

Abstract
Methods for facile site‐selective modifications of proteins are in high demand. We have recently shown that a flavin transferase can be used for site‐specific covalent attachment of a chromo‐ and fluorogenic flavin (FMN) to any targeted protein. Although this Flavin‐tag method resulted in efficient labeling of proteins in vitro, labelling in E. coli cells resulted in partial flavin incorporation. It was also restricted in the type of installed label with only one type of flavin, FMN, being incorporated. Here, we report on an extension of the Flavin‐tag method that addresses previous limitations. We demonstrate that co‐expression of FAD synthetase improves the flavin incorporation efficiency, allowing complete flavin‐labeling of a target protein in E. coli cells. Furthermore, we have found that various flavin derivatives and even a nicotinamide can be covalently attached to a target protein, rendering this method even more versatile and valuable.
Keywords: ApbE, FAD synthetases, flavin-tags, FMN derivatives, protein labeling
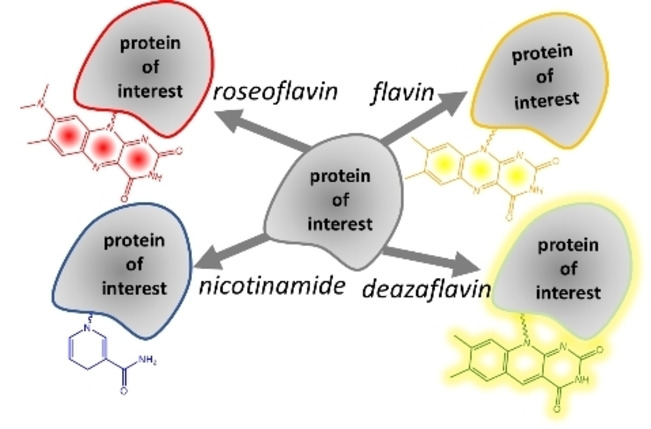
Flavin‐tag 2.0: The Flavin‐tag method can be used for labelling of proteins with various chromo‐ and fluorophores and redox‐active probes, including flavin (yellow), roseoflavin (red), 5‐deazaflavin (fluorescent), and nicotinamide.
Introduction
Site‐specific protein labeling plays a core role in investigating protein function at molecular level. Site‐specific protein labeling is, essentially, artificially modifying proteins at the post‐translational stage by covalent attachment of a chromo‐ or fluorophore. In recent years, enzymatically catalyzed protein modifications have become popular as they provide a versatile platform for efficient bioconjugation methods that only require mild reaction conditions while being extremely specific and attaining a high modification efficiency.[ 1 , 2 , 3 , 4 ] Many of these methods are based on the coupling action of an enzyme on a specific peptide, coined “tag”. Exemplary methods include biotin ligase (AP tag), [5] transglutaminase (Q tag), [6] sortase A, [7] lipoic acid ligase (LAP tag), [8] phosphopantetheinyl transferase (ACP or PCP tags), [9] E2 small ubiquitin‐like modifier‐conjugating enzyme Ubc9 (LACE tag), [10] and flavin transferase (Flavin‐tag). [11]
We have recently introduced the Flavin‐tag method. [11] This protein labeling method is based on the covalent enzymatic tethering of flavin mononucleotide (FMN) to a threonine residue of a short target peptide through a phosphate ester bond (Scheme 1). This covalent flavinylation is catalyzed by the flavin transferase from V. cholerae (ApbE).[ 12 , 13 ] This methodology, applicable to both in vitro and in vivo settings, only requires a flavin transferase as labelling enzyme, FAD as label precursor, and a tagged target protein and can be performed under mild conditions. Nevertheless, the described Flavin‐tag method had some limitations. In particular, the relatively low incorporation of FMN in the recombinant target protein in living cells could limit the application in bioorthogonal systems. Improvement of the Flavin‐tag method can significantly expand the application of this labeling method. Motivated by a desire to enhance the flavinylation efficiency of recombinant proteins in Escherichia coli cells, we set out to improve the incorporation efficiency, as well as to diversify the label scope of the Flavin‐tag method.
Scheme 1.
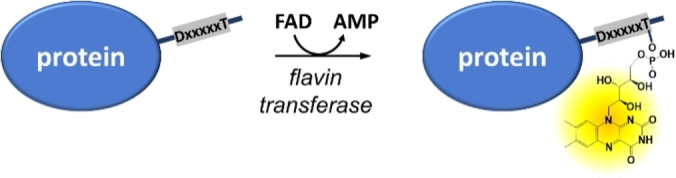
The Flavin‐tag method: a specific threonine‐containing sequence is recognized by flavin transferase (ApbE) for covalent flavin attachment (threonyl‐FMN) at the expense of FAD. [11]
Covalent tethering of FMN by action of a flavin transferase relies on FAD as substrate. In cells, the flavin cofactor FAD is synthesized in a two‐step process from riboflavin. First, FMN is formed from riboflavin by action of a flavokinase. FMN is further converted into FAD by FMN adenylyltransferase activity (Figure 1).[ 14 , 15 ] Some bifunctional FAD synthetases possess both activities.[ 14 , 16 , 17 , 18 ] To boost the intracellular level of FAD, we have explored the use of the bifunctional and well‐studied FAD synthetase from Corynebacterium ammoniagenes (FADS). FADS is an efficient enzyme in producing FAD from its riboflavin precursor and also accepts riboflavin derivatives for the chemical synthesis of FAD analogs.[ 16 , 19 ] Except for its use in vivo to promote incorporation of FMN on the Flavin‐tag, we also used FADS as biocatalyst to prepare FAD derivatives. Specifically, FADS was found to be able to convert all tested riboflavin analogs into their corresponding FAD derivatives (Figure 1). These FAD derivatives could subsequently be used as unnatural substrates of the flavin transferase ApbE, resulting in the covalent modification of Flavin‐tagged target proteins with the corresponding unnatural FMN analogs.
Figure 1.
