

Abstract
The open‐shell iron pentacarbonyl cation [Fe(CO)5].+ was isolated by deelectronation, i.e., the single‐electron oxidation of the parent neutral Fe(CO)5 using [phenazineF].+ as the [Al(ORF)4]− and [F‐{Al(ORF)3}2]− salt (RF=C(CF3)3; phenazineF=perfluoro‐5,10‐bis(perfluorophenyl)‐5,10‐dihydrophenazine). [Fe(CO)5].+[Al(ORF)4]− was fully characterized (scXRD analysis, IR, NMR, EPR, 57Fe spectroscopy, CV and SQUID magnetization study) and, apart from being a compound of fundamental interest, may serve as a precursor for low‐valent iron coordination chemistry.
Keywords: Carbonyl Ligands, Iron, Radical Ions, Weakly Coordinating Anions
The open‐shell iron pentacarbonyl cation [Fe(CO)5].+ was synthesized and isolated as a stable salt for the first time. The strategy involved the use of a strong deelectronator, i.e., single‐electron oxidant, in combination with weakly coordinating anions (WCAs). The iron carbonyl complex is a novel example of low‐valent iron and has thus been fully characterized. Isolation was achieved using standard laboratory conditions in combination with fluorinated benzenes as solvents.
Transition metal carbonyls (TMCs) are textbook compounds of fundamental interest. Using TMCs, every chemistry student learns the basic concepts of organometallic chemistry, such as the 18‐valence‐electron (VE) rule, Hoffmann's isolobal principle, redox states of transition metals, and of course the ‐donor and ‐backbonding concept. Discovered in 1889, the famous liquid Ni(CO)4 was the first example of a homoleptic carbonyl compound. [1] Almost equally long known, and first described by Mond as well, is the iron pentacarbonyl Fe(CO)5. [2] TMC anions (e.g. [Fe(CO)4]2−) have been reported since the 1930s.[ 3 , 4 ] Yet, it took more than 70 years to discover the first homoleptic TMC cation (TMCC) [Mn(CO)6]+. [5] Since the ‐backbonding component is strongly reduced in TMCCs, they are rather electrophilic and the majority of past reports have generated TMCCs by using super acidic media,[ 6 , 7 , 8 , 9 , 10 , 11 ] such as SbF5, HF‐SbF5 or HSO3F, and by employing weakly coordinating anions (WCAs) to stabilize these species.[ 12 , 13 , 14 , 15 , 16 ] Super acids only allowed access to diamagnetic, mainly 18 VE TMCCs. By using the deelectronators[ 17 , 18 ] Ag+, [NO]+ and Ag+/0.5 X2 (X=I), in combination with the aluminate WCAs [Al(ORF)4]− and [F‐{Al(ORF)3}2]− (RF=C(CF3)3), our group succeeded to generate salts of the first open‐shell mononuclear TMCCs [M(CO)6].+ (M=Cr, Mo, W) and, just recently reported the first nickel carbonyl cation [Ni(CO)4].+.[ 19 , 20 , 21 ] Even TMCCs exceeding the coordination number (CN) six, i.e., [M(CO)7]+ (M=Nb, Ta), could be stabilized in condensed phase by these WCAs. [22]
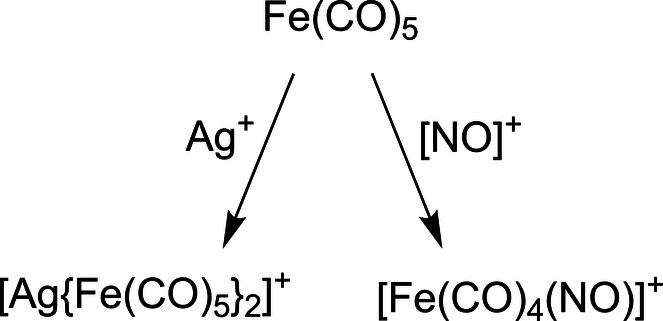
