

Abstract
Protein persulfides (R‐S‐SH) have emerged as a common post‐translational modification. Detection and quantitation of protein persulfides requires trapping with alkylating agents. Here we show that alkylating agents differ dramatically in their ability to conserve the persulfide's sulfur–sulfur bond for subsequent detection by mass spectrometry. The two alkylating agents most commonly used in cell biology and biochemistry, N‐ethylmaleimide and iodoacetamide, are found to be unsuitable for the purpose of conserving persulfides under biologically relevant conditions. The resulting persulfide adducts (R‐S‐S‐Alk) rapidly convert into the corresponding thioethers (R‐S‐Alk) by donating sulfur to ambient nucleophilic acceptors. In contrast, certain other alkylating agents, in particular monobromobimane and N‐t‐butyl‐iodoacetamide, generate stable alkylated persulfides. We propose that the nature of the alkylating agent determines the ability of the disulfide bond (R‐S‐S‐Alk) to tautomerize into the thiosulfoxide (R‐(S=S)‐Alk), and/or the ability of nucleophiles to remove the sulfane sulfur atom from the thiosulfoxide.
Keywords: Alkylation, Desulfurization, Protein Persulfidation, Sulfane Sulfur, Thiosulfoxide
Detection and quantitation of protein persulfides requires trapping with alkylating agents. Persulfide adducts derived from commonly used alkylating agents are unstable under biologically relevant conditions. The adducts convert to thioethers by losing sulfane sulfur to nucleophilic acceptors. Alkylating agents that form stable persulfide adducts and are suitable for persulfide trapping in biological samples are also reported.
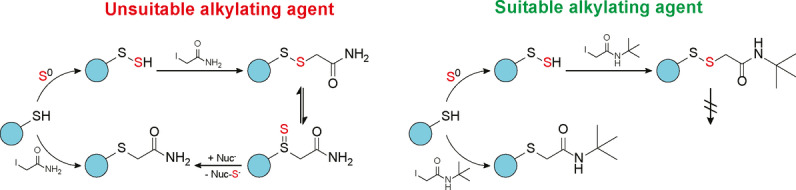
Introduction
Protein S‐persulfidation is the conversion of protein thiols (R‐SH) to protein persulfides (R‐S‐SH). Growing evidence shows that protein persulfidation is a common post‐translational modification.[ 1 , 2 , 3 ] It affects a large variety of proteins, and can be found across all of biology, from bacteria to man. [1] There is also evidence suggesting that protein persulfidation plays important roles in regulating protein function.[ 3 , 4 , 5 , 6 , 7 ] To examine the physiological role of protein persulfides, it is crucial to detect and quantify these modifications. However, the unstable nature of persulfides makes their detection and quantification challenging. In fact, the phenomenon of wide‐spread protein persulfidation has only been recognized within the last decade, exactly because these modifications are typically lost during standard sample preparation workflows. In principle, derivatization with electrophilic alkylating agents can be used to stabilize persulfides as disulfides (R‐S‐S‐Alk), to enable their detection, for instance, by mass spectrometry.[ 8 , 9 , 10 ] In previous studies, different kinds of alkylating agents have been used to alkylate persulfides, often yielding inconsistent outcomes. Quantitation of either low molecular weight persulfides or protein persulfides led to very different results depending on the specific alkylating agent used.[ 9 , 11 ]
In this study we systematically investigated the question of how different alkylating agents react with a protein persulfide. We first developed an in vitro model system that allows us to generate a defined persulfide on a defined protein and follow the fate of the persulfide by whole protein mass spectrometry. Our work led us to the following conclusions: i) Some alkylating agents, in particular monobromobimane and N‐t‐butyl‐iodoacetamide, generate alkylated persulfides (R‐S‐S‐Alk) that are stable and detectable. These agents are most suitable for the detection and quantitation of protein persulfides. ii) Other alkylating agents, in particular iodoacetamide and N‐ethylmaleimide, generate alkylated persulfides that rapidly convert into the corresponding thioether (R‐S‐Alk) by losing sulfur to ambient nucleophiles. These agents are unfit for the purpose of detecting protein persulfides in the presence of thiophilic nucleophiles. iii) Yet other alkylating agents generate alkylated persulfides of intermediate stability, and typically lead to an underestimation of actual persulfide levels, to different extent, depending on the specific alkylating agent. iv) The differences in R‐S–S‐Alk stability are probably caused by the influence of the alkylating agent side chain on the disulfide‐thiosulfoxide tautomeric equilibrium and/or on the sulfur transfer reaction between the thiosulfoxide and ambient nucleophiles. v) Differences in R‐S‐S‐Alk stability also apply to the detection of low molecular weight persulfides. vi) Differences in R‐S‐S‐Alk stability strongly influence the detection and quantification of protein persulfidation in biological samples.
