

Abstract
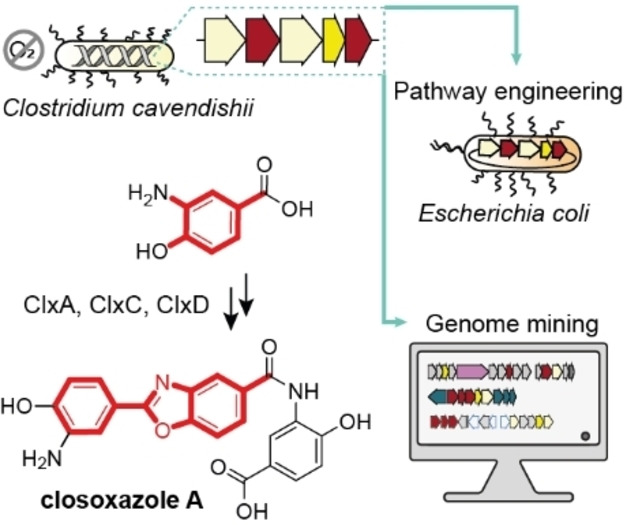
Benzoxazole scaffolds feature prominently in diverse synthetic and natural product‐derived pharmaceuticals. Our understanding of their bacterial biosynthesis is, however, limited to ortho‐substituted heterocycles from actinomycetes. We report an overlooked biosynthetic pathway in anaerobic bacteria (typified in Clostridium cavendishii) that expands the benzoxazole chemical space to meta‐substituted heterocycles and heralds a distribution beyond Actinobacteria. The first benzoxazoles from the anaerobic realm (closoxazole A and B) were elucidated by NMR and chemical synthesis. By genome editing in the native producer, heterologous expression in Escherichia coli, and systematic pathway dissection we show that closoxazole biosynthesis invokes an unprecedented precursor usage (3‐amino‐4‐hydroxybenzoate) and manner of assembly. Synthetic utility was demonstrated by the precursor‐directed biosynthesis of a tafamidis analogue. A bioinformatic survey reveals the pervasiveness of related gene clusters in diverse bacterial phyla.
Keywords: Biosynthesis, Genome Mining, Heterocycles, Natural Products, Total Synthesis
A novel biosynthetic route to a medically relevant benzoxazole scaffold was discovered in anaerobic bacteria. Genome editing, heterologous expression, and biotransformation studies show that the pathway invokes specialized precursors, backbone assembly, and heterocyclization to form closoxazoles. The biocatalysts involved display synthetic utility, and prompted the genomics‐driven identification of cryptic gene clusters in diverse bacterial phyla.
Introduction
Owing to their broad range of biological activities, benzoxazoles are among the most prominent classes of pharmaceutically relevant heterocycles. [1] As a nucleotide bioisoster, the benzoxazole scaffold is privileged for binding to various targets and biopolymers. [2] One medicinally used benzoxazole derivative is tafamidis (1), which delays neurodegeneration and other severe diseases linked to transthyretin amyloidosis. [3] Moreover, numerous benzoxazole derivatives have been explored as leads for development of anti‐tuberculosis (2) [4] or anti‐inflammatory drugs (3, 4). [4b] Apart from these valuable therapeutics and drug candidates designed by medicinal chemists,[ 1 , 5 ] it is noteworthy that nature has evolved specialized metabolic pathways to assemble structurally and functionally diverse benzoxazoles. [6] Prominent examples of such natural products have been isolated mainly from marine‐ and soil‐derived actinomycetes; nocarbenzoxazoles (F, G) (5, 6), which selectively inhibit various tumor cell lines, [7] the cytotoxic and antibacterial caboxamycin (7), [8] the antibiotic calcimycin (8), [9] the antitumoral agent nataxazole (9), [10] and the effective anti‐leishmanial agent A3385 (10) (Figure 1A). [11]
Figure 1.
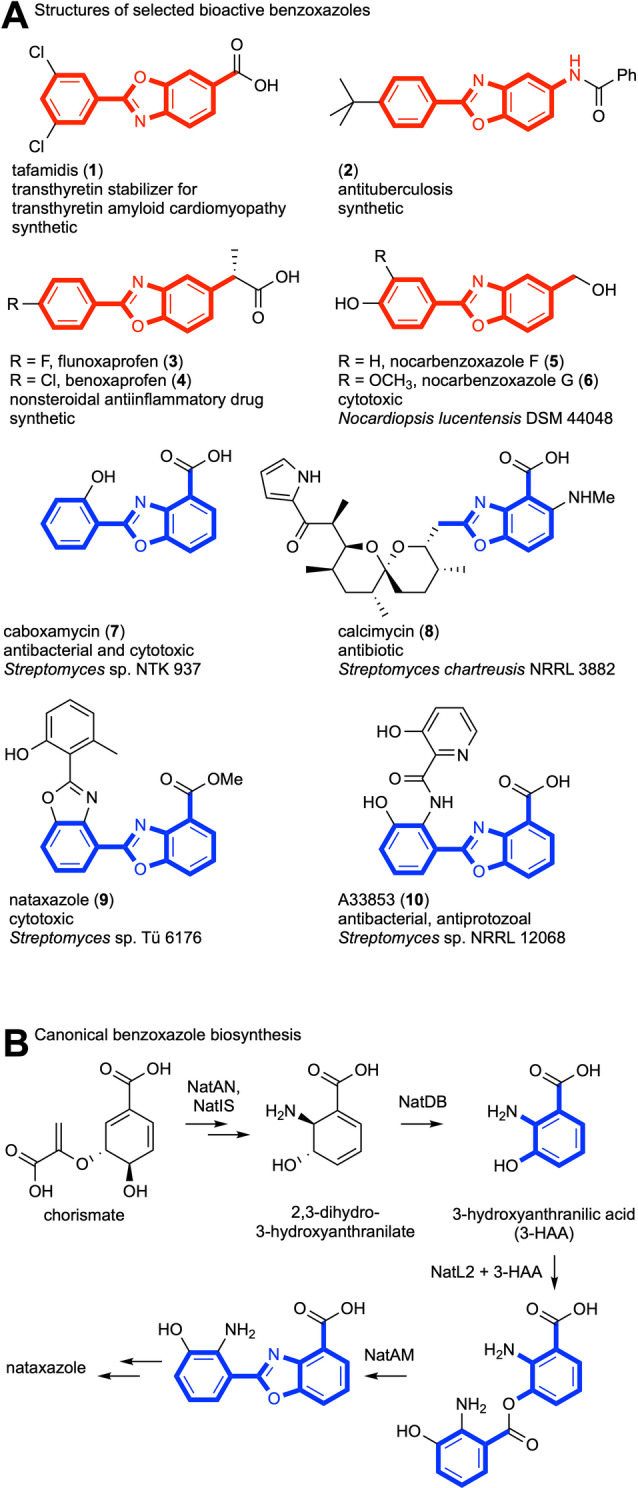
Structures of selected bioactive benzoxazoles, and benzoxazole formation in nataxazole biosynthesis. A) Bioactive benzoxazoles with meta‐ (red) and ortho‐substituted (blue) heterocycles. In the case of natural products, the bacterial producer strain is named. B) Benzoxazole ring formation in nataxazole biosynthesis from fusion of two 3‐HAA units leads to an ortho‐substituted heterocycle.