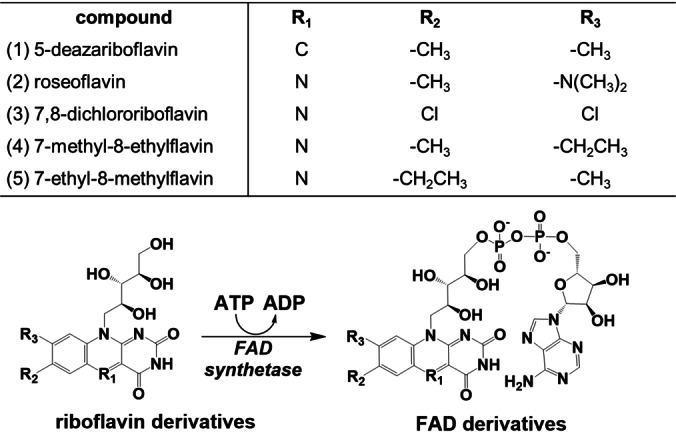
Enzymatic conversion of riboflavin (derivatives) into FMN/FAD (derivatives). Overview of modifications in the isoalloxazine moiety of the flavins used in this study. FADS was used for converting 5‐deazariboflavin (1), roseoflavin (2), 7,8‐dichlororiboflavin (3), 7‐methyl‐8‐ethylflavin (4), and 7‐ethyl‐8‐methylflavin (5) into the corresponding FAD derivatives.
Results and Discussion
Boosting flavinylation by co‐expressing FADS and ApbE
In our previous paper, where the Flavin‐tag method was first described, we demonstrated that FMN could be incorporated in Flavin‐tagged proteins in E. coli cells. [11] However, flavin incorporation was far from complete reaching at most 45 % flavinylation using optimal conditions. As part of our efforts to create a more effective flavinylation system for the site‐specific labeling of recombinant proteins in E. coli, we explored approaches to boost the efficiency of flavinylation. First, we tested whether addition of riboflavin in the medium would improve the amount of flavinylated protein. Adding up to 120 mg/L riboflavin to the TB medium did not lead to a significant increase in FMN incorporation (Figure S1). We speculated that low levels of FAD in E. coli might be the limiting factor for the flavinylation of recombinant Flavin‐tagged proteins. It has been shown that FADS can increase the productivity of FAD from riboflavin in C. ammoniagenes cells. [16] We hypothesized that overexpressing FADS in E. coli would result in a higher intracellular level of FAD. ApbE could take advantage of the overproduced FAD to flavinylate Flavin‐tagged recombinant proteins. To test our hypothesis, the genes encoding for full‐length FADS (amino acids 1–338, 36.8 kDa) and truncated ApbE (amino acids 19–334, 35.1 kDa) [12] were cloned into a standard pRSF‐Duet‐1 vector. Upon induction of heterologous protein co‐expression, both enzymes were found to be produced in soluble form in E. coli BL21‐AI (Figure S2A). First, we assessed the flavinylation of maltose binding protein (MBP) equipped with a N‐ or C‐terminal Flavin‐tag (MBP‐NF1 and MBP‐CF1, respectively). MBP‐NF1 and MBP‐CF1 were co‐expressed with FADS and ApbE. The resulting recombinant MBP was purified as yellow protein fraction and analyzed by mass spectrometry. Gratifyingly, for both MBP variants, ESI‐MS analysis revealed only peaks corresponding to the FMN‐modified proteins. Proteins without a flavin attached were not detected. This revealed that full modification of MPB‐NF1/CF1 occurred in vivo when expressing at 30 °C (Figure 2A and Figure S3). Similar results were obtained when expressing the MPB variants at 24 °C and 37 °C. Only when expressing at 17 °C flavin incorporation was incomplete (Figure 2A). The low expression level of ApbE and FADS at 17 °C may be the main reason for the decreased flavinylation of the target protein (Figure S2B).
Figure 2.

The flavinylation efficiency in E. coli cells. (A) The effect of expression temperature on FMN incorporation efficiency in E. coli using MBP as target protein. (B) Flavinylation efficiency of Flavin‐tagged ADH and SUMO in E. coli. N=N‐terminal tag, C=C‐terminal tag, F1‐tag=GVDGLSGATLTS.
To further confirm that the combination of ApbE and FADS could generate a 100 % FMN‐incorporation of recombinant proteins in E. coli, additional experiments were carried out with two other proteins: ADH and SUMO, carrying N‐terminal and C‐terminal Flavin‐tags. ADH is an engineered thermostable alcohol dehydrogenase from Candida magnoliae DSMZ 70638 (ADH). [20] Previously, Flavin‐tagged versions of this ADH were analyzed and found to have similar enzyme activity. [11] The SUMO protein is structurally related to ubiquitin and known to be well‐expressed as recombinant protein. All four Flavin‐tagged proteins were found to be fully flavinylated when the cells were grown at 30 °C for 24 h (Figure 2B). These results nicely demonstrate that co‐expressing FADS indeed boosts flavin incorporation of Flavin‐tagged proteins. Furthermore, the results demonstrated that fully modified target proteins can be obtained in E. coli by which in vitro reactions can be avoided, saving time and resources needed for purification of target proteins and the required flavin transferase.
Synthesis of FAD derivatives
A covalently attached FMN, tethered via a Flavin‐tag, exhibits attractive features such as being a chromophore (intense yellow color) and fluorescent. Still, it would be attractive when the Flavin‐tag can be used to label proteins with other probes, which alternative spectral features. Therefore, we explored the use of FAD derivatives as substrate for the flavin transferase, AbpE. Total synthesis of FAD derivatives is not really feasible as it is a rather complex molecule. As the ADP part should not be changed, we focused on the riboflavin moiety of FAD. Synthesis procedures for various riboflavin derivatives have been reported in literature.[ 21 , 22 ] Previous studies have shown that FAD synthetase exhibits a broad specificity for riboflavin substrates. For example, Iamurri et al. demonstrated that a truncated C‐terminal RFK domain from FADS could convert 7,8‐dichlororiboflavin, 8‐aminoriboflavin, and 5‐deazariboflavin to their corresponding FMN derivatives. [19] Hence, we set out to investigate the ability of full‐length recombinant FADS to convert riboflavin derivatives into their corresponding FAD analogs. For this, we selected five riboflavin analogs (5‐deazariboflavin (1), roseoflavin (2), 7,8‐dichlororiboflavin (3), 7‐methyl‐8‐ethylflavin (4), and 7‐ethyl‐8‐methylflavin (5)) whether FADS could convert these flavins into the corresponding FAD derivatives (Figure 1). We found that all riboflavin analogs were accepted by FADS for the synthesis of the respective FAD derivatives (Figure S4). Incubating each riboflavin analog (100 μM) with 15 μM FADS resulted in the complete conversion within 16 h at 25 °C (Figure S5). The products were purified and isolated from the reaction mixture by reverse‐phase liquid chromatography followed by freeze‐drying. The purified compounds show the expected flavin absorption spectra (Figure S6). To further confirm that the products are the expected FAD derivatives, the purified compounds were analyzed by the negative‐ion mass spectrometry. The observed molecular weights were in perfect agreement with the calculated molecular weights of the FAD form of the riboflavin analogs (Figure S7). This demonstrates that FADS can successfully synthesize such FAD derivatives from the corresponding riboflavin precursors.