Results and Discussion
We started out by developing an in vitro model system of protein persulfidation. To this end we recombinantly expressed and purified a variant of human thioredoxin‐1 (Trx1). In this variant, previously named Trx1(CSAAA) [12] (henceforth abbreviated Trx1CS), Cys‐35 is replaced by serine and three additional cysteines (62, 69 and 73) are replaced by alanines, so that the protein retains a single cysteine residue (Cys‐32), which is exposed on its surface. In wild type Trx1, Cys‐32 acts as the nucleophile in disulfide bond reduction. Furthermore, the C‐terminus carries both a streptavidin binding peptide (SBP) and a hexahistidine tag (Figure S1A). The mass of the purified protein precisely matched the predicted mass, except for the fact that about half of the protein lacks the N‐terminal methionine residue (Figure S1B, upper left panel), indicating that the recombinant protein is partially processed by E. coli methionine aminopeptidase. A cysteine‐less control protein, Trx1SS, also conformed to the calculated mass, plus/minus methionine (Figure S1B, upper right panel). As expected, alkylation with monobromobimane (MBB) shifted the mass of Trx1CS, but not the mass of the cysteine‐less variant (Figure S1B, lower panels). We then looked for a procedure that would allow us to persulfidate the single cysteine residue with high efficiency and minimal side products.
We first asked if incubation with polysulfides would lead to protein thiol persulfidation, because it has been suggested previously that polysulfides can donate sulfane sulfur (S0) atoms directly to thiols. [13] To this end, we immobilized Trx1CS on streptavidin beads and incubated it with glutathione trisulfide (GS34SSG). In order to trap and preserve potentially formed persulfides we quenched the reaction with either of two alkylating agents, MBB or iodoacetamide (IAM). The main product (≈80 %) was glutathionylated protein (P‐S‐SG), and a minor product (≈20 %) was P‐S‐34S‐SG. The choice of alkylating reagent did not make a difference for the outcome (Figure 1A). Incubation with glutathione tetrasulfide (GSSSSG) led to the same products in similar proportions, ≈85 % P‐S‐SG and ≈15 % P‐S‐S‐SG. Again, the choice of alkylating reagent did not make a difference for the outcome (Figure S1C). A control experiment with glutathione disulfide (GSSG) exclusively yielded P‐S‐SG (Figure S1D). These results show that RSSxSR‐type polysulfides do not transfer single S0 atoms to thiols, at least not under the given conditions. Instead, the protein thiolate preferentially attacks the polysulfide at the “outer” sulfur atom to become glutathionylated. Only a small proportion of protein thiolate attacks at the “inner” (i.e., central) sulfur atom to form a protein‐glutathione trisulfide (P‐S‐S‐SG) (Figure S1E). Preferential attack on the “outer” sulfur is consistent with a SN2 mechanism that favors the best leaving group: the one with the most acidic conjugate acid (GSS x H is more acidic than GSH). [14]
Figure 1.

Development of a protein persulfidation model system. A)–D) Whole protein mass spectra of Trx1CS (13.5 μM) treated with A) GS34SSG (300 μM), B) H2O2 (50 μM) plus Na2 34S (500 μM), C) MPST (10 μM) plus 3MP (40 μM), or D) Na2S2 (300 μM), and subsequently alkylated with either 1 mM MBB (left panels) or IAM (right panels). Peaks corresponding to unmodified or thiol‐alkylated proteins are highlighted in blue and peaks corresponding to per‐/polysulfide‐alkylated proteins are highlighted in yellow.
We then tried to achieve protein persulfidation through combined treatment with hydrogen peroxide (H2O2) and sodium sulfide (Na2 34S). The reaction between the protein thiol and H2O2 is expected to generate a sulfenic acid (P‐SOH) which then reacts with sulfide to yield a persulfide. However, this procedure only generated hyperoxidized persulfides (i.e., perthiosulfonic acid; P‐S‐34SO3H) (Figure 1B). This observation is in line with the idea that persulfides are easily oxidized at the outer sulfur atom, thus protecting the inner sulfur atom against irreversible oxidation.[ 1 , 15 , 16 , 17 ] Indeed, a direct comparison of thiol oxidation products in the presence or absence of sulfide supports this notion (Figure S1F).
We then tried an enzymatic strategy to generate protein persulfides. To this end, we used recombinant mercaptopyruvate sulfur transferase (MPST). MPST desulfurizes 3‐mercaptopyruvate (3MP) to generate an enzyme‐bound persulfide which can be transferred to various substrates. [18] Indeed, MPST persulfidated Trx1CS (Figure 1C), and the reaction was dependent on the active site thiol of MPST (Figure S1G). Interestingly, we observed more alkylated persulfides with MBB than with IAM (Figure 1C). The detection of alkylated protein polysulfides as minor products (P‐S‐S‐S‐Alk and P‐S‐S‐S‐S‐Alk) indicates that MPST can repeatedly add S0 atoms to existing per‐ and polysulfides in a sequential manner.
Finally, we explored the use of sodium disulfide (Na2S2) as a persulfidation agent. This compound should react with thiols to directly form a persulfide, with sulfide as the leaving group. A brief treatment (3 min) with Na2S2 was highly efficient in generating P‐S‐S‐Alk as the main product, with P‐S‐S‐S‐Alk and P‐S‐S‐S‐S‐Alk as minor products, but all of these could only be recovered if MBB was used as the alkylating agent (Figure 1D, left panel). Remarkably, IAM did not recover these products at all (Figure 1D, right panel), but instead only generated alkylated thiols (P‐S‐Alk). Of note, when alkylating agents are omitted, persulfides rapidly react further to generate di‐ and trisulfide intermolecular crosslinks (Figure S1H), confirming that alkylation is a necessary step to preserve and assess protein persulfidation.
