

Abstract
We recently disclosed a dehydrogenative double C−H bond activation reaction in the unusual pincer‐type rhodium‐germyl complex [(ArMes)2ClGeRh] (ArMes=C6H3‐2,6‐(C6H2‐2,4,6‐Me3)2). Herein we investigate the catalytic applications of this Rh/Ge system in several transformations, namely trans‐semihydrogenation of internal alkynes, trans‐isomerization of olefins and hydrosilylation of alkynes. We have compared the activity and selectivity of this catalyst against other common rhodium precursors, as well as related sterically hindered rhodium complexes, being the one with the germyl fragment superior in terms of selectivity towards E‐isomers. To increase this selectivity, a tandem catalytic protocol that incorporates the use of a heterogeneous catalyst for the trans‐semihydrogenation of internal alkynes has been devised. Kinetic mechanistic investigations provide important information regarding the individual catalytic cycles that comprise the overall trans‐semihydrogenation of internal alkynes.
Keywords: germanium, rhodium, semihydrogenation, kinetic studies, tandem catalysis
Main group catalysis: Herein we investigate the catalytic potential of a rhodium/germyl system for the selective semi‐hydrogenation of internal alkynes towards trans‐olefins. Among the rhodium complexes studied, only the germyl complex mediates cis/trans‐isomerization and thus allows access to the less common isomer derived from partial hydrogenation. A whole mechanistic picture is provided based on kinetic analysis.

Introduction
Ambiphilic ligands containing both an electron donor and an acceptor group have become the focus of frontier research in organometallic chemistry and catalysis owing to the remarkable reactivity that they confer to transition metals. [1] Among those, low‐valent main group elements with reduced HOMO‐LUMO gaps constitute paramount examples of biphilic centres (single‐site ambiphiles). [2] Heavier tetrylenes, that is, heavier group 14 divalent elements (:ER2; E=Si, Ge, Sn, Pb), pertain to such class of compounds and their combination with transition metals offers widespread possibilities. [3] Recent examples of this fruitful partnership include multi‐site oxidative addition of strong bonds [4] or the unusual reactivity of unsaturated substrates across the transition metal‐tetrylene motif. [5] Nonetheless, the use of tetrylenes as non‐innocent ligands for transition metals is not restricted to stoichiometric reactions. Aside from the successful application of base‐stabilized tetrylenes as strongly σ‐donating ligands, [6] efficient turnover has also been built on the direct synergisms between a transition metal and a base‐free tetrel site. A foremost example derives from the work of Tilley on the hydrosilylation of olefins by a ruthenium silylene complex. [7] This system circumvents the traditional Chalk‐Harrod type mechanisms and instead operates through the direct addition of a silylene Si–H bond to the olefin, with implications in other catalytic processes. [8] More recently, Esteruelas and Cabeza have described an osmium germylene complex that mediates the dehydrogenation of formic acid by an Os/Ge synergistic pathway. [9]
Our group has recently capitalized on this capacity for transition metal‐tetrel bimetallic cooperation to disclose an unusual dehydrogenative double C−H bond activation process. This transformation, which involves the formation of compound 3, is triggered by thermolysis of compound 2 (Figure 1). [10] The latter was obtained by halide abstraction from [(ArMes)2ClGeRh] (1, ArMes=C6H3‐2,6‐(C6H2‐2,4,6‐Me3)2), [11] which in turn derives from the reaction between [RhCl(COD)]2 (COD=1,5‐cyclooctadiene) and [(ArMes2)2Ge:]. During this process, the dynamic binding capacity of the germanium site [12] was evinced by reversible rearrangement between germylene and germyl forms, with concomitant formation of Ge−Cl, Ge−H and Ge−C bonds in concert with the rhodium centre. These multiple bond‐breaking and bond‐forming events have now led us to interrogate the potential capacity of compound 2 as a catalyst.
Figure 1.

Relevant Rh/Ge complexes (A) and other benchmark Rh species (B) used as catalysts in this work.
We have first turned our attention to the semihydrogenation of alkynes, a transformation of industrial relevance where controlling chemo‐ and regioselectivity is key. [13] The use of Lindlar's catalyst remains the most popular method to access Z‐alkenes, [14] the isomers that are obtained in the majority of organometallic homogeneous catalysed processes. [15] In contrast, an equally versatile and efficient catalytic strategy for the synthesis of E‐alkenes is yet lacking despite numerous advances. [16] Among the most recent approaches, bimetallic catalysts have shown good results for the selective semi‐hydrogenation towards E‐alkenes. [17] In particular, Lu and co‐workers have demonstrated that a Rh/Ga heterobimetallic catalyst anchored to a metal‐organic framework promotes the formation of E‐alkenes. Conversely, the analogous system lacking the P‐block metal is unselective and promotes instead complete hydrogenation towards the alkane. [18] Encouraged by these results, here we investigate the potential of 2 as a catalyst for the selective semihydrogenation of alkynes and related transformations, focusing on the mechanism by which this complex operates.
Results and Discussion
Catalysis studies. Initial investigations focused on the hydrogenation of diphenylacetylene (DPA) using the germyl‐rhodium complex 2 as catalyst in 10 mol% loading and working under mild conditions (H2, 1 bar; 80 °C). For convenience, instead of using 2 directly we employed a mixture of its chloride precursor 1 and NaBArF ([BArF]−=[B(C6H2‐3,5‐(CF3)2)4]− (see Experimental Section for details). Reactions were carried out in J. Young NMR tubes in C6D6 with continuous sample shaking to facilitate hydrogen diffusion into the solution. At short times most of the hydrogenated product (88 %) corresponded to cis‐stilbene (Table 1, entry 1). However, at longer reaction times the stereoselectivity shifted to the unusual E‐isomer (73 %) whereas the only other observable product is the corresponding alkane due to complete hydrogenation (entry 2). For comparison, we tested the catalytic activity of two other commonly used catalysts, more precisely the [RhCl(COD)]2 dimer (COD=1,5‐cyclooctadiene) and Wilkinson's catalyst, [RhCl(PPh3)3] (entries 4 and 5, respectively). While the former led to an almost equimolar mixture of Z‐isomer and alkane, the latter readily converted DPA into its fully hydrogenated derivative. It is clear that the presence of the germyl fragment is necessary to accomplish the semi‐hydrogenation of the alkyne towards the E‐isomer. The hydrogenation of other internal alkynes revealed that the less‐common E‐isomer prevails over the Z‐isomer. However, chemoselectivity is only limited since variable amounts of the corresponding alkane are also observed in all cases, in fact usually as the major product. Owing to the propensity of colloidal rhodium to act as a hydrogenation catalyst, we evaluated its active participation by a mercury test (entry 3), which did not alter the reaction outcome and thus discarded this hypothesis.
Table 1.
