed in serum by LC-MS at 12 weeks (Figures S7B and S7C). Neither Δ9-THC nor cotreatment with Δ9-THC and genistein affected serum lipid profiles or blood pressure (Figures S7D and S7E). After 12 weeks of HFD, the mice were euthanized, and the atherosclerotic lesion area was examined by cross-sections of the aortic root and an en face analysis of the thoracic aorta. In the cross-sectional analysis, mice treated with Δ9-THC showed significantly increased plaque size, and cotreatment with genistein ameliorated plaque size when stained with oil red O (Figures 6L and 6M). The improvement of plaque formation by genistein was also observed in en face analysis of the thoracic aorta stained with oil red O (Figures S7F and S7G). Macrophage recruitment in the aortic root was analyzed using aortic plaque stained with an anti-CD68 antibody. The CD68-positive area was significantly increased by Δ9-THC administration and rescued by cotreatment with genistein (Figures 6N and 6O). These results suggest that Δ9-THC exacerbated atherosclerosis formation and macrophage recruitment in atherosclerotic plaques, which were ameliorated by genistein cotreatment.
An Apoe−/− mouse model was also employed to investigate the effects of Δ9-THC and cotreatment with genistein on atherosclerosis. Partial carotid artery ligation (PCAL) was performed in Apoe−/− mice at 10–16 weeks. After PCAL, the Apoe−/− mice were divided into three groups: (1) vehicle control, (2) Δ9-THC, and (3) Δ9-THC plus genistein (n = 5/group). All mice were fed a HFD for 10 days (Figure 6P). At the end of the treatment protocol, the Apoe−/− mice were euthanized and subjected to histological analysis. The ligated carotid artery showed increased fat deposition with oil red O staining compared with the unligated, contralateral vessel (Figure S6J). H&E staining suggested neointimal thickening, fat deposition, and increased macrophage recruitment in ligated carotid atherosclerotic plaques (Figure 6Q). Oil red O staining found that the plaque area was increased by Δ9-THC treatment, whereas Δ9-THC and genistein cotreatment ameliorated the plaque size (Figure 6R). Macrophage recruitment was tested using the macrophage-specific F4/80 antibody and CD68 antibody (Figures S6K and S6L). In the ligated carotid artery, the F4/80-positive area was significantly increased by Δ9-THC administration and decreased by cotreatment with genistein. Immunohistochemical analysis with CD68-specific antibody revealed that the CD68-positive area was significantly increased by Δ9-THC administration and reduced by cotreatment with genistein. Consistent with the Ldlr−/− model findings, the Apoe−/− PCAL mouse model found increased plaque size and increased macrophage recruitment with Δ9-THC, and genistein cotreatment ameliorated these indices of atherosclerosis in ligated carotid arteries.
Genistein binds to the CB1 receptor and does not attenuate Δ9-THC neurobehavioral effects in vivo
After finding that genistein attenuated the vascular dysfunction caused by Δ9-THC in both in vitro and in vivo studies, we found direct evidence of genistein-binding CB1 using fluorescently labeled genistein (BODIPY517/547-genistein) (Figures 7A and 7B). In CB1 radioligand binding assays, BODIPY517/547-genistein had an IC50 of 375 nM on human CB1 receptors and labeled nearly all hiPSC-ECs cells after 12 h incubation (Figure 7C). BODIPY517/547-genistein cotreatment attenuates Δ9-THC-induced inflammation-related gene expression and NF-κB phosphorylation and ameliorates oxidative stress protective-related gene expression, SOD expression, and glutathione concentration in mouse serum (Figures S6A–S6I). In vivo binding was investigated using intravenously injected BODIPY517/547-genistein in C57BL/6J mice. After 48 h, BODIPY517/547-genistein was detected in the abdominal viscera, thoracic aorta, heart, and lungs but was minimally detected in the brain (Figures 7D–7G). This is consistent with a previous report that shows genistein has poor blood-brain barrier (BBB) penetration (Yang et al., 2012).
Figure 7. In vitro validation, in vivo distribution, and confirmation of in vivo binding to the CB1 receptor using fluorescently labeled genistein.
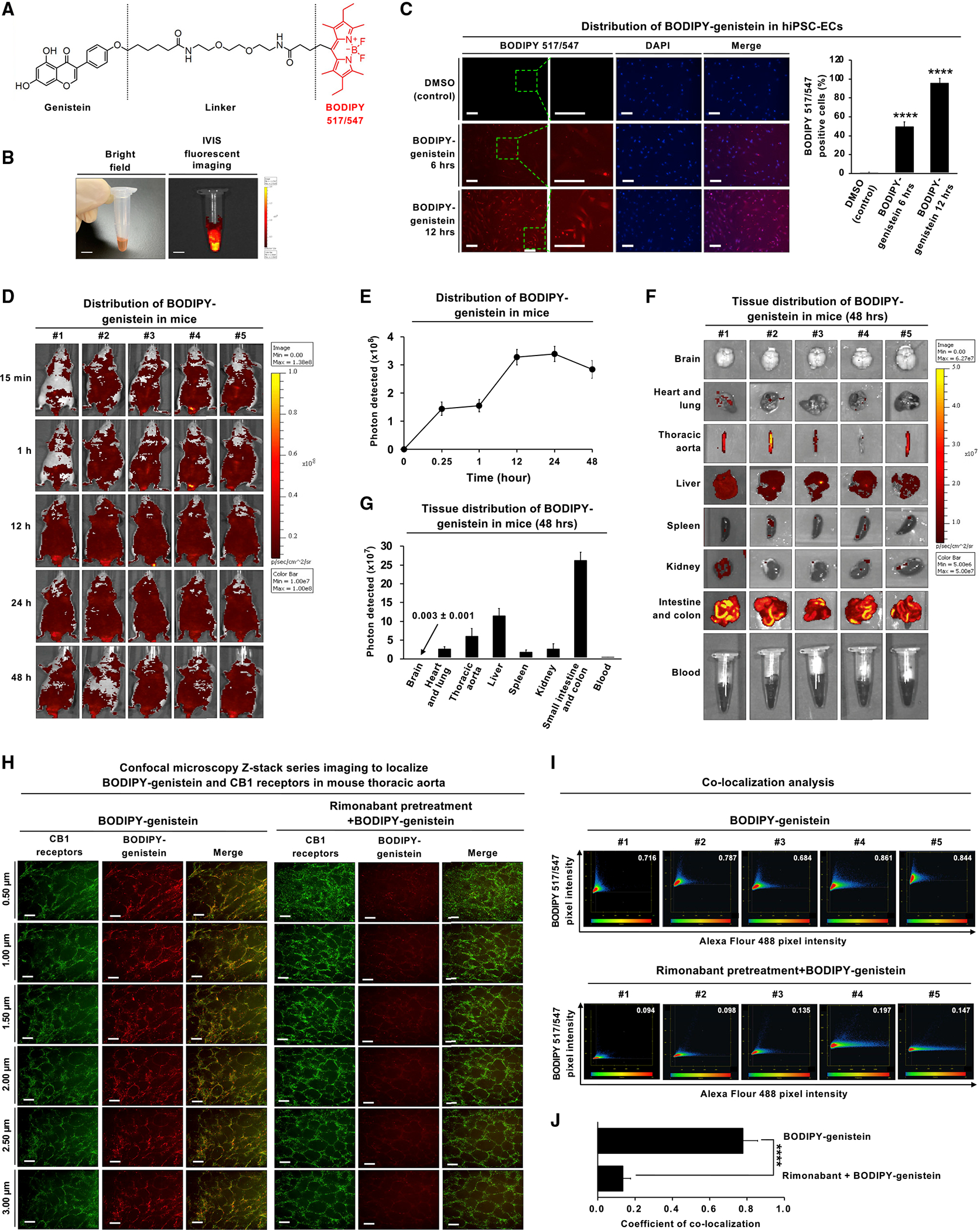
(A) Chemical structure of BODIPY517/547-genistein. The core structures of the BODIPY fluorophore are highlighted in red.
(B) Bright-field image (left panel) and fluorescent image (right panel) of BODIPY517/547-genistein were detected by the IVIS Spectrum in vivo imaging system. The scale bar,5 mm.
(C) The cellular distribution of BODIPY517/547-genistein in hiPSC-ECs. The cells were treated with BODIPY517/547-genistein (5 μM) for 6 or 12 h and subjected to fluorescence microscopy for BODIPY517/547-genistein (red fluorescence) and DAPI (blue fluorescence). Representative fluorescence images of the cells with the inset showing higher magnification as indicated with the green-boxed area. The scale bar,100 μm.
(D) The fluorescence images of BODIPY517/547-genistein after intravenous injection into male nude mice. Adult male BALB/c nude mice (n = 5) were intravenously injected with BODIPY517/547-genistein at a dose of 5 mg/kg. The in vivo fluorescence images were detected by the IVIS Spectrum at indicated time points. All mice were sacrificed at 48 h, and major organs were collected and then fixed using formalin.
(E) The tissue distribution of BODIPY517/547-genistein in mice over 48 h. Fluorescence images were quantified using the IVIS imaging system. The fluorescence intensity peaked at 24 h. The fluorescence images of BODIPY517/547-genistein were quantified by ImageJ software.
(F) Images of the brain, heart and lung, thoracic aorta, liver, spleen, kidney, intestine, colon, and blood were obtained from n = 5 mice. BODIPY517/547-genistein was detected predominately in the intestine and abdominal viscera but with minimal signal in the brain.
(G) BODIPY517/547-genistein accumulated in the small intestine, colon, liver, thoracic artery, heart, and lungbut was minimally detected in the brain. Fluorescence images were quantified using the ImageJ software.
(H) En face immunofluorescence staining was performed to observe the localization of BODIPY517/547-genistein and CB1 receptors in vascular endothelial cells from the mouse thoracic aorta. Mice were either injected with BODIPY517/547-genistein alone or pre-treated with 100 nM rimonabant for 30 min prior to injection of BODIPY517/547-genistein. Mouse aortas were isolated and permeabilized with Triton X100. The aortas were incubated with anti-CB1 antibody Alexa Fluor 488, green. Fluorescent z stack images were collected at 0.5 μm steps by laser scan confocal microscopy. En face immunofluorescence images of BODIPY517/547-genistein (red fluorescence) and CB1 receptor (Alexa Fluor 488, green fluorescence) in vascular endothelial cells after treatment with BODIPY517/547-genistein for 48 h. Scale bar, 20 μm.
