

Abstract
This review outlines recent developments in protein engineering of stereo‐ and regioselective enzymes, which are of prime interest in organic and pharmaceutical chemistry as well as biotechnology. The widespread application of enzymes was hampered for decades due to limited enantio‐, diastereo‐ and regioselectivity, which was the reason why most organic chemists were not interested in biocatalysis. This attitude began to change with the advent of semi‐rational directed evolution methods based on focused saturation mutagenesis at sites lining the binding pocket. Screening constitutes the labor‐intensive step (bottleneck), which is the reason why various research groups are continuing to develop techniques for the generation of small and smart mutant libraries. Rational enzyme design, traditionally an alternative to directed evolution, provides small collections of mutants which require minimal screening. This approach first focused on thermostabilization, and did not enter the field of stereoselectivity until later. Computational guides such as the Rosetta algorithms, HotSpot Wizard metric, and machine learning (ML) contribute significantly to decision making. The newest advancements show that semi‐rational directed evolution such as CAST/ISM and rational enzyme design no longer develop on separate tracks, instead, they have started to merge. Indeed, researchers utilizing the two approaches have learned from each other. Today, the toolbox of organic chemists includes enzymes, primarily because the possibility of controlling stereoselectivity by protein engineering has ensured reliability when facing synthetic challenges. This review was also written with the hope that undergraduate and graduate education will include enzymes more so than in the past.
Keywords: enzyme stereoselectivity, process engineering, rational enzyme design, saturation mutagenesis, semi-rational directed evolution
Directed evolution of enzymes did not attract the attention of organic chemists until the control of stereoselectivity was successfully achieved 25 years ago. Traditionally, semi‐rational directed evolution based on focused saturation mutagenesis at sites lining the binding pocket and rational enzyme design were considered to be two different approaches, especially when evolving stereoselectivity. However, because the former has become more and more rational, the two approaches have merged with the development of focused rational iterative site‐specific mutagenesis (FRISM).

Introduction
Over millions of years, Nature has evolved enzymes that function as selective catalysts in all living organisms. About a century ago, industrial chemists started to discover that enzymes can be exploited as catalysts in reactions of some unnatural compounds. [1] Unfortunately, their widespread use in organic and pharmaceutical chemistry was severely limited by the often observed low or wrong stereoselectivity as well as narrow substrate scope. [2] Consequently, organic chemists were not “fond” of enzymes and chose not to include them in most undergraduate or graduate chemical courses.
The advent of directed evolution in the period 1986–1997 marked the beginning of a fundamental change in attitude, as evidenced by seminal papers on increasing enzyme thermostability (kanamycin nucleotidyltransferase), [3] robustness toward hostile organic solvents (protease), [4] resistance to antibiotics (β‐lactamase), [5] and especially enhancing enantioselectivity (lipase). [6] Iterative cycles of mutagenesis, expression and screening (or selection) form the basis of all techniques of directed evolution, which means that the best mutant of the first cycle is used as a template for mutagenesis in the next cycle, and so on. [2] The primary bottleneck is the screening step, especially when aiming to enhance or invert enantioselectivity, for which high‐throughput ee‐assays had to be invented.[ 2d , 6 ] Prior to these historical landmarks, Michael Smith had developed the molecular biological method of genetically substituting the amino acid at any residue of a protein for any one of the other 19 canonical amino acids, [7] which in principle set the stage for so‐called rational design of enzymes as an alternative to directed evolution. Accordingly, a limited number of predictions of point mutations are made and then the small collection of variants are tested experimentally. This exciting new research field originally first focused on thermostability, [8] although enzyme immobilization as part of process engineering was another technique for stabilization.[ 1d , 9 ] Rational enzyme design for increasing enzyme thermostability and resistance to hostile organic solvents was guided by the concept of introducing salt bridges on the protein surface, constructing disulfide bonds for rigidification, performing molecular dynamics (MD) simulations, and applying phylogenetic analyses (consensus). [10] As the field progressed, the accumulating knowledge of mutational effects provided further support. In parallel, increasing the robustness of enzymes was also studied by directed evolution. [11] However, organic chemists were not impressed by any of these advances because they were interested in stereoselectivity for which many synthetic transition metal catalysts were being developed with impressive success. [12]
In the early phase of rational protein engineering, controlling stereoselectivity was considered to be a daunting task. [13] Predicting the necessary exchange of an amino acid at a given residue for enhancing or even inverting enantioselectivity appeared to be a mission impossible, not to speak of predicting several mutations. What residues should be rationally chosen, and which amino acid exchange should be implemented at each residue?
Being aware of these difficulties, my group initially chose for the problem of manipulating stereoselectivity, not rational design, but directed evolution utilizing mutagenesis based on error‐prone polymerase chain reaction (epPCR), [6] a random mutagenesis technique. [14] The hydrolytic kinetic resolution of 2‐methyl decanoic acid p‐nitrophenyl ester, catalyzed by the lipase from Pseudomonas aeruginosa (PAL), served as the model experimental platform and the selectivity factor E as the degree of enantioselectivity (E=relative rate of reaction of one enantiomeric substrate with respect to the other). Following 4 rounds of epPCR, the enantioselectivity factor increased 10‐fold from E=1.4 (S enantiomer) to E=11, at the time a breakthrough, but far from industrially relevant. [6] Since subsequent epPCR experiments proved to be disappointing (E=13), my doctoral student Steffi Wilensek suggested focused saturation mutagenesis at sites lining the binding pocket using so‐called NNK codon degeneracy (20 canonical amino acids as building blocks), the first time that this technique was applied to stereoselectivity.[ 13 , 15 ] It worked, but the breakthrough came later (E=594) with the systematization in the form of Combinatorial Active‐site Saturation Test (CAST). It was a crucial move from “blind” to semi‐rational directed evolution of stereoselectivity.[ 2d , 15 ] Iterative Saturation Mutagenesis (ISM) was also established, which simply meant that the best mutant of the first generation was used as a template for saturation mutagenesis at a different site in the second cycle, and so on.[ 2d , 16 , 17 ] Thus, CAST/ISM as a focused technique is fundamentally different from iterative epPCR, or iterative DNA shuffling, a recombinant technique in which homologous genes or genes of several mutants are disassembled into pieces (oligonucleotides) and then reassembled by PCR. [5] It is also different from using a mutator strain, which like epPCR is a random mutagenesis method. [18]
The early CAST/ISM studies were the beginning of methodology development in directed evolution with the purpose of providing semi‐rational methods for mutant library construction, which eventually enabled a dramatic reduction of the screening labor. [2d] During the last few years, we and others intensified efforts to produce smaller and smarter mutant libraries. These new developments are summarized below, the advancements serving as an inspiration for merging semi‐rational directed evolution and rational enzyme design.