Neutral, mononuclear, binary iron carbonyl Fe(CO)5 is an air‐ and light‐sensitive liquid at room temperature. [2] Irradiation with ultraviolet light results in the formation of Fe2(CO)9. [23] Reduction of neutral Fe(CO)5 with sodium amalgam leads to Na2Fe(CO)4. [24] Oxidation in super acidic media under CO pressure generates the dicationic [Fe(CO)6]2+ complex as the [Sb2F11]− salt. [10] These mononuclear, binary iron carbonyl compounds have been known for decades and all obey the 18 VE rule. On the other hand, the [Fe(CO)5].+ cation has an open shell and was exclusively studied in the gas phase by mass spectrometry, with even gas phase reactivity being observed.[ 25 , 26 , 27 , 28 , 29 , 30 , 31 , 32 , 33 , 34 , 35 , 36 , 37 , 38 , 39 , 40 ] Only by co‐doping of tiny amounts of Fe(CO)5 to other TMCs and γ‐irradiation at 77 K, the EPR signature of the [Fe(CO)5].+ cation and its krypton‐complex could be studied.[ 41 , 42 ] To the best of our knowledge, the isolation of a stable salt of this cation has been unknown in the condensed phase. Previous efforts from our group to isolate [Fe(CO)5].+ have resulted in the synthesis of novel transition metal complexes: Attempted deelectronation of Fe(CO)5 by Ag+[Al(ORF)4]− led to coordination and formation of the silver complex [Ag{Fe(CO)5}2]+. [43] The analogous reaction with [NO]+[F‐{Al(ORF)3}2]− led to the substitution of CO and the isolation of the first heteroleptic iron carbonyl‐nitrosyl cation complexes [Eq. 1]. [44]
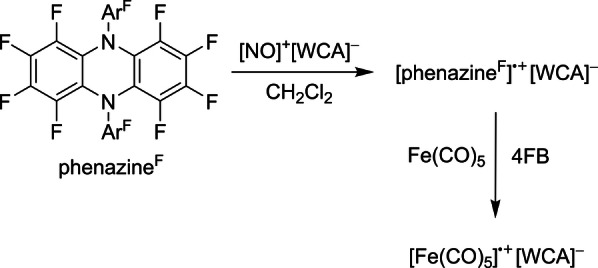
Thus, in order to truly deelectronate Fe(CO)5 to the respective 17 VE species, a strong and innocent deelectronator was needed. The recently described salt [phenazineF].+ [Al(ORF)4]−, with a high formal potential E°′ of 1.29 V vs. Fc+/Fc in ortho‐difluorobenzene (oDFB), seemed to be the ideal candidate. [45] Hence, when treating a stock solution of Fe(CO)5 in oDFB with one equivalent of [phenazineF].+[Al(ORF)4]− at ambient temperature, an immediate color change from the characteristic purple of [phenazineF].+ to dark green was observed. Green and red crystals were obtained by layering the reaction mixture with n‐pentane; their structures were determined by scXRD analysis [46] (cf. ESI Figure S8/S9). The deelectronation of FeCO5 resulted in the green solvent adduct [Fe(CO)5 oDFB].+ with a square pyramidal structure almost in C 4V symmetry. The fluorine atoms of an incorporated oDFB solvent molecule form secondary bonds to the carbon atoms of the carbonyl moieties in between 2.639(2) and 2.931(8) Å. The observed red minor component is a novel, bromide‐bridged, dinuclear iron carbonyl cation [Fe2(CO)8Br]+[F‐{Al(ORF)3}2]−, including the fluoride‐bridged anion as a typical [47] decomposition product of [Al(ORF)4]−. The unwanted bromine impurity, which is a frequently observed problem, stems from the synthesis of [phenazineF].+[Al(ORF)4]− through the deelectronation of neutral phenazineF using the synergistic Ag+/0.5 Br2 system. [45] However, the formal potential of [NO]+ vs. Fc+/Fc was measured to be higher than that of phenazineF, if using 1,2,3,4‐tetrafluorobenzene (4FB) or CH2Cl2 as a solvent. [48] Accordingly, phenazineF can be deelectronated with [NO]+[Al(ORF)4]− in 4FB, and also in CH2Cl2, eliminating the risk of the AgBr impurity [Eq. 2]. Additionally, it was possible to generate [phenazineF].+[F‐{Al(ORF)3}2]− by using the [NO]+ salt of the even better WCA [F‐{Al(ORF)3}2]− (cf. Supporting Information Figure S4 for the scXRD structure [46] ). PhenazineF deelectronation in CH2Cl2, as shown in Equation (2), has become our standard method to isolate pure and solvent‐free salts of [phenazineF].+[WCA]−, and was used from this point on (ArF=C6F5).
