

Abstract
Multidrug-resistant tuberculosis (MDR-TB) accounts for one third of the annual deaths due to antimicrobial resistance1. Drug resistance-conferring mutations frequently cause fitness costs in bacteria2–5. Experimental work indicates that these drug resistance-related fitness costs might be mitigated by compensatory mutations6–10. However, the clinical relevance of compensatory evolution remains poorly understood. Here we show that, in the country of Georgia, during a 6-year nationwide study, 63% of MDR-TB was due to patient-to-patient transmission. Compensatory mutations and patient incarceration were independently associated with transmission. Furthermore, compensatory mutations were overrepresented among isolates from incarcerated individuals that also frequently spilled over into the non-incarcerated population. As a result, up to 31% of MDR-TB in Georgia was directly or indirectly linked to prisons. We conclude that prisons fuel the epidemic of MDR-TB in Georgia by acting as ecological drivers of fitness-compensated strains with high transmission potential.
Growing antimicrobial resistance is a threat to global public health and the economy1. In 2019, an estimated 464,000 new cases of human tuberculosis (TB) were caused by rifampicin-resistant Mycobacterium tuberculosis (Mtb) worldwide, of which 78% were MDR-TB strains, resistant to the two first-line antibiotics, isoniazid and rifampicin11. However, the number of MDR-TB-associated fatalities is small compared to the annual total of 1.4 million deaths due to TB in general1,11. Moreover, only 3.3% of the 10 million annual new TB cases in the world are caused by MDR Mtb variants, and this proportion has remained stable despite TB being treated with antibiotics for decades12. Based on these observations, it was suggested that MDR-TB might be generally less transmissible due to fitness costs of MDR13, and, as a consequence, MDR-TB was predicted to remain a localized problem14. Indeed, several geographical hotspots of MDR-TB exist, with the countries of the former Soviet Union being heavily affected, for reasons not well understood. For instance, in the country of Georgia, 12% of all new TB cases in 2019 were caused by MDR strains15. Recent studies have also demonstrated that most drug resistance in TB is due to transmission, as opposed to de novo evolution within patients16–19.
In Mtb, drug resistance is predominantly conferred by chromosomal mutations in the genes encoding the drug target4. Rifampicin resistance is caused by mutations in the gene rpoB, encoding the β subunit of the bacterial RNA polymerase. In vitro data demonstrated a fitness deficit for rifampicin-resistant Mtb5. In contrast, analysis of paired clinical isolates from patients who acquired rifampicin resistance during treatment (mediated by the same rpoB mutations as assessed previously in vitro) revealed that some of these strains did not carry any detectable fitness deficit in vitro. It was hypothesized that these clinical strains acquired secondary, fitness-deficit compensating mutations. Genome analyses of experimentally evolved Mtb laboratory strains, together with a collection of rifampicin-resistant clinical strains, revealed the presence of compensatory mutations in the RNA polymerase10. Subsequent work conducted in several bacterial species6,7,20 showed that secondary mutations in the RNA polymerase restored the transcriptional activity of the enzyme. However, whether these compensatory mutations influence the transmission fitness of Mtb in human populations remains to be established. Although several studies in human populations assessed the effect of compensatory mutations on the transmissibility of MDR-TB, findings have been inconsistent16,21–23. Moreover, these previous studies relied on small datasets and did not control for confounding factors.
In this study, we assessed associations among various bacterial factors and corresponding patient data with measures of MDR-TB transmission inferred with the R package phybreak24. For this, we used a nationwide collection of 1,613 MDR-TB whole-genome sequences from Georgia, isolated between 2011 and 2016 (Fig. 1 and Supplementary Fig. 1). For the remainder of this article, our use of the term MDR will include pre-XDR (pre-XDR: MDR + resistance to either aminoglycosides or fluoroquinolones) and XDR (XDR: MDR + fluoroquinolone and aminoglycoside resistance) cases, unless otherwise stated. The dataset represents 70% of all culture-confirmed MDR-TB cases isolated in Georgia in this timeframe (Supplementary Table 1 and Supplementary Fig. 2). The accession numbers of all included genomes are listed in Supplementary Table 2.
Fig. 1 |. Phylogeny of 1,613 MdR M. tuberculosis strains.
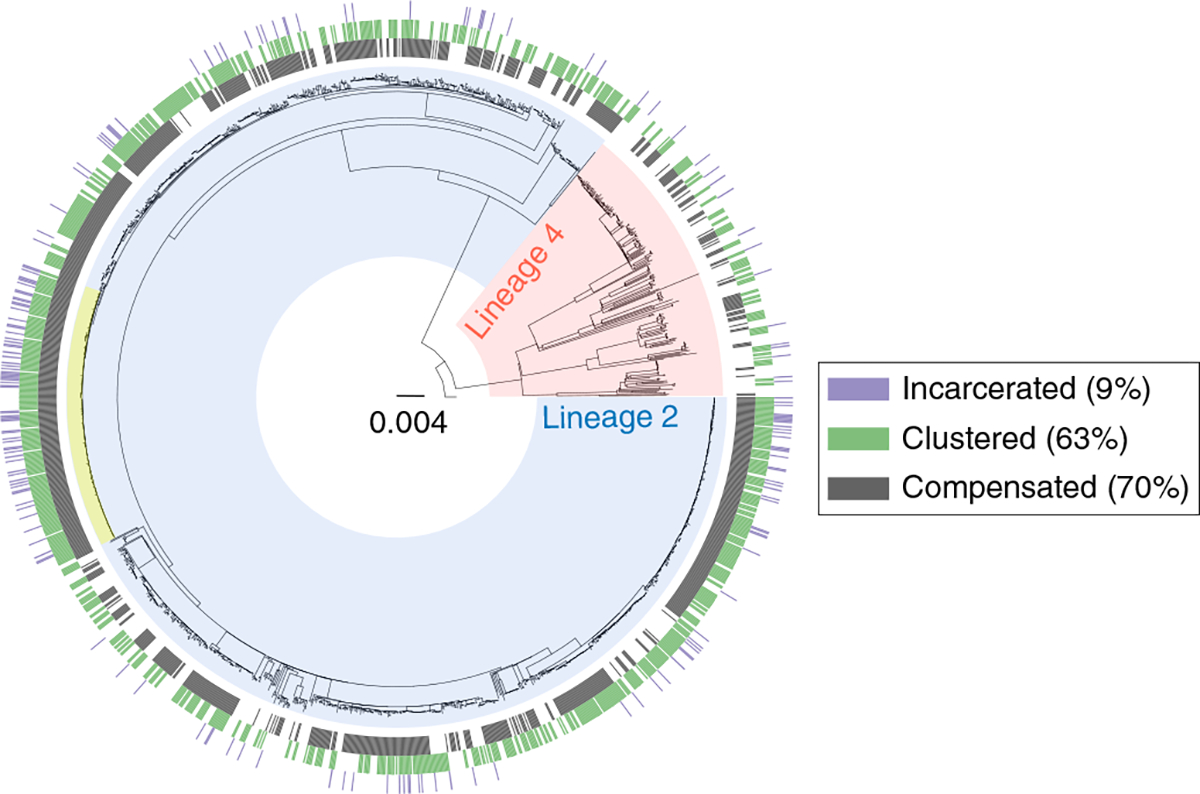
The blue clade corresponds to Lineage 2 (Beijing sublineage); the red clade corresponds to Lineage 4. The phylogeny is rooted on M. canettii. Scale bar indicates substitutions per site. The largest transmission cluster is highlighted in yellow (n = 183 strains). The outer rings indicate the status of incarceration (purple), compensation (dark gray) and clustering (green).