Hydrogenation of alkynes catalysed by 2 and other Rh precursors.
|
| ||||||||
|---|---|---|---|---|---|---|---|---|
|
Entry[a] |
Substrate |
Cat. |
t [h] |
Additive |
Yield [%][b] |
|
||
|
R |
R’ |
Z |
E |
Alkane |
||||
|
1 |
Ph |
Ph |
2 |
3 |
– |
88 |
1 |
11 |
|
2 |
Ph |
Ph |
2 |
20 |
– |
0 |
73 |
27 |
|
3 |
Ph |
Ph |
2 |
20 |
Hg[c] |
0 |
75 |
25 |
|
4 |
Ph |
Ph |
5 [d] |
24 |
– |
42 |
0 |
58 |
|
5 |
Ph |
Ph |
6 [e] |
18 |
– |
0 |
0 |
100 |
|
6 |
Me |
Me |
2 |
1 |
– |
0 |
0 |
100 |
|
7 |
Ph |
Me |
2 |
24 |
– |
0 |
32 |
68 |
|
8 |
Ph |
TMS |
2 |
48 |
– |
0 |
48 |
52 |
|
9 |
TMS |
TMS |
2 |
24 |
– |
0 |
0 |
0 |
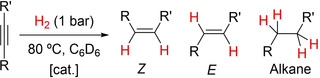
[a] Reaction conditions: alkyne (0.03 mmol), H2 (1 bar), catalyst (0.003 mmol; 10 mol%), C6D6 (0.5 mL). [b] Yields determined by 1H NMR spectroscopy using C6Me6 as internal standard. Conversion of DPA is full in all cases except for entry 9, where no conversion is detected. [c] A drop of Hg was added before heating the mixture to 80 °C; [d] [RhCl(COD)]2 dimer (10 mol%) as catalyst. [e] Wilkinson's catalyst ([RhCl(PPh3)3], 10 mol%).
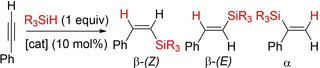
We then wondered if the tendency towards E‐isomer formation in the semi‐hydrogenation of alkynes was maintained in the somewhat related hydrosilylation reaction. To test this notion, we performed some preliminary catalytic runs for the hydrosilylation of phenylacetylene (Table 2). We had to use a terminal alkyne because no reaction took place with internal ones (including DPA), most likely due to steric congestion caused by the terphenyl substituents on germanium. The hydrosilylation of terminal alkynes is of great relevance for the silicon industry and has been intensively investigated in the last decades. [19] Once more, controlling regio‐ and stereoselectivity has been the focus of intense research on this topic. [20] In principle, three types of isomers can be produced, namely, α, β‐(E) and β‐(Z) (Table 2). While the selective synthesis of β‐isomers is well‐documented, that is, the products derived from the anti‐Markovnikov addition, particularly for the β‐(E)‐isomer, [21] the selective formation of the α‐isomer is more unusual. [22]
Table 2.
Preliminary hydrosilylation reactions of phenylacetylene.
|
| |||||||||
|---|---|---|---|---|---|---|---|---|---|
|
Entry[a] |
Cat. |
R |
t |
T |
Solvent |
Conversion[b] |
Selectivity[b] [%] |
||
|
|
|
|
[h] |
[°C] |
|
[%] |
β‐(Z) |
β‐(E) |
α |
|
1 |
2 |
Et |
48 |
80 |
C6D6 |
79 |
– |
56 |
23 |
|
2 |
2 |
Et |
48 |
80 |
Toluene |
85 |
– |
44 |
41 |
|
3 |
2 |
Et |
48 |
80 |
THF |
58 |
– |
44 |
14 |
|
4 |
2 |
Et |
48 |
80 |
CH2Cl2 |
85 |
– |
56 |
29 |
|
5 |
2 |
Et |
48 |
80 |
Acetone |
100 |
– |
72 |
28 |
|
6 |
5 |
Et |
48 |
80 |
C6D6 |
60 |
– |
50 |
9 |
|
7 |
6 |
Et |
48 |
80 |
C6D6 |
74 |
– |
61 |
1 |
|
8 |
7 |
Et |
48 |
80 |
C6D6 |
58 |
– |
41 |
5 |
|
9 |
8 |
Et |
48 |
80 |
C6D6 |
60 |
– |
52 |
18 |
|
10 |
9 |
Et |
48 |
80 |
C6D6 |
64 |
– |
46 |
18 |
|
11 |
10 |
Et |
48 |
80 |
C6D6 |
37 |
– |
24 |
13 |
|
12 |
2 |
Ph |
96 |
100 |
C6D6 |
64 |
– |
– |
64 |
|
13 |
5 |
Ph |
96 |
100 |
C6D6 |
70 |
– |
– |
14 |
|
14 |
6 |
Ph |
96 |
100 |
C6D6 |
84 |
16 |
– |
2 |
|
15 |
7 |
Ph |
96 |
100 |
C6D6 |
100 |
4 |
– |
7 |
|
16 |
8 |
Ph |
96 |
100 |
C6D6 |
50 |
26 |
– |
14 |
|
17 |
9 |
Ph |
96 |
100 |
C6D6 |
100 |
23 |
– |
9 |
|
18 |
10 |
Ph |
96 |
100 |
C6D6 |
73 |
23 |
– |
35 |
|
19 |
2 |
OEt |
20 |
80 |
C6D6 |
100 |
– |
55 |
45 |
[a] Reaction conditions: phenylacetylene (0.03 mmol), hydrosylane (0.03 mmol) or piperidine (0.03 mmol), catalyst (0.003 mmol; 10 mol%), solvent (0.5 mL). [b] Yields determined by 1H NMR spectroscopy using C6Me6 as internal standard.
Hydrosilylation of phenylacetylene with Et3SiH in the presence of catalyst 2 resulted again in the formation of the corresponding trans‐olefin (β‐(E)‐isomer) as the major product, although with a non‐negligible amount of the α‐isomer (Table 2, entry 1). While the cis‐olefin (β‐(Z)‐isomer) was not detected under any of the attempted conditions, the β‐(E)/α ratio span from 3.1 to 1.1 under different solvents (entries 1–5). Using triethoxysilane (Si(OEt)3H) did not drastically alter selectivity but employing triphenylsilane (Ph3SiH) resulted in the regioselective formation of the α‐isomer (entry 12), with no apparent production of any β‐vinylsilanes. We attribute this remarkable and unusual Markovnikov selectivity to the greater steric pressure exerted by Ph3SiH in conjunction with the bulkiness of catalyst 2. In fact, conversion only reached 64 % in this case despite using more forcing conditions (96 h, 100 °C).