In conclusion, Na2S2 treatment was found to be the simplest and most efficient method to generate protein persulfides. Intriguingly, the choice of alkylation agent turned out to be critical for detecting persulfides. MBB is found to be highly efficient in preserving persulfides, while IAM appears to be incapable of doing so.
Intrigued by the apparent difference between MBB and IAM in their ability to preserve protein persulfides, we investigated the utility of additional alkylating agents. Using the Na2S2‐based protein persulfidation system, we tested β‐(4‐hydroxyphenyl)ethyl iodoacetamide (HPE‐IAM), iodoacetic acid (IAA) and N‐ethyl maleimide (NEM). Both HPE‐IAM and IAA preserved a substantial fraction of persulfides (Figure 2A, B). In contrast, NEM was completely ineffective (Figure 2C). A direct side‐by‐side comparison of all five tested alkylating agents showed that MBB is the best preserver of persulfides, HPE‐IAM and IAA exhibit intermediate effectiveness, IAM is nearly ineffective, and NEM is completely ineffective (Figure 2D). The difference between IAM and HPE‐IAM was also observed when MPST was used to persulfidate Trx1CS (Figure S2), hence showing that the observed differences between alkylating agents are not dependent on the method of persulfidation.
Figure 2.

Alkylating agents differ widely in their ability to conserve protein persulfides. A–C) Whole protein mass spectra of Trx1CS (13.5 μM) persulfidated with Na2S2 (300 μM) and alkylated with A) HPE‐IAM, B) IAA, or C) NEM (1 mM each). Peaks corresponding to unmodified or thiol‐alkylated proteins are highlighted in blue and peaks corresponding to per‐/polysulfide‐alkylated proteins are highlighted in yellow. D) Relative proportion of alkylated persulfide recovered with different alkylating agents (n=2–5, error bars represent SD).
We then focused on the difference between IAM and HPE‐IAM, as both compounds use the same reactive group and only differ by a relatively remote modification that is not expected to impact reactivity with thiols. Indeed, previous studies suggest that both compounds exhibit similar thiol reactivity.[ 19 , 20 ] A time course experiment revealed that HPE‐IAM mediated alkylation of persulfides (P‐S‐S‐Alk) is already maximal at the shortest measurable time point, directly after addition of the alkylating agent, as it does not further increase during prolonged incubation (Figure S3A, upper panels). In contrast, treatment with IAM immediately (i.e., at the shortest measurable time point) leads to complete thiol alkylation (P‐S‐Alk) and does not preserve persulfides at any time point (Figure S3A, lower panels).
Recently, it has been suggested that polysulfides are susceptible to alkaline hydrolysis and that phenolic compounds can inhibit such hydrolysis. [21] We therefore asked whether unmodified and/or alkylated persulfides are susceptible to hydrolysis and if the phenol side chain of HPE‐IAM would suppress their hydrolysis. To this end, we compared the persulfide preserving effect of IAM and HPE‐IAM at different pH. For both alkylating agents the recovery of P‐S‐S‐Alk conjugates was improved at higher pH (Figures 3A and S3B). This finding contradicts the idea that alkaline hydrolysis destroys P‐S‐SH and/or P‐S‐S‐Alk. In contrast to previously published studies on polysulfides, [21] a large excess of tyrosine failed to enhance the recovery of P‐S‐S‐Alk (Figure S3C). In conclusion, our results do not suggest a role for hydrolysis, nor do they suggest a specific influence of phenolic compounds. The fact that the difference between IAM and HPE‐IAM is already established at the earliest time point of incubation (Figure S3A) also speaks against hydrolysis.
Figure 3.

Alkylated persulfides differ in stability. A) Whole protein mass spectra of Trx1CS (13.5 μM) persulfidated with Na2S2 (300 μM) and alkylated with 1 mM of either IAM (left panel) or HPE‐IAM (right panel), at three different pH values. B) Whole protein mass spectra of Trx1CS persulfidated with Na2S2 and alkylated with either IAM (left panel) or HPE‐IAM (middle panel) or an equimolar mixture of IAM and HPE‐IAM (right panel) (1 mM each). Peaks corresponding to IAM‐modified proteins are highlighted in red and peaks corresponding to HPE‐IAM‐modified proteins are highlighted in blue. C) Quantification of the relative abundance of product species obtained in (B). D) Whole protein mass spectra of Trx1CS persulfidated with Na2S2, alkylated with HPE‐IAM and then reacted with either NEM or IAM (1 mM each). A)–D) Peaks corresponding to unmodified or thiol‐alkylated proteins are highlighted in blue and peaks corresponding to per‐/polysulfide‐alkylated proteins are highlighted in yellow.
Next, we considered the possibility that IAM may somehow attack and destroy alkylated persulfides. Therefore, we asked if IAM would destroy persulfides previously alkylated with HPE‐IAM (P‐S‐S‐carbamidomethyl‐HPE; P‐S‐S‐CAM‐HPE). Simultaneous exposure of the protein to both IAM and HPE‐IAM revealed that both compounds react independently of each other (Figures 3B, C). Likewise, sequential treatment of the protein, i.e. HPE‐IAM followed by IAM or NEM, showed that neither IAM nor NEM is capable of destroying P‐S‐S‐CAM‐HPE adducts (Figures 3D and S3D). In conclusion, already alkylated persulfides are stable in the presence of unreacted IAM or NEM.