We first predicted the drug resistance profiles of the sequenced Mtb strains by screening for the presence of known drug resistance markers (Supplementary Table 3) and found that the epidemic of drug-resistant TB in Georgia is driven by strains resistant to many drugs, with most being even more resistant than regular MDR strains (Supplementary Fig. 3 and Supplementary Table 4). Of the 1,613 strains analyzed, 699 strains (43%) were pre-XDR, 605 (38%) were MDR and 309 (19%) were XDR (Supplementary Table 4). The pre-XDR and XDR resistance profiles are strongly associated with treatment failure25,26. Apart from the mutations defining the drug resistance class, all strains carried a median of two (25th percentile = 2 and 75th percentile = 2) additional resistance mutations. Control measures in Georgia will be hampered by the predominance of pre-XDR phenotypes and the high prevalence of additional resistance mutations; for example, 92% of the strains harbored streptomycin resistance-conferring mutations (Supplementary Table 4). Treatment regimens, including the novel and repurposed drugs bedaquiline, pretomanid, delamanid and linezolid, will be necessary to combat the epidemic of MDR-TB in Georgia27. On that note, we identified 22 strains with mutations in the promoter region or coding sequence of the transcriptional repressor Rv0678/mmpR. Mutations in Rv0678/mmpR are implicated in bedaquiline/clofazimine cross-resistance28, and bedaquiline has been used in Georgia on a compassionate basis since 2011 (ref. 29).
To identify putative compensatory mutations, we screened the RNA polymerase genes rpoA, rpoB and rpoC for the presence of non-synonymous mutations. After filtering for phylogenetic markers, we identified a total of 71 distinct substitutions (Fig. 2 and Supplementary Tables 5 and 6). Most compensatory mutations evolved between one and five times in the dataset and were shared only among a limited number of strains (Fig. 2). However, several mutations were shared by many strains and independently evolved multiple times (Fig. 2), indicating a strong selective benefit to MDR-TB strains harboring these mutations.
Fig. 2 |. Putative compensatory mutations identified in the three subunits of the RNA polymerase.
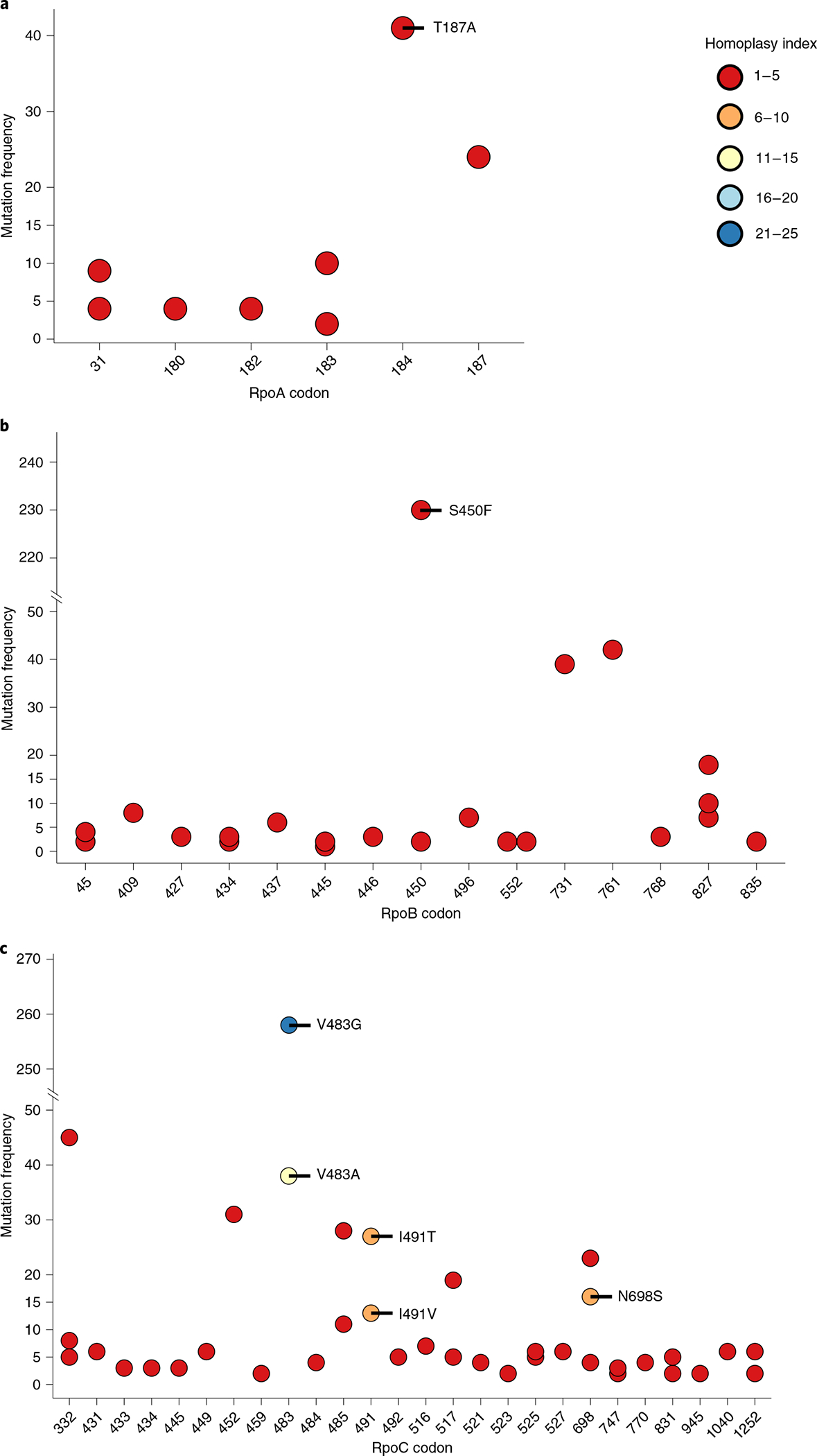
Mutations remaining after filtering for phylogenetic markers. The homoplasy index indicates the number of independent evolution events of the mutation in question. The frequency of the mutation indicates the number of strains harboring the respective mutation.