Semi‐Rational Methods in Directed Evolution
Following the realization that recursive cycles of epPCR do not lead to high‐quality mutant libraries, certainly not when wanting to influence stereoselectivity, [13] we reasoned early on that it is necessary to sharpen the CAST/ISM tools, as summarized in a recent review. [2d] Today, the screening effort of a CAST library typically amounts to 1000 to 2000 transformants, while exploratory on‐plate assaying for activity can lead to further reductions. The guidelines for implementing CAST/ISM‐based semi‐rational enzyme directed evolution are as follows, which are similar to those of rational enzyme design:
X‐ray data or homology model
Mechanistic information
Previous mutational data
Phylogenetic analysis (consensus technique)
Docking and molecular dynamics (MD) computations
Machine learning (ML)
The goal of CAST/ISM is to manipulate the shape of the binding pocket so that the substrate of interest will be transformed stereo‐ and/or regioselectively as desired by the operator (Figure 1).[ 2d , 16 , 17 ] This is reminiscent of Emil Fischer's famous lock‐and‐key hypothesis [19] and Linus Pauling's extension thereof, according to which interactions between substrate and appropriate protein residues lower the energy of the transition states. [20] This has been impressively visualized by X‐ray structures of evolved stereocomplementary mutants of an epoxide hydrolase in comparison with the wildtype (WT) structure, and explained by MD computations. [21] Saturation mutagenesis at a single residue is generally called site‐saturation mutagenesis (SSM), while simultaneous randomization at more than one residue has been dubbed combinatorial saturation mutagenesis (CSM). [2d] In the CAST/ISM concept, not all residues identified by the above guidelines need to be considered (Figure 1b). When choosing 2, 3 or 4 CAST sites, then 2, 6 or 24 upward pathways, respectively, are theoretically possible (Figure 1c). They do not necessarily terminate with the same multi‐mutational variant. Not all pathways need to be explored, as was done in a model study, [22] but some upward climbs are better than others. Tips on how to escape from possible local minima include the utilization of an inferior mutant as a template in the subsequent round of saturation mutagenesis. [22]
Figure 1.
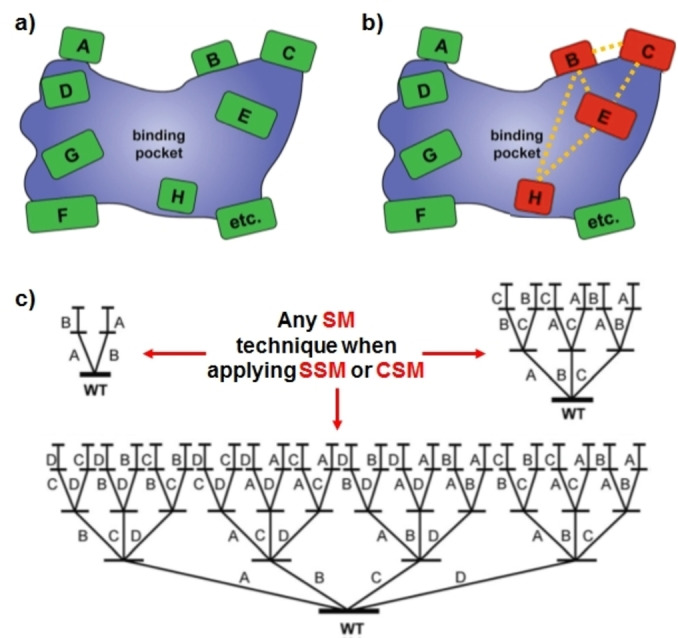
CAST/ISM schemes. [2d] a) Schematic representation of CAST sites. b) Illustration indicating that only a limited number of CAST sites (red‐marked) generally need to be considered for obtaining excellent results. c) Schematic representation of ISM showing the number of upward pathways when choosing 2, 3 or 4 CAST sites, each composed of a single or of several residues. SM: Saturation mutagenesis: SSM: Site‐saturation mutagenesis; CSM: Combinatorial saturation mutagenesis.
Thus far, nothing has been noted about statistical aspects relating to the degree of oversampling in the screening step, yet the mathematics are, in fact, at the heart of CAST/ISM. As an organic chemist, I wondered early on whether the traditional use of NNK codon degeneracy encoding all 20 canonical amino acids is really necessary, or whether a much smaller number, a so‐called reduced amino acid alphabet, would suffice. But why should lower structural diversity of the building blocks have any advantage? For example, why not use NDT codon degeneracy, which encodes only 12 amino acids, representing a cocktail of charged/non‐charged, aromatic/non‐aromatic, and small/large amino acids? In order to illuminate this issue, we exploited the Patrick/Firth statistical algorithm [23] for calculating the degree of oversampling to reach 95 % library coverage (or any other %) by developing the user‐friendly CASTER computer aid. [17] It can then be seen that the screening effort for a given %‐library coverage is drastically reduced. Alternatively, the Nov‐metric can be used to estimate the n th best mutant. [24] An extensive statistical study recently demonstrated that our original hypothesis is correct, namely that when applying saturation mutagenesis, it is better to reduce diversity on the basis of an appropriate reduced amino acid alphabet and to screen for maximum library coverage, rather than employing NNK codons and screening only a minute part of the respective protein sequence space. [25] Indeed, we and others had already started to utilize all kinds of codon degeneracies with reduction of structural diversity. [26] In the extreme case, a reduced amino acid alphabet can consist of a single amino acid. The respective codon can then be applied when randomizing at a site comprising several residues, or even a single residue. Along this line, a revealing study showed that only 384 transformants had to be screened for enantioselectivity in order to ensure 95 % coverage of the respective CAST library. [27] This low number is comparable to the screening effort in many studies which rely on rational enzyme design, which again suggests that the two approaches to protein engineering are merging.