We wonder if this unusual selectivity would be shared by other rhodium catalysts or would rather be specific of our Rh/Ge system. To examine this further, beside the common precursors 5 and 6, we also tested the activity of other rhodium complexes of considerable steric constraints that share some similarities with catalyst 2 (see Figure 1). Compound 7 is a highly electron rich Rh(I) complex that we previously used to design metallic frustrated Lewis pairs (FLPs). [23] Compound 8 contain a bulky CptBu ligand as well as chelating bulky phosphine, [24] while 9 and 10 are stabilized by π‐type coordination with flanking aryl rings of a terphenyl fragment, [25] as occurs in catalyst 2. While all these rhodium complexes revealed comparable activity and selectivity to that determined for 2 during hydrosilylation using Et3SiH (entries 6–11), its highly unusual α‐selectivity when using SiPh3H found no competitor (entries 13–18). Only for catalyst 5 the α‐isomer was the major product, though in rather low yield (14 % after 96 h at 100 °C)
As for the mechanism by which E‐isomers are obtained with catalyst 2, not surprisingly, our initial experiments on the hydrogenation of DPA provided evidence for the stepwise formation of trans‐stilbene, after initial suprafacial hydrogenation to cis‐stilbene, followed by isomerization (entries 1 and 2, Table 1). We decided to explore whether catalyst 2 could mediate cis/trans olefin isomerization in the absence of dihydrogen (Table 3). Heating a benzene solution of cis‐stilbene in the presence of compound 2 (10 mol%) yielded indeed full conversion into the corresponding trans‐olefin despite the absence of H2, which contrasts with most previously reported systems that require its presence.[ 16f , 16g , 18b , 26 ] Other cis‐olefins were also isomerized with good yields and, contrary to our expectations, the introduction of a bulky tert‐butyl group in CH3CH=CH( t Bu) compared to CH3CH=CHCH2CH3 enhanced the isomerization rate (entries 3 and 4). As control experiments, we attempted the same transformation using [RhCl(COD)]2 (4) and Wilkinson's catalyst (6) as the rhodium precursor and under otherwise identical conditions (entries 5 and 6). However, no trace of the trans‐isomer was detected. Only the electron‐rich catalyst 7 exhibits reduced activity for isomerization under our experimental conditions, though to a considerable lesser extent (<10 %, entry 7). However, rhodium catalysts 8–10 (entries 8–10), despite of providing a similar congested environment around the transition metal, in the cases of 9 and 10 even with analogous terphenyl coordination, do not reveal any reactivity. These results suggest that the presence of the germyl fragment in catalyst 2 is important to facilitate isomerization.
Table 3.
Isomerisation of Z‐olefins catalysed by rhodium complexes.
|
| ||||||
|---|---|---|---|---|---|---|
|
Entry[a] |
Substrate |
Cat. |
t |
T |
Yield[b] |
|
|
|
R |
R’ |
|
[h] |
[°C] |
[%] |
|
1 |
Ph |
Ph |
2 |
24 |
80 |
100 |
|
2 |
Ph |
Me |
2 |
12 |
80 |
77 |
|
3 |
Me |
Et |
2 |
24 |
120 |
75 |
|
4 |
Me |
t Bu |
2 |
6 |
80 |
100 |
|
5 |
Ph |
Ph |
5 |
24 |
80 |
0 |
|
6 |
Ph |
Ph |
6 |
24 |
80 |
0 |
|
7 |
Ph |
Ph |
7 |
24 |
80 |
<10 |
|
8 |
Ph |
Ph |
8 |
24 |
80 |
0 |
|
9 |
Ph |
Ph |
9 |
24 |
80 |
0 |
|
10 |
Ph |
Ph |
10 |
24 |
80 |
0 |

[a] Reaction conditions: alkene (0.03 mmol), catalyst (0.003 mmol; 10 mol%), C6D6 (0.5 mL). [b] Yields determined by 1H NMR spectroscopy using C6Me6 as internal standard.
Since catalyst 2 was not selective enough to produce E‐alkenes from internal alkynes due to partial overreduction to the corresponding alkanes, we decided to examine an alternative tandem catalytic protocol. We hypothesized that a combination of Lindlar's catalyst and compound 2 in a two‐step/one pot procedure could yield the corresponding E‐alkene without double hydrogenation to alkanes. A similar strategy has been previously used by Furukawa and Komatsu using two heterogeneous catalysts, more precisely Pd3Pb/SiO2 (semihydrogenation) and RhSb/SiO2 (isomerization). [27] However, to the best of our knowledge, this is the first time that a related approach is carried out with a mixed heterogeneous/homogeneous system for this transformation. In a typical process the alkyne is cis‐hydrogenated by Lindlar's catalyst and then, to avoid overreduction of the alkenes, the H2 atmosphere is replaced by argon and complex 2 is added to promote isomerization to the corresponding trans‐alkene. Table 4 collects the preliminary results of this one‐pot/two‐step procedure to access E‐olefins in moderate to good yields. Beyond the synthetic utility of the proposed method, it represents a compelling example of the benefits to improve catalytic performance derived from combining heterogeneous and homogeneous catalysts in one‐pot.
Table 4.
Tandem alkyne semihydrogenation/alkene isomerization mediated by the combination of Lindlar's catalyst and complex 2 in one‐pot/two‐steps.
|
| |||||||||
|---|---|---|---|---|---|---|---|---|---|
|
Entry[a] |
Substrate |
Step |
H2 [bar] |
t |
T |
Yield [%][b] |
|||
|
|
R |
R’ |
|
|
[h] |
[°C] |
Z |
E |
Alkane |
|
1 |
Ph |
Ph |
1 |
1.5 |
10 |
80 |
66 |
16 |
18 |
|
2 |
– |
43 |
80 |
0 |
81 |
19 |
|||
|
2 |
Ph |
Me |
1 |
1.5 |
3 |
80 |
81 |
19 |
0 |
|
2 |
– |
12 |
100 |
19 |
81 |
0 |
|||
|
3 |
Me |
Me |
1 |
1.0 |
2.5 |
25 |
77 |
11 |
12 |
|
2 |
– |
28 |
80 |
25 |
55 |
20 |
|||
|
4 |
Me |
nPr |
1 |
1.5 |
4 |
60 |
80 |
20 |
0 |
|
2 |
– |
28 |
100 |
25 |
75 |
0 |
|||
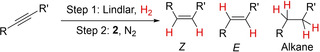
[a] Reaction conditions: alkyne (0.03 mmol), H2, Lindlard's catalyst (10 to 50 mol%), C6D6 (0.5 mL); then H2 substituted by N2 and 2 added (0.003 mmol; 10 mol%). [b] Yields determined by 1H NMR spectroscopy using C6Me6 as internal standard.
Mechanistic investigations and catalyst speciation. As stated above, our main focus with these investigations is on the mechanism by which mixed transition metal/tetrel complexes can affect bond activation and, eventually, catalysis. With this goal, we carried out some mechanistic investigations. We have already discussed that the overall semihydrogenation of DPA proceeds by two consecutive catalytic cycles, first the cis‐semihydrogenation of diphenylacetylene followed by a trans‐isomerization of cis‐stilbene. 1H NMR monitoring of product distribution along a catalytic reaction provides further visual evidence (Figure 2A). This assumption was also confirmed by the ability of 2 to mediate the trans‐isomerization of cis‐stilbene under the same catalytic conditions used for hydrogenation reactions. Interestingly, the latter process works equally well in the absence of H2 (Table 3), which further confirmed that the cis‐semihydrogenation and trans‐isomerization are processes independent from one another and thus amenable to individual kinetic analysis.
Figure 2.