We next used the whole-genome sequences to identify transmission clusters based on the genetic distance between two given strains to determine whether MDR Mtb strains evolved de novo in patients or stemmed from transmission. We identified 212 transmission clusters with a median size of two strains (25th percentile = 2, 75th percentile = 4 and interquartile range (IQR) = 2), resulting in 63% (n = 1,018) of strains being clustered (Fig. 1). The high proportion of strains in clusters indicates frequent transmission of MDR-TB in Georgia. We detected a cluster containing at least 183 pre-XDR strains, which is one of the largest transmission clusters of pre-XDR strains reported to date (Fig. 1). Moreover, we detected an XDR-TB transmission cluster with at least eight members, harboring the putative bedaquiline/clofazimine resistance mutation Rv0678 F93L30, potentially indicating transmitted bedaquiline/clofazimine resistance.
To test if compensatory mutations are associated with the transmission of MDR Mtb in Georgia, we performed multivariable Poisson regression on a subset of 1,263 strains for which complete epidemiological records were available (Supplementary Table 2), with the number of secondary cases generated per individual patient during the study timeframe as the outcome variable. The number of secondary cases generated per patient was inferred from the phybreak24 infector–infectee relationships (Supplementary Table 7). For the phybreak analysis, we used a set of priors for the generation and sampling time intervals inferred by fitting a gamma distribution to real-world data on the time course of Mtb infections from the pre-antibiotic era31. We performed a sensitivity analysis using different sets of priors (Supplementary Table 8), resulting in similar infector–infectee relationships (Supplementary Tables 9–13) and multivariable Poisson regression results (Supplementary Tables 14–18). We found that compensatory mutations in the RNA polymerase were associated with the number of secondary cases generated per patient (adjusted incidence rate ratio (IRRadj) = 1.34, 95% confidence interval (CI95) = 1.05–1.71, P = 0.019; Table 1 (posterior probability (PP) > 0.5) and Supplementary Tables 14–18). Other predictors of transmission success of MDR-TB strains included a patient being incarcerated (IRRadj = 1.42, CI95 = 1.11–1.81, P = 0.005; Table 1 (PP > 0.5) and Supplementary Tables 14–18), in line with the known role of prisons in the epidemic of MDR Mtb in the former Soviet Union32–34. Age was negatively associated with the number of secondary cases generated (IRRadj = 0.98, CI95 = 0.97–0.99, P < 0.001; Table 1 (PP > 0.5) and Supplementary Tables 14–18), as was female sex (IRRadj = 0.73, CI95 = 0.55–0.95, P = 0.022; Table 1 (PP > 0.5) and Supplementary Tables 14–18), consistent with the epidemiology of TB in middle- and high-burden countries35. In concordance with previous reports21,36, the Lineage 2/Beijing family of Mtb was associated with transmission (IRRadj = 2.24, CI95 = 1.48–3.53, P < 0.001; Table 1 (PP > 0.5) and Supplementary Tables 14–18), supporting the notion that Lineage 2 strains might suffer from smaller drug resistance-related fitness costs5. To control for the possible overrepresentation of prison samples, we repeated this analysis excluding all incarcerated individuals (nincarcerated = 171 and nnon-incarcerated = 1,092), but the results remained unchanged (Table 1 and Supplementary Tables 14–18).
Table 1 |.
Estimated association among bacterial and patient factors and the rate of secondary cases generated
| Dependent variable: rate of secondary cases | ||||||
|---|---|---|---|---|---|---|
| All isolates with metadata (PP > 0.5), n = 1,263 | Excluding isolates from incarcerated individuals (PP > 0.5), n = 1,092 | All isolates with metadata, n = 1,263 | ||||
| Explanatory variables | Levels | Total | Univariable IRR (CI95, P value) | Multivariable aIRRadJ (CI95, P value) | Multivariable aIRRadJ (CI95, P value) | Multivariable aIRRadJ (CI95, P value) |
| Putative compensatory mutation in rpoA, rpoB and rpoC | 0 | 389 (30.80) | – | – | – | – |
| 1 | 874 (69.20) | 1.68 (1.34–2.12, P < 0.001) | 1.34 (1.05–1.71, P = 0.019) | 1.30 (1.01–1.69, P = 0.046) | 1.37 (1.11–1.70, P = 0.003) | |
| Incarcerated individual | 0 | 1092 (86.46) | – | – | – | – |
| 1 | 171 (13.54) | 2.14 (1.71–2.64, P < 0.001) | 1.42 (1.11–1.81, P = 0.005) | – | 1.51 (1.22–1.87, P < 0.001) | |
| Lineage 2 strain | 0 | 180 (14.25) | – | – | – | – |
| 1 | 1083 (85.75) | 2.75 (1.88–4.23, P < 0.001) | 2.24 (1.48–3.53, P < 0.001) | 2.44 (1.53–4.12, P < 0.001) | 2.64 (1.82–3.98, P < 0.001) | |
| Age | Mean (s.d.) | 38.54 (13.75) | 0.98 (0.97–0.99, P < 0.001) | 0.98 (0.97–0.99, P < 0.001) | 0.98 (0.97–0.99, P < 0.001) | 0.98 (0.97–0.99, P < 0.001) |
| Female sex | 0 | 981 (77.67) | – | – | – | – |
| 1 | 282 (22.33) | 0.67 (0.52–0.86, P = 0.002) | 0.73 (0.55–0.95, P = 0.022) | 0.74 (0.56–0.97, P = 0.033) | 0.96 (0.77–1.19, P = 0.735) | |
| Number of additional drug resistance mutations | Mean (s.d.) | 2.02 (0.68) | 1.18 (1.03–1.36, P = 0.020) | 1.04 (0.87–1.24, P = 0.686) | 1.05 (0.86–1.26, P = 0.647) | 1.00 (0.85–1.16, P = 0.983) |
| Drug resistance profile | MDR | 512 (40.54) | – | – | – | – |
| Pre-XDR | 534 (42.28) | 1.15 (0.93–1.42, P = 0.192) | 0.91 (0.73–1.14, P = 0.423) | 0.94 (0.73–1.22, P = 0.645) | 0.91 (0.75–1.10, P = 0.314) | |
| XDR | 217 (17.18) | 1.25 (0.95–1.61, P = 0.101) | 1.17 (0.88–1.54, P = 0.266) | 1.14 (0.84–1.55, P = 0.396) | 0.98 (0.76–1.25, P = 0.859) | |
| TB diagnosis in the past | 0 | 716 (56.69) | – | – | – | – |
| 1 | 547 (43.31) | 0.99 (0.82–1.19, P = 0.913) | 0.94 (0.77–1.15, P = 0.544) | 0.89 (0.71–1.12, P = 0.341) | 0.95 (0.80–1.13, P = 0.566) | |
IRRs were estimated by multivariable Poisson regression adjusting for the presence of putative compensatory mutations, incarceration status, Mtb lineage, patient age, patient sex, number of additional drug resistance mutations, drug resistance profile and TB diagnosis in the past. We corrected for unequal observation time of the isolates by including the isolation time as a category. For the 6-year study period, we included 12 half-year categories.
