

Abstract
Purpose:
Despite the established role of EGFR tyrosine kinase inhibitors (TKIs) in EGFR-mutated NSCLC, drug resistance inevitably ensues, with a paucity of treatment options especially in EGFRT790M-negative resistance.
Experimental Design:
We performed whole-exome and transcriptome analysis of 59 patients with first- and second-generation EGFR TKI-resistant metastatic EGFR-mutated NSCLC to characterize and compare molecular alterations mediating resistance in T790M-positive (T790M+) and -negative (T790M−) disease.
Results:
Transcriptomic analysis revealed ubiquitous loss of adenocarcinoma lineage gene expression in T790M− tumors, orthogonally validated using multiplex IHC. There was enrichment of genomic features such as TP53 alterations, 3q chromosomal amplifications, whole-genome doubling and nonaging mutational signatures in T790M− tumors. Almost half of resistant tumors were further classified as immunehot, with clinical outcomes conditional on immune cell-infiltration state and T790M status. Finally, using a Bayesian statistical approach, we explored how T790M− and T790M+ disease might be predicted using comprehensive genomic and transcriptomic profiles of treatment-naïve patients.
Conclusions:
Our results illustrate the interplay between genetic alterations, cell lineage plasticity, and immune microenvironment in shaping divergent TKI resistance and outcome trajectories in EGFR-mutated NSCLC. Genomic and transcriptomic profiling may facilitate the design of bespoke therapeutic approaches tailored to a tumor's adaptive potential.
Translational Relevance.
Understanding resistance to targeted therapy requires in-depth analysis at multiple levels (single gene, chromosome, and transcriptome). Our results illustrate the interplay between genetic alterations, cell lineage plasticity, and the tumor microenvironment in shaping divergent TKI resistance and outcome trajectories in EGFR-mutated NSCLC. Transcriptomic analysis revealed ubiquitous loss of adenocarcinoma lineage gene expression in T790M− tumors. TP53 alterations, 3q chromosomal amplifications, whole-genome doubling and nonaging mutational signatures were also enriched in T790M− tumors. Genomic and transcriptomic profiling may facilitate the design of bespoke therapeutic approaches tailored to a tumor's adaptive potential.
Introduction
Activating driver mutations in the EGFR gene represent the most common therapeutically actionable alterations in non–small cell lung cancer (NSCLC). EGFR tyrosine kinase inhibitors (TKIs) are standard of care in the first-line setting for advanced or metastatic EGFR-mutated NSCLC. The upfront treatment paradigm consists of first-generation (1G; erlotinib, gefitinib), second-generation (2G; afatinib, dacomitinib) or third-generation (3G; osimertinib) EGFR TKI, either alone or in combination with other therapies. However, despite high response rates of up to 80%, drug resistance inevitably ensues after a median period of 10 to 17 months (1–4).
Recent trials have demonstrated improved outcomes with upfront combination platinum-doublet chemotherapy and EGFR TKI (5, 6), combination anti-angiogenic agents and EGFR TKI (7, 8), and dacomitinib or osimertinib alone compared with 1G EGFR TKI (9, 10). An observational study has also shown that a sequential treatment strategy with afatinib followed by osimertinib may result in improved outcomes in a significant proportion of patients (11, 12). Importantly, first-line osimertinib is still not reimbursed in many countries, and a lack of cost-effectiveness for osimertinib has been demonstrated in many diverse health care systems, including the United States (13–16). Together, this illustrates the increasing complexity in the selection and sequencing of treatment in EGFR-mutated NSCLC, with ongoing debate over optimal first-line therapy. Ultimately, improved patient selection incorporating clinical and molecular characteristics will be crucial to optimize therapeutic strategies (17).
The most common mechanism of resistance for 1G and 2G EGFR TKI is the EGFRT790M gatekeeper mutation, which emerges in 50% to 60% of patients (18–20). Osimertinib, which has high selectivity for T790M, was originally developed and approved for the treatment of T790M-positive (T790M+) resistance (21). Resistance mechanisms in remaining patients, as well as in 3G EGFR TKI-treated patients, are thought to be diverse, consisting of bypass signaling pathway activation such as MET or HER2 amplification, rare phenotypic change via small-cell transformation or epithelial–mesenchymal transition (22, 23), and alterations in other oncogenic drivers such as BRAF, KRAS, and RET, or the EGFR C797S mutation (24, 25). Collectively, T790M-negative (T790M−) EGFR TKI resistance remains a patient population with significant unmet medical need. Current therapeutic options are predominantly limited to cytotoxic chemotherapy, with low-response rates observed with immune checkpoint inhibitors (26). Although aforementioned previous studies have identified putative genomic alterations implicated in resistance, the approaches have largely been limited to genomic profiling with targeted panels (23, 24), or transcriptomic profiling without accompanying genomic analysis (27). Importantly, there has been a paucity of unbiased and integrative molecular profiling studies at the time of resistance for joint characterization of genomic and transcriptomic features underpinning the resistant state.
Here, through whole-exome and transcriptome analysis of 59 patients with 1G/2G TKI-resistant EGFR-mutated NSCLC, we present the first integrative genomic and transcriptomic description of the EGFR TKI resistance landscape. We first analyzed the extent that specific genetic alterations are recurrently associated with the development of resistance, reassessing the statistical evidence for many previously reported genetic resistance mechanisms beyond EGFRT790M mutations. By further contrasting the T790M+ (63%) and T790M− (37%) cohorts, we delineated the genomic and transcriptomic alterations underlying these two distinct resistance states. Strikingly, this revealed frequent and extensive de-differentiation and lineage plasticity in the T790M− cases. In addition, we explored the immune landscape of EGFR TKI resistance, demonstrating that many T790M− samples are “hot” tumors with shorter time to progression on first-line EGFR TKI, suggesting a role for targeting and modulating the immune microenvironment in this group of EGFR TKI-resistant tumors.
Materials and Methods
Patient cohort, sample collection, and clinical outcomes
All patients had stage IV NSCLC with an activating EGFR mutation at the point of biopsy and were resistant to a 1G/2G EGFR TKI (afatinib, erlotinib or gefitinib) at the National Cancer Center Singapore. All patients were enrolled on the Individualized Molecular Profiling for Allocation to Clinical Trials (IMPACT) study between February 2012 and September 2015 and provided written informed consent for fresh-frozen tissue collection and analysis. The IMPACT study protocol was approved by the SingHealth Centralized Institutional Review Board (CIRB ref No. 2011/441/B) in accordance with ethical principles outlined in the Belmont Report. Tumor tissue was collected via percutaneous biopsy or surgical resection. All tumor samples were collected before any 3G EGFR TKI therapy. Patient demographics, baseline disease features, treatment details, and patient outcomes were collected from the medical record. Time to progression (TTP) was defined as the time from first TKI treatment to the date of progressive disease (PD). Time-to-treatment failure (TTF) was defined as the duration of the first TKI treatment. For patient A452, initial TKI was ceased due to liver toxicity, and TTP/TTF was taken from the second TKI treatment. OS was defined as the time from diagnosis of stage IV disease to death. For comparative analysis, we used an in-house treatment-naïve lung adenocarcinoma cohort (28) with whole-exome sequencing (WES) and mRNA sequencing data; genomic and transcriptomic data were processed uniformly in both cohorts.
