

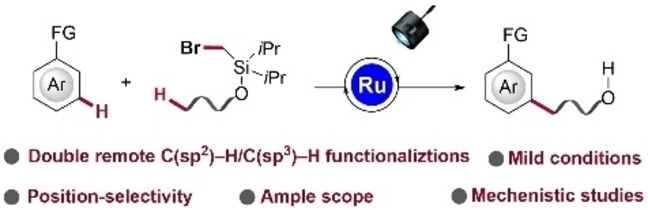
Abstract
Distal C(sp2)−H and C(sp3)−H functionalizations have recently emerged as step‐economical tools for molecular synthesis. However, while the C(sp2)−C(sp3) construction is of fundamental importance, its formation through double remote C(sp2)−H/C(sp3)−H activation has proven elusive. By merging the ruthenium‐catalyzed meta‐C(sp2)−H functionalization with an aliphatic hydrogen atom transfer (HAT) process, we, herein, describe the catalyzed twofold remote C(sp2)−H/C(sp3)−H functionalizations via photo‐induced ruthenium‐mediated radical relay. Thus, meta‐C(sp2)−H arene bonds and remote C(sp3)−H alkane bonds were activated by a single catalyst in a single operation. This process was accomplished at room temperature by visible light—notably without exogenous photocatalysts. Experimental and computational theory studies uncovered a manifold comprising ortho‐C−H activation, single‐electron‐transfer (SET), 1,n‐HAT (n=5–7) and σ‐activation by means of a single ruthenium(II) catalyst.
Keywords: C−H Activation, Hydrogen Atom Transfer, meta-Functionalization, Photocatalysis, Ruthenium
The ruthenium‐catalyzed double remote C(sp2)−H/C(sp3)−H functionalizations via radical relay have been accomplished by mild visible‐light photocatalysis without an exogenous photocatalyst, featuring meta‐arene and hydrogen atom transfer (HAT) functionalizations with a single catalyst.
Introduction
C−H bonds are the ubiquitous backbone of organic compounds. The direct and site‐selective functionalization of C−H bonds to amend molecular complexity are tremendously attractive on grounds of step and atom economy. While the activation of C−H bonds, adjacent to a functional groups, has been common, [1] distal C−H functionalization is significantly more difficult by challenges that include the intrinsic inertness as well as regioselectivity due to subtle differences of bond dissociation energies of multiple C−H bonds. Encouragingly, significant progress has been achieved for remote C−H functionalization in the last decade.[ 2 , 3 , 4 , 5 , 6 , 7 , 8 , 9 , 10 , 11 , 12 ] Elegant approaches, such as the use of transient mediators, [3] templates, [4] non‐covalent interactions, [5] ruthenium‐catalyzed σ‐activation, [6] as well as non‐directed [7] and/or metal‐free methods, [8] are now available for the remote arene C(sp2)−H activation (Scheme 1a). Meanwhile, cyclometallation [9] and metal carbene [10] pathways as well as radical strategies involving hydrogen atom transfer (HAT) processes[ 11 , 12 ] are amenable for the remote C(sp3)−H functionalization (Scheme 1b). However, while the molecules containing C(sp2)−C(sp3) bonds are omnipresent in petrochemical, agrochemical, pharmaceutical and food industries, the C(sp2)−C(sp3) bond construction through double remote C(sp2)−H/C(sp3)−H activation has thus far proven elusive.
Scheme 1.
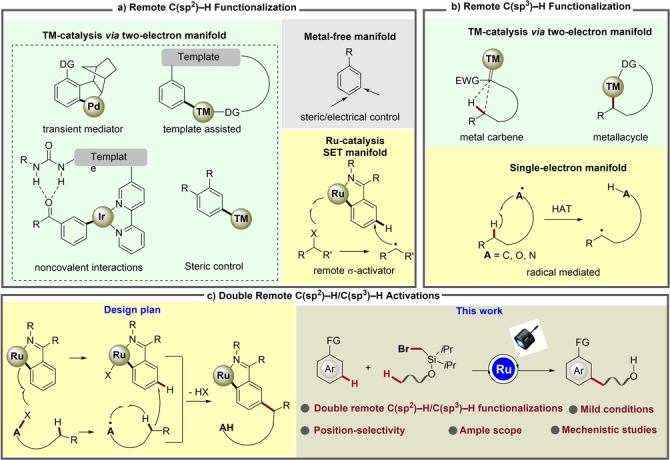
Design of transition‐metal‐catalyzed double remote C(sp2)−H/C(sp3)−H functionalizations via radical relay.
From a mechanistic viewpoint of the arene C(sp2)−H activation, the ruthenium‐catalyzed meta‐C(sp2)−H functionalization involving a single electron transfer process is unique and unusual as compared to other transition metals, which typically rely on two‐electron manifolds (Scheme 1a). In recent years, Ackermann, and Greaney, among others have investigated ruthenium‐catalyzed arene meta‐C−H alkylations. [6] This transformation proceeds via single‐electron‐transfer (SET) process between alkyl halide RX and an in situ generated ruthenacycle(II), generating a ruthenium(III) species and an alkyl radical. Then, the alkyl radical attacks the ruthenacycle(III) at the position para to the C−Ru bond, followed by an elimination of HX to deliver the meta‐alkylated product.
In contrast, benefiting from the rapid development of a mild method for the generation of heteroatom‐ or carbon‐centered radical species,[ 11 , 12 , 13 , 14 ] the HAT process has emerged as a versatile tool for remote C(sp3)−H functionalization. This process generally occurs under mild conditions with high efficiency and regioselectivity, exhibiting a selectivity trend of tertiary C−H sites over secondary and primary ones. This is complementary to that observed for transition metal‐catalyzed distal C(sp3)−H activation via metallacycle or carbene‐insertion pathway, which largely require high temperatures, directing groups, and featuring preference for primary C−H bonds (Scheme 1b).