We next hypothesized that environments facilitating transmission of Mtb (for example, prisons; Table 1) also facilitate the evolution and persistence of compensatory mutations. Every new infection leads to cell division events during the growth of the bacterial population. Each cell division (that is, genome replication) offers the possibility to acquire a compensatory mutation. Clones carrying a compensatory mutation might then outgrow the uncompensated population within a single patient. In accordance with previous reports33,34,37,38, our results showed that prisons in Georgia were associated with the number of secondary cases generated (Table 1). These results indicate that Georgian prisons are environments conducive for Mtb transmission. In support of our hypothesis, we also found that compensatory mutations were overrepresented in strains isolated from incarcerated individuals (82% of prison isolates versus 67% of isolates from non-incarcerated individuals harbored compensatory mutations; P < 0.001, two-tailed χ2 test; Supplementary Fig. 4). To test the hypothesis that prisons act as a source for newly compensated MDR-TB strains, we compared the number of compensatory mutations per genotype among incarcerated and non-incarcerated individuals. Incarcerated individuals tended to harbor more compensatory mutations per genotype than non-incarcerated individuals, but the difference was not statistically significant (0.71 versus 0.62; P = 0.19, two-tailed χ2 test). Given our initial hypothesis that transmission-conducive settings select for highly transmissible, fitness-compensated strains, we could also imagine that prison isolates carry additional compensatory mutations in other genes than the canonical targets: rpoA, rpoB and rpoC. In support of this possibility, the mean number of secondary cases generated by incarcerated individuals harboring a compensated strain, which clustered with at least one incarcerated individual, was larger compared to non-incarcerated individuals with a compensated strain that clustered only with non-incarcerated individuals (0.78 versus 0.55; P = 0.04, Wilcoxon rank-sum test). Moreover, clusters containing incarcerated individuals were larger than clusters containing only non-incarcerated individuals (median size of clusters: 4 versus 2; P < 0.001, Wilcoxon rank-sum test; Supplementary Fig. 5).
To further investigate the number of secondary cases derived from incarcerated and non-incarcerated individuals, we used phybreak to estimate the most likely index cases of every transmission cluster24. We classified the clusters derived from incarcerated and non-incarcerated individuals based on the index case and calculated secondary case rates and secondary case rate ratios, by incarceration status and presence of compensatory mutations. We observed that transmission clusters classified as being founded by incarcerated individuals had higher secondary case rates among non-incarcerated individuals, compared to clusters founded by non-incarcerated individuals (secondary case rate ratio = 10.6, CI95 = 8.7–13.1, P < 0.01; Supplementary Table 19). This was particularly true for clusters founded by incarcerated individuals infected with a strain carrying a compensatory mutation in rpoA, rpoB and rpoC (secondary case rate ratio among strains carrying a compensatory mutation in rpoA, rpoB and rpoC = 13.6, CI95 = 10.8–17.1, P < 0.01; Supplementary Table 19). However, in the absence of any compensatory mutations in rpoA, rpoB and rpoC, the secondary case rates among non-incarcerated individuals did not differ significantly between transmission clusters derived from incarcerated or non-incarcerated individuals (secondary case rate ratio among uncompensated strains = 1.0, CI95 = 0.3–2.4, P = 0.94; Supplementary Table 19).
Lastly, we aimed to quantify the spill-over of MDR-TB strains from prisons to the general public. For this, we analyzed the transmission networks (Fig. 3 and Supplementary Fig. 6), identified events where incarcerated individuals infected non-incarcerated individuals and counted all non-incarcerated individuals downstream of the initial transmission event (Supplementary Fig. 7). Although the prison population declined during the study period, we were able to document multiple spill-over events from prisons and the establishment of these clones among the general public (Fig. 3a,b and Supplementary Fig. 7). Overall, we identified 42 transmission events, where incarcerated individuals directly infected non-incarcerated individuals, 83% (35/42) of which carried compensatory mutations in rpoA, rpoB and rpoC. The non-incarcerated individuals, infected by incarcerated individuals, subsequently infected 159 non-incarcerated individuals (Supplementary Fig. 7). In total, 20% of all cases among non-incarcerated individuals sampled in this study originated from incarcerated individuals. Extrapolating to the whole population, based on the World Health Organization (WHO)-notified, culture-confirmed MDR-TB cases (n = 2,292; Supplementary Table 1), we estimated that, during the 6-year study period, 31% (n = 705) of MDR-TB cases were directly or indirectly linked to prisons.
Fig. 3 |. Spill-over of MdR-TB from prisons into the general public.
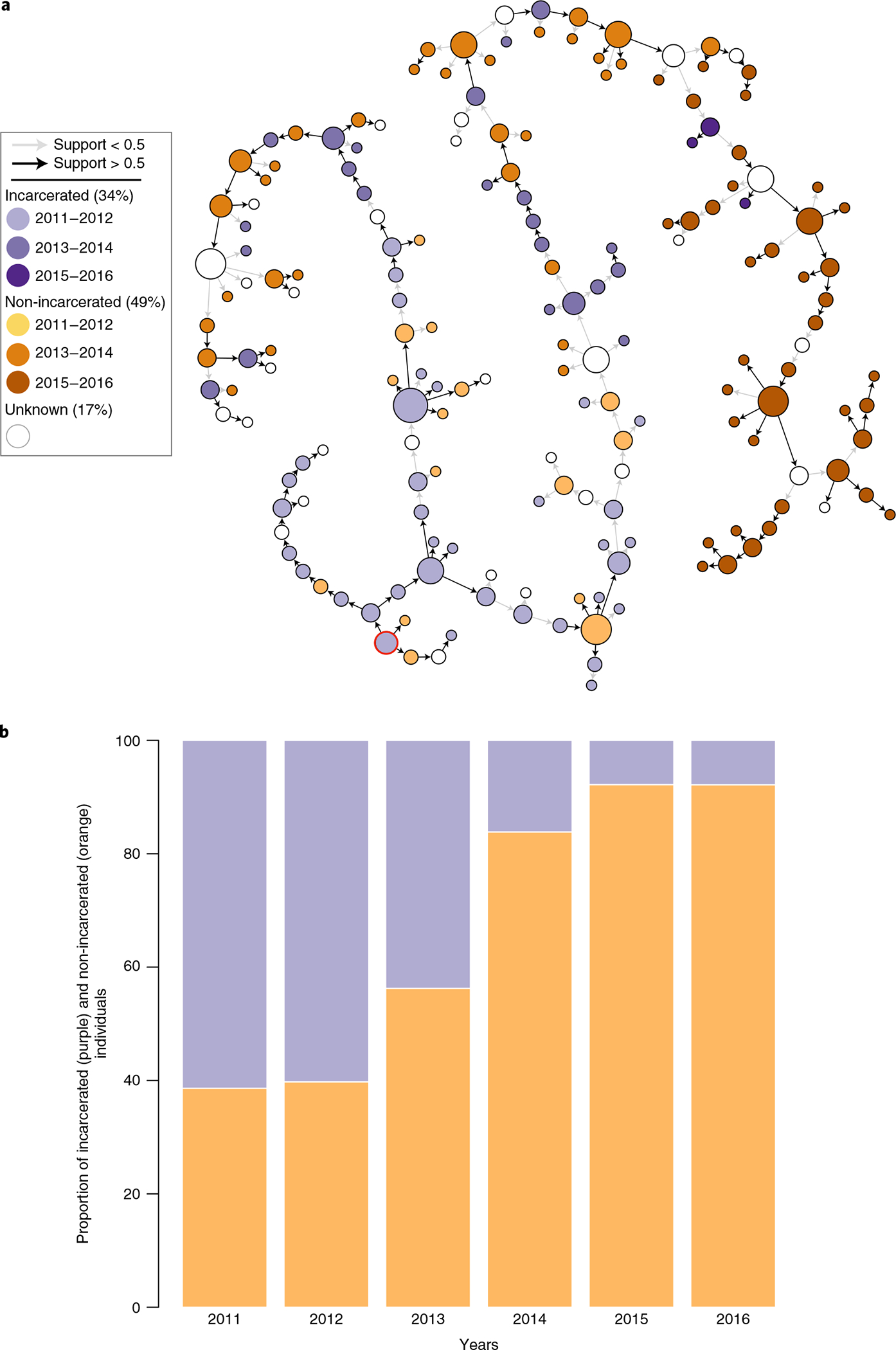
a, Illustration of the transmission chain for the largest transmission cluster (n = 183). Isolates from incarcerated individuals have purple-colored nodes; isolates from non-incarcerated individuals have orange-colored nodes. Darker colors represent more recently sampled isolates. The most likely index case of the cluster is circled in red. Arrows indicate the directionality of the transmission events. High-confidence transmission events with probabilities >0.5 have a black arrow. b, Proportion of isolates from incarcerated and non-incarcerated individuals across the years in clusters containing at least one incarcerated individual. Orange represents non-incarcerated individuals; purple represents incarcerated individuals.