IHC and multiplex immunofluorescence
For histopathology review, specimens were handled using standard histopathological techniques, being formalin-fixed paraffin-embedded (FFPE), sectioned and stained with hematoxylin–eosin (H&E), and reviewed by two independent pathologists. FFPE tissue sections were processed according to a standard IHC protocol (29). mIF was performed using an Opal Multiplex fIHC kit (PerkinElmer Inc.), as previously described (29, 30). Slides were labeled with TTF1 (clone SPT24, Leica Biosystems), napsin A (clone IP64, Leica Biosystems), and L858R (clone 43B2, Cell Signaling Technology) for a histology panel, and similarly PD-L1 (clone E1L3N, Cell Signaling Technology), CD8 (clone 4B11, Leica Biosystems), CD68 (clone PGM1, Dako Agilent Technologies Inc.), Foxp3 (clone 236A/E7, Abcam), and CD39 (clone ENTPD1, OriGene Technologies Inc.) for an immune panel. Briefly, FFPE tissue sections were cut onto Bond Plus slides (Leica Biosystems Richmond) and heated at 60°C for 20 minutes. Tissue slides were then subjected to deparaffinization, rehydration, and heat-induced epitope retrieval (HIER) using a Leica Bond Max autostainer (Leica Biosystems Melbourne), before endogenous peroxidase blocking (Leica Biosystems Newcastle). Slides were incubated with a single clone of primary antibody followed by application of polymeric horseradish peroxidase–conjugated secondary antibodies (Leica Biosystems Newcastle). An appropriate Opal fluorophore-conjugated TSA (PerkinElmer) was then added at 1:100 dilution. Slides were rinsed with washing buffer after each step. Following TSA deposition, slides were again subjected to HIER to strip the tissue-bound primary/secondary antibody complexes and ready for labeling of the next marker. These steps were repeated until all markers were labeled and finally added with spectral DAPI (PerkinElmer) at 1:10 dilution. Slides were mounted in ProLong Diamond Anti-fade Mountant (Molecular Probes, Life Technologies) and cured in the dark at room temperature for 24 hours. Ten images (viable tumor regions were selected randomly by pathologists) were acquired for each case using a Vectra 3 pathology imaging system microscope (PerkinElmer, Inc.) and analyzed using inForm software (version 2.4.2; PerkinElmer, Inc.).
WES
One hundred to 400 ng of genomic DNA was sheared using Covaris to a size of 300 to 400 bp and subjected to library preparation using the NEBNext Ultra DNA Library Prep kit (New England Biolabs). In each library, six samples were pooled together and hybridized using SeqCap EZ Human Exome Library v3.0 kit (Nimblegen, Roche Diagnostics). Captured regions were washed, purified, amplified, and subjected to 2 × 151 sequencing on the Hiseq 4000 to obtain a median coverage of ×112 (range, ×73 to ×155). DNA from either paired peripheral blood mononuclear cells, saliva or normal tissue (Supplementary Table S1) of the same patient were sequenced as a germline control to obtain a median coverage of ×107 (range, ×70 to ×163). For patient A003 and A152, sequencing was carried out on an older platform with single-indexed adapters. Additional information on the mutation calling and copy-number variation identification is provided in the Supplementary Appendix. As WES data were only available for the treatment-naïve cohort, copy-number variations were detected using whole-exome data for cohort comparisons. Given the higher resolution of SNP array (due to the segmentation algorithm), genes with significant differences by copy-number alteration (homozygous deletion and ≥2 copy amplification) were further interrogated using SNP array data to confirm statistical significance.
Total RNA sequencing
Total RNA was extracted from the patient samples using the Qiagen AllPrep universal kit. One microgram of RNA from samples with RIN number of at least 3.2 was used for library preparation using TruSeq Stranded Total RNA Library Prep Gold. Multiple samples were barcoded using TruSeq RNA Single Indexes. Twelve samples were pooled and sequenced on one lane of HiSeq High output v3, 2 × 100 cycles, using Hiseq 2000 to obtain an average of 22.4 millions read (range, 13.8 to 24.7 millions). An in-house pipeline was used to perform RNA-seq quantification (https://github.com/gis-rpd/pipelines) for both the EGFR TKI-resistant and our treatment-naïve cohorts (LCCS). Briefly, raw RNA-seq sequences were aligned to human transcriptome (hg19 build) with STAR. Transcription of genes was quantified using the RSEM software. Gene expression profiles were quantile-normalized before analysis and visualization. For TCGA-LUAD RNA-seq data, we downloaded the upper-quartile normalized (FPKM-UQ) expression matrix from GDC (31). Mutations were annotated using mutations provided by the TCGA MC3 dataset (32).
Gene deconvolution in the TKI-resistant tumors was carried out using TUMERIC (33). To obtain purities with less bias, we estimated purities of each tumor using RNA-seq data with ESTIMATE (34) in addition to the SNP-array–based ASCAT and exome-based Sequenza (described above). As not all tumors have all types of sequencing data available, we imputed missing data based on a principal component analysis model using imputePCA function from R package missMDA (35). The purities estimates were then quantile normalized. We took the mean of the tumor purities across three methods as for TUMERIC deconvolution. Briefly, gene expression, log2(fpkm+1), was regressed against tumor purity using non-negative least squares regression. Regression lines were extrapolated to 0% and 100% purity to estimate expression in stromal and cancer cells, respectively. Tumors' tissues not from lung or have abnormally high expression of neuroendocrine and squamous carcinoma markers are excluded from deconvolution (Supplementary Fig. S1A) to ensure that our results are robust to tissues of origin and tumor histology. We carried out deconvolution in T790M+ and T790M− separately and calculated the differences between cancer and stroma comparing the two groups. Then, we permutated T790M labels to generate a null distribution to obtain P values defined as the probability of observing the differences given the null distribution. Details on test statistics can be found in the Supplementary Appendix.
Assignment of molecular subtypes were by carried out as described previously (36). Briefly, Pearson correlation was calculated with respect to the gene expression centroids provided (37) for each tumor. The subtype with the highest correlation was then assigned as the molecular subtype for each tumor. For clustering of immune subtype, we used R package CancerSubtypes (38) to conduct consensus k-mean clustering (function ExecuteCC, k = 2, repeats n = 1,000). Clustering was performed using the z-score standardized expression matrix of expanded immune GEP geneset consisting of CD38, CTLA4, and ENTPD1 in addition to the original 18 genes (39). Immune GEP score was calculated as the weighted sum of normalized expression values using Log2(FPKM+1) with weights (39). We used TIDE to compute three immune-suppressive genes signatures, namely MDSC (myeloid-derived suppressor cell), TAMs (tumor-associated macrophages) M2, and CAF. Only MDSC and TAM M2 showed strong differences comparing IH T790M+ and IH T790M−. CAF is shown in Supplementary Fig. S2.
Probabilistic forecasting of T790M resistance status following TKI treatment
We focused on genomic and transcriptomic features with significantly different prevalence in T790M+ and T790M− TKI-resistant patients (χ2 test). For TP53 alterations, we included both SNVs and genomic loss (Supplementary Fig. S3). The T790M− associated immune infiltration subtype was not included in this analysis because the prevalence of this feature had not been estimated for treatment-naïve tumors. To determine whether T790M− associated features were likely present before treatment, or alternatively acquired on/following TKI treatment, we compared (two-tailed Binomial test) the prevalence of features in the TKI-resistant and EGFR-mutated treatment-naïve tumors (Supplementary Table S2). This analysis identified three features (EGFR exon 19 deletion, WGD, TP53 alteration) with evidence of all alterations being present before treatment (P > 0.83). The prior odds for development of T790M+ disease was set to 1.8:1 (0.64:0.36). Assuming conditional independence of the three features given T790M status, we computed Bayes factors and posterior probabilities for developing T790M+ disease given each feature combination.
Statistical analyses
Patient characteristics were summarized using descriptive statistics. Kaplan–Meier survival analysis was performed to determine TTP and OS. Survival performance based on EGFRT790M status was tested using Cox proportional hazard model. Hazard ratio with 95% confidence intervals and associated log-rank P value was calculated. Proportion tests between groups were calculated using Pearson the 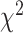 -test (with N-1 correction; ref. 40). Comparisons of gene expression and mutational signatures were conducted using t tests.
-test (with N-1 correction; ref. 40). Comparisons of gene expression and mutational signatures were conducted using t tests.
Data and code availability
Raw sequencing data have been deposited in the European Genome-phenome Archive (EGA, EGAS00001005389, http://www.ebi.ac.uk/ega/). Computational methods have been uploaded to a public git (https://gitlab.com/gis_khipin/ccr_tki_resistance_2021). Clinical records, somatic mutations, copy-number variations, gene expression data, and histological images from our study are hosted in OncoSG (https://src.gisapps.org/OncoSG/) that is publicly available.