Based on this analysis and within our program on ruthenium‐catalyzed remote functionalizations, we questioned whether it would be possible to establish a double remote C(sp2)−H/C(sp3)−H activation by the combination of the ruthenium‐catalyzed meta‐C(sp2)−H functionalization with an aliphatic HAT process (Scheme 1c). We, thus, envisioned that a suitable alkyl radical precursor reacts with ruthenacycle(II) to generate ruthenacycle(III) and a heteroatom or carbon‐centered radical species, following by an intramolecular HAT process to produce a new carbon‐centered radical species, which subsequently adds to ruthenacycle(III) at the para‐position of the C−Ru bond to form a doubly remote C(sp2)−C(sp3) bond. As a result of our studies, we, herein, report the transition metal‐catalyzed double remote C(sp2)−H/C(sp3)−H functionalizations via radical relay catalysis by user‐friendly [RuX2(p‐cymene)]2 (Scheme 1c). [15] The ruthenium‐catalysis was accomplished under exceedingly mild visible‐light‐mediated conditions without an exogenous photocatalysts, featuring high levels of positional selectivity and ample substrate scope.
Results and Discussion
Optimization of the Reaction Conditions
At the outset of our studies, phenylpyridine (1 a) was employed as the substrate. After preliminary experimentation, alcohol 2 a with an easily installed and removable silylmethyl bromide was selected as the HAT substrate (Table 1). The reaction in 1,4‐dioxane with K2CO3 as the base and Ru(OAc)2(p‐cymene) as the catalyst gave a low conversion after 24 h at 80 °C (Entry 1). When PPh3 was added as a ligand, the desired twofold remote C(sp2)−H/C(sp3)−H functionalization product 3 was formed in 28 % yield (Entry 2). However, a non‐negligible number of isomeric byproducts were also observed. (p‐CF3C6H4)3P instead of PPh3 did not afford a satisfactory yield and selectivity (Entry 3). Therefore, we next focused on testing photoconditions at room temperature with [RuCl2(p‐cymene)]2 as the precatalyst. While MesCO2H as the additive gave no desired product 3 (Entry 4), (n‐BuO)2PO2H afforded 3 in 34 % isolated yield with high selectivity (Entry 5). Further tests showed that (PhO)2P(O)OH was ideal, giving meta‐product 3 in 73 % isolated yield (Entry 6). [16] Thereby, only traces of other isomers of less than 5 % were observed. Alternative solvents, such as DCE and DMAc, led to significantly lower efficacies (Entries 7 and 8). The transformation could not occur under base‐free conditions or when using KOAc in lieu of K2CO3 (Entry 9). An improved yield of 82 % was obtained when adjusting the stoichiometry of substrate 2 a (Entry 10). While [RuCl2(benzene)]2 was also an effective catalyst, other ruthenium complexes, such as Ru(OAc)2(p‐cymene), RuCl3⋅3 H2O and Ru3(CO)12, failed to efficiently mediate the double remote functionalization (Entries 11–14). Control experiments verified the essential role of the light irradiation (Entries 15–17). Notably, the silyl methyl iodide 4, which was previously ideal for remote C(sp3)−H functionalization of alcohols by inter alia Gevorgyan, [12] was found to be less effective (Entry 18). Alcohols with a less sterically congested dimethyl silyl motif afforded a rather low yield and poor selectivity (see Supporting Information).
Table 1.
Optimization of the reaction conditions.[a]
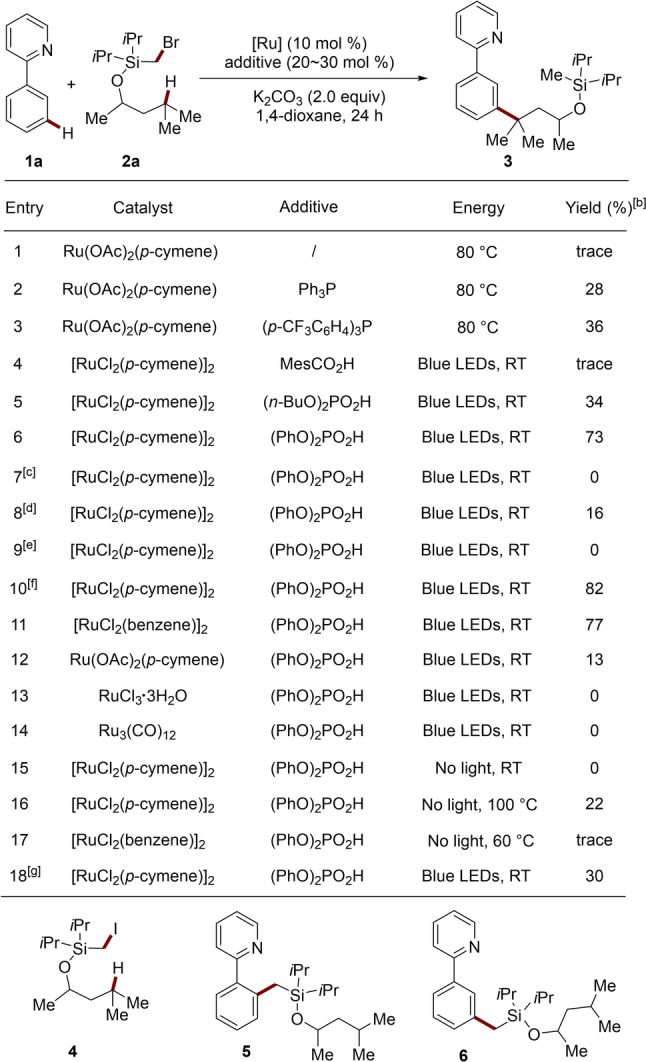
[a] Reaction conditions: 1 a (0.5 mmol), 2 a (0.75 mmol), [Ru] (0.05 mmol), additive (0.1 mmol for PPh3 and (p‐CF3C6H4)3P, 0.15 mmol for the others), K2CO3 (1.0 mmol), 1,4‐dioxane (2.0 mL), RT=30–35 °C. [b] Yield of isolated products. [c] Using DCE as solvent. [d] Using DMAc as solvent. [e] Using KOAc as the base. [f] 2 a (1.0 mmol). [g] Using 4 instead of 2 a.