The previous experiments showed that various alkylated persulfides are unstable, to different degrees, depending on the specific alkylating agent. In particular, the comparison between IAA and IAM (Figures 1 and 2 and Figure S4) revealed a striking difference between the carboxylic acid and the corresponding amide. This example prompted us to consider the role of pi systems in β‐position to a disulfide. Indeed, all tested alkylating agents harbor a double bond in β‐position, a carbonyl group in the cases of IAM (and its derivatives) and NEM, and a carbon‐carbon double bond in the case of MBB (Figure 4A). It has been proposed that a carbonyl group in β‐position to a disulfide bond promotes formation of the thiosulfoxide, [13] although direct evidence for this notion seems to be lacking. We therefore asked if the instability of certain alkylated persulfides is related to their intrinsic tendency to form the thiosulfoxide, which may be easily desulfurized by ambient nucleophiles to generate the thioether (Figure 4B).
Figure 4.

The concept of disulfide–thiosulfoxide tautomerism predicts differences in the stability of alkylated persulfides. A) Structures of alkylated persulfides (R‐S‐S‐Alk) as formed with the alkylating agents NEM, IAM, IAM‐HPE, IAA and MBB (from left to right). B) Proposed mechanism of sulfur loss from alkylated persulfides: Disulfide‐thiosulfoxide tautomerism of alkylated persulfides is coupled to nucleophile‐mediated desulfurization of the thiosulfoxide, generating the thioether. C) Proposed influence of the bulkiness of the substituent in γ‐position on the efficiency of tautomerization and/or desulfurization. D) Whole protein mass spectra of Trx1CS (13.5 μM) persulfidated with Na2S2 (300 μM) and alkylated with Ethyl‐IAM (upper left panel), PE‐IAM (upper right panel) or N‐t‐butyl‐IAM (lower left panel) (1 mM each). Peaks corresponding to unmodified or thiol‐alkylated proteins are highlighted in blue and peaks corresponding to per‐/polysulfide‐alkylated proteins are highlighted in yellow. E) Relative proportion of recovered R‐S‐S‐Alk species for different IAM derivatives arranged in order of increasing γ‐substituent bulkiness.
It has been reported previously that the isomerization of allyl‐substituted disulfides to the corresponding thiosulfoxide is kinetically accessible at moderate temperatures[ 22 , 23 , 24 , 25 , 26 ] and that bulky groups at the γ position result in a reduced rate of subsequent desulfurization reactions.[ 22 , 25 ] We wondered if increasing the steric bulk next to the β‐carbonyl group would improve the ability of IAM derivatives to preserve persulfides, perhaps by perturbing the disulfide‐thiosulfoxide equilibrium (Figure 4C). We therefore tested additional IAM derivatives, namely ethyl‐, phenethyl (PE)‐, and t‐butyl‐IAM (Figure 4D), for comparison with IAM and HPE‐IAM. Indeed, we find a clear correlation between steric hindrance and the successful recovery of persulfides (Figure 4E).
The proposed disulfide–thiosulfoxide tautomerism (Figure 4B) predicts that sulfur loss is driven by the presence of suitable (thiophilic) nucleophiles. In our previous experiments we performed alkylation in the presence of unreacted Na2S2 and of HS− (generated as a by‐product of the persulfidation reaction), both of which are potent thiophilic nucleophiles. Hence, the removal of potential nucleophiles prior to alkylation should stabilize P‐S‐S‐Alk conjugates that are otherwise unstable. To test this notion, we reduced the quantity of potential nucleophiles by washing immobilized persulfidated Trx1CS prior to alkylation. Indeed, the washing step improved the recovery of IAM‐alkylated persulfides (Figure S5A), in support of the hypothesis.
To better understand the role of nucleophiles in relation to the proposed disulfide‐thiosulfoxide tautomerism we investigated the effect of cyanide. Cyanide can be expected to react differently with the disulfide and thiosulfoxide tautomeric forms of alkylated persulfides. On the one hand, alkylated persulfides in the disulfide form should be susceptible to cleavage at the disulfide bond. Disulfide cleavage by cyanide (CN−) will remove the alkylating group from the protein and leave behind a free thiol and/or a cyanylated thiol (Figure 5A, left side). On the other hand, the thiosulfoxide tautomer should be susceptible to desulfurization by cyanide. [23] The reaction with CN− should therefore generate thiocyanate (SCN−) and a thioether, i.e., an alkylated thiol (Figure 5A, right side). To test these predictions, we exposed MBB‐alkylated protein persulfides (thoroughly washed to remove any other potential nucleophiles) to CN− (Figure 5B). A substantial fraction of alkylated protein persulfide (P‐S‐S‐Bim) was converted to the cyanylated protein thiol (P‐SCN) and there was no increase in the formation of P‐S‐Bim. To generate significant amounts of IAM‐alkylated persulfides, we made use of the fact that these can be stabilized to some extent by elevated pH. After persulfidation and thorough washing, we performed alkylation at pH 9.5. After further washing, the pH was adjusted to 7.4 and the alkylated persulfide exposed to CN−. The alkylated protein persulfide (P‐S‐S‐CAM) was almost entirely converted to its desulfurization product (P‐S‐CAM) (Figure 5C). The same result was obtained when CN− treatment was performed at pH 9.5 (Figure S5B), showing that conversion of the alkylated persulfide to the thioether (i.e., alkylated thiol) is primarily caused by the presence of the nucleophile. To further validate these findings, we also monitored the formation of SCN− by GC/MS. Cyanide treatment of IAM‐alkylated persulfides generated a similar amount of SCN− as was released from non‐alkylated persulfides, while MBB‐ and t‐butyl‐IAM‐alkylated persulfides released much less SCN− (Figure 5D). Similar results were obtained when we used the fluorogenic sulfur acceptor SSP4 instead of CN− (Figure 5E).