(A) Extent of the reaction in toluene‐d 8 at 80 °C as determined by 1H NMR ([cat]0=11.92 mM, [dpa]0=0.0596 M, p(H2)0=1 bar). (B) Plotting of ln(v0)=f(ln[H2]) for initial pressures in hydrogen of 0.5, 1 and 1,5 bars. (C) Variable Time Normalization Analysis for a partial order of 0.5 in catalyst). (D) Variable Time Normalization Analysis for a partial order of 0 in diphenylacetylene.
We first decided to examine catalyst speciation in solution. We have previously demonstrated that complex 2 quantitatively evolves into 3 (see Figure 1) upon heating at 80 °C for seven hours. [10] It seems obvious that this species must be present under catalytic conditions. To evaluate the catalytic competence of 2 vs 3 we monitored by 1H NMR, using C6Me6 as internal standard, the hydrogenation of DPA under our standard catalytic conditions with both rhodium complexes. The evolution of the disappearance of DPA and the formation of the hydrogenated products was identical within the experimental error in both cases (see Figures S26). In line with a careful monitoring of the reaction by 1H NMR, a fast conversion of compound 2 into compound 3 is observed in the conditions of the reaction. For instance, after 1 h at 80 °C, only complex 3 is present in solution and thus we attribute it as the main resting state.
As stated above, we have generated compound 2 (and thus 3) in situ from the reaction of the chloride complex 1 and NaBArF. We performed an additional experiment to examine the rate of the reaction when catalysed from an isolated sample of complex 3 versus its in situ generation (Figure S26). Once more, we found no difference which indicates that there is no apparent effect on the presence of excess NaBArF or produced sodium chloride.
Moreover, regarding catalyst speciciation and in accordance with our previous work on this Rh/Ge system, [10] our NMR investigations herein have as well revealed that a prolongated standing of 3 in toluene at 80 °C yields variable amounts of complex 4 through a dehydrogenation reaction. Even though the formation of 4 is not observed or only detected in very small amounts during the reaction time (<5 % with respect to total Rh/Ge catalyst), the capacity of 4 to catalyse the trans‐semihydrogenation of DPA has also been studied and compared to the activity of complex 3 (see Figure S27). After 8 h under our standard conditions (1 bar, tol‐d8 , 80 °C), we measured a cis‐stilbene/trans‐stilbene/alkane of 32 : 37 : 24 for 4, while a 41 : 26 : 13 ratio was observed for catalyst 3. Despite the dehydrogenated compound 4 being slightly more active than 3, it is also less selective towards the formation of the E‐alkene due to a higher propensity to achieve total hydrogenation. This is also confirmed after full conversion of DPA after prolonged reaction times which lead to trans‐stilbene/alkane ratios of 67 : 32 and 75 : 23 for 4 and 3, respectively. Moreover, we discard the direct participation of 4 also on the basis of its irreversible formation from 3 and the fact that it does not accumulate over the course of the catalytic reactions. We have also investigated the reactivity between 4 and D2 but we have not observed any isotopic labelling of the benzylic positions of the terphenyl fragments which, once more, speak against the reversible formation of 3 from 4 and thus on the actual role of the later during catalysis.
We also performed some stoichiometric experiments between complexes 2 and 3 and the substrates present in the catalytic runs, namely, dihydrogen, DPA and two isomers of stilbene in independent reactions (Scheme 1). However, no potential intermediates could be detected in any of the cases which supports the notion of germyl complex 3 as the main resting state during catalysis. Although the proportion of 2 and 3 varies due to the conversion of the former into the latter (completed in less than one hour), the overall amount of both of them remain constant as observed by 1H NMR monitoring (Figure 2A).
Scheme 1.

Attempted stoichiometric reactions of complexes 2 and 3 with dihydrogen, diphenylacetylene and stilbene.
Cis‐semihydrogenation. Previous experimental and theoretical investigations on the cis‐hydrogenation of alkynes catalysed by transition metal (TM) complexes have proposed several mechanisms disclosing the same elementary steps, namely hydrogen complexation/oxidative addition (or heterolysis), alkyne coordination/migratory insertion in the TM−H bond and C−H reductive elimination (or σ‐metathesis). Nevertheless, their order of occurrence within the catalytic cycles are highly dependent of the nature of the active species. For instance, oxidative addition of dihydrogen may occur before alkyne coordination,[ 18a , 18b , 28 ] after alkyne coordination but before its insertion [23] or following the latter process.[ 16e , 16h ]
The partial orders with respect to the concentrations of catalyst, alkyne and hydrogen are valuable elements to provide insights about the reaction profile. To obtain this information we performed Variable Time Normalization Analysis (Bures’ VTNA method) [29] in toluene‐d8 at 80 °C (Figures 2C and 2D; see Supporting Information for more details). These studies evinced a first order in catalyst and a zero order in diphenylacetylene. [12] Also, a first order with respect to hydrogen concentration was determined by classical initial rates method. The plotting of ln(v0) vs ln[H2] for different initial pressures of hydrogen (0.5–1.5 bar) led to a straight line with an approximate slope of 1 (Figure 2B). Thus, the rate law for this pseudo second order process can be stated as below in Equation 1. [30]
| (1) |
with kobs(80 °C)=0.2637±0.0176 mol−1 L s−1. As substantiated by the non‐zero orders in catalyst and hydrogen, both participate in the reaction coordinate before or during the rate limiting step. This scenario has also been supported by isotopic studies in which slower rates were observed when hydrogen was substituted by deuterium gas. It is not unusual that rate determining oxidative addition of H2 in related transition metal catalysed processes exhibits apparent inverse kinetic isotopic effect (KIE <1), as a direct consequence of a pre‐equilibrium involving the complexation of hydrogen prior to H−H bond cleavage with an associated inverse Equilibrium Isotope Effect. [31] Herein, a normal primary KIE of 3.52±0.26 was found (see Figure S24 and S25), which is also consistent, for instance, with a study reported by McIndoe and co‐workers where a KIE of 2.1 was measured for a Rh‐catalysed partial hydrogenation of alkyne. [25] Besides, this value is also larger than what would be expected if migratory insertion or reductive elimination were rate determining. [32] As example, a KIE of 1.64 was reported by Lu and co‐workers during the semihydrogenation of alkynes catalysed by a Rhodium‐Gallium heterobimetallic complex in which the rate determining step corresponds to alkyne migratory insertion. [23] Altogether, this data suggests a direct oxidative addition of dihydrogen as the rate limiting step, without the existence of any pre‐equilibrium with a putative σ‐H2 complex.
With this information on hand, we propose the catalytic cycle depicted in Scheme 2, right. Thus, the cis‐hydrogenation studied in this work proceeds through a monometallic cycle where 3 is first oxidized by hydrogen to generate the rhodium(III) trihydride intermediate A by rate determining oxidative addition ( (i) in Scheme 2). [33] The latter most likely undergoes syn‐insertion of the alkyne into a Rh−H bond to yield intermediate B (step (ii) ) where the metallic fragment and the newly incorporated hydrogen atom are in cis‐disposition. Finally, the C(sp 2)‐H reductive elimination (step (iii) ) occurs with retention of configuration and affords the selective formation of cis‐stilbene.
Scheme 2.