Reaction Robustness
With the optimized reaction conditions in hand, the viable substrate scope of the ruthenium‐catalyzed twofold remote C(sp2)−H/C(sp3)−H activation was first examined with differently substituted arylpyridines 1 (Scheme 2). A range of arenes bearing electron‐withdrawing or electron‐donating groups could thus be selectively alkylated at the meta‐position with high efficacy (8–21). Functional groups, including fluoro (8, 9), ether (11–13), ester (14) and TMS (15) were well tolerated. Other heteroarenes, including pyrimidines (22, 23) and pyrazoles (24, 25), were identified as suitable orienting groups for the photo‐induced ruthenium‐catalyzed double remote C(sp2)−H/C(sp3)−H functionalizations. In contrast, substrates featuring oxazoline were not converted under the photochemical conditions. Instead, the desired product 26 could be obtained under thermal reaction conditions. Notably, the silyl auxiliary was easily removed within a one‐pot procedure with the aid of TBAF to afford the corresponding OH‐free alcohols 7, 10 and 17.
Scheme 2.
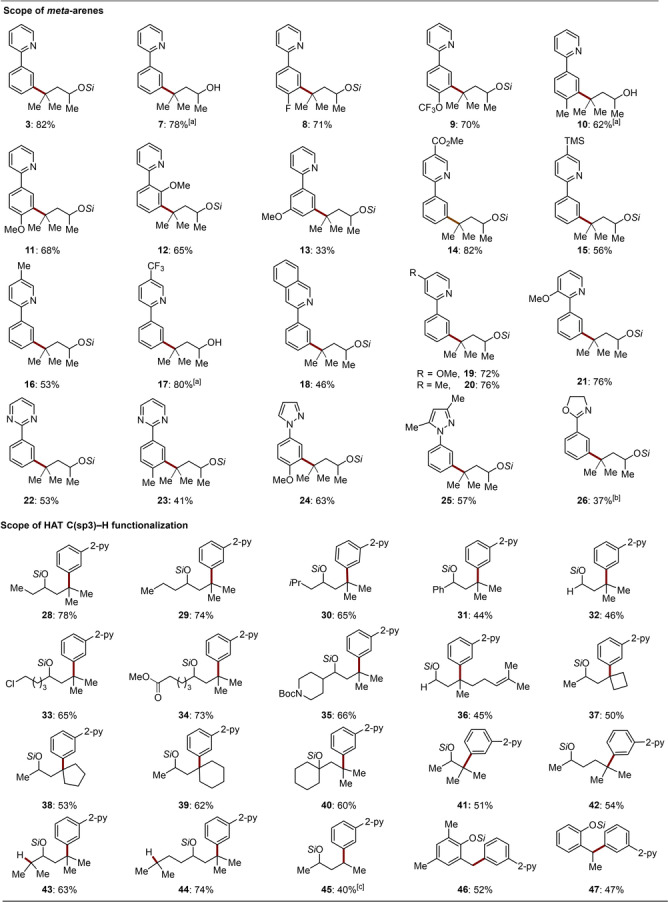
Ruthenium‐catalyzed double remote C(sp2)−H/C(sp3)−H functionalization. Reaction conditions: Arene (0.5 mmol), alcohol derivative (1.0 mmol), [RuCl2(p‐cymene)]2 (0.025 mmol), (PhO)2PO2H (0.15 mmol), K2CO3 (1.0 mmol), 1,4‐dioxane (2.0 mL), blue light, RT, N2, 24–48 h. [a] Work‐up with TBAF (1.0 M in THF, 4.0 mL), see Supporting Information. [b] Arene (0.5 mmol), alcohol derivative (1.0 mmol), Ru(OAc)2(p‐cymene)2 (0.05 mmol), K2CO3 (1.0 mmol), 1,4‐dioxane (2.0 mL), 90 °C, N2, 24 h. [c] Mixture.
Thereafter, the scope of the double remote C(sp2)−H/C(sp3)−H functionalizations was further examined with various tethered silylethers (Scheme 2). Alcohols possessing phenyl (31), chloro (33), alkoxycarbonyl (34), and amino (35) substituted chains efficiently furnished the desired products with perfect regioselectivities. Primary alcohol smoothly underwent the γ‐3°C−H arylation reaction in 46 % yield (32). Citronellol derivative was identified as a feasible substrate, leaving intact the otherwise reactive double bond (36). Cyclic alcohols, featuring 4‐ to 6‐membered rings, also reacted well (37–39). Furthermore, γ‐arylation of C−H bonds of tertiary alcohol proceeded efficiently (40). It is noteworthy that this transformation was not limited to γ‐3°C−H activation. Indeed, substrates with tertiary β‐H and δ‐H proved also amenable, delivering the corresponding products 41–42 in a selective fashion. For the substrates that possess competitive β‐H and δ‐H sites, the reaction preferentially occurred at the γ‐C−H sites to deliver products 43 and 44. These findings are in good agreement with the previously observation that a silicon‐based tether displays a preference for a 1,6‐HAT over a 1,5‐HAT or a 1,7‐HAT for tertiary sites with similar BDE. A mixture of products 45 was obtained for the alcohol without remote tertiary C−H site. Gratifyingly, 2‐alkylphenols were also suitable substrates for the remote C−H functionalization for the first time, affording the benzylic arylation products 46 and 47.
Late‐Stage Derivatization
A gram‐scale synthesis of product 3 was successfully performed to demonstrate the scalability of our approach, giving 1.13 g of the desired product 3 in 74 % yield (Scheme 3a). Furthermore, the double remote functionalization could be further cascaded. Thus, the merger of twofold remote C−H functionalization with a ruthenium‐catalyzed ortho‐C−H arylation (48), allylation (49), benzylation (50), methylation (51) or alkylation (52) proved to viable, notably without changing the nature of the catalyst or of the solvent (Scheme 3b). Thereby, a sequential threefold C−H transformation was achieved in a sustainable one‐pot fashion, involving C(sp2)−H/C(sp3)−H/C(sp2)−H functionalizations. The pyridine ring could be efficiently converted into a variety of heterocycles, such as pyrrole 53, polycyclic pyridinium salt 54 and indazole 56 (Scheme 3c), the structures of which are prevalent in bioactive compounds or light‐emitting materials. In addition, the pyrazolyl motif was efficiently removed to deliver synthetically meaningful aniline 57 (Scheme 3c).
Scheme 3.