Results
Patient characteristics
Tumor biopsies obtained from 59 patients with advanced EGFR-mutated NSCLC who had previously experienced PD on 1G/2G TKIs were interrogated (Fig. 1A; Supplementary Fig. S4A). EGFRT790M was detected in 38 (63%) patients (Fig. 1C). Median TTP on 1G/2G TKI was 10.3 months (range, 1.3–75.8 months), with 7.4 versus 13.6 months in the T790M− versus T790M+ groups, respectively (HR, 1.64; 95% CI, 0.95–2.83; P = 0.07; Supplementary Fig. S5). There were no differences in exposure to prior therapy between the T790M+ and T790M− groups, including EGFR TKI as last line of therapy (65.8% vs. 61.9%, P = 0.76), prior use of chemotherapy (76% vs. 66%, P = 0.75, Wilcoxon test), or duration of chemotherapy exposure (mean 215 days vs. 175 days, P = 0.75, Supplementary Fig. S4B). Consistent with previous reports (20), microscopic histologic transformation was rare, with one case of squamous transformation (A450; adenocarcinoma at baseline) and another case of mixed adenocarcinoma small-cell histology (A092) observed both at baseline and post resistance.
Figure 1.

Genomic correlates of EGFR TKI resistance. A, Treatment histories for individual patients; each bar color represents a treatment type. B, Sequencing experiments conducted in this study. C, Genomic landscape of EGFR TKI resistance. Mutations shown are previously proposed mechanisms (EGFR, ERBB2 and MET amplification; MDM2, RB1, PIK3CA, PTEN, PIK3CB alterations) or alterations in >5 patients that were significantly different between T790M+ and T790M− cohorts (*, P < 0.10; **, P < 0.05). Significant differences between T790M+ and T790M− tumors are highlighted in bold red color. WGD: whole-genome doubling. Other EGFR: any other EGFR mutations besides L858R and exon 19 indel. D, Left, relative contribution of aging signature mutations comparing T790M+ and T790M− cohorts. Right, the absolute number of aging mutations (adjusted for tumor purity) are similar between T790M+ and T790M− tumors. E, (Top) Recurrent focal copy-number events (red: amplification; blue: deletion). Bottom, P values comparing proportion of samples with chromosomal arm events between T790M+ and T790M− cohorts. Chromosome 3q gain (highlighted in red) is the only significant event.
Genomic landscape of T790M+ and T790M− disease states
We first evaluated differences in prevalence of somatic genomic alterations (apart from EGFRT790M) in our cohort compared with a treatment-naïve cohort of 38 patients with advanced EGFR-mutated NSCLC with WES data. TP53 SNV alterations were more prevalent in the TKI resistant (63%) compared with the treatment-naïve (45%) cohort (Supplementary Fig. S6). Furthermore, we detected overall lower rates of genomic amplification for TERT and three genes encoded at 14q22 (PTGDR, SAV1, and SOS2) in treatment-resistant as compared with treatment-naïve tumors. We next sought to determine previously reported and identify potentially novel therapeutically tractable resistance mechanisms, through a comparison between T790M+ and T790M− TKI-resistant samples. MET alterations were seen in five (8%) patients and predominantly in the T790M− cohort (P = 0.03, Fig. 1C). In one patient (A056), a splice site deletion causing MET exon 14 skipping (METex14) was detected, with loss of transcription at exon 14 verified with RNAseq data (Supplementary Fig. S7; ref. 41). This patient also demonstrated chromosomal arm 7p deletion (with loss of EGFR mutation) and 12q amplification (containing MDM2 and CDK4, commonly amplified with METex14; ref. 42). Similarly, we did not detect the primary exon 19 deletion/L858R mutation in another patient who was T790M− (A058), suggesting loss of activating EGFR mutation as a bona fide rare mechanism of acquired resistance (43, 44). PIK3CA mutations (n = 10, 17%) and HER2 amplification (n = 4, 7%) were frequent but with no difference based on T790M status (P = 0.69 and P = 0.54, respectively), although the frequency of PIK3CA alterations (17%) was overall higher in resistant tumors compared with patients who had late-stage treatment-naïve EGFR-mutated tumors (5%, P = 0.09, Supplementary Table S3). Of the PIK3CA mutations, it is noteworthy that most were clonal (n = 5/7) and annotated as likely pathogenic (n = 6/7) by ClinVar (45). Taken together, this suggests a role for PI3K signaling in mediating TKI resistance independent of T790M status. Given the potential role of co-mutations in KEAP1 and NFE2L2 in conferring resistance to therapy (46), alterations in these genes were also evaluated. However, we did not detect any NFE2L2 mutations in the cohort, and we only detected one KEAP1 mutation in a T790M+ tumor (A449).
TP53 alterations are early clonal events found in approximately half of EGFR-mutated tumors (23, 47, 48). We found significant enrichment of TP53 alterations in T790M− compared with T790M+ patients (86% vs. 50% P = 0.01), of which the majority (n = 36/37, 97%) were timed to be early clonal events. In addition, MDM2 and YEATS4 amplifications were more frequent in T790M+ patients and mutually exclusive with TP53 mutations (P = 0.004). Both genes are co-located at the same chromosomal location and negatively regulate TP53 (49, 50). As previously reported by ourselves and others (47, 51), a higher number of mutations in known cancer driver genes was strongly associated with shorter TTP on multivariate analysis (HR, 1.41; 95% CI, 1.09–1.81; P = 0.008; Supplementary Fig. S8A). In particular, concurrent RB1/TP53 alterations (n = 5) were strongly associated with TTP independent of T790M status (multivariable Cox model; HR, 8.80; 95% CI, 2.74–28.32; P < 0.001; Supplementary Fig. S8B), consistent with a recent report (52).
Copy-number analysis uncovered that WGD was common and comparable with the rate observed in treatment-naïve EGFR-mutated tumors (88% vs. 89% of tumors). However, of the seven (12%) patients without WGD, all were T790M+ (
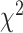 -test, P = 0.04) and had an EGFR exon 19 deletion (Fig. 1C), suggesting absence of WGD is predictive for emergence of T790M+-resistant disease. There was no difference in the genome instability index (GII; median 52% vs. 55%, respectively) between T790M+ and T790M− tumors, nor was TP53 co-mutation associated with increased GII. Tumor mutational burden (TMB) was higher in T790M− than T790M+ patients (median TMB 2.36 vs. 1.66 mutations per Mb, P = 0.02, Fig. 1C; Supplementary Fig. S9A), likely due to a greater number of smokers in the T790M− cohort (29% vs. 5%, P = 0.04; Supplementary Fig. S9C).
-test, P = 0.04) and had an EGFR exon 19 deletion (Fig. 1C), suggesting absence of WGD is predictive for emergence of T790M+-resistant disease. There was no difference in the genome instability index (GII; median 52% vs. 55%, respectively) between T790M+ and T790M− tumors, nor was TP53 co-mutation associated with increased GII. Tumor mutational burden (TMB) was higher in T790M− than T790M+ patients (median TMB 2.36 vs. 1.66 mutations per Mb, P = 0.02, Fig. 1C; Supplementary Fig. S9A), likely due to a greater number of smokers in the T790M− cohort (29% vs. 5%, P = 0.04; Supplementary Fig. S9C).
Enrichment of 3q amplifications in T790M-negative tumors
We next examined for recurrent focal amplifications and deletion events associated with T790M status—revealing amplificaitons in 3q23 (containing genes such as PIK3CB, MRAS, and FOXL2) in T790M− patients, and at 14q21 (containing FOXA1 and NKX2–1) in T790M+ patients (Supplementary Fig. S10). Strikingly, 3q arm amplification was highly frequent in T790M− (57%) compared with T790M+ (13%) patients, and the only statistically significant arm level event (Fig. 1E, 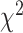 -test P ≤ 0.001). We confirmed higher expression of chromosome 3q genes (including SOX2) in the patients with 3q arm gain (Supplementary Fig. S11C). To understand the extent to which these chromosomal level alterations may be acquired after TKI treatment, we compared again with the treatment-naïve cohort. In the treatment-naïve cohort, 3q gain was present in seven out of 38 (18%, Supplementary Fig. S3) tumors. This was significantly lower than T790M− (P = 0.003) but similar to T790M+ (P = 0.53) patients, suggesting that 3q amplification may be acquired specifically in T790M− tumors. Interestingly, chromosome 3q harbors squamous lineage transcription factors TP63 and SOX2, and is a predominant feature of lung squamous cell carcinomas but not previously associated with lung adenocarcinoma (53).