With the aim of harnessing bacterial benzoxazole biosynthesis, considerable effort has been devoted to the study of the requisite biosynthetic pathways. Functional analyses at the genetic and biochemical levels have revealed that the aryl‐benzoxazole cores of 7, 9 and 10 are formed from condensation of 3‐hydroxyanthranilic acid (3‐HAA) building blocks with a second aromatic acid (3‐HAA or other) by ATP‐dependent coenzyme A ligases, followed by enzymatic cyclocondensation (Figure 1B).[ 6b , 6c , 6d ] This heterocyclase‐catalyzed step proceeds via an unstable ester intermediate. [12] A third aromatic building block is sometimes linked by means of an acyl transferase that is related to ketoacylsynthases.[ 6b , 6c ] Although these characterized enzymes show synthetic utility for benzoxazole production, [13] the currently known pathways are restricted to ortho‐substituted heterocycles, thereby excluding the meta‐substitution pattern of drug candidates and clinically used benzoxazoles, such as the synthetic 1 and 2 and the naturally occurring 5 and 6. Despite the pharmaceutical relevance of these compounds, biocatalysts giving rise to this privileged heterocyclic scaffold have not been found.
Here, we report the discovery of a novel family of benzoxazoles (closoxazoles) produced by the anaerobic bacterium Clostridium cavendishii DSM 21758 and provide the first insights into biosynthesis of the meta‐substituted benzoxazole scaffold. We show that closoxazole biosynthesis markedly differs from hitherto studied benzoxazole biosynthesis pathways in terms of the building blocks incorporated, the manner of backbone formation, and the subsequent heterocyclization. We create an expression platform that enables the heterologous production of meta‐substituted benzoxazoles. Finally, we demonstrate that putative benzoxazole biosynthesis gene clusters are widely distributed in the genomes of bacteria belonging to diverse phyla.
Results and Discussion
Discovery of Novel Benzoxazoles Produced by an Anaerobic Bacterium
In the course of genome mining to explore the untapped biosynthetic potential of anaerobic bacteria, our attention was drawn to C. cavendishii DSM 21758. [14] Analysis of the genome of this strain using antiSMASH [15] revealed a remarkably high number of biosynthetic gene clusters (BGCs) tentatively coding for specialized metabolism. This encouraged us to examine the metabolic profile of C. cavendishii. We cultivated the strain as resting cultures in an anaerobic chamber in various liquid media, extracted the cultures with ethyl acetate and monitored their metabolic profiles by high‐performance liquid chromatography with diode array detection coupled to high‐resolution mass spectrometry (HPLC‐DAD‐HRMS) (Figure 2A and S1). We detected four metabolites displaying pronounced UV/Vis spectra (λ max=307–326 nm) that are characteristic for conjugated π‐systems, like benzoxazole chromophores (Figure S2).[ 7a , 10 ] While UV/Vis and HRMS data indicated that all compounds are congeners, signature fragment ions were common to the MS/MS fragmentation patterns of 11 and the later eluting pair, but were absent in that of 12 (Figure S3).
Figure 2.

Isolation and structure elucidation of the closoxazoles. A) HPLC profiles of crude ethyl acetate extracts from cultures of C. cavendishii and C. cavendishii Rif1. Asterisks (*) indicate signals corresponding to putative congeners of closoxazole A. B) Structures with 2D NMR couplings of the main compound 11 and the second candidate structure 14. Compound 12 represents a congener of 11. C) Synthetic route to the reference compounds 11 and 14. a) Dimethylformamide (DMF), Boc2O, 70 °C, 3 h. b) DMF, 4‐methyl benzyl bromide (4‐MeBnBr), K2CO3, then water, then MeOH, NaOH, 70 °C, 4 h then acetic acid. c) Dichloromethane (DCM), N,N‐diisopropylethylamine (DIPEA), then SOCl2, 20 °C, 10 min, then DIPEA, Br‐aniline (18), 20 °C, 12 h. d) CuI, Cs2CO3, phenanthroline, MeCN, microwave. e) DCM, TFA, 65 °C, 3.5 h then MeOH, NaOH. f) DMF, Boc2O, 8 h, then H2O, then MeOH, NaOH, 70 °C, 1 h then HCl, then DCM, DIPEA then SOCl2, 20 °C, 10 min, then DIPEA, 17, 20 °C, 1.5 h. g) DCM, TFA, 70 min, 65 °C. Bn‐protected 17 was synthesized from 15 as follows: DMF, K2CO3, 4‐MeBnBr, 70 °C, then DCM, TFA, 12 h, 40 °C (for details see Supporting Information). HPLC profiles and selected region of 13C NMR spectra of the isolated natural product 11 and both reference compounds 11 and 14.
Isolation of sufficient amounts of the compounds of interest for structural elucidation was hampered by their extremely low production rates. These low titers could not be increased by variation of cultivation conditions or by conventional ways to optimize extraction, such as pH adjustment and addition of absorber resin. We therefore attempted to upregulate expression of the biosynthesis genes by a method based on the selection of mutants resistant to RNA polymerase‐targeting antibiotics. [16] This led to the generation of a spontaneous rifampicin‐resistant mutant of C. cavendishii, C. cavendishii Rif1, producing approximately 1.5‐fold higher titers of the compounds compared to the wild type (Figure 2A). When large‐scale batch fermentations (20 L) of C. cavendishii Rif1 were performed, however, compound production was almost completely abolished. As a result, we resorted to scaling up cultivation of C. cavendishii Rif1 in the anaerobic chamber, and individually extracted 500 mL cultures totaling 27 L. The pooled organic extract was pre‐purified using size‐exclusion chromatography (Sephadex LH‐20), then purified by preparative HPLC to give 1 mg of compound 11 and 800 μg of compound 12 (approximately 37 μg L−1 and 30 μg L−1 yield, respectively).