Figure 5.

Labile alkylated persulfides lose sulfur to nucleophilic acceptors. A) Expected reactions of cyanide with the two different tautomeric forms of alkylated persulfides. B) Whole protein mass spectra of Trx1CS (13.5 μM) persulfidated with Na2S2 (300 μM), alkylated with MBB (1 mM), and then treated with buffer (left panel) or KCN (0.5 mM) (middle panel). Quantification of relative species abundance (right panel). C) Whole protein mass spectra of Trx1CS persulfidated with Na2S2, alkylated with IAM (1 mM) at pH 9.5, and then treated with buffer (left panel) or KCN (0.5 mM) (middle panel). Quantification of relative species abundance (right panel). B), C) Peaks corresponding to thiol‐alkylated proteins are highlighted in blue and peaks corresponding to per‐/polysulfide‐alkylated proteins are highlighted in yellow. In addition, peaks corresponding to Trx1CS‐SCN are highlighted in red and peaks corresponding to Trx1CS‐SH are highlighted in orange. D) GC‐MS based quantification of thiocyanate (SCN−) released from alkylated persulfides by treatment with cyanide, n=2. E) Quantification of sulfane sulfur released from alkylated persulfides by treatment with the fluorogenic sulfane sulfur acceptor SSP4 (100 μM), n=3. Error bars represent SD, * P≤0.05; ** P≤0.01, based on a two‐tailed unpaired t‐test.
The above results suggested that β‐carbonyl groups can promote sulfur loss from alkylated protein persulfide bonds, depending on additional factors, including the bulk in γ‐position and the availability of ambient nucleophiles. We therefore asked if β‐carbonyl‐driven sulfur loss can also be demonstrated for small model compounds. To this end, we prepared 2‐mercaptoacetamide disulfide (CAM‐SS‐CAM) and 2‐thioethanolamine disulfide (cystamine, Cyst‐SS‐Cyst) (Figure S6A). We found that the disulfide containing β‐carbonyl groups donates sulfur to cyanide, but not the disulfide lacking β‐carbonyl groups (Figure 6A). Correspondingly, the thioether was formed from CAM‐SS‐CAM, but not from cystamine (Figure S6B). Instead, cysteamine's disulfide bond was cleaved by cyanide to form cyanylated 2‐thioethanolamine (Cyst‐SCN) (Figure S6B, right panel). We also prepared disulfide‐linked dimers of 3‐mercaptopyruvate (3MP) and 3‐mercaptopropionic acid (3MPA) (Figure S6C). Again, the disulfide containing β‐carbonyl groups lost more sulfur to cyanide than the corresponding disulfide lacking β‐carbonyl groups (Figure 6B). In conclusion, β‐carbonyl‐driven sulfur loss from disulfides appears to be a general phenomenon, not being restricted to alkylated protein persulfides.
Figure 6.

β‐carbonyl‐driven desulfurization can also be observed with small molecules. A) Quantification of SCN− released from disulfides CAM‐S‐S‐CAM and cystamine (25 mM each) upon exposure to cyanide (250 mM) (left panel), n=3. Proposed reaction schemes for CAM‐S‐S‐CAM (i) and for cystamine (ii) (right panel). B) Quantification of SCN− released from the disulfide forms of 3MP and 3MPA (2.5 mM each) upon exposure to cyanide (50 mM) (left panel), n=3. Proposed reaction schemes for 3MP (i) and for 3MPA (ii) (right panel). Error bars represent SD, * P≤0.05; ** P≤0.01, based on a two‐tailed unpaired t‐test.
To further address the generality of our findings we persulfidated the free amino acid L‐cysteine with H2S2 and then reacted it with either MBB or NEM. The Cys‐S‐S‐Alk adduct was formed in the presence of MBB, while the Cys‐S‐Alk adduct was formed in the presence of NEM (Figure S7A). This result speaks in favor of generality, as the free amino acid persulfide reacted in a similar manner as the persulfidated cysteine side chain in the protein.