Synthesis and analysis of FMN derivatives‐modified MBP
Encouraged by the successful synthesis of the FAD derivatives using FADS, we further examined their utility as site‐specific chromogenic and fluorescent probes for recombinant proteins carrying a Flavin‐tag. We used FAD derivatives to modify MBP. MBP variants with an N‐terminal or C‐terminal Flavin‐tag (MBP‐NF1/CF1) were expressed, purified and subsequently used in in vitro flavinylation reactions. It was found that all five FAD derivatives were substrates of ApbE and the respective FMN moieties were efficiently attached to the Flavin‐tags of MBP. Fully modified protein was observed when a 60 μM solution of MBP‐NF1/CF1 was treated with 1 mM FAD or a FAD derivative and 20 μM ApbE for 4 hours, in 50 mM Tris‐HCl (pH 8.0) containing 10 mM Mg2+. The modified MBP was purified using Ni2+ affinity chromatography and was analyzed by ESI‐MS (Figure 3). The MBP variants displayed different colors due to the different spectral features of the attached flavins. For example, roseo‐FMN modified MBP had a clear red color.
Figure 3.

Electrospray ionization mass analysis of MBP‐NF1 (a‐g) and MBP‐CF1 (h‐n) that were modified with different FMN derivatives. The control shows the mass of the purified MBP without treatment, while the rest of the panels show the mass of the protein after incubation with FAD derivatives and ApbE. The mass difference corresponds to the mass of FMN derivatives. N=N‐terminal tag, C=C‐terminal tag, F1‐tag=GVDGLSGATLTS.
The influence of FAD derivatives and tag location on in vitro coupling efficiency
Next, the modification efficiency of different FAD derivatives with Flavin‐tagged MBP was investigated. To determine the efficiency of flavinylation, we monitored the covalent incorporation of FMN derivatives over time (Table 1 and Figure S8). Reactions were initiated by adding ApbE (1.0 mg/mL) to the reaction mixtures containing 1.0 mM FAD derivative, 10 mM Mg2+, and 3.0 mg/mL Flavin‐tagged MBP. Samples were taken at distinct time intervals and analyzed by in‐gel fluorescence. All the FAD derivatives were tested for the flavin incorporation with the Flavin‐tag at the C‐ or N‐terminus of MBP. The efficiency of covalent flavinylation of MBP‐NF1 was somewhat higher compared to the C‐terminal variant (t1/2 of 0.02‐0.46 h vs. 0.09‐1.47 h, respectively) (Table 1), indicating that the Flavin‐tag located at the N‐terminus is more prone to flavinylation. Surprisingly, ApbE displays a similar incorporation efficiency with all tested FAD derivatives and is, therefore, capable of incorporating all tested FMN derivatives to the Flavin‐tag. The differences in the isoalloxazine moiety of these FAD derivatives only slightly affect the reaction rate. The crystal structure of ApbE (PDB:6NXI) explains the broad substrate scope. Inspection of the active site of ApbE reveals that the FAD binding pocket is composed of two subpockets: an adenosine binding pocket and an isoalloxazine binding pocket (Figure S9). The bound adenosine moiety is located in the inside of the overall FAD binding pocket, and is stabilized by hydrogen bonds formed with the main chain carbonyl oxygens of residues I259, A110 and interactions with the side chains of residues D112 and I259 (Figure S9B). [13] Binding is further stabilized by a hydrogen bond that is formed between the side chain of the catalytic residue H257 and the 3’‐OH of the ribose moiety (Figure S9B). The bound isoalloxazine part is relatively exposed, and seems to be mainly bound in the binding pocket through π–π interactions with residue Y69. The binding of FAD derivatives, as well as the positioning of the catalytic histidine H257 do not seem to be influenced by substitutions on the isoalloxazine ring. A subsequent substrate acceptance test further supported our analysis. We tested whether ApbE could also use NADH as substrate, which also contains an ADP moiety but has a nicotinamide riboside moiety instead of a riboflavin moiety when compared with FAD. Intriguingly, ApbE was also able to covalently modify Flavin‐tagged MBP with NADH, incorporating nicotinamide mononucleotide (NMN). Except for the typical absorbance at 340 nm of the attached NMN, mass spectrometry of the protein also confirmed the expected mass of the target protein with a tethered NMN (Figure S10).
Table 1.
The influence of FAD derivatives and tag location on rate of flavinylation (t1/2).[a]
|
FAD derivative |
NF1 [t1/2 , h] |
CF1 [t1/2 , h] |
|---|---|---|
|
FAD |
0.11 ±0.01 |
0.42 ±0.06 |
|
5‐deaza‐FAD |
0.04 ±0.01 |
0.14 ±0.02 |
|
roseo‐FAD |
0.03 ±0.01 |
0.08 ±0.01 |
|
7,8‐dichloro‐FAD |
0.10 ±0.02 |
0.29 ±0.03 |
|
7‐methyl‐8‐ethyl‐FAD |
0.46 ±0.04 |
1.47 ±0.19 |
|
7‐ethyl‐8‐methyl‐FAD |
0.02 ±0.004 |
0.09 ±0.01 |
[a] The reactions were performed in 50 mM Tris‐HCl buffer, pH 8.0 and at fixed concentrations of reactants and enzyme: 1.0 mM FAD derivative, 10 mM Mg2+, 3.0 mg/mL MBP and 1.0 mg/mL ApbE.