From HRMS data of 11 (m/z 406.1031 [M+H]+) and 12 (m/z 390.1076 [M+H]+), the molecular formulae of C21H16N3O6 and C21H16N3O5 were deduced, respectively. Since 11 was the predominant compound, we focused on its structural elucidation first. The number of carbon atoms of 11 was corroborated by 13C NMR and DEPT 135 NMR data. The proton NMR spectrum shows nine signals resonating at δ 6.83–8.31 ppm that were assigned to three trisubstituted benzene rings based on H,H‐COSY, HSQC and HMBC measurements. 13C signals at δ 167.1, 165.1, and 164.8 ppm indicate the presence of three carboxyl moieties. Their positions were determined based on HMBC correlations of H‐4/6 with C‐8, H‐10/14 with C‐2, and H‐18/20 with C‐21. Taking into account the molecular composition, the chemical shifts of the quaternary carbons of the aromatic ring systems, and the HMBC correlations, 11 possesses 3‐amino‐4‐hydroxybenzoic acid (3,4‐AHBA, 13) moieties connected via a benzoxazole ring and an amide bond.
Structural Confirmation by Total Synthesis
Since the exact connection of the partial structure of 11 could not be unambiguously assigned by interpretation of the observed HMBC couplings alone, we synthesized authentic reference compounds, the constitutional isomers 11 and 14 (Figure 2B). Starting from 3,4‐AHBA (13), we prepared tert‐butyloxycarbonyl (Boc)‐protected amine 15, Boc‐ and benzyl (Bn)‐protected ether 16, and the Bn‐protected ester 17. Amide linkage between methyl 3‐amino‐4‐bromobenzoate (18) and benzoic acid 16 gave amide 19, and subsequent microwave‐assisted, copper‐catalyzed cyclization with concomitant Boc deprotection yielded benzoxazole 20. [17] Coupling of 20 with benzoic acid 16 and subsequent removal of protection groups with trifluoroacetic acid (TFA) and NaOH gave benzoxazole 14 (total yield 2.6 %, Figure 2C). The second possible structure, benzoxazole 11, was obtained by modification of benzoxazole ester 20. Amide protection and ester hydrolysis followed by coupling with Bn‐protected ester 17 gave benzoxazole amide 21. TFA‐mediated global cleavage of the protecting groups provided the target molecule (11) (total yield 2.5 %, Figure 2C). Comparison of NMR, HPLC retention times, and MS/MS spectra showed that the structures of synthetic 11 and 11 from C. cavendishii are identical (Figures 2C and S5, Table S5). Accordingly, 11 is a benzoxazole composed of three 3,4‐AHBA units in which the first 3,4‐AHBA moiety is connected via its carboxyl to form a benzoxazole ring with the second 3,4‐AHBA moiety. The carboxyl group of the benzoxazole moiety is connected via an amide bond to the third 3,4‐AHBA molecule.
By comparison of NMR, HRMS, and MS/MS data we concluded that 11 and 12 share a molecular framework, yet instead of the 3,4‐aminohydroxy substituents at the benzoxazole‐fused phenyl ring of 11, compound 12 has a 4‐amino group. MS/MS fragmentation patterns indicate that the later eluting pair of metabolites are N‐ or O‐acylated congeners of 11 (Figure S3). It should be highlighted that these new compounds, which we named closoxazole A (11) and closoxazole B (12), represent the first known benzoxazole natural products isolated from anaerobic bacteria and outside the phylum Actinobacteria. The topology of these compounds markedly differs from that of previously reported benzoxazole metabolites in that it is meta‐substituted, as is often found in medicinally relevant benzoxazoles (Figure 1A).
Identification of the Genetic Origin of Closoxazole Biosynthesis
The substitution patterns of 11 and 12 indicate that the benzoxazole moieties are derived from 3,4‐AHBA and p‐aminobenzoic acid (PABA) building blocks. This is remarkable because in all hitherto reported biosynthetic pathways to benzoxazoles in bacteria (Streptomyces spp.), 3‐HAA moieties are fused to a second 3‐HAA moiety, salicylic acid or a polyketide.[ 6b , 6d , 12 ] Evidently, the biosynthesis of the closoxazoles deviates from this scheme and, accordingly, the genome sequence of C. cavendishii lacks a complete set of canonical benzoxazole biosynthesis genes. However, we noticed that a cryptic BGC (now named clx, Figure 3A, Table S6), predicted by antiSMASH [15] to be involved in β‐lactone biosynthesis, [18] harbors two genes (clxE and clxB) that encode enzymes tentatively involved in 3,4‐AHBA formation, a putative 2‐amino‐3,7‐dideoxy‐d‐threo‐hept‐6‐ulosonate synthase (ClxE) and a putative 3,4‐AHBA‐synthase (ClxB). [19] Located in the same locus are two genes (clxA and clxC) that code for members of the ANL (acyl‐CoA synthetases, nonribosomal peptide synthetase adenylation domains and luciferase enzymes) superfamily. A HHpred [20] search indicated that the deduced gene products are similar to NatL2, a ligase that catalyzes the ATP‐dependent activation and ligation of 3‐HAA building blocks in Streptomyces spp. [12] Lastly, clxD encodes a putative amidohydrolase superfamily member that is, however, unrelated to amidohydrolases that catalyze heterocycle formation in Streptomyces spp., e.g. NatAM (only 16 % sequence similarity). [12]
Figure 3.