Our study demonstrates that most MDR-TB cases in the country of Georgia are due to ongoing transmission of highly drug-resistant Mtb strains. Moreover, our results complement previous experimental findings by confirming that compensatory mutations in the RNA polymerase of Mtb contribute to the transmission fitness of MDR-TB strains in a human population. The strong association between MDR-TB and incarceration is consistent with previous studies conducted in the former Soviet republics32–34. In addition, our study allowed us to quantify the effect of incarceration on the transmission of MDR-TB and further highlights the role of prisons in the epidemic of MDR-TB. Unfortunately, the limited metadata did not allow us to assess transmission in other marginalized populations, such as drug users and the homeless. Although the overall number of incarcerated individuals in Georgia declined during the study period, our analyses indicate that MDR-TB strains frequently spill over from prisons into the general public. This might be due either to the large proportion of incarcerated individuals who are lost to follow-up after release from prison, who might continue to transmit, or to the transmission from incarcerated individuals to employees working in the prison system. Moreover, our findings revealed a link between incarceration and compensatory evolution and suggest that forcing people into a crowded environment, highly conducive of TB transmission, might influence the evolutionary trajectories toward highly drug-resistant and fitness-compensated bacteria. Combined, these observations suggest that prisons serve as ecological drivers of compensatory mutations and might amplify the presence of these mutations among non-incarcerated individuals, highlighting the importance of infection control among the highly vulnerable prison population.
Online content
Any methods, additional references, Nature Research reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41591-021-01358-x.
Methods
Sample set and associated metadata.
The dataset consisted of 2,063 strains, including all culture-confirmed Mtb isolates demonstrating at least an MDR phenotype, collected between 2011 and 2016 and stored by the National Center for Tuberculosis and Lung Disease in Tbilisi, Georgia. The isolates were re-cultured and processed for DNA extraction for whole-genome sequencing. A total of 450 strains were excluded from further analysis owing to failed sequencing, no MDR genotype, large numbers of unfixed positions (potential mixed infections/cross-contamination) or strains with multiple differing metadata entries (Supplementary Fig. 1). The final dataset consisted of 1,613 whole-genome sequences of MDR Mtb strains, representing 70% of all culture-confirmed MDR Mtb strains isolated between 2011 and 2016. The per-year sampling coverages are summarized in Supplementary Table 1. The per-year lineage proportions remained stable (Supplementary Fig. 8). Limited anonymized patient data, including sex, age, year released from prison, current incarceration status and prior TB diagnosis, were available (Supplementary Table 2). For the purpose of this study, prison isolates were defined as follows: patient was incarcerated at the time of sample collection or patient was released during the study timeframe (2011–2016), and the sample was collected at the latest within the 6 months of the first year after release. For 22% of the analyzed strains, no matching metadata were available; for all other strains, complete records were available.
Ethical approval.
To be able to quantify TB transmission accurately, this study had to be population based and nationwide—that is, all culture-confirmed MDR-TB cases were included during all of the 6-year study period from the whole country of Georgia. Because, every year, thousands of patients with suspected TB are seen in 67 TB clinics distributed across the country, the logistics required for obtaining formal informed patient consent were prohibitive. As only pseudonymized samples and only limited pseudonymized patient data available at the National TB Reference Center were used, and because the project was of particular interest to the Georgian Ministry of Health and the National TB Control Program, we obtained formal exemption from the Georgian Ministry of Health for the need for individualized informed patient consent. The corresponding study protocol was then also formally reviewed and approved by the national ethics review boards in both Georgia and Switzerland.
Statistical analyses.
Identification of covariates associated with the number of secondary cases per patient was performed by multivariable Poisson regression. We tested the assumption of linearity between the continuous variables (age and number of drug resistance mutations, in addition to the resistance class (MDR, pre-XDR and XDR) determining mutations) and the log of the rate graphically. To investigate whether super-spreaders could be driving the associations in the Poisson regression model, we excluded all isolates with an outdegree >1 (n = 103) and repeated the regression analysis. The same explanatory variables, except for incarceration, remained associated with the outdegree (data not shown). Secondary case rates and secondary case rate ratios were calculated using the R package epitools (v0.5–10.1). Two-tailed χ2 tests were used to analyze the association of the number of compensatory mutations with incarceration as well as to compare the number of compensatory mutations per genotype. Wilcoxon rank-sum tests were used to assess differences in mean cluster sizes and mean number of secondary cases generated between clusters containing incarcerated individuals and clusters containing only non-incarcerated individuals. A two-sided z-proportion test was used to assess differences in lineage proportions during the sampling time frame. All analyses were performed using R (v3.6.2) unless otherwise stated.
Whole-genome sequencing.