Another development in directed evolution and in rational enzyme design concerns the use of Machine Learning (ML) and Deep Learning (DL), which in recent times have pervaded the life sciences for a variety of different applications. [28] ML involves the creation of algorithms that enable computers to learn, so that defined tasks can be performed more easily for specific practical applications. ML includes Artificial Neural Networks, often called Deep Neural Networks which have intermediate information layers. These are part of DL. From the viewpoint of synthetic organic chemistry, the most important applications of ML/DL concern stereo‐ and regioselectivity of enzymes. [28b] The first attempt in this respect was made in 2012 with a study of the hydrolytic kinetic resolution of a rac‐glycidyl phenyl ether, catalyzed by mutants of the epoxide hydrolase from Aspergillus niger (ANEH), their sequence being predicted by application of the Adaptive Substituent Reordering Algorithm (ASRA). [29] It is quite different from traditional structure‐activity relationships (QSAR), because only patterns of regularity apparent in the input data are needed.[ 29 , 30 ] In all ML studies, the quality of the input data is crucial. In the ANEH study utilizing ASRA, data from an earlier experimental CAST/ISM investigation was used, which had led to the enhancement of enantioselectivity from E∼5 to E=115. ASRA predicted new variants with stereoselectivity factors of E>50, which was substantiated experimentally. [29] Nevertheless, it still needs to be applied in more challenging cases.
The second ML‐based study of enzyme enantioselectivity was reported in 2019, [31] utilizing the previously developed metric innov'SAR as applied to the same ANEH‐catalyzed kinetic resolution. This algorithm is based on an innovative sequence‐activity relationship approach, in which Fourier transform (FT) plays an essential role. Choices for suggesting amino acid exchange events are based on the electronic and steric properties of the respective amino acids, guided by structural and mechanistic data and by the consensus technique. Variants with extremely high enantioselectivity factors were predicted. For example, one variant was suggested to have a never before reported sequence, that was expected to enable a selectivity factor of E=250. This prediction was experimentally validated (E=253) (Figure 2). [31]
Figure 2.

The innov'SAR workflow used in the ML‐based prediction of enantioselective ANEH variants as catalysts in the hydrolytic kinetic resolution of glycidyl phenyl ether (fitness defined by the enantioselectivity factor E). [31]
The relative merits of ASRA versus innov'SAR were recently summarized, but more studies are necessary before final conclusions can be made. [28b] As this review was being finished, the third study appeared describing the use of ML in manipulating enzyme stereoselectivity, this time involving a Fe/β‐ketoglutarate dependent halogenase for late‐stage functionalization of pharmaceutically important soraphens. [32]
Current Methods of Rational Enzyme Design
Studies reporting rational and semi‐rational enzyme design for enhancing protein stability [8] have continued to the present day, but will not be reviewed here. A number of de novo protein design algorithms have been developed for a variety of applications, the Rosetta computer package [33] and HotSpot Wizard algorithms [34] being two particularly prominent examples, which nowadays include ML/DL techniques. Rosetta involves a multi‐step computational procedure based on:
Protein structure prediction
QM energy refinement
Sequence design
Additional guidance in decision making by ML/DL and other techniques
An impressive example is a stereoselective Diels–Alder cycloaddition (Figure 3). First, over 200 Rosetta‐predictions were made for possible mutants, based on QM computations. Then, 84 were expressed and tested in the shown model reaction. [35] Mutations that enable H‐bond interactions at the acceptor and donor residues were designed for improving binding and others were added for increasing activity. Two variants showed Diels–Alder activity, and one of them corresponded to the predicted high enantioselectivity (>95 %ee; ee=enantiomeric excess) and complete endo‐selectivity. The authors stated that the relatively low success rate indicates that the rational approach needs further improvements. [35] It would be great to design exo‐selectivity.
Figure 3.

Biocatalytic stereoselective Diels–Alder cycloaddition achieved by rational design using Rosetta algorithm. [35] Reproduced with permission from Ref. [35]. Copyright 2010, The American Association for the Advancement of Science.
In other cases, Rosetta‐based rational design was combined with semi‐rational CAST‐like mutagenesis, as in the biocatalytic Kemp elimination. [36] Promiscuous transformations catalyzed by mutants of artificial metalloenzymes continue to be enabled primarily by semi‐rational mutagenesis at sites lining the binding pocket,[ 2f , 37 ] more recently also guided by ML/DL. [28d] Rosetta undergoes continuous improvements. For example, protein backbone manipulation by RosettaRemodel is based on a unified interface to the Rosetta modeling suite, with which the control over numerous aspects of flexible backbone protein design computations are possible. [38a] The design challenges include loop insertion/deletion, disulfide engineering, domain assembly, loop remodeling, motif drafting and de novo structure modeling (Figure 4). An informative review of an approach to computer‐based enzyme design which unites the newest developments in the areas of computational chemistry and biology appeared a decade ago, which includes the utility of Rosetta. [38b]
Figure 4.

The utility of RosettaRemodel for a variety of protein backbone manipulations. [38a] The crystal structure of the model protein G (PDB ID: 1PGA) served as the starting point of the different cases. The colored regions indicate places of predictions made by RosettaRemodel.
As noted above, HotSpot Wizard is another outstanding technique for rational enzyme design. [34] The respective workflow is user‐friendly. Tips for selecting substitutions at hotspots identified by other protein engineering strategies such as sequence consensus are provided.
In addition to Rosetta and Hotspot Wizard, which can be combined with Machine Learning (ML), other important techniques of use in rational enzyme design include alanine scanning (or other amino acid scans), in which all or a limited number of residues are subjected to such amino acid exchanges. [39] This allows the identification of potential hotspots at which further mutagenesis can be performed, while occasionally some mutations already lead to an improved catalytic profile which can then be combined. Related is the concept of mutability scanning, according to which not just one amino acid but several are introduced at a select number of hotspots. [40] A fingerprint of positive, neutral and detrimental effects at each residue becomes visible. However, this can be quite labor intensive. Therefore, only a limited number of such amino acid exchange events are implemented, based on consensus data, previous mutational experience and ML. In an impressive application, amorpha‐4,11‐diene synthase served as the enzyme, which is responsible the first (rate‐determining) step in the biosynthesis of antimalarial artemisinin. [41] Up to 16 residues surrounding the binding pocket were first considered, but this was reduced drastically by mutability landscaping, guided by consideration of sequences of other terpene synthases (consensus). Among the 258 produced variants which were screened for activity, the double mutant T399S/H448A enhanced k cat 5‐fold. [41] In principle, this could be improved even more if the sequence and mutational information were to be used in a follow‐up ISM‐study.