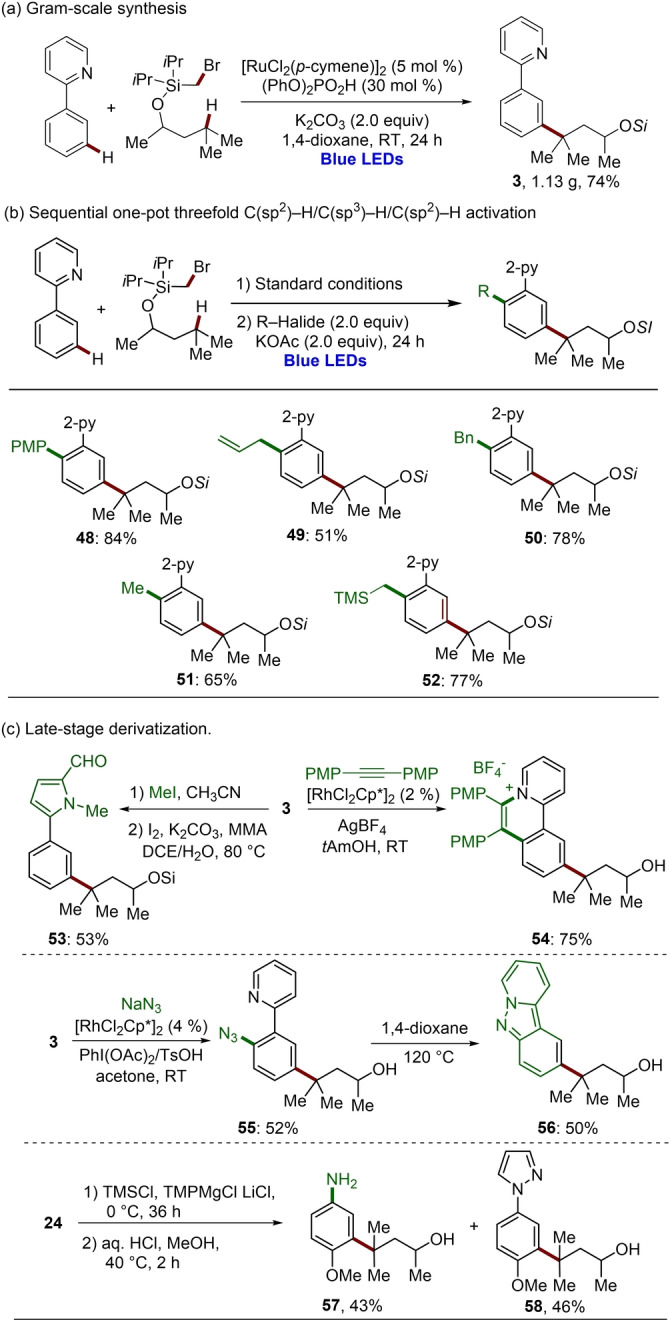
Gram‐scale synthesis and late‐stage derivatization.
Mechanistic Studies
Since photo‐induced ligand dissociation have been widely reported, we wondered whether p‐cymene dissociation occurred from the ruthenium(II) complex during the catalysis. Hence, the arene‐ligand‐free ruthenacycle 59 was used. Indeed, the reaction selectively yielded the desired product 3 in high yield (Scheme 4a). In addition, monitoring the reaction by GC‐MS showed that a significant amount of free p‐cymene was released by the blue light irradiation in the initial phase of the reaction (Scheme 4b). UV/Vis absorption spectra of [RuCl2(p‐cymene)]2 featured broad absorbance in the region of 400–500 nm, which is in good agreement of the catalytic activity under blue‐light irradiation (Scheme 4c). To further elucidate the role of the blue light, an on‐off‐experiment was conducted. Interestingly, the result demonstrated that the formation of the product 3 was only slightly suppressed in the absence of light during the latter phase of the transformation (Scheme 4d). Then, the reaction was conducted under the irradiation of blue light for only 2 h, and subsequently the light was switched off for 22 h, affording the desired product in 65 % yield (Scheme 4e). [17] These results show that continuous irradiation is not necessary, but is required initially for the decoordination of the p‐cymene. This observation contrasts with the previously reported photo‐induced ruthenium‐catalyzed ortho‐arylation of phenylpyridines. [18]
Scheme 4.
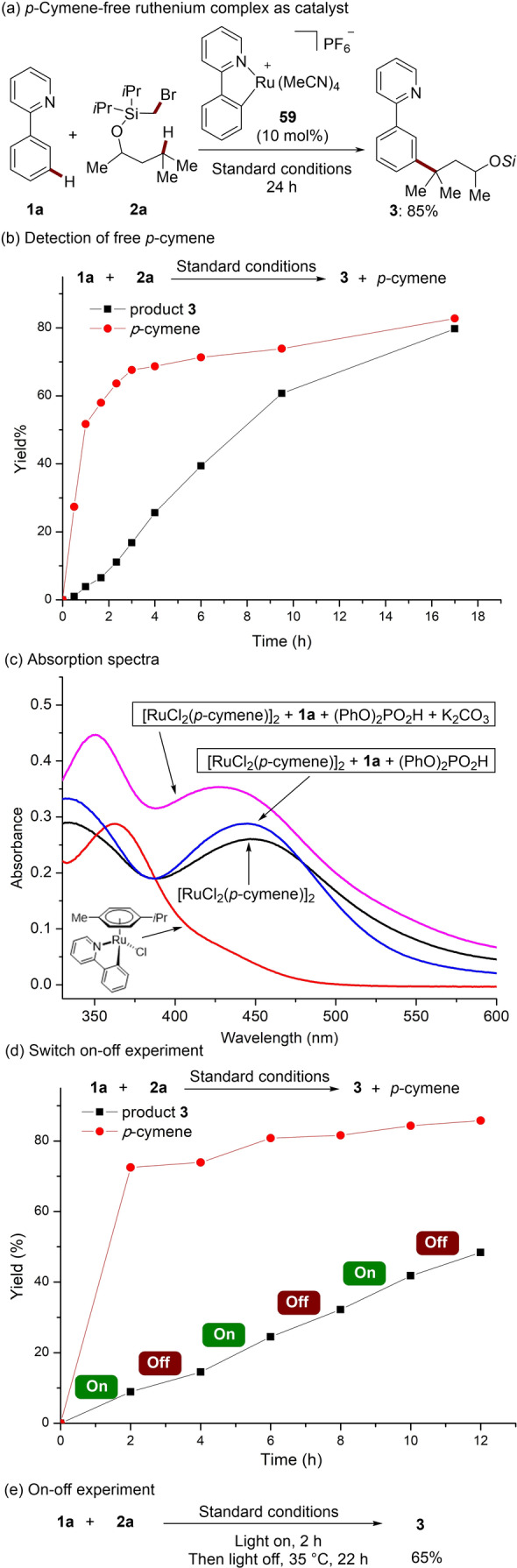
Summary of key mechanistic findings.