-test P ≤ 0.001). We confirmed higher expression of chromosome 3q genes (including SOX2) in the patients with 3q arm gain (Supplementary Fig. S11C). To understand the extent to which these chromosomal level alterations may be acquired after TKI treatment, we compared again with the treatment-naïve cohort. In the treatment-naïve cohort, 3q gain was present in seven out of 38 (18%, Supplementary Fig. S3) tumors. This was significantly lower than T790M− (P = 0.003) but similar to T790M+ (P = 0.53) patients, suggesting that 3q amplification may be acquired specifically in T790M− tumors. Interestingly, chromosome 3q harbors squamous lineage transcription factors TP63 and SOX2, and is a predominant feature of lung squamous cell carcinomas but not previously associated with lung adenocarcinoma (53).
Mutational signatures in T790M+ and T790M− tumors
Next, we investigated mutational processes associated with EGFR TKI resistance. Well-established mutational signatures in lung adenocarcinoma were identified (Fig. 1C), namely aging, APOBEC, DNA double-strand break repair, smoking, and DNA mismatch repair. EGFRT790M-resistant tumors showed higher relative contributions of the aging signature compared with T790M−, consistent with the highest probability nucleotide change of A[C>T]G in trinucleotide context of EGFRT790M point mutation (Supplementary Figs. S12 and S13). Conversely, the proportional contribution of nonaging signatures (smoking, APOBEC and DNA repair) was significantly higher in T790M− compared with T790M+ tumors (P = 0.003; Fig. 1D; Supplementary Fig. S13), implicating alternative mutational processes in driving T790M− resistance. Overall, this suggests that T790M resistance may be less likely to emerge in tumors with greater proportions of active nonaging mutational signatures, for example, smoking/APOBEC signatures.
Transcriptomic analysis reveals ubiquitous loss of adenocarcinoma lineage markers in T790M− tumors
We next interrogated the transcriptomic profiles of the TKI-resistant tumors, classifying the tumors based on the three known transcriptomic subtypes of lung adenocarcinoma: terminal respiratory unit (TRU), proximal proliferative (PP), and proximal inflammatory (PI; refs. 37, 54). 15/29 (52%) T790M+ tumors were classified as TRU, comparable with the frequency observed (47%) in treatment-naïve EGFR-mutated tumors (Fig. 2A). In striking contrast, 15/15 (100%) T790M− tumors were classified as non-TRU (either PI or PP), suggesting fundamental transcriptomic differences underpinning T790M+ and T790M− resistance mechanisms.
Figure 2.

Tumor transcriptomic correlates of EGFR TKI resistance. A, Proportions of molecular transcriptomic subtype assigned to EGFR TKI-resistant tumors and treatment-naïve EGFR-mutated tumors. B, Volcano plot of log10(P values) against log-fold change in cancer cell expression of all genes tested. Lung adenocarcinoma markers (NAPSA, NKX2–1, SFTA2, and SFTA2) and other pulmonary differentiation markers are highlighted in red (Supplementary Table S4). C, Illustration of tumor transcriptome deconvolution approach for napsin-A (NAPSA). NAPSA gene expression is strongly correlated with purity in T790M− but not T790M+ tumors. NAPSA expression is inferred for a tumor with 0% (stroma) and 100% (cancer) tumor purity. Only lung tumor tissue and samples without abnormal high expression of squamous or neuroendocrine related genes are used for (B) and (C). D, Multiplex IHC staining of NAPSA, NKX2–1, and L858R (Surrogate for cancer cells). Images shown represent two tumors with striking difference in the IHC staining. Bottom, Bar–violin plots compare the median IHC intensity in NAPSA and NKX2–1 in all cells stained for L858R for each individual tumor with IHC data available. E, Cancer cell expression of lung adenocarcinoma markers comparing T790M+ and T790M− cohorts. F, Bulk tumor expression of lung adenocarcinoma markers comparing treatment-naïve EGFR-mutated and EGFR wild-type tumors across three public cohorts.
We estimated the fraction of cancer cells (purity) of each tumor sample using WES data, and used a transcriptome deconvolution approach, TUMERIC (33), to estimate and compare gene expression in cancer versus stromal (nonmalignant) cells in T790M+ and T790M− tumors (Fig. 2B and C). Remarkably, we inferred ubiquitous and near complete loss of expression of lung adenocarcinoma marker genes, such as NAPSA, NKX2–1, SFTA2, and SFTA3 in cancer cells of T790M− tumors (Fig. 2B and E). In addition, we observed increased expression of histological marker genes of either squamous cell or neuroendocrine carcinoma in a small subset of T790M− tumors (n = 4, 27%; Supplementary Fig. S1A). These findings were validated orthogonally using multiplex immunofluorescence (mIF), confirming decreased NAPSA and NKX2–1 (TTF-1) expression in cancer cells of T790M− compared with T790M+ tumors (Fig. 2D; Supplementary Fig. S14). Strikingly, analysis across three treatment-naïve NSCLC adenocarcinoma cohorts showed that low adenocarcinoma marker gene expression (NAPSA and NKX2–1) was extremely rare and only observed in EGFR wild-type tumors (Fig. 2F). Collectively, these data highlight a previously underappreciated degree of acquired lineage plasticity after TKI resistance particularly in T790M− tumors, co-occurring with 3q amplification and nonaging mutational signature processes, potentially facilitating EGFR independent signaling mechanisms.
Tumor immune contexture in TKI-resistant EGFR-mutated NSCLC
The tumor microenvironment (TME) is increasingly known to play a critical role in NSCLC (55). Given the lack of efficacy of checkpoint inhibitors in EGFR-mutated NSCLC, we sought to characterize the immune landscape associated with EGFR TKI resistance, initially stratifying tumors based on the “T-cell inflamed gene expression profile” (GEP) signature (39). Broadly, tumors were divided using consensus k-means clustering (Fig. 3A), revealing an immunecold (n = 24) and immunehot (n = 20) cluster, similarly observed in previous analyses of treatment-naïve NSCLC tumors (56). Using multiplex immunofluorescence, we verified that gene expression levels indicative of T cells (CD8A) and expression of PD-L1 was highly correlated to the protein level (Supplementary Fig. S15A). Both were also increased in immunehot compared with immunecold tumors without co-localization (Supplementary Fig. S16). Notably, the immunehot subtype was enriched in T790M− tumors compared with T790M+ tumors (n = 10, 67% vs. n = 10, 34%, P = 0.04). These findings appear distinct to the recently characterized Treg-mediated noninflamed TME in a small series of surgically resected EGFR-mutated LUAD (57).
Figure 3.

Stromal transcriptomic correlates of EGFR TKI resistance. A, Relative expression of genes used in the immune GEP calculation (39). Patients were clustered into two groups using consensus k-mean clustering and sorted by T790M status followed by time to progression (TTP). B, Comparison of immune-suppressive cells correlation index (derived using TIDE) and expression levels of immune checkpoint genes (PD-L1 gene expression shown here) between immune subtypes. Immune GEP score was calculated using the method from Cristescu et al. (39). Horizontal line demarcates GEP score of −0.318, defined as the cutoff for high GEP in the original article. Pairwise comparison test was carried out using Games–Howell test and P values were adjusted using Benjamin–Hochberg procedure. C, Kaplan–Meier curve of EGFR TKI TTP comparing different immune subtypes.