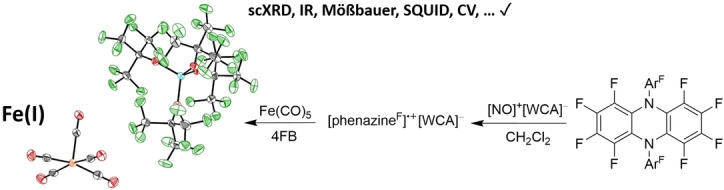
The solvent oDFB was substituted with the less basic 4FB to isolate an undistorted [Fe(CO)5].+ salt. The deelectronation was carried out analogously to the synthesis of [Fe(CO)5 oDFB].+. Neutral Fe(CO)5 reacted with [phenazineF].+[Al(ORF)4]− instantaneously at ambient temperature, resulting in a color change to dark green. Green crystals of [Fe(CO)5].+[Al(ORF)4]− (1) suitable for scXRD analysis [46] were grown by layering the 4FB solution with n‐pentane in 70 % yield (Figure 1a). Analogous to [Fe(CO)5 oDFB].+, the structure is square pyramidal, exhibiting nearly undistorted C 4v symmetry (τ 5=0.05). [49] Hence, [Fe(CO)5].+ is isostructural to the [Mn(CO)5]⋅ radical.[ 50 , 51 ] The equatorial Fe−C bond lengths average to 1.872(5) Å and the axial bond length is 1.910(4) Å, analogous to [Fe(CO)5 oDFB].+. The individual equatorial Fe−C bond lengths are within of the average and differ from the axial Fe−C bond length. The average C−O bond length is 1.120(6) Å, analogous to [Fe(CO)5 oDFB].+. As expected, both [Fe(CO)5 oDFB].+ and [Fe(CO)5].+ exhibit longer Fe−C and shorter C−O bond lengths as the neutral Fe(CO)5. [52] When compared to [Fe(CO)3(PPh3)2][PF6], which exhibits a distorted C 4v symmetry, the [Fe(CO)5].+ cation has longer Fe−C and similar C−O bond lengths. [53] By using [phenazineF].+[F‐{Al(ORF)3}2]−, it was also possible to generate [Fe(CO)5].+[F‐{Al(ORF)3}2]− (2, cf. ESI Figure S21 for the scXRD structure). In 2, [Fe(CO)5].+ exhibits an average equatorial Fe−C bond length of 1.872(4) Å and an axial bond length of 1.903(4) Å.
Figure 1.

a) Molecular structure of [Fe(CO)5].+[Al(ORF)4]− (1) (P212121, R 1=5.4 %, wR 2=9.0 %); thermal ellipsoids set at 50 % probability. b) Experimental ZnSe‐ATR FT‐IR spectrum of solid 1. c) Experimental and simulated Q‐band (34 GHz), continuous‐wave EPR spectra of solid 1 at 100 K, a potential impurity is marked by an asterisk. d) The carbonyl region of experimental ZnSe‐ATR FT‐IR spectrum of solid 1 (black), 1 in 4FB (gray), solid 2 (green) and calculated (@BP86‐(D3BJ)/def2‐TZVPP) 13C IR spectrum of the superposition of all possible isotopomers weighed by their natural abundance (blue). Double or more 13C substitution were neglected.
1 was further characterized by 19F‐, 27Al‐, 13C NMR (only anion peaks visible) and pXRD revealing 1 to be the only crystalline phase (cf. ESI Figure S16, S17). Continuous‐wave (cw) EPR measurements of solid 1 at the X‐band (cf. ESI Figure S23) and at the Q‐band (Figure 1c) found a rhombic g‐tensor of g=(2.011, 2.059, 2.080) after a global simulation of the X‐ and Q‐band data recorded at 100 K. The obtained g‐values are in good agreement with the values calculated for the experimental crystal structure by DFT at the B3LYP/EPR‐II(CP(PPP) for Fe) level of theory g=(2.000, 2.060, 2.063). Additionally, an EPR spectrum of 1 in oDFB was measured (cf. ESI Figure S24). The simulation of the observed spectrum indicated the presence of two components. The first of which is similar to that of solid uncoordinated [Fe(CO)5].+, while the second is shifted towards lower g‐values and can be assigned to the solvent adduct [Fe(CO)5 oDFB].+(cf. ESI Figure S25).