Computational Studies
To gain further insights into the details of the reaction mechanism, theory studies by means of density functional theory (DFT) calculations were conducted at the PBE0‐D3(BJ)/6‐311+G(d,p)‐SDD‐SMD(1,4‐Dioxane)//B3LYP−D3(BJ)/6‐1G(d)‐LANL2DZ level of theory (Figure 1). The excitation of bromoalkane‐coordinated ruthenacycle complex int2 followed by intersystem crossing (ISC) leads to a long‐lived triplet complex int2′. Next, inner‐sphere electron transfer (ISET) to bromoalkane occurs through transition state TS1 to form radical A and ruthenium(III) intermediate int3. DFT results also showed that int2 could be directly converted into int3 in the absence of light excitation with a low barrier of only 19.3 kcal mol−1. This is in good agreement with our experimental observation that the reaction kept going after discontinuation of the light irradiation (see above). Radical A easily converted to the tertiary carbon‐centered radical B via intramolecular HAT. In addition, the radical attach product is significantly stabilized as the singlet metallacycle int4 by 20 kcal mol−1 compared to corresponding triplet species int4′.
Figure 1.

Computed free energy surface. Computational methods: PBE0‐D3(BJ)/6–311+G(d,p)‐SDD‐SMD(1,4‐Dioxane)//B3LYP‐D3(BJ)/6‐31G(d)‐LANL2DZ.
Proposed Mechanism
Based on our detailed experimental and computational findings, a plausible catalytic cycle commences by ortho C−H ruthenation and dissociation of p‐cymene, thereby generating the catalytically competent complex C (Figure 2). Subsequently, single electron transfer (SET) occurs between intermediate C and substrate 2 a to generate the ruthenium(III) intermediate E and a silly methyl radical A. The newly formed radical A undergoes a HAT process to deliver the more stable distal carbon‐centered radical B, followed by the immediate addition to intermediate E at the para‐position to the C−Ru bond. Elimination of HBr by K2CO3 and ligand exchange delivers the double remote, meta‐C(sp2)−H/C(sp3)−H functionalization product 3, while at the same time regenerating the catalytically active species.
Figure 2.
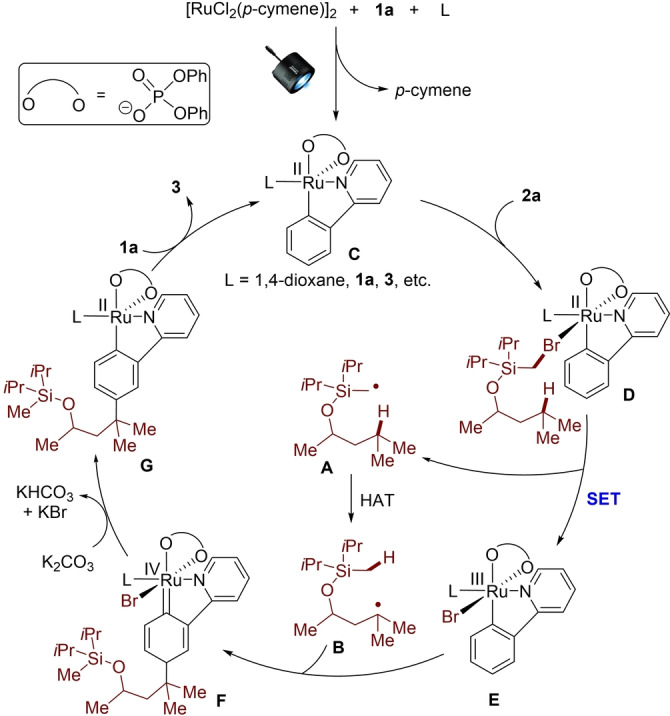
Proposed catalytic cycle.
Conclusion
In summary, we have disclosed the merger of the ruthenium‐catalyzed meta‐C(sp2)−H functionalization with an alkane hydrogen atom transfer (HAT) process. Thereby, we have established the twofold remote C(sp2)−H/C(sp3)−H functionalizations via photo‐induced radical relay, wherein an arene meta‐C(sp2)−H bond and a remote C(sp3)−H bond of alkyl alcohols are selectively functionalized in a single operation by a single catalyst. The ruthenium catalysis was accomplished under exceedingly mild conditions at room temperature, while tolerating a broad range of otherwise sensitive functional groups. Mechanistic studies by experiment and theory were suggestive of an initial p‐cymene decoordination from the ruthenium precatalyst to generate the catalytically competent ruthenacycle(II) complex. Given the current topical interest in modern radical HAT chemistry, we hope that our findings will inspire further studies towards multifold remote C(sp2)−H/C(sp3)−H functionalization manifolds.
Conflict of interest
The authors declare no conflict of interest.
1.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
The authors gratefully acknowledge support from the DFG Gottfried‐Wilhelm‐Leibniz award and SPP1807 (L.A.), and the Alexander‐von‐Humboldt Foundation (fellowship to Y.W.) is gratefully acknowledged. Open Access funding enabled and organized by Projekt DEAL.
Y. Wang, S. Chen, X. Chen, A. Zangarelli, L. Ackermann, Angew. Chem. Int. Ed. 2022, 61, e202205562; Angew. Chem. 2022, 134, e202205562.
Data Availability Statement
The data that support the findings of this study are available in the Supporting Information of this article.