Determination of physicochemical properties of modified MBP by FMN derivatives
To gain a better insight into the spectral properties of MBP equipped with different flavins, we measured their absorption spectra (Figure 4A). This revealed that all modified MBP variants displayed features identical to those of the corresponding FMN derivatives free in solution. The FMN derivatives had been prepared using the engineered riboflavin kinase from C. ammoniagenes (CaRFK) and the respective riboflavin derivatives (Figures S11 and S12).
Figure 4.
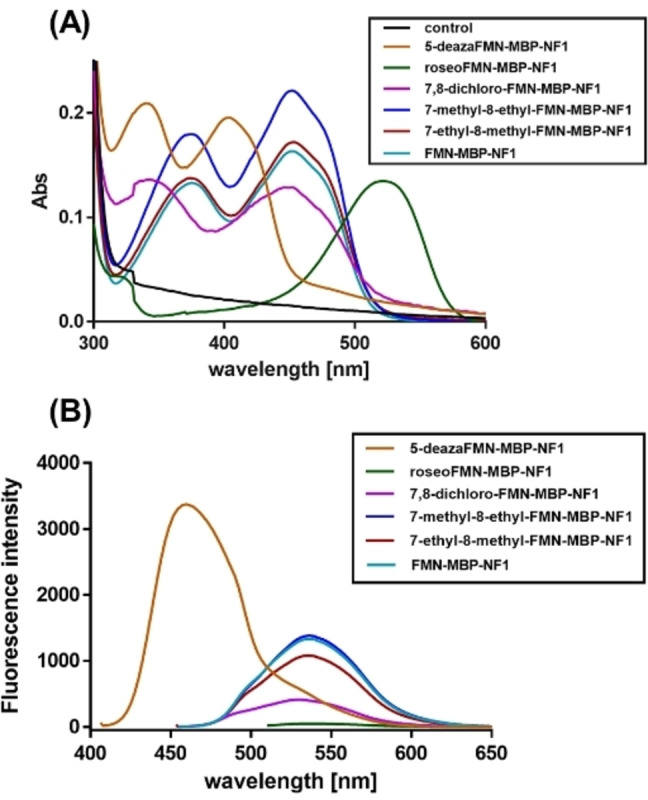
Spectral properties of MBPs containing different FMN derivatives. (A) UV/Vis absorption spectra of flavin‐labelled MBP‐NF1 variants. (B) Emission spectra of flavin‐MBP‐NF1 variants were obtained by exciting the samples at an excitation wavelength based on the maximum absorption of these FMN derivatives and measuring fluorescence emission spectra (410‐700 nm).
To determine the fluorescence quantum yields (Φf) of MBP‐NF1 variants, we monitored the fluorescence at a specific excitation wavelength, which was based on the maximum absorption wavelength of the FMN derivative‐modified MBP‐NF1 variants. The Φf of flavin‐modified MBP was calculated by comparing the integrated fluorescence intensity of the MBP variant with that of fluorescein, which was used as a reference standard (Φf=0.91). [23] All the flavin‐modified MBP variants exhibited fluorescence with excitation and emission wavelengths similar to the flavins free in solution (Figure 4B, Figure S12B, and Table 2). However, the Φf of flavin‐labeled MBP was consistently lower (25‐50 %) than the fluorescence quantum yields of the flavins when free in solution. We speculate that the decrease in fluorescence of the covalently attached flavins is due to quenching by the protein microenvironment. The quantum yield of FMN (Φf=0.21) is in good agreement with the previously reported value (Φf=0.22). [24] Similar fluorescence quantum yields as those for FMN (Φf=0.21) were determined for 7‐ethyl‐8‐methyl‐FMN (Φf=0.19) and for 7‐methyl‐8‐ethyl‐FMN (Φf=0.23). A significantly larger quantum yield, Φf=0.85, was found for 5‐deaza‐FMN. This shows that incorporation of 5‐deazaflavin using the Flavin‐tag results in a highly fluorescent protein. A poor fluorescence signal was detected for roseo‐FMN, in agreement with the known literature on this natural flavin derivative.[ 25 , 26 ]
Table 2.
Fluorescence and redox properties of FMN derivatives and MBP‐NF1 labeled with these FMN derivatives.
|
FMN derivative |
Quantum yield free/bound[a] |
λex [nm] free/bound |
λem [nm] free/bound |
Em [mV] free/bound[b] |
|---|---|---|---|---|
|
FMN |
0.21/0.09 |
450/446 |
536/536 |
−207/−221±9 |
|
5‐deaza‐FMN |
0.85/0.38 |
400/400 |
458/458 |
< −325/< −325 |
|
roseo‐FMN |
0.003/0.001 |
510/520 |
536/536 |
−225±20/−236±1 |
|
7‐methyl‐8‐ethyl‐FMN |
0.23/0.07 |
447/446 |
533/533 |
−208±1/−205±7 |
|
7‐ethyl‐8‐methyl‐FMN |
0.19/0.08 |
447/446 |
534/534 |
−223±11/−213±12 |
|
7,8‐dichloro‐FMN |
0.05/0.03 |
444/445 |
533/533 |
−102±2/‐92±2 |
[a] Standard deviations of two independent measurements was below 10 %. [b] The reactions were carried out in 50 mM phosphate (pH 7.0) buffer, containing 400 μM xanthine, 5 μM benzyl viologen, 50 mM glucose, 5 μg/mL catalase, 50 μg/mL glucose oxidase. Standard deviation of two independent measurements is given.