(I) Scatter plots of co-localization analysis in vascular endothelial cells from mouse thoracic aorta at 48 h (left panel). The x axis represents CB1 receptors (Alexa Fluor 488, green fluorescence), and the y axis represents BODIPY517/547-genistein (red fluorescence).
(J) Quantification of BODIPY517/547-genistein co-localized with CB1 receptors. The co-localized signal is colored orange. Error bars represent mean ± SEM. *p < 0.05 versus vehicle; **p < 0.01 versus vehicle; ***p < 0.001 versus vehicle; ****p < 0.0001 versus vehicle; ns, not significant versus vehicle.
We next investigated whether genistein is co-localized with the CB1 receptor in vivo using confocal microscopy. Thoracic aortas from mice injected with BODIPY517/547-genistein were permeabilized and stained with an anti-CB1 receptor antibody labeled with Alexa Fluor 488. The mice were imaged with confocal microscopy, and z stacked images revealed co-localization of Alexa Fluor-488-labeled CB1 receptor and BODIPY517/547-genistein in aortic vascular cells (Figures 7H and 7I). The specificity of genistein binding was interrogated by injecting the selective CB1 antagonist rimonabant before BODIPY517/547-genistein. Rimonabant has a stronger binding affinity for the CB1 receptor and reduced BODIPY517/547-genistein binding and fluorescence significantly (0.78 ± 0.08 versus 0.13 ± 0.04, p < 0.00001) (Figure 7J).
Despite BODIPY517/547 having excellent bioavailability (Kwon et al., 2021), genistein has poor BBB penetration, which compelled us to interrogate the neurological effects of Δ9-THC in the context of genistein co-administration. The neurobehavioral effects of Δ9-THC include decreased mobility, analgesia, hypothermia, and sedation. These are described as the Billy Martin tetrad (Little et al., 1988; Fride et al., 2006; Wiley and Martin, 2003). The sedative and analgesic effects of Δ9-THC may have therapeutic benefit (Gurley et al., 1998). While protecting against the detrimental vascular effects, genistein may antagonize the neurobehavioral tetrad effects of Δ9-THC. Therefore, the neurological effects of Δ9-THC and genistein co-administration were tested in C57BL/6J mice treated with vehicle, genistein, Δ9-THC, or in combination. The tetrad effects were assessed using an activity chamber for mobility, a hot plate for analgesia, a rectal probe for hypothermia, and the bar test for sedation. Δ9-THC caused decreased mobility, analgesia, hypothermia, and sedation in C57BL/6J mice (Figures S7H–S7K). Genistein alone did not affect tetrad effects, and genistein could not attenuate the Δ9-THC neurobehavioral effects in cotreatment.
DISCUSSION
With expanding legalization, marijuana use is expected to increase (Cerdá et al., 2020). The long-term cardiovascular implications of cannabis are unclear and may take decades to percolate up through epidemiological studies (Pope et al., 2004). Recently, De-Fillipis et al. found in a cohort of 2,097 patients that marijuana is associated with MI in young adults when controlling for demographic factors (DeFilippis et al., 2018). In this study, the UK Biobank data of 34,878 participants also found marijuana to be associated with MI. Several studies have also identified that the CB1 receptor mediates increased oxidative stress and inflammation as implicated in diabetic retinopathy, cardiomyopathy, and endothelial dysfunction (Bayazit et al., 2017; Mukhopadhyay et al., 2010; El-Remessy et al., 2011; Rajesh et al., 2010b, 2012). CB1 activation occurs via the MAP kinase pathway, which causes oxidative stress, inflammation, and cell death in human coronary artery endothelial cells (Liu et al., 2000; Pertwee et al., 2010; Rajesh et al., 2010b; Park et al., 2011). Here, we confirmed that Δ9-THC activation of CB1 causes inflammation and oxidative stress via MAP kinase, TNF-α, and NF-kB pathways (Figures 6C–6E, S2H,S7l, and S7M). Now, for the first time, we show the link between Δ9-THC-mediated vascular inflammation and oxidative stress, endothelial dysfunction, and atherosclerosis using two independent mouse models. (Figures 6K–6R and S6J–S6P). Using in silico compound screening combined with wet bench validation, we discovered that genistein, a neutral CB1 antagonist, attenuates Δ9-THC-induced endothelial dysfunction.
Δ9-THC and CBD are the two principal components of marijuana. Our focus is on Δ9-THC. While CBD can undergo intramolecular cyclization to form Δ9-THC, CBD does not exert toxicity, and CBD might have therapeutic benefits (Pacher et al., 2018; Rajesh et al., 2010a). Δ9-THC and CBD exert divergent responses, and because both are components of marijuana, their combined effects on the cardiovascular system merit further investigation. CB1 antagonists may have therapeutic effects, but the psychiatric side effects have largely thwarted their translation to the clinic (Onakpoya et al., 2016; Moreira et al., 2009). In this study, we discovered that genistein, a neutral CB1 antagonist, attenuated the adverse effects of Δ9-THC on the vasculature without the psychiatric side effects seen in some other CB1 antagonists. Taken together, the in vivo binding assay and neurobehavioral analysis suggest that genistein inhibits CB1 activity peripherally and has a lower CNS penetration (Gleason et al., 2009).
A pan-assay interference compound (PAIN) or invalid metabolic panacea (IMP) is a chemical compound that gives false-positive results in biochemical assays through various mechanisms (Baell and Holloway, 2010). Genistein is a PAIN and/or an IMP (Bisson et al., 2016). As such, it can aggregate, act as a membrane disruptor, and interfere with luciferase in luminescence-reporting assays. Genistein has a protective effect against Δ9-THC, but we cannot exclude the possibility that genistein may function via other additional mechanisms independent of the CB1 receptor pathway.
The enthusiasm for using Δ9-THC medically in such applications as sedation, analgesia, or anti-nausea (Hill et al., 2017; Badowski, 2017; Cotter, 2009; Baron, 2015) should also be tempered by the awareness of increased cardiovascular risk. This study shows that Δ9-THC might cause adverse cardiovascular effects based on collective data from the UK Biobank, hiPSC-ECs, murine model, and increased atherogenic inflammatory cytokines found in the plasma of marijuana smokers. Clinicians must balance the therapeutic benefits of marijuana with the potential for adverse cardiovascular consequences mediated by Δ9-THC oxidative stress and inflammation. Because of its limited bioavailability in the brain, genistein could mitigate the adverse cardiovascular effects of Δ9-THC and preserve the therapeutic benefits of marijuana that are thought to be centrally mediated.
In conclusion, our findings indicate that Δ9-THC induces inflammation, oxidative stress, endothelial dysfunction, and atherosclerosis through the CB1-dependent signaling pathway. Thus, CB1 antagonists could attenuate Δ9-THC-induced inflammation and may prevent atherosclerosis, but the psychiatric effects are concerning. We show that the neutral CB1 antagonist, genistein, can attenuate the adverse effects of Δ9-THC on the cardiovascular system while preserving the beneficial central effects of Δ9-THC. Further exploration of compounds with homology to genistein is expected to yield more potent CB1 antagonists with similar safety profiles as genistein.
Limitations of the study
In the present study, genistein was discovered to be a ligand for the CB1 receptor using the SWEATLEAD database (Moshiri et al., 2020), which has been validated with virtual-ligand receptor modeling using the Schrödinger suite. We have also conducted wet bench experiments to validate this finding using hiPSC-ECs and a mouse model. iPSCs are a powerful system for modeling disease because they can provide a limitless source of tissue. However, iPSC-ECs are immature, having low eNOS expression and are a mixture of arterial, venous, and lymphatic ECs. Therefore, it was imperative to validate the effects observed in the iPSC-ECs with primary endothelial cells and murine models.
The clinical data from the UK Biobank and the Olink inflammation panel show an increased incidence of MI and acute upregulation of inflammatory cytokines associated with CVD. The Olink data inflammatory markers are associated with MI and are correlative. The UK Biobank study was carried out between 2000 and 2016 when marijuana smoking was illegal. Despite concerns about participants not disclosing illicit cannabis use, an increased incidence of cardiovascular events with a worse prognosis was observed among cannabis users. While cannabis is an independent risk factor when controlled for age and BMI, the confounder of tobacco abuse could not be addressed due to the sample size of the UK Biobank cohort and individuals underreporting illicit cannabis use. The study by DeFilippis et al. showed a significant association between marijuana and CVD in patients presenting with index MI (DeFilippis et al., 2018). The confounder of cigarette smoking is also noted in the DeFilippis et al. study. However, when adjusted for baseline characteristics including cigarette smoking, the DeFilippis et al. study showed that the hazard ratio for marijuana and cardiovascular events was higher than smoking traditional cigarettes alone.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to the lead contact, Dr. Joseph C. Wu (joewu@stanford.edu).
Material availability
This study did not generate new reagents.
Data and code availability
Raw data from the Olink and Luminex analysis were deposited on Medeley https://data.mendeley.com/datasets/s6j8h9yy57/1 and are publicly available as of the date of publication. The RNA sequencing data was deposited at the NCBI (GSE198578). Any additional information required for reanalysis is available upon request.
EXPERIMENTAL MODELS AND SUBJECT DETAILS
Calculation of incidence of myocardial infarction (MI) using UK Biobank
We first determined the number of individuals in the UK Biobank who were cannabis users versus non-cannabis users (http://biobank.ctsu.ox.ac.uk/crystal/field.cgi?id=20453). We identified individuals who responded “no” to having never used cannabis as non-cannabis users. The remaining responses were then segregated based on cannabis frequency use. We identified individuals who responded “yes” to cannabis use more than once a month as cannabis users. We then identified the number of cannabis users and non-cannabis reported as having a previous episode of myocardial infarction (MI). The phenotype of MI was defined according to previously published analyses and criteria. The incidence of MI in cannabis and non-cannabis users was then determined by taking the ratio of cannabis users who developed MI to the total number of cannabis users and the ratio of non-cannabis users who developed MI to the total number of non-cannabis users. To determine the incidence of MI in cannabis and non-cannabis users under the age of 50, we followed the same procedure as described above but only analyzed cannabis and non-cannabis users under the age of 50 at the time of the survey.
