

Abstract
Purpose:
We describe the first-in-human dose-escalation trial for ALRN-6924, a stabilized, cell-permeating peptide that disrupts p53 inhibition by mouse double minute 2 (MDM2) and MDMX to induce cell-cycle arrest or apoptosis in TP53-wild-type (WT) tumors.
Patients and Methods:
Two schedules were evaluated for safety, pharmacokinetics, pharmacodynamics, and antitumor effects in patients with solid tumors or lymphomas. In arm A, patients received ALRN-6924 by intravenous infusion once-weekly for 3 weeks every 28 days; arm B was twice-weekly for 2 weeks every 21 days.
Results:
Seventy-one patients were enrolled: 41 in arm A (0.16–4.4 mg/kg) and 30 in arm B (0.32–2.7 mg/kg). ALRN-6924 showed dose-dependent pharmacokinetics and increased serum levels of MIC-1, a biomarker of p53 activation. The most frequent treatment-related adverse events were gastrointestinal side effects, fatigue, anemia, and headache. In arm A, at 4.4 mg/kg, dose-limiting toxicities (DLT) were grade 3 (G3) hypotension, G3 alkaline phosphatase elevation, G3 anemia, and G4 neutropenia in one patient each. At the MTD in arm A of 3.1 mg/kg, G3 fatigue was observed in one patient. No DLTs were observed in arm B. No G3/G4 thrombocytopenia was observed in any patient. Seven patients had infusion-related reactions; 3 discontinued treatment. In 41 efficacy-evaluable patients with TP53-WT disease across both schedules the disease control rate was 59%. Two patients had confirmed complete responses, 2 had confirmed partial responses, and 20 had stable disease. Six patients were treated for >1 year. The recommended phase 2 dose was schedule A, 3.1 mg/kg.
Conclusions:
ALRN-6924 was well tolerated and demonstrated antitumor activity.
Translational Relevance.
The p53 tumor-suppressor protein and its endogenous regulator mouse double minute 2 (MDM2) have been a focus of cancer research for over 30 years, with several small-molecule inhibitors of MDM2 evaluated in clinical trials. However, no inhibitors of MDMX, the second regulator of p53, have been tested clinically, due in part to the challenge of developing small-molecule inhibitors of the p53–MDMX interface. Stapled peptides are a new therapeutic modality capable of disrupting protein–protein interactions in cells. ALRN-6924, a dual MDM2/MDMX inhibitor, is the first member of this new modality to reach the clinic. We evaluated the safety, pharmacokinetics, pharmacodynamics, and antitumor effects of ALRN-6924 in patients with solid tumors or lymphomas, and show that ALRN-6924 was well tolerated with encouraging antitumor activity, including durable complete and partial responses with more than half of evaluable patients achieving disease control. Strikingly, the hematopoietic toxicity typically observed for selective MDM2 inhibitors was nearly absent for ALRN-6924, suggesting that dual MDM2/MDMX inhibition may allow for more complete p53 activation with a toxicity profile that would allow for combination therapy and use as a chemoprotection agent.
Introduction
Commonly referred to as “the guardian of the genome,” p53 plays a central role in the mechanisms that defend the human body from cancer (1). The natural tumor-suppressor function of p53 involves responding to DNA damage by arresting cell division, allowing for repair, and if repair is unsuccessful, inducing an apoptotic response. To subvert this antitumor functionality, cancer cells neutralize p53 by deletion, mutation, degradation, or sequestration (2, 3). The latter two mechanisms are mediated by protein–protein interactions between p53 and its negative regulators mouse double minute 2 (MDM2), which binds and destroys p53, and MDMX, which binds and sequesters p53 (4, 5). Mutation of the TP53 gene that encodes the p53 tumor-suppressor protein has been observed in approximately 50% of adult and 4% of childhood cancers, highlighting the importance of p53 signaling (6). Conversely, many human cancers retain wild-type TP53, making reactivation of the natural p53 pathway by targeted inhibition of MDM2 and MDMX in TP53 wild-type cancer cells an appealing strategy for therapeutic intervention.
MDM2 is an E3 ubiquitin-protein ligase that engages the transactivation domain of p53 by a helix-in-groove interaction (7). Whereas the MDM2–p53 interaction results in p53 ubiquitination and degradation, an analogous helix-in-groove interaction between MDMX and p53 thwarts transcriptional activity by complex formation. Although MDMX does not possess E3 ligase activity, it enhances MDM2-mediated degradation of p53 (8). Importantly, both MDM2 and MDMX are essential to negative regulation and homeostasis of p53 at the organism level, as evidenced by the embryonic lethality of MDM2 or MDMX deletion in mice (9, 10). In human cancer, the finding of suppression of p53 by overexpression of MDM2, whether by gene amplification or protein upregulation, inspired the development of small-molecule MDM2 inhibitors to restore the p53 tumor-suppressor pathway (11). However, sequestration of drug-induced p53 by MDMX was found to be a mechanism of resistance to selective MDM2 inhibition (12). Although MDM2 inhibitors have now advanced to clinical trials (13–15), clinically viable small molecules that target the corresponding groove on MDMX remain elusive. Given the hematopoietic toxicity—especially thrombocytopenia—observed upon selective MDM2 inhibition in the clinic (16) and the role of MDMX in p53 suppression and in resistance to MDM2 inhibitor treatment, the development of dual MDM2- and MDMX-directed agents offers a path to more complete inhibition of negative p53 regulation. Yet, such agents have not been clinically tested.
Here, we report the results of the first phase 1 trial of ALRN-6924, a stabilized, cell-permeating α-helical peptide that mimics the transactivation domain α-helix of p53 to bind with high affinity to both MDMX and MDM2. ALRN-6924 represents a new class of inhibitors of intracellular protein–protein interactions, referred to as stapled peptides, that contain a bridging hydrocarbon linker to maintain the α-helical shape of bioactive peptides and simultaneously confer proteolytic resistance and cell permeability (17–19). Although early stapled peptide prototypes showed inconsistent results in vitro under standard serum-containing tissue culture conditions (20), detailed optimization of p53 stapled peptides to improve their biological and biophysical properties led to the development of ALRN-6924, which consistently demonstrated on-mechanism activity in vitro under standard serum-containing tissue culture conditions as well as antitumor activity in mouse models of TP53 wild-type hematologic malignancies and solid tumors, providing proof-of-concept for clinical translation (21–23). To evaluate the safety and preliminary activity of ALRN-6924 in human cancer, we conducted a phase 1 dose-escalation study of 71 patients with advanced solid tumors or lymphomas. Now, we report the tolerability, pharmacokinetics, pharmacodynamics, and clinical activity of this first-in-class representative of the novel stapled peptide therapeutic modality.
Patients and Methods
Patient selection
Patients 18 years or older with advanced solid tumors or lymphomas were recruited to participate in this phase I, open-label, multicenter, dose-escalation trial at eight centers throughout the United States. Patients with solid tumors were required to have at least one target lesion per RECIST 1.1, and patients with lymphomas were required to have at least one measurable lesion per Revised International Working Group Response Criteria (IWG) 2014, following the exhaustion of standard-of-care treatment options. Eligibility criteria also included Eastern Cooperative Oncology Group performance status of 0–1, life expectancy ≥3 months, and adequate hematologic, hepatic, coagulation, and renal function (see protocol in Supplementary Information for detailed eligibility criteria). Women with childbearing potential were required to have a negative serum or urine pregnancy test during screening. The shorter of 4 weeks or 5 half-lives was required for the washout of all prior therapeutic agents unless no drug–drug interaction was anticipated and the patient had unequivocally experienced disease progression during the most recent line of therapy.
Mandatory tumor TP53 mutation status evaluation was conducted in fresh or archival (≤1 year old) tumor specimens using next-generation sequencing. However, for the three lowest dose levels of arm A, patients were enrolled irrespective of TP53 mutation status. At higher dose levels, patients were required to have wild-type TP53 based on either central or local laboratory assessment. Patients known to be human papillomavirus positive were also excluded from enrollment because viral E6 protein can mediate p53 degradation. Patients without archived tissue and for whom a biopsy posed a significant risk were not enrolled.
On the basis of low central nervous system penetration of ALRN-6924 prototypes observed in preclinical studies, patients with primary central nervous system tumors and patients with known brain metastasis were excluded unless these metastases had been treated and been clinically stable for at least 30 days. Patients with cardiovascular risk factors, including the New York Heart Association class III or IV heart failure, uncontrolled arrythmias or hypertension, and acute coronary syndromes within 6 months, as well as patients with active or uncontrolled infections, including HIV/AIDS and hepatitis B and C, were excluded. Any previous primary malignancy must have been in remission for at least 2 years, except for non-melanoma skin cancers, carcinomas in situ, or squamous intraepithelial neoplasms. Patients with pulmonary embolism, deep venous thrombosis, gastrointestinal hemorrhage in the past 6 months, hereditary angioedema of any severity, or history of severe or life-threatening angioedema due to any cause were also excluded. Finally, use of concomitant medications that are predominantly cleared by organic anion transporter polypeptide (OATP) hepatobiliary transporters OATP1B1 and OATP1B3 was prohibited within 48 hours of ALRN-6924 infusion because ALRN-6924 is primarily cleared through hepatobiliary elimination and inhibits these transporters.