Genetic origin and biosynthesis of the closoxazoles. A) Top: Closoxazole BGC of C. cavendishii (see also Table S6). Bottom: Scheme depicting the targeted knock‐out of clxA in the genome of C. cavendishii Rif1 by CRISPR/Cas9n‐mediated genome editing, alongside mutant verification by colony PCR and subsequent EcoRV restriction analysis of the resulting PCR fragment (unedited: 1012 bp; ΔclxA: 552 and 460 bp). BS: biosynthesis. WT: wild type. B) Extracted ion chromatograms (EIC) corresponding to 11 and 12 are shown for crude ethyl acetate extracts of C. cavendishii Rif1, C. cavendishii Rif1ΔclxA, E. coli pET28a‐clxA–E (heterologous expression) and E. coli pET28a (vector control). EICs of the calculated exact mass of the compounds are shown with m/z ±5 ppm. C) EICs corresponding to closoxazoles and proposed biosynthetic intermediates detected in crude ethyl acetate extracts from E. coli strains expressing the listed combinations of clx genes along with the respective synthetic reference. (†) not related. D) Proposed closoxazole biosynthesis pathway in C. cavendishii and precursor‐directed biosynthesis in E. coli. Exogenous supply of the alternative starter unit 3,5‐dichlorobenzoic acid (3,5‐DCA) results in the production of the bioactive tafamidis analogue (25).
Based on these findings, we sought to determine whether the closoxazoles originate from the clx genetic locus by creating a targeted knock‐out. It should be emphasized that specific genetic modifications are particularly challenging in Clostridium species, [21] and there are no reports on the genetic tractability of C. cavendishii. To generate the desired mutant, we adapted the CRISPR‐Cas9n genome editing approach that we previously applied to study biosynthetic pathways in Ruminiclostridium cellulolyticum. [22] By analogy, we constructed a CRISPR‐Cas9n‐based genome editing vector [23] designed to incorporate a nonsense mutation (STOP+EcoRV) in the ligase gene clxA, resulting in the targeted mutant C. cavendishii Rif1 ΔclxA (Figure 3A). We noted the absence of 11, its putative congeners, and 12 from this mutant‘s metabolic profile, implicating a crucial role of clxA in closoxazole biosynthesis (Figures 3B and S6).
Heterologous Production of Closoxazoles
To further dissect the role of the clx locus in closoxazole biosynthesis, we set out to heterologously reconstitute production in Escherichia coli. This approach would help to determine the essential biosynthesis genes, to decipher their functions, and to facilitate benzoxazole production in vivo. Our analysis using the Enzyme Function Initiative (EFI)‐Genome Neighborhood Tool [24] suggested that clxA–E are the minimal set of genes required for closoxazole production (for details see Supporting Information). We therefore cloned clxA–E into the pET28a expression vector and introduced the construct into E. coli BL21. The metabolic profile of the resulting strain, E. coli pET28a‐clxA–E, showed production of 11 and 12 (Figures 3B and S7). Moreover, we obtained 1 mg of 11 from a 5 L E. coli culture, which equates to an over 5‐fold increase in yield compared to that of the native producer. In order to test whether the five genes are collectively required for biosynthesis of 11 and 12, we generated five expression strains, each lacking one of the genes. Their metabolic profiles indicated that production of 11 and 12 is abolished or decreased when any of the genes is missing (Figure S8). Consequently the β‐lactone BGC predicted by antiSMASH [15] has been misannotated and instead codes for a previously unknown pathway to meta‐substituted benzoxazoles.
Dissection of Closoxazole Backbone Assembly
The successful production of 11 and 12 by E. coli enabled us to gain further insights into closoxazole assembly. Specifically, we dissected the pathway in vivo by heterologously expressing various combinations of the clx biosynthesis genes. First, we expressed in E. coli the genes (clxB and clxE) tentatively coding for 3,4‐AHBA biosynthesis. By LC‐HRMS analysis and comparison of retention time and HRMS with a synthetic reference, we found that E. coli pET‐clxBE produces 3,4‐AHBA. Notably, 3,4‐AHBA cannot be detected in extracts of E. coli strains harboring either the empty vector or expressing only one of the two genes (clxB or clxE) (Figures 3C and S9). Accordingly, biosynthesis of 11 and 12 could be complemented by provision of exogenous 3,4‐AHBA to a strain lacking both clxB and clxE (E. coli pET‐clxACD) (Figure 3C). These experiments unequivocally prove that 3,4‐AHBA is produced by ClxB and ClxE and is a precursor for closoxazole biosynthesis.
With the enzymes responsible for the biosynthesis of 3,4‐AHBA established, we endeavored to identify those that subsequently activate and fuse the 3,4‐AHBA building blocks. We proposed the predicted ligases ClxA and ClxC as the likely candidates and therefore analyzed the metabolic profiles of strains lacking either clxA (E. coli pET‐clxBCDE) or clxC (E. coli pET‐clxABDE). Similar to C. cavendishii Rif1 ΔclxA, closoxazole production is greatly diminished in E. coli pET‐clxBCDE (Figures 3C and S8), confirming the importance of ClxA for closoxazole formation. In the absence of ClxC (E. coli pET‐clxABDE), lower levels of 11 and 12 are produced compared to E. coli pET‐clxA–E, and a new metabolite (22) with m/z 271.0713 [M+H]+ accumulates. The calculated sum formula (C14H10N2O4) and UV/Vis absorption spectrum of 22 indicate that it is a benzoxazole composed of two 3,4‐AHBA moieties (Figures 3C and S10), which was confirmed by comparison with a synthetic reference (Figures 3C, S11, S12). Taken together, these data infer that ClxA catalyzes the initial linkage of two 3,4‐AHBA molecules, while ClxC catalyzes the downstream reaction, specifically the fusion of benzoxazole 22 to a third 3,4‐AHBA unit. The continued production of 11 and 12 by E. coli pET‐clxABDE is likely enabled by the broad substrate tolerance of ClxA and/or other ANL superfamily members encoded in the E. coli genome. In line with this, we detected formation of 11 when cultures of E. coli pET28a and E. coli pET‐clxA were supplemented with 3,4‐AHBA and synthetic 22, with the signal corresponding to 11 being markedly enhanced when ClxA is present (Figure S13).
With ClxA assigned as the enzyme responsible for the activation of 3,4‐AHBA, we proceeded to compare it to its counterparts that activate 3‐HAA during biosynthesis of ortho‐substituted benzoxazoles. Therefore, we performed a multiple sequence alignment of ClxA, NatL2 and BomJ, and found their sequences to be grossly similar. However, key residues implicated in nucleotide and 3‐HAA binding by NatL2 are not conserved in ClxA (Figure S14).