Proposed mechanisms for cis‐semihydrogenation of alkynes and trans‐isomerization of alkenes leading to the overall trans‐semihydrogenation of alkynes catalysed by 3. R.d.s.=rate‐determining‐step.
Trans ‐isomerization. We also investigated by kinetic methods the trans‐isomerization of cis‐stilbene in the presence of the Rh‐germyl catalyst 2 and its derivative 3. To be consistent with the kinetic investigations ran for the cis‐hydrogenation of diphenylacetylene, the isomerization of cis‐stilbene to trans‐stilbene has been studied in toluene‐d8 under the same range of concentrations. As stated previously, trans‐isomerization catalysed by 2 or 3 does not require hydrogen, though thermal activation remains necessary and no reaction takes place below 60 °C. As observed for the hydrogenation of alkynes, the resting state of the catalyst can be clearly identified by 1H NMR monitoring as a mixture of 2 and 3 that eventually evolves to the latter. Here also, no trace of 4 was observed in the reaction time. Similarly, the kinetic law has been experimentally determined using the VTNA method described above, while in this peculiar case a classical kinetic analysis was not conclusive (Figures S38, S39 and S40, see Supporting Information for more details). The plotting of [trans‐stilbene] vs ([cat]n ⋅ time) and [trans‐stilbene] vs ([cis‐stilbene]n ⋅ time) show good overlays for partial orders of 1 in catalyst and alkene. [34] Thus, the rate law can be described as an apparent second order reaction ruled by Equation 2.
| (2) |
with kobs(80 °C)=3.88 ⋅ 10−3 ±0.02 ⋅ 10−3 L ⋅ mol−1 ⋅ s−1. We also evaluated herein kinetic isotopic effects. To do that, cis‐stilbene‐d 2 was prepared by treating DPA with D2 in the presence of Lindlar's catalyst. The isomerization of cis‐stilbene‐d 2 is considerably slower than with its non‐deuterated isotopologue, revealing a strong kinetic isotopic effect of 7.12. Such a normal primary KIE is consistent with a turnover limiting migratory insertion of the alkene involving cleavage of the Rh−H bond. Interestingly, the measured KIE represents quite a large value compared to what is usually featured in the literature. [30] Nonetheless, in most cases the smaller KIE values measured for other related processes are usually a direct consequence of the existence of an inverse Equilibrium Isotope Effect. This may again suggest the absence of a pre‐equilibrium associated to alkyne coordination as it was the case in the foregoing activation of H2 during cis‐semihydrogenation.
To further support the notion of a rate‐limiting migratory insertion event, we carried out an Eyring analysis between 80 °C and 100 °C. This experiments afforded a good estimation of the activation parameters with an enthalpy of activation of ΔHǂ=20.7±0.5 kcal ⋅ mol−1 and an entropy of activation of ΔSǂ=−10.5±1.4 cal ⋅ K−1 ⋅ mol−1. The large negative contribution of the entropy clearly pinpoints an associative elementary step and is highly suggestive of a rate determining insertion. A positive value would have been expected for C−H reductive elimination as the alternative rate‐limiting‐step. It is noteworthy to mention that an even higher absolute value (≈35 cal ⋅ K−1 ⋅ mol−1) was found in a similar system and was attributed to a highly ordered metallacycle transition state by means of theoretical and experimental studies. [23] A smaller value may suggest a less concerted mechanism. The small enthalpy of activation, about 14 kcal ⋅ mol−1 lower than for the reported gallium‐rhodium catalyst developed by Lu and co‐workers, [25] illustrates the prospects of designing other germanium‐rhodium catalysts.
Altogether these results support a mechanism involving the initial and turnover limiting migratory insertion of the alkene into the RhI‐H bond ((iv) in Scheme 2 ) to yield intermediate C. This insertion is followed by a rotation around the C(sp 3)−C(sp 3) bond of the substrate leading to the generation of trans‐stilbene via C−H reductive elimination (step (v) ).
Conclusion
In summary, the catalytic studies discussed in the previous sections demonstrate the prospects of combining main group and transition metals in unusual architectures. At variance with other common rhodium catalysts, the presence of the germyl fragment with two bulky terphenyl substituents as the only embracing ligand for the rhodium centre permits the latter to mediate cis/trans‐olefin isomerization. In fact, the unusual semihydrogenation of internal alkynes towards trans‐alkenes has been achieved with moderate to good selectivity. This selectivity towards E‐isomers was increased by a tandem catalytic one‐pot/two‐step procedure combining Lindlar's catalyst and our rhodium/germyl system. Kinetic mechanistic investigations provide an overall mechanistic picture with two independent cycles for cis‐semihydrogenation and trans‐isomerization. Our mechanistic investigations do not support at this stage the direct participation of the germanium centre in the reported bond activation events. However, its presence is essential for the observed selectivities through alkyne isomerization and also for hydrosilylation using SiPh3H, likely due to a combination of the strongly σ‐donating character and the high steric profile of the substituents on germanium. Besides, the evaluation of activation parameters for trans‐isomerization advises pursuing other related structures for this and related transformations.
Experimental Section
General considerations. All manipulations were carried out using standard Schlenk and glove‐box techniques, under an atmosphere of argon and of high purity nitrogen, respectively. All solvents were dried, stored over 4 Å molecular sieves, and degassed prior to use. N‐pentane (C5H12) was distilled under nitrogen over sodium. [D6]Benzene was distilled under argon over sodium/benzophenone. Compounds [(ArMes2)2Ge:] (ArMes=C6H3‐2,6‐(C6H2‐2,4,6‐Me3)2), [11] [RhCl(COD)]2 (COD=1,5‐ciclooctadinene), [35] 7, [23] 8, [24] 9, [25] 10 [25] and NaBArF[36] were prepared as described previously. Compounds 1 and 2 were recently reported by our group. [10] Other chemicals were commercially available and used as received. Solution NMR spectra were recorded on Bruker AMX‐300, DRX‐400 and DRX‐500 spectrometers. Spectra were referenced to external SiMe4 (δ: 0 ppm) using the residual proton solvent peaks as internal standards (1H NMR experiments), or the characteristic resonances of the solvent nuclei (13C NMR experiments), while 31P was referenced to H3PO4. Spectral assignments were made by routine one‐ and two‐dimensional NMR experiments (1H, 13C{1H}, 31P{1H}, COSY, NOESY, HSQC and HMBC) where appropriate. Spectroscopic NMR resonances due to BArF anion: 1H: δ 8.37 (s, 8H, o‐Ar) and 7.67 (s, 4H, p‐Ar) and 13C: 162.3 (q, 1 J CB=50 Hz, ipso‐Ar), 135.1 (s, o‐Ar) and 117.7 (s, p‐Ar).
General procedure for the hydrosilylation of phenylsilane. In a J‐Young NMR tube, to a solution of precursor [(ArMes)2ClGeRh] (2.5 mg, 0.003 mmol) in C6D6 (0.5 mL) was added an excess of NaBArF (10 mg, 0.011 mmol). The tube was sealed with a Teflon screw‐cap to ensure air tightness and was vigorously shaken before being allowed to stand overnight at room temperature. The formation of catalyst 1 was monitored by 1H NMR. After total conversion, phenylacetylene (3.5 μL, 0.03 mmol) and the corresponding silane (0.03 mmol) were added. Finally, the J‐Young tube was placed in an oil bath at 80 °C or 100 °C (NB: no reaction occurs at room‐temperature). The extent of the reaction was monitored by 1H NMR.