References
- 1.For selected reviews of C−H activation, see:
- 1a. Guillemard L., Kaplaneris N., Ackermann L., Johansson M., Nat. Rev. Chem. 2021, 5, 522–545; [DOI] [PubMed] [Google Scholar]
- 1b. Prabagar B., Yang Y., Shi Z., Chem. Soc. Rev. 2021, 38, 3242–3272; [DOI] [PubMed] [Google Scholar]
- 1c. Rogge T., Kaplaneris N., Chatani N., Kim J., Chang S., Punji B., Schafer L. L., Musaev D. G., Wencel-Delord J., Roberts C. A., Sarpong R., Wilson Z. E., Brimble M. A., Johansson M. J., Ackermann L., Nat. Rev. Methods Primer 2021, 1, 43; [Google Scholar]
- 1d. Gandeepan P., Müller T., Zell D., Cera G., Warratz S., Ackermann L., Chem. Rev. 2019, 119, 2192–2452; [DOI] [PubMed] [Google Scholar]
- 1e. Davies D. L., Macgregor S. A., McMullin C. L., Chem. Rev. 2017, 117, 8649–8709; [DOI] [PubMed] [Google Scholar]
- 1f. Gensch T., Hopkinson M. N., Glorius F., Wencel-Delord J., Chem. Soc. Rev. 2016, 45, 2900–2936; [DOI] [PubMed] [Google Scholar]
- 1g. Liu C., Yuan J., Gao M., Tang S., Li W., Shi R., Lei A., Chem. Rev. 2015, 115, 12138–12204; [DOI] [PubMed] [Google Scholar]
- 1h. Colby D. A., Bergman R. G., Ellman J. A., Chem. Rev. 2010, 110, 624–655; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1i. Yeung C. S., Dong V. M., Chem. Rev. 2011, 111, 1215–1292. [DOI] [PubMed] [Google Scholar]
- 2.For selected reviews of remote C−H activation, see:
- 2a. Lam N. Y. S., Fan Z., Wu K., Park H. S., Sham S. Y., Strassfeld D. A., Yu J.-Q., J. Am. Chem. Soc. 2022, 144, 2793–2803; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2b. Dutta U., Maiti D., Acc. Chem. Res. 2022, 55, 354–372; [DOI] [PubMed] [Google Scholar]
- 2c. Sinha S. K., Guin S., Maiti S., Biswas J. P., Porey S., Maiti D., Chem. Rev. 2022, 122, 5682; [DOI] [PubMed] [Google Scholar]
- 2d. Korvorapun K., Samanta R. C., Rogge T., Ackermann L., Synthesis 2021, 53, 2911–2934; [Google Scholar]
- 2e. Dutta U., Maiti S., Bhattacharya T., Maiti D., Science 2021, 372, eabd5992; [DOI] [PubMed] [Google Scholar]
- 2f. Guo W., Wang Q., Zhu J., Chem. Soc. Rev. 2021, 50, 7359–7377; [DOI] [PubMed] [Google Scholar]
- 2g. Mihai M. T., Genov G. R., Phipps R. J., Chem. Soc. Rev. 2018, 47, 149–171; [DOI] [PubMed] [Google Scholar]
- 2h. Meng G., Lam N. Y. S., Lucas E. L., Saint-Denis T. G., Verma P., Chekshin N., Yu J. Q., J. Am. Chem. Soc. 2020, 142, 10571–10591; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2i. Sarkar S., Cheung K. P. S., Gevorgyan V., Chem. Sci. 2020, 11, 12974–12993; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2j. Wang J., Dong G., Chem. Rev. 2019, 119, 7478–7528; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2k. Leitch J. A., Frost C. G., Chem. Soc. Rev. 2017, 46, 7145–7153; [DOI] [PubMed] [Google Scholar]
- 2l. Della Ca’ N., Fontana M., Motti E., Catellani M., Acc. Chem. Res. 2016, 49, 1389–1400; [DOI] [PubMed] [Google Scholar]
- 2m. Davies H. M. L., Morton D., Chem. Soc. Rev. 2011, 40, 1857–1869; [DOI] [PubMed] [Google Scholar]
- 2n. Mkhalid I. A. I., Barnard J. H., Marder T. B., Murphy J. M., Hartwig J. F., Chem. Rev. 2010, 110, 890–931. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Shi H., Lu Y., Weng J., Bay K. L., Chen X., Tanaka K., Verma P., Houk K. N., Yu J.-Q., Nat. Chem. 2020, 12, 399–404; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3b. Wang X.-C., Gong W., Fang L.-Z., Zhu R.-Y., Li S., Engle K. M., Yu J.-Q., Nature 2015, 519, 334–338; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3c. Ye J., Lautens M., Nat. Chem. 2015, 7, 863–870; [DOI] [PubMed] [Google Scholar]
- 3d. Dong Z., Wang J., Dong G., J. Am. Chem. Soc. 2015, 137, 5887–5890; [DOI] [PubMed] [Google Scholar]
- 3e. Ding Q., Ye S., Cheng G., Wang P., Farmer M. E., Yu J.-Q., J. Am. Chem. Soc. 2017, 139, 417–425; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3f. Wang P., Farmer M. E., Yu J.-Q., Angew. Chem. Int. Ed. 2017, 56, 5125–5129; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 5207–5211; [Google Scholar]
- 3g. Catellani M., Top. Organomet. Chem. 2005, 14, 21–35. [Google Scholar]
- 4.
- 4a. Saha A., Guin S., Ali W., Bhattacharya T., Sasmal S., Goswami N., Prakash G., Sinha S. K., Chandrashekar H. B., Panda S., Anjana S. S., Maiti D., J. Am. Chem. Soc. 2022, 144, 1929–1940; [DOI] [PubMed] [Google Scholar]
- 4b. Bag S., Mondal S. K. A., Jayarajan R., Dutta U., Porey S., Sunoj R. B., Maiti D., J. Am. Chem. Soc. 2020, 142, 12453–12466; [DOI] [PubMed] [Google Scholar]
- 4c. Porey S., Zhang X., Bhowmick S., Kumar Singh V., Guin S., Paton R. S., Maiti D., J. Am. Chem. Soc. 2020, 142, 3762–3774; [DOI] [PubMed] [Google Scholar]
- 4d. Shi H., Herron A. N., Shao Y., Shao Q., Yu J.-Q., Nature 2018, 558, 581–585; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4e. Bag S., Jayarajan R., Mondal R., Maiti D., Angew. Chem. Int. Ed. 2017, 56, 3182–3186; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 3230–3234; [Google Scholar]
- 4f. Li S., Cai L., Ji H., Yang L., Li G., Nat. Commun. 2016, 7, 10443–10450; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4g. Tang R.-Y., Li G., Yu J.-Q., Nature 2014, 507, 215–220; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4h. Leow D., Li G., Mei T.-S., Yu J.-Q., Nature 2012, 486, 518–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.