Indeed, a strategy comprising rational mutability landscaping in combination with semi‐rational ISM was applied to the known F87A mutant of P450‐BM3 in the quest to achieve regio‐ and diastereoselective C16α‐ and C16β‐hydroxylation of testosterone and 4 other steroids with high activity. [42a] Based on previous mutational data, 5 residues were identified as hotspots, and only 95 variants with polar/non‐polar and small/large amino acid substitutions were produced, leading to important insights. For example, variant A82 L was found to enhance conversion 3‐fold, while polar amino acids diminish activity, and A82W shifts regio‐ and diastereoselectivity toward formation of the 16β‐product (3 %→41 %). Moreover, MD simulations showed at which residues steric clashes with the steroid occurred, and also indicated dynamic effects. Then rational ISM followed with a screening effort as low as 767 transformants for assaying individual libraries, leading to variants with considerably better catalytic profiles than earlier attempts of rational design of 16C‐selectivity. [42b] In the latest steroid study, the concept of complete deconvolution was applied to an earlier quadruple mutant showing a different regioselectivity, flanked by MD computations. [42c] Previously, my group had practiced complete deconvolution of multi‐mutational variants as a unique way to learn lessons in protein engineering, since it allows the construction of fitness pathway landscapes at each evolutionary stage. The focus was always on a single catalytic parameter such as stereoselectivity. In the new study, three parameters were involved for the first time, regio‐ and diastereoselectivity as well as activity, which all contribute to overall fitness. [42c]
Prior to these rational and semi‐rational protein engineering studies, [42] rational enzyme design had been applied to the standard P450‐BM3 monooxygenase using (4R)‐limonene as a chiral substrate. [43] WT delivers a mixture of products at low conversion. The aim was to increase activity for C7‐hydroxylation with formation of perillyl alcohol. The rational design campaign involved several steps, starting with screening of a previous mutant library and performing MD computations, leading to the decision to test the introduction of hydrophobic amino acids alanine, valine, phenylalanine, leucine and isoleucine at various residues. A collection of 48 single and double mutants at residues 87 and 328 were tested, and variant A328V was found to form 27 % perillyl alcohol. Using this mutation, rational mutagenesis at positions 87 and 328 was checked again with introduction of hydrophobic amino acids, but no improvements were noted. Following more MD simulations, mutations regarding hydrophobic amino acids were added to mutant A328V, leading to 60 % of the desired product. In the third round, a similar strategy resulted in the further addition of A328V/L43F allowing for 97 % conversion to perillyl alcohol. Like ISM, the overall strategy is focused, not random iterative mutagenesis, except that only small collections of mutants had to be generated and screened. [43]
Along the same line of reasoning, rational enzyme design was implemented for the generation of more active mutants of deacetoxycephalosporin C synthase from Streptomyces clavuligerus. [44a] As the authors noted, they were inspired by ISM and therefore dubbed the technique Iterative Combinatorial Mutagenesis (ICM), which led to distinctly enhanced activity toward penicillin G. The starting template was a known quadruple mutant C155Y/Y184H/V275I/C281Y, evolved by the same authors using random epPCR and DNA shuffling and screening of ∼10,500 clones, which had shown a 41‐fold increase in k cat/K M relative to WT. In the ICM study, three rounds of targeted mutagenesis were performed in which a total of only 24 new mutants had to be screened. The best mutant resulted in an 87‐fold increase in k cat/K M relative to WT. [44a] The criteria (guidelines) for predicting the 24 new mutants were not offered. MD computations were also not performed, but reference to earlier mutations was made, which was probably helpful. Nevertheless, the practical results are of notable significance in the pharmaceutical industry. A related rational design strategy was used in biocatalytic Pictet–Spengler reactions with formation of pharmaceutically important 1‐aryltetrahydro‐β‐carbolines. [44b]
Based on recent developments of CAST/ISM, in which smaller and smaller saturation mutagenesis libraries ensure higher efficiency, [2d] it can be concluded that a sharp separation line between advanced semi‐rational directed evolution and strict rational enzyme design does not exist. This is also apparent with the development of a technique dubbed Focused Rational Iterative Site‐specific Mutagenesis (FRISM). [45] It was directly inspired by CAST/ISM, and continues along previous rational enzyme design strategies.[ 35 , 43 , 44a ] The systematization of FRISM is shown in Figure 5. [2d] Upon focusing first on site 1 using a limited number of the 20 canonical amino acids as building blocks, a set of variants are predicted and assayed in the laboratory, the best one then being used similarly in the second step at site 2. Each site can be composed of one or more residues. The same procedure is applied for the opposite order of genetic events, first site 2 and then site 1. Since the two pathways are not necessarily expected to deliver the same final variant, the scheme in Figure 5a is not symmetrical. When 3 sites are chosen, 3!=6 pathways are possible, two being illustrated in Figure 5b. [2d] In contrast to the ICM investigation, [44a] in the first FRISM study a recipe was offered for rationally choosing a limited number of residues as hotspots and guides for deciding on specific amino acid exchanges thereat.[ 2d , 45 ]
Figure 5.

Illustration of Focused Rational Iterative Site‐specific Mutagenesis (FRISM).[2d] a) FRISM with 2 mutational sites; b) ISM with 3 mutational sites, 2 of the 6 possible pathways being shown.