Timing and Substrate of Heterocyclization
With roles established for four of the five enzymes encoded in the clx BGC (ClxB and ClxE in formation of the 3,4‐AHBA precursor, and ClxA and ClxC in precursor activation and fusion), we deemed the remaining uncharacterized enzyme (ClxD) as likely catalyzing the heterocyclization step in closoxazole biosynthesis. When we scrutinized the metabolic profile of E. coli pET‐clxABCE, it became apparent that the absence of the putative amidohydrolase ClxD causes the accumulation of two new metabolites, 23 with m/z 289.0819 [M+H]+ (Figure 3C), and 24 with m/z 424.1143 [M+H]+ (Figure S15) instead of 11 and 12. The deduced sum formulas C14H12N2O5 (23) and C21H17N3O7 (24) fit to the composition of conjugates composed of two (23) and three (24) 3,4‐AHBA units, respectively. We confirmed the identities of the dimer (23) and the trimer (24) by LC‐HRMS and MS/MS comparison with synthetic reference compounds (Figures 3C, S12, S16–S17). Conclusively, these data show that ClxD is required for oxazole ring formation. In nataxazole biosynthesis, the ClxD homolog NatAM can possibly catalyze a second heterocyclization. [13] Similarly, when we supplemented E. coli pET‐clxD with 11, we detected a new metabolite, of which HRMS, MS/MS and retention time are consistent with a compound derived from 11 that harbors a second benzoxazole heterocycle (Figure S18, S19).
To test if 23 and 24 could be transformed into the corresponding benzoxazoles by ClxD, we individually added synthetic 23 and 24 to cultures of E. coli pET‐clxD. Since trimer 24 is not converted into 11 (Figure S20), it may be considered to be a shunt product. In contrast, dimer 23 is a true biosynthetic intermediate, being readily cyclized by ClxD into benzoxazole 22 (Figure 3C), which is ultimately fused to a third 3,4‐AHBA unit by ClxC to yield 11. Notably, 23 is absent in the 3,4‐AHBA‐producing E. coli pET‐clxBE, excluding the possibility that 23 is decomposed to 3,4‐AHBA and re‐assembled by endogenous ANL superfamily enzymes of E. coli (Figure S21).
To clarify the precise functions of the two adenylating enzymes ClxA and ClxC in the assembly of 23 and 24, we created strains lacking both clxD and either clxA or clxC. Unsurprisingly, 23 and 24 are not produced in the absence of clxA and clxD (E. coli pET‐clxBCE). The strain lacking both clxC and clxD (E. coli pET‐clxABE), however, shows the same chemotype as E. coli pET‐clxABCE (Figure S22). Thus, we conclude ClxA is able to form conjugates composed of two (23) and three (24) 3,4‐AHBA building blocks, but in the native pathway the third 3,4‐AHBA unit is linked to 23 primarily by ClxC.
Incorporation of Alternative Starter Units
The structure of 12 implies that ClxA can use PABA, likely recruited from primary metabolism, as an alternative building block to 3,4‐AHBA. To test this possibility, we supplemented E. coli pET‐clxACD with a mixture of 3,4‐AHBA and deuterated PABA (PABA‐d2). The 2 amu mass shift (m/z 392.1210 [M+H]+) consistent with incorporation of one PABA‐d2 unit into 12 (Figure S23) was detected in the resulting metabolite profile. Furthermore, we noted that E. coli pET‐clxABDE produces a minor metabolite with m/z 255.0764 [M+H]+ (Figure S24) and a deduced sum formula (C14H10N2O3) that corresponds to a benzoxazole derived from PABA and 3,4‐AHBA, the plausible pathway intermediate en route to 12. Upon supplementation of the E. coli pET‐clxABDE culture with PABA‐d2, the biosynthetic intermediate underwent the expected mass shift of 2 amu (m/z 257.0882 [M+H]+, Figure S23). Conclusively, these data confirm that PABA is accepted by ClxA, ultimately leading to the formation of 12.
Inspired by the substrate tolerance of ClxA, we probed the scope of enzymatic meta‐substituted benzoxazole assembly by performing a precursor‐directed biosynthesis experiment. With the goal of accessing the tafamidis analogue 25, which is known as a synthetic transthyretin amyloid fibril inhibitor, [3c] we supplemented E. coli pET‐clxA–E with 3,5‐dichlorobenzoic acid. We isolated the desired compound (production rate 2.5 mg L−1) and unequivocally confirmed its identity by HRMS (Figure S25) and NMR analysis. This proof‐of‐concept experiment demonstrates that the closoxazole biosynthesis enzymes can be used to access pharmaceutically valuable meta‐substituted benzoxazoles.
Model for Alternative Benzoxazole Assembly in Anaerobes
Based on the results of our mutational, heterologous expression, precursor supplementation, and isotope labeling experiments, we propose a model for the biosynthesis of meta‐substituted benzoxazole natural products (Figure 3D). Broadly speaking, the steps of closoxazole biosynthesis by C. cavendishii mirror those of benzoxazole biosynthesis by Streptomyces spp. (canonical), consisting of precursor formation, activation and fusion of building blocks, benzoxazole formation and modification. Nevertheless, we noted numerous striking differences between the two assembly strategies.
In the canonical biosynthetic pathway, 3‐HAA serves as the initial building block and is fused with a second 3‐HAA or other aromatic acid. 3‐HAA is proposed to be derived from chorismate.[ 6b , 6c , 6d ] In contrast, the closoxazole pathway uses 3,4‐AHBA as the initial building block, which is provided by ClxB and ClxE, presumably independent of the shikimate pathway. [19] 3,4‐AHBA is condensed with a second aromatic acid (PABA or 3,4‐AHBA) by the ligase ClxA, followed by ClxD‐mediated benzoxazole formation. Consequently, the 3,4‐AHBA‐derived closoxazoles feature a meta‐substituted benzoxazole core rather than the ortho‐substitution pattern of canonical benzoxazoles.[ 6b , 6c , 6d ]
Furthermore, it appears that the enzymatic cyclocondensation reactions that form the benzoxazole moieties in the canonical pathways and in closoxazole biosynthesis proceed via different mechanisms. In nataxazole biosynthesis, it is known that the ligase NatL2 introduces an ester linkage between two 3‐HAA molecules, followed by heterocyclization catalyzed by the amidohydrolase NatAM. [12] In contrast, the two respective homologs in the closoxazole pathway, ClxA and ClxD, probably catalyze heterocycle formation via an amide linkage.