We then used a published computational method (TIDE; ref. 58) to further elucidate infiltrating immune cell subsets associated with TKI resistance. This revealed greater inferred levels of MDSCs (P = 0.04, t test), but lower levels of TAM M2 (P = 0.003, t test), in immunehot T790M− compared with immunehot T790M+ tumors (Fig. 3B). There was also significantly higher expression of PD-L1, FOXP3, and IDO1 in the immunehot T790M− compared with immunehot T790M+ tumors (Fig. 3B, multiplex immunofluorescence staining for PD-L1 and FOXP3 in Supplementary Fig. S15B and S15C). We next investigated whether the immune phenotype at the time of resistance, related to duration on prior 1G/2G TKI. Interestingly, immunehot T790M− tumors had the shortest TTP overall (Fig. 3C), of which half (5/10 patients) had a TTP of less than 3 months. In contrast, immunehot T790M+ tumors had longest TTP overall (median TTP 20.6 months; range, 8.2 to 76.8 months), with significantly longer TTP compared with immunecold T790M+ tumors (median TTP 4.1 months; range, 1.3 to 13 months; HR, 11.78; P = 0.004; 95% CI, 3.01–46.2; P = 0.001; Fig. 3C). Consistent with meta-analyses that have highlighted the lack of efficacy of single-agent immune checkpoint inhibitors in EGFR-mutated NSCLC (59), 7/8 (88%, 4/8 “hot,” 2/8 “cold,” 2/8 unknown) patients experienced PD as best response (Supplementary Table S5). However, one immunehot T790M+ patient (A096) received combination nivolumab–ipilimumab immune checkpoint inhibitor therapy on a clinical trial (60), and achieved stable disease for 8.9 months. Taken together, our data suggest a potential role for inflammatory chemokines, for example, CXCL9—likely driven by MDSCs—in mediating T790M− TKI resistance. Furthermore, our data highlight marked heterogeneity in composition of the TME in GEP “hot” tumors, illustrating the need for more detailed interrogation of the immune milieu to delineate specific immune targets.
A probabilistic data-driven forecasting model for T790M resistance
Although third-generation EGFR TKIs are increasingly adopted in the first-line setting, this clinical practice is driven in part by the inability to predict the resistance trajectories of individual patients. Having identified novel molecular features (3q amplication, transcriptomic subtype, loss of adenocarcinoma lineage markers and inflamed TME) associated with different EGFR TKI resistant states, we sought to develop a model to predict for the emergence of T790M. We surmised that these genomic, chromosome level and transcriptomic features could either be present at baseline or represent changes acquired over the course of treatment (Fig. 4A). To explore this further, we identified three truncal features that were likely present before the onset of 1G/2G TKI treatment: EGFR exon 19 deletion, absence of WGD, and TP53 alterations (Fig. 4A). Using a Bayesian approach, individual patients could be stratified into groups with very different odds of developing EGFRT790M resistance based on their pre-treatment molecular genotype (Fig. 4B). For instance, the probability of developing T790M resistance ranged from 87.2% to 97.9% in non-WGD tumors, with the potential implication that a sequential approach with first/second-generation to third-generation EGFR TKI might be a viable clinical strategy for these patients (11% of patients in our cohort). The predictive power of these features needs to be further validated in larger cohorts. However, these results illustrate how data-driven treatment algorithms can be derived through real-world evidence, and may potentially help define optimal sequencing strategies for individual patients.
Figure 4.

Data-driven TKI treatment algorithm. A, Genomic and transcriptomic alterations with distinct frequencies in patients with T790M+ and T790M− disease. The observed prevalence of each alteration in T790M+ and T790M− groups as well as patients with treatment-naïve late-stage EGFR-mutated tumors are shown. Testing the null-hypothesis that frequencies of individual alteration types are not different between treatment-naïve and resistant cohorts, alterations were divided into either likely pre-existing or likely TKI-treatment acquired alterations. B, The expected patient prevalence for each (n = 8) combination/genotype of the three inferred pre-existing alterations. The posterior probability was estimated for each genotype using Bayesian updating. C, Summary of molecular features that modify probability of T790M (left), and potential to use baseline clinical and molecular features, as well as adaptive changes to determine optimal therapeutic strategy (right).
Discussion
Our study presents the first comprehensive and integrative analysis of the genomic and transcriptomic landscape of EGFR TKI resistance. Strikingly, our data reveal a degree of lineage plasticity hitherto underappreciated. Although histologic transformation has been reported in 1% to 3% of patients post TKI resistance, we demonstrate ubiquitous loss of adenocarcinoma markers (napsin-A and TTF-1), coupled with strong enrichment for non-TRU subtypes (PI and PP), in T790M− tumors. Although the lack of paired baseline samples was a limitation in our study, the comparison with treatment-naïve EGFR-mutated NSCLC suggests that loss of adenocarcinoma lineage markers particularly in T790M− disease likely represents an early de-differentiation event induced by chronic EGFR TKI exposure. Other genomic alterations more significantly represented in T790M− disease were TP53 mutations (86% vs. 50%), 3q amplification (57% vs. 13%), and MET alterations (19% vs. 3%), which further contribute to lineage plasticity and T790M− drug resistance.
Of particular clinical interest is the immunehot subset in the T790M− cohort, representing a group of patients with significantly shorter TTP, characterized by high GEP score, PD-L1 overexpression, and a chemokine-rich immunosuppressive microenvironment. Consistent with our findings, retrospective analyses have suggested a relationship between high PD-L1 expression and lower response rates and PFS (61, 62), implicating an “inflamed” TME in mediating primary resistance to EGFR TKI. Recently, inhibition of EGFR signaling has been found to deplete Treg and increase IFN gamma signaling (63), supporting the link between an “inflamed” TME as an adaptive change that can potentially impair response to targeted therapies. Our data further suggest that the “inflamed” TME may occur at the time of primary or secondary resistance and be variably composed of CD8 T cells (tumor antigen specific and/or bystander), Treg and MDSC (64). Finally, the observation of high expression of IDO1 especially in the T790M− immunehot tumors coupled with overexpression of kynurenine (KYNU; Fig. 2B) implicates the IDO pathway in sustaining Treg activation and the immunosuppressive milieu in a subset of tumors. Recently, through single-cell RNA-seq of a series of oncogene-driven NSCLC tumors, Maynard and colleagues (27) similarly highlighted the significance of the IDO pathway, immune microenvironment, and alveolar regeneration cell signature in response to targeted therapy. Our data extend these observations and illustrate how therapy-induced adaptive cell states may be influenced by genomic alterations and suggest a complex interplay between EGFR dependency and lineage plasticity in cancer cells, immune cell populations, and chemokines. Prospective studies are underway to better elucidate the role of such immune mediators. Immune checkpoint inhibition, including combination anti–PD-1 and anti–CTLA-4 therapy, has shown a distinct lack of efficacy from clinical trials in the TKI-resistant setting (26, 60). In addition to ongoing efforts to evaluate immunosuppressive targets of the adenosine axis such as the adenosine 2A receptor (A2AR), CD39, and CD73 (65), rational targets for future clinical trials may include IDO, MDSC-depleting strategies such as bevacizumab or selective inhibition of PI3K-gamma.
With the expanding number of EGFR TKIs and alternative treatment options now available for treatment of EGFR-mutated NSCLC, one foremost priority is to tailor the optimal sequence of therapies for each individual patient. Through depicting the transcriptomic profiles of the treatment-naïve and T790M-resistant states, our study reveals novel molecular features (transcriptomic subtype, loss of adenocarcinoma lineage markers and inflamed TME) that may underpin the quality of responses in oncogene-driven NSCLC: whether in primary versus secondary TKI resistance, or with differences in PFS and RR in the first-line setting compared with later lines (4, 66, 67). Our study further cautions against sole reliance on plasma-based genomic profiling and underscores the importance of tissue-based analysis in depicting expanded genomic and transcriptomic features, for example, the TRU-inflamed phenotype unique to Asian LUAD (28). We anticipate that the accuracy of the data-driven modeling and forecasting will be further enhanced through building an iterative, prospective knowledge bank that links comprehensive catalogues of genomic and transcriptomic profiles with high-resolution annotation of clinical outcomes both at baseline and each subsequent progression. In addition, such high-granularity data will allow for the comparison of resistance profiles of T790M− tumors after 1/2G EGFR TKI versus 3G EGFR TKI in future studies, and provide further insights into the distinct spectrum of resistance alterations observed thus far.
Other considerations in the interpretation of our findings, include the concomitant or intervening use of chemotherapy in a subset of patients before sample collection, and the relatively modest size of the overall cohort. Nevertheless, the use of chemotherapy and last line of therapy before sample collection was well balanced comparing the T790M+ and T790M− cohorts (Supplementary Fig. S4B). Further analysis also revealed consistent findings when patients with the use of chemotherapy as the last line of therapy before sample collection were excluded, in particular regarding the tumor immune contexture (Supplementary Fig. S17 and Supplementary Table S10).