Determination of the redox potential of FMN derivatives and FMN derivatives‐labeled MBP
Flavin analogs with substituents in the 5, 7, and 8 positions are also valuable for systematically altering the redox potential (Em) of the cofactor.[ 27 , 28 ] Although the studied FMN derivatives are very similar to that of FMN, their redox properties are modulated by the substitution. Hence, the redox potentials of the free FMN derivatives and FMN‐labeled MBP were determined at pH 7 using the method described by Massey which involves the use of reference redox dyes.[ 29 , 30 , 31 ] As shown in Table 2, the redox potentials of FMN, roseo‐FMN, 7‐methyl‐8‐ethyl‐FMN, and 7‐ethyl‐8‐methyl‐FMN are in the same range (from −207 to −225 mV). 7,8‐Dichloro‐FMN displayed the highest Em (−102 mV) while 5‐deaza‐FMN displays the lowest redox potential (below −325 mV) (Table 2 and Figure S13). The Em values of the flavins attached to MBP are very similar to the corresponding free FMN derivatives. The results show that the flavin‐attachment does hardly affect the redox properties of the flavin moiety.
Conclusion
In summary, we have improved the efficiency of the Flavin‐tag protein labeling method. For boosting flavin labeling in E. coli, the co‐expression of FADS (resulting in higher intracellular FAD levels) leads to complete labeling of Flavin‐tagged recombinant proteins. Flavin labeling was demonstrated using several structurally and functionally distinct proteins (MBP, ADH, and SUMO). Complete flavinylation could be achieved with having the Flavin‐tag at the C‐ or N‐terminus. Thus, fully flavin‐labeled protein can be easily obtained in living cells by increasing the FAD synthetic capability of the host. It is worth noting that we have previously demonstrated that the Flavin tag method can also be used to attach a flavin in a loop region of a target protein. [11] Thus, the Flavin‐tag method is not restricted to the N‐ or C‐terminus of a protein.
As an extension to the Flavin‐tag method, we have established that also flavin derivatives can be attached to recombinant proteins using the small Flavin‐tag peptide and the flavin transferase, ApbE. In previous work, only FAD was used to label Flavin‐tagged proteins with a covalently attached FMN. [11] In this work, we enzymatically synthesized several FAD derivatives, with substituents in the isoalloxazine moiety, and demonstrated that these are accepted as substrates by ApbE as well. The resulting flavinylated proteins, depending on the type of flavin, exhibit highly different absorbance, fluorescence and redox properties (Scheme 2). For example, recombinant proteins can now be equipped with a stably bound roseoflavin label, which results in a bright red‐colored protein. Alternatively, the use of 5‐deaza‐FAD results in a highly fluorescent 5‐deaza‐FMN‐containing protein. Intriguingly, we have discovered that even NADH can be used for incorporation a covalently tethered nicotinamide moiety to a target protein. This extension in possibilities of labeling Flavin‐tagged proteins will further expand the application of the Flavin‐tag method as an efficient protein labeling method.
Scheme 2.
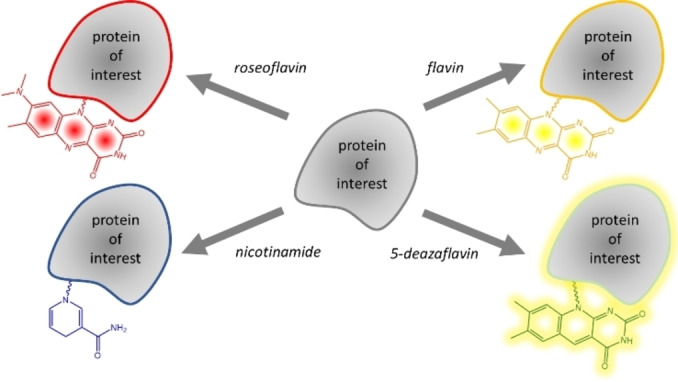
Flavin‐tag labeling allow installing different chromo‐ and fluorophores, including flavin (yellow), roseoflavin (red), 5‐deazaflavin (fluorescent), and nicotinamide.
Experimental Section
Chemicals and strains: Ni SepharoseTM 6 fast flow was purchased from GE Healthcare. T4 ligase and the restriction enzyme BsaI were purchased from New England Biolabs. E. coli NEB 10‐beta and BL21(DE3) (NEB, Ipswich, MA, USA) strains were used as the host for cloning and protein expression. Roseoflavin was ordered from Toronto Research Chemicals. 5‐Deazariboflavin was chemically synthesized using a previously published protocol (Figure S14). [22] All other chemicals were ordered from Sigma‐Aldrich.
Plasmid construction: The E. coli codon‐optimized mbp gene with N‐terminus and C‐terminus Flavin‐tag (F1: GVDGLSGATLTS) were synthesized by Twist Bioscience. MBP constructs carrying N‐terminal or C‐terminal Flavin‐tags were cloned into a pBAD−N‐His6x vector using the Golden Gate method. [32] The genes apbe and FADS were cloned into multiple cloning site 1 (MCS‐1; Hind III and NcoI) and MCS‐2 (NdeI and Pac I) of pRSF‐Duet‐1, respectively. The plasmids were isolated and sent for sequencing (GATC, Germany) to confirm the correct ligation of the genes. All the protein sequences, primers, and vectors used for this work are shown in Tables S1–S3.
Expression and purification: The recombinant strain E. coli NEB 10‐beta, carrying the apbe or mbp gene, was incubated at 24 °C for 24 h in 200 mL TB medium containing 50 μg/mL ampicillin. L‐arabinose was added (0.02 % w/v) when the OD600 was around 1.0. Cells were harvested by centrifugation (6,000 rpm, 20 minutes, 4 °C, Beckman‐Coulter centrifuge), and the pellet was resuspended in lysis buffer (50 mM Tris‐HCl buffer, pH 8) containing 100 mM MgCl2, 1 μg/mL DNase, and 0.1 mM phenylmethylsulfonyl fluoride. Cells were lysed by ultrasonic treatment, and the extract was cleared by centrifugation (12,000 rpm, 30 minutes, 4 °C). The His6x–tagged proteins were purified using 4 mL HisTrap Ni‐Sepharose HP columns (GE Healthcare Lifesciences, USA) and desalted with a HiPrep 26/10 Desalting column (GE Healthcare Lifesciences), using 50 mM Tris‐HCl buffer, pH 8. Enzyme aliquots were frozen using liquid nitrogen and stored at −80 °C until further use. The enzyme concentration was determined by measuring the absorbance at 280 nm and using the extinction coefficient of 32,890 M−1cm−1 (ApbE) and 66,350 M−1cm−1 (MBP) (calculated based on protein sequence using Expasy server).