Sequencing libraries were prepared using the Illumina Nextera XT kit and subjected to massive parallel sequencing on the Illumina HiSeq 2500 platform, whereby 31–138-bp paired-end reads were generated. All sequencing runs were performed at the core sequencing facility of the ETHZ/University of Basel in Basel, Switzerland. The reads were processed using an in-house pipeline as described previously39. Briefly, Trimmomatic (v0.33) was used to clip adapters and filter for quality, whereby <20-bp reads were discarded. Overlapping paired-end reads were merged with SeqPrep (https://github.com/jstjohn/SeqPrep). The resulting reads were mapped to a reconstructed hypothetical ancestor of the Mtb complex10 with BWA (v0.7.12); duplicate reads were marked with Picard (v2.1.1) (https://github.com/broadinstitute/picard) with the MarkDuplicates module. To enhance mapping of reads in the vicinity of indels, local realignments were performed with the GATK (v3.4.0) modules RealignerTargetCreator and IndelRealigner40. Pileups were generated with SAMtools (v1.2)41, and single-nucleotide variants (SNVs) were subsequently called with VarScan (v2.4.1)42, applying the following thresholds: minimum mapping / mimimum base quality of 20, minimum read depth 7× at a given position. SNVs were called if at least five reads supported the alternative allele without strand bias. A given SNV was considered fixed if its frequency reached 90%, and a position was called as ancestral if the frequency was below 10%. The effect of the SNV was inferred using SnpEff (v4.11)43 using the Mtb H37Rv reference annotation (NC_000962.3). SNVs lying in regions that share ≥50-bp sequence identity with other regions in the genome were excluded39. SNVs in ‘PPE/PGRS’ genes, regions annotated as ‘maturase’, ‘phage’ or ‘insertion sequence’, as well as regions that were previously identified to contain repetitive regions, were excluded.
The unfixed position outliers were defined as having >3× the IQR of the ratio between fixed and unfixed positions, and the IQR was calculated separately for Lineage 2 and Lineage 4 strains.
Variable position alignment and phylogenetic analysis.
Variable SNV pseudo-alignments were generated by concatenating all quality filtered SNVs in the dataset. A position was encoded as an X in the alignment if it was covered by fewer than seven reads or if it fell into one of the excluded regions (see above) or if it was unfixed. If a position was not covered, it was encoded as a gap. Only positions that had less than 90% encoded as X or gaps were investigated. A position was considered as variable if at least one isolate in the dataset had a fixed SNV at the position in question. Two separate alignments were produced: one including genes known to be involved in drug resistance and a separate alignment excluding variable positions in drug resistance-related genes. The former alignment was used for genetic distance-based transmission cluster inference (see below), and the latter was used to infer a maximum likelihood phylogeny using RAxML (v8.2.8)44. The phylogeny was inferred using the general time-reversible model of sequence evolution and rooted on Mycobacterium canettii (SRR011186). Strains were classified into main- and sub-lineages based on the presence of previously established markers45.
Drug resistance profile prediction.
We collated a list of high-confidence drug resistance mutations (mutations and sources summarized in Supplementary Table 3), which we used to screen the genomes. In addition, all non-synonymous substitutions in ethA, pncA and Rv0678/mmpR were regarded as conferring resistance to ethionamide, pyrazinamide and bedaquiline, respectively.
Identification of compensatory mutations.
We screened the genes rpoA, rpoB and rpoC, encoding the DNA-dependent RNA polymerase, for the presence of non-synonymous mutations. This resulted in a list of rifampicin resistance-conferring, phylogenetic and putative compensatory mutations. Per definition, compensatory mutations must co-occur with rifampicin resistance-conferring mutations but are never found on their own. To remove phylogenetic markers, we collated a list of non-synonymous mutations in rpoA, rpoB and rpoC identified in rifampicin-susceptible strains (defined as harboring none of the high-confidence rifampicin resistance-conferring mutations listed in Supplementary Table 3), using a large collection of published genomes39 and an in-house collection of genomes, including drug-susceptible strains from Georgia. After filtering for phylogenetic and rifampicin resistance-conferring mutations, every mutation identified in rpoA, rpoB and rpoC was assumed to be a secondary and compensatory mutation if it met one of the following criteria: the mutation occurred multiple times independently; more than one distinct amino acid substitution affected the same codon; the mutation was present in the rifampicin resistance-determining region consisting of RpoB codons 426–452 in addition to a known rifampicin resistance-conferring mutation; the mutation was present in the RNA exit tunnel consisting of RpoA codons 172–192 and RpoC codons 423–563 (refs. 7,46); or the mutation was reported previously10,16,21. Furthermore, strains harboring two mutations affecting the same codon in rpoB were assumed to be compensated if one of the mutations conferred rifampicin resistance on its own—for example, rpoB c.1349T>C (S450L confers rifampicin resistance; Supplementary Table 3) and rpoB c.1350G>C (S450S); combined, these two mutations result in the substitution RpoB S450F.
Genetic distance-based transmission cluster definition.
The likelihood of two strains being members of a transmission chain decreases with the number of genetic differences between two strains. Previous analyses have demonstrated that two Mtb strains, isolated from patients with a proven epidemiologic link, rarely differ by more than five mutations from each other47. A distance matrix, based on pairwise SNV distances between any given two strains, was inferred using the variable position pseudo-alignment including variable positions in drug resistance-related genes with custom scripts (https://git.scicore.unibas.ch/TBRU/tacos). Insertions and deletions were considered as missing data. Agglomerative clustering was performed using the R package cluster (v2.0.6) with the agnes function using the unweighted pair group average method. A threshold of five SNVs, on average, was used as a cutoff for likely patient-to-patient transmission47. The function hclust was used to cut the tree at a height of five SNVs.
Transmission networks, index case inference and infector–infectee relationships.
Transmission graphs, index cases and infector–infectee relationships were inferred using the R package phybreak (v0.2.0)24 running under R (v3.3.1). phybreak infers consensus transmission trees by combining transmission models, within-host dynamics, case observation and mutation rate using Bayesian inference combined with Markov chain Monte Carlo (MCMC) sampling of the posterior distribution of model parameters, transmission and phylogenetic trees. Priors for the mean of the sampling time for both distributions were inferred from collated data on the time course of Mtb infections31 by fitting a gamma distribution with the R package fitdistrplus (v1.0–11). The mutation rate prior was set at 1 mutation per genome and year47,48. We ran 20 independent MCMC chains with a burn-in set at 10,000 cycles, and the sampling of the independent chains was set at 50,000 cycles to ensure that most estimated parameters reached an effective sample size >200 (ref. 49). We excluded infector–infectee relationships for which the effective sample size did not reach at least 200. This frequently occurred when sampling dates of cluster members in small clusters were identical or within a short time period. As a sensitivity analysis, we repeated the phybreak analysis with different sets of priors for the generation/sampling time distributions and mutation rate (Supplementary Table 8). The infector–infectee relationships based on the different phybreak runs are summarized in Supplementary Tables 7 and 9–13, and the results of the regression analyses are summarized in Supplementary Tables 14–18. The Python package NetworkX (v2.2) was used to transform the infector–infectee relationships inferred by phybreak into a network. The software Gephi (v0.9.2) was then used to plot and annotate the networks.
Number of compensatory mutations per genotype.