The solid‐state ATR‐IR spectrum of 1 shows characteristic CO bands at 2128, 2113 and 2082 cm−1 (Figure 1b, d/Table 1; ATR=attenuated total reflection). The solid‐state spectrum of the oDFB solvent adduct [Fe(CO)5 oDFB].+‐[Al(ORF)4]− has even more bands: Compared to solvent‐free 1, the stretch at 2113 cm−1 in pure 1 splits further into 2110 and 2116 cm−1 in the adduct (Table 1, cf. ESI Figure S6). By contrast, the spectrum of solid 2 − with the less coordinating WCA— is simpler, showing only one strong band at 2116 cm−1 and a weaker one at 2084 cm−1 (Figure 1d). Solution ATR‐IR spectra of 1 in oDFB and 4FB (Figure 1d, Table 1) are comparable to that of solid 2 and show only one strong CO band at 2112 / 2115 cm−1 and weak ones at 2090 / 2084 cm−1, respectively. Gas phase calculations of the undistorted C 4v‐[Fe(CO)5].+ at the BP86‐(D3BJ)/def2‐TZVPP level of theory suggest the presence of two sets of degenerate e‐stretches, both at 2102 cm−1, in addition to an a 1‐mode at 2106 cm−1 that could superimpose to one strong band in the experimental spectrum. Hence, the spectrum of solid 2 as well as the solution spectra of 1 agree with the expectation, given that the lower frequency band at 2082…2090 cm−1 is assigned to a mixture of the axial and equatorial 13CO‐isotopomers of the cation. Support comes from BP86‐(D3BJ)/def2‐TZVPP calculations in which all possible 13C isotopomers were weighed by their natural abundance (Figure 1d, Table 1, cf. related work in [54] ). The appearance of an additional band at 2128 cm−1 in the carbonyl region of 1 is probably caused by solid‐state interionic interactions with the slightly more coordinating anion [Al(ORF)4]− that may easily distort the square pyramidal cation. Note, calculations at the B3LYP‐(D3BJ)/def2‐TZVPP level of theory revealed a low‐lying C 2v transition state with an activation barrier of H°≠=20.05 and G°≠=19.62 kJ mol−1. Along this path, the distortion of the symmetry and lifting of degeneracies should be facile. To substantiate this claim, we calculated the vibrational CO frequencies of [Fe(CO)5].+ in the presence of negative point charges q with a charge of −0.3e placed at the positions of anion F‐atoms in the solid state that are closer than the sum of the van der Waals radii (see ESI, Table S24). This mimics the influence of the partial charge envelope typically residing on the fluorine atoms of −CF3 groups of the aluminate anion (cf. our related work in [55] ). As expected, the negative point charges q break the symmetry and split the degenerate CO bands and cause the appearance of additional bands (Table 1, IR calc. C 4v+q). In addition, we note that the main CO stretch of [Fe(CO)5].+ in oDFB solution at 2112 cm−1 is slightly red‐shifted compared to 4FB (2115 cm−1), possibly due to the more electron rich solvent.
Table 1.
Experimental and calculated spectroscopic and structural data of [Fe(CO)5 oDFB].+[Al(ORF)4]−, 1 and 2. All calculations were done at the BP86‐(D3BJ)/def2‐TZVPP level of theory.
|
|
IR[a] solid (CO) [cm−1] |
IR[a] solution (CO) [cm−1] |
IR calc. (C 4v) (CO) [cm−1] |
IR[b] calc. (13CO) [cm−1] |
IR calc. (C 4v+q)[f] (CO) [cm−1] |
avg. d(M−C) Å |
avg. d(CO) Å |
|---|---|---|---|---|---|---|---|
|
[Fe(CO)5 oDFB].+ [Al(ORF)4]− |
2083 (vw)[c] 2110 (w) 2116 (w) 2128 (vw) |
2090 (vw)[c,d] 2112 (s)[d] |
– |
– |
– |
ax.: 1.907(3) eq.: 1.874(5) |
ax.: 1.105(3) eq.: 1.118(6) |
|
[Fe(CO)5].+ [Al(ORF)4]− (1) |
2082 (vw)[c] 2113 (s) 2128 (w) |
2084 (vw)[c,e] 2115 (s)[e] |
2102 (100), , E 2102 (100), , E 2106 (48), , A1 |
2071 (2)[b] 2102 (100)[b] |
2063 (100) 2075 (75) 2099 (22) 2141 (26) 2179 (6) |
ax.: 1.910(4) eq.: 1.872(5) |
ax.: 1.115(5) eq.: 1.126(6) |
|
[Fe(CO)5].+ [F‐{Al(ORF)3}2]− (2) |
2084 (vw)[c] 2116 (s) |
– |
– |
– |
– |
ax.: 1.903(4) eq.: 1.872(4) |
ax.: 1.118(4) eq.: 1.118(5) |
[a] v: very, s: strong, w: weak. [b] Superposition of the CO‐stretches calculated for a C 4v ground state including the relative contributions of the 13C natural abundance isotope. Individual contributions: 13CO axial C 4v: A 1 2061 (43), E 2102 (100), B 1 2120 (0), A 1 2168 (3) and 13CO equat. 2065 (73), 2102 (100), 2106 (50), 2115 (19), 2164 (3). [c] Probably the 13CO stretch. [d] Measured in oDFB. [e] Measured in 4FB. [f] Calculated CO bands in the presence of −0.3e point charges obtained from scXRD.