To conclude, we demonstrate that understanding resistance to targeted therapy requires in depth analysis at multiple levels (single gene, chromosome, and transcriptome; Fig. 4). We report novel genomic and transcriptomic associations with T790M status, including EGFR mutation type, concomitant alterations, mutational signatures, transcriptional subtypes, and immune signatures (Fig. 4C). These molecular features are likely correlates of lineage plasticity and propensity for resistance adaptation, both of which remain incompletely understood. Nevertheless, the potential ability to foretell a tumor's evolutionary trajectory and predict for T790M status has important clinical implications, including optimal sequencing of EGFR-directed therapies and the application of high-precision diagnostic tools to detect resistance early. Our study underscores the expanding role for genomic and transcriptomic profiling of EGFR-mutated NSCLC longitudinally, to facilitate the design of bespoke therapeutic approaches tailored to a tumor's adaptive potential.
Authors' Disclosures
K.P. Chua reports grants from Genome Institute of Singapore during the conduct of the study and reports employment with Pacific Biosciences Singapore as a field application scientist since January 2020; the majority of the work in the article before the revision was completed before employment with PacBio. A.C. Tan reports personal fees from ThermoFisher and Amgen outside the submitted work. G.G.Y. Lai reports grants from MSD Pharma, BMS, Roche, AstraZeneca, and Pfizer during the conduct of the study; personal fees from Amgen; and other support from MSD Pharma outside the submitted work. C.W. Too reports personal fees from Sirtex, Boston Scientific, and from Starmed outside the submitted work. R. Kanesvaran reports personal fees from Pfizer, Astellas, Johnson & Johnson, Bayer, AstraZeneca, MSD, BMS, Eisai, Amgen, and Ipsen outside the submitted work. T. Rajasekaran reports other support from AstraZeneca, Novartis, and Ipsen during the conduct of the study. A.S.M. Teo reports grants from Genome Institute of Singapore during the conduct of the study. M.J. Lim reports grants from GIS during the conduct of the study. N.G. Iyer reports personal fees from PairX Therapeutics, Invitrocue PLC, Agilent, and other support from AstraZeneca outside the submitted work. A.M. Hillmer reports grants from National Medical Research Council, Singapore during the conduct of the study. D.S. Tan reports grants from GlaxoSmithKline, National Cancer Center Research Fund, and National Medical Research Council (Singapore) during the conduct of the study as well as personal fees from Novartis, Bayer, Boehringer Ingelheim, AstraZeneca, Amgen, Janssen, and C4 Therapeutics outside the submitted work. No disclosures were reported by the other authors.
Supplementary Material
Acknowledgments
Funded by grants from the National Medical Research Council (NMRC; Singapore; NMRC/TCR/007-NCC/2013; NMRC/OFLCG/002–2018; NMRC/OFIRG/0064/2017; OFYIRG16nov013), the NMRC clinician-scientist award (NMRC/CSA/007/2016, to D.S.W. Tan), the Trailblazer Foundation, Singapore Millennium Foundation, Agency for Science, Research and Technology, Singapore (A*STAR), National Research Foundation (NRF) Fellowship, Singapore (NRF-NRFF2015–04, to W.L. Tam), and the National Cancer Center Research Fund, which jointly supported the infrastructure of the Lung Cancer Consortium Singapore. This research is supported in part by Agency for Science, Technology and Research (ASTAR) under its CDAP program (grant no. 1727600057). A.C. Tan is the recipient of an International Association for the Study of Lung Cancer (IASLC) Fellowship 2018–2020. The computational work for this article was partially performed on resources of the National Supercomputing Center, Singapore (https://www.nscc.sg).
The publication costs of this article were defrayed in part by the payment of publication fees. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Footnotes
Note: Supplementary data for this article are available at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/).
Authors' Contributions
K.P. Chua: Data curation, formal analysis, validation, investigation, visualization, methodology, writing–original draft, writing–review and editing. Y.H.F. Teng: Data curation, formal analysis, validation, investigation, visualization, methodology, writing–original draft, writing–review and editing. A.C. Tan: Data curation, formal analysis, validation, investigation, visualization, methodology, writing–original draft, writing–review and editing. A. Takano: Resources, data curation, formal analysis, validation, investigation, methodology, writing–review and editing. J.J.S. Alvarez: Data curation, formal analysis, validation, investigation. R. Nahar: Data curation, formal analysis, validation, investigation. N. Rohatgi: Data curation, formal analysis, validation, investigation. G.G.Y. Lai: Data curation, formal analysis, validation, investigation. Z.W. Aung: Data curation, formal analysis, validation, investigation. J.P.S. Yeong: Data curation, formal analysis, validation, investigation. K.H. Lim: Data curation, formal analysis, validation, investigation. M.M. Naeini: Data curation, formal analysis, validation, investigation. I. Kassam: Data curation, formal analysis, validation, investigation. A. Jain: Data curation, formal analysis, validation, investigation. W.L. Tan: Data curation, formal analysis, validation, investigation. A. Gogna: Data curation, formal analysis, validation, investigation. C.W. Too: Data curation, formal analysis, validation, investigation. R. Kanesvaran: Data curation, formal analysis, validation, investigation. Q.S. Ng: Data curation, formal analysis, validation, investigation. M.K. Ang: Data curation, formal analysis, validation, investigation. T. Rajasekaran: Data curation, formal analysis, validation, investigation. D. Anantham: Data curation, formal analysis, validation, investigation. G.C. Phua: Data curation, formal analysis, validation, investigation. B.S. Tan: Data curation, formal analysis, validation, investigation. Y.Y. Lee: Data curation, formal analysis, validation, investigation. L. Wang: Data curation, formal analysis, validation, investigation. A.S.M. Teo: Data curation, formal analysis, validation, investigation. A.J. Khng: Data curation, formal analysis, validation, investigation. M.J. Lim: Data curation, formal analysis, validation, investigation. L. Suteja: Data curation, formal analysis, validation, investigation. C.K. Toh: Data curation, formal analysis, validation, investigation. W.-T. Lim: Data curation, formal analysis, validation, investigation. N.G. Iyer: Data curation, formal analysis, validation, investigation. W.L. Tam: Data curation, formal analysis, validation, investigation. E.-H. Tan: Data curation, formal analysis, funding acquisition, validation, investigation. W. Zhai: Data curation, formal analysis, validation, investigation. A.M. Hillmer: Data curation, formal analysis, validation, investigation. A.J. Skanderup: Resources, data curation, formal analysis, supervision, validation, investigation, visualization, methodology, writing–original draft, project administration, writing–review and editing. D.S.W. Tan: Conceptualization, resources, data curation, formal analysis, supervision, funding acquisition, validation, investigation, visualization, methodology, writing–original draft, project administration, writing–review and editing.