Developing a logistic regression model to predict the risk of developing MI
To develop a logistic regression model, we created a dataset from the UK Biobank of individuals who had developed MI versus those who had not developed MI. Each individual in the dataset was then annotated with information regarding their cannabis use, sex, BMI, and age. The dataset was then used to fit a logistic regression model using the “glm()” function, which fits generalized linear models including logistic regressions, in R version 3.5.3. Coefficients of the model included cannabis use, sex, BMI, and age. Using the p-value, we then identified coefficients that had a statistically significant predictive factor on the development of MI. The analyses were performed using the UK Biobank data as of April 2021.
Plasma protein analysis
Twenty recreational marijuana smokers were previously recruited to smoke a single marijuana cigarette and had serial blood draws to assess for Δ9-THC levels in plasma (Lynch et al., 2019). Patient demographics were previously reported (Lynch et al., 2019). Participants abstained from using marijuana for 24 hr before testing. Two participants were excluded from subsequent plasma analyses because they did not consent to the study. The remaining participants gave consent. Plasma was isolated from 18 individuals using sodium citrate tubes at 15 min intervals from 0 min to 180 min and frozen at −80 °C. The Olink Target 96 Inflammation panel was used to identify changes in the production of 92 common inflammatory cytokines (Sun et al., 2018) from the blood samples at 0 min, 90 min, and 180 min. The Olink platform is based on proximity extension assay (PEA) following target recognition by antibodies and was performed by the Stanford Human Immune Monitoring Core following the manufacturer’s protocols. Internal and external controls were included to establish detection limits. Data were measured as the Olink normalized protein extension (NPX) values, and proteins with missing values or below the limits of detection in ≥33% of samples were excluded. Differential protein abundance was compared using limma (Ritchie et al., 2015) with subject and time as factors. Proteins with adjusted p values ≤0.1 at 90 min vs. 0 min were considered statistically significant.
Cell culture
We obtained the following: human umbilical vein endothelial cells (HUVECs), human coronary artery endothelial cells (HCAECs), normal human cardiac fibroblasts-ventricular (NHCF-V) cells, human erythroleukemia (HEL 92.1.7), and human neuroblastoma cells (SK-N-FI) from the American Type Culture Collection (ATCC). The human embryonic stem cell-derived cardiomyocytes (hESC-CMs) were differentiated from the Wicell H7 line. The hiPSC-derived neurons were provided by Dr. Thomas Sudhof (Department of Molecular and Cellular Physiology, Stanford University). TNFα and genistein treatment of hiPSC-ECs were performed on hiPSCs from healthy control line 1 or CRISPRi knockdown of CNR1. The hiPSCs were differentiated into hiPSC-ECs and treated with vehicle, 10 ng/ml TNFα, or 10 μM genistein. Gene expression was quantified using quantitative real-time PCR (qRT-PCR).
Differentiation of hiPSC-ECs
hiPSC lines were obtained from the Stanford Cardiovascular Institute Biobank. Patients were enrolled under the Stanford Institutional Review Board and StemCell Research Oversight Committee guidelines. All volunteers gave consent. hiPSCs were generated from peripheral blood mononuclear cells (PBMCs) from 4 anonymous healthy individuals (age range from 26 to 51; 3 males and 1 female) using Sendai virus (CytoTuneTM-iPS 2.0 Sendai Reprogramming Kit, Thermo Fisher Scientific) (Key Resource Table). hiPSCs were dissociated using 0.5 mM EDTA/PBS (Innovative Cell Technologies, CA) and plated as single cells in E8 medium supplemented with ROCK inhibitor Y-27632 (Selleck Chemicals) on Matrigel to a final density of 20,000–30,000 cells/cm2. hiPSC-ECs were differentiated using a protocol previously described in our lab (Paik et al., 2018). Briefly, the hiPSC monolayers were cultured to 85% cell confluency. Cells were then treated for 2 days with 6 μM CHIR99021 (Selleck Chemicals) in RPMI medium plus B27 supplement without insulin (Life Technologies). On day 2, cells were treated for 2 days with 2 μM CHIR99021 in RPMI plus B27 supplement without insulin. On day 4, cells were treated with 10 ng/ml recombinant human fibroblast growth factor-2 (rhFGF-2) (PeproTech) and 20 ng/ml vascular endothelial growth factor (VEGF) (R&D Systems) for EC expansion for 8 days. On day 12, cells were harvested by Trypsin solution (Sigma-Aldrich) and sorted using anti-CD144 MicroBeads (Thermo Fisher Scientific). The differentiation efficiency was calculated by the number of CD144-positive cells. hiPSC-ECs were cultured in gelatin-coated (Sigma Aldrich) 6-well plates in EGM-2 medium (Lonza).
hiPSC-derived engineered heart tissues
Beating cardiomyocytes generated from hiPSC lines SCVI 854 and SCVI 951 (at day 22 of differentiation culture) were washed twice with PBS and digested with TrypLE™ Select (Thermo Fisher Scientific, A1217703) for 10 min at 37 °C (5% CO2). The dissociated cells were washed (PBS without Ca2+ or Mg2+; centrifugation at 300 3 g for 5 min) and re-suspended in RPMI/B27 medium and cell concentration adjusted to 1 × 106 cells/ml. Similarly, hiPSC-ECs from both lines frozen at passage 1 (P1) were thawed and expanded on 0.2% gelatin-coated dishes with EGM-2 medium. The cell types were mixed in a 7:3 ratio to a final cell concentration of 2 × 106 cells/ml. Fibrin-based human engineered heart tissues (EHTs) were generated using commercially available Teflon spacers and EHT silicone racks (EHT Technologies GmbH). In brief, casting molds were generated with 2% agarose in sterile DI water in 24-well plates (Nunc, 122475). Teflon spacers were introduced into the molten agarose to form the casting mold. Cells (final concentration 2 × 106 cells/ml) were mixed with 100 μl/ml Matrigel (BD Bioscience, 256235), 5 mg/ml bovine fibrinogen (200 mg/ml in NaCl 0.9% [Sigma, F4753], with 50:50 RPMI/B27 and EGM-2 medium mix). EHTs were generated with 80 μl per EHT (1 × 106 cells) and 3 U/ml thrombin. The cell mix was pipetted into agarose casting molds, and silicone posts were carefully dipped into the gelation mix. After fibrin polymerization (37 °C, 2 hr), the silicone racks with attached fibrin gels were transferred to new 24-well plates and cultured at 37 °C, 5% CO2. The culture medium was changed every 3 days and consisted of RPMI/B27 and EGM-2 (80:20), 1% penicillin/streptomycin, and 15 μg/ml aprotinin. After 5 days in culture, human EHTs displayed spontaneous coherent, regular beating deflecting the silicone posts, thus allowing for video-optical contraction analysis.
Evaluation of vascular function in small animal models
Seven-week-old male C57BL/6J mice were obtained from Jackson Laboratory (Bar Harbor, ME, USA). All small animal experiments were performed by protocols approved by the Institutional Animal Care and Use Committee (IACUC) at Stanford University. The mice were treated with vehicle, Δ9-THC, or Δ9-THC plus genistein (n=5/group) for 30 days and then sacrificed. Organs were collected, weighed, and fixed with 4% paraformaldehyde (PFA). The thoracic aortas (TAs) were surgically dissected out and cleaned from the surrounding connective tissue for all groups. The TAs were transferred to ice-cold Krebs’-Henseleit solution (in mM: 133 NaCl, 4.6 KCl, 2.5 CaCl2, 16.3 NaHCO3, 1.75 NaH2PO4, 0.6 MgSO4, 10 glucose) and cut into segments of 2 mm. The artery segments were mounted in wire-myograph chambers for isometric tension recordings (DanishMyo Technology [DMT]).The chambers contained Krebs’-Henseleit solution. All chambers were aerated with 95% O2 / 5% CO2, and the temperature was kept constant at 37 °C to mimic physiological conditions. After a manual stabilization and normalization procedure, we used the PowerLab4/25-Chart7 acquisition systems (AD Instruments Ltd) to record force and convert it into tension. The TA segments were pre-constricted using either 1–3 μM 5HT serotonin (5HT) or U46619. A cumulative application of acetylcholine (1–10 μM) was performed, and the induced vascular dilatations were measured. Subsequently, concentration-response relationship curves were established for each group of 3 mice. Time-matched dimethylsulfoxide (DMSO) application was performed in a separate chamber as the control for all experiments.
LDL receptor knockout mice model
LDL receptor knockout mice (B6.129S7-Ldlrtm1Her/J) were purchased from the Jackson Laboratory (Bar Harbor, ME, USA). Mice were housed in specific pathogen-free animal facilities maintained by the Stanford University School of Medicine. The diet was switched to a high-fat diet (HFD; Research Diets, Inc) containing 40% fat and 1.25% cholesterol (modified D12079B) from 9–12 weeks of age for 12 weeks to accelerate atherosclerotic plaque formation. Δ9-THC (1 mg/kg/d) or vehicle (90% saline, 5% ethanol, 5% Cremophor) was administered using osmotic pumps (Alzet, model 2006). To achieve continuous administration of Δ9-THC, the mice underwent implantation of the first pumps one day before HFD was started, and in 6 weeks, they received the second pumps. Genistein (50 mg/kg/d) or vehicle (corn oil 100 μL/d, Sigma-Aldrich, C8267) was orally administered daily using a feeding needle (Fine Science Tools, 18065–20). Mice were euthanized at 12 weeks of HFD, and the aorta, heart, liver, and serum were harvested. Serum lipid profiles were measured at Stanford Animal Diagnostic Lab. Blood pressure was measured with CODA Non-Invasive Blood Pressure System (Kent Scientific). All experiment procedures were performed in compliance with guidelines set forth by the National Institutes of Health Office of Laboratory Animal Welfare and approved by the Stanford APLAC protocol.