1 was further investigated by zero‐field 57Fe Mössbauer spectroscopy, exhibiting an isomer shift, , of 0.17 mms−1 and a quadrupole splitting, Q, of 0.53 mms−1 (Figure 2). Those values are compared to the neutral, pentacoordinated Fe(CO)5, the tetracoordinated, dianionic [Fe(CO)4]2−, with its formal −2 oxidation state, and the hexacoordinated, dicationic FeII complex [Fe(CO)6]2+ (Table 2, cf. ESI Figure S28/S29 for spectra). Typically, in 57Fe Mössbauer spectroscopy, higher formal oxidation states are expected to lead to lower, more negative isomer shifts. Yet, except for [Fe(CO)6]2+, the opposite trend can be observed with a lowering of the isomer shift from the formal FeI to Fe‐II. In order to explain this unusual progression, the Fe−C and C−O bond lengths have been considered. The Fe−C bond length decreases together with the isomer shift and the C−O bond length increases. Hence, through enhanced π‐backbonding, more electron density is moved towards the Fe−C bond and the CO‐ligand for the neutral or reduced species. Thus, the physical oxidation state of iron, reflecting its reduced electron density at the core, becomes higher, leading to lower isomer shifts for Fe(CO)5 and [Fe(CO)4]2−. Although such a trend is surprising, similar observations have been reported.[ 58 , 59 ]
Figure 2.

Zero‐field 57Fe Mössbauer spectrum of a solid sample of 1 recorded at 77 K (left). The gray circles are experimental data and the solid black line represents the numerical fit. Cyclic voltammogram of 1 (10 mM) in 4FB with [NBu4][Al(ORF)4] as conducting salt (right). Measurements were done at sweep rates of 20, 50 and 100 mV s−1. The half‐wave potential was found to be independent of the sweep rate.
Table 2.
Comparison of isomer shift and quadrupole splitting with Fe−C/C−O bond lengths of homoleptic, mononuclear iron carbonyls.
|
|
[Fe(CO)4]2− |
Fe(CO)5 |
[Fe(CO)5].+ |
[Fe(CO)6]2+ |
|---|---|---|---|---|
|
[mms−1] |
−0.16(1) |
−0.08(1) |
0.17(1) |
−0.001(6)[c] |
|
Q [mms−1] |
0.19(1) |
2.55(1) |
0.53(1) |
– |
|
avg. d(Fe−C) [Å] |
1.747(4)[a] |
1.815(5)[b] |
1.882(5) |
1.911(5)[d] |
|
avg. d(C−O) [Å] |
1.175(5)[a] |
1.142(9)[b] |
1.124(6) |
1.104(5)[d] |
Magnetic measurements of two independently synthesized solid samples of 1 were performed by variable‐temperature SQUID experiments, revealing a nearly temperature‐independent magnetic moment of μ eff=1.83/1.84 μB (cf. ESI Figure S27) between 25 and 300 K. These values agree well with the EPR measurement of solid 1, underlining the doublet ground state. Additionally, the magnetic moment μ eff=2.13 μB (2.12 μB without diamagnetic contributions) of 1 in 4FB was measured by Evans’ NMR method (cf. ESI). The cyclic voltammogram of 1 in 4FB revealed a half‐wave potential of 0.86 V vs. Fc+/Fc and is independent of the sweep rate (Figure 2). This measured value is in agreement with a previously reported deelectronation potential. [60] The solvent window of 4FB only showed the 0/+1 transition from neutral Fe(CO)5 to cationic [Fe(CO)5].+ (cf. ESI Figure S18).