For the overexpression of bifunctional FAD synthetase from C. ammoniagenes (FADS, NCBI#: D37967.1) and the riboflavin kinase (CaRFK), which was composed of the C‐terminal kinase domain of the FAD synthetase from C. ammoniagenes, E. coli NEB10 beta cells were transformed with the pBAD‐NHis‐FADS plasmid or pBAD‐NHis‐CaRFK and grown in 200 mL TB medium containing 50 μg/mL ampicillin at 24 °C for 24 h. The expression was induced by adding 0.02 % L‐arabinose at an OD600 of 0.8‐1.0. The purification of FADS and CaRFK was carried out the same method as that of ApbE and MBP. Purity was checked with SDS‐PAGE analysis, and protein concentrations were measured by measuring the absorbance at 280 nm and using the extinction coefficient of 28,420 M−1cm−1 (FADS) and 14,440 M−1cm−1 (CaRFK) (calculated based on protein sequence using Expasy server).
For the co‐expression of ApbE with MBP, ADH, and SUMO, E. coli BL21 (AI) cells were transformed with the plasmid carrying the apbe gene and the plasmids carrying mbp, adh, or sumo gene. The cells were grown in 30 mL TB medium containing 50 μg/mL kanamycin and ampicillin. The expression was induced by adding 0.5 mM IPTG and 0.02 % L‐arabinose at an OD600 of 0.8–1.0. Expression continued overnight at 37 °C, at 30 °C and 24 °C for 24 hours, and 17 °C for 48 hours. Cells were harvested by centrifugation and purified as described above for ApbE. The purified protein concentration was measured by using Pierce™ Rapid Gold BCA Protein Assay Kit (Thermoscientific). The samples were diluted to 1 μM before carrying out the mass spectrometry experiment for the binding efficiency analysis.
Size exclusion chromatography: SUMO was cleaved from SUMO‐ApbE by adding SUMO protease at 4 °C overnight. Then samples were used for gel filtration analysis on an ÄKTA purifier (GE Healthcare Lifesciences, USA). Samples were applied to a Superdex 200 increase 10/300 GL column (GE Healthcare Lifesciences, USA). The column was equilibrated with 50 mM Tris‐HCl (pH 8) buffer containing 200 mM NaCl.
HPLC analysis: High‐performance liquid chromatography (HPLC) was used to monitor the enzymatic synthesis of the FAD and FMN derivatives. Samples were taken after 16 h to check the conversion of flavin analogs. The HPLC method was described in previous work.[ 19 , 33 ] Briefly, 100 μL reaction mixture were quenched with 30 μL 100 % formic acid (FA), followed by incubation on ice for 5 min. Then, the samples were spun down at 13,000 g at 4 °C for 5 min to remove the denatured enzyme and were neutralized with 20 μL 1.6 mM NaOH. The supernatant (100 μL) was used for HPLC analysis (JASCO, UV‐2075 Plus). Samples were separated on a C18 (Gemini® 5 μm C18 110 Å, LC Column 250×4.6 mm) column by applying a linear gradient of 50 mM ammonium acetate pH 6.0 with 5 % acetonitrile (solvent A) and 100 % acetonitrile (solvent B). Roseo‐FAD/FMN/flavin at: t=0 min/100:0 (A : B), t=20 min/75 : 30 (A : B), t=30 min/5 : 95 (A : B), t=35 min/5 : 95 (A : B), t=40 min/75 : 30 (A : B), t=45 min/100:0 (A : B). The separation was monitored over time at 510 nm for roseo‐FAD/FMN/flavin. 7,8‐Dichloro‐FAD/FMN/flavin, 7‐methyl‐8‐ethyl‐FAD/FMN/flavin, 7‐ethyl‐8‐methyl‐FAD/FMN/flavin, 5‐deaza‐FAD/FMN/flavin, at: t=0 min/100:0 (A : B), t=16 min/80 : 20 (A : B), t=19 min/5 : 95 (A : B), t=22 min/5 : 95 (A : B), t=26 min/95 : 5 (A : B), t=28 min/100:0 (A : B). The separation was monitored over time at 400 nm for 5‐deaza‐FAD/FMN/flavin, and 450 nm for others.
Chemoenzymatic synthesis of FAD and FMN derivatives: The FAD and FMN derivatives were synthesized by FADS using riboflavin analogs. FADS (15 μM) or CaRFK (10 μM) was added to the reaction mixtures containing 100 μM flavin analogs in 50 mM Tris‐HCl, pH 8, 15 mM MgCl2, 3 mM ATP. The reaction mixtures with a total volume of 50 mL were incubated at 25 °C for 16 h. The reaction mixtures were first analyzed by HPLC to determine whether the reaction was complete. The fully converted reaction mixture was spun down at 12,000 g (Centrifuge 5920 R, Eppendorf) for 20 minutes, then the supernatant was applied to a Reveleris C18‐WP Flash cartridge column. FAD derivatives were eluted with 5 % ethanol in deionized water, and as described above, purity was verified by HPLC, as described above. The product was concentrated by freeze‐drying. FAD and FMN derivatives were either kept at −20 °C for long‐term storage or at 4 °C for short‐term storage. The formation of FAD derivatives and FMN derivatives was confirmed by LCMS.
Fluorescence analysis of in vitro flavinylation efficiency: ApbE catalyzes the flavin transfer to its target proteins, using FAD as substrate, producing AMP. Flavinylation of the target protein can be measured by SDS‐PAGE followed by exposure of gels to UV light. By this method, the fluorescence of FMN can be measured, offering a relatively simple and reliable method to detect covalently bound flavin. Standard in vitro flavinylation[ 12 , 34 ] was performed in 50 mM Tris‐HCl buffer at pH 8, containing 1 mM FAD derivatives, 10 mM MgCl2, target protein (3 mg/mL) and ApbE (1 mg/mL). The reactions were typically carried out by incubation at 30 °C. To monitor flavinylation in time, the in vitro reaction was initiated by adding ApbE, samples were taken at distinct time points and were immediately quenched by adding 4x SDS‐PAGE loading buffer. The samples were subsequently applied to a 12 % polyacrylamide gel, and the in‐gel fluorescent bands were detected using the imaging system (FUJIFILM‐LAS‐3000). After that, the gels were stained with InstantBlue.
Purification and identification of modified target protein from in vitro reaction mixture: Based on the in‐gel fluorescence analysis result, the final reaction mixture was applied to 0.5 mL HisTrap Ni‐Sepharose HP columns (GE Healthcare Lifesciences, USA) and desalted by the buffer exchange (50 mM Tris‐HCl buffer pH 8) with a 10 kDa Amicon® Ultra 0.5 mL Centrifugal Filters. The purified samples were used for the ESI‐MS analyses to identify the final modified proteins.
ESI‐MS analysis: Electrospray Ionization Mass spectrometry (ESI‐MS) was used to verify successfully synthesized FAD derivatives, FMN derivatives, and FMN derivatives modified MBP. The measurements were performed using the ACQUITY UPLC H‐Class® System (Waters) coupled to a Xevo® G2 quadrupole/time‐of‐flight (QToF) mass spectrometer (Waters) equipped with a PDA detector.
ESI‐MS analysis of modified proteins: The eluent system employed was a combination of solvent A (0.1 % formic acid in water) and solvent B (0.1 % formic acid in acetonitrile) at a flow rate of 0.3 mL/min. Protein samples were separated on an Acquity BEH C4 (150×2.1 mm, 1.7 μm, Waters) column operated at 40 °C. Protein samples were diluted to 1 μM prior to analysis. The sample injection volume was 3 μL. Protein was eluted using solvents A and B as the mobile phase, using the following gradient: linear from 5 to 70 % B (v/v) from 0–10 min, 70 to 95 % B from 10–11 min, kept at 95 % B from 11–13 min, returning to 20 % B in 2 min, re‐equilibration to 5 % B from 15–20 min. Mass spectra were obtained in the ESI‐positive ion mode. Obtained charge density spectra were deconvoluted using the MagTran software. The mass spectra images were generated using the software MagTran. The concentration of modified MBP‐NF1 was measured by using the extinction coefficient of these FMN derivatives[ 19 , 35 , 36 ] (Table S4).
ESI‐MS analysis of FMN and FAD derivatives: Adduct formation was analyzed using an Acquity UPLC® BEH C18 (50×2.1 mm, 1.7 μm, Waters) column. The mobile phase consisted of solvent A (0.1 % NH4Cl in water) and solvent B (0.1 % NH4Cl in acetonitrile). Compounds were eluted at a flow rate of 0.3 mL/min. The analysis was performed by injecting 3 μL of FMN and FAD derivatives solutions (300 μM). The gradient varied linearly from 1 to 99 % B (v/v) from 0–10.5 min, kept at 95 % B from 10.5‐11 min, returning to 1 % B in 1 min, re‐equilibration to 1 % B from 12–15 min. The mass spectrometer detected negative ions over the mass range m/z 300–1,000. Chromatograms were also recorded at a wavelength of 510 nm (roseo‐FMN/FAD) or 400 nm (5‐deaza‐FMN/FAD) or 450 nm (7,8‐dichloro‐FMN/FAD, 7‐methyl‐8‐ethyl‐FMN/FAD, and 7‐ethyl‐8‐methyl‐FMN/FAD).
Fluorescence quantum yield analysis: For quantum yield (Φf) measurements, FMN derivatives, MBP modified with FMN derivatives, and fluorescein (as a reference standard) were diluted in PBS pH 7.4 and 0.1 M NaOH buffer, respectively. Fluorescence spectra of individual samples were recorded by exciting the sample at the maximum absorbance wavelength as shown in Table 2, using a JASCO FP‐8300 spectrofluorometer and measuring the fluorescence emission in the range of 410–700 nm. The integrated fluorescence intensity was calculated from the fluorescence spectra. The formula (1) was used to calculate the fluorescence quantum yield:
| (1) |
of unknown samples, where F is the integrated intensity (areas), fx is an absorption factor (fx =1–10−Ax , where A=absorbance <0.1), n is the refractive index (RI) of the solvent (PBS pH 7.4 or 0.1 M NaOH), and is the quantum yield of the reference fluorophore fluorescein ( =0.91).[ 37 , 38 ]
Redox potential determination: The reductive titration of FMN derivatives and FMN derivatives modified MBP‐NF1 variants were performed spectroscopically according to the xanthine/xanthine oxidase electron delivering system as described by Massey.[ 29 , 30 , 31 ] The reaction was performed in 50 mM potassium phosphate, pH 7.0, at 25 °C containing 5 μM benzyl viologen as a mediator, 50 mM glucose, 5 μg/mL catalase, 50 μg/mL glucose oxidase, 400 μM xanthine, and xanthine oxidase in catalytic amounts. Anaerobic conditions were established with argon layered upon the top of the cuvette for 10 min. The spectra were collected for 60 min. Anthraquinone‐2‐sulfonate (AQS) (E0 =−225 mV), Neutral Red (E0 =−325 mV), and Nile Blue (E 0=−117 mV) were used as dyes for redox potential determination. The Em values of FMN derivatives and modified MBP were determined using the Nernst equation. [29]
Conflict of interest
The authors declare no conflict of interest.
1.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
We thank Jeroen Drenth and Ivana Marić for the suggestions and manuscript preparation, and Milos Trajkovic for synthesis of 5‐deazariboflavin. Y. Tong was supported by the China Scholarship Council (Ph.D. fellowship).
Y. Tong, M. R. Loonstra, M. W. Fraaije, ChemBioChem 2022, 23, e202200144.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1. Zhang Y., Park K. Y., Suazo K. F., Distefano M. D., Chem. Soc. Rev. 2018, 47, 9106–9136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rashidian M., Dozier J. K., Distefano M. D., Bioconjugate Chem. 2013, 24, 1277–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chen X., Wu Y. W., Org. Biomol. Chem. 2016, 14, 5417–5439. [DOI] [PubMed] [Google Scholar]
- 4. Rabuka D., Curr. Opin. Chem. Biol. 2010, 14, 790–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chen I., Howarth M., Lin W., Ting A. Y., Nat. Methods 2005, 2, 99–104. [DOI] [PubMed] [Google Scholar]
- 6. Lin C. W., Ting A. Y., J. Am. Chem. Soc. 2006, 128, 4542–4543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Popp M. W., Antos J. M., Grotenbreg G. M., Spooner E., Ploegh H. L., Nat. Chem. Biol. 2007, 3, 707–708. [DOI] [PubMed] [Google Scholar]
- 8. Fernández-Suárez M., Baruah H., Martínez-Hernández L., Xie K. T., Baskin J. M., Bertozzi C. R., Ting A. Y., Nat. Biotechnol. 2007, 25, 1483–1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhou Z., Cironi P., Lin A. J., Xu Y., Hrvatin S., Golan D. E., Silver P. A., Walsh C. T., Yin J., ACS Chem. Biol. 2007, 2, 337–346. [DOI] [PubMed] [Google Scholar]
- 10. Hofmann R., Akimoto G., Wucherpfennig T. G., Zeymer C., Bode J. W., Nat. Chem. 2020, 12, 1008–1015. [DOI] [PubMed] [Google Scholar]
- 11. Tong Y., Lee M., Drenth J., Fraaije M. W., Bioconjugate Chem. 2021, 32, 1559–1563. [DOI] [PubMed] [Google Scholar]
- 12. Bertsova Y. V., Fadeeva M. S., Kostyrko V. A., Serebryakova M. V., Baykov A. A., Bogachev A. V., J. Biol. Chem. 2013, 288, 14276–14286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fang X., Osipiuk J., Chakravarthy S., Yuan M., Menzer W. M., Nissen D., Liang P., Raba D. A., Tuz K., Howard A. J., Joachimiak A., Minh D. D. L., Juarez O., J. Biol. Chem. 2019, 294, 13800–13810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Manstein D. J., Pai E. F., J. Biol. Chem. 1986, 261, 16169–16173. [PubMed] [Google Scholar]
- 15. Nakagawa S., Igarashi A., Ohta T., Aisaka K., Hagihara T., Fujio T., Biosci. Biotechnol. Biochem. 1995, 59, 694–702. [DOI] [PubMed] [Google Scholar]
- 16. Hagihara T., Fujio T., Aisaka K., Appl. Microbiol. Biotechnol. 1995, 42, 724–729. [DOI] [PubMed] [Google Scholar]
- 17. Grill S., Busenbender S., Pfeiffer M., Köhler U., Mack M., J. Bacteriol. 2008, 190, 1546–1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wang W., Kim R., Yokota H., Kim S. H., Proteins Struct. Funct. Genet. 2005, 58, 246–248. [DOI] [PubMed] [Google Scholar]
- 19. Iamurri S. M., Daugherty A. B., Edmondson D. E., Lutz S., Protein Eng. Des. Sel. 2013, 26, 791–795. [DOI] [PubMed] [Google Scholar]
- 20. Aalbers F. S., Fürst M. J. L. J., Rovida S., Trajkovic M., Rubén Gómez Castellanos J., Bartsch S., Vogel A., Mattevi A., Fraaije M. W., eLife 2020, 9, 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hossain M. S., Le C. Q., Joseph E., Nguyen T. Q., Johnson-Winters K., Foss F. W., Org. Biomol. Chem. 2015, 13, 5082–5085. [DOI] [PubMed] [Google Scholar]
- 22. Su Q., Boucher P. A., Rokita S. E., Angew. Chem. Int. Ed. 2017, 56, 10862–10866; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 11002–11006. [Google Scholar]
- 23. Magde D., Wong R., Seybold P. G., Photochem. Photobiol. 2002, 75, 327. [DOI] [PubMed] [Google Scholar]
- 24. Islam S. D. M., Penzkofer A., Hegemann P., Chem. Phys. 2003, 291, 97–114. [Google Scholar]
- 25. Zirak P., Penzkofer A., Mathes T., Hegemann P., Chem. Phys. 2009, 358, 111–122. [Google Scholar]
- 26. Song P., Walker E. B., Vierstra R. D., Poff K. L., Photochem. Photobiol. 1980, 32, 393–398. [Google Scholar]
- 27. Jacobson F., Walsh C., Biochemistry 1984, 979–988. [Google Scholar]
- 28. Edmondson D. E., Ghisla S., Flavin Flavoproteins 1999, 76, 71–76. [Google Scholar]
- 29. Maklashina E., Cecchini G., Bio-Protocol 2020, 10, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Flavins and Flavoproteins 1990 (Eds.: B. Curti, S. Ronchi, G. Zanetti) Walter de Gruyter, 1991, pp. 59–66.
- 31.S. L. Christgen, S. M. Becker, D. F. Becker, Methods for Determining the Reduction Potentials of Flavin Enzymes, Elsevier, 2019. [DOI] [PubMed]
- 32. Engler C., Kandzia R., Marillonnet S., PLoS One 2008, 3, e3647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Drenth J., Trajkovic M., Fraaije M. W., ACS Catal. 2019, 9, 6435–6443. [Google Scholar]
- 34. Zhang L., Trncik C., Andrade S. L. A., Einsle O., Biochim. Biophys. Acta Bioenerg. 2017, 1858, 95–102. [DOI] [PubMed] [Google Scholar]
- 35.F. Müller, in Chemistry and Biochemistry of Flavoenzymes, Taylor & Francis, 1991, p. 105.
- 36. Spencer R., Fisher J., Walsh C., Biochemistry 1976, 15, 1043–1053. [DOI] [PubMed] [Google Scholar]
- 37. Ko S., Ko S., Jeon H., Yoon S., Kyung M., Yun H., Na J. H., Jung S. T., J. Agric. Food Chem. 2020, 68, 5873–5879. [DOI] [PubMed] [Google Scholar]
- 38. Brouwer A. M., Pure Appl. Chem. 2011, 83, 2213–2228. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.