The final step of closoxazole biosynthesis involves the attachment of another 3,4‐AHBA moiety to the benzoxazole core structure at its meta‐position. This reaction is catalyzed by ClxC, which activates the benzoxazole carboxy group, not the 3,4‐AHBA moiety. This behavior differs from that of characterized ANL superfamily members in the biosynthesis of canonical benzoxazoles, [12] where each building block is proposed to be individually activated by a distinct ligase.[ 6b , 6c , 6d ] In this way, ClxC is an outlier among the characterized benzoxazole‐forming ANL superfamily enzymes.
Yet another remarkable observation is that closoxazole biosynthesis is independent of any thiotemplate mechanism, unlike canonical pathways to benzoxazoles.[ 6b , 6c , 6d ] It should also be noted that the collective action of the enzyme types involved in the biosynthesis of 3,4‐AHBA‐derived benzoxazoles is unprecedented, explaining the misannotation of the closoxazole BGC by automated bioinformatic analysis. We expect that our discovery of this overlooked pathway will contribute to the ongoing refinement of BGC prediction tools, thereby supporting future natural product discovery efforts.
Putative Benzoxazole Biosynthesis Gene Clusters in Diverse Bacterial Phyla
In light of the newly discovered closoxazole BGC clx, we set out to determine if additional benzoxazole BGCs are overlooked in bacterial genomes. To this end, we created a sequence similarity network (SSN) using the EFI‐Enzyme Similarity Tool [24] and employing the heterocyclase ClxD as the genetic handle. Although ClxD and the heterocyclases involved in ortho‐substituted benzoxazole formation are members of the amidohydrolase superfamily, they share very low sequence similarity. As a result, canonical benzoxazole heterocyclases, e.g. NatAM [12] were absent in an initial ClxD‐based SSN. With the goal of encompassing all known benzoxazole‐forming amidohydrolases, we created a hybrid SSN based on both NatAM and ClxD (for details, see Supporting Information). Since building block activation is required for heterocyclization, we constrained the search to ClxD/NatAM homologs that are encoded in the vicinity of genes encoding enzymes possibly involved in amide bond formation/esterification. This hybrid SSN consists of 243 nodes (Figure 4A), each representing a ClxD/NatAM homolog found in a locus encoding putative activation enzymes. Thus, these loci were deemed BGCs putatively involved in biosynthesis of benzoxazole‐containing natural products.
Figure 4.
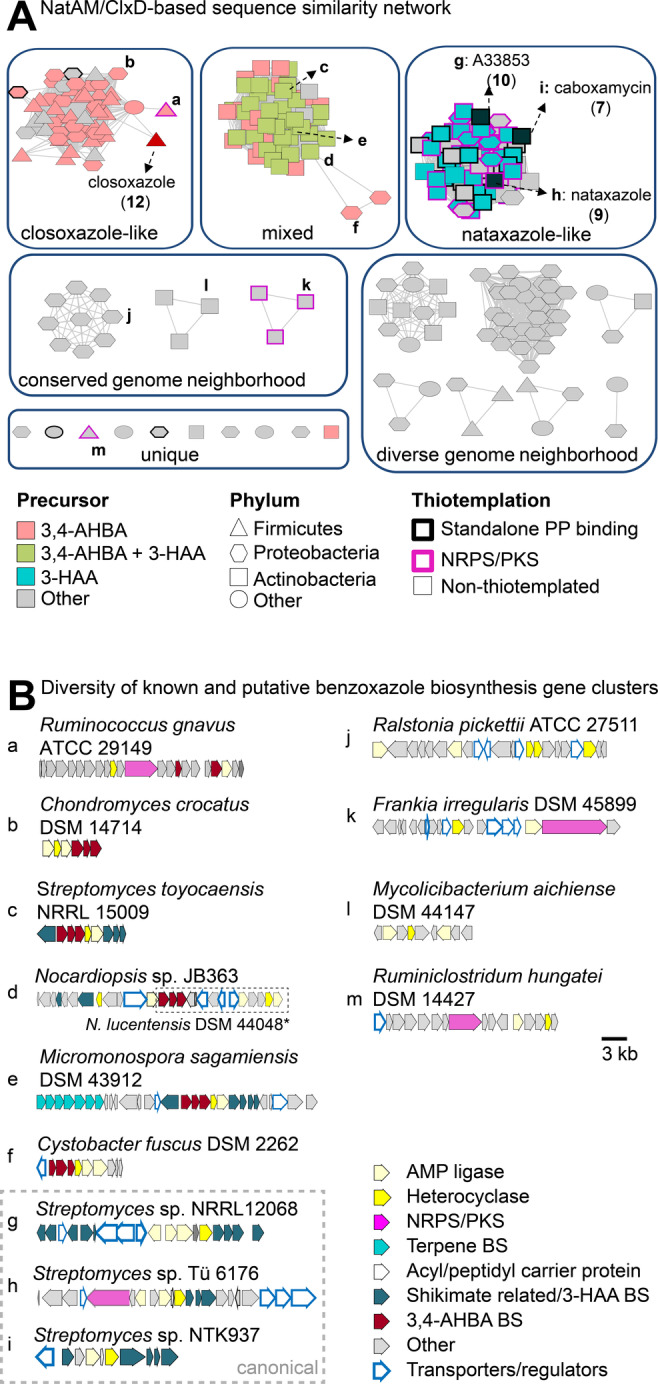
Genome mining for putative benzoxazole biosynthetisis gene clusters. A) Sequence similarity network based on ClxD and NatAM homologs encoded in the vicinity of genes encoding enzymes putatively involved in building block activation. B) Diversity of reported and putative benzoxazole BGCs selected based on architecture diversity and/or ecological or medicinal significance of the potential producer (e.g. R. picketti is an emerging human pathogen and F. irregularis is a plant symbiont). Letters correspond to the node labels in (A). Grey genes marked as “other” also include genes encoding putative tailoring enzymes. The interested reader is directed to the respective protein identifiers in Table S7 to further explore the genome neighborhoods. The conserved subset of genes (noc) in the genome of the nocarbenzoxazole producer N. lucentensis DSM 44048 is highlighted with a dashed box. (*) not represented in sequence similarity network. AMP: adenosine monophosphate. BS: biosynthesis. NRPS: nonribosomal peptide synthetase. PKS: polyketide synthase.