Mouse model of partial carotid artery ligation
Apolipoprotein E knockout (Apoe−/−) mice (C57BL/6-ApoEem1Narl/Nail) were purchased from the National Laboratory Animal Center (Taipei, Taiwan). An accelerated carotid atherosclerosis model was generated by performing partial carotid artery ligation (PCAL) in Apoe−/− mice fed a HFD (Nam et al., 2009). Briefly, anesthesia was induced in Apoe−/− mice by intraperitoneal injection of 2,2,2-tribromeethanol (250 mg/kg i.p.). After shaving, a ventral midline incision (4 to 5 mm) was made in the neck. The left carotid artery (LCA) was exposed by blunt dissection. Three of four caudal branches of LCA including the left external carotid artery, left internal carotid artery, and occipital artery were ligated while the superior thyroid artery was left intact. The incision was then closed, and mice were monitored in a chamber on a heating pad following surgery until fully recovered. One day after PCAL, the mice were treated with vehicle, Δ9-THC, or Δ9-THC plus genistein (n=5/group) for 10 days. After sacrificing and pressure perfusion, the aorta and the carotid arteries were isolated and fixed in 4% PFA for 10 min. The aorta and carotids were immersed in a 30% sucrose solution for 24 hr and subsequently incubated in a 1:1 ratio of an optimal cutting temperature (OCT) compound and 30% sucrose solution for 1 h. The samples were embedded in OCT. Frozen embedded samples were sectioned with a cryostat in 8 μm thickness and stained with an Oil Red O Staining Kit (ScienCell Research Laboratories, CA, USA) according to manufacturer’s instructions. The plaque areas were calculated using ImageJ software. The extent of the carotid atherosclerosis was presented as plaque areas quantified on each carotid artery section.
In vivo biodistribution of BODIPY517/547-genistein
To evaluate the in vivo biodistribution, BODIPY517/547-genistein (50 mg/kg) or rimonabant 100 nM followed 30 min later by BODIPY517/547-genistein (50 mg/kg) was intravenously injected into adult male BALB/c nude mice (n=5) and observed via the IVIS Spectrum In Vivo Imaging System (Xenogen) at 0.25, 1, 12, 24, and 48 h after injection. After 48 h, mice were sacrificed, and the main organs were collected for IVIS observation, including the brain, heart, lung, thoracic aorta, liver, spleen, kidney, intestine, colon, and whole blood. Regions of photon intensity were quantified using the IVIS Image software.
METHOD DETAILS
High-throughput virtual screening assay
The structures of 4 selective CB1 antagonists (rimonabant, otenabant, AM251, and DBPR-211) were used as lead compound candidates for high-throughput virtual screening. The selective CB1 antagonists were stripped of salts, and query structures were generated by BROOD (version 3.0, OpenEye Scientific Software). ROCS (version 2.3, OpenEye Scientific Software) was used for the high-throughput virtual screening with the SWEETLEAD cheminformatics database as the chemical library (https://simtk.org/projects/sweetlead). The top hits were ranked by combo Tanimoto and were selected from each CB1 antagonist.
Knockdown of CB1 using siRNA or CRISPR interference (CRISPRi) technology
The plasmids and SmartPool siRNA (Dharmacon) were transfected with Lipofectamine 2000 according to the manufacturer’s protocol. Briefly, 50% confluentcells in 6-well plates were transfected with plasmid or siRNA in 1 mL of serum-free medium for 6 h at 37 °C. Then, 1 mL of mediumcontaining 20% FBS was added to the transfection mixture. After 48 h, cells were treated with Δ9-THC for 48 h, and the mRNA expression of various genes was measured by qPCR. mRNA expression was normalized to GAPDH. The single guide RNA sequences for CRISPRi were adapted from Human Genome-wide CRISPRi-v2 Libraries (Addgene #83969). The lentivirus expressing sgRNAs was generated with pMD2.G (Addgene #12259), psPAX2 (Addgene #12260), and a sgRNA plasmid (Addgene #60955) in HEK293T packaging cells (Takara Bio, 632180 Lenti-X™ 293T Cell Line), and infected into the CRISPRi hiPSCs (Mandegar et al., 2016). Then, sgRNA-positive cells were selected with puromycin (0.5 μg/ml) for 14 days. The hiPSCs were subsequently differentiated into endothelial cells. The CRISPRi system was induced by treating with doxycycline (2 μM) for 48 h. Afterward, cells were treated with Δ9-THC for 48 h, and the mRNA expression of various genes was measured by qPCRanalysis and normalized to GAPDH.
Molecular docking analysis
Protein-ligand docking studies were performed on the crystal structure of the human CB1 receptor (PDB: 5TGZ). The X-ray structure of CB1-AM6538 was taken from the Protein Data Bank (PDB ID: 5TGZ) (Friesner et al., 2006). The Schrodinger suite was used to prepare the protein by adding hydrogens and performing a restrained minimization using the default settings. The default force field, OPLS_2005, was used for all minimizations. The Glide module was used to dock the flexible ligands to the rigid binding site without constraints using the default settings (Halgren et al., 2004). The resulting GlideScores were used to estimate and rank the binding energy of the ligands.
Cell viability assay
Cells were seeded at 3,000 cells/well in 96-well plates, maintained for 14–16 h, and then treated with test compounds. After 48 h, cells were washed with PBS, and then the medium containing CellTiter-Glo® Reagent (Promega) was diluted 1:1 with culture medium for 30 min at room temperature. The relative luminescence units (RLU) were recorded with a plate reader (Promega GloMax Multi Detection System). The half-maximal inhibition concentration (IC50) of cell viability was calculated by SigmaPlot software (Systat Software).
ROS-Glo™ H2O2 assay
Cells were seeded at 3,000 cells/well in 96-well plates, maintained for 14–16 h, and then treated with the test compound. The H2O2 substrate solution was added to the supernatant. The plate was returned to the incubator for an additional 6 h of treatment. ROS-Glo Detection Solution was added to each well and then incubated for 20 min at room temperature. Reactive oxygen species (ROS) activity was measured with a plate reader (Promega GloMax Multi Detection System).
Monocyte adhesion assay
U937 monocytes were fluorescence-labeled using Calcein-AM (100 μM, 1 × 106 cells; R&D Systems). Endothelial cells were seeded at 3,000 cells/well in 96-well plates and maintained for 14–16 h, then treated with test compounds. After 48 h, monocytes were added and co-cultured with hiPSC-ECs for 30 min at 37 °C. The wells were washed three times with PBS to remove non-adherentcells. Cells were viewed with fluorescence microscopy, and spectrofluorometric quantification was performed at 492 nm (excitation) and 535 nm (emission) on a 96-well plate reader (BioteckCytation 5).
Tube formation assay
Cells were seeded in 6-well plates and maintained for 14–16 h, and treated with test compounds. After 48 h, trypsinized cells were cultured at a density of 1×105 on Matrigel basement membrane matrix, and tube formation was analyzed after 12 h. Cumulative tube length was quantified from microscopic images taken from 3 random fields for each condition. Quantification was performed with Wimasis image analysis software.
Quantitative reverse transcription-polymerase chain reaction (q-PCR)
Total RNA was isolated by Trizol reagent. Reverse transcription reaction was performed by 2 μg of total RNA, which was reverse transcribed into cDNA using oligo-dT. qPCR analysis was performed with cDNA samples using the ABI Prism 7900 Sequence Detection System (Applied Biosystems). mRNA expression was normalized to GAPDH.
Western blot analysis
Cells were lysed using RIPA buffer, and lysates were centrifuged at 13,000 rpm for 15 min at 4°C and then subjected to SDS-PAGE using polyacrylamide gels. Western blot was done as described previously (Lee et al., 2019). Immunoblotting was performed using specific antibodies to evaluate the expression of different proteins.
CellROX Deep Red oxidative stress assay
Cells were seeded at 3,000 cells/well in 96-well plates and maintained for 14–16 h, then treated with test compounds. After 48 h, CellROX® Reagent was added at a final concentration of 5 μM, and the plate was returned to the incubator for 30 min. The medium was removed and washed times with PBS. The cells were fixed with 3.7% formaldehyde, stained with DAPI for 15 min, and were viewed using a Zeiss LSM 780 microscope. The results were analyzed using LSM version software.
Plaque-size quantification in LDL receptor knockout mice
Atherosclerotic lesions were quantified by cross-sectional analysis of the aortic root and en face analysis of the thoracic aorta (Yun et al., 2017; Baba et al., 2021). The heart, thoracic aorta, and abdominal aorta were collected and fixed in 4% PFA overnight. For cross-sectioning of the aortic root, the samples were cut at the mid-ventricle level and above the aortic root, incubated in 40% sucrose overnight, and then embedded in OCT. The OCT-embedded aortas were sectioned using a cryostat, and 20-μm sections were obtained at the aortic root level in which three valve leaflets are observed. For Oil Red O staining of the aortic-root sections, the sections were pretreated with 100% propylene glycol (1,2-Propanediol; Sigma-Aldrich, 398039) for 2 min, incubated with 0.5% Oil Red O solution (Sigma-Aldrich, O0625) for 10 min at room temperature, washed with 85% propylene glycol and distilled water, and then counterstained with Mayer’s Modified Hematoxylin solution. For en face analysis of the aorta, the thoracic aortas were pretreated with 100% propylene glycol for 15 min, incubated with 0.5% oil red O solution for 3 h at room temperature, washed with 85% propylene glycol, and then washed with PBS. After staining, aortas were placed between 2 transparent sheets and imaged with a stereomicroscope and a digital camera.