We aimed to assess whether there were more compensatory mutations among genotypes associated with incarcerated compared to non-incarcerated individuals. A genotype was defined as follows. Every unclustered strain and every transmission cluster represents an opportunity to acquire a compensatory mutation and was counted as an individual genotype. We divided the dataset into an incarcerated and a non-incarcerated cohort. Every cluster containing at least one incarcerated individual was counted as a prison cluster/genotype. The number of compensatory mutations per genotype among incarcerated individuals and non-incarcerated individuals was given as follows:
where Cinc denoted the number of compensatory mutations per genotype linked to the incarcerated population, and Cnon-inc denoted the number of compensatory mutations per genotype among the non-incarcerated population. The variables denote the following: Iucl, set of unclustered incarcerated individuals/genotypes; Icomp, set of incarcerated individuals/genotypes harboring compensated MDR-TB strains; ClI, set of clusters/genotypes containing at least one incarcerated individual; Clcomp, set of clusters/genotypes harboring compensatory mutations; NIucl, set of unclustered non-incarcerated individuals/genotypes; NIcomp, set of non-incarcerated individuals/genotypes harboring compensated MDR-TB strains; and ClNI, clusters containing only non-incarcerated individuals/genotypes. To calculate Cnon-inc, we sampled 181 non-incarcerated individuals, corresponding to the total number of incarcerated individuals in the dataset, and repeated the sampling 10,000 times. We reported the mean number of compensatory mutations per genotype.
Secondary case rates, case rate ratios and estimation of prison effect.
For this, we first used the phybreak output to estimate the most likely index case (PP > 0.5) and classify the cluster as derived from either an incarcerated or a non-incarcerated individual. To obtain the secondary case rates among non-incarcerated individuals infected by incarcerated individuals, we divided the total number of secondary cases among non-incarcerated individuals infected by incarcerated individuals by the sum of all prison-related index cases (the number of prison-derived clusters plus all unique cases among incarcerated individuals). We performed the same calculation for clusters derived from non-incarcerated individuals. The rate ratio was obtained by dividing the two rates. We further subclassified the secondary cases among non-incarcerated individuals by their compensation status (compensatory mutation yes/no). Secondary case rates and secondary case rate ratios were calculated using the R package epitools (v0.5–10.1). To quantify the effect of prisons on the MDR-TB epidemic in Georgia, we used the phybreak infector–infectee relationships to identify transmission events where an incarcerated individual infected a non-incarcerated individual (PP > 0.5). We then followed the transmission chain from the non-incarcerated individual infected by an incarcerated individual and counted all downstream cases (NNIds, PP > 0.0; Supplementary Fig. 7).
We used the formula below to estimate the burden of MDR-TB attributable to prisons. The extrapolated number of incarcerated individuals (NIe) in the 6-year study period is given by:
where NIe denotes the extrapolated number of incarcerated individuals during the 6-year sampling time frame; NI is the number of incarcerated individuals identified; NNI is the number of non-incarcerated individuals; and Ntot is the WHO-notified, culture-confirmed MDR-TB cases.
The extrapolated number of non-incarcerated individuals (NNIe) is given by:
The proportion of cases among non-incarcerated individuals directly or indirectly linked to incarcerated individuals (PNI-I) is given by:
The extrapolated number of cases among non-incarcerated individuals directly or indirectly associated to incarcerated individuals (NNI-Ie) is given by:
Reporting Summary.
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The raw sequences were deposited at the European Nucleotide Archive under BioProject ID PRJEB39561. Accession numbers are listed in Supplementary Table 2. Metadata associated with the genomes are provided in Supplementary Table 2.
Supplementary Material
Acknowledgements
Calculations were performed at the sciCORE (http://scicore.unibas.ch/) scientific computing core facility at the University of Basel. We would like to thank D. Klinkenberg (Dutch National Institute for Public Health and the Environment) for help with the phybreak analysis and D. Brites for feedback on the manuscript. We also thank J. Andrews and the two other anonymous reviewers for their excellent comments that helped us improve our manuscript. Funding: This work was supported by the Swiss National Science Foundation (grants 310030_188888, IZRJZ3_164171, IZLSZ3_170834 and CRSII5_177163, all to S.G.), the European Research Council (309540-EVODRTB and 883582-ECOEVODRTB, both to S.G.) and SystemsX.ch (to S.G. and C.B.).
Footnotes
Competing interests
The authors declare no competing interests.
Additional information
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41591-021-01358-x.
Peer review information Nature Medicine thanks Jason Andrews and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Editor recognition statement: Alison Farrell was the primary editor on this article and managed its editorial process and peer review in collaboration with the rest of the editorial team.
Reprints and permissions information is available at www.nature.com/reprints.
References
- 1.O’Neill J Antimicrobial Resistance: Tackling a Crisis for the Health and Wealth of Nations. http://www.jpiamr.eu/wp-content/uploads/2014/12/AMR-Review-Paper-Tackling-a-crisis-for-the-health-and-wealth-of-nations_1-2.pdf (Joint Programming Initiative on Antimicrobial Resistance, 2014). [Google Scholar]
- 2.Andersson DI & Levin BR The biological cost of antibiotic resistance. Curr. Opin. Microbiol. 2, 489–493 (1999). [DOI] [PubMed] [Google Scholar]
- 3.Andersson DI & Hughes D Antibiotic resistance and its cost: is it possible to reverse resistance? Nat. Rev. Microbiol. 8, 260–271 (2010). [DOI] [PubMed] [Google Scholar]
- 4.Gygli SM, Borrell S, Trauner A & Gagneux S Antimicrobial resistance in Mycobacterium tuberculosis: mechanistic and evolutionary perspectives. FEMS Microbiol. Rev. 41, 354–373 (2017). [DOI] [PubMed] [Google Scholar]
- 5.Gagneux S et al. The competitive cost of antibiotic resistance in Mycobacterium tuberculosis. Science 312, 1944–1946 (2006). [DOI] [PubMed] [Google Scholar]
- 6.Reynolds MG Compensatory evolution in rifampin-resistant Escherichia coli. Genetics 156, 1471–1481 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Song T et al. Fitness costs of rifampicin resistance in Mycobacterium tuberculosis are amplified under conditions of nutrient starvation and compensated by mutation in the β′ subunit of RNA polymerase. Mol. Microbiol. 91, 1106–1119 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maisnier-Patin S, Berg OG, Liljas L & Andersson DI Compensatory adaptation to the deleterious effect of antibiotic resistance in Salmonella typhimurium. Mol. Microbiol. 46, 355–366 (2002). [DOI] [PubMed] [Google Scholar]
- 9.Qi Q, Preston GM & MacLean RC Linking system-wide impacts of RNA polymerase mutations to the fitness cost of rifampin resistance in Pseudomonas aeruginosa. mBio 5, 1–12 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Comas I et al. Whole-genome sequencing of rifampicin-resistant Mycobacterium tuberculosis strains identifies compensatory mutations in RNA polymerase genes. Nat. Genet. 44, 106–110 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.World Health Organization. Global Tuberculosis Report 2020. https://www.who.int/publications/i/item/9789240013131 (World Health Organization, 2020). [Google Scholar]