- 5a. Kuninobu Y., Ida H., Nishi M., Kanai M., Nat. Chem. 2015, 7, 712–717; [DOI] [PubMed] [Google Scholar]
- 5b. Hoque M. E., Bisht R., Haldar C., Chattopadhyay B., J. Am. Chem. Soc. 2017, 139, 7745–7748; [DOI] [PubMed] [Google Scholar]
- 5c. Davis H. J., Genov G. R., Phipps R. J., Angew. Chem. Int. Ed. 2017, 56, 13351–13355; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 13536–13540; [Google Scholar]
- 5d. Davis H. J., Mihai M. T., Phipps R. J., J. Am. Chem. Soc. 2016, 138, 12759–12762. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Wei W., Yu H., Zangarelli A., Ackermann L., Chem. Sci. 2021, 12, 8073–8078; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6b. Gou X.-Y., Li Y., Luan Y.-Y., Shi W.-Y., Wang C.-T., An Y., Zhang B.-S., Liang Y.-M., ACS Catal. 2021, 11, 4263–4270; [Google Scholar]
- 6c. Korvorapun K., Moselage M., Struwe J., Rogge T., Messinis A. M., Ackermann L., Angew. Chem. Int. Ed. 2020, 59, 18795–18803; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 18956–18965; [Google Scholar]
- 6d. Korvorapun K., Kuniyil R., Ackermann L., ACS Catal. 2020, 10, 435–440; [Google Scholar]
- 6e. Wang X.-G., Li Y., Liu H.-C., Zhang B.-S., Gou X.-Y., Wang Q., Ma J.-W., Liang Y.-M., J. Am. Chem. Soc. 2019, 141, 13914–13922; [DOI] [PubMed] [Google Scholar]
- 6f. Sagadevan A., Greaney M. F., Angew. Chem. Int. Ed. 2019, 58, 9826–9830; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 9931–9935; [Google Scholar]
- 6g. Gandeepan P., Koeller J., Korvorapun K., Mohr J., Ackermann L., Angew. Chem. Int. Ed. 2019, 58, 9820–9825; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 9925–9930; [Google Scholar]
- 6h. Korvorapun K., Kaplaneris N., Rogge T., Warratz S., Stückl A. C., Ackermann L., ACS Catal. 2018, 8, 886–892; [Google Scholar]
- 6i. Li J., Korvorapun K., De Sarkar S., Rogge T., Burns D. J., Warratz S., Ackermann L., Nat. Commun. 2017, 8, 15430; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6j. Leitch J. A., McMullin C. L., Mahon M. F., Bhonoah Y., Frost C. G., ACS Catal. 2017, 7, 2616–2623; [Google Scholar]
- 6k. Li B., Fang S.-L., Huang D.-Y., Shi B.-F., Org. Lett. 2017, 19, 3950–3953; [DOI] [PubMed] [Google Scholar]
- 6l. Ruan Z., Zhang S.-K., Zhu C., Ruth P. N., Stalke D., Ackermann L., Angew. Chem. Int. Ed. 2017, 56, 2045–2049; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 2077–2081; [Google Scholar]
- 6m. Li J., Warratz S., Zell D., De Sarkar S., Ishikawa E. E., Ackermann L., J. Am. Chem. Soc. 2015, 137, 13894–13901; [DOI] [PubMed] [Google Scholar]
- 6n. Hofmann N., Ackermann L., J. Am. Chem. Soc. 2013, 135, 5877–5884; [DOI] [PubMed] [Google Scholar]
- 6o. Ackermann L., Novák P., Vicente R., Hofmann N., Angew. Chem. Int. Ed. 2009, 48, 6045–6048; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 6161–6164. [Google Scholar]
- 7.
- 7a. Saito Y., Segawa Y., Itami K., J. Am. Chem. Soc. 2015, 137, 5193–5198; [DOI] [PubMed] [Google Scholar]
- 7b. Ishiyama T., Takagi J., Ishida K., Miyaura N., Anastasi N. R., Hartwig J. F., J. Am. Chem. Soc. 2002, 124, 390–391; [DOI] [PubMed] [Google Scholar]
- 7c. Cho J.-Y., Tse M. K., Holmes D., Maleczka R. E., Smith M. R., Science 2002, 295, 305–308. [DOI] [PubMed] [Google Scholar]
- 8. Berger F., Plutschack M. B., Riegger J., Yu W., Speicher S., Ho M., Frank N., Ritter T., Nature 2019, 567, 223–228. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Liu L., Liu Y.-H., Shi B.-F., Chem. Sci. 2020, 11, 290–294; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9b. He C., Whitehurst Q. G., Gaunt M. J., Chem 2019, 5, 1031–1058; [Google Scholar]
- 9c. Jin L., Wang J., Dong G., Angew. Chem. Int. Ed. 2018, 57, 12352–12355; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 12532–12535; [Google Scholar]
- 9d. He J., Wasa M., Chan K. S. L., Shao Q., Yu J.-Q., Chem. Rev. 2017, 117, 8754–8786; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9e. Li S.-H., Zhu R.-Y., Xiao K.-J., Yu J.-Q., Angew. Chem. Int. Ed. 2016, 55, 4317–4321; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 4389–4393; [Google Scholar]
- 9f. Ano Y., Tobisu M., Chatani N., J. Am. Chem. Soc. 2011, 133, 12984–12986; [DOI] [PubMed] [Google Scholar]
- 9g. Xu J.-W., Zhang Z.-Z., Rao W.-H., Shi B.-F., J. Am. Chem. Soc. 2016, 138, 10750–10753. [DOI] [PubMed] [Google Scholar]
- 10. Davies H. M. L., Manning J. R., Nature 2008, 451, 417–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.