To test the relevance of our findings in relation to biological samples, we compared the performance of MBB and NEM as protein persulfide trapping agents in cells using a “switch assay” (Figure 7A). Following incubation of intact cells with either MBB or NEM, we osmotically ruptured the cells to release cytosolic proteins (and to minimize the release of disulfide‐containing proteins from the secretory pathway) in the presence of the alkylating agent. Alkylated proteins were precipitated, washed and treated with DTT (to convert alkylated persulfides into free thiols). Finally, the newly generated thiols were conjugated to a fluorescent dye. We find that alkylation with MBB yields significantly more protein labeling than alkylation with NEM (Figure 7B), a result that is fully in line with the notion that MBB preserves the persulfide's S−S bond, while NEM facilitates its destruction. This result further supports the notion that the choice of alkylating agent is critical for efficient protein persulfide trapping in biological samples (which always contain thiophilic nucleophiles).
Figure 7.

Alkylating agents differ in their ability to preserve protein persulfides in cells. A) Concept of using a comparative “switch assay” to visualize protein persulfides in cellular lysates. MBB trapped persulfides can be fluorescently labeled after reduction, while NEM trapped persulfides are destroyed. In contrast, the labeling of conventional protein disulfide bonds does not depend on the choice of alkylating agent. B) Representative gel visualizing fluorescence (upper left panel) or Coomassie staining (lower left panel). Quantification of overall fluorescence in each lane (right panel). Fluorescence intensity was normalized to the Coomassie stain intensity, n=3. Error bars represent SD. ** P≤0.01, based on a two‐tailed unpaired t‐test.
Conclusion
Having established an in vitro protein persulfidation model system, we noted marked differences in the ability of different alkylating agents to preserve persulfides in their alkylated form (P‐S‐S‐Alk). This finding is broadly in line with a previous study aiming to quantitate alkylated persulfides in cellular lysates and obtaining different results with different alkylating agents. [9] Assessing several alkylating agents, in the presence of nucleophiles, we made the following observations: i) MBB recovered almost 100 % of the protein in the persulfidated state; ii) NEM did not recover any persulfidated protein; iii) IAM was almost completely inefficient; iv) IAA was of intermediate efficiency; and v) various IAM derivatives with N‐substituents were more efficient than IAM, the bulkiness of the substituent correlating with increased recovery, N‐t‐butyl‐IAM being almost as efficient as MBB. These findings raised the question of how these pronounced differences come about.
Investigating the stability of alkylated persulfides at different pH, we did not find evidence for hydrolysis to play a role, nor did we find evidence supporting the notion that unreacted alkylating agents can destroy already alkylated persulfides. Instead, we found evidence in favor of the notion that alkylated persulfides can convert into thioethers (i.e., alkylated thiols) by losing one sulfur atom to ambient nucleophiles. Apparently, the tendency of an alkylated persulfide to lose sulfur depends not only on the presence of ambient nucleophiles, but also on the exact chemical structure of the alkylating agent. Indeed, we identified two structural factors that promote sulfur loss from alkylated persulfides, namely i) the presence of a β‐carbonyl, and ii) the absence of a bulky substituent in γ‐position.
We reasoned that the selective loss of one sulfur atom from the disulfide bond between the protein and the alkylating agent must depend on the tautomeric rearrangement of the disulfide bond into a thiosulfoxide, as only the thiosulfoxide would be able to donate a sulfur atom and leave behind a single sulfur atom, bound as a thioether. This raises the question of how disulfide‐thiosulfoxide tautomerization occurs mechanistically. It is known that β‐alkenyl (allylic) disulfides undergo 2,3‐sigmatropic rearrangement leading directly to thiosulfoxides. [25] While one can theorize a similar type of 2,3‐rearrangement reaction with a β‐carbonyl disulfide, we consider it unlikely. Even in the event that it did occur, the resulting intermediate would need to undergo an additional 1,3‐shift, either before, or after desulfurization, to yield the thioether product (Figure S8). A second mechanistic possibility is migration of the S−C bond to the “internal” sulfur with concomitant expansion of that sulfur's coordination number to 4 (Figure 8). This is reminiscent of one reaction pathway that coordinatively unsaturated carbenes can follow to stabilize themselves. [27] However, the actual mechanism of tautomerization as relevant to alkylated persulfides remains an open question and is beyond the scope of this study. In any case, and irrespective of the actual mechanism of tautomerization, it can be assumed that the chemical nature of the alkylating agent either influences the disulfide‐thiosulfoxide tautomer equilibrium, or the ability of nucleophiles to remove the sulfur from the thiosulfoxide, or both processes.
Figure 8.

Possible mechanism of alkylating agent‐driven sulfur loss. (i) The thiosulfoxide may form by migration of the S‐C bond to the ‘internal’ sulfur with concomitant expansion of that sulfur's coordination number to 4. (ii) Shared sulfane sulfur–carbonyl oxygen proton binding may support the formation and stabilization of the thiosulfoxide. (iii) The cationic intermediate is expected to lose sulfur to nucleophiles more favorably than the corresponding non‐protonated species. The figure also points out the four factors promoting sulfur loss as identified in this study, i.e., two factors intrinsic to the alkylating agent (a carbonyl in β‐position and small substituents in γ‐position) and two extrinsic factors (proton and nucleophile availability).