Immunofluorescence staining on vascular endothelial cells in mouse thoracic aortas
Adult male BALB/c nude mice (n=5) were treated with BODIPY517/547-genistein (50 mg/kg) for 48 h and then sacrificed. En face immunofluorescence staining of vascular endothelial cells in mice was performed as previously described (Li et al., 2019). Briefly, the thoracic aortas were isolated and permeabilized by Triton X-100 for 10 min at room temperature. Next, tissues were blocked with 10% normal goat serum in TBS buffer containing 2.5% polysorbate 20 for 1 h at room temperature. Tissues were then exposed to an anti-CB1 antibody (Cayman Chemical) for 12 h at 4 °C and then exposed to Alexa Fluor 488 secondary antibody for 1 h at room temperature. The tissues were then placed on a coverslip with the luminal surface downward and moved slowly to the antifade mounting solution previously dropped on the coverslip. Fluorescence images were viewed by a confocal microscope system (Zeiss LSM 780), and tissues were observed through a series of z-stack images. The results of co-localized pixels were analyzed using LSM version software.
Radioligand binding assay for the human CB1 receptor
The radioligand binding assay on the human CB1 receptor was performed by Eurofins Discovery (St Charles, MO). Briefly, human recombinant Chem-1 cells were used, and cells were incubated with 2 nM [3H] SR141716A, 2.4 nM [3H] WIN-55,212–2, or 0.5 nM [3H] Estradiol for 90 min at 37 °C. Non-specific binding was estimated in the presence of 10 μM CP-55,940, 10 μM R(+)- WIN-55,212–2, or 1 μM diethylstilbestrol. Test compounds were prepared in assay buffer (20 mM HEPES, pH 7.0, 0.5% BSA) with serial dilution. Membranes were filtered and washed four times, and the filters were counted to determine specifically bound radioligand. IC50 values were determined by a non-linear, least squares regression analysis using the MathIQTM statistical software (ID Business Solutions Ltd., UK). The inhibition constants (Ki) values were calculated by the Cheng-Prussoff equation using the observed IC50 of the tested compound, the concentration of radioligand employed in the assay, and the historical values for the dissociation constants of the ligand.
Radioligand binding assay for the mouse CB1 receptor
The radioligand assay for the mouse receptor was performed by Gifford Biosciences Limited (Birmingham, UK).
Membrane preparation
Frozen tissues were homogenized in ice-cold lysis buffer (50 mM Tris-HCl; 5 mM MgCl2; 5 mM EDTA; protease inhibitor cocktail) and centrifuged at 100 × g for 2 min at 4 °C to remove large debris. Supernatants were centrifuged at 13,000 × g for 10 min at 4 °C to isolate pellets containing the membranes. The membranes were resuspended in wash buffer (50 mM Tris-HCl; 5 mM MgCl2; 5 mM EDTA), centrifuged at 13,000 × g for 10 min at 4 °C, and the pellet was resuspended in 0.4 ml wash buffer containing 10% sucrose and stored at 80 °C. Protein content was analyzed using the Sigma® BCA assay. On the day of the assay, the membrane preparations were thawed, and the pellet resuspended in the final assay buffer.
Assay protocol
Filtration binding assays were carried out in 96-well plates in a final volume of 250 μL per well. Each well had 150 μL membranes, 50 μL of the competing unlabeled compound or buffer, and 50 μL radioligand solution ([3H]CP55940; 108.45 Ci/mmol (PerkinElmer, 2795349). The plate was incubated at 25 °C for 60 min with gentle agitation. The incubation was stopped by vacuum filtration onto presoaked (buffer with 1% BSA) GF/C filters using a 96-well FilterMate™ harvester and subsequently washed five times with ice-cold wash buffer. Filters were then dried under a warm air stream, sealed in polyethylene, scintillation cocktail added, and the radioactivity counted in a Wallac® TriLux1450 MicroBeta counter.
Data analysis
For each drug concentration, non-specific binding was subtracted from total binding to give specific binding. Data were fitted using the non-linear curve fitting routines in Prism® (Graphpad Software Inc) to obtain IC50 values. Ki was calculated from the IC50 values using the Cheng-Prusoff equation.
Mouse plasma cytokine analysis
This assay was performed by the Human Immune Monitoring Center at Stanford University using a Mouse 48 plex ProcartaPlex™ kit (Thermo Fisher Scientific, EPX480–20834-901) according to the manufacturer’s instructions. Briefly, beads were added to a 96-well plate and washed in a Biotek ELx405 washer. Mouse EDTA Plasma samples were diluted 3-fold, added to the plate containing the mixed antibody-linked beads, and incubated overnight at 4 °C with shaking. Following overnight incubation, plates were washed in a Biotek ELx405 washer. Then biotinylated detection antibody was added for 1 h at ambient temperature (22°C) with shaking, followed by washing and the addition of streptavidin-PE, incubation for 30 min, and a final washing. Each sample was measured in duplicate on a Flex Map FM3D instrument with a lower bound of 50 beads per sample per cytokine and with the aid of custom Assay Chex control beads (Radix BioSolutions). Output MFI data were transformed to concentration in pg/ml using standards serially diluted and run in the assay. The resulting concentrations for cytokines/chemokines measured are reported here, and values below the detection limits in ≥33% of samples were excluded. Select cytokines’ differential protein abundance was compared using a two-sample Wilcoxon test in R v.3.6.3.
Neurobehavioral testing (Billy Martin tetrad)
Animals
Group housed, 6–7 weeks old, male C57Bl/6J mice from Jackson Laboratory were used in the study. Mice were ordered at 5–6 weeks old and handled for 3 days before testing. The mice were housed in a Stanford University animal facility with a 12:12 h light/dark cycle (8:30 am light off, 8:30 am light on) and had free access to water and food. All behavioral tests were conducted during the animals’ dark cycle. All procedures followed National Institute of Health guidelines and were approved by the Institutional Administrative Panel on Laboratory Animal Care (APLAC).
Dosing
The mice were randomized into different dosing groups based on their body weight. Δ9-THC (20 mg/kg i.p.) and genistein (50 mg/kg p.o.) were used in the study. The vehicle for Δ9-THC was 5% ethanol, 5% Cremophor EL, and 90% saline (1:1:18 ratio). The vehicle for genistein was corn oil. Vehicle, Δ9-THC, and genistein were dosed at the same time according to their treatment groups: Group 1: n=8 Veh i.p.+ Veh p.o.; Group 2: n=9 THC (20 mg/kg i.p.) + Veh p.o.; Group 3: n=8 Veh i.p.+ Genistein (50 mg/kg p.o.); and Group 4: n=9 THC (20 mg/kg i.p.) + Genistein (50 mg/kg p.o.).
Body temperature measurement and open field activity chamber
Body temperature and locomotor activity were measured on the same day. The body temperature was taken using a Rodent Rectal Temperature Probe-For Mouse with lubricant (Animal Temperature Controller, World Precision Instrument, Model ACT-2000) before dosing and 60 min post-dose. The locomotor assessment took place in an Open Field Activity Arena (Med Associates Inc., St. Albans, VT. Model ENV-515) mounted with three planes of infrared detectors within a specially designed sound-attenuating chamber (Med Associates Inc., St. Albans, VT. MED-017M-027). The arena was 43 cm (L) × 43 cm (W) × 30 cm (H) and the sound-attenuating chamber was 74 cm (L) × 60 cm (W) × 60 cm (H). The mice were dosed and immediately placed in the corner of the testing arena. They were allowed to explore the arena for 10 min under tracking by an automated tracking system. Parameters, including distance moved, velocity, rearing, and time spent in the periphery and center of the arena, were analyzed. The periphery was defined as the zone 5 cm away from the arena wall. The arena was cleaned with a 1% Virkon solution at the end of each trial.
Hot plate test
A Hot Plate apparatus (IITC Inc. Model 39) set to 55 °C was used in the study. Mice were tested before dosing and 20 min post-dosing. The mice were placed on the hot plate and covered by a transparent glass cylinder that was 25 cm in height and 12 cm in diameter. A 30 sec maximum trial duration was used during the test to minimize the exposure of animals to painful stimuli and prevent permanent tissue damage from the heat. A remote foot-switch pad was used to control the start/stop/reset function. The latency to hind paw licking, flicking, or jumping was recorded.
Bar test
The Bar test is the most commonly used test in psychopharmacological induced catalepsy, a condition characterized by a lack of response to external stimuli and muscular rigidity, wherein the limbs remain in whatever position they are placed (Sanberg et al., 1988). A typical catalepsy test involves putting an animal in an unnatural position and recording the time it takes for the animal to correct itself. A small metal rod (0.7 cm diameter, 10 cm long) was suspended 3–5 cm above the table, and the forepaws of the mouse were placed on the bar. The total duration the mice spent with one or both paws on the bar in a catalepsy state was recorded for a total of 60 sec. If the mouse jumped over the bar, it was placed in the original position on the bar a maximum of 4 times. A score of zero second was assigned to the mice if they jumped over the bar a fifth time. Mice were tested once before dosing and 60 min post-dose.
Synthesis of BODIPY517/547-genistein
All the reagents were commercially available and used without further purification unless indicated otherwise. All solvents were anhydrous grade unless indicated otherwise. All non-aqueous reactions were carried out in oven-dried glassware under a slight positive argon pressure unless noted otherwise. Reactions were magnetically stirred and monitored by thin-layer chromatography on silica gel. Flash chromatography was performed on silica gel of 60–200μm particle size. Yields were reported for spectroscopically pure compounds. Melting points were recorded on a Fargo MP-2D melting point apparatus. 1H, 13C, and 31P NMR spectra were recorded on Bruker AV 600 (600 MHz), Bruker AV 500 (500 MHz), and Bruker AVIII 400 (400 MHz) spectrometers. Chemical shifts are given in δ values relative totetramethylsilane (TMS, δH = 0); coupling constants J are given in Hz. Internal standards were CDCl3 (δH = 7.24), CD3OD (δH = 3.31) or DMSO-d6 (δH = 2.49) for 1H NMR spectra and CDCl3 (δC = 77.0, central line of triplet), CD3OD (δC = 49.0, central line of septet) or DMSO-d6 (δC = 39.5, central line of septet) for 13C NMR spectra. The splitting patterns are reported as s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet), br (broad), and dd (double of doublets). High-resolution electrospray ionization (ESI) and fast atom bombardment (FAB) mass spectra were recorded on a JMS-T100LP AccuTOF LC-plus 4G mass spectrometer.