Before patient recruitment began at each participating site, the protocol was approved by its institutional review board. All patients provided written informed consent before undergoing study-related procedures, including the molecular assessment of tumor specimens. This trial was conducted in accordance with current United States FDA regulations, ICH Good Clinical Practice Guidelines, the principles of the Declaration of Helsinki, and local ethical and legal requirements. This trial is registered on ClinicalTrials.gov (NCT02264613).
Study design and treatment plan
A standard 3+3 dose-escalation design was used to independently assess two single-agent dosing regimens: patients in treatment arm A received ALRN-6924 by intravenous infusion on days 1, 8, and 15 of a 28-day cycle, whereas those in treatment arm B received ALRN-6924 on days 1, 4, 8, and 11 of a 21-day cycle. Enrollment began with arm A, and arm B was introduced once the third dose level on arm A was reached. Subsequently, patients were assigned to one of the two dosing regimens based on the availability of enrollment slots. Doses were doubled until ≥1 of 3 patients in a cohort experienced any grade ≥2 treatment-emergent adverse event (TEAE) that was possibly related to the study drug, at which time a modified Fibonacci sequence was implemented (24). Intra-patient dose escalation was allowed.
The primary endpoints of the study were TEAEs, dose-limiting toxicities (DLT), and safety assessments. Secondary endpoints included overall response rate, time to response, duration of response, disease control rate, and duration of clinical benefit. Pharmacologic secondary endpoints included pharmacokinetic parameters of ALRN-6924 and its primary metabolite; serum levels of macrophage inhibitory cytokine-1 (MIC-1), an established secreted biomarker of p53 activation; and immunogenicity as measured by anti-drug antibody levels in blood.
Treatment continued until unacceptable toxicity, disease progression, or patient or physician decision to end treatment. Patients experiencing clinical benefit could continue the study despite radiographic disease progression.
Toxicity assessments
Adverse events were recorded at each visit and graded by National Cancer Institute Common Terminology Criteria for Adverse Events, version 4.03. DLTs were defined as any grade ≥3 non-hematologic adverse event, excluding the following: fatigue, nausea, emesis, diarrhea, or mucositis that responded to supportive treatment within 48 hours; electrolyte aberrations that responded to correction within 24 hours; and grade ≤3 infusion-related reactions that did not require hospitalization. Hematologic DLTs included grade 4 thrombocytopenia of any duration, grade 3 thrombocytopenia lasting ≥7 days or associated with clinically significant bleeding, grade 4 neutropenia lasting for ≥3 days, or any grade ≥3 febrile neutropenia.
Pharmacokinetics and pharmacodynamics
The pharmacokinetic profiles of ALRN-6924 and its active metabolite ALRN-8714 in patient plasma were assessed using a validated liquid chromatography method with tandem mass spectrometric detection. Blood samples were obtained within 1 hour before the start and at the end of infusion (±5 minutes) and at 0.5, 1, 2, 4, 8, 24, and 48 hours after infusion on cycle 1 day 1 in both arm A and arm B. In addition, selective sampling was done on subsequent dosing days in subsequent cycles of therapy in both regimens to evaluate accumulation of exposure, dose-to-dose variability, and changes in exposure due to possible induction or inhibition of clearance mechanisms. Pharmacokinetic parameters were calculated by non-compartmental analysis using Phoenix WinNonlin v.8.1 and included maximum plasma concentration (Cmax), area under the plasma concentration curve (AUC), and half-life calculations.
The pharmacodynamic profile of MIC-1 was assessed in patient serum samples using a validated, quantitative sandwich ELISA. Blood samples for serum MIC-1 levels were obtained in parallel with plasma pharmacokinetic samples at the collection times noted above.
Exploratory analyses included correlation of antitumor activity with baseline protein and gene expression levels of p53, MDM2, MDMX, and related biomarkers in tumor biopsy tissue. Fresh or archival (≤1 year old) formalin-fixed tumor specimens collected for next-generation sequencing of TP53 were further used for IHC analysis of protein biomarkers, and nucleic acid extracts for next-generation sequencing were further used for qRT-PCR analysis of gene expression levels (Supplementary Methods and Supplementary Table S1).
Response assessment
Tumor response was assessed every 2 cycles (8 weeks) for patients in arm A and every 3 cycles (9 weeks) for patients in arm B. Response was determined by the investigators in accordance with RECIST 1.1 for patients with solid tumors (25) and IWG 2014 for lymphoma patients (26). Patients were considered eligible for response assessment if they had received at least one dose of ALRN-6924 and had undergone post-baseline radiological tumor assessment or exhibited clinical or objective progression as determined by the investigator and excluded patients with TP53-mutant tumors and patients in the lowest dose levels (<0.8 mg/kg). Data cutoff date for the response analysis was May 10, 2020.
Drug supply and administration
ALRN-6924 was supplied and stored in single-use glass vials as a refrigerated (2°C to 8°C) liquid product dissolved at 15 mg/mL in 20 mmol/L sodium phosphate, 240 mmol/L trehalose, and 300 ppm polysorbate 20, pH 7.5. A weight-adjusted dose of the ALRN-6924 drug product was diluted into 250 mL, 5% dextrose in water for intravenous administration to patients via a 1-hour infusion, except for dose level 7–2 in arm A, which was infused over 2 hours.
Results
Patient characteristics
From October 2014 to January 2017, 71 patients were enrolled, with 41 (58%) allocated to treatment arm A and 30 (42%) allocated to treatment arm B. At the time of data cutoff for analysis (April 13, 2018), 66 (93.0%) of 71 patients had discontinued study treatment: 38 in treatment arm A and 28 in arm B. Key demographics and baseline characteristics are shown in Table 1 and were similar for those in treatment arms A and B. Overall, the median age was 61.0 years, and the percentages of men and women were similar (49.3% and 50.7%, respectively), and most patients were Caucasian (74.6%). The patient population was heavily pretreated, with 67.6% of patients having received more than two prior lines of systemic therapy, including 43.7% of patients with at least five lines of prior therapy. Though not an exclusion factor, no patients had prior therapy with an investigational MDM2 inhibitor. Of the 71 patients enrolled, 67 had solid tumors, and 4 patients had lymphomas. Patients in the study had 24 different tumor types; these included thymoma (n = 5), peripheral T-cell lymphoma (PTCL; n = 1), colorectal cancer (n = 1), sarcoma (n = 17), Merkel cell carcinoma (n = 4), renal cell carcinoma (n = 2), ovarian cancer (n = 2), adenoid cystic carcinoma (n = 5), hepatocellular carcinoma (n = 1), melanoma (n = 6), salivary gland cancer (n = 3), breast cancer (n = 3), and other cancers (n = 21). Patients were enrolled irrespective of TP53 status in the three lowest dose levels (n = 12) of arm A, which preceded enrollment in arm B; therefore, the TP53-wild-type (WT) confirmation rate was higher for patients enrolled in arm B (100%) than in arm A (82.9%), or 90.1% (64/71) overall. Other patients had either mutant (n = 2) or indeterminate (n = 5) TP53 status.
Table 1.
Demographic characteristics of patients overall and in treatment arms A and B.
| Characteristics | Arm A (n = 41) | Arm B (n = 30) | Total (n = 71) |
|---|---|---|---|
| Median age (y; range) | 62 (25–78) | 58 (31–76) | 61 (25–78) |
| Sex, n (%) | |||
| Male | 20 (48.8) | 15 (50.0) | 35 (49.3) |
| Female | 21 (51.2) | 15 (50.0) | 36 (50.7) |
| Race, n (%) | |||
| Caucasian | 28 (68.3) | 25 (83.3) | 53 (74.6) |
| African American | 9 (22.0) | 4 (13.3) | 13 (18.3) |
| Asian | 1 (2.4) | 0 | 1 (1.4) |
| Other | 3 (7.3) | 1 (3.3) | 4 (5.6) |
| Ethnicity, n (%) | |||
| Hispanic or Latino | 4 (9.8) | 1 (3.3) | 5 (7.0) |
| Not Hispanic or Latino | 34 (82.9) | 26 (86.7) | 60 (84.5) |
| Unknown | 3 (7.3) | 3 (10) | 6 (8.5) |
| ECOG PS, n (%) | |||
| 0 | 13 (31.7) | 9 (30.0) | 22 (31.0) |
| 1 | 28 (68.3) | 21 (70.0) | 49 (69.0) |
| Prior lines of treatment, n (%) | |||
| 1 or 2 | 9 (22.0) | 14 (46.7) | 23 (32.4) |
| 3 or 4 | 11 (26.8) | 6 (20.0) | 17 (23.9) |
| ≥5 | 21 (51.2) | 10 (33.3) | 31 (43.7) |
| Tumor TP53-WT, n (%) | 34 (82.9) | 30 (100) | 64 (90.1) |
Abbreviation: ECOG PS, Eastern Cooperative Oncology Group performance status.