- 12.Dye C Doomsday postponed? Preventing and reversing epidemics of drug-resistant tuberculosis. Nat. Rev. Microbiol. 7, 81–87 (2009). [DOI] [PubMed] [Google Scholar]
- 13.Dye C & Espinal MA Will tuberculosis become resistant to all antibiotics? Proc. R. Soc. B Biol. Sci. 268, 45–52 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dye C, Williams BG, Espinal MA & Raviglione MC Erasing the world’s slow stain: strategies to beat multidrug-resistant tuberculosis. Science 295, 2042–2046 (2002). [DOI] [PubMed] [Google Scholar]
- 15.World Health Organization. Global Tuberculosis Report 2019. https://www.who.int/teams/global-tuberculosis-programme/tb-reports/global-report-2019 (World Health Organization, 2019). [Google Scholar]
- 16.Casali N et al. Evolution and transmission of drug-resistant tuberculosis in a Russian population. Nat. Genet. 46, 279–286 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eldholm V et al. Four decades of transmission of a multidrug-resistant Mycobacterium tuberculosis outbreak strain. Nat. Commun. 6, 7119 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gagneux S et al. Impact of bacterial genetics on the transmission of isoniazid-resistant Mycobacterium tuberculosis. PLoS Pathog. 2, e61 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang C et al. Transmission of multidrug-resistant Mycobacterium tuberculosis in Shanghai, China: a retrospective observational study using whole-genome sequencing and epidemiological investigation. Lancet Infect. Dis 3099, 1–10 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brandis G, Wrande M, Liljas L & Hughes D Fitness-compensatory mutations in rifampicin-resistant RNA polymerase. Mol. Microbiol. 85, 142–151 (2012). [DOI] [PubMed] [Google Scholar]
- 21.Merker M et al. Compensatory evolution drives multidrug-resistant tuberculosis in Central Asia. eLife 7, 1–31 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu Q et al. Have compensatory mutations facilitated the current epidemic of multidrug-resistant tuberculosis? Emerg. Microbes Infect. 7, 98 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.de Vos M et al. Putative compensatory mutations in the rpoC gene of rifampin-resistant Mycobacterium tuberculosis are associated with ongoing transmission. Antimicrob. Agents Chemother. 57, 827–832 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Klinkenberg D, Backer JA, Didelot X, Colijn C & Wallinga J Simultaneous inference of phylogenetic and transmission trees in infectious disease outbreaks. PLoS Comput. Biol. 13, e1005495 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ahmad N et al. Treatment correlates of successful outcomes in pulmonary multidrug-resistant tuberculosis: an individual patient data meta-analysis. Lancet 392, 821–834 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lomtadze N et al. Prevalence and risk factors for multidrug-resistant tuberculosis in the Republic of Georgia: a population-based study. Int. J. Tuberc. Lung Dis. 13, 68–73 (2009). [PMC free article] [PubMed] [Google Scholar]
- 27.World Health Organization. Companion Handbook to the WHO Guidelines for the Programmatic Management of Drug-resistant Tuberculosis. https://www.who.int/tb/publications/pmdt_companionhandbook/en/ (World Health Organization, 2014). [PubMed] [Google Scholar]
- 28.Ismail NA et al. Defining bedaquiline susceptibility, resistance, cross-resistance and associated genetic determinants: a retrospective cohort study. EBioMedicine 28, 136–142 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.World Health Organization. Extensive Review of Tuberculosis Prevention, Control and Care in Georgia 6–14 November 2014. https://www.euro.who.int/en/health-topics/communicable-diseases/tuberculosis/publications/2016/extensive-review-of-tuberculosis-prevention,-control-and-care-in-georgia-614-november-2014 (World Health Organization, 2014). [Google Scholar]
- 30.Nimmo C et al. Population-level emergence of bedaquiline and clofazimine resistance-associated variants among patients with drug-resistant tuberculosis in southern Africa: a phenotypic and phylogenetic analysis. Lancet Microbe 1, e165–e174 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Behr MA, Edelstein PH & Ramakrishnan L Revisiting the timetable of tuberculosis. BMJ 362, k2738 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stuckler D, Basu S, McKee M & King L Mass incarceration can explain population increases in TB and multidrug-resistant TB in European and central Asian countries. Proc. Natl Acad. Sci. USA 105, 13280–13285 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kenyon G Russia’s prisons fuel drug-resistant tuberculosis. Lancet Infect. Dis. 9, 594 (2009). [Google Scholar]
- 34.Aerts A et al. Pulmonary tuberculosis in prisons of the ex-USSR state Georgia: results of a nation-wide prevalence survey among sentenced inmates. Int. J. Tuberc. Lung Dis. 4, 1104–1110 (2000). [PubMed] [Google Scholar]
- 35.Yates TA et al. The transmission of Mycobacterium tuberculosis in high burden settings. Lancet Infect. Dis. 16, 227–238 (2016). [DOI] [PubMed] [Google Scholar]
- 36.Niemann S et al. Mycobacterium tuberculosis Beijing lineage favors the spread of multidrug-resistant tuberculosis in the Republic of Georgia. J. Clin. Microbiol. 48, 3544–3550 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Baussano I et al. Tuberculosis incidence in prisons: a systematic review. PLoS Med. 7, 1–10 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gegia M, Kalandadze I, Madzgharashvili M & Furin J Developing a human rights-based program for tuberculosis control in Georgian prisons. Health Hum. Rights 13, 9 (2011). [PMC free article] [PubMed] [Google Scholar]
- 39.Menardo F et al. Treemmer: a tool to reduce large phylogenetic datasets with minimal loss of diversity. BMC Bioinformatics 19, 164 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McKenna A et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li H et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Koboldt DC et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 22, 568–576 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cingolani P et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff. Fly 6, 80–92 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stamatakis A RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Coll F et al. A robust SNP barcode for typing Mycobacterium tuberculosis complex strains. Nat. Commun. 5, 4812 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stefan MA, Ugur FS & Garcia GA Source of the fitness defect in rifamycin-resistant Mycobacterium tuberculosis RNA polymerase and the mechanism of compensation by mutations in the β′ subunit. Antimicrob. Agents Chemother. 62, e00164–18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Walker TM et al. Whole-genome sequencing to delineate Mycobacterium tuberculosis outbreaks: a retrospective observational study. Lancet Infect. Dis. 13, 137–146 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Menardo F, Duchêne S, Brites D & Gagneux S The molecular clock of Mycobacterium tuberculosis. PLoS Pathog. 15, e1008067 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lanfear R, Hua X & Warren DL Estimating the effective sample size of tree topologies from Bayesian phylogenetic analyses. Genome Biol. Evol. 8, 2319–2332 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw sequences were deposited at the European Nucleotide Archive under BioProject ID PRJEB39561. Accession numbers are listed in Supplementary Table 2. Metadata associated with the genomes are provided in Supplementary Table 2.