Glutathione levels in the serum of mice treated with Δ9-THC or genistein
Male C57BL/6J mice were treated with Δ9-THC, genistein, or their combination every day for 30 days (n=5/group). According to the manufacturer’s instructions, all mice were sacrificed on day 30, and plasma was collected and analyzed by the Glutathione colorimetric assay kit (BioVision, K261). Briefly, a serial dilution of reduced glutathione (GSH) and oxidized glutathione (GSSG) stock solutions was prepared as standards. Glutathione level was measured by the periodic reaction of 5,5-dithio-bis-(2-nitrobenzoic acid) (DTNB). The plate reader measured the absorbance with a wavelength of 405 nm after 10 min with ELISA. The detected reduced and oxidized GSH levels were normalized to the total serum content of the respective serum samples. Total glutathione level (GSH+GSSG) and the ratio of reduced GSH to oxidized GSH (GSH/GSSG ratio) were calculated.
LC-MS analysis of Δ9-THC in mouse plasma
Mice were treated with vehicle, genistein, Δ9-THC, or combination for 30 days and then sacrificed. Blood was collected with a cardiac puncture and plasma isolated using 5 mM EDTA. A 100 μl aliquot of plasma was transferred to a 1.5 ml microtube. The LC-MS analysis was performed by the Forensic and Clinical Toxicology Center at National Taiwan University. Briefly, protein precipitation was performed to remove the interference of proteins in serum samples by adding 20 μL 50% methanol (MeOH), and 60 μL IS solution in acetonitrile to 20 μL serum samples. 40 μL of supernatants were collected after centrifugation at 12,000 RCF for 10 min and dried with steam nitrogen. The resulting residues were reconstituted with 80 μL 50% MeOH and transferred to autosampler vials before LC-MS analysis. Analytical standards were prepared in MeOH as a stock solution at a concentration of 1000 μg/ml. Calibrators were prepared with series dilutions and spiked into a blank sample matrix. Calibration curves were constructed in a serum blank matrix with analytical ranges from 3.125 to 100 ng/ml. LC-MS analysis was performed with Agilent 1290 LC-Sciex 6500 QqQ system. A Biphenyl column was employed as an analytical column (100 × 2.1 mm, 2.6 μm, Phenomenex, Torrance, CA).
GTPase-GloTM assay
CB1 and Gi1 were purified as described previously (Krishna Kumar et al., 2019). The GTP turnover assay was performed using a modified protocol of the GTPase-GloTM assay (Promega) as described previously (Gregorio et al., 2017) with the following modification. The final reaction buffer consisted of 20 mM HEPES pH 7.5, 100 mM sodium chloride, 10 mM magnesium chloride, 100 μM TCEP MNG/CHS (0.01%/0.001%), and 10 μM GTP. Before the reaction was started, CB1 (250 nM) was incubated in reaction buffer, at room temperature, in the presence of 20 μM agonist (CP 55,490), 20 μM inverse agonists (rimonabant or taranabant), or 100 μM genistein. Simultaneously, Gi1 protein 250 μM (in DDM) was incubated with 1 % MNG on ice. After 60 min, Gi1 was added to the reaction buffer to a final concentration of 250 nM together with CB1 to a final concentration of 125 nM. After incubation for 1 hr, reconstituted GTPase-Glo reagent supplemented with 10 mM ADP was added to the sample and incubated for 30 min at room temperature. Luminescence was measured after the addition of detection reagent and incubation for 10 min at room temperature using a SpectraMax Paradigm plate reader. Percentage GTP turnover was calculated with receptor alone as 0 and CP 55490 as 100%. The curves were analyzed using GraphPad Prism.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data shown were from at least three independent biological replicates. Quantitative data were presented as mean with standard error of the mean (SEM). The SPSS program (SPSS Inc.) or Prism was used for statistical analysis, including Student’s t-test, one-way ANOVA with multiple testing, and Tukey correction for multiple testing where appropriate. Statistical significance defined as p<0.05 (* or #), p<0.01 (** or ##), and p<0.001 (***or ###).
Supplementary Material
Highlights.
Marijuana use is associated with MI in UK Biobank database
Δ9-THC caused inflammation and oxidative stress in endothelial cells
Δ9-THC caused vascular dysfunction in mice models
Genistein, a CB1 antagonist, attenuated Δ9-THC-induced atherosclerosis
ACKNOWLEDGMENTS
We thank the Wu laboratory members for critical discussion and feedback. We are grateful to Thomas Sudhof for providing hiPSC-derived neural cells for this project. Bruce Conklin (Gladstone Institute) kindly provided a CRISPRi iPSC line (Gen1C) (Mandegar et al., 2016). Tony Chour and Evgenios Neofytou helped with tissue sectioning and H&E staining. June-Wha Rhee helped with generating CB1-deficient cells by CRISPRi and cannabis user recruitment. Yael Rosenberg Hasson and the Stanford Human Immune Monitoring Core-Immunoassays Team assisted with the Olink assay. Ludmilla Alexandrova from Stanford University mass spectrometry provided advice on an assay for the analysis of oxidative stress. Lejla Pepcic assisted with echocardiographic analysis. Detlef Obal helped obtain clinical samples and conducted mouse dissection. Cong-Kai Luo and Dai-Jung Chung (National Taiwan University) assisted with the immunofluorescence staining. Po-An Hu and Tzong-Shyuan Lee (Graduate Institute and Department of Physiology, National Taiwan University) assisted with the animal study in Apoe−/− mice. Mehrdad Shamloo and Nay Saw from the Stanford Behavioral and Functional Neurosciences Laboratory are supported by NIH equipment grant (1S10OD030452–01) and assisted with the design and execution of the Billy Martin tetrad testing. This work was supported by the Stanford Cardiovascular Institute, Tobacco-Related Disease Research Program (TRDRP) 27IR-0012, American Heart Association 17MERIT33610009, Steven M. Gootter Foundation, and Leducq Foundation 18CVD05 (to J.C.W.); Taiwan Ministry of Science and Technology MOST108–2636-B-002–007, MOST109–2636-B-002–007, MOST110–2636-B-002–016, and MOST111–2636-B-002–017 (to T.T.W.); Clinician Scientist Training Program, Detweiler Traveling Fellowship from the Royal College of Physicians and Surgeons (M.C.); and TRDRP Postdoctoral fellowship T31FT1758 (M.N.). The graphical abstract and Figure S7 (L&M) were created with BioRender.com.
Footnotes
DECLARATION OF INTERESTS
J.C.W. is a co-founder and SAB of Greenstone Biosciences, but the work was done independently. B.K.K. is a co-founder and SAB of ConfometRx, but the work was done independently.
INCLUSION AND DIVERSITY
We worked to include sex balance in the recruitment of human and nonhuman subjects. While citing references in this manuscript, we actively promoted sex and gender balance in our reference list.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.cell.2022.04.005.
REFERENCES
- Akira S, Uematsu S, and Takeuchi O (2006). Pathogen recognition and innate immunity. Cell 124, 783–801. [DOI] [PubMed] [Google Scholar]
- Ardigo D, Assimes TL, Fortmann SP, Go AS, Hlatky M, Hytopoulos E, Iribarren C, Tsao PS, Tabibiazar R, and Quertermous T; ADVANCE Investigators (2007). Circulating chemokines accurately identify individuals with clinically significant atherosclerotic heart disease. Physiol. Genomics 31, 402–409. [DOI] [PubMed] [Google Scholar]
- Atakan Z (2012). Cannabis, a complex plant: different compounds and different effects on individuals. Ther. Adv. Psychopharmacol. 2, 241–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba O, Huang LH, Elvington A, Szpakowska M, Sultan D, Heo GS, Zhang X, Luehmann H, Detering L, Chevigne A, et al. (2021). CXCR4-binding positron emission tomography tracers link monocyte recruitment and endothelial injury in murine atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 41, 822–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badowski ME (2017). A review of oral cannabinoids and medical marijuana for the treatment of chemotherapy-induced nausea and vomiting: a focus on pharmacokinetic variability and pharmacodynamics. Cancer Chemother. Pharmacol. 80, 441–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baell JB, and Holloway GA (2010). New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 53, 2719–2740. [DOI] [PubMed] [Google Scholar]
- Baron EP (2015). Comprehensive review of medicinal marijuana, cannabinoids, and therapeutic implications in medicine and headache: what a long strange trip it’s been. Headache 55, 885–916. [DOI] [PubMed] [Google Scholar]
- Bayazit H, Selek S, Karababa IF, Cicek E, and Aksoy N (2017). Evaluation of oxidant/antioxidant status and cytokine levels in patients with cannabis use disorder. Clin. Psychopharmacol. Neurosci. 15, 237–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisson J, Mcalpine JB, Friesen JB, Chen SN, Graham J, and Pauli GF (2016). Can invalid bioactives undermine natural product-based drug discovery? J. Med. Chem. 59, 1671–1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boesten LS, Zadelaar AS, Van Nieuwkoop A, Gijbels MJ, De Winther MP, Havekes LM, and Van Vlijmen BJ (2005). Tumor necrosis factor-alpha promotes atherosclerotic lesion progression in APOE*3-Leiden transgenic mice. Cardiovasc. Res. 66, 179–185. [DOI] [PubMed] [Google Scholar]
- Brizzi MF, Formato L, Dentelli P, Rosso A, Pavan M, Garbarino G, Pegoraro M, Camussi G, and Pegoraro L (2001). Interleukin-3 stimulates migration and proliferation of vascular smooth muscle cells: a potential role in atherogenesis. Circulation 103, 549–554. [DOI] [PubMed] [Google Scholar]