Patients were enrolled on the basis of either central or local laboratory TP53 assessment. Twenty-nine patients underwent both local and central laboratory testing, and 5 of these 29 patients (17.2%) had discrepant results (all TP53-WT on local testing and TP53-mutant on central testing), possibly due to differences in gene coverage between local and central laboratory assays and instances in which different biopsy samples (from primary tumor or metastases) were analyzed by the central and local laboratories. Details are provided in Supplementary Table S2. These patients who were TP53-WT on local testing but TP53-mutant on central laboratory assessment were excluded from the response-evaluable cohort.
Dose escalation and toxicity
As shown in Fig. 1, arm A began enrollment at 0.16 mg/kg, and arm B began enrollment once the third dose level in arm A (DL-3A) was completed. Enrollment proceeded in dose increments of 100% until grade ≥2 drug-related TEAEs were observed in DL-4A (fatigue) and in DL-3B (neutropenia); subsequent dose escalations in DL-5A and DL-4B continued with a modified Fibonacci sequence (i.e., 67%, 50%, 40%, and 33%) up to 4.4 mg/kg once-weekly in arm A and 2.7 mg/kg twice-weekly in arm B. Because of hypotension observed in patients during infusion at DL-7A, DL-7A-2 was introduced to evaluate a 2-hour infusion time with dexamethasone and fluids administered in cycles 1 and 2. All other patients received ALRN-6924 via a 1-hour infusion.
Figure 1.

Study schema. Patients in arm A received ALRN-6924 on days 1, 8, and 15 of a 28-day cycle, whereas those in arm B received ALRN-6924 on days 1, 4, 8, and 11 of a 21-day cycle.
All 71 enrolled patients received at least one dose of ALRN-6924 and were included in the safety analysis population. All 71 patients experienced at least one TEAE. Common TEAEs (defined as ≥10% occurrences) across both treatment arms are shown in Supplementary Table S3. These TEAEs were primarily grade 1 and 2 (60.6%), and the most frequent TEAEs regardless of relationship to study treatment included nausea (73.2%), fatigue (60.6%), vomiting (46.5%), anemia and headache (31.0% each), decreased appetite and constipation (29.6% each), and diarrhea (19.7%). In general, there were no marked differences in the incidence of TEAEs between arm A and arm B and no apparent dose-related trends. There were no fatal TEAEs.
Table 2 shows all observed grade ≥3 drug-related TEAEs. Overall, these occurred in 22.5% of patients (16/71). The rate of drug-related grade ≥3 TEAEs was similar in arms A and B. Only 2 patients experienced drug-related grade 4 TEAEs: 1 patient with neutropenia in each arm; no grade 5 TEAEs were observed.
Table 2.
Drug-related grade ≥3 treatment-emergent adverse events.
| Arm A | Arm B | Total | |
|---|---|---|---|
| n = 41 | n = 30 | n = 71 | |
| n (%) | n (%) | n (%) | |
| Patients with at least one grade ≥3 TEAE event | 10 (24.4) | 6 (20.0) | 16 (22.5) |
| Adverse event | |||
| Fatigue | 2 (4.9) | 1 (3.3) | 3 (4.2) |
| Anemia | 1 (2.4) | 1 (3.3) | 2 (2.8) |
| Neutropenia | 1 (2.4) | 1 (3.3) | 2 (2.8) |
| Nausea | 1 (2.4) | 1 (3.3) | 2 (2.8) |
| Infusion-related reaction | 2 (4.9) | 0 | 2 (2.8) |
| Hyponatremia | 1 (2.4) | 1 (3.3) | 2 (2.8) |
| Hypotension | 2 (4.9) | 0 | 2 (2.8) |
| Diarrhea | 0 | 1 (3.3) | 1 (1.4) |
| Vomiting | 0 | 1 (3.3) | 1 (1.4) |
| Asthenia | 1 (2.4) | 0 | 1 (1.4) |
| Aspartate aminotransferase increased | 1 (2.4) | 0 | 1 (1.4) |
| Alkaline phosphatase increased | 1 (2.4) | 0 | 1 (1.4) |
| Hypoxia | 1 (2.4) | 0 | 1 (1.4) |
Abbreviation: TEAE, treatment-emergent adverse event.
Protocol-defined DLTs were experienced by 5 patients in treatment arm A. Dose escalation for this regimen proceeded to the 3.1 mg/kg dose level, wherein 9 patients were treated and only 1 experienced a DLT (i.e., grade 3 fatigue that did not respond to supportive treatment within 48 hours). Dose escalation then continued to 4.4 mg/kg, where four DLTs were noted among the 11 patients across both 1- and 2-hour ALRN-6924 infusions, including grade 3 anemia (n = 1), grade 3 hypotension (n = 1), grade 4 neutropenia (n = 1), and grade 3 elevated alkaline phosphatase level (n = 1). The patient with a DLT of grade 3 fatigue recovered following dose delay within the 7 days before the next clinical visit and continued treatment after dose reduction. The patient with grade 3 anemia continued treatment without dose reduction and without clinical symptoms or complications and recovered within 34 days. The patients experiencing DLTs of grade 3 hypotension and grade 4 neutropenia discontinued treatment and recovered within 5 and 7 days, respectively. The patient with a DLT of grade 3 elevated alkaline phosphatase level discontinued treatment and did not recover, and a subsequent liver biopsy revealed diffuse breast cancer metastases. On the basis of these DLTs, ALRN-6924 administered at 4.4 mg/kg once-weekly in arm A was considered to exceed the MTD and was not further explored. The safety, pharmacokinetic, pharmacodynamic, and antitumor activity of ALRN-6924 at doses up to 3.1 mg/kg was considered favorable for further exploration as a single-agent in specific tumor types and in combination studies with other anticancer agents. Although no DLTs were observed in arm B, and the efficacy profile in arm B was similar to that in arm A (vide infra), the twice-weekly regimen was considered less convenient than the once-weekly schedule and was not pursued.
A total of 11 serious adverse events (SAE) were reported in 7 patients in arm A and 1 patient in arm B. Of these serious adverse events, 3 in 2 patients were considered treatment related. One patient had grade 3 hypotension and shortness of breath at the 3.1 mg/kg dose level in arm A, likely due to a drug–drug interaction. This patient presented with hypotension the day after ALRN-6924 dosing following self-administration of the antihypertensive agent telmisartan, a prohibited concomitant medication. Telmisartan is primarily cleared by hepatobiliary transporter OATP1B3, for which ALRN-6924 is an inhibitor (27). Pharmacokinetic samples from this patient showed elevated plasma levels of telmisartan from the self-administered dose. The second patient had two events of grade 3 hypotension at the 4.4 mg/kg dose level in arm A, a dose level determined to be above the MTD. Both events occurred at the time of infusion and no apparent drug–drug interaction was identified. The second event resulted in discontinuation of study treatment and was considered a DLT as discussed above. To mitigate potential infusion-related hypotension as a safety concern, the clinical protocol was amended to specify that intravenous fluids (saline) or oral fluids (500–1,000 mL) should be administered at the end of the infusion.
Five patients (7.0%) discontinued treatment due to at least one drug-related TEAE, all in arm A. The TEAEs leading to treatment discontinuation were infusion-related reactions (n = 3), hypotension (n = 1), and neutropenia (n = 1).
The rates of dose reductions were 6/41 (14.6%) in arm A and 3/30 (10%) in arm B. In arm A, the dose of ALRN-6924 was reduced in response to mild to moderate fatigue, nausea, and vomiting in 5 patients across dose levels from 2.1 to 4.4 mg/kg, and in 1 patient with grade 2 chest pain at 2.1 mg/kg. In arm B, 1 patient with grade 3 diarrhea at the 0.8 mg/kg dose level was dose reduced, as was 1 patient with grade 3 nausea at 2.0 mg/kg and 1 patient with grade 1 fatigue and grade 1 nausea at 2.7 mg/kg. Notably, the observed toxicities were acute, with no evidence of cumulative hematologic or non-hematologic toxicities. Ophthalmic examination was not specified in the ALRN-6924 clinical trial, and no SAEs related to eye disorders were reported.
Pharmacokinetic and pharmacodynamic studies
Noncompartmental pharmacokinetic parameters and fold-change from baseline in serum MIC-1 levels are shown in Table 3. Following a single intravenous infusion, ALRN-6924 achieved Cmax at the end of infusion, and the mean Cmax values ranged dose proportionally from 3.4 to 96.4 μg/mL over the dose range of 0.16 to 4.4 mg/kg (Fig. 2A). The mean AUC across all collection time points (AUCall) increased slightly more than dose proportionally from 8.0 to 1,200 μg*h/mL over this same dose range, whereas the mean half-life also increased with increasing dose levels and ranged from 1.5 to 6.8 hours (see Supplementary Information). The mean apparent clearance decreased with increasing dose and ranged from 3.70 to 23.5 mL/h/kg, whereas volume of distribution ranged from 25.7 to 51.1 mL/kg independent of dose level. Plasma pharmacokinetic results for individual patients on subsequent dosing days and subsequent cycles of therapy showed no accumulation of exposure, reproducible dose-to-dose exposure, and no indication of changes in exposure due to possible induction or inhibition of clearance mechanisms (data not shown). Noncompartmental pharmacokinetic parameters for 7 patients with complete pharmacokinetic sample collection at the 3.1 mg/kg dose level are shown in Fig. 2B. At the recommended phase II dose (RP2D), ALRN-6924 exhibited a Cmax of 71.2 μg/mL, AUCall of 705 μg*h/mL, and half-life of 5.4 hours, with a coefficient of variation of 20% and 26% for Cmax and AUCall, respectively. No ALRN-6924–specific anti-drug antibodies were detected in any patient following administration of ALRN-6924.