References
- 1. Mok TS, Wu Y-L, Thongprasert S, Yang C-H, Chu D-T, Saijo N, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med 2009;361:947–57. [DOI] [PubMed] [Google Scholar]
- 2. Park K, Tan E-H, O'Byrne K, Zhang L, Boyer M, Mok T, et al. Afatinib versus gefitinib as first-line treatment of patients with EGFR mutation-positive non–small cell lung cancer (LUX-Lung 7): a phase 2B, open-label, randomised controlled trial. Lancet Oncol 2016;17:577–89. [DOI] [PubMed] [Google Scholar]
- 3. Rosell R, Carcereny E, Gervais R, Vergnenegre A, Massuti B, Felip E, et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non–small cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol 2012;13:239–46. [DOI] [PubMed] [Google Scholar]
- 4. Soria J-C, Ohe Y, Vansteenkiste J, Reungwetwattana T, Chewaskulyong B, Lee KH, et al. Osimertinib in untreated EGFR-mutated advanced non–small cell lung cancer. N Engl J Med 2018;378:113–25. [DOI] [PubMed] [Google Scholar]
- 5. Noronha V, Patil VM, Joshi A, Menon N, Chougule A, Mahajan A, et al. Gefitinib versus gefitinib plus pemetrexed and carboplatin chemotherapy in EGFR-mutated lung cancer. J Clin Oncol 2020;38:124–36. [DOI] [PubMed] [Google Scholar]
- 6. Hosomi Y, Morita S, Sugawara S, Kato T, Fukuhara T, Gemma A, et al. Gefitinib alone versus gefitinib plus chemotherapy for non–small cell lung cancer with mutated epidermal growth factor receptor: NEJ009 Study. J Clin Oncol 2020;38:115–23. [DOI] [PubMed] [Google Scholar]
- 7. Nakagawa K, Garon EB, Seto T, Nishio M, Ponce Aix S, Paz-Ares L, et al. Ramucirumab plus erlotinib in patients with untreated, EGFR-mutated, advanced non–small cell lung cancer (RELAY): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol 2019;20:1655–69. [DOI] [PubMed] [Google Scholar]
- 8. Yu HA, Schoenfeld AJ, Makhnin A, Kim R, Rizvi H, Tsui D, et al. Effect of osimertinib and bevacizumab on progression-free survival for patients with metastatic EGFR-mutant lung cancers: a phase 1/2 single-group open-label trial. JAMA Oncol 2020;6:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ramalingam SS, Vansteenkiste J, Planchard D, Cho BC, Gray JE, Ohe Y, et al. Overall survival with osimertinib in untreated, EGFR-mutated advanced NSCLC. N Engl J Med. 2020;382:41–50. [DOI] [PubMed] [Google Scholar]
- 10. Wu Y-L, Cheng Y, Zhou X, Lee KH, Nakagawa K, Niho S, et al. Dacomitinib versus gefitinib as first-line treatment for patients with EGFR-mutation-positive non–small cell lung cancer (ARCHER 1050): a randomised, open-label, phase 3 trial. Lancet Oncol 2017;18:1454–66. [DOI] [PubMed] [Google Scholar]
- 11. Feinberg B, Halmos B, Gucalp R, Tang W, Moehring B, Hochmair MJ. Making the case for EGFR TKI sequencing in EGFR mutation-positive NSCLC: a GioTag study US patient analysis. Future Oncol 2020;16:1585–95. [DOI] [PubMed] [Google Scholar]
- 12. Hochmair MJ, Morabito A, Hao D, Yang C-T, Soo RA, Yang JC-H, et al. Sequential afatinib and osimertinib in patients with EGFR mutation-positive non–small cell lung cancer: updated analysis of the observational GioTag study. Future Oncol 2019;15:2905–14. [DOI] [PubMed] [Google Scholar]
- 13. Aguiar PN, Haaland B, Park W, San Tan P, Del Giglio A, de Lima Lopes G. Cost-effectiveness of osimertinib in the first-line treatment of patients with egfr-mutated advanced non-small cell lung cancer. JAMA Oncol 2018;4:1080–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Aziz MIA, Foo WYX, Toh CK, Lim W-T, Ng K. Cost-effectiveness analysis of osimertinib for first-line treatment of locally advanced or metastatic EGFR mutation positive non–small cell lung cancer in Singapore. J Med Econ 2020;1–10. [DOI] [PubMed] [Google Scholar]
- 15. Holleman MS, Al MJ, Zaim R, Groen HJM, Uyl-de Groot CA. Cost-effectiveness analysis of the first-line EGFR-TKIs in patients with non–small cell lung cancer harbouring EGFR mutations. Eur J Health Econ 2020;21:153–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ezeife DA, Kirk V, Chew DS, Nixon NA, Lee R, Le LW, et al. Economic analysis of osimertinib in previously untreated EGFR-mutant advanced non–small cell lung cancer in Canada. Lung Cancer 2018;125:1–7. [DOI] [PubMed] [Google Scholar]
- 17. Tan AC, Teh YL, Lai GGY, Tan DSW. Third-generation EGFR TKI landscape for metastatic EGFR mutant non–small cell lung cancer (NSCLC). Expert Rev Anticancer Ther 2019;19:1–5. [DOI] [PubMed] [Google Scholar]
- 18. Campo M, Gerber D, Gainor JF, Heist RS, Temel JS, Shaw AT, et al. Acquired resistance to first-line afatinib and the challenges of prearranged progression biopsies. J Thorac Oncol 2016;11:2022–6. [DOI] [PubMed] [Google Scholar]
- 19. Sequist LV, Waltman BA, Dias-Santagata D, Digumarthy S, Turke AB, Fidias P, et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med 2011;3:75ra26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yu HA, Arcila ME, Rekhtman N, Sima CS, Zakowski MF, Pao W, et al. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin Cancer Res 2013;19:2240–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mok TS, Wu Y-L, Ahn M-J, Garassino MC, Kim HR, Ramalingam SS, et al. Osimertinib or platinum-pemetrexed in EGFR T790M-positive lung cancer. N Engl J Med 2017;376:629–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Westover D, Zugazagoitia J, Cho BC, Lovly CM, Paz-Ares L. Mechanisms of acquired resistance to first- and second-generation EGFR tyrosine kinase inhibitors. Ann Oncol 2018;29:i10–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yu HA, Suzawa K, Jordan E, Zehir A, Ni A, Kim R, et al. Concurrent alterations in egfr-mutant lung cancers associated with resistance to EGFR kinase inhibitors and characterization of MTOR as a mediator of resistance. Clin Cancer Res 2018;24:3108–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Oxnard GR, Hu Y, Mileham KF, Husain H, Costa DB, Tracy P, et al. Assessment of resistance mechanisms and clinical implications in patients with egfr t790m-positive lung cancer and acquired resistance to osimertinib. JAMA Oncol 2018;4:1527–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ramalingam SS, Cheng Y, Zhou C, Ohe Y, Imamura F, Cho BC, et al. LBA50Mechanisms of acquired resistance to first-line osimertinib: preliminary data from the phase III FLAURA study. Ann Oncol 2018;29:VIII740. [Google Scholar]
- 26. Lee CK, Man J, Lord S, Links M, Gebski V, Mok T, et al. Checkpoint inhibitors in metastatic EGFR-mutated non–small cell lung cancer-a meta-analysis. J Thorac Oncol 2017;12:403–7. [DOI] [PubMed] [Google Scholar]
- 27. Maynard A, McCoach CE, Rotow JK, Harris L, Haderk F, Kerr DL, et al. Therapy-induced evolution of human lung cancer revealed by single-cell RNA sequencing. Cell 2020;182:1232–51.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chen J, Yang H, Teo ASM, Amer LB, Sherbaf FG, Tan CQ, et al. Genomic landscape of lung adenocarcinoma in East Asians. Nat Genet 2020;52:177–86. [DOI] [PubMed] [Google Scholar]
- 29. Yeong J, Lim JCT, Lee B, Li H, Chia N, Ong CCH, et al. High densities of tumor-associated plasma cells predict improved prognosis in triple negative breast cancer. Front Immunol 2018;9:1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lim JCT, Yeong JPS, Lim CJ, Ong CCH, Wong SC, Chew VSP, et al. An automated staining protocol for seven-colour immunofluorescence of human tissue sections for diagnostic and prognostic use. Pathology 2018;50:333–41. [DOI] [PubMed] [Google Scholar]
- 31. Grossman RL, Heath AP, Ferretti V, Varmus HE, Lowy DR, Kibbe WA, et al. Toward a shared vision for cancer genomic data. N Engl J Med 2016;375:1109–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ellrott K, Bailey MH, Saksena G, Covington KR, Kandoth C, Stewart C, et al. Scalable open science approach for mutation calling of tumor exomes using multiple genomic pipelines. Cell Syst 2018;6:271281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ghoshdastider U, Rohatgi N, Mojtabavi Naeini M, Baruah P, Revkov E, Guo YA, et al. Pan-cancer analysis of ligand-receptor cross-talk in the tumor microenvironment. Cancer Res 2021;81:1802–12. [DOI] [PubMed] [Google Scholar]