Two of our findings support the notion that the presence of a β‐carbonyl group promotes sulfur loss: i) Only the one alkylating agent without a β‐carbonyl, MBB, conserved persulfides to full extent, and ii) sulfur loss from small molecule analogues depended on the presence of the β‐carbonyl (Figure 6). Furthermore, we observed that alkylated persulfides are stabilized against sulfur loss by increased pH (Figure 3A) and bulky γ‐substituents (Figure 4E). Consistent with these observations is an acid‐dependent mechanism, where shared sulfane sulfur‐carbonyl oxygen proton binding may support the formation and/or stabilization of the thiosulfoxide (Figure 8). Such a cationic intermediate should also lose sulfur to nucleophiles more favorably than the corresponding non‐protonated species, which is likely to be more prevalent in the absence of a β‐carbonyl (Figure 8). Bulky γ‐substituents can be expected to hinder nucleophilic attack on the thiosulfoxide for steric reasons; however, we cannot exclude that they may also perturb the disulfide/thiosulfoxide tautomerization in the first place. Finally, the higher stability of IAA‐alkylated versus IAM‐alkylated persulfides (Figure S4) may be explained by the negative charge at the carboxyl group. The carboxyl group prevents formation of an intermediate with net positive charge and may create electrostatic repulsion with incoming nucleophiles.
The fact that our findings have been obtained for a specific thiol on a specific protein raises the question of generality. How representative are our findings beyond the Trx1CS model protein? It is conceivable that the specific protein environment of a cysteine can influence the stability of the corresponding alkylated persulfide, so some variability may be expected. Nevertheless, the pronounced difference between MBB and NEM alkylation was also observed for the free amino acid (Figure S7A) and for the collective set of proteins in a cell lysate (Figure 7B), suggesting that the stability of alkylated protein persulfides is mostly determined by the nature of the alkylating agent, and to a lesser extent by its protein microenvironment.
In any case, the type and concentration of ambient nucleophiles appears to be a key factor in determining the rate of sulfur loss from intrinsically labile alkylated persulfides. Trapping of persulfides with IAM was improved when the persulfidated protein was washed before alkylation (Figure S5A). Also, some P‐S‐S‐CAM could be detected when MPST/3MP was used for persulfidation (Figure 1C), but not when Na2S2 was used (Figure 1D). This may be explained by differences in nucleophile properties. Disulfide and sulfide ions are very efficient sulfur acceptors, while 3MP and pyruvate are expected to be less efficient in attacking thiosulfoxides.
A paucity of ambient nucleophiles may explain why trapping of persulfides with IAM (or NEM) is possible under certain conditions. Small molecule persulfides have previously been trapped as IAM adducts.[ 28 , 29 ] These studies performed alkylation in organic solvents, meaning that abundant external nucleophiles were not present to promote desulfurization. Gpx3 and papain persulfides have been trapped with IAM in aqueous solution. [30] In this case, alkylation was performed after gel filtration, again suggesting the absence of abundant external nucleophiles.
In the context of intact cells or cell lysates, rapid alkylation of protein persulfides cannot be performed in the absence of nucleophiles. Indeed, thiophilic nucleophiles (incl. GSH and other thiols) are omnipresent within cells, thus the formation and/or stabilization of thiosulfoxides will inevitably lead to sulfur loss. Hence, the choice of alkylating agent is critical. The most commonly used thiol alkylating agents, IAM and NEM, appear to be unfit for the purpose of persulfide detection in biological settings, or in any other nucleophile‐rich environment. Most other alkylating agents (including HPE‐IAM) are expected to lead to underestimates of persulfide levels. We recommend the use of MBB or N‐t‐butyl‐IAM in future studies.
Conflict of interest
The authors declare no conflict of interest.
1.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
We gratefully acknowledge technical support by Nicole Lübbehusen and Thomas Ruppert (ZMBH core facility for mass spectrometry and proteomics). We also thank Virginie Crétet Malak for technical assistance and Dr. Brandán Pedre for providing recombinant MPST. We acknowledge funding by the German Research Council (DFG Priority Program SPP2306 and Collaborative Research Center TRR186, to T.P.D.) and by the European Commission (742039, to T.P.D.). Open Access funding enabled and organized by Projekt DEAL.
D. Schilling, U. Barayeu, R. R. Steimbach, D. Talwar, A. K. Miller, T. P. Dick, Angew. Chem. Int. Ed. 2022, 61, e202203684; Angew. Chem. 2022, 134, e202203684.
Data Availability Statement
The data that support the findings of this study are available in the Supporting Information of this article.