General procedure for the hydrogenation of internal alkynes. In a J‐Young NMR tube, to a solution of precursor [(ArMes)2ClGeRh] (2.5 mg, 0.003 mmol) in C6D6 (0.5 mL) was added an excess of NaBArF (10 mg, 0.011 mmol). The tube was sealed with a Teflon screw‐cap to ensure air tightness and was vigorously shaken before being allowed to stand overnight at room temperature. The formation of catalyst 1 was monitored by 1H NMR. After total conversion, the corresponding alkyne (0.03 mmol) and H2 (1 bar) were added. Finally, the J‐Young tube was placed in an oil bath at 80 °C or 120 °C (NB: no reaction occurs at room‐temperature). The tube was continuously shaken over ca. 10 min time periods to avoid hydrogen diffusion limitations (discarded by 1H NMR analysis). The extent of the reaction was monitored by 1H NMR.
General procedure for the trans‐isomerization of cis‐alkenes. In a J‐Young NMR tube, to a solution of precursor [(ArMes)2ClGeRh] (2.5 mg, 0.003 mmol) in C6D6 (0.5 mL) was added an excess of NaBArF (10 mg, 0.011 mmol). The tube was sealed with a Teflon screw‐cap to ensure air tightness and was vigorously shaken before being allowed to stand overnight at room temperature. The formation of catalyst 1 was monitored by 1H NMR. After total conversion, the corresponding cis‐alkene (0.03 mmol) was added. Finally, the J‐Young tube was placed in an oil bath at 80 °C (NB: no reaction occurs at room‐temperature). The extent of the reaction was monitored by 1H NMR.
General procedure for the one pot sequential cis‐hydrogenation/trans‐isomerization of internal alkynes. A J‐Young NMR tube was charged with Lindlar's catalyst (10 to 50 mol%). The corresponding internal alkyne (0.03 mmol, 1 equiv.) and hexamethylbenzene (0.03 mmol, 1 equiv.) were incorporated by adding 0.1 mL of their freshly prepared solutions (0.3 mM) in C6D6. Next, the total volume was adjusted to 0.5 mL by addition of 0.3 mL of C6D6 and the tube was sealed with a Teflon screw‐cap to ensure air‐tightness. The solution was degassed using freeze‐pump technique (3 cycles) prior loading of the NMR tube with hydrogen (1 bar) at room temperature at the conditions stated in Table 4. The extent of the reaction was monitored by 1H NMR allowing to determine the proportion of cis‐ and trans‐alkene (stemming from the semi‐hydrogenation) and alkane (resulting from full hydrogenation). Afterwards, hydrogen was removed by freeze‐pump technique (3 cycles) before precursor [(ArMes)2ClGeRh] (2.5 mg, 0.003 mmol, 10 mol%) and NaBArF (20 mg, 0.0226 mmol) were added. The solution was let stand overnight at room temperature to ensure the complete formation of 1 (as assessed by 1H NMR). Finally, the reaction mixture was heated under the conditions stated in Table 4 and the extent of the trans‐isomerization was monitored by 1H NMR.
Conflict of interest
The authors declare no conflict of interest.
1.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
This work has been supported by the European Research Council (ERC Starting Grant, CoopCat, 756575) and Junta de Andalucia (P18‐FR‐4688). SB acknowledges Marie Skłodowska‐Curie program for a postdoctoral fellowship (project 101023461). Dr. Marta Roselló is acknowledged for helpful discussions.
S. Bajo, C. A. Theulier, J. Campos, ChemCatChem 2022, 14, e202200157.
Data Availability Statement
The data that support the findings of this study are available in the supplementary material of this article.
References
- 1.
- 1a. Devillard M., Bouhadir G., Bourissou D., Angew. Chem. Int. Ed. 2015, 54, 730–732; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 740–742; [Google Scholar]
- 1b. Bouhadir G., Bourissou D., Chem. Soc. Rev. 2016, 45, 1065–1079; [DOI] [PubMed] [Google Scholar]
- 1c. You D., Gabbai F. P., Trends Chem. 2019, 1, 485–496. [Google Scholar]
- 2.
- 2a. Power P. P., Nature 2010, 463, 171–177; [DOI] [PubMed] [Google Scholar]
- 2b. Power P. P., Chem. Rec. 2012, 12, 238–255; [DOI] [PubMed] [Google Scholar]
- 2c. Yadav S., Saha S., Sen S. S., ChemCatChem 2016, 8, 486–501. [Google Scholar]
- 3. Somerville R. J., Campos J., Eur. J. Inorg. Chem. 2021, 2021, 3488–3498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.
- 4a. Hadlington T. J., Szilvási T., Driess M., Angew. Chem. Int. Ed. 2017, 56, 7470–7474; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 26, 7578–7582; [Google Scholar]
- 4b. Hadlington T. J., Szilvási T., Driess M., J. Am. Chem. Soc. 2019, 141, 3304–3314; [DOI] [PubMed] [Google Scholar]
- 4c. Takahashi S., Bellan E., Baceiredo A., Saffon-Merceron N., Massou S., Nakata N., Hashizume D., Branchadell V., Kato T., Angew. Chem. Int. Ed. 2019, 58, 10310–10314; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 10416–10420; [Google Scholar]
- 4d. Juckel M. M., Hicks J., Jiang D., Zhao L., Frenking G., Jones C., Chem. Commun. 2017, 53, 12692–12695; [DOI] [PubMed] [Google Scholar]
- 4e. Hayes P. G., Waterman R., Glaser P. B., Tilley T. D., Organometallics 2009, 28, 5082–5089. [Google Scholar]
- 5.
- 5a. Price J. S., Emslie D. J. H., Britten J. F., Angew. Chem. Int. Ed. 2017, 56, 6223–6227; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 6319–6323; [Google Scholar]
- 5b. Price J. S., Emslie D. J. H., Chem. Sci. 2019, 10, 10853–10869; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5c. Whited M. T., Zhang J., Conley A. M., Ma S., Janzen D. E., Kohen D., Angew. Chem. Int. Ed. 2021, 60, 1615–1619; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 1639–1643. [Google Scholar]
- 6.
- 6a. Asay M., Jones C., Driess M., Chem. Rev. 2011, 111, 354–396; [DOI] [PubMed] [Google Scholar]
- 6b. Alvarez-Rodriguez L., Cabeza J. A., Garcia-Alvarez P., Polo D., Coord. Chem. Rev. 2015, 300, 1–28; [Google Scholar]
- 6c. Zhou Y.-P., Driess M., Angew. Chem. Int. Ed. 2019, 58, 3715–3728; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 3753–3766; [Google Scholar]
- 6d. Cabeza J. A., Garcia-Alvarez P., Laglera-Gandara C. J., Eur. J. Inorg. Chem. 2020, 2020, 784–795. [Google Scholar]
- 7.