Table 3.
Noncompartmental pharmacokinetic parameters and fold-change from baseline in serum MIC-1 levels (mean; % CV) for patients with complete sample collection following a single 1-hour intravenous infusion on cycle 1 day 1 at select doses.
| 0.16 mg/kg | 0.32 mg/kg | 0.53 mg/kg | 0.64 mg/kg | 1.25 mg/kg | 2.1 mg/kg | 3.1 mg/kg | 4.4 mg/kg | |
|---|---|---|---|---|---|---|---|---|
| N | 5 | 3 | 5 | 4 | 2 | 6 | 7 | 8 |
| AUCall (μg*h/mL) | 8.02 (52.7) | 17.8 (20.4) | 42.8 (68.9) | 55.6 (25.3) | 164 (35.2) | 434 (53.2) | 705 (26.4) | 1200 (14.5) |
| C max (μg/mL) | 3.37 (26.5) | 5.56 (5.8) | 10.1 (40.8) | 13.4 (16.6) | 27.7 (22.5) | 45.2 (40.4) | 71.2 (19.6) | 96.4 (13.1) |
| t 1/2 (h) | 1.5 (64.4) | 1.97 (54.1) | 2.57 (49.7) | 2.72 (52.9) | 2.22 (6.9) | 4.63 (40.5) | 5.36 (38.6) | 6.84 (25.6) |
| CL (mL/h/kg) | 23.5 (43.1) | 17.4 (30.7) | 16.6 (56.3) | 12.1 (25.5) | 8.10 (35.1) | 6.49 (63.1) | 4.74 (40.5) | 3.70 (15.4) |
| V z (mL/kg) | 42.2 (46.6) | 44.3 (31.2) | 50.8 (40.7) | 51.1 (76.9) | 25.7 (28.5) | 41.8 (62.8) | 33.8 (26.9) | 36.8 (36.4) |
| MIC-1 at 8 h | n/d | 7.7 (100) | 6.0 (28) | n/d | 8.9 (14) | 10.5 (77) | 9.6 (73) | 9.6 (39) |
| MIC-1 at 24 h | 2.9 (102) | 1.7 (40) | 3.1 (42) | 4.8 (75) | 16.3 (32) | 21.6 (87) | 19.9 (67) | 26.3 (57) |
| MIC-1 at 48 h | 1.4 (51) | 1.1 (12) | 1.5 (38) | 1.6 (35) | 2.6 (31) | 8.1 (91) | 14.0 (79) | 24.2 (59) |
Abbreviations: AUCall, area under the curve for all measurable sampling times; CL, clearance; Cmax, maximum concentration; CV, coefficient of variation; h, hour(s); N, number; n/d, not determined; t1/2, half-life; Vz, volume of distribution.
Figure 2.
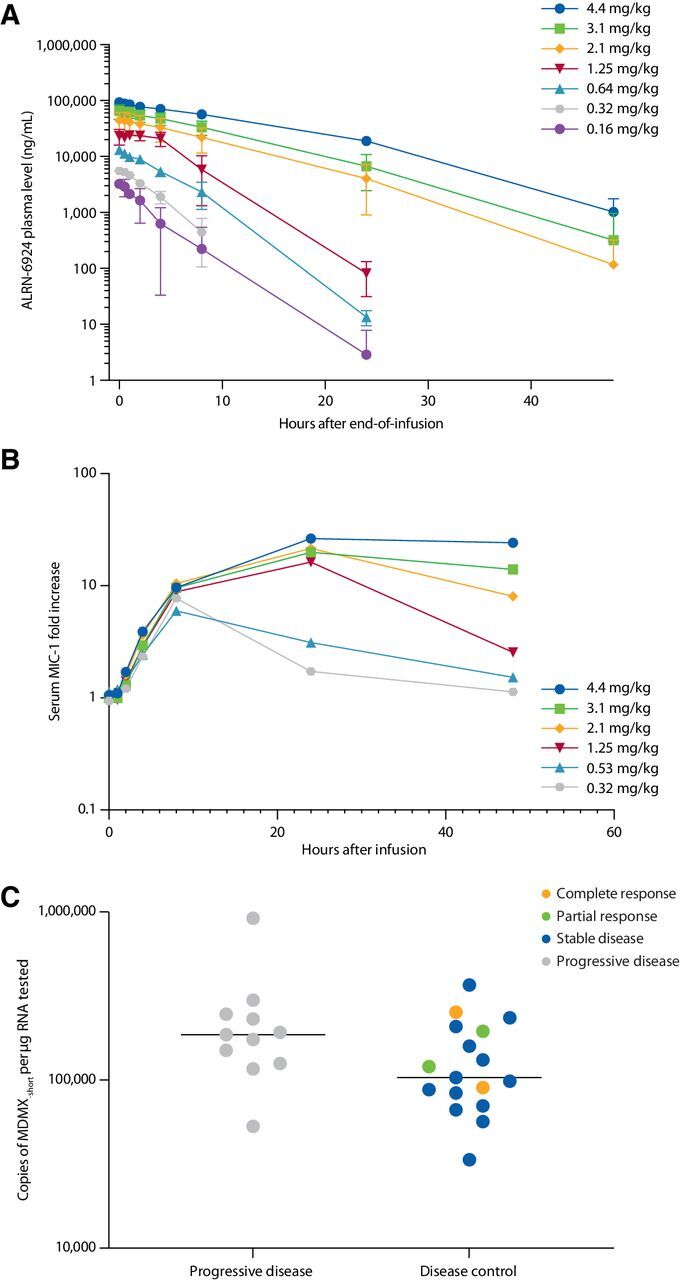
Pharmacokinetics and pharmacodynamics, and association of antitumor activity with MDM2 amplification and MDMX-short splice variant expression. A, Plasma levels of ALRN-6924 following a single 1-hour intravenous infusion. For clarity, only dose levels in arm A are shown. B, Mean increase from baseline in serum macrophage inhibitory cytokine 1 levels following a single dose of ALRN-6924. For clarity, only select dose levels are shown. C, Expression levels of the MDMX-short splice variant in pretreatment tumor biopsy specimens for 28/41 efficacy-evaluable patients versus best overall response. Line demarks median value. Two-tailed P = 0.099 (not significant).
Pharmacodynamic activity of ALRN-6924 was demonstrated by dose-related elevations of serum MIC-1, which is an established biomarker for activation of the p53 pathway (Fig. 2C). A 10-fold increase in MIC-1 levels was observed by 8 hours after a single dose starting at 0.8 mg/kg, with a gradual decline to baseline after 24 hours at doses of 1.25 mg/kg and below, whereas doses of 2.1 mg/kg and above demonstrated a sustained elevation of MIC-1 up to 48 hours post-dose.
Antitumor activity
The efficacy-evaluable population included patients who received at least one dose of ALRN-6924 at dose levels ≥0.8 mg/kg, had confirmed TP53-WT tumors, and had undergone post-baseline radiological tumor assessment or exhibited clinical or objective progression as determined by the investigator. A total of 41 of the 71 enrolled patients were evaluable for response (a CONSORT diagram is shown in Supplementary Fig. S1). The best change in target lesion size is shown in Fig. 3A. Of the 41 evaluable patients, 24 patients (59%) demonstrated disease control by RECIST 1.1 or IWG 2014 criteria. Four patients achieved a response: 2 patients (1 with PTCL and one with Merkel cell carcinoma) achieved a confirmed complete response (CR) and 2 patients (1 with colorectal cancer and 1 with liposarcoma) demonstrated a confirmed partial response (PR). Twenty patients (49%) achieved stable disease (SD), with 55% of the patients with SD experiencing shrinkage of target lesions. The overall disease control rate was 59% (24/41), and among the 24 patients with disease control, the median duration of clinical benefit was 7.5 months at the time of data cutoff. A swimmer plot of treatment duration is shown in Fig. 3B.
Figure 3.

A, Best overall response for efficacy-evaluable patients treated with ALRN-6924. The graph shows best response per the RECIST 1.1 (or Revised International Working Group Response Criteria 2014 for patients with lymphoma) in 39 of 41 efficacy-evaluable patients. Two efficacy-evaluable patients with non-radiological evidence of disease progression are not shown. Response-evaluable patients with TP53-WT tumors with MDM2 amplification are designated with an asterisk (*). B, Swimmer plot of treatment duration for 41 efficacy-evaluable patients in arms A and B.