The kinetic resolution employing a racemic alcohol and a racemic acid ester was performed, catalyzed by the lipase from Candida antarctica B (CALB). [45] This means that two diastereomeric products are possible, each with two chirality possibilities (enantio‐preference), forming four different stereoisomers. All four pathways were successfully explored, leading to >95 % diastereoselectivity and >95 % enantioselectivity, which required a total screening effort of less than 100 mutants! This was achieved by considering the binding pocket as being composed of an ester‐ and an alcohol‐cavity, in conjunction with known mechanistic and X‐ray structural data. Then, mainly steric factors supported by MD computations were assessed as guides for predicting and testing the limited number of designed mutants. [45]
In rapid succession, FRISM has been applied and extended using different enzyme types, each time guides for making rational choices being offered. [46] Prominent examples concern a photodecarboxylase, [46a] and a glycosyltransferase for synthetically difficult regioselective glucosylation of poly‐hydroxy compounds. [46b] Interestingly, around the same time a study appeared in which a glycosyltransferase was also engineered for substrate acceptance (plant flavonoids) using CAST/ISM, the authors noting that the use of FRISM would not have provided their best quadruple mutant. [47] Advanced versions of CAST/ISM and FRISM are thus complementary rational techniques, each with similar computational supports. Other computational advances likewise deserve mention. [48] This includes rational enzyme design of access tunnels.[ 48g , 49 ]
Conclusions and Perspectives
For many years, the two fields of protein engineering, rational enzyme design and directed evolution, seemed to move forward on two different parallel tracks. However, when research began to focus on making stereo‐ and/or regioselective enzymes suitable for organic chemistry,[ 16 , 17 ] a sharp division between semi‐rational saturation mutagenesis and rational design never reflected the true state of affairs. Indeed, the semi‐rational technique of CAST/ISM has become more and more rational as time went by, requiring considerably less screening. [2d] FRISM appears to be a successful fusion of the two approaches to protein engineering,[ 45 , 46 ] although more research is necessary for final evaluations. An excellent review illuminating challenges from an industrial viewpoint has appeared, which includes patent issues. [2c]
Finally, the recent finding that high‐fidelity chemical gene synthesis of small and medium‐sized mutant libraries is possible in parallel on Si‐chips may well constitute another future perspective, provided the prices continue to go down. [50] This does not mean that researchers are relieved from designing mutant libraries. The users of this commercial option should make sure that the company provides quantitative evidence of the actual quality by complete sequencing. It allows operators to spend less of their valuable time in the laboratory performing routine work, and more for rationally designing mutants with the help of the progressive techniques summarized in this review.
Conflict of interest
The author declares no conflict of interest.
Biographical Information
Manfred T. Reetz obtained his doctoral degree in 1969 under the direction of Ulrich Schöllkopf at Göttingen University in Germany followed by a postdoctoral stay with Reinhard W. Hoffmann at Marburg University. He then performed independent research in various institutions in Germany and the USA, including directorship of the Max‐Planck‐Institut für Kohlenforschung in Mülheim for two decades, where he pioneered the concept of directed evolution of stereo‐ and regioselective enzymes as catalysts in organic and pharmaceutical chemistry. After his first retirement in 2011, he continued research at Marburg University and at the Tianjin Institute of Industrial Biotechnology, Chinese Academy of Sciences, and returned to Mülheim in 2017.

Acknowledgements
The author thanks the Max‐Planck‐Society for support. Open Access funding enabled and organized by Projekt DEAL.
M. Reetz, ChemBioChem 2022, 23, e202200049.
References
- 1.Recent reviews of enzymes in organic chemistry and biotechnology:
- 1a. Simic S., Zukic E., Schmermund L., Faber K., Winkler C. K., Kroutil W., Chem. Rev. 2022, 122, 1052–1126; [DOI] [PubMed] [Google Scholar]
- 1b. Winkler C. K., Schrittwieser J. H., Kroutil W., ACS Cent. Sci. 2021, 7, 55–71; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1c. Hanefeld U., Hollmann F., Paul C. E., Chem. Soc. Rev. 2022, 51, 594–627; [DOI] [PubMed] [Google Scholar]
- 1d. Sheldon R. A., Woodley J. M., Chem. Rev. 2018, 118, 801–838; [DOI] [PubMed] [Google Scholar]
- 1e. Woodley J. M., Appl. Microbiol. Biotechnol. 2019, 103, 4733–4739; [DOI] [PubMed] [Google Scholar]
- 1f. Green A. P., Turner N. J., Perspect. Sci. 2016, 9, 42–48; [Google Scholar]
- 1g. Bell E. L., Finnigan W., France S. P., Green A. P., Hayes M. A., Hepworth L. J., Lovelock S. L., Niikura H., Osuna S., Romero E., Ryan K. S., Turner N. J., Flitsch S. L., Nat. Rev. 2021, 1, 46, (Methods Primers). [Google Scholar]
- 2.Select reviews of directed evolution of enzymes:
- 2a. Zeymer C., Hilvert D., Annu. Rev. Biochem. 2018, 87, 131–157; [DOI] [PubMed] [Google Scholar]
- 2b. Arnold F. H., Chem. Int. Ed. 2019, 58, 14420–14426, (Nobel Lecture); [DOI] [PubMed] [Google Scholar]
- 2c. Hauer B., ACS Catal. 2020, 10, 8418–8427; [Google Scholar]
- 2d. Qu G., Li A., Acevedo-Rocha C. G., Sun Z., Reetz M. T., Angew. Chem. Int. Ed. 2020, 59, 13204–13231; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 13304–13333; [Google Scholar]
- 2e. Wang Y., Xue P., Cao M., Yu T., Lane S. T., Zhao H., Chem. Rev. 2021, 121, 12384–12444; [DOI] [PubMed] [Google Scholar]
- 2f. Yang Y., Arnold F. H., Acc. Chem. Res. 2021, 54, 1209–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Liao H., McKenzie T., Hageman R., Proc. Natl. Acad. Sci. USA 1986, 83, 576–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chen K. Q., Arnold F. H., Proc. Natl. Acad. Sci. USA 1993, 90, 5618–5622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.
- 5a. Stemmer W. P. C., Nature 1994, 370, 389–391; [DOI] [PubMed] [Google Scholar]
- 5b. Crameri A., Raillard S.-A., Bermudez E., Stemmer W. P. C., Nature 1998, 391, 288–291. [DOI] [PubMed] [Google Scholar]
- 6. Reetz M. T., Zonta A., Schimossek K., Liebeton K., Jaeger K.-E., Angew. Chem. Int. Ed. Engl. 1997, 36, 2830–2832. [Google Scholar]
- 7. Smith M., Angew. Chem. Int. Ed. Engl. 1994, 33, 1214–1221, (Nobel Lecture). [Google Scholar]
- 8.Seminal review of rational design of protein thermostabilization: Eijsink V. G. H., et al., J. Biotechnol. 2004, 113, 105–120.15380651 [Google Scholar]
- 9.