References
- 1. Zivanovic J., Kouroussis E., Kohl J. B., Adhikari B., Bursac B., Schott-Roux S., Petrovic D., Lj Miljkovic J., Thomas-Lopez D., Jung Y., Miler M., Mitchell S., Milosevic V., Gomes J. E., Benhar M., Gonzalez-Zorn B., Ivanovic-Burmazovic I., Torregrossa R., Mitchell J. R., Whiteman M., Schwarz G., Snyder S. H., Paul B. D., Carroll K. S., Filipovic M. R., Cell Metab. 2020, 31, 207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Paul B. D., Snyder S. H., Nat. Rev. Mol. Cell Biol. 2012, 13, 499–507. [DOI] [PubMed] [Google Scholar]
- 3. Filipovic M. R., Zivanovic J., Alvarez B., Banerjee R., Chem. Rev. 2018, 118, 1253–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yang G., Zhao K., Ju Y., Mani S., Cao Q., Puukila S., Khaper N., Wu L., Wang R., Antioxid. Redox Signaling 2013, 18, 1906–1919. [DOI] [PubMed] [Google Scholar]
- 5. Mustafa A. K., Sikka G., Gazi S. K., Steppan J., Jung S. M., Bhunia A. K., Barodka V. M., Gazi F. K., Barrow R. K., Wang R., Amzel L. M., Berkowitz D. E., Snyder S. H., Circ. Res. 2011, 109, 1259–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sen N., Paul B. D., Gadalla M. M., Mustafa A. K., Sen T., Xu R., Kim S., Snyder S. H., Mol. Cell 2012, 45, 13–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bestetti S., Medraño-Fernandez I., Galli M., Ghitti M., Bienert G. P., Musco G., Orsi A., Rubartelli A., Sitia R., Sci. Adv. 2022, 4, 10.1126/sciadv.aar5770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Longen S., Richter F., Köhler Y., Wittig I., Beck K.-F., Pfeilschifter J., Sci. Rep. 2016, 6, 29808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Li X., Day N. J., Feng S., Gaffrey M. J., Lin T.-D., Paurus V. L., Monroe M. E., Moore R. J., Yang B., Xian M., Qian W.-J., Redox Biol. 2021, 46, 102111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fu L., Liu K., He J., Tian C., Yu X., Yang J., Antioxid. Redox Signaling 2020, 33, 1061–1076. [DOI] [PubMed] [Google Scholar]
- 11. Bogdándi V., Ida T., Sutton T. R., Bianco C., Ditrói T., Koster G., Henthorn H. A., Minnion M., Toscano J. P., van der Vliet A., Pluth M. D., Feelisch M., Fukuto J. M., Akaike T., Nagy P., Br. J. Pharmacol. 2019, 176, 646–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Schwertassek U., Balmer Y., Gutscher M., Weingarten L., Preuss M., Engelhard J., Winkler M., Dick T. P., EMBO J. 2007, 26, 3086–3097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Toohey J. I., Cooper A. J. L., Molecules 2014, 19, 12789–12813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Benchoam D., Semelak J. A., Cuevasanta E., Mastrogiovanni M., Grassano J. S., Ferrer-Sueta G., Zeida A., Trujillo M., Möller M. N., Estrin D. A., Alvarez B., J. Biol. Chem. 2020, 295, 15466–15481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Heppner D. E., Hristova M., Ida T., Mijuskovic A., Dustin C. M., Bogdándi V., Fukuto J. M., Dick T. P., Nagy P., Li J., Akaike T., van der Vliet A., Redox Biol. 2018, 14, 379–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Millikin R., Bianco C. L., White C., Saund S. S., Henriquez S., Sosa V., Akaike T., Kumagai Y., Soeda S., Toscano J. P., Lin J., Fukuto J. M., Free Radical Biol. Med. 2016, 97, 136–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ono K., Akaike T., Sawa T., Kumagai Y., Wink D. A., Tantillo D. J., Hobbs A. J., Nagy P., Xian M., Lin J., Fukuto J. M., Free Radical Biol. Med. 2014, 77, 82–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pedre B., Dick T. P., Biol. Chem. 2021, 402, 223–237. [DOI] [PubMed] [Google Scholar]
- 19. Lin C., Mihal K. A., Krueger R. J., Anal. Biochem. 1989, 179, 389–395. [DOI] [PubMed] [Google Scholar]
- 20. Bernhard S. A., MacQuarrie R. A., Biochemistry 1971, 10, 2456–2466. [DOI] [PubMed] [Google Scholar]
- 21. Hamid H. A., Tanaka A., Ida T., Nishimura A., Matsunaga T., Fujii S., Morita M., Sawa T., Fukuto J. M., Nagy P., Tsutsumi R., Motohashi H., Ihara H., Akaike T., Redox Biol. 2019, 21, 101096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Moore C. G., Trego B. R., Tetrahedron 1962, 18, 205–218. [Google Scholar]
- 23. Baechler R. D., Hummel J. P., Mislow Kurt, J. Am. Chem. Soc. 1973, 95, 4442–4444. [Google Scholar]
- 24. Baechler R. D., Daley S. K., Daly B., McGlynn K., Tetrahedron Lett. 1978, 19, 105–108. [Google Scholar]
- 25. Hoefle G., Baldwin J. E., J. Am. Chem. Soc. 1971, 93, 6307–6308. [Google Scholar]
- 26. Kutney G. W., Turnbull K., Chem. Rev. 1982, 82, 333–357. [Google Scholar]
- 27. Anslyn E. V., Dougherty D. A., Modern Physical Organic Chemistry, University Science Books, Sausalito, 2006. [Google Scholar]
- 28. Park C.-M., Johnson B. A., Duan J., Park J.-J., Day J. J., Gang D., Qian W.-J., Xian M., Org. Lett. 2016, 18, 904–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kang J., Xu S., Radford M. N., Zhang W., Kelly S. S., Day J. J., Xian M., Angew. Chem. Int. Ed. 2018, 57, 5893–5897; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 5995–5999. [Google Scholar]
- 30. Pan J., Carroll K. S., ACS Chem. Biol. 2013, 8, 1110–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Data Availability Statement
The data that support the findings of this study are available in the Supporting Information of this article.