Overall, 7 of 71 patients received ≥15 cycles of treatment, and 6 patients remained on treatment for over 1 year, including 5 patients who continued treatment beyond the data cutoff. A patient with PTCL achieved a durable CR at cycle 6 of the 2.1 mg/kg dose level in arm A, and at the time of data cutoff, the patient had remained on treatment for 52 months. The patient with Merkel cell carcinoma showed a CR at the sixth cycle of treatment at the 2.7 mg/kg dose level in arm B, and the CR lasted for 6.5 months; this patient continued to receive clinical benefit and remained on treatment for 26 months. A patient with colorectal cancer who was treated at the 0.8 mg/kg dose level in arm B experienced continuous tumor shrinkage that met the criteria for PR at cycle 15 and remained on treatment for 16 months. A patient with liposarcoma achieved a durable PR at the 4.4 mg/kg dose level in arm A at cycle 8 and remained on treatment for 32 months. Two patients with thymoma achieved durable SD, one at the 2.1 mg/kg dose level in arm A for 57 months and another at the 2.7 mg/kg dose level in arm B for 17 months.
Association between molecular alterations and antitumor activity
Genomic sequencing results from the Ion Torrent PGM platform (Knight Diagnostic Laboratories) were available for all 4 patients with objective responses. The patient with rectal cancer who experienced a PR had a KRAS G12D mutation and a subclonal PIK3CA P381S variant. The patient with Merkel cell cancer who had a CR had a variant of unknown significance in ALK (N1394H). The other 2 patients, one with liposarcoma and the other with PTCL, had no mutations in the 36 cancer-related genes sequenced.
We enrolled 10 TP53-WT patients with MDM2 amplification as determined by central or local laboratory next-generation sequencing; of these, 7 were in the efficacy-evaluable population (dose ≥0.8 mg/kg), including 4 patients with liposarcoma and 1 patient each with bladder carcinoma, gastric adenocarcinoma, and breast cancer. Among these 7 MDM2-amplified, efficacy-evaluable patients, 5 patients (71%) patients achieved disease control, including 1 with PR (liposarcoma) and 4 with SD as best overall response (Fig. 3A).
Anti-tumor activity was compared with pretreatment tumor biopsy levels of the full-length MDMX gene transcript (MDMX-FL) and a splice variant that skips exon 6 to encode a truncated isoform, MDMX-short. Nucleic acid extracts derived from tumor tissue for next-generation sequencing of TP53 were available in sufficient quantity for gene expression analysis for 28 of 41 efficacy-evaluable patients, including all 4 patients with CR or PR, 13 with SD as their best response, and 11 with progressive disease (PD) as their best response. As shown in Fig. 2C, across the range of tumor types evaluated in this phase 1 study, a trend was observed in which patients with lower expression levels of MDMX-short were more likely to achieve disease control (CR, PR, or SD as best overall response) than were patients with higher MDMX-short levels; however, this association did not reach statistical significance.
Additional biomarker analyses showed no trend between ALRN-6924 antitumor activity and protein levels of p53, MDM2, or MDMX-FL by either IHC analysis of available formalin-fixed pretreatment biopsy samples or by gene expression levels of TP53, MDM2, or MDMX-FL (data not shown). MDMX-short protein levels were not evaluated by IHC.
Discussion
This study is the first clinical trial of ALRN-6924, a stabilized, cell-permeating, α-helical stapled peptide, and is also the first clinical trial of a dual MDM2/MDMX inhibitor. Two single-agent dosing regimens were evaluated to assess safety and antitumor activity on a once-weekly or twice-weekly dosing schedule, with these treatment regimens, including a 14-day (arm A) or 10-day (arm B) break between the last dose of one cycle and the first dose of the next, to allow recovery from potential toxicities. ALRN-6924 showed a good safety profile, with mild to moderate nausea, fatigue, and vomiting as the most common drug-related toxicities, and yielded disease control and objective responses in patients with a variety of tumor types.
Over the past decade, several MDM2 inhibitors have entered clinical trials, including RG7112 (RO5045337), idasanutlin (RG7388), AMG-232 (KRT-232), APG-115, CGM097, siremadlin (HDM201), milademetan (DS-3032b), MK-8242, and SAR405838 (13, 14, 28–35). A striking finding in our study was the limited myelosuppressive effect of ALRN-6924, in contrast with clinical trial reports from small molecule MDM2 inhibitors (Supplementary Table S4). In reports from other MDM2 inhibitors, neutropenia and thrombocytopenia were often the DLTs as detailed in a recent review by Konopleva and colleagues (28). MDM2 is involved in normal hematopoiesis, and treatment with MDM2 antagonists can lead to on-target hematopoietic defects (36, 37). Of all 37 normal tissues reported in the Protein Atlas (38, 39), MDMX expression is highest in bone marrow cells, which may contribute to the lower degree of bone marrow toxicity observed with a dual MDM2/MDMX inhibitor. We speculate that high MDMX levels may act as a pharmacokinetic “sink” to sequester ALRN-6924 in bone marrow cells and minimize its inhibition of MDM2, thereby minimizing MDM2-mediated ubiquitination and degradation of p53 in bone marrow cells. This concept remains a topic of continued research (40).
ALRN-6924 demonstrated favorable pharmacokinetic properties, with reproducible patient-to-patient values for key pharmacokinetic parameters as judged by the narrow coefficients of variation for Cmax and AUCall, 20%, and 26%, respectively. This narrow range of variability in exposure contrasts with the inherent variability associated with uptake of oral drugs, allowing target therapeutic exposures to be more readily achieved with intravenous administration of ALRN-6924 without unexpected high exposure leading to toxicities seen with oral agents, in particular small-molecule MDM2 inhibitors, which are oral.
Because ALRN-6924 activates p53, which is a transcription factor, the pharmacodynamic response to ALRN-6924 is moderated by the unique nature of its target. Serum levels of MIC-1, a protein under transcriptional control of p53, rapidly increased after ALRN-6924 dosing, demonstrating activation of the p53 pathway in tissues in response to the drug. MIC-1 levels were sustained for over 48 hours at the RP2D, well after the drug was mostly cleared from circulation, indicating a long-acting pharmacodynamic effect of p53 activation by ALRN-6924. This multi-day pharmacodynamic effect, considered in tandem with the disease control rate and objective responses observed on both once-weekly and twice-weekly dosing regimens, indicates that intermittent intravenous dosing of ALRN-6924 can produce satisfactory antitumor activity, and more frequent administration is not necessarily needed despite a plasma half-life of 5.4 hours at the RP2D.
The once-weekly dosing of ALRN-6924 at 3.1 mg/kg was chosen as the RP2D based on observed safety, tolerability, and antitumor activity. No DLTs were observed in arm B, and although the per-cycle drug exposure and favorable risk/benefit profile were similar for the two regimens at the highest tolerated doses tested, the once-weekly infusion schedule was considered more convenient by patients and investigators.
Encouraging single-agent activity, including durable CRs and PRs, was observed in a number of patients with different tumor types despite the heavily pretreated nature of this patient population. Furthermore, more than half of the evaluable patients achieved disease control. The durable CRs seen in Merkel cell carcinoma and PTCL highlight these diseases as potential therapeutic opportunities. The PR observed in the patient with colorectal cancer harboring KRAS mutation emphasizes that although TP53 mutations are known to be enriched in metastatic colorectal cancer compared with primary tumors; over 40% of patients with colorectal cancer have TP53-WT tumors, so targeting MDM2/MDMX may hold promise (41, 42). Notably, a previous study with SAR405838 and pimasertib also showed some activity, with prolonged SD in colorectal cancer patients with a KRAS mutation and TP53-WT (43).
Liposarcoma has been considered a disease of interest for MDM2 inhibitors, as MDM2 amplifications have been reported in over 60% of liposarcomas (44). Furthermore, in dedifferentiated liposarcoma, MDM2 amplification and MDM2 overexpression was associated with shorter time to recurrence after surgical resection and reduced overall survival in patients receiving systemic therapy (45). In the phase I trial reported here, we observed a PR in 1 of 5 evaluable patients with liposarcoma and stable disease in the other 4. PRs have also been reported in liposarcoma with other MDM2 inhibitors, including MK-8242 and milademetan (DS-3032B or RAIN-32; refs. 29, 30), emphasizing that targeting MDM2 or MDM2/MDMX in liposarcoma alone or in combination therapy may be another therapeutic opportunity.