The alignment score of the hybrid SSN was determined empirically such that orthologs group together, forming three main groups of putative BGCs that differ mainly in the genes involved in precursor supply (Figure 4B): benzoxazole formation from 3,4‐AHBA, 3‐HAA or a mixture of both. There are also various putative BGCs (43 %) that do not to contain homologs of any genes involved in 3‐HAA or 3,4‐AHBA formation. This could be due to i) the use of uncharacterized precursors for which the responsible biosynthesis genes are not predictable, ii) the precursor biosynthetic genes not being located within the BGC, or (iii) the precursors of the cognate natural products originating from primary metabolism, as exemplified by PABA in closoxazole biosynthesis. If legitimate, these BGCs hint to an even greater structural variety of benzoxazoles derived from various precursors, particularly since various loci harbor genes encoding enzymes often associated with secondary metabolism, like methyltransferases, prenyltransferases, oxidoreductases and polyketide synthases.
Since the amidohydrolase superfamily is large and functionally diverse, it follows that not all of the enzymes represented in the hybrid SSN are necessarily involved in benzoxazole formation. Indeed, the hybrid SSN includes MxcM, a NatAM homolog implicated in imidazoline formation. [25] The hybrid SSN also does not provide a complete overview of all benzoxazole‐forming enzymes, given that the calcimycin producer S. chartreusis NRRL3882 is not represented. This arose because the cognate BGC lacks an amidohydrolase‐encoding gene [6a] and raises the intriguing possibility that at least one further mechanism of benzoxazole formation remains to be uncovered.
Among the putative BGCs belonging to the mixed group, we noticed one in the genome of Nocardiopsis sp. JB363 that encodes a ClxD homolog, with a part (noc) of the BGC being common to the genome of the nocarbenzoxazole producer, N. lucentensis DSM 44048 (Figure 4B). [7a] This observation piqued our interest given that the genetic basis for nocarbenzoxazole production has remained elusive and, other than closoxazole, it is the only bacterially produced meta‐substituted benzoxazole known. Drawing on our knowledge of the function of ClxA‐E in closoxazole biosynthesis and taking the structure of nocarbenzoxazole G (6) into account, we could envision a putative biosynthetic route to the meta‐substituted benzoxazole core of nocarbenzoxazole G (for further details see Supporting Information, Table S8 and Figure S75).
Intriguingly, previously only known from Actinobacteria, we found putative benzoxazole BGCs in the genomes of bacteria from different phyla including Proteobacteria, Firmicutes, and Spirochaetae. Overall, our findings point to an untapped biosynthetic potential for benzoxazole‐containing natural products within the bacterial kingdom.
Conclusion
The benzoxazole heterocycle is a crucial structural motif of various synthetic and natural bioactive molecules.[ 1 , 5 ] With the discovery of closoxazoles and the characterization of their biosynthetic origin, we reveal a pathway for the formation of the pharmacologically relevant meta‐substituted benzoxazole scaffold, in which both the building blocks and their assembly deviate from previously reported biosynthetic pathways to benzoxazoles. The successful heterologous production of 3,4‐AHBA‐derived benzoxazoles represents an important addition to the biocatalytic toolbox, exemplified by the precursor‐directed biosynthesis of a medically relevant halogenated benzoxazole that was previously inaccessible by reported biosynthetic enzymes. Also, our discovery of a benzoxazole produced by a Clostridium sp. supports previous findings of anaerobes as a promising source of natural products and non‐canonical pathways.[ 22b , 26 ] Finally, we provide a first bioinformatic survey of putative benzoxazole BGCs that may guide future natural product discovery efforts.
Experimental Section
For Experimental Details, see Supporting Information.
Conflict of interest
The authors declare no conflict of interest.
1.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
We thank A. Perner for MS and MS/MS measurements, H. Heinecke for NMR measurements, and M. Cyrulies for initial fermentation attempts. Helpful discussions with K. Ishida, J. Krabbe and F. Kloss are gratefully acknowledged. This work was financially supported by the Deutsche Forschungsgemeinschaft (Leibniz Award to C.H.) Open Access funding enabled and organized by Projekt DEAL.
T. Horch, E. M. Molloy, F. Bredy, V. G. Haensch, K. Scherlach, K. L. Dunbar, J. Franke, C. Hertweck, Angew. Chem. Int. Ed. 2022, 61, e202205409; Angew. Chem. 2022, 134, e202205409.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1. Wong X. K., Yeong K. Y., ChemMedChem 2021, 16, 3237–3262. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Boyer J., Arnoult E., Médebielle M., Guillemont J., Unge J., Jochmans D., J. Med. Chem. 2011, 54, 7974–7985; [DOI] [PubMed] [Google Scholar]
- 2b. Erol M., Celik I., Temiz-Arpaci O., Kaynak-Onurdag F., Okten S., J. Biomol. Struct. Dyn. 2021, 39, 3080–3091. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Coelho T., Maia L. F., da Silva A. M., Cruz M. W., Planté-Bordeneuve V., Suhr O. B., Conceiçao I., Schmidt H. H.-J., Trigo P., Kelly J. W., Labaudinière R., Chan J., Packman J., Grogan D. R., J. Neurol. 2013, 260, 2802–2814; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3b. Ando Y., Sekijima Y., Obayashi K., Yamashita T., Ueda M., Misumi Y., Morita H., Machii K., Ohta M., Takata A., Ikeda S.-i., J. Neurol. Sci. 2016, 362, 266–271; [DOI] [PubMed] [Google Scholar]
- 3c. Razavi H., Palaninathan S. K., Powers E. T., Wiseman R. L., Purkey H. E., Mohamedmohaideen N. N., Deechongkit S., Chiang K. P., Dendle M. T., Sacchettini J. C., Kelly J. W., Angew. Chem. Int. Ed. 2003, 42, 2758–2761; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2003, 115, 2864–2867. [Google Scholar]
- 4.
- 4a. Arisoy M., Temiz-Arpaci O., Kaynak-Onurdag F., Ozgen S., Z. Naturforsch. Teil C 2013, 68, 453–460; [PubMed] [Google Scholar]
- 4b. Šlachtová V., Brulíková L., ChemistrySelect 2018, 3, 4653–4662. [Google Scholar]
- 5. Kakkar S., Tahlan S., Lim S. M., Ramasamy K., Mani V., Shah S. A. A., Narasimhan B., Chem. Cent. J. 2018, 12, 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.