- 9a. Immobilization of Enzymes and Cells (Ed.: J. M. Guisan), Humana, Totowa, 2013;
- 9b. Cavalcante F. T. T., Cavalcante A. L. G., de Sousa I. G., Neto F. S., dos Santos J. C. S., Catalysts 2021, 11, 1222; [Google Scholar]
- 9c. Janson N., Heinks T., Beuel T., Alam S., Höhne M., Bornscheuer U. T., Fischer von Mollard G., Sewald N., ChemCatChem 2022, 14, e202101485; [Google Scholar]
- 9d. Morellon-Sterling R., Carballares D., Arana-Pena A., Siar E.-H., Ait Braham S., Fernandez-Lafuente R., ACS Sustainable Chem. Eng. 2021, 9, 7508–7518; [Google Scholar]
- 9e. Rodriguez-Nunez K., Bernal C., Martinez R., Int. J. Biol. Macromol. 2021, 170, 61–70. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Steipe B., Plückthun A., Steinbacher S., J. Mol. Biol. 1994, 240, 188–192; [DOI] [PubMed] [Google Scholar]
- 10b. Janda J. O., Busch M., Kück F., Porfenenko M., Merkl R., BMC Bioinf. 2012, 13, 55; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10c. Sternke M., Tripp K. W., Barrick D., Proc. Natl. Acad. Sci. USA 2019, 116, 11275–11284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.
- 11a. Arnold F. H., Acc. Chem. Res. 1998, 31, 125–131; [Google Scholar]
- 11b. Arnold F. H., Proc. Natl. Acad. Sci. USA 1998, 95, 2035–2036; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11c. Romero P. A., Arnold F. H., Nat. Rev. Mol. Cell Biol. 2009, 10, 866–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.
- 12a. Knowles W. S., Angew. Chem. Int. Ed. 2002, 41, 1998–2007, (Nobel Lecture); [PubMed] [Google Scholar]
- 12b. Noyori R., Angew. Chem. Int. Ed. 2002, 41, 2008–2022, (Nobel Lecture); [PubMed] [Google Scholar]
- 12c. Sharpless K. B., Angew. Chem. Int. Ed. 2002, 41, 2024–2032, (Nobel Lecture). [PubMed] [Google Scholar]
- 13.S. Wilensek, Dissertation, Ruhr-Universität-Bochum/Germany, 2001.
- 14.
- 14a. Leung D. W., Chen E., Goeddel D. V., Technique 1989, 1, 11–15; [Google Scholar]
- 14b. Cadwell R. C., Joyce G. F., PCR Methods Appl. 1994, 3, S136–S140. [DOI] [PubMed] [Google Scholar]
- 15. Reetz M. T., Wilensek S., Zha D., Jaeger K.-E., Angew. Chem. Int. Ed. 2001, 40, 3589–3591; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2001, 113, 3701–3703. [Google Scholar]
- 16. Reetz M. T., Bocola M., Carballeira J. D., Zha D., Vogel A., Angew. Chem. Int. Ed. 2005, 44, 4192–4196; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2005, 117, 4264–4268. [Google Scholar]
- 17.
- 17a. Reetz M. T., Carballeira J. D., Nat. Protoc. 2007, 2, 891–903; [DOI] [PubMed] [Google Scholar]
- 17b. Qu G., Sun Z., Reetz M. T., Iterative Saturation Mutagenesis for Semi-rational Enzyme Design, in Protein Engineering: Tools and Applications (Eds.: Zhao H., Lee S. Y., Stephanopoulus G.), Wiley-VCH, Weinheim, 2021. [Google Scholar]
- 18.
- 18a. Carr R., Alexeeva M., Enright A., Eve T. S. C., Dawson M. J., Turner N. J., Angew. Chem. Int. Ed. 2003, 42, 4807–4810; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2003, 115, 4955–4958; [Google Scholar]
- 18b. Batista V. F., Galman J. L., Pinto D. C. G. A., Silva A. M. S., Turner N. J., ACS Catal. 2018, 8, 11889–11907. [Google Scholar]
- 19.
- 19a. Fischer E., Dtsch. Chem. Ges. 1894, 27, 2984–2993; [Google Scholar]
- 19b. Lichtenthaler F. W., Angew. Chem. Int. Ed. Engl. 1995, 33, 2364–2374. [Google Scholar]
- 20.
- 20a. Pauling L., Nature 1948, 161, 707–709; [DOI] [PubMed] [Google Scholar]
- 20b. Amyes T. L., Richard J. P., Biochemistry 2013, 52, 2021–2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sun Z., Lonsdale R., Wu L., Li G., Li A., Wang J., Zhou J., Reetz M. T., ACS Catal. 2016, 6, 1590–1597. [Google Scholar]
- 22. Gumulya Y., Sanchis J., Reetz M. T., ChemBioChem 2012, 13, 1060–1066. [DOI] [PubMed] [Google Scholar]
- 23.
- 23a. Firth A. E., Patrick W. M., Bioinformatics 2005, 21, 3314–3315; [DOI] [PubMed] [Google Scholar]
- 23b. Firth A. E., Patrick W. M., Nucleic Acids Res. 2008, 36, W281–W285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nov Y., Appl. Environ. Microbiol. 2012, 78, 258–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Li A., Qu G., Sun Z., Reetz M. T., ACS Catal. 2019, 9, 7769–7778. [Google Scholar]
- 26.