An intriguing correlation was observed between antitumor activity and pretreatment expression levels in patient tumors for an MDMX splice variant that skips exon 6 to encode a truncated isoform, MDMX-short. High expression of MDMX-short has been associated with faster progression and poorer survival in several cancers (46). Although the exact mechanism of this association remains controversial, one theory suggests that alternate splicing is a mechanism for posttranslational regulation of MDMX-FL protein levels, such that the MDMX-short splicing event leads to nonsense-mediated mRNA decay and a truncated protein that is also rapidly degraded, yielding lower levels of MDMX-FL protein expression (47). Therefore, tumor cells with low levels of MDMX-short expression may have higher MDMX-FL expression and greater dependence on MDMX for p53-dependent and p53-independent mechanisms for survival (48). Analysis of the 28 efficacy-evaluable patients for whom a sufficient sample was available revealed that across the range of tumor types and doses evaluated in this phase 1 study, patients with lower expression levels of MDMX-short were more likely to achieve disease control (CR, PR, or SD as best overall response) than were patients with higher MDMX-short levels. Although the number of patients tested in this assay was small and the results did not reach statistical significance, the trend suggests that MDMX inhibition by ALRN-6924 provides greater antitumor activity against tumors with lower MDMX-short levels. Alternative MDMX splicing is one of several mechanisms that can affect the function of p53, but with further study, this biomarker may be useful to select patients with a higher likelihood of response to ALRN-6924 therapy.
The disease control rate observed in this study suggests that p53 activation by ALRN-6924 may induce cell-cycle arrest in TP53-WT tumor tissues, whereas the favorable tolerability profile, especially the virtual absence of thrombocytopenia and neutropenia that is otherwise common for MDM2 inhibitors, indicates that ALRN-6924 does not induce profound levels of apoptosis in healthy tissues such as bone marrow, even at the highest tested doses. This further suggests that besides its use as an anticancer drug, ALRN-6924 may be an ideal agent for treatment of normal, healthy TP53-WT tissues, such as bone marrow, when administered at lower doses more likely to induce cell-cycle arrest, resulting in protection from the toxic effects of chemotherapy. This concept, known as cyclotherapy (49), has been proposed for MDM2 inhibitors (50, 51) and was recently demonstrated in the clinic for a CDK4/6 inhibitor (52). Because approximately half of all cancers carry inactivating mutations in the TP53 gene, which renders TP53-mutant cancer cells insensitive to the effects of ALRN-6924, prophylactic treatment with ALRN-6924 before chemotherapy may induce cell-cycle arrest in normal tissues with wild-type TP53 while leaving the rapidly proliferating TP53-mutant cancer cells susceptible to S- or M-phase poisons, such as topoisomerase inhibitors (which are most effective in S phase) or drugs targeting the mitotic spindle, such as taxanes (which are most effective during mitosis; ref. 53).
In conclusion, this study demonstrated the safety of first-in-class dual MDM2/MDMX inhibitor ALRN-6924. ALRN-6924 yielded disease control and objective responses in patients with a variety of TP53-WT tumors. The limited myelosuppression observed has provided rationale for ongoing clinical trials with ALRN-6924 as a chemoprotective drug for patients with TP53-mutant cancers.
Authors' Disclosures
M.R. Patel reports other support from Aileron during the conduct of the study. T.M. Bauer reports grants from Aileron during the conduct of the study; as well as personal fees from Pfizer, Lilly, Bayer, and BMS outside the submitted work. G.S. Falchook reports grants from AIleron during the conduct of the study. G.S. Falchook also reports royalties from Wolters Kluwer; advisory role grants (to institution) from Fujifilm, Silicon, Navire, and Turning Point; advisory role grants (self) from EMD Serono; travel expenses from Bristol Myers Squibb, EMD Serono, Fujifilm, Millennium, and Sarah Cannon Research Institute; speakers honorarium from Total Health Conferencing and Rocky Mountain Oncology Society; and research funding (to institution) from 3-V Biosciences, Abbisko, AbbVie, ADC Therapeutics, Aileron, American Society of Clinical Oncology, Amgen, ARMO/Eli Lilly, AstraZeneca, BeiGene, Bioatla, Bioinvent, Biothera, Bicycle, Celldex, Celgene, Ciclomed, Curegenix, Curis, Cyteir, Daiichi, DelMar, eFFECTOR, Eli Lilly, EMD Serono, Epizyme, Erasca, Exelixis, Fujifilm, Genmab, GlaxoSmithKline, Hutchison MediPharma, IGM Biosciences, Ignyta, ImmunoGen/MacroGenics, Incyte, Jacobio, Jounce, Kolltan, Loxo/Bayer, MedImmune, Millennium, Merck, miRNA Therapeutics, National Institutes of Health, Navire, Novartis, OncoMed, Oncorus, Oncothyreon, Poseida, Precision Oncology, Prelude, PureTech, RasCal, Regeneron, Rgenix, Ribon, Sapience, Silicon, Strategia, Syndax, Synthorx/Sanofi, Taiho, Takeda, Tarveda, Teneobio, Tesaro, Tocagen, Turning Point Therapeutics, U.T. MD Anderson Cancer Center, Vegenics, and Xencor outside the submitted work. G.I. Shapiro reports other support from Aileron during the conduct of the study. G.I. Shapiro also reports grants from Eli Lilly and Merck & Co.; grants and personal fees from Merck KGaA/EMD Serono and Sierra Oncology; personal fees from Pfizer, G1 Therapeutics, Roche, Bicycle Therapeutics, Fusion Pharmaceuticals, Cybrexa Therapeutics, Astex, Almac, Ipsen, Bayer, Angiex, Daiichi Sankyo, Seattle Genetics, Boehringer Ingelheim, ImmunoMet, Asana, Artios, Atrin, Concarlo Holdings, Syros, Zentalis, CytomX Therapeutics, and Blueprint Medicines outside the submitted work. G.I. Shapiro also has a patent for dosage regimen for sapacitabine and seliciclib pending and issued to Cyclacel Pharmaceuticals and Geoffrey Shapiro and compositions and methods for predicting response and resistance to CDK4/6 inhibition pending to L. Cornell and G.I. Shapiro. J.R. Infante reports other support from Aileron Therapeutics during the conduct of the study, as well as personal fees from Janssen Pharmaceuticals outside the submitted work. G. Rabinowits reports other support from Castle, Sanofi-Genzyme, Regeneron, EMD Serono, Pfizer, and Merck outside the submitted work. D.S. Hong reports grants from AbbVie, Adaptimmune, Adlai Nortye, Amgen, AstraZeneca, Bayer, Bristol Myers Squibb, Daiichi Sankyo, Eisai, Eli Lilly, EMD Serono, Erasca, Fate Therapeutics, Genentech, GlaxoSmithKline, Ignyta, Infinity, Kite, Kyowa, LOXO, Merck, Medimmune, Millennium, Mirati, miRNA, Molecular Templates, Mologen, Navier, NCI-CTEP, Novartis, Numab, Pfizer, Seattle Genetics, Takeda, Turning Point Therapeutics, Verstatem, and VM Oncology during the conduct of the study. D.S. Hong also reports other support from AACR, ASCO, Celgene, Eli Lilly, Genmab, GlaxoSmithKline, LOXO, miRNA, Phillips, SITC, Molecular Match, OncoResponse, Presagia Inc., and AstraZeneca; personal fees and other support from Amgen, Bayer, Genentech, Janssen, Pfizer, and Takeda; and personal fees from Alpha Insights, Acuta, Axiom, Adaptimmune, Baxter, Boxer Capital, COG, Ecor1, Genentech, GLG, Group H, Guidepoint, HCW Precision, Infinity, Merrimack, Medscape, Numab, Prime Oncology, Seattle Genetics, ST Cube, Tavistock, Trieza Therapeutics, and WebMD outside the submitted work. J.S. Wang reports other support from Florida Cancer Specialists during the conduct of the study, as well as other support from Syndax, H3 Biomedicine, AstraZeneca, Phoenix Molecular Designs, Olema, LSK BioPartners, Taiho, Celgene, Teneobio, Prelude, Acerta, Portola, LOXO, Forty Seven, Nurix, Novartis, Aevi Genomics, Ignyta, Hutchinson MediPharma, BioNTech, Stemline, GlaxoSmithKline, Agenus, Boehringer Ingelheim, Moderna, TopAlliance, Macrogenics, Jacobio, Merck, Birdie, CytomX, Janssen, Klus, Revolution, Kymab, Bicycle, Cyteir, Daiichi Sankyo, Qilu Puget Sound, Vigeo, Black Diamond, Xencor, Clovis, Ribon, Synthorx, Relay, Vedanta, StingThera, ORIC, Artios, Genentech, Treadwell, Mabspace, IGM, PureTech, Erasca, and BioTheryX outside the submitted work. U. Steidl reports grants from Bayer; personal fees from Celgene/BMS, Pieris, Vor Biopharma, and Trillium Therapeutics; grants and personal fees from Aileron and Novartis; and personal fees and other support from Stelexis outside the submitted work. V. Vukovic reports employment with Aileron Therapeutics Inc. D.A. Annis reports personal fees from Aileron Therapeutics, Inc. during the conduct of the study. M. Aivado reports other support from Aileron Therapeutics during the conduct of the study, as well as other support from Aileron Therapeutics outside the submitted work; M. Aivado also has a patent for peptidomimetic macrocycles and uses thereof issued. F. Meric-Bernstam reports grants from Aileron Therapeutics during the conduct of the study; F. Meric Bernstam also reports personal fees from AbbVie, Aduro BioTech, Alkermes, AstraZeneca, DebioPharm, eFFECTOR Therapeutics, F. Hoffman-La Roche Ltd., Genentech, IBM Watson, Infinity Pharmaceuticals, Jackson Laboratory, Kolon Life Science, OrgiMed, PACT Pharma, Parexel International, Pfizer Inc., Samsung Bioepis, Seattle Genetics, Tyra Biosciences, Xencor, Zymeworks, Black Diamond, Eisai, Immunomedics, Inflection Biosciences, Karyopharm Therapeutics, Mersana Therapeutics, Puma Biotechnology, Silverback Therapeutics, Spectrum Pharmaceuticals, Zentalis, and Chugai Pharmaceuticals, as well as grants from AstraZeneca, Bayer Healthcare Pharmaceutical, Calithera Biosciences, Curis, CytomX, Daiichi Sankyo, Debiopharm, eFFECTOR Therapeutics, Genentech, Guardant Health, Klus Pharma, Takeda Pharmaceutical, Novartis, Puma Biotechnology, and Taiho outside the submitted work. No disclosures were reported by the other authors.