- 6a. Wu Q., Liang J., Lin S., Zhou X., Bai L., Deng Z., Wang Z., Antimicrob. Agents Chemother. 2011, 55, 974–982; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6b. Lv M., Zhao J., Deng Z., Yu Y., Chem. Biol. 2015, 22, 1313–1324; [DOI] [PubMed] [Google Scholar]
- 6c. Cano-Prieto C., García-Salcedo R., Sánchez-Hidalgo M., Braña A. F., Fiedler H. P., Méndez C., Salas J. A., Olano C., ChemBioChem 2015, 16, 1461–1473; [DOI] [PubMed] [Google Scholar]
- 6d. Losada A. A., Cano-Prieto C., García-Salcedo R., Braña A. F., Méndez C., Salas J. A., Olano C., Microb. Biotechnol. 2017, 10, 873–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.
- 7a. Sun M., Zhang X., Hao H., Li W., Lu C., J. Nat. Prod. 2015, 78, 2123–2127; [DOI] [PubMed] [Google Scholar]
- 7b. Kim T., Lee S.-A., Noh T., Choi P., Choi S.-J., Song B. G., Kim Y., Park Y.-T., Huh G., Kim Y.-J., Ham J., J. Nat. Prod. 2019, 82, 1325–1330. [DOI] [PubMed] [Google Scholar]
- 8. Hohmann C., Schneider K., Bruntner C., Irran E., Nicholson G., Bull A. T., Jones A. L., Brown R., Stach J. E. M., Goodfellow M., Beil W., Krämer M., Imhoff J. F., Süssmuth R. D., Fiedler H.-P., J. Antibiot. 2009, 62, 99–104. [DOI] [PubMed] [Google Scholar]
- 9. Chaney M. O., Demarco P. V., Jones N. D., Occolowitz J. L., J. Am. Chem. Soc. 1974, 96, 1932–1933. [DOI] [PubMed] [Google Scholar]
- 10. Sommer P. S. M., Almeida R. C., Schneider K., Beil W., Süssmuth R. D., Fiedler H.-P., J. Antibiot. 2008, 61, 683–686. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Michel K. H., Boeck L. D., Hoehn M. M., Jones N. D., Chaney M. O., J. Antibiot. 1984, 37, 441–445; [DOI] [PubMed] [Google Scholar]
- 11b. Tipparaju S. K., Joyasawal S., Pieroni M., Kaiser M., Brun R., Kozikowski A. P., J. Med. Chem. 2008, 51, 7344–7347. [DOI] [PubMed] [Google Scholar]
- 12. Song H., Rao C., Deng Z., Yu Y., Naismith J. H., Angew. Chem. Int. Ed. 2020, 59, 6054–6061; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 6110–6117. [Google Scholar]
- 13. Ouyang H., Hong J., Malroy J., Zhu X., ACS Synth. Biol. 2021, 10, 2151–2158. [DOI] [PubMed] [Google Scholar]
- 14. Bowman K. S., Dupré R. E., Rainey F. A., Moe W. M., Int. J. Syst. Evol. Microbiol. 2010, 60, 358–363. [DOI] [PubMed] [Google Scholar]
- 15. Blin K., Shaw S., Steinke K., Villebro R., Ziemert N., Lee S. Y., Medema M. H., Weber T., Nucleic Acids Res. 2019, 47, W81–W87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ochi K., Hosaka T., Appl. Microbiol. Biotechnol. 2013, 97, 87–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Viirre R. D., Evindar G., Batey R. A., J. Org. Chem. 2008, 73, 3452–3459. [DOI] [PubMed] [Google Scholar]
- 18. Robinson S. L., Christenson J. K., Wackett L. P., Nat. Prod. Rep. 2019, 36, 458–475. [DOI] [PubMed] [Google Scholar]
- 19. Suzuki H., Ohnishi Y., Furusho Y., Sakuda S., Horinouchi S., J. Biol. Chem. 2006, 281, 36944–36951. [DOI] [PubMed] [Google Scholar]
- 20. Zimmermann L., Stephens A., Nam S.-Z., Rau D., Kübler J., Lozajic M., Gabler F., Söding J., Lupas A. N., Alva V., J. Mol. Biol. 2018, 430, 2237–2243. [DOI] [PubMed] [Google Scholar]
- 21. Joseph R. C., Kim N. M., Sandoval N. R., Front. Microbiol. 2018, 9, 154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.
- 22a. Dunbar K. L., Büttner H., Molloy E. M., Dell M., Kumpfmüller J., Hertweck C., Angew. Chem. Int. Ed. 2018, 57, 14080–14084; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 14276–14280; [Google Scholar]
- 22b. Molloy E. M., Dell M., Hänsch V. G., Dunbar K. L., Feldmann R., Oberheide A., Seyfarth L., Kumpfmüller J., Horch T., Arndt H. D., Hertweck C., Angew. Chem. Int. Ed. 2021, 60, 10670–10679; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 10765–10774; [Google Scholar]
- 22c. Dunbar K. L., Dell M., Molloy E. M., Kloss F., Hertweck C., Angew. Chem. Int. Ed. 2019, 58, 13014–13018; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 13148–13152. [Google Scholar]
- 23. Xu T., Li Y., Shi Z., Hemme C. L., Li Y., Zhu Y., Van Nostrand J. D., He Z., Zhou J., Appl. Environ. Microbiol. 2015, 81, 4423–4431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zallot R., Oberg N., Gerlt J. A., Biochemistry 2019, 58, 4169–4182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Winand L., Vollmann D. J., Hentschel J., Nett M., Catalysts 2021, 11, 892. [Google Scholar]
- 26.
- 26a. Dunbar K. L., Dell M., Molloy E. M., Büttner H., Kumpfmüller J., Hertweck C., Angew. Chem. Int. Ed. 2021, 60, 4104–4109; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 4150–4155; [Google Scholar]
- 26b. Li J. S., Barber C. C., Zhang W., J. Ind. Microbiol. Biotechnol. 2019, 46, 375–383; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26c. Li J. S., Du Y., Gu D., Cai W., Green A., Ng S., Leung A., Del Rio Flores A., Zhang W., Org. Lett. 2020, 22, 8204–8209. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.