In conclusion, we report the isolation and characterization of [Fe(CO)5].+ as the [Al(ORF)4]− and [F‐{Al(ORF)3}2]− salt. These compounds can be isolated using standard laboratory equipment and are stable at room temperature. In addition, a new and simple, side‐product free synthetic route to the deelectronator [phenazineF].+ is presented and its compatibility with the even less coordinating anion [F‐{Al(ORF)3}2]− is shown. Preliminary results suggest that the carbonyl ligands in [Fe(CO)5].+ can be exchanged. This is not surprising, since open‐shell complexes exhibit more rapid ligand exchange, and substitution chemistry has already been studied in 17 VE Fe species.[ 61 , 62 , 63 , 64 ] Thus, the [Fe(CO)5].+ cation marks a conveniently accessible precursor for further coordination chemistry of low‐valent iron.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Acknowledgements
We would like to thank Dr. Thilo Ludwig for measuring powder‐XRD, Dr. Harald Scherer and Fadime Bitgül for NMR measurements, Dr. Burkhard Butschke and Philipp Dabringhaus for help with scXRD measurements. We thank Prof. Dr. Peter Roesky for supply of Fe(CO)5 and [Fe(CO)4]2−. This work was supported by the German Research Foundation (DFG)—Project number 417643975 (S.R.) and KR2046/30‐2 and 37‐1 (I.K). Additionally, we acknowledge the support by the state of Baden‐Württemberg through bwHPC, the German Research Foundation (DFG) through grant no INST 40/467‐1 and 575‐1 FUGG (JUSTUS1 and 2 cluster) and the FAU Erlangen‐Nürnberg for generous financial support. Open Access funding enabled and organized by Projekt DEAL.
J. M. Rall, M. Schorpp, M. Keilwerth, M. Mayländer, C. Friedmann, M. Daub, S. Richert, K. Meyer, I. Krossing, Angew. Chem. Int. Ed. 2022, 61, e202204080; Angew. Chem. 2022, 134, e202204080.
References
- 1. Mond L., Langer C., Quincke F., J. Chem. Soc. 1890, 57, 749–753. [Google Scholar]
- 2. Mond L., Langer C., J. Chem. Soc. Dalton Trans. 1891, 59, 1090–1093. [Google Scholar]
- 3. Ellis J. E., Organometallics 2003, 22, 3322–3338. [Google Scholar]
- 4. Hieber W., Leutert F., Ber. Dtsch. Chem. Ges. 1931, 64, 2832–2839. [Google Scholar]
- 5. Fischer E. F., Fichtel K., Öfele K., Chem. Ber. 1962, 95, 249–252. [Google Scholar]
- 6. Willner H., Aubke F., Inorg. Chem. 1990, 29, 2195–2200. [Google Scholar]
- 7. Willner H., Schaebs J., Hwang G., Mistry F., Jones R., Trotter J., Aubke F., J. Am. Chem. Soc. 1992, 114, 8972–8980. [Google Scholar]
- 8. Hwang G., Wang C., Willner H., Bodenbinder M., Aubke F., Can. J. Chem. 1993, 71, 1532–1536. [Google Scholar]
- 9. Bodenbinder M., Balzer-Jöllenbeck G., Willner H., Batchelor R. J., Einstein F. W. B., Wang C., Aubke F., Inorg. Chem. 1996, 35, 82–92. [DOI] [PubMed] [Google Scholar]
- 10. Bley B., Willner H., Aubke F., Inorg. Chem. 1997, 36, 158–160. [Google Scholar]
- 11. Bach C., Willner H., Wang C., Rettig S. J., Trotter J., Aubke F., Angew. Chem. Int. Ed. Engl. 1996, 35, 1974–1976; [Google Scholar]; Angew. Chem. 1996, 108, 2104–2106. [Google Scholar]
- 12. Hurlburt P. K., Anderson O. P., Strauss S. H., J. Am. Chem. Soc. 1991, 113, 6277–6278. [Google Scholar]
- 13. Hurlburt P. K., Rack J. J., Dec S. F., Anderson O. P., Strauss S. H., Inorg. Chem. 1993, 32, 373–374. [Google Scholar]
- 14. Hurlburt P. K., Rack J. J., Luck J. S., Dec S. F., Webb J. D., Anderson O. P., Strauss S. H., J. Am. Chem. Soc. 1994, 116, 10003–10014. [Google Scholar]
- 15. Rack J. J., Webb J. D., Strauss S. H., Inorg. Chem. 1996, 35, 277–278. [DOI] [PubMed] [Google Scholar]
- 16. Ivanova S. M., Ivanov S. V., Miller S. M., Anderson O. P., Solntsev K. A., Strauss S. H., Inorg. Chem. 1999, 38, 3756–3757. [Google Scholar]
- 17.