- 7a. Glaser P. B., Tilley T. D., J. Am. Chem. Soc. 2003, 125, 13640–13641; [DOI] [PubMed] [Google Scholar]
- 7b. Fasulo M. E., Lipke M. C., Tilley T. D., Chem. Sci. 2013, 4, 3882–3887. [Google Scholar]
- 8. Schneider N., Finger M., Haferkemper C., Bellemin-Laponnaz S., Hofmann P., Gade L. H., Angew. Chem. Int. Ed. 2009, 48, 1609–1613; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 1637–1641. [Google Scholar]
- 9. Buil M. L., Cabeza J. A., Esteruelas M. A., Izquierdo S., Laglera-Gándara C. J., Nicasio A. I., Oñate E., Inorg. Chem. 2021, 60, 16860–16870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bajo S., Alcaide M. M., López-Serrano J., Campos J., Chem. Eur. J. 2021, 27, 16422–16428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Simons R. S., Pu L., Olmstead M. M., Power P. P., Organometallics 1997, 16, 1920–1925. [Google Scholar]
- 12.
- 12a. Pu L., Twamley B., Haubrich S. T., Olmstead M. M., Mork B. V., Simons R. S., Power P. P., J. Am. Chem. Soc. 2000, 122, 650–656; [Google Scholar]
- 12b. Filippou A. C., Weidemann N., Philippopoulos A. I., Schnakenburg G., Angew. Chem. Int. Ed. 2006, 45, 5987–5991; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 6133–6137; [Google Scholar]
- 12c. Matsumoto T., Nakaya Y., Itakura N., Tatsumi K., J. Am. Chem. Soc. 2008, 130, 2458–2459; [DOI] [PubMed] [Google Scholar]
- 12d. Matsumoto T., Itakura N., Nakaya Y., Tatsumi K., Chem. Commun. 2011, 47, 1030–1032. [DOI] [PubMed] [Google Scholar]
- 13.
- 13a. Molnár Á., Sárkány A., Varga M., J. Mol. Catal. A 2001, 173, 185–221; [Google Scholar]
- 13b. Swamy K. C. K., Reddy A. S., Sandeep K., Kalyani A., Tetrahedron Lett. 2018, 59, 419–429; [Google Scholar]
- 13c. Decker D., Drexler H.-J., Heller D., Beweries T., Catal. Sci. Technol. 2020, 10, 6449–6463. [Google Scholar]
- 14. Lindlar H., Helv. Chim. Acta 1952, 35, 446–450. [Google Scholar]
- 15.See for example:
- 15a. Van Laren M. W., Elsevier C. J., Angew. Chem. Int. Ed. 1999, 38, 3715–3717; [DOI] [PubMed] [Google Scholar]
- 15b. Hauwert P., Maestri G., Sprengers J. W., Catellani M., Elsevier C. J., Angew. Chem. Int. Ed. 2008, 47, 3223–3226; [DOI] [PubMed] [Google Scholar]
- 15c. La Pierre H. S., Arnold J., Toste F. D., Angew. Chem. Int. Ed. 2011, 50, 3900–3903; [DOI] [PubMed] [Google Scholar]
- 15d. Warsink S., Bosman S., Weigand J. J., Elsevier C. J., Appl. Organomet. Chem. 2011, 25, 276–282; [Google Scholar]
- 15e. Drost R. M., Bouwens T., van Leest N. P., de Bruin B., Elsevier C. J., ACS Catal. 2014, 4, 1349–1357; [Google Scholar]
- 15f. Drost R. M., Broere D. L. J., Hoogenboom J., de Baan S. N., Lutz M., de Bruin B., Elsevier C. J., Eur. J. Inorg. Chem. 2015, 2015, 982–996; [Google Scholar]
- 15g. Chen C., Huang Y., Zhang Z., Dong X.-Q., Zhang X., Chem. Commun. 2017, 53, 4612–4615; [DOI] [PubMed] [Google Scholar]
- 15h. Korytia-ková E., Thiel N. O., Pape F., Teichert J. F., Chem. Commun. 2017, 53, 732–735; [DOI] [PubMed] [Google Scholar]
- 15i. Brzozowska A., Azofra L. M., Zubar V., Atodiresei I., Cavallo L., Rueping M., El-Sepelgy O., ACS Catal. 2018, 8, 4103–4109; [Google Scholar]
- 15j. Johnson C., Albrecht M., Catal. Sci. Technol. 2018, 8, 2779–2783; [Google Scholar]
- 15k. Osborn J. A., Jardine F. H., Young J. F., Wilkinson G., J. Chem. Soc. A 1966, 1711–1732; [Google Scholar]
- 15l. Tseng K−N. T., Kampf J. W., Szymczak N. K., J. Am. Chem. Soc. 2016, 138, 10378–10381. [DOI] [PubMed] [Google Scholar]
- 16.
- 16a. Schleyer D., Niessen H. G., Bargon J., New J. Chem. 2001, 25, 423–426; [Google Scholar]
- 16b. Radkowski K., Sundararaju B., Fürstner A., Angew. Chem. Int. Ed. 2013, 52, 355–360; [DOI] [PubMed] [Google Scholar]
- 16c. Srimani D., Diskin-Posner Y., Ben-David Y., Milstein D., Angew. Chem. Int. Ed. 2013, 52, 14131–14134; [DOI] [PubMed] [Google Scholar]
- 16d. Tokmic K., Fout A. R., J. Am. Chem. Soc. 2016, 138, 13700–13705; [DOI] [PubMed] [Google Scholar]
- 16e. Murugesan K., Bheeter C. B., Linnebank P. R., Spannenberg A., Reek J. N. H., Jagadeesh R. V., Beller M., ChemSusChem 2019, 12, 3363–3369; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16f. Yadav S., Dutta I., Saha S., Das S., Pati S. K., Choudhury J., Bera J. K., Organometallics 2020, 39, 3212–3223; [Google Scholar]
- 16g. Guthertz A., Leutzsch M., Wolf L. M., Gupta P., Rummelt S. M., Goddard R., Farès C., Thiel W., Fürstner A., J. Am. Chem. Soc. 2018, 140, 3156–3169; [DOI] [PubMed] [Google Scholar]
- 16h. Biberger T., Gordon C. P., Leutzsch M., Peil S., Guthertz A., Copéret C., Fürstner A., Angew. Chem. Int. Ed. 2019, 58, 8845–8850; [DOI] [PubMed] [Google Scholar]
- 16i. Thiel N. O., Kaewmee B., Tran Ngoc T., Teichert J. F., Chem. Eur. J. 2020, 26, 1597–1603; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16j. Hale D. J., Ferguson M. J., Turculet L., ACS Catal. 2022, 12, 146–155. [Google Scholar]
- 17.