- 26a. Reetz M. T., Wu S., Chem. Commun. 2008, 5499–5501; [DOI] [PubMed] [Google Scholar]
- 26b. Sun Z., Wikmark Y., Bäckvall J.-E., Reetz M. T., Chem. Eur. J. 2016, 22, 5046–5054. [DOI] [PubMed] [Google Scholar]
- 27. Acevedo-Rocha C. G., Li A., D'Amore L., Hoebenreich S., Sanchis J., Lubrano P., Matteo M. P., Garcia-Borras M., Osuna S., Reetz M. T., Nat. Commun. 2021, 12, 1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Selected articles and reviews on ML/DL in protein science:
- 28a. LeCun Y., Bengio Y., Hinton G., Nature 2015, 521, 436–444; [DOI] [PubMed] [Google Scholar]
- 28b. Li G., Dong Y., Reetz M. T., Adv. Synth. Catal. 2019, 361, 2377–2386; [Google Scholar]
- 28c. Mazurenko S., Prokop Z., Damborsky J., ACS Catal. 2020, 10, 1210–1223; [Google Scholar]
- 28d. Yang K. K., Wu Z., Arnold F. H., Nat. Methods 2019, 16, 687–694; [DOI] [PubMed] [Google Scholar]
- 28e. Humphreys I. R., al et, Science 2021, 374: eabm4805; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28f. Wittmann B. J., Yue Y., Arnold F. H., Cell Systems 2021, 12, 1026–1045, e7; [DOI] [PubMed] [Google Scholar]
- 28g. Qiu Y., Hu J., Wie G.-W., Nat. Comput. 2021, 1, 809–818; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28h. Baek M., Baker D., Nat. Methods 2022, 19, 11–26; [DOI] [PubMed] [Google Scholar]
- 28i. McKenna A., Dubey S. P. N., J. Biomed. Inf. 2022, 128, 10416; [DOI] [PubMed] [Google Scholar]
- 28j. Bishop C. M., Pattern Recognition and Machine Learning, Springer, New York, 2016; [Google Scholar]
- 28k. Muggleton S., King R. D., Stenberg M. J. E., Prot. Eng. Des. Sci. 1992, 5, 647–657. [DOI] [PubMed] [Google Scholar]
- 29. Feng X., Sanchis J., Reetz M. T., Rabitz H., Chem. Eur. J. 2012, 18, 5646–565. [DOI] [PubMed] [Google Scholar]
- 30. McAllister S. R., Feng X., P. A. DiMaggio, Jr. , Floudas C. A., Rabinowitz J. D., Rabitz H., Bioorg. Med. Chem. Lett. 2008, 18, 5967–5970. [DOI] [PubMed] [Google Scholar]
- 31. Cadet F., Fontaine N., Li G., Sanchis J., Chong M. N., Pandjaitan R., Vetrivel I., Offmann B., Reetz M. T., Sci. Rep. 2018, 8, 16757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Büchler J., Malca S. H., Patsch D., Voss M., Turner N. J., Bornscheuer U. T., Allemann O., Le Chapelain C., Lumbroso A., Loiseleur O., Buller R., Nat. Commun. 2022, 13, 371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.
- 33a. Alford R. F., Leaver-Fay A., Jeliazkov J. R., O'Meara M. J., DiMaio F. P., Park H., Shapovalov M. V., Renfrew P. D., Mulligan V. K., Kappel K., Labonte J. W., Pacella M. S., Bonneau R., Bradley P., R. L. Dunbrack, Jr. , Das R., Baker D., Kuhlman B., Kortemme T., Gray J. J., J. Chem. Theory Comput. 2017, 13, 3031–3048; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33b. Anishchenko I., Pellock S. J., Chidyausiku T. M., Ramelot T. A., Ovchinnikov S., Hao J., Bofna K., Norn C., Kang A., Bera A. K., DiMaio F., Carter L., Chow C. M., Montelione G. T., Baker D., Nature 2021, 600, 547–552; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33c. Du Z., Su H., Wang W., Ye L., Wei H., Peng Z., Anishchenko I., Baker D., Yang J., Nat. Protoc. 2021, 16, 5634–5651. [DOI] [PubMed] [Google Scholar]
- 34.Reviews and key articles on HotSpot Wizard, FireProt and related rational design techniques:
- 34a. Marques S. M., Planas-Iglesias J., Damborsky J., Curr. Opin. Struct. Biol. 2021, 69, 19–34; [DOI] [PubMed] [Google Scholar]
- 34b. Musil M., Khan R. T., Beier A., Stourac J., Konegger H., Damborsky J., Bednar D., Briefings Bioinf. 2021, 22, 1–11; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34c. Musil M., Stourac J., Bendl J., Brezovsky J., Prokop Z., Zendulka J., Martinek T., Bednar D., Damborsky J., Nucleic Acids Res. 2017, W393–W399; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34d. Xia Y., Li X., Yang L., Lou X., Shen W., Cao Y., Peplowsky L., Chen X., Appl. Microbiol. Biotechnol. 2021, 105, 7309–7319; HotSpot Wizard web server: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34e. Sumbalova L., Stourac J., Martinek T., Bednar D., Damborsky J., Nucleic Acids Res. 2018, 46, W356–W362; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34f. Wei R., von Haugwitz G., Pfaff L., Mican J., Badenhorst C. P. S., Liu W., Weber G., Austin H. P., Bednar D., Damborsky J., Bornscheuer U. T., ACS Catal. 2022, 12, 3382–3396; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34g. Schenkmayerova A., Pinto G. P., Toul M., Marek M., Hernychova L., Planas-Iglesias J., Liskova V. D., Pluskal D., Vasina M., Emond S., Dörr M., Chaloupkova R., Bednar D., Prokop Z., Hollfelder F., Bornscheuer U. T., Damborsky J., Nat. Commun. 2021, 12, 3616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Siegel J. B., Zanghellini A., Lovick H. M., Kiss G., Lambert A. R., St. Clair J. L., Gallaher J. L., Hilvert D., Gelb M. H., Stoddard B. L., Houk K. N., Michael F. E., Baker D., Science 2010, 329, 309–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Key studies on biocatalytic Kemp elimination:
- 36a. Röthlisberger D., Khersonsky O., Wollacott A. M., Jiang L., DeChancie J., Betker J., Gallaher J. L., Althoff E. A., Zanghellini A., Dym O., Albeck S., Houk K. N., Tawfik D. S., Baker D., Nature 2008, 453, 190–195; [DOI] [PubMed] [Google Scholar]
- 36b. Khersonsky O., Röthlisberger D., Wollacott A. M., Murphy P., Dym O., Albeck S., Kiss G., Houk K. N., Baker D., Tawfik D. S., J. Mol. Biol. 2011, 407, 391–412; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36c. Blomberg R., Kries H., Pinkas D. M., Mittal P. R. E., Grütter M. G., Privett H. K., Mayo S. L., Hilvert D., Nature 2013, 503, 418–421. [DOI] [PubMed] [Google Scholar]
- 37.
- 37a. Chalkley M. J., Mann S. I., DeGrado W. F., Nat. Methods 2022, 6, 31–50; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37b. Baiyoumy A., Vallapurackel J., Schwizer F., Heinisch T., Kardashliev T., Held M., Panke S., Ward T. R., ACS Catal. 2021, 11, 10705–10712; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37c. Leveson-Gower R. B., Mayer C., Roelfes G., Nat. Chem. Rev. 2019, 3, 687–705; [Google Scholar]
- 37d. Ren X., Fasan R., Curr. Opin. Green Sustain. Chem. 2021, 31, 100494; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37e. Reetz M. T., Acc. Chem. Res. 2019, 52, 336–344. [DOI] [PubMed] [Google Scholar]
- 38.