- 34. Yoshihara K, Shahmoradgoli M, Martínez E, Vegesna R, Kim H, Torres-Garcia W, et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat Commun 2013;4:2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Josse J, Husson F. missMDA: a package for handling missing values in multivariate data analysis. J Stat Software 2016;70:1–31. [Google Scholar]
- 36. Collisson EA, Campbell JD, Brooks AN, Berger AH, Lee W, Chmielecki J, et al. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014;511:543–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wilkerson MD, Yin X, Walter V, Zhao N, Cabanski CR, Hayward MC, et al. Differential pathogenesis of lung adenocarcinoma subtypes involving sequence mutations, copy number, chromosomal instability, and methylation. PLoS ONE 2012;7:e36530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Xu T, Le TD, Liu L, Su N, Wang R, Sun B, et al. CancerSubtypes: an R/Bioconductor package for molecular cancer subtype identification, validation and visualization. Bioinformatics 2017;33:3131–3. [DOI] [PubMed] [Google Scholar]
- 39. Cristescu R, Mogg R, Ayers M, Albright A, Murphy E, Yearley J, et al. Pan-tumor genomic biomarkers for PD-1 checkpoint blockade-based immunotherapy. Science 2018;362:eaar3593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Campbell I. Chi-squared and Fisher-Irwin tests of two-by-two tables with small sample recommendations. Stat Med 2007;26:3661–75. [DOI] [PubMed] [Google Scholar]
- 41. Suzawa K, Offin M, Lu D, Kurzatkowski C, Vojnic M, Smith RS, et al. Activation of KRAS mediates resistance to targeted therapy in MET Exon 14-mutant non–small cell lung cancer. Clin Cancer Res 2019;25:1248–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Frampton GM, Ali SM, Rosenzweig M, Chmielecki J, Lu X, Bauer TM, et al. Activation of MET via diverse exon 14 splicing alterations occurs in multiple tumor types and confers clinical sensitivity to MET inhibitors. Cancer Discov 2015;5:850–9. [DOI] [PubMed] [Google Scholar]
- 43. Nosaki K, Satouchi M, Kurata T, Yoshida T, Okamoto I, Katakami N, et al. Re-biopsy status among non–small cell lung cancer patients in Japan: a retrospective study. Lung Cancer 2016;101:1–8. [DOI] [PubMed] [Google Scholar]
- 44. Tabara K, Kanda R, Sonoda K, Kubo T, Murakami Y, Kawahara A, et al. Loss of activating EGFR mutant gene contributes to acquired resistance to EGFR tyrosine kinase inhibitors in lung cancer cells. PLoS ONE 2012;7:e41017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. McGranahan N, Favero F, de Bruin EC, Birkbak NJ, Szallasi Z, Swanton C. Clonal status of actionable driver events and the timing of mutational processes in cancer evolution. Sci Transl Med 2015;7:283ra54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hellyer JA, Stehr H, Das M, Padda SK, Ramchandran K, Neal JW, et al. Impact of KEAP1/NFE2L2/CUL3 mutations on duration of response to EGFR tyrosine kinase inhibitors in EGFR mutated non–small cell lung cancer. Lung Cancer 2019;134:42–5. [DOI] [PubMed] [Google Scholar]
- 47. Nahar R, Zhai W, Zhang T, Takano A, Khng AJ, Lee YY, et al. Elucidating the genomic architecture of Asian EGFR-mutant lung adenocarcinoma through multi-region exome sequencing. Nat Commun 2018;9:216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Jamal-Hanjani M, Wilson GA, McGranahan N, Birkbak NJ, Watkins TBK, Veeriah S, et al. Tracking the evolution of non–small cell lung cancer. N Engl J Med 2017;376:2109–21. [DOI] [PubMed] [Google Scholar]
- 49. Marine J-C, Lozano G. Mdm2-mediated ubiquitylation: p53 and beyond. Cell Death Differ 2010;17:93–102. [DOI] [PubMed] [Google Scholar]
- 50. Pikor LA, Lockwood WW, Thu KL, Vucic EA, Chari R, Gazdar AF, et al. YEATS4 is a novel oncogene amplified in non–small cell lung cancer that regulates the p53 pathway. Cancer Res 2013;73:7301–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Offin M, Rizvi H, Tenet M, Ni A, Sanchez-Vega F, Li BT, et al. Tumor mutation burden and efficacy of EGFR-tyrosine kinase inhibitors in patients with EGFR-mutant lung cancers. Clin Cancer Res 2019;25:1063–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kim Y, Lee B, Shim JH, Lee S-H, Park W-Y, Choi Y-L, et al. Concurrent genetic alterations predict the progression to target therapy in EGFR-mutated advanced NSCLC. J Thorac Oncol 2019;14:193–202. [DOI] [PubMed] [Google Scholar]
- 53. Hammerman PS, Lawrence MS, Voet D, Jing R, Cibulskis K, Sivachenko A, et al. Comprehensive genomic characterization of squamous cell lung cancers. Nature 2012;489:519–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014;511:543–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Altorki NK, Markowitz GJ, Gao D, Port JL, Saxena A, Stiles B, et al. The lung microenvironment: an important regulator of tumour growth and metastasis. Nat Rev Cancer 2019;19:9–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Rosenthal R, Cadieux EL, Salgado R, Bakir MA, Moore DA, Hiley CT, et al. Neoantigen-directed immune escape in lung cancer evolution. Nature 2019;567:479–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Santaniello A, Napolitano F, Servetto A, De Placido P, Silvestris N, Bianco C, et al. Tumour microenvironment and immune evasion in EGFR addicted NSCLC: hurdles and possibilities. Cancers 2019;11:1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Jiang P, Gu S, Pan D, Fu J, Sahu A, Hu X, et al. Signatures of T-cell dysfunction and exclusion predict cancer immunotherapy response. Nat Med 2018;24:1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Lee CK, Man J, Lord S, Cooper W, Links M, Gebski V, et al. Clinical and molecular characteristics associated with survival among patients treated with checkpoint inhibitors for advanced non–small cell lung carcinoma. JAMA Oncol 2018;4:210–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Lai G, Alvarez J, Tan A, Zhou S, Suteja L, Lim T, et al. OA01.06 Randomised phase 2 study of nivolumab (N) versus nivolumab and ipilimumab (NI) combination in EGFR mutant NSCLC. World Conference on Lung Cancer 2020. 2021. [DOI] [PMC free article] [PubMed]
- 61. Su S, Dong Z-Y, Xie Z, Yan L-X, Li Y-F, Su J, et al. Strong programmed death ligand 1 expression predicts poor response and de novo resistance to EGFR tyrosine kinase inhibitors among NSCLC patients with EGFR mutation. J Thorac Oncol 2018;13:1668–75. [DOI] [PubMed] [Google Scholar]
- 62. Hsu K-H, Huang Y-H, Tseng J-S, Chen K-C, Ku W-H, Su K-Y, et al. High PD-L1 expression correlates with primary resistance to EGFR-TKIs in treatment naïve advanced EGFR-mutant lung adenocarcinoma patients. Lung Cancer 2019;127:37–43. [DOI] [PubMed] [Google Scholar]
- 63. Sugiyama E, Togashi Y, Takeuchi Y, Shinya S, Tada Y, Kataoka K, et al. Blockade of EGFR improves responsiveness to PD-1 blockade in EGFR-mutated non–small cell lung cancer. Sci Immunol 2020;5:eaav3937. [DOI] [PubMed] [Google Scholar]
- 64. Becker JH, Gao Y, Soucheray M, Pulido I, Kikuchi E, Rodríguez ML, et al. CXCR7 reactivates ERK signaling to promote resistance to egfr kinase inhibitors in NSCLC. Cancer Res 2019;79:4439–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Passarelli A, Aieta M, Sgambato A, Gridelli C. Targeting immunometabolism mediated by CD73 pathway in EGFR-mutated non–small cell lung cancer: a new hope for overcoming immune resistance. Front Immunol 2020;11:1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Mok TS, Wu Y-L, Ahn M-J, Garassino MC, Kim HR, Ramalingam SS, et al. Osimertinib or platinum–pemetrexed in EGFR T790M-positive lung cancer. N Engl J Med 2017;376:629–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Wolf J, Seto T, Han J-Y, Reguart N, Garon EB, Groen HJM, et al. Capmatinib in MET exon 14-mutated or MET-amplified non–small cell lung cancer. N Engl J Med 2020;383:944–57. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw sequencing data have been deposited in the European Genome-phenome Archive (EGA, EGAS00001005389, http://www.ebi.ac.uk/ega/). Computational methods have been uploaded to a public git (https://gitlab.com/gis_khipin/ccr_tki_resistance_2021). Clinical records, somatic mutations, copy-number variations, gene expression data, and histological images from our study are hosted in OncoSG (https://src.gisapps.org/OncoSG/) that is publicly available.