- 17a.We use the elementary steps electronation and deelectronation in their strict sense, i.e., addition or removal of e−. Thus, a classically termed “oxidant” is addressed as a “deelectronator” and a “reductant” as an “electronator”, if only a single electron transfer as the elementary step takes place. This particle-based terminology is related to the acid-base picture, where the terms deprotonation and protonation describe the transfer of a proton between two partners, i.e., deelectronation is the electron-based equivalent to a deprotonation. This terminology has been discussed in ref. [17b] and ref. [18] from our work on the protoelectric potential map and is transferred to describe these types of reactions more accurately;
- 17b. Radtke V., Himmel D., Pütz K., Goll S. K., Krossing I., Chem. Eur. J. 2014, 20, 4194–4211. [DOI] [PubMed] [Google Scholar]
- 18. Himmel D., Radtke V., Butschke B., Krossing I., Angew. Chem. Int. Ed. 2018, 57, 4386–4411; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 4471–4498. [Google Scholar]
- 19. Bohnenberger J., Feuerstein W., Himmel D., Daub M., Breher F., Krossing I., Nat. Commun. 2019, 10, 624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bohnenberger J., Schmitt M., Feuerstein W., Krummenacher I., Butschke B., Czajka J., Malinowski P. J., Breher F., Krossing I., Chem. Sci. 2020, 11, 3592–3603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Schmitt M., Mayländer M., Goost J., Richert S., Krossing I., Angew. Chem. Int. Ed. 2021, 60, 14800–14805; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 14926–14931. [Google Scholar]
- 22. Unkrig W., Schmitt M., Kratzert D., Himmel D., Krossing I., Nat. Chem. 2020, 12, 647–653. [DOI] [PubMed] [Google Scholar]
- 23. Dewar J., Jones H. O., Proc. R. Soc. London Ser. A 1905, 76, 558–576. [Google Scholar]
- 24. Cooke J., Manning P., J. Am. Chem. Soc. 1970, 92, 6080–6082. [Google Scholar]
- 25. Vilesov I. F., Kurbatov L. B., Dokl. Akad. Nauk SSSR 1961, 140, 1364–1367. [Google Scholar]
- 26. Winters R. E., Kiser R. W., Inorg. Chem. 1964, 3, 699–702. [Google Scholar]
- 27. Bañares L., Baumert T., Bergt M., Kiefer B., Gerber G., J. Chem. Phys. 1998, 108, 5799–5811. [Google Scholar]
- 28. Wang G., Cui J., Chi C., Zhou X., Li Z. H., Xing X., Zhou M., Chem. Sci. 2012, 3, 3272–3279. [Google Scholar]
- 29. Russell E. M., Cudjoe E., Mastromatteo M. E., Kercher J. P., Sztáray B., Bodi A., J. Phys. Chem. A 2013, 117, 4556–4563. [DOI] [PubMed] [Google Scholar]
- 30. Lacko M., Papp P., Wnorowski K., Matejčík Š., Eur. Phys. J. D 2015, 69, 84–93. [Google Scholar]
- 31. Allison J., Ridge D. P., J. Am. Chem. Soc. 1979, 101, 4998–5009. [Google Scholar]
- 32. Foster M. S., Beauchamp J. L., J. Am. Chem. Soc. 1975, 97, 4808–4814. [Google Scholar]
- 33. Foffani A., Pignataro S., Cantone B., Grasso F., Z. Phys. Chem. 1965, 45, 79–88. [Google Scholar]
- 34. Bidinosti D. R., McIntyre N. S., Can. J. Chem. 1967, 45, 641–648. [Google Scholar]
- 35. Junk G. A., Svec H. J., Z. Naturforsch. 1968, 23b, 1–9. [Google Scholar]
- 36. Lloyd D. R., Schlag E. W., Inorg. Chem. 1969, 8, 2544–2555. [Google Scholar]
- 37. Distefano G., J. Res. Natl. Bur. Stand. 1970, 74, 233–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Norwood K., Ali A., Flesch G. D., Ng C. Y., J. Am. Chem. Soc. 1990, 112, 7502–7508. [Google Scholar]
- 39. Horning S. R., Vincenti M., Cooks R. G., J. Am. Chem. Soc. 1990, 112, 119–126. [Google Scholar]
- 40. Schultz R. H., Crellin K. C., Armentrout P. B., J. Am. Chem. Soc. 1991, 113, 8590–8601. [Google Scholar]
- 41. Fairhurst S. A., Morton J. R., Perutz R. N., Preston K. F., Organometallics 1984, 3, 1389–1391. [Google Scholar]
- 42. Lionel T., Morton J. R., Preston K. F., J. Chem. Phys. 1982, 76, 234–239. [Google Scholar]
- 43. Malinowski P. J., Krossing I., Angew. Chem. Int. Ed. 2014, 53, 13460–13462; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 13678–13680. [Google Scholar]
- 44. Bohnenberger J., Krossing I., Angew. Chem. Int. Ed. 2020, 59, 5581–5585; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 5629–5633. [Google Scholar]
- 45. Schorpp M., Heizmann T., Schmucker M., Rein S., Weber S., Krossing I., Angew. Chem. Int. Ed. 2020, 59, 9453–9459; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 9540–9546. [Google Scholar]
- 46.Deposition Numbers 2144641, 2144643, 2144645, 2144646, 2144650 contain the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service.