- 11a. Nakafuku K. M., Zhang Z., Wappes E. A., Stateman L. M., Chen A. D., Nagib D. A., Nat. Chem. 2020, 12, 697–704; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11b. Chen H., Fan W., Yuan X.-A., Yu S., Nat. Commun. 2019, 10, 4743–4751; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11c. Jiang H., Studer A., Angew. Chem. Int. Ed. 2018, 57, 1692–1696; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 1708–1712; [Google Scholar]
- 11d. Dauncey E. M., Morcillo S. P., Douglas J. J., Sheikh N. S., Leonori D., Angew. Chem. Int. Ed. 2018, 57, 744–748; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 752–756; [Google Scholar]
- 11e. Wu X., Wang M., Huan L., Wang D., Wang J., Zhu C., Angew. Chem. Int. Ed. 2018, 57, 1640–1644; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 1656–1660; [Google Scholar]
- 11f. Hu A., Guo J.-J., Pan H., Tang H., Gao Z., Zuo Z., J. Am. Chem. Soc. 2018, 140, 1612–1616; [DOI] [PubMed] [Google Scholar]
- 11g. Friese F. W., Mück-Lichtenfeld C., Studer A., Nat. Commun. 2018, 9, 2808; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11h. Shu W., Nevado C., Angew. Chem. Int. Ed. 2017, 56, 1881–1884; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 1907–1910; [Google Scholar]
- 11i. Wappes E. A., Nakafuku K. M., Nagib D. A., J. Am. Chem. Soc. 2017, 139, 10204–10207; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11j. Choi G. J., Zhu Q., Miller D. C., Gu C. J., Knowles R. R., Nature 2016, 539, 268; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11k. Chu J. C. K., Rovis T., Nature 2016, 539, 272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.
- 12a. Kurandina D., Yadagiri D., Rivas M., Kavun A., Chuentragool P., Hayama K., Gevorgyan V., J. Am. Chem. Soc. 2019, 141, 8104–8109; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12b. Chuentragool P., Yadagiri D., Morita T., Sarkar S., Parasram M., Wang Y., Gevorgyan V., Angew. Chem. Int. Ed. 2019, 58, 1794–1798; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 1808–1812; [Google Scholar]
- 12c. Cao Z., Li J., Sun Y., Zhang H., Mo X., Cao X., Zhang G., Chem. Sci. 2021, 12, 4836–4840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.
- 13a. Holmberg-Douglas N., Nicewicz D. A., Chem. Rev. 2022, 122, 1925–2016; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13b. Capaldo L., Ravelli D., Fagnoni M., Chem. Rev. 2022, 122, 1875–1924; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13c. Cheung K. P. S., Sarkar S., Gevorgyan V., Chem. Rev. 2022, 122, 1543–1625; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13d. Juliá F., Constantin T., Leonori D., Chem. Rev. 2022, 122, 2292–2352; [DOI] [PubMed] [Google Scholar]
- 13e. Chan A. Y., Perry L. B., Bissonnette N. B., Buksh B. F., Edwards G. A., Frye L. I., Garry O. L., Lavagnino M. N., Li B. X., Liang Y., Mao E., Millet A., Oakley J. V., Reed N. L., Sakai H. A., Seath C. P., MacMillan D. W. C., Chem. Rev. 2022, 122, 1485–1542; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13f. Crespi S., Fagnoni M., Chem. Rev. 2021, 121, 9790–9833; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13g. Skubi K. L., Blum T. R., Yoon T. P., Chem. Rev. 2016, 116, 10035–10074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.
- 14a. Stateman L. M., Dare R. M., Paneque A. N., Nagib D. A., Chem 2022, 8, 210–224; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14b. Shu W., García-Domínguez A., Quirós M. T., Mondal R., Cárdenas D. J., Nevado C., J. Am. Chem. Soc. 2021, 143, 4085–4089; [DOI] [PubMed] [Google Scholar]
- 14c. Campbell M. W., Yuan M., Polites V. C., Gutierrez O., Molander G. A., J. Am. Chem. Soc. 2021, 143, 3901–3910; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14d. Guo W., Wang Q., Zhu J., Angew. Chem. Int. Ed. 2021, 60, 9820–9825; [Google Scholar]
- 14e. Zhang X., Smith R. T., Le C., McCarver S. J., Shireman B. T., Carruthers N. I., MacMillan D. W. C., Nature 2020, 580, 220–226; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14f. Zhang W., Wang F., McCann S., Wang D., Chen P., Stahl S. S., Liu G., Nature 2016, 553, 1014–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.During the submission of our manuscript, Liang reported on an elegant double remote functionalizations via dual catalysis by a ruthenium and a photoredox catalyst, Liu H.-C., Kong X., Gong X.-P., Li Y., Niu Z.-J., Gou X.-Y., Li X.-S., Wang Y.-Z., Shi W.-Y., Huang Y.-C., Liu X.-Y., Liang Y.-M., Chem. Sci. 2022, 13, 5382–5389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.For the initial use of (PhO)2P(O)OH as additive in catalyzed C−H activations, see: Ackermann L., Vicente R., Althammer A., Org. Lett. 2008, 10, 2299–2302. [DOI] [PubMed] [Google Scholar]
- 17.For examples of aromatic C(sp2)−H functionalization catalyzed by p-cymene-free ruthenium catalysts, see:
- 17a. Simonetti M., Cannas D. M., Just-Baringo X., Vitorica-Yrezabal I. J., Larrosa I., Nat. Chem. 2018, 10, 724–731; [DOI] [PubMed] [Google Scholar]
- 17b. Ackermann L., Althammer A., Born R., Synlett 2007, 2833–2836. [Google Scholar]
- 18.
- 18a. Sagadevan A., Charitou A., Wang F., Ivanova M., Vuagnat M., Greaney M. F., Chem. Sci. 2020, 11, 4439–4443; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18b. Korvorapun K., Struwe J., Kuniyil R., Zangarelli A., Casnati A., Waeterschoot M., Ackermann L., Angew. Chem. Int. Ed. 2020, 59, 18103–18109; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 18259–18265. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Data Availability Statement
The data that support the findings of this study are available in the Supporting Information of this article.