- Bycroft C, Freeman C, Petkova D, Band G, Elliott LT, Sharp K, Motyer A, Vukcevic D, Delaneau O, O’Connell J, et al. (2018). The UK biobank resource with deep phenotyping and genomic data. Nature 562, 203–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavusoglu E, Marmur JD, Yanamadala S, Chopra V, Hegde S, Nazli A, Singh KP, Zhang M, and Eng C (2015). Elevated baseline plasma IL-8 levels are an independent predictor of long-term all-cause mortality in patients with acute coronary syndrome. Atherosclerosis 242, 589–594. [DOI] [PubMed] [Google Scholar]
- Cerdá M, Mauro C, Hamilton A, Levy NS, Santaella-Tenorio J, Hasin D, Wall MM, Keyes KM, and Martins SS (2020). Association Between recreational marijuana legalization in the United States and changes in marijuana use and cannabis use disorder From 2008 to 2016. JAMA Psychiatry 77, 165–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang TT, Yang HY, Chen C, and Chen JW (2020). CCL4 inhibition in atherosclerosis: effects on plaquestability, endothelial cell adhesiveness, and macrophages activation. Int. J. Mol. Sci. 21, 6567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen PY, Qin L, Li G, Wang Z, Dahlman JE, Malagon-Lopez J, Gujja S, Cilfone NA, Kauffman KJ, Sun L, et al. (2019). Endothelial TGF-beta signalling drives vascular inflammation and atherosclerosis. Nat. Metab. 1, 912–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi JH, Cheong C, Dandamudi DB, Park CG, Rodriguez A, Mehandru S, Velinzon K, Jung IH, Yoo JY, Oh GT, and Steinman RM (2011). Flt3 signaling-dependent dendritic cells protect against atherosclerosis. Immunity 35, 819–831. [DOI] [PubMed] [Google Scholar]
- Cinar R, Iyer MR, and Kunos G (2020). The therapeutic potential of second and third generation CB1R antagonists. Pharmacol. Ther. 208, 107477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotter J (2009). Efficacy of crude marijuana and synthetic Delta-9-tetrahydro cannabinol as treatment for chemotherapy-induced nausea and vomiting: A systematic literature review. Oncol. Nurs. Forum 36, 345–352. [DOI] [PubMed] [Google Scholar]
- Damås JK, Smith C, Øie E, Fevang B, Halvorsen B, Waehre T, Boullier A, Breland U, Yndestad A, Ovchinnikova O, et al. (2007). Enhanced expression of the homeostatic chemokines CCL19 and CCL21 in clinical and experimental atherosclerosis: possible pathogenic role in plaque destabilization. Arterioscler. Thromb. Vasc. Biol. 27, 614–620. [DOI] [PubMed] [Google Scholar]
- Defilippis EM, Singh A, Divakaran S, Gupta A, Collins BL, Biery D, Qamar A, Fatima A, Ramsis M, Pipilas D, et al. (2018). Cocaine and marijuana use among young adults with myocardial infarction. J. Am. Coll. Cardiol. 71, 2540–2551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Després JP, Golay A, and Sjöström L; Rimonabant in Obesity-Lipids Study Group (2005). Effects of rimonabant on metabolic risk factors in overweight patients with dyslipidemia. N. Engl. J. Med. 353, 2121–2134. [DOI] [PubMed] [Google Scholar]
- DiMarzo V, Fontana A, Cadas H, Schinelli S, Cimino G, Schwartz JC, and Piomelli D (1994). Formation and inactivation of endogenous cannabinoid anandamide in central neurons. Nature 372, 686–691. [DOI] [PubMed] [Google Scholar]
- El-Remessy AB, Rajesh M, Mukhopadhyay P, Horvath B, Patel V, Al-Gayyar MM, Pillai BA, and Pacher P (2011). Cannabinoid 1 receptor activation contributes to vascular inflammation and cell death in a mouse model of diabetic retinopathy and a human retinal cell line. Diabetologia 54, 1567–1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Förstermann U, and Sessa WC (2012). Nitric oxide synthases: regulation and function. Eur. Heart J. 33, 829–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fride E, Perchuk A, Hall FS, Uhl GR, and Onaivi ES (2006). Behavioral methods in cannabinoid research. Methods Mol. Med. 123, 269–290. [DOI] [PubMed] [Google Scholar]
- Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood JR, Halgren TA, Sanschagrin PC, and Mainz DT (2006). Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J.Med.Chem. 49, 6177–6196. [DOI] [PubMed] [Google Scholar]
- Gleason CE, Carlsson CM, Barnet JH, Meade SA, Setchell KD, Atwood CS, Johnson SC, Ries ML, and Asthana S (2009). A preliminary study of the safety, feasibility and cognitive efficacy of soy isoflavone supplements in older men and women. Age Ageing 38, 86–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregorio GG, Masureel M, Hilger D, Terry DS, Juette M, Zhao H, Zhou Z, Perez-Aguilar JM, Hauge M, Mathiasen S, et al. (2017). Single-molecule analysis of ligand efficacy in beta2AR-G-protein activation. Nature 547, 68–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurley RJ, Aranow R, and Katz M (1998). Medicinal marijuana: a comprehensive review. J.Psychoactive Drugs 30, 137–147. [DOI] [PubMed] [Google Scholar]
- Halgren TA, Murphy RB, Friesner RA, Beard HS, Frye LL, Pollard WT, and Banks JL (2004). Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 47, 1750–1759. [DOI] [PubMed] [Google Scholar]
- Heitzer T, Schlinzig T, Krohn K, Meinertz T, and Münzel T (2001). Endothelial dysfunction, oxidative stress, and risk of cardiovascular events in patients with coronary artery disease. Circulation 104, 2673–2678. [DOI] [PubMed] [Google Scholar]
- Heller EA, Liu E, Tager AM, Yuan Q, Lin AY, Ahluwalia N, Jones K, Koehn SL, Lok VM, Aikawa E, et al. (2006). Chemokine CXCL10 promotes atherogenesis by modulating the local balance of effector and regulatory T cells. Circulation 113, 2301–2312. [DOI] [PubMed] [Google Scholar]
- Hill KP, Palastro MD, Johnson B, and Ditre JW (2017). Cannabis and pain: A clinical review. Cannabis Cannabinoid Res. 2, 96–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman AF, and Lupica CR (2013). Synaptic targets of Δ9-tetrahydrocannabinol in the central nervous system. Cold Spring Harb. Perspect. Med. 3, a012237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huestis MA, and Cone EJ (2004). Relationship of Delta 9-tetrahydro cannabinol concentrations in oral fluid and plasma after controlled administration of smoked cannabis. J. Anal. Toxicol. 28, 394–399. [DOI] [PubMed] [Google Scholar]
- Ibsen MS, Connor M, and Glass M (2017). Cannabinoid CB1 and CB2 receptor signaling and bias. Cannabis Cannabinoid Res. 2, 48–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia Z, Babu PV, Si H, Nallasamy P, Zhu H, Zhen W, Misra HP, Li Y, and Liu D (2013). Genistein inhibits TNF-alpha-induced endothelial inflammation through the protein kinase pathway A and improves vascular inflammation in C57BL/6 mice. Int. J. Cardiol. 168, 2637–2645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein TW, and Newton CA (2007). Therapeutic potential of cannabinoid-based drugs. Adv.Exp.Med.Biol. 601, 395–413. [DOI] [PubMed] [Google Scholar]
- KrishnaKumar K, Shalev-Benami M, Robertson MJ, Hu H, Banister SD, Hollingsworth SA, Latorraca NR, Kato HE, Hilger D, Maeda S, et al. (2019). Structure of a signaling cannabinoid receptor 1-G protein complex. Cell 176, 448–458.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon YD, Byun Y, and Kim HK (2021). 18F-labelled BODIPY dye as a dual imaging agent: Radiofluorination and applications in PET and optical imaging. Nucl. Med. Biol. 93, 22–36. [DOI] [PubMed] [Google Scholar]
- Lau E, Paik DT, and Wu JC (2019). Systems-wide approaches in induced pluripotent stem cell models. Annu. Rev. Pathol. 14, 395–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Termglinchan V, Diecke S, Itzhaki I, Lam CK, Garg P, Lau E, Greenhaw M, Seeger T, Wu H, et al. (2019). Activation of PDGF pathway links LMNA mutation to dilated cardiomyopathy. Nature 572, 335–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Liu ZH, Chen JW, Shu XY, Shen Y, Ding FH, Zhang RY, Shen WF, Lu L, and Wang XQ (2019). Using enface immunofluorescence staining to observe vascular endothelial cells directly. J. Vis. Exp. 150. [DOI] [PubMed] [Google Scholar]
- Little PJ, Compton DR, Johnson MR, Melvin LS, and Martin BR (1988). Pharmacology and stereo selectivity of structurally novel cannabinoids in mice. J.Pharmacol. Exp.Ther. 247, 1046–1051. [PubMed] [Google Scholar]
- Liu J, Gao B, Mirshahi F, Sanyal AJ, Khanolkar AD, Makriyannis A, and Kunos G (2000). Functional CB1 cannabinoid receptors in human vascular endothelial cells. Biochem. J. 346, 835–840. [PMC free article] [PubMed] [Google Scholar]
- Lynch KL, Luo YR, Hooshfar S, and Yun C (2019). Correlation of breath and blood Δ9-tetrahydro cannabinol concentrations and release kinetics following controlled administration of smoked cannabis. Clin. Chem. 65, 1171–1179. [DOI] [PubMed] [Google Scholar]
- Mackie K (2006). Cannabinoid receptors as therapeutic targets. Annu. Rev. Pharmacol. Toxicol. 46, 101–122. [DOI] [PubMed] [Google Scholar]
- Mandegar MA, Huebsch N, Frolov EB, Shin E, Truong A, Olvera MP, Chan AH, Miyaoka Y, Holmes K, Spencer CI, et al. (2016). CRISPR interference efficiently induces specific and reversible gene silencing in human iPSCs. Cell Stem Cell 18, 541–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattioli I, Sebald A, Bucher C, Charles RP, Nakano H, Doi T, Kracht M, and Schmitz ML (2004). Transient and selective NF-kappa B p65 serine 536 phosphorylation induced by T cell costimulation is mediated by I kappa B kinase beta and controls the kinetics of p65 nuclear import. J. Immunol. 172, 6336–6344. [DOI] [PubMed] [Google Scholar]