- 17a. Karunananda M. K., Mankad N. P., J. Am. Chem. Soc. 2015, 137, 14598–14601; [DOI] [PubMed] [Google Scholar]
- 17b. Zhang Y., Karunananda M. K., Yu H.-C., Clark K. J., Williams W., Mankad N. P., Ess D. H., ACS Catal. 2019, 9, 2657–2663; [Google Scholar]
- 17c. Higashida K., Mashima K., Chem. Lett. 2016, 45, 866–868; [Google Scholar]
- 17d. Takemoto S., Kitamura M., Saruwatari S., Isono A., Takada Y., Nishimori R., Tsujiwaki M., Sakaue N., Matsuzaka H., Dalton Trans. 2019, 48, 1161–1165; [DOI] [PubMed] [Google Scholar]
- 17e. Gramigna K. M., Dickie D. A., Foxman B. M., Thomas C. M., ACS Catal. 2019, 9, 3153–3164; [Google Scholar]
- 17f. Ramirez B. L., Lu C. C., J. Am. Chem. Soc. 2020, 142, 5396–5407. [DOI] [PubMed] [Google Scholar]
- 18.
- 18a. Desai S. P., Ye J., Zheng J., Ferrandon M. S., Webber T. E., Platero-Prats A. E., Duan J., Garcia-Holley P., Camaioni D. M., Chapman K. W., Delferro M., Farha O. K., Fulton J. L., Gagliardi L., Lercher J. A., Penn R. L., Stein A., Lu C. C., J. Am. Chem. Soc. 2018, 140, 15309–15318; [DOI] [PubMed] [Google Scholar]
- 18b. Desai S. P., Ye J., Islamoglu T., Farha O. K., Lu C. C., Organometallics 2019, 38, 3466–3473. [Google Scholar]
- 19. He P., Hu M.-Y., Zhang X.-Y., Zhu S.-F., Synthesis 2022, 54, 49–66. [Google Scholar]
- 20.
- 20a. Marciniec B., Maciejewski H., Pietraszuk C., Pawluć P., In Hydrosilyaltion: A Comprehensive Review on Recent Advances (Ed.: Marciniec B.) Springer, Berlin, 2009; [Google Scholar]
- 20b. Almeida L. D., Wang H., Junge K., Cui X., Beller M., Angew. Chem. Int. Ed. 2021, 60, 550–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.
- 21a. Mutoh Y., Yamamoto K., Mohara Y., Saito S., Chem. Rec. 2021, 21, 3429–3441; [DOI] [PubMed] [Google Scholar]
- 21b. Sun J., Deng L., ACS Catal. 2016, 6, 290–300; [Google Scholar]
- 21c. Zaranek M., Marciniec B., Pawluć P., Org. Chem. Front. 2016, 3, 1337–1344; [Google Scholar]
- 21d. Wen H., Liu G., Huang Z., Coord. Chem. Rev. 2019, 386, 138–153. [Google Scholar]
- 22.
- 22a. Rivero-Crespo M. A., Leyva-Pérez A., Corma A., Chem. Eur. J. 2017, 23, 1702–1708; [DOI] [PubMed] [Google Scholar]
- 22b. Zhang S., Ibrahim J. J., Yang Y., Org. Lett. 2018, 20, 6265–6269; [DOI] [PubMed] [Google Scholar]
- 22c. Guo J., Lu Z., Angew. Chem. Int. Ed. 2016, 55, 10835–10838; [DOI] [PubMed] [Google Scholar]
- 22d. Trost B. M., Ball Z. T., J. Am. Chem. Soc. 2001, 123, 12726–12727; [DOI] [PubMed] [Google Scholar]
- 22e. Hu M., He P., Qiao T., Sun W., Li W., Lian J., Li J., Zhu S., J. Am. Chem. Soc. 2020, 142, 16894–16902; [DOI] [PubMed] [Google Scholar]
- 22f. Wang D., Lai Y., Wang P., Leng X., Xiao J., Deng L., J. Am. Chem. Soc. 2021, 143, 12847–12856. [DOI] [PubMed] [Google Scholar]
- 23. Alférez M. G., Hidalgo N., Moreno J. J., Campos J., J. Angew. Chem. Int. Ed. 2020, 59, 20863–20867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Espada M. F., Esqueda A. C., Campos J., Rubio M., López-Serrano J., Álvarez E., Maya C., Carmona E., Organometallics 2018, 37, 11–21. [Google Scholar]
- 25. Moreno J. J., Espada M. F., Krüger E., López-Serrano J., Campos J., Carmona E., Eur. J. Inorg. Chem. 2018, 2309–2321. [Google Scholar]
- 26.
- 26a. Azcarate I., Costentin C., Robert M., Savéant J.-M., J. Am. Chem. Soc. 2016, 138, 16639–16644; [DOI] [PubMed] [Google Scholar]
- 26b. Ramirez B. L., Lu C. C., J. Am. Chem. Soc. 2020, 142, 5396–5407. [DOI] [PubMed] [Google Scholar]
- 27.
- 27a. Furukawa S., Yokoyama A., Komatsu T., ACS Catal. 2014, 4, 3581–3585; [Google Scholar]
- 27b. Furukawa S., Komatsu T., ACS Catal. 2016, 6, 2121–2125. [Google Scholar]
- 28. Luo J., Oliver A. G., McIndoe J. S., Dalton Trans. 2013, 42, 11312–11318. [DOI] [PubMed] [Google Scholar]
- 29.
- 29a. Burés J., Angew. Chem. Int. Ed. 2016, 55, 2028–2031; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29b. Burés J., Angew. Chem. Int. Ed. 2016, 55, 16084–16087; [DOI] [PubMed] [Google Scholar]
- 29c. Martínez-Carrión A., Howlett M. G., Alamillo-Ferrer C., Clayton A. D., Bourne R. A., Codina A., Vidal-Ferran A., Adams R. W., Burés J., Angew. Chem. Int. Ed. 2019, 58, 10189–10193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.See Supplementary Information for more details concerning the experimental resolution of the kinetic law.
- 31.
- 31a. Janak K. E., Parkin G., Organometallics 2003, 22, 4378–4380; [Google Scholar]
- 31b. Zhu G., Janak K. E., Parkin G., Chem. Commun. 2006, 23, 2501–2503. [DOI] [PubMed] [Google Scholar]
- 32.
- 32a. Rosenberg E., Polyhedron 1989, 8, 383–405; [Google Scholar]
- 32b. Gómez-Gallego M., Sierra M. A., Chem. Rev. 2011, 111, 4857–4963. [DOI] [PubMed] [Google Scholar]
- 33.These species are known from the literature to perform hydrogenation of unsaturated substrate. See: Bianchini C., Meli A., Peruzzini M., Vizza F., Albinati A., Organometallics 1990, 9, 2283–2291. [Google Scholar]
- 34.Even better fits were found for broken orders of 1.2 in catalyst and 0.8 in alkene, an effect that we attribute to the “elasticity” of the catalysis. See: Burés J., Top. Catal. 2017, 60, 631–633. [Google Scholar]
- 35. Giordano G., Crabtree R. H., Inorg. Synth. 1979, 19, 218. [Google Scholar]
- 36. Martínez-Martínez A. J., Weller A. S., Dalton Trans. 2019, 48, 3551–3554. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Data Availability Statement
The data that support the findings of this study are available in the supplementary material of this article.