- 47. Martens A., Weis P., Krummer M. C., Kreuzer M., Meierhöfer A., Meier S. C., Bohnenberger J., Scherer H., Riddlestone I., Krossing I., Chem. Sci. 2018, 9, 7058–7068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Connelly N. G., Geiger W. E., Chem. Rev. 1996, 96, 877–910. [DOI] [PubMed] [Google Scholar]
- 49. Addison A. W., Rao T. N., Reedijk J., van Rijn J., Verschoor G. C., J. Chem. Soc. Dalton Trans. 1984, 1349–1356. [Google Scholar]
- 50. Church S. P., Poliakoff M., Timney J. A., Turner J. J., J. Am. Chem. Soc. 1981, 103, 7515–7520. [Google Scholar]
- 51. Zhou M., Andrews L., Bauschlicher C. W., Chem. Rev. 2001, 101, 1931–1961. [DOI] [PubMed] [Google Scholar]
- 52. Farrugia L. J., Evans C., J. Phys. Chem. A 2005, 109, 8834–8848. [DOI] [PubMed] [Google Scholar]
- 53. MacNeil J. H., Chiverton A. C., Fortier S., Baird M. C., Hynes R. C., Williams A. J., Preston K. F., Ziegler T., J. Am. Chem. Soc. 1991, 113, 9834–9842. [Google Scholar]
- 54. Morales-Cerrada R., Fliedel C., Daran J. C., Gayet F., Ladmiral V., Ameduri B., Poli R., Chem. Eur. J. 2019, 25, 296–308. [DOI] [PubMed] [Google Scholar]
- 55. Meier S. C., Himmel D., Krossing I., Chem. Eur. J. 2018, 24, 19348–19360. [DOI] [PubMed] [Google Scholar]
- 56. Teller R. G., Finke R. G., Collman J. P., Chin H. B., Bau R., J. Am. Chem. Soc. 1977, 99, 1104–1111. [Google Scholar]
- 57. Bernhardt E., Bley B., Wartchow R., Willner H., Bill E., Kuhn P., Sham I. H. T., Bodenbinder M., Bröchler R., Aubke F., J. Am. Chem. Soc. 1999, 121, 7188–7200. [Google Scholar]
- 58. Keilwerth M., Hohenberger J., Heinemann F. W., Sutter J., Scheurer A., Fang H., Bill E., Neese F., Ye S., Meyer K., J. Am. Chem. Soc. 2019, 141, 17217–17235. [DOI] [PubMed] [Google Scholar]
- 59. Chalkley M. J., Peters J. C., Angew. Chem. Int. Ed. 2016, 55, 11995–11998; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 12174–12177. [Google Scholar]
- 60. Pickett C. J., Pletcher D., J. Chem. Soc. Dalton Trans. 1976, 636–638. [Google Scholar]
- 61. Heninger M., Pernot P., Mestdagh H., Boissel P., Lemaire J., Marx R., Mauclaire G., Int. J. Mass Spectrom. 2000, 199, 267–285. [Google Scholar]
- 62. Le Caër S., Heninger M., Mestdagh H., Chem. Phys. Lett. 2002, 352, 393–400. [DOI] [PubMed] [Google Scholar]
- 63. Therien M. J., Ni C.-L., Anson F. C., Osteryoung J. G., Trogler W. C., J. Am. Chem. Soc. 1986, 108, 4037–4042. [Google Scholar]
- 64. Trogler W. C., Organometallic Radical Processes, Elsevier, Amsterdam, 1990. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information