

Abstract
Purpose:
We recently reported the development of a cell-free DNA (cfDNA) targeted methylation (TM)-based sequencing approach for a multi-cancer early detection (MCED) test that includes cancer signal origin prediction. Here, we evaluated the prognostic significance of cancer detection by the MCED test using longitudinal follow-up data.
Experimental Design:
As part of a Circulating Cell-free Genome Atlas (CCGA) substudy, plasma cfDNA samples were sequenced using a TM approach, and machine learning classifiers predicted cancer status and cancer signal origin. Overall survival (OS) of cancer participants in the first 3 years of follow-up was evaluated in relation to cancer detection by the MCED test and clinical characteristics.
Results:
Cancers not detected by the MCED test had significantly better OS (P < 0.0001) than cancers detected, even after accounting for other covariates, including clinical stage and method of clinical diagnosis (i.e., standard-of-care screening or clinical presentation with signs/symptoms). Additionally, cancers not detected by the MCED test had better OS than was expected when data were adjusted for age, stage, and cancer type from the Surveillance, Epidemiology, and End Results (SEER) program. In cancers with current screening options, the MCED test also differentiated more aggressive cancers from less aggressive cancers (P < 0.0001).
Conclusions:
Cancer detection by the MCED test was prognostic beyond clinical stage and method of diagnosis. Cancers not detected by the MCED test had better prognosis than cancers detected and SEER-based expected survival. Cancer detection and prognosis may be linked by the underlying biological factor of tumor fraction in cfDNA.
Translational Relevance.
Screening tests for early cancer detection may be subject to the risk of overdiagnosis—identification of indolent cancers that may not become symptomatic or cause harm during the patient's lifetime, and therefore may not require intervention. We recently reported the development of a cell-free DNA targeted methylation sequencing assay with machine learning classifiers for multi-cancer detection and cancer signal origin (i.e., tissue of origin) prediction. Longitudinal follow-up data were utilized here to evaluate prognostic significance of cancer detection. Our data demonstrated that this multi-cancer early detection (MCED) test detected clinically significant cancers, and that detection was prognostic beyond clinical stage and method of clinical diagnosis. Cancers not detected by the MCED test tended to be less aggressive. Together, these results suggest that addition of this MCED test to existing screening paradigms may not add to overdiagnosis, while detecting more clinically significant cancers.
Introduction
Previous studies demonstrated the prognostic value of circulating tumor DNA levels in the plasma of cancer patients (1–12). Cancer-related mutations, copy-number variants, and abnormal methylation patterns detected in baseline cfDNA have been associated with overall survival (OS) and/or progression-free survival in various cancer types (1–7, 13). In addition, decreasing cfDNA tumor content with treatment is a potential biomarker for good response and prognosis (8–10), while its rebound or persistence after treatment heralds cancer recurrence (3, 11, 14). Furthermore, methylation markers in cfDNA have been shown to detect cancer (15) and differentiate cancers into high-risk and low-risk groups with distinct survival outcomes (12). Thus, we hypothesize that cfDNA-based cancer detection could potentially be a surrogate for cancer biology associated with prognosis and survival.
Early detection and intervention can alter the course of cancer, improve patient outcomes, and reduce cancer-related mortality (16, 17). Screening as a means to early detection, however, is recommended by the United States Preventive Services Task Force (USPSTF) for only a subset of common cancers in the United States (breast, colorectal, cervix, lung, and—on an individualized basis—prostate). Most malignancies lack recommended screening programs partly because their prevalence in the population is too low for the benefits of tumor-specific screening to outweigh the risks (18). To extend the benefits of single-cancer screens to a broader population, and to maximize the potential public health benefit, an effective multi-cancer test should detect as many cancer types as possible at very high specificity (>99%), and accurately predict the cancer signal origin (i.e., tissue of origin).
To support the development of a MCED test, we conducted the CCGA (NCT02889978) study (19). Previously, we reported that targeted methylation analysis of plasma cfDNA simultaneously detected multiple cancer types at >99% specificity, and predicted the cancer signal origin with >90% accuracy (19). Here, by following CCGA cancer participants over time, we reported OS of cancer participants in the first 3 years of follow-up, and explored how cfDNA signal was associated with cancer prognosis across multiple cancer types.
Materials and Methods
Study design
CCGA (NCT02889978) is a prospective, multicenter, case–control, observational study with longitudinal follow-up. Deidentified pretreatment plasma samples were collected from 15,254 participants with (n = 8,584, 56%) or without (n = 6,670; 44%) cancer from 142 sites in North America (19). Annual follow-up is ongoing in participants with and without cancer for up to 5 years. For participants with cancer, longitudinal data were collected, including vital status, treatment, current cancer status, and new cancer diagnoses. Outcome assessment was performed through electronic medical record and/or phone contact with the participant. Reported here was a survival analysis for cancer participants from the second prespecified substudy of CCGA (19) that had vital status collected through November 6, 2020.
All participants were required to provide written informed consent. The study was approved by the Institutional Review Board or an independent ethics committee at each participating trial site, and conducted in accordance with the International Conference on Harmonization for Good Clinical Practice guidelines and the Declaration of Helsinki.
Sample processing and analysis
Processing was performed as described in Liu et al. (19). Briefly, up to eight 10-mL tubes of peripheral blood were collected from all participants included in this analysis using Streck Cell-free DNA BCT at participating sites and shipped to Brooks Life Sciences. Whole blood was isolated into plasma (up to 4.2 mL per tube) and buffy coat and stored at −80°C at Brooks Life Sciences until processing. A tube was considered evaluable if it met all the following criteria: the parent tube was a Streck tube with volume
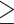 3 mL; time from sample collection to plasma isolation was
3 mL; time from sample collection to plasma isolation was
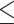 5 days; the Streck tube was free of any fatal deviations; and the plasma tube had high-quