

Abstract
In synthetic method development, the most rewarding path is seldom a straight line. While our initial entry into pentafluorosulfanyl (SF5) chemistry did not go according to plan (due to inaccessibility of reagents such as SF5Cl at the time), a “detour” led us to establish mild and inexpensive oxidative fluorination conditions that made aryl‐SF5 compound synthesis more accessible. The method involved the use of potassium fluoride and trichloroisocyanuric acid (TCICA)—a common swimming pool disinfectant—as opposed to previously employed reagents such as F2, XeF2, HF, and Cl2. Thereafter, curiosity led us to explore applications of TCICA/KF as a more general approach to the synthesis of fluorinated Group 15, 16, and 17 heteroatoms in organic scaffolds; this, in turn, prompted SC‐XRD, VT‐NMR, computational, and physical organic studies. Ultimately, it was discovered that TCICA/KF can be used to synthesize SF5Cl, enabling SF5 chemistry in an unexpected way.
Keywords: Gas-Reagent-Free, Inorganic Fluorine Chemistry, Oxidative Fluorination, Pentafluorosulfanyl, Trichloroisocyanuric Acid
Oxidative fluorination of various Group 15, 16, and 17 heteroatoms in organic scaffolds was made significantly more accessible in recent years using potassium fluoride and trichloroisocyanuric acid (TCICA)—a common swimming pool disinfectant. Behind‐the‐scenes insight and discussion on discoveries, applications, and opportunities associated with this “TCICA/KF approach” are chronicled herein.
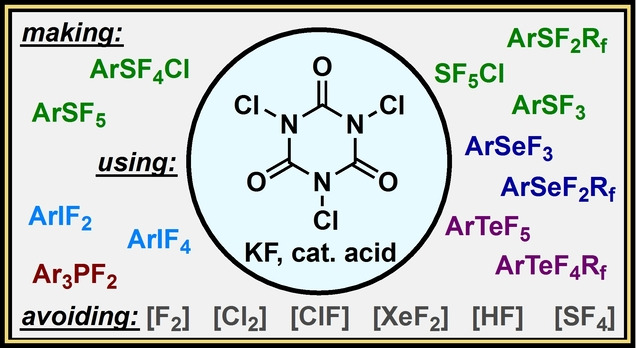
1. Introduction
Traditionally, oxidative fluorination connotes the use of harsh fluorinating reagents or otherwise unfriendly reaction conditions, though this stigma seems to be gradually disappearing. In the realm of organic fluorine chemistry, consider applications of the so‐called “N−F reagents” in the construction of C−F bonds over the last few decades. [1] Whether reagents such as Selectfluor, NFSI, or N‐fluoropyridinium salts are used directly in C−F bond formation,[ 2 , 3 ] or by relaying fluorine to high oxidation state transition metal[ 4 , 5 ] or main group fluorinating reagents,[ 6 , 7 ] N−F reagents have essentially allowed the broader community to explore the chemistry of “tamed F2” with a bench‐stable salt. Other strategies for C−F bond formation involve the combination of mild exogenous oxidants and metal fluorides; [8] these approaches occasionally drive down overall cost and even open oxidative fluorination as a plausible route to applications such as 18F‐labeling.[ 9 , 10 ] In the realm of inorganic fluorine chemistry, on the other hand, the user‐friendliness of reagents and methods available to forge heteroatom–fluorine bonds—especially with respect to polyfluorination—is not yet on par.
One could argue the reason stems from a philosophical disparity of organic vs. inorganic approaches to fluorine chemistry, as the training and skillset of inorganic fluorine chemists (not to mention crucial investments in equipment, PPE, and infrastructure) make the use of hazardous reagents and reaction conditions seem less intimidating, despite their inherent dangers. While the inorganic chemists undoubtedly sow the fields of fluorine chemistry through exploring fundamentally new functional groups, reactivities, and reagents by any means, the organic chemists tend to pursue the practical advancements that make this research area safer and more accessible to the broader chemical community. The emphasis on practicality is arguably responsible for the most significant periods of growth in the field, yet this view can be myopic and subject to induction periods before such practicality is realized. Thus, at the interface of these disciplines is where fluorine chemistry truly blossoms.
Forming multiple heteroatom–fluorine bonds is situated comfortably at this interface but likely has received less attention from organic chemists due to the more “obvious” relevance of C−F bond formation in the modern world. However, the recognized utility of stable, highly fluorinated functional groups such as the pentafluorosulfanyl (SF5) group [11] and applications of scaffolds such as (difluoro)aryl‐λ3‐iodanes (aryl‐IF2 compounds) as versatile reagents [12] (e.g., in forming C–F bonds) indicate that making heteroatom–fluorine bond formation more accessible could trigger growth spurts in various fields. Accordingly, in the spirit of exploratory chemistry and through the lens of practicality, we detail our recent development of a safe, simple, and versatile approach to oxidative fluorination of heteroatoms herein. We highlight several instances whereby forming one or more heteroatom–fluorine bonds with Group 15, 16, or 17 elements no longer requires the use of difficult‐to‐handle reagents such as F2, XeF2, HF, SF4, COF2, Cl2, etc. Instead, the same transformations are accomplished using only trichloroisocyanuric acid 1 (TCICA)—a common disinfectant used in swimming pools [13] —and potassium fluoride (KF) in the presence or absence of catalytic acid (Figure 1). This TCICA/KF approach has addressed several challenging oxidative fluorination problems over a series of publications and, in our eyes, may serve a timely liaison in realizing a greater potential of fluorinated heteroatom moieties as substituents, in method development, and in reagent design.
Figure 1.

Story arc of our unexpected foray into oxidative fluorination of heteroatoms with the TCICA/KF approach.
2. From CF3 to the SF5 Group
Our initial involvement in this research area (ca. 2017) was fueled by an interest in the SF5 group. This functional group has been presented in the literature as a surprisingly stable, large, lipophilic, and highly electron‐withdrawing substituent‐of‐interest in studies related to medicinal chemistry, agrochemistry, and materials. Although its size is between that of a CF3 group and a bulky aliphatic substituent (e.g., the tert‐butyl group), [14] its similar but exaggerated properties in comparison to the CF3 group has led the SF5 group to be coined the “super trifluoromethyl group”. [15] Yet, while several studies have demonstrated advantages, unique properties, and applications of the SF5 group sporadically over the last 20 years,[ 16 , 17 , 18 ] there remained little practicality associated with its synthesis/installation. Most progress during this era was centered on developing lengthy building block approaches from the few commercially available sources of “SF5”, primarily nitro‐substituted arenes. Conceptually, this presents an opportunity for modern synthetic fluorine chemists and a clear space for innovation and expansion. Since the Togni laboratory had invested over a decade of research on the CF3 group, [19] an entrance into developing methodologies surrounding this “super CF3 group” seemed just as logical as it was alluring.
Our first idea was to consider easier ways to harness the SF5 radical, as there is precedent for its utility in radical addition reactions, [20] and this could pave new late‐stage functionalization avenues. Recently, some have examined the reduction of SF6, [21] which is an attractive approach but also carries inherent challenges, including presumed overreduction to SF4,[ 22 , 23 ] which is a notoriously toxic gas. [24] Concomitantly, we considered another approach involving the design/synthesis of a more user‐friendly SF5 transfer reagent. Even if this were inevitably accomplished with harsh reagents, the hard synthesis would be worth the effort if it made SF5 chemistry easy for the broader community (after all, even Selectfluor is made with F2 in an industrial setting [25] ). It also seemed intuitive to generate, for instance, a compound with a weak heteroatom–SF5 bond from a compound with a weaker halogen–SF5 bond (a top‐down approach), as opposed to creating such a motif using the bottom‐up approach of oxidative fluorination. While we entertained many ideas for scaffolds that seemed like good reagent candidates according to DFT calculations, with virtually no access to reagents such as SF5Cl at the time (it is a corrosive gas often in limited supply, depending on what part of the world one is in), we never really gained momentum toward this end.
In general, the accessibility problems that needed to be overcome appeared far more fundamental than would allow for us to waltz into reagent design and late‐stage SF5 installation. For instance, arene‐SF5 synthesis was considered the most developed area, but even this required the use of corrosive gases such as Cl2 in the state‐of‐the‐art method (while Cl2 may be fine in some industrial settings, not all laboratories are equipped to handle it properly, especially on scale). A more realistic short‐term goal came to light given 1) the state of affairs in the field and 2) the tenure of the postdocs working on it at the time. That new goal was to make any aspect of SF5 synthesis more accessible and led us to figure out how to make aryl‐SF5 compounds without needing hazardous fluorinating reagents or corrosive gases.
3. Making the SF5 Group More Accessible on Arenes
The state‐of‐the‐art method in aryl‐SF5 synthesis was reported by Umemoto: [26] it is a two‐step approach that requires synthesis of a key aryl tetrafluorosulfanyl‐λ6‐chloride (aryl‐SF4Cl) intermediate using Cl2/KF, which can then be converted to the desired aryl‐SF5 compound via Cl−F exchange. Due to significant contributions from the laboratories of Umemoto, [27] Shibata,[ 28 , 29 ] Dolbier, [30] and Beier, [31] several possible approaches to Cl−F exchange on aryl‐SF4Cl compounds had been established in the literature. However, an alternative to the oxidative fluorination step had not yet materialized. Considering aryl‐SF4Cl compounds were also shown to be desirable intermediates beyond SF5 chemistry (e.g., in the synthesis of structures of the Ar‐SF4‐R type [32] ), we focused on better ways to access them. And with this more realistic goal in mind, we pondered: how can we replace Cl2 with a user‐friendly reagent that could serve as a chlorine source and a powerful oxidant? As an instance of being in the right place at the right time, our laboratory was routinely working with large amounts of trichloroisocyanuric acid (TCICA). In contrast to Cl2, TCICA is a bench‐stable, easy‐to‐handle solid that is sold commercially as a common disinfectant for swimming pools. [13]
The primary use of TCICA in our laboratory was for simplified, large‐scale syntheses of iodine(III) trifluoromethylation reagents. [33] Oxidative chlorination of an aryl iodide was achieved using TCICA, and Cl−F exchange was accomplished using KF in a subsequent step. Building on this idea, it only took two days of screening to reveal that TCICA and KF—often with the aid of catalytic acid (Lewis or Brønsted, most often trifluoroacetic acid)—could effectively generate trans‐phenyl‐SF4Cl from diphenyl disulfide, thus completely obviating the reliance on Cl2. This was an exciting result as, overnight, aryl‐SF5 synthesis became considerably more accessible for us (Figure 2).
Figure 2.

Reaction discovery, representative substrate scope, select applications, and opportunities for development surrounding the TCICA/KF approach to the synthesis of aryl‐SF5, aryl‐SF4R, and aryl‐SF3 compounds.
The complete details of the screening, reaction development, and investigation of scope (see 2–10 for select examples) are compiled in our original report, [34] but a few aspects are worth mentioning here. For one, a similar scope of aryl‐SF4Cl compounds originally synthesized from diaryl disulfides with Cl2/KF were accessible using TCICA/KF. Additionally, as TCICA is a milder reagent than Cl2, our approach exhibited functional group tolerance that either was not or cannot be demonstrated with Cl2/KF. For instance, even though strong electron‐donating groups presented issues with background reactivity under TCICA conditions (similar to Cl2/KF), certain esters, amides, azides, and heteroaromatics were compatible, allowing us to access novel aryl‐ and heteroaryl‐SF4Cl compounds. Fortunately, most of these “new” aryl‐SF4Cl compounds were converted to aryl‐SF5 (11, 12) or aryl‐SF4‐R (13, 14) congeners using established literature procedures and notably required no intermediate purification beyond extraction from the TCICA/KF reaction mixture (though it was possible, in some cases, to crystallize aryl‐SF4Cl compounds under inert atmosphere for structural studies [35] ). However, we noted that beyond pyridine, pyrimidine, and quinoline ring systems, accomplishing Cl−F exchange on triazoles (15), tetrazoles (16), indazoles (17), and other heteroaryl‐SF4Cl compounds remains challenging.
The demonstrated functional group tolerance of esters under TCICA/KF conditions also generated hypothetically faster routes, e.g., to phenol and benzoic acid derivatives. Whether during workup immediately following the Cl−F exchange reaction or as a separate step, a mild saponification reaction with LiOH could yield an alcohol/acid in two steps from a diaryl disulfide that previously took four or five steps to make from SF5‐substituted nitrobenzenes. [36] Note that we only reported the synthesis of phenol 12, but benzoic acid derivatives can be produced just as easily via saponification following synthesis of the SF5 group from an intermediate such as 6.
There was another finding that also echoed loudly in our future work: the TCICA/KF protocol was equally as sensitive to ortho‐substitution patterns as the Cl2/KF method. Just as Umemoto had previously discovered/leveraged in the development of the nucleophilic fluorinating reagent Fluolead, [37] ortho‐substituents larger than fluorine atoms greatly inhibit oxidation of the sulfur atom beyond the SIV oxidation state, providing access to aryl‐SF3 compounds in good yields. This made us curious about what other fluorinated groups might be accessible under TCICA/KF conditions.
Our approach to reaction discovery thereafter spawned multiple projects in a short period due to a random, exploratory, and fun form of screening that resembled a Monte Carlo simulation. Where “reaction screening” conventionally implies varying conditions to achieve a high yield on a specific type of transformation, we embarked upon a different type of screening that involved applying the TCICA/KF method (optimized on diaryl disulfides) to any heteroatom‐containing substrates that came to mind, with no expectations regarding yields or often the types of transformations we would hope to achieve. This increased element of randomness in reaction discovery by varying the substrates with the method (as opposed to the methods with the substrate) allowed us to rapidly determine situations where the TCICA/KF protocol could do anything interesting and, subsequently, how to develop and triage the work based on implications of the results. In essence, we surveyed the TCICA/KF potential surface and found as many wells as we could before ascertaining how deep they were.
4. Diaryl Dichalcogenides and the Turn toward TeF5
The first choice of substrates to survey beyond diaryl disulfides followed a logical curiosity: If diaryl disulfides convert to aryl‐SF4Cl or aryl‐SF3 compounds under TCICA/KF conditions, what is the result of subjecting diaryl diselenides to TCICA/KF? We found diphenyl diselenide in our inventory, tried it under TCICA/KF conditions, and obtained trifluoro(phenyl)‐λ4‐selane (phenyl‐SeF3, 18) selectively in 95 % yield without any further optimization; there was no trace of products in the SeVI oxidation state. This is an interesting (though unsurprising) divergence from the behavior of its sulfur congener, and so we kept moving down the periodic table: How do diaryl ditellurides behave under TCICA/KF conditions? Commercially available diphenyl ditelluride was subjected to TCICA/KF conditions and afforded another intriguing result—phenyl pentafluoro‐λ6‐tellane (Ph‐TeF5, 19) formation in high yield. All three chalcogen derivatives provided products with different degrees of fluorination and/or with different oxidation states (Figure 3).
Figure 3.

Reaction discovery, representative substrate scope, structural/spectroscopic analyses, and opportunities for development surrounding the TCICA/KF approach to the synthesis of aryl‐SeF3, aryl‐TeF5, and aryl‐TeF4CF3 compounds.
An isolation attempt of each phenyl‐SeF3 and phenyl‐TeF5 revealed that the former was more sensitive to hydrolytic decomposition, and phenyl‐TeF5 was more stable than the aryl‐SF4Cl compounds. Although limited examples of both aryl‐SeF3 and aryl‐TeF5 compounds exist in the literature (each synthesized on a small scale using XeF2 [38] ), the relative stability of phenyl‐TeF5 in our hands indicated that following this line of inquiry would be more appealing from an operational standpoint. Accordingly, we disclosed the phenyl‐SeF3 result in our original report [34] and decided to pursue aryl‐TeF5 compounds in a separate, detailed study. [39] After re‐optimization of TCICA/KF conditions, we discovered the substrate scope of converting diaryl ditellurides to aryl‐TeF5 compounds (19–26) was similar to the sulfur chemistry, in that primarily electron‐withdrawing or electron‐neutral substituents were well tolerated (this is a common theme throughout). Moreover, many of the products were isolated after extraction and concentration on the rotary evaporator, as opposed to requiring a glovebox and PFA‐ or PTFE‐walled vessels (we would recommend careful measures here anyway, especially with lower‐molecular‐weight aryl‐TeF5 compounds, which emit a pungent odor reminiscent of hot, rotten garlic). Additionally, we synthesized phenyl‐TeF5 easily on a two‐gram scale (78 % yield).
As phenyl‐TeF5 was previously reported to be a difluorinating reagent for alkenes [40] (but the experiments were only conducted in NMR tubes following in situ generation), we decided to investigate its capabilities as a “reagent” initially by first repeating the literature protocols with, for the first time, isolated phenyl‐TeF5. However, we found that the reactions were not reproducible. In our report, we detailed several control experiments, but our conclusion was that the previously reported behavior of phenyl‐TeF5 as a difluorinating reagent was questionable. Rather, the observed reactivity may have been from unreacted XeF2 in the NMR tubes. This finding inspired us to look at the reactivity of “pure” phenyl‐TeF5 under a variety of conditions. Although we successfully reproduced previously reported fluoride substitution reactions (by Janzen [41] ) with alcohols, secondary amines, azides, and water, the integrity of the TeF5 group oddly held up to several other added nucleophiles, photochemical conditions, etc. in our hands.
This also prompted us to look at the TeF5 group more closely as a substituent, e.g., in the solid state. Upon obtaining six new X‐ray crystal structures of compounds containing this group (exhibiting distorted octahedral geometries around the Te atom), we observed a few trends. Also, given our interest in solid‐state structures containing SF5 groups, we were able to draw more meaningful comparisons (27 vs. 28). Differences in bond lengths and angles were compiled/discussed in our original reports, but one of the interesting takeaway messages pertains to the differences in volume. Where the SF5 group was determined to be 1.5–1.6 times larger in volume than the CF3 group, [42] the TeF5 group, depending on the method of calculation, was determined to be 1.3–1.5 times larger in volume than the SF5 group. The hydrolytic instability of the TeF5 group may curtail applications for these molecules in medicinal chemistry or agrochemistry. However, its notable size, greasiness, and electron‐withdrawing effect may inspire investigation of its applications to certain materials (29), in the synthesis of new fluorinated tellurium compounds, or conceivably in studies pertaining to superacid chemistry.
5. Effects of Perfluoroalkyl Substituents on Organochalcogen Reactivity
As other members of the laboratory isolated phenyl‐TeCF3 as a byproduct in an independent project, [43] we became interested in the behavior of aryl(trifluoromethyl)tellanes vis‐à‐vis oxidative fluorination. Despite the steric bulk of the CF3 group, the TCICA/KF reaction produced the TeVI product—tetrafluoro(phenyl)(trifluoromethyl)‐λ6‐tellane (phenyl‐TeF4CF3)—selectively in 89 % yield. The stability/reactivity of phenyl‐TeF4CF3 was not studied in detail as per phenyl‐TeF5, but we noted its sensitivity to hydrolysis upon direct contact with water. As this motif was previously unknown, a derivative (30) was synthesized in order to obtain an X‐ray crystal structure (Figure 3). The TeF4CF3 group exhibited a trans arrangement and the expected distorted octahedral geometry around the Te center and—festooned with fluorine atoms—was determined to be 1.4–1.5 times larger in volume than the TeF5 group.
Working our way back up the periodic table, we asked, “How would the corresponding selenium and sulfur compounds with perfluoroalkyl substituents react?” Under TCICA/KF conditions, we obtained both difluoro(phenyl)(trifluoromethyl)‐λ4‐sulfane and difluoro(phenyl)(trifluoromethyl)‐λ4‐selane (phenyl‐SF2CF3 and phenyl‐SeF2CF3) from their SII and SeII precursors. The fact that tetrafluoro(phenyl)(trifluoromethyl)‐λ6‐sulfane (phenyl‐SeF4CF3), as opposed to phenyl‐TeF4CF3, is not formed is due to the well‐known reduced tendency of main group elements of the fourth (and sixth) period to reach their highest possible oxidation state. [44] On the other hand, it is somewhat surprising that tetrafluoro(phenyl)(trifluoromethyl)‐λ6‐sulfane (phenyl‐SF4CF3) is not observed. This might be due to a combination of the presence of a strongly electron‐withdrawing CF3 substituent and the insufficient oxidizing ability of TCICA. Admittedly, we were hoping we could access the SVI product, as the SF4CF3 group is known to be stable and is arguably harder to access than the SF5 group, in some cases. [45] However, several attempts were unsuccessful while phenyl‐SF2CF3 was obtained in high yield instead.
Very little was known about the SF2CF3 and SeF2CF3 groups (again, only limited examples were synthesized on small scale using XeF2 [46] ). So, after re‐optimizing TCICA/KF conditions and exploring a range of substrates (31–38), we studied these substituents in more detail (Figure 4). [47] Although the SF2CF3 and SeF2CF3 groups both proved sensitive to hydrolysis, we were able to employ an artful tactic developed by Taft and co‐workers to estimate Hammett parameters by 19F NMR spectroscopy.[ 48 , 49 ] Additionally, we obtained SC‐XRD data revealing that both the SF2CF3 and SeF2CF3 groups exhibited slightly distorted seesaw geometries, as anticipated. In our original report, we compared several features not only between these groups, but also with their aryl‐SCF3 and aryl‐SeCF3 starting materials, as well as a higher SeF2CF2CF3 analog (39).
Figure 4.

Reaction discovery, representative substrate scope, Hammett and structural analyses, and opportunities for development surrounding the TCICA/KF approach to the synthesis of aryl‐SF2Rf and aryl‐SeF2Rf compounds.
Next, as the SF2CF3 group was far less stable than the SF4CF3 group, [45] we began to wonder how it would behave as a reagent. It was not farfetched to see the connection between SF2CF3 and SF3 groups, though whether using the former as a fluorinating reagent over the latter opens new reaction pathways or has any advantages at all remains to be seen. This facet was not explored in depth; however, we were able to obtain a promising initial result indicating that aryl‐SF2CF3 compounds are capable of deoxygenative fluorination, as evidenced by the conversion of benzyl alcohol 40 to benzyl fluoride 41 in 83 % yield. This result is preliminary, but it is our hope that it inspires others to examine possible applications of these compounds as reagents for more interesting reactions than this one.
Finally, another important question we asked was, “Do we absolutely need a perfluoroalkyl substituent on the chalcogen atom—what about just alkyl?” We addressed the influence of fluorine substitution systematically by comparing the reactivities of substrates containing SCF3 vs. SCF2H vs. SCFH2 vs. SCH3 moieties under TCICA/KF conditions. Although phenyl‐SCF2H converted to phenyl‐SF2CF2H selectively and in 96 % yield, the monofluoromethyl‐ and methyl‐substituted sulfur compounds underwent exothermic perchlorination of the alkyl group in addition to oxidative fluorination. What is more, in these reaction mixtures, some aryl‐SF3 and aryl‐SF4Cl compounds became visible in the 19F NMR spectra that indicated S−C(sp3) bond cleavage had occurred.
6. A Lateral Move to Organoiodine
While we made our way down and up the periodic table fluorinating Group 16 heteroatoms, another contemporary project in the laboratory was drawing isolobal analogies between organotellurium and organoiodine compounds.[ 50 , 51 ] This, in part, inspired our side‐step to studying aryl iodides. Additionally, the synthesis of fluoroiodanes from aryl iodides with ortho‐substituents capable of coordinating to the iodine centers was known using TCICA and KF. [33] What happens under TCICA/KF conditions in the absence of a coordinating ortho‐substituent? We envisioned a route to difluoro(aryl)‐λ3‐iodanes (aryl‐IF2 compounds), though, based on existing literature, we were under the impression the hydrolytic stability of the products varied greatly depending on arene substitution. We started our screen with 4‐iodotoluene due to the popularity of its IF2 congener as a fluorinating reagent,[ 52 , 53 ] and did, indeed, observe the aryl‐IF2 compound in the crude mixture. The product was observed in low yield (≈20 %), with a precipitate forming in the NMR tube during analysis and no easy way to track the material balance.
Accordingly, we modified our screening tactic to involve “any iodoarene in the cabinet that contains fluorine” to be able to track the material balance by 19F NMR analysis; the most interesting result upfront came from comparing ortho‐ vs. meta‐ vs. para‐trifluoromethyl‐iodobenzene derivatives under TCICA/KF conditions. We found that the ortho‐trifluoromethyl‐substituted starting material produced a significantly higher yield of the aryl‐IF2 product by 19F NMR analysis, and there was no evidence of a precipitate rapidly forming in the NMR tube, which was the case with the other two samples. Without completing a thorough analysis, we jumped to the conclusion that ortho‐substituted aryl‐IF2 compounds could be more stable than their isomers and explored the reaction scope (42–51). We also drew an analogy to the putative “shielding” effect of the ortho‐substituents on the sulfur atom in Fluolead, [37] leaving it less susceptible to nucleophilic attack than most aryl‐SF3 compounds.
This result sparked an interesting physical organic inquiry regarding the influence of the ortho‐substituent on the stability of the IF2 moiety (Figure 5). [54] Starting with DFT calculations, it was clear ortho‐substitution had an influence on the preferred conformation, so we ambitiously pursued an eight‐step synthesis from 52 to probe molecule 53 for dynamic NMR studies. To our satisfaction, 53 was successfully employed to determine the rotational barrier (E a) of the IF2 moiety about the C−I bond in the presence of an ortho‐substituent. Note that DFT calculations indicated the dynamic process in our case was far more likely from C−I bond rotation rather than pseudorotation (though previous dynamic NMR studies from Ochiai [55] and Reich and Cooperman [56] are noteworthy regarding other aryl iodane pseudorotation mechanisms). Additionally, we obtained an X‐ray crystal structure of 53 that exhibited near perpendicularity of the T‐shaped IF2 moiety relative to the arene, but it is difficult to separate effects of ortho‐substitution from crystal packing effects.
Figure 5.

Reaction discovery, representative substrate scopes, structural analyses, and opportunities for development surrounding the TCICA/KF approach to the synthesis of aryl‐IF2 and aryl‐IF4 compounds.
It was clear ortho‐substituents had an influence. Yet, the study thus far still did not provide convincing answers regarding the role the ortho‐substituent plays in aryl‐IF2 hydrolytic stability or the magnitude of its effect. Careful, controlled hydrolysis kinetic studies seemed like a reasonable next step.
7. Substituent‐Controlled Access to Fluorinated IIII‐ or IV‐Compounds
By revisiting meta‐ and para‐substituted iodoarene substrates and preparing samples under rigorously dry conditions for the first time, we noticed something we had overlooked at the outset. In the absence of an ortho‐substituent, the reaction proceeded to form the more elusive tetrafluoro(aryl)‐λ5‐iodanes (aryl‐IF4 compounds)! Aryl‐IF4 compounds are less hydrolytically stable in solution than aryl‐IF2 compounds (we observed this during our kinetic experiments). Therefore, the precipitate forming in the NMR tubes during our “sloppy” sample preparation early in our investigation was likely composed of more iodylarene than iodosoarene. Finally, the role of the ortho‐substituent on iodoarenes under TCICA/KF conditions was understood—inhibiting further oxidation of IIII to IV.
With more rigorous technique and a slight optimization of reaction conditions, we examined several aryl‐IF4 compounds (54–58) in addition to our scope of ortho‐substituted aryl‐IF2 derivatives (Figure 5). We also proceeded to examine a few solid‐state structures; however, much of the original structural characterization was carried out more thoroughly by Frohn [57] and Seppelt. [58] Furthermore, with this new access to IV, we wondered whether we could make an exception to the rule that ortho‐substituents lead to IIII compounds by incorporating a coordinating ortho‐substituent. Immediately, the fluoroiodanes often made in our laboratory came to mind. Evidently, when we let fluoroiodane 59 stir for 48 h in the presence of TCICA/KF, IV derivative 60 was obtained (and also characterized by SC‐XRD).
Considering aryl‐IF4 compounds and other fluorinated IV compounds were historically more difficult to synthesize than aryl‐IF2 compounds (requiring SF4 gas [58] ), their potential as reagents or in catalysis remains largely unrealized.
8. Circling Back to SF5 Chemistry
Over a two‐year stint, curiosity had led us to explore oxidative fluorination of sulfur, selenium, and tellurium, as well as iodine, on various organic frameworks. We also briefly explored applications of TCICA/KF with Group 15 heteroatoms (e.g., in the difluorination of triarylphosphines and triarylarsines). [59] These results are not discussed herein, as we concede that the TCICA/KF approach may not be the best way to access a vast majority of fluorinated phosphorus‐, arsenic‐, and antimony‐based compounds (a deoxygenative fluorination approach seems generally more prudent [60] ). But, in any case, while traversing Groups 15–17 of the periodic table, we never really lost interest in SF5 chemistry—where our journey began.
We realized that we (and more recently others[ 61 , 62 , 63 ]) made SF5 synthesis more accessible on arenes, but we had yet to address the problem of general accessibility of the SF5 group on aliphatic molecules. An opportunity arose in early 2018 when we discovered that the TCICA/KF approach could be applied to elemental sulfur for the first gas‐reagent‐free synthesis of SF5Cl (Figure 6). This result was originally disclosed in our patent application (with and without catalytic TFA). [64] It was a remarkable breakthrough as it is by far the most accessible synthesis of SF5Cl [65] —one of the few known sources of SF5 radicals—and creates a possibility for more synthetic chemists to make/use it safely in‐house. Such an exciting result, however, laid dormant for a while, as we recognized that even though we avoided hazardous gas reagents during the synthesis, the product itself was a toxic gas. There was still an important problem to solve. As we were developing our own strategy for user‐friendly synthesis/use of SF5Cl on an as‐needed basis, Qing reported an elegant solution in 2021 establishing a protocol that allows extraction of SF5Cl into hexanes solution directly (≈0.10–0.15 M solution) so that SF5Cl never needs to be handled as a gas. [66] Between our discovery of a gas‐reagent‐free SF5Cl synthesis and Qing's optimization of the extraction procedure, exploring new applications in SF5 radical chemistry and even designing/synthesizing new possible SF5 transfer reagents now seem within reach. Moreover, several transformations have been developed in the past few years with SF5Cl that may become even more accessible with this ability to synthesize the reagent easily in house.[ 67 , 68 , 69 , 70 , 71 ]
Figure 6.

Reaction discovery, prior art, and opportunities for development surrounding the first gas‐reagent‐free synthesis of SF5Cl.
9. Concluding Remarks
The development of the TCICA/KF approach began as a practical advancement for aryl‐SF5 synthesis, but quickly transcended this to make both SF5 chemistry and oxidative fluorination of heteroatoms accessible from a much broader perspective. In most cases, we view our TCICA/KF protocol as a timely advancement in making useful products more available, but in some cases, it is debatable we have developed a practical method to make some impractical things. Regardless, this user‐friendly protocol has generated opportunities to explore entirely novel or underexplored fluorinated functional groups, which has allowed us to obtain fundamental knowledge about their structures, properties, stabilities, and reactivities. Moreover, in a relatively short time span, we have also begun to see exciting applications of TCICA/KF appear in the literature from several other laboratories following our initial report (for some examples, see Figure 7).[ 61 , 62 , 66 , 72 , 73 , 74 ]
Figure 7.

Highlighting examples of recent applications of the TCICA/KF combination to oxidative fluorination in other laboratories.[ 61 , 62 , 66 , 73 ]
As the exploratory inorganic fluorine chemistry detailed herein was founded on a pragmatic approach to synthesis at the inception, we hope our efforts sit comfortably enough to the organic–inorganic interface of fluorine chemistry to see applications of molecules containing heteroatom–fluorine bonds continue to blossom.
Conflict of interest
The authors declare no conflict of interest.
Biographical Information
Yannick Kraemer is from San Ramon, California (USA) and obtained a BS in chemistry from the University of California, Santa Cruz (2020). He is currently a graduate student conducting research in the CRP Lab at the University of California, Davis.

Biographical Information
Emily Nicole Bergman is from Aliso Viejo, California (USA). She is currently an undergraduate student in the CRP Lab at the University of California, Davis pursuing a degree in Applied Chemistry: Forensic Chemistry. She completed her first two years of undergraduate education at Irvine Valley College.

Biographical Information
Antonio Togni, born in the Italian part of Switzerland, studied chemistry and obtained his PhD at ETH Zürich. After a postdoctoral stay at Caltech in John E. Bercaw's group (1983–1984), he joined the Central Research Laboratories of the former Ciba‐Geigy Ltd. In 1992, he returned to his alma mater as a non‐tenure‐track assistant professor and he became a full professor in 1999. This is also about the time when he started slowly moving away from his original main research interests in organometallic chemistry and asymmetric catalysis towards fluorine chemistry. Since January 2021, he is a happy retiree who quit research, but temporarily continues teaching.

Biographical Information
Cody Ross Pitts is from Waterbury, Connecticut (USA). He obtained a BS in chemistry with minors in physics and musical theater from Monmouth University (2010), completed his PhD at Johns Hopkins University with Thomas Lectka (2011–2017), conducted research as an ETH Zürich postdoctoral fellow with Antonio Togni (2017–2019), and trained as an NIH postdoctoral fellow at Scripps Research with Phil S. Baran (2019–2021). He recently began his assistant professorship at the University of California, Davis, starting a research program rooted in synthetic and physical organic chemistry.

Acknowledgements
The authors thank ETH Zürich for an ETH Postdoctoral Fellowship (C.R.P.), the University of California, Davis, and all co‐authors who helped make the work highlighted in this review possible. Open access funding provided by Eidgenössische Technische Hochschule Zürich.
In memory of Professor Jack Dunitz
Y. Kraemer, E. N. Bergman, A. Togni, C. R. Pitts, Angew. Chem. Int. Ed. 2022, 61, e202205088; Angew. Chem. 2022, 134, e202205088.
Contributor Information
Prof. Dr. Antonio Togni, Email: atogni@ethz.ch.
Prof. Dr. Cody Ross Pitts, Email: crpitts@ucdavis.edu.
References
- 1. Umemoto T., Yang Y., Hammond G. B., Beilstein J. Org. Chem. 2021, 17, 1752–1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Champagne P. A., Desroches J., Hamel J.-D., Vandamme M., Paquin J.-F., Chem. Rev. 2015, 115, 9073–9174. [DOI] [PubMed] [Google Scholar]
- 3. Bume D. D., Harry S. A., Lectka T., Pitts C. R., J. Org. Chem. 2018, 83, 8803–8814. [DOI] [PubMed] [Google Scholar]
- 4. Furuya T., Benitez D., Tkatchouk E., Strom A. E., Tang P., Goddard W. A. III, Ritter T., J. Am. Chem. Soc. 2010, 132, 3793–3807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Racowski J. M., Gary J. B., Sanford M. S., Angew. Chem. Int. Ed. 2012, 51, 3414–3417; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 3470–3473. [Google Scholar]
- 6. Ye C., Twamley B., Shreeve J., Org. Lett. 2005, 7, 3961–3964. [DOI] [PubMed] [Google Scholar]
- 7. Planas O., Wang F., Leutzsch M., Cornella J., Science 2020, 367, 313–317. [DOI] [PubMed] [Google Scholar]
- 8. Liu W., Groves J. T., Acc. Chem. Res. 2015, 48, 1727–1735. [DOI] [PubMed] [Google Scholar]
- 9. Taylor N. J., Emer E., Preshlock S., Schedler M., Tredwell M., Verhoog S., Mercier J., Genicot C., Gouverneur V., J. Am. Chem. Soc. 2017, 139, 8267–8276. [DOI] [PubMed] [Google Scholar]
- 10. Halder H., Ritter T., J. Org. Chem. 2021, 86, 13873–13884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Savoie P. R., Welch J. T., Chem. Rev. 2015, 115, 1130–1190. [DOI] [PubMed] [Google Scholar]
- 12. Kohlhepp S. V., Gulder T., Chem. Soc. Rev. 2016, 45, 6270–6288. [DOI] [PubMed] [Google Scholar]
- 13. Tilstam U., Weinmann H., Org. Process Res. Dev. 2002, 6, 384–393. [Google Scholar]
- 14. Altomonte S., Baillie G. L., Ross R. A., Riley J., Zanda M., RSC Adv. 2014, 4, 20164–20176. [Google Scholar]
- 15. Kirsch P., Modern Fluoroorganic Chemistry: Synthesis, Reactivity, Applications, Wiley-VCH, Weinheim, 2004. [Google Scholar]
- 16. Stump B., Eberle C., Schweizer W. B., Kaiser M., Brun R., Krauth-Siegel R. L., Lentz D., Diederich F., ChemBioChem 2009, 10, 79–83. [DOI] [PubMed] [Google Scholar]
- 17. Joliton A., Plancher J.-M., Carreira E. M., Angew. Chem. Int. Ed. 2016, 55, 2113–2117; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 2153–2157. [Google Scholar]
- 18. Sowaileh M. F., Hazlitt R. A., Colby D. A., ChemMedChem 2017, 12, 1481–1490. [DOI] [PubMed] [Google Scholar]
- 19. Charpentier J., Früh N., Togni A., Chem. Rev. 2015, 115, 650–682. [DOI] [PubMed] [Google Scholar]
- 20. Aït-Mohand S., W. R. Dolbier, Jr. , Org. Lett. 2002, 4, 3013–3015. [DOI] [PubMed] [Google Scholar]
- 21. Rombach D., Wagenknecht H.-A., Angew. Chem. Int. Ed. 2020, 59, 300–303; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 306–310. [Google Scholar]
- 22. Rombach D., Wagenknecht H.-A., ChemCatChem 2018, 10, 2955–2961. [Google Scholar]
- 23. McTeague T. A., Jamison T. F., Angew. Chem. Int. Ed. 2016, 55, 15072–15075; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 15296–15299. [Google Scholar]
- 24. Kraut A., Lilis R., Br. J. Ind. Med. 1990, 47, 829–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Banks R. E., Mohialdin-Khaffaf S. N., Lal G. S., Sharif I., Syyret R. G., J. Chem. Soc. Chem. Commun. 1992, 595–596. [Google Scholar]
- 26. Umemoto T., Garrick L. M., Saito N., Beilstein J. Org. Chem. 2012, 8, 461–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Umemoto T., WO2008118787, 2008.
- 28. Cui B., Kosobokov M., Matsuzaki K., Tokunaga E., Shibata N., Chem. Commun. 2017, 53, 5997–6000. [DOI] [PubMed] [Google Scholar]
- 29. Cui B., Jia S., Tokunaga E., Saito N., Shibata N., Chem. Commun. 2017, 53, 12738–12741. [DOI] [PubMed] [Google Scholar]
- 30. Kanishchev O. S., W. R. Dolbier, Jr. , Angew. Chem. Int. Ed. 2015, 54, 280–284; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 282–286. [Google Scholar]
- 31. Lummer K., Ponomarenko M. V., Röschenthaler G.-V., Bremer M., Beier P., J. Fluorine Chem. 2014, 157, 79–83. [Google Scholar]
- 32. Zhong L., Savoie P. R., Filatov A. S., Welch J. T., Angew. Chem. Int. Ed. 2014, 53, 526–529; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 536–539. [Google Scholar]
- 33. Matoušek V., Pietrasiak E., Schwenk R., Togni A., J. Org. Chem. 2013, 78, 6763–6768. [DOI] [PubMed] [Google Scholar]
- 34. Pitts C. R., Bornemann D., Liebing P., Santschi N., Togni A., Angew. Chem. Int. Ed. 2019, 58, 1950–1954; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 1970–1974. [Google Scholar]
- 35. Liebing P., Pitts C. R., Reimann M., Trapp N., Rombach D., Bornemann D., Kaupp M., Togni A., Chem. Eur. J. 2021, 27, 6086–6093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kirsch P., Bremer M., Heckmeier M., Tarumi K., Angew. Chem. Int. Ed. 1999, 38, 1989–1992; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 1999, 111, 2174–2178. [Google Scholar]
- 37. Umemoto T., Singh R. P., Xu Y., Saito N., J. Am. Chem. Soc. 2010, 132, 18199–18205. [DOI] [PubMed] [Google Scholar]
- 38. Alam K., Janzen A. F., J. Fluorine Chem. 1985, 27, 467–469. [Google Scholar]
- 39. Bornemann D., Pitts C. R., Ziegler C. J., Pietrasiak E., Trapp N., Santschi N., Togni A., Angew. Chem. Int. Ed. 2019, 58, 12604–12608; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 12734–12738. [Google Scholar]
- 40. Lermontov S. A., Zavorin S. I., Bakhtin I. V., Pushin A. N., Zefirov N. S., Stang P. J., J. Fluorine Chem. 1998, 87, 75–83. [Google Scholar]
- 41. Jansen A. F., Alam K., Blackburn B. J., J. Fluorine Chem. 1989, 42, 173–178. [Google Scholar]
- 42. Hansch C., Leo A., Unger S. H., Kim K. H., Nikaitani D., Lien E. J., J. Med. Chem. 1973, 16, 1207–1216. [DOI] [PubMed] [Google Scholar]
- 43. Pietrasiak E., Baxter A. F., Jelier B., Santschi N., Togni A., Helv. Chim. Acta 2019, 102, e1900079. [Google Scholar]
- 44.
- 44a. Holleman A. F., Wiberg E., Wiberg N., Eagleson M., Brewer W. D., Aylett B. J., Inorganic Chemistry, Academic Press, San Diego, 2001, chap. IX, p. 290 and chap. XIII, p. 579. See also: [Google Scholar]
- 44b. Holleman A. F., Wiberg N., Anorganische Chemie, Vol. 1, 103 rd ed., de Gruyter, Berlin, 2017, chap. XIII, p. 705. [Google Scholar]
- 45. Kirsch P., Hahn A., Eur. J. Org. Chem. 2006, 1125–1131. [Google Scholar]
- 46. Yagupol'skii Y. L., Savina T. I., Zh. Org. Khim. 1979, 15, 438–439. [Google Scholar]
- 47. Brüning F., Pitts C. R., Kalim J., Bornemann D., Ghiazza C., de Montmillon J., Trapp N., Billard T., Togni A., Angew. Chem. Int. Ed. 2019, 58, 18937–18941; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 19113–19117. [Google Scholar]
- 48. Taft R. W., Price E., Fox I. R., Lewis I. C., Andersen K. K., Davis G. T., J. Am. Chem. Soc. 1963, 85, 709–724. [Google Scholar]
- 49. Taft R. W., Price E., Fox I. R., Lewis I. C., Andersen K. K., Davis G. T., J. Am. Chem. Soc. 1963, 85, 3146–3156. [Google Scholar]
- 50. Pietrasiak E., Togni A., Organometallics 2017, 36, 3750–3757. [Google Scholar]
- 51. Pietrasiak E., Gordon C. P., Copéret C., Togni A., Phys. Chem. Chem. Phys. 2020, 22, 2319–2326. [DOI] [PubMed] [Google Scholar]
- 52. Molnár I. G., Gilmour R., J. Am. Chem. Soc. 2016, 138, 5004–5007. [DOI] [PubMed] [Google Scholar]
- 53. Sarie J. C., Thiehoff C., Mudd R. J., Daniliuc C. G., Kehr G., Gilmour R., J. Org. Chem. 2017, 82, 11792–11798. [DOI] [PubMed] [Google Scholar]
- 54. Häfliger J., Pitts C. R., Bornemann D., Käser R., Santschi N., Charpentier J., Otth E., Trapp N., Verel R., Lüthi H. P., Togni A., Chem. Sci. 2019, 10, 7251–7259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ochiai M., Takaoka Y., Masaki Y., Nagao Y., Shiro M., J. Am. Chem. Soc. 1990, 112, 5677–5678. [Google Scholar]
- 56. Reich H. J., Cooperman C. S., J. Am. Chem. Soc. 1973, 95, 5077–5078. [Google Scholar]
- 57. Frohn H. J., Görg S., Henkel G., Läge M., Z. Anorg. Allg. Chem. 1995, 621, 1251–1256. [Google Scholar]
- 58. Hoyer S., Seppelt K., J. Fluorine Chem. 2004, 125, 989–996. [Google Scholar]
- 59. Bornemann D., Brüning F., Bartalucci N., Wettstein L., Pitts C. R., Helv. Chim. Acta 2021, 104, e2000218. [Google Scholar]
- 60. Bornemann D., Pitts C. R., Wettstein L., Brüning F., Küng S., Guan L., Trapp N., Grützmacher H., Togni A., Angew. Chem. Int. Ed. 2020, 59, 22790–22795; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 22982–22988. [Google Scholar]
- 61. Wang L., Cornella J., Angew. Chem. Int. Ed. 2020, 59, 23510–23515; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 23716–23721. [Google Scholar]
- 62. Wang L., Ni S., Cornella J., Synthesis 2021, 53, 4308–4312. [Google Scholar]
- 63. Magre M., Ni S., Cornella J., Angew. Chem. Int. Ed. 2022, 10.1002/anie.202200904; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2022, 10.1002/ange.202200904. [DOI] [Google Scholar]
- 64. Pitts C. R., Santschi N., Togni A., WO2019229103, 2019.
- 65. Winter R., WO2009152385A2, 2009.
- 66. Shou J.-Y., Xu X.-H., Qing F.-L., Angew. Chem. Int. Ed. 2021, 60, 15271–15275; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 15399–15403. [Google Scholar]
- 67. Cloutier M., Roudias M., Paquin J.-F., Org. Lett. 2019, 21, 3866–3870. [DOI] [PubMed] [Google Scholar]
- 68. Feng F.-F., Ma J.-A., Cahard D., J. Org. Chem. 2021, 86, 13808–13816. [DOI] [PubMed] [Google Scholar]
- 69. Lefebvre G., Charron O., Cossy J., Meyer C., Org. Lett. 2021, 23, 5491–5495. [DOI] [PubMed] [Google Scholar]
- 70. Debrauwer V., Leito I., Lokov M., Tshepelevitsh S., Parmentier M., Blanchard N., Bizet V., ACS Org. Inorg. Au 2021, 1, 43–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Birepinte M., Champagne P. A., Paquin J.-F., Angew. Chem. Int. Ed. 2022, 61, e202112575; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2022, 134, e202112575. [DOI] [PubMed] [Google Scholar]
- 72. Saidalimu I., Liang Y., Niina K., Tanagawa K., Saito N., Shibata N., Org. Chem. Front. 2019, 6, 1157–1161. [Google Scholar]
- 73. Surjadinata G., Hunter L., Matesic L., Pascali G., J. Flow Chem. 2021, 11, 107–115. [Google Scholar]
- 74. Taponard A., Jarrosson T., Khrouz L., Médebielle M., Broggi J., Tlili A., Angew. Chem. Int. Ed. 2022, 10.1002/anie.202204623; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2022, 10.1002/ange.202204623. [DOI] [Google Scholar]