Supplementary Material
A link to the protocol as referenced in this manuscript.
Experimental Methods for MDMX Gene Expression Analysis from Patient Tumor Specimens
Table S1 shows QuantStudio 7 Flex Real-Time PCR System Conditions
Table S2 shows TP53 Status and Concordance Between Local and Central Laboratory Results
All treatment-emergent adverse events that occurred in {greater than or equal to}10% of patients regardless of relationship with ALRN-6924 (no grade 4 treatment-emergent adverse events occurred in {greater than or equal to}10% of patients; no grade 5 events were observed). G1, grade 1; G2, grade 2; G3, grade 3; CTCAE, Common Terminology Criteria for Adverse Events.
Caption: The efficacy evaluable population was defined as those patients who met all the eligibility criteria: 1) received at least one dose of ALRN-6924 at a dose level at least 0·8 mg/kg per infusion; 2) were TP53 wild type or indeterminate (as assessed by the central laboratory or local laboratory, if not assessed by the central laboratory); and 3) had at least one post-baseline evaluation or exhibited clinical or objective progression as determined by the investigator.
Table S4 shows Hematological Toxicities in MDM2 Inhibitor Phase 1 All-comers Trials
Acknowledgments
We thank the research data managers, coordinators, and nurses, including Krystle Luna, Mohammad Ghalib, and Imran Chaudhary, for their contributions. We thank Stephanie Deming and Ashli Nguyen-Villarreal in the Research Medical Library at The University of Texas MD Anderson Cancer Center for editorial assistance, and Eric Smith for graphical assistance. This study was funded by Aileron Therapeutics.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
Note: Supplementary data for this article are available at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/).
Authors' Contributions
M.N. Saleh: Data curation, formal analysis, writing–original draft, writing–review and editing, patient enrollment. M.R. Patel: Data curation, writing–review and editing, patient enrollment. T.M. Bauer: Data curation, writing–review and editing, patient enrollment. S. Goel: Data curation, writing–review and editing, patient enrollment. G.S. Falchook: Data curation, writing–review and editing, patient enrollment. G.I. Shapiro: Data curation, writing–review and editing, patient enrollment. K.Y. Chung: Data curation, writing–review and editing, patient enrollment. J.R. Infante: Data curation, writing–review and editing, patient enrollment. R.M. Conry: Data curation, writing–review and editing, patient enrollment. G. Rabinowits: Writing-review and editing. D.S. Hong: Data curation, writing–review and editing, patient enrollment. J.S. Wang: Data curation, writing–review and editing, patient enrollment. U. Steidl: Formal analysis, writing–review and editing. G. Naik: Formal analysis, writing–original draft, writing–review and editing. V. Guerlavais: Writing-review and editing. V. Vukovic: Formal analysis, writing–review and editing. D.A. Annis: Formal analysis, writing–original draft, writing–review and editing. M. Aivado: Data curation, writing–review and editing. F. Meric-Bernstam: Data curation, formal analysis, writing–original draft, writing–review and editing, patient enrollment.
References
- 1. Vousden KH, Lane DP. p53 in health and disease. Nat Rev Mol Cell Biol 2007;8:275–83. [DOI] [PubMed] [Google Scholar]
- 2. Nigro JM, Baker SJ, Preisinger AC, Jessup JM, Hostetter R, Cleary K, et al. Mutations in the p53 gene occur in diverse human tumour types. Nature 1989;342:705–8. [DOI] [PubMed] [Google Scholar]
- 3. Wade M, Li YC, Wahl GM. MDM2, MDMX and p53 in oncogenesis and cancer therapy. Nat Rev Cancer 2013;13:83–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Honda R, Tanaka H, Yasuda H. Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Lett 1997;420:25–7. [DOI] [PubMed] [Google Scholar]
- 5. Shvarts A, Steegenga WT, Riteco N, van Laar T, Dekker P, Bazuine M, et al. MDMX: a novel p53-binding protein with some functional properties of MDM2. EMBO J 1996;15:5349–57. [PMC free article] [PubMed] [Google Scholar]
- 6. Grobner SN, Worst BC, Weischenfeldt J, Buchhalter I, Kleinheinz K, Rudneva VA, et al. The landscape of genomic alterations across childhood cancers. Nature 2018;555:321–7. [DOI] [PubMed] [Google Scholar]
- 7. Kussie PH, Gorina S, Marechal V, Elenbaas B, Moreau J, Levine AJ, et al. Structure of the MDM2 oncoprotein bound to the p53 tumor-suppressor transactivation domain. Science 1996;274:948–53. [DOI] [PubMed] [Google Scholar]
- 8. Linares LK, Hengstermann A, Ciechanover A, Muller S, Scheffner M. HdmX stimulates Hdm2-mediated ubiquitination and degradation of p53. PNAS 2003;100:12009–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Montes de Oca Luna R, Wagner DS, Lozano G. Rescue of early embryonic lethality in mdm2-deficient mice by deletion of p53. Nature 1995;378:203–6. [DOI] [PubMed] [Google Scholar]
- 10. Parant JM, Reinke V, Mims B, Lozano G. Organization, expression, and localization of the murine mdmx gene and pseudogene. Gene 2001;270:277–83. [DOI] [PubMed] [Google Scholar]
- 11. Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004;303:844–8. [DOI] [PubMed] [Google Scholar]
- 12. Patton JT, Mayo LD, Singhi AD, Gudkov AV, Stark GR, Jackson MW. Levels of HdmX expression dictate the sensitivity of normal and transformed cells to Nutlin-3. Cancer Res 2006;66:3169–76. [DOI] [PubMed] [Google Scholar]
- 13. Andreeff M, Kelly KR, Yee K, Assouline S, Strair R, Popplewell L, et al. Results of the phase I trial of RG7112, a small-molecule MDM2 antagonist in leukemia. Clin Cancer Res 2016;22:868–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. de Jonge M, de Weger VA, Dickson MA, Langenberg M, Le Cesne A, Wagner AJ, et al. A phase I study of SAR405838, a novel human double minute 2 (HDM2) antagonist, in patients with solid tumours. Eur J Cancer 2017;76:144–51. [DOI] [PubMed] [Google Scholar]
- 15. Ravandi F, Gojo I, Patnaik MM, Minden MD, Kantarjian H, Johnson-Levonas AO, et al. A phase I trial of the human double minute 2 inhibitor (MK-8242) in patients with refractory/recurrent acute myelogenous leukemia (AML). Leuk Res 2016;48:92–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Iancu-Rubin C, Mosoyan G, Glenn K, Gordon RE, Nichols GL, Hoffman R. Activation of p53 by the MDM2 inhibitor RG7112 impairs thrombopoiesis. Exp Hematol 2014;42:137–45. [DOI] [PubMed] [Google Scholar]
- 17. Schafmeister CE, Po J, Verdine GL. An all-hydrocarbon cross-linking system for enhancing the helicity and metabolic stability of peptides. J Am Chem Soc 2000;122:5891–2. [Google Scholar]
- 18. Bernal F, Tyler AF, Korsmeyer SJ, Walensky LD, Verdine GL. Reactivation of the p53 tumor suppressor pathway by a stapled p53 peptide. J Am Chem Soc 2007;129:2456–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Verdine GL, Hilinski GJ. Stapled peptides for intracellular drug targets. Methods Enzymol 2012;503:3–33. [DOI] [PubMed] [Google Scholar]
- 20. Okamoto T, Zobel K, Fedorova A, Quan C, Yang H, Fairbrother WJ, et al. Stabilizing the pro-apoptotic BimBH3 helix (BimSAHB) does not necessarily enhance affinity or biological activity. ACS Chem Biol 2013;8:297–302. [DOI] [PubMed] [Google Scholar]
- 21. Chang YS, Graves B, Guerlavais V, Tovar C, Packman K, To KH, et al. Stapled alpha-helical peptide drug development: a potent dual inhibitor of MDM2 and MDMX for p53-dependent cancer therapy. PNAS 2013;110:E3445–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Carvajal LA, Neriah DB, Senecal A, Benard L, Thiruthuvanathan V, Yatsenko T, et al. Dual inhibition of MDMX and MDM2 as a therapeutic strategy in leukemia. Sci Transl Med 2018;10:eaao3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ng SY, Yoshida N, Christie AL, Ghandi M, Dharia NV, Dempster J, et al. Targetable vulnerabilities in T- and NK-cell lymphomas identified through preclinical models. Nat Commun 2018;9:2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Penel N, Kramar A. What does a modified-Fibonacci dose-escalation actually correspond to? BMC Med Res Methodol 2012;12:103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 2009;45:228–47. [DOI] [PubMed] [Google Scholar]