- Mckellar GE, Mccarey DW, Sattar N, and Mcinnes IB (2009). Role for TNF in atherosclerosis? Lessons from autoimmune disease. Nat. Rev. Cardiol. 6, 410–417. [DOI] [PubMed] [Google Scholar]
- Mestas J, and Ley K (2008). Monocyte-endothelial cell interactions in the development of atherosclerosis. Trends Cardiovasc. Med. 18, 228–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreira FA, Grieb M, and Lutz B (2009). Central side-effects of therapies based on CB1 cannabinoid receptor agonists and antagonists: focus on anxiety and depression. Best Pract. Res. Clin. Endocrinol. Metab. 23, 133–144. [DOI] [PubMed] [Google Scholar]
- Moshiri J, Constant DA, Liu B, Mateo R, Kearnes S, Novick P, Prasad R, Nagamine C, Pande V, and Kirkegaard K (2020). A targeted computational screen of the SWEETLEAD database reveals FDA-approved compounds with anti-dengue viral activity. mBio 11. e02839–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Movahed P, Evilevitch V, Andersson TL, Jönsson BA, Wollmer P, Zygmunt PM, and Högestätt ED (2005). Vascular effects of anandamide and N-acylvanillylamines in the human forearm and skin microcirculation. Br. J. Pharmacol. 146, 171–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay P, Rajesh M, Bátkai S, Patel V, Kashiwaya Y, Liaudet L, Evgenov OV, Mackie K, Haskó G, and Pacher P (2010). CB1 cannabinoid receptors promote oxidative stress and cell death in murine models of doxorubicin-induced cardiomyopathy and in human cardiomyocytes. Cardiovasc. Res. 85, 773–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagarkatti P, Pandey R, Rieder SA, Hegde VL, and Nagarkatti M (2009). Cannabinoids as novel anti-inflammatory drugs. Future Med.Chem. 1, 1333–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam D, Ni CW, Rezvan A, Suo J, Budzyn K, Llanos A, Harrison D, Giddens D, and Jo H (2009). Partial carotid ligation is a model of acutely induced disturbed flow, leading to rapid endothelial dysfunction and atherosclerosis. Am. J. Physiol. Heart Circ. Physiol. 297, H1535–H1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onakpoya IJ, Heneghan CJ, and Aronson JK (2016). Worldwide withdrawal of medicinal products because of adverse drug reactions: a systematic review and analysis. Crit. Rev. Toxicol. 46, 477–489. [DOI] [PubMed] [Google Scholar]
- Pacher P, and Mechoulam R (2011). Is lipid signaling through cannabinoid 2 receptors part of a protective system? Prog. Lipid Res. 50, 193–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacher P, Steffens S, Haskó G, Schindler TH, and Kunos G (2018). Cardiovascular effects of marijuana and synthetic cannabinoids: the good, the bad, and the ugly. Nat. Rev. Cardiol. 15, 151–166. [DOI] [PubMed] [Google Scholar]
- Paik DT, Tian L, Lee J, Sayed N, Chen IY, Rhee S, Rhee JW, Kim Y, Wirka RC, Buikema JW, et al. (2018). Large-scale single-cell RNA-Seq reveals molecular signatures of heterogeneous populations of human induced pluripotent stem cell-derived endothelial cells. Circ. Res. 123, 443–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paramel GV, Karadimou G, Eremo AG, Ljungberg LU, Hedin U, Olofsson PS, Folkersen L, Paulsson-Berne G, Sirsjö A, and Fransén K (2020). Expression of CARD8 in human atherosclerosis and its regulation of inflammatory proteins in human endothelial cells. Sci. Rep. 10, 19108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JG, Yoo JY, Jeong SJ, Choi JH, Lee MR, Lee MN, Hwa Lee J, Kim HC, Jo H, Yu DY, et al. (2011). Peroxiredoxin 2 deficiency exacerbates atherosclerosis in apolipoprotein E-deficient mice. Circ.Res. 109, 739–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertwee RG, Howlett AC, Abood ME, Alexander SP, DiMarzo V, Elphick MR, Greasley PJ, Hansen HS, Kunos G, Mackie K, et al. (2010). International Union of Basic and Clinical Pharmacology. LXXIX. Cannabinoid receptors and their ligands: beyond CB₁ and CB₂. Pharmacol. Rev. 62, 588–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pope CA 3rd, Burnett RT, Thurston GD, Thun MJ, Calle EE, Krewski D, and Godleski JJ (2004). Cardiovascular mortality and long-term exposure to particulate air pollution: epidemiological evidence of general pathophysiological pathways of disease. Circulation 109, 71–77. [DOI] [PubMed] [Google Scholar]
- Rajesh M, Bátkai S, Kechrid M, Mukhopadhyay P, Lee WS, Horváth B, Holovac E, Cinar R, Liaudet L, Mackie K, et al. (2012). Cannabinoid 1 receptor promotes cardiac dysfunction, oxidative stress, inflammation, and fibrosis in diabetic cardiomyopathy. Diabetes 61, 716–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajesh M, Mukhopadhyay P, Bátkai S, Patel V, Saito K, Matsumoto S, Kashiwaya Y, Horváth B, Mukhopadhyay B, Becker L, et al. (2010a). Cannabidiol attenuates cardiac dysfunction, oxidative stress, fibrosis, and inflammatory and cell death signaling pathways in diabetic cardiomyopathy. J. Am. Coll. Cardiol. 56, 2115–2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajesh M, Mukhopadhyay P, Haskó G, Liaudet L, Mackie K, and Pacher P (2010b). Cannabinoid-1 receptor activation induces reactive oxygen species-dependent and -independent mitogen-activated protein kinase activation and cell death in human coronary artery endothelial cells. Br. J. Pharmacol. 160, 688–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridker PM, Libby P, Macfadyen JG, Thuren T, Ballantyne C, Fonseca F, Koenig W, Shimokawa H, Everett BM, and Glynn RJ (2018). Modulation of the interleukin-6 signalling pathway and incidence rates of atherosclerotic events and all-cause mortality: analyses from the Canakinumab Anti-Inflammatory Thrombosis Outcomes Study (CANTOS). Eur. Heart J. 39, 3499–3507. [DOI] [PubMed] [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, and Smyth GK (2015). Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha RN, Jana M, and Pahan K (2007). MAPK p38 regulates transcriptional activity of NF-kappaB in primary human astrocytes via acetylation of p65. J. Immunol. 179, 7101–7109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanberg PR, Bunsey MD, Giordano M, and Norman AB (1988). The catalepsy test: its ups and downs. Behav.Neurosci. 102, 748–759. [DOI] [PubMed] [Google Scholar]
- Shi Y, Inoue H, Wu JC, and Yamanaka S (2017). Induced pluripotent stem cell technology: a decade of progress. Nat. Rev. Drug Discov. 16, 115–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugamura K, Sugiyama S, Nozaki T, Matsuzawa Y, Izumiya Y, Miyata K, Nakayama M, Kaikita K, Obata T, Takeya M, and Ogawa H (2009). Activated endocannabinoid system in coronary artery disease and antiinflammatory effects of cannabinoid 1 receptor blockade on macrophages. Circulation 119, 28–36. [DOI] [PubMed] [Google Scholar]
- Sun BB, Maranville JC, Peters JE, Stacey D, Staley JR, Blackshaw J, Burgess S, Jiang T, Paige E, Surendran P, et al. (2018). Genomic atlas of the human plasma proteome. Nature 558, 73–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szentes V, Gazdag M, Szokodi I, and Dézsi CA (2018). The role of CXCR3 and associated chemokines in the development of atherosclerosis and During myocardial infarction. Front. Immunol. 9, 1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas G, Kloner RA, and Rezkalla S (2014). Adverse cardiovascular, cerebrovascular, and peripheral vascular effects of marijuana inhalation: what cardiologists need to know. Am. J. Cardiol. 113, 187–190. [DOI] [PubMed] [Google Scholar]
- Wang X, Derakhshandeh R, Liu J, Narayan S, Nabavizadeh P, Le S, Danforth OM, Pinnamaneni K, Rodriguez HJ, Luu E, et al. (2016). One minute of marijuana second hand smoke exposure substantially impairs vascular endothelial function. J. Am. Heart Assoc. 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsh P, Murray HM, Ford I, Trompet S, DeCraen AJ, Jukema JW, Stott DJ, Mcinnes IB, Packard CJ, Westendorp RG, et al. (2011). Circulating interleukin-10 and risk of cardiovascular events: a prospective study in the elderly at risk. Arterioscler.Thromb.Vasc.Biol. 31, 2338–2344. [DOI] [PubMed] [Google Scholar]
- Whiting PF, Wolff RF, Deshpande S, DiNisio M, Duffy S, Hernandez AV, Keurentjes JC, Lang S, Misso K, Ryder S, et al. (2015). Cannabinoids for medical use: a systematic review and meta-analysis. JAMA 313, 2456–2473. [DOI] [PubMed] [Google Scholar]
- Wiley JL, and Martin BR (2003). Cannabinoid pharmacological properties common to other centrally acting drugs. Eur. J. Pharmacol. 471, 185–193. [DOI] [PubMed] [Google Scholar]
- Yang Z, Kulkarni K, Zhu W, and Hu M (2012). Bioavailability and pharmacokinetics of genistein: mechanistic studies on its ADME. Anticancer. Agents Med. Chem. 12, 1264–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun TJ, Lee JS, Shim D, Choi JH, and Cheong C (2017). Isolation and characterization of aortic dendritic cells and lymphocytes in atherosclerosis. Methods Mol. Biol. 1559, 419–437. [DOI] [PubMed] [Google Scholar]
- Zernecke A, and Weber C (2010). Chemokines in the vascular inflammatory response of atherosclerosis. Cardiovasc. Res. 86, 192–201. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw data from the Olink and Luminex analysis were deposited on Medeley https://data.mendeley.com/datasets/s6j8h9yy57/1 and are publicly available as of the date of publication. The RNA sequencing data was deposited at the NCBI (GSE198578). Any additional information required for reanalysis is available upon request.