- 38a. Huang P.-S., Ban Y.-E. A., Richter F., Andre I., Vernon R., Schief W. R., Baker D., PLoS One 2011, 6, e24109; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38b. Kiss G., Celebi-Ölcüm N., Moretti R., Baker D., Houk K. N., Angew. Chem. Int. Ed. 2013, 52, 5700–5725; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 5810–5836. [Google Scholar]
- 39. Morrison K. L., Weiss G. A., Curr. Opin. Chem. Biol. 2001, 5, 302–307. [DOI] [PubMed] [Google Scholar]
- 40.
- 40a. van der Meer J. Y., Biewenga L., Poelarends G. J., ChemBioChem 2016, 17, 1792–1799; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40b. van der Meer J. Y., Poddar H., Baas B. J., Miao Y., Rahimi M., Kunzendorf A., van Merkerk R., Tepper P. G., Geertsema E. M., Thunnissen A. M., Quax W. J., Poelarends G. J., Nat. Commun. 2016, 7, 10911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Abdallah I. I., van Merkerk R., Klumpenaar E., Quax W. J., Sci. Rep. 2018, 8, 9961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.
- 42a. Acevedo-Rocha C. G., Gamble C., Lonsdale R., Li A., Nett N., Hoebenreich S., Lingnau J. B., Wirtz C., Fares C., Hinrichs H., Deege A., Mulholland A. J., Nov Y., Leys D., McLean K. J., Munro A. W., Reetz M. T., ACS Catal. 2018, 8, 3395–3410; [Google Scholar]
- 42b. Venkataraman H., de Beer S. B. A., van Bergen L. A. H., van Essen N., Geerke D. P., Vermeulen N. P. E., Commandeur J. N. M., ChemBioChem 2012, 13, 520–523; [DOI] [PubMed] [Google Scholar]
- 42c. Acevedo-Rocha C. G., Li A., D'Amore L., Hoebenreich S., Sanchis J., Lubrano P., Ferla M. P., Garcia-Borras M., Osuna S., Reetz M. T., Nat. Commun. 2021, 12, 1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Seifert A., Antonovici M., Hauer B., Pleiss J., ChemBioChem 2011, 12, 1346–1351. [DOI] [PubMed] [Google Scholar]
- 44.
- 44a. Ji J., Fan K., Tian X., Zhang X., Zhang Y., Yang K., Appl. Environ. Microbiol. 2012, 78, 7809–7812; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44b. Eger E., Schrittwieser J. H., Wetzl D., Iding H., Kuhn B., Kroutil W., Chem. Eur. J. 2020, 26, 16281–16285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Xu J., Cen Y., Singh W., Fan J., Wu L., Lin X., Zhou J., Huang M., Wu Q., Reetz M. T., Wu Q., J. Am. Chem. Soc. 2019, 141, 7934–7945. [DOI] [PubMed] [Google Scholar]
- 46.
- 46a. Xu J., Fan J., Lou Y., Xu Q., Wang Z., Li D., Zhou H., Lin X., Wu Q., Nat. Commun. 2021, 12, 3983; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46b. Li J., Qu G., Shang N., Chen P., Men Y., Liu W., Mei Z., Sun Y., Sun Z., Green Synth. Catal. 2021, 2, 45–53; [Google Scholar]
- 46c. Li D., Han T., Xue J., Xu W., Xu J., Wu Q., Angew. Chem. Int. Ed. 2021, 60, 20695–20699; [DOI] [PubMed] [Google Scholar]
- 46d. Ma N., Fang W., Liu C., Qin X., Wang X., Jin L., Wang B., Cong Z., ACS Catal. 2021, 11, 8449–8455. [Google Scholar]
- 47. Wen Z., Zhang Z.-M., Zhong L., Fan J., Li M., Ma Y., Zhou Y., Zhang W., Guo B., Chen B., Wang J.-B., ACS Catal. 2021, 11, 14781–14790. [Google Scholar]
- 48.
- 48a. Wijma H. H., Floor R. J., Bjelic S., Marrink S. J., Baker D., Janssen D. B., Angew. Chem. Int. Ed. 2015, 54, 3726–3730; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 3797–3801; [Google Scholar]
- 48b. Maria-Solano M. A., Kinateder T., Iglesias-Fernandez J., Sterner R., Osuna S., ACS Catal. 2021, 11, 13733–13743; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48c. Chovancova E., Pavelka A., Benes P., Strnad O., Sustr V., Klvana M., Medek P., Biedermannova L., Sochor J., Damborsky J., PLoS Comput. Biol. 2012, 8, e1002708; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48d. Rehem F. B. H., Tyler T. J., Xie J., Yap K., Durek T., Craik D. J., ChemBioChem 2021, 22, 2079–2086; [DOI] [PubMed] [Google Scholar]
- 48e. Arabnejad H., Bombino E., Colpa D. I., Jekel P. A., Traijkovic M., Wijma H. J., Janssen D. B., ChemBioChem 2020, 21, 1893–1904; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48f. Kokkonen P., Bednar D., Pinto G., Damborsky J., Biotechnol. Adv. 2019, 37, 107386; [DOI] [PubMed] [Google Scholar]
- 48g. Zhou J., Panaitu A. E., Grigoryan G., Proc. Natl. Acad. Sci. USA 2020, 117, 1059–1068; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48h. Cui Y., Chen Y., Liu X., Dong S., Tian Yu'e, Qiao Y., Mitra R., Han J., Li C., Han X., Liu W., Chen Q., Wei W., Wang X., Du W., Tang S., Xiang H., Liu H., Liang Y., Houk K. N., Wei B., ACS Catal. 2021, 11, 13401350; [Google Scholar]
- 48i. Park A., Park S., ACS Catal. 2022, 12, 2397–2402. [Google Scholar]
- 49.M. Bzowka, K. Mitusinska, A. Raczynska, T. Skalski, A. Samol, W. Bagrowska, T. Magdziarz, A. Gora, bioRxiv: 10.1101/2021.12.08.471815. [DOI] [PMC free article] [PubMed]
- 50. Li A., Sun Z., Reetz M. T., ChemBioChem 2018, 19, 2023–2032. [DOI] [PubMed] [Google Scholar]