- 26. Cheson BD, Fisher RI, Barrington SF, Cavalli F, Schwartz LH, Zucca E, et al. Recommendations for initial evaluation, staging, and response assessment of Hodgkin and non-Hodgkin lymphoma: the Lugano classification. J Clin Oncol 2014;32:3059–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ishiguro N, Maeda K, Kishimoto W, Saito A, Harada A, Ebner T, et al. Predominant contribution of OATP1B3 to the hepatic uptake of telmisartan, an angiotensin II receptor antagonist, in humans. Drug Metab Dispos 2006;34:1109–15. [DOI] [PubMed] [Google Scholar]
- 28. Konopleva M, Martinelli G, Daver N, Papayannidis C, Wei A, Higgins B, et al. MDM2 inhibition: an important step forward in cancer therapy. Leukemia 2020;34:2858–74. [DOI] [PubMed] [Google Scholar]
- 29. Wagner AJ, Banerji U, Mahipal A, Somaiah N, Hirsch H, Fancourt C, et al. Phase I trial of the human double minute 2 inhibitor MK-8242 in patients with advanced solid tumors. J Clin Oncol 2017;35:1304–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bauer TM, Gounder MM, Weise AM, Schwartz GK, Carvajal RD, Kumar P, et al. A phase 1 study of MDM2 inhibitor DS-3032b in patients with well/de-differentiated liposarcoma (WD/DD LPS), solid tumors (ST) and lymphomas (L). J Clin Oncol 2018;36:11514. [Google Scholar]
- 31. Siu LL, Italiano A, Miller WH, Blay J-Y, Gietema JA, Bang Y-J, et al. Phase 1 dose escalation, food effect, and biomarker study of RG7388, a more potent second-generation MDM2 antagonist, in patients (pts) with solid tumors. J Clin Oncol 2014;32:2535. [Google Scholar]
- 32. Yee K, Martinelli G, Vey N, Dickinson MJ, Seiter K, Assouline S, et al. Phase 1/1b Study of RG7388, a potent MDM2 antagonist, in acute myelogenous leukemia (AML) patients (Pts). Blood 2014;124:116. [Google Scholar]
- 33. Rasco DW, Lakhani NJ, Li Y, Men L, Wang H, Ji J, et al. A phase I study of a novel MDM2 antagonist APG-115 in patients with advanced solid tumors. J Clin Oncol 2019;37:3126. [Google Scholar]
- 34. Stein E, Chromik J, DeAngelo DJ, Chatterjee M, Noppeney R, Fd V, et al. Abstract CT152: Phase I dose- and regimen-finding study of NVP-HDM201 in pts with advanced TP53wt acute leukemias. Cancer Res 2017;77:CT152–CT. [Google Scholar]
- 35. Gluck WL, Gounder MM, Frank R, Eskens F, Blay JY, Cassier PA, et al. Phase 1 study of the MDM2 inhibitor AMG 232 in patients with advanced P53 wild-type solid tumors or multiple myeloma. Invest New Drugs 2020;38:831–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mahfoudhi E, Lordier L, Marty C, Pan J, Roy A, Roy L, et al. P53 activation inhibits all types of hematopoietic progenitors and all stages of megakaryopoiesis. Oncotarget 2016;7:31980–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tisato V, Voltan R, Gonelli A, Secchiero P, Zauli G. MDM2/X inhibitors under clinical evaluation: perspectives for the management of hematological malignancies and pediatric cancer. J Hematol Oncol 2017;10:133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, et al. Proteomics. Tissue-based map of the human proteome. Science 2015;347:1260419. [DOI] [PubMed] [Google Scholar]
- 39. The Human Protein Atlas. Available from: http://www.proteinatlas.org.
- 40. Carvajal LA, Ren J-G, Guerlavais V, Steidl UG, Aivado M, Annis A. ALRN-6924, a dual inhibitor of MDMX and MDM2 that causes minimal thrombocytopenia in patients, disrupts different stages of thrombopoiesis compared to Mdm2-only inhibition European Hematology Association 24th Congress. Amsterdam; 2019. Available from: https://library.ehaweb.org/eha/2019/24th/266137. [Google Scholar]
- 41. Vakiani E, Janakiraman M, Shen R, Sinha R, Zeng Z, Shia J, et al. Comparative genomic analysis of primary versus metastatic colorectal carcinomas. J Clin Oncol 2012;30:2956–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012;487:330–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hata AN, Rowley S, Archibald HL, Gomez-Caraballo M, Siddiqui FM, Ji F, et al. Synergistic activity and heterogeneous acquired resistance of combined MDM2 and MEK inhibition in KRAS mutant cancers. Oncogene 2017;36:6581–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kato S, Ross JS, Gay L, Dayyani F, Roszik J, Subbiah V, et al. Analysis of MDM2 amplification: next-generation sequencing of patients with diverse malignancies. JCO Precision Oncology 2018;2018:PO.17.00235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bill KLJ, Seligson ND, Hays JL, Awasthi A, Demoret B, Stets CW, et al. Degree of MDM2 amplification affects clinical outcomes in dedifferentiated liposarcoma. Oncologist 2019;24:989–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bardot B, Bouarich-Bourimi R, Leemput J, Lejour V, Hamon A, Plancke L, et al. Mice engineered for an obligatory Mdm4 exon skipping express higher levels of the Mdm4-S isoform but exhibit increased p53 activity. Oncogene 2015;34:2943–8. [DOI] [PubMed] [Google Scholar]
- 47. Valianatos G, Valcikova B, Growkova K, Verlande A, Mlcochova J, Radova L, et al. A small-molecule drug promoting miRNA processing induces alternative splicing of MdmX transcript and rescues p53 activity in human cancer cells overexpressing MdmX protein. PLoS ONE 2017;12:e0185801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lenos K, Grawenda AM, Lodder K, Kuijjer ML, Teunisse AF, Repapi E, et al. Alternate splicing of the p53 inhibitor HDMX offers a superior prognostic biomarker than p53 mutation in human cancer. Cancer Res 2012;72:4074–84. [DOI] [PubMed] [Google Scholar]
- 49. Choong ML, Yang H, Lee MA, Lane DP. Specific activation of the p53 pathway by low dose actinomycin D: a new route to p53 based cyclotherapy. Cell Cycle 2009;8:2810–8. [DOI] [PubMed] [Google Scholar]
- 50. Brown CJ, Quah ST, Jong J, Goh AM, Chiam PC, Khoo KH, et al. Stapled peptides with improved potency and specificity that activate p53. ACS Chem Biol 2013;8:506–12. [DOI] [PubMed] [Google Scholar]
- 51. Carvajal D, Tovar C, Yang H, Vu BT, Heimbrook DC, Vassilev LT. Activation of p53 by MDM2 antagonists can protect proliferating cells from mitotic inhibitors. Cancer Res 2005;65:1918–24. [DOI] [PubMed] [Google Scholar]
- 52. Weiss JM, Csoszi T, Maglakelidze M, Hoyer RJ, Beck JT, Domine Gomez M, et al. Myelopreservation with the CDK4/6 inhibitor trilaciclib in patients with small-cell lung cancer receiving first-line chemotherapy: a phase Ib/randomized phase II trial. Ann Oncol 2019;30:1613–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Dobbelstein M, Levine AJ. Mdm2: open questions. Cancer Sci 2020;111:2203–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A link to the protocol as referenced in this manuscript.
Experimental Methods for MDMX Gene Expression Analysis from Patient Tumor Specimens
Table S1 shows QuantStudio 7 Flex Real-Time PCR System Conditions
Table S2 shows TP53 Status and Concordance Between Local and Central Laboratory Results
All treatment-emergent adverse events that occurred in {greater than or equal to}10% of patients regardless of relationship with ALRN-6924 (no grade 4 treatment-emergent adverse events occurred in {greater than or equal to}10% of patients; no grade 5 events were observed). G1, grade 1; G2, grade 2; G3, grade 3; CTCAE, Common Terminology Criteria for Adverse Events.
Caption: The efficacy evaluable population was defined as those patients who met all the eligibility criteria: 1) received at least one dose of ALRN-6924 at a dose level at least 0·8 mg/kg per infusion; 2) were TP53 wild type or indeterminate (as assessed by the central laboratory or local laboratory, if not assessed by the central laboratory); and 3) had at least one post-baseline evaluation or exhibited clinical or objective progression as determined by the investigator.
Table S4 shows Hematological Toxicities in MDM2 Inhibitor Phase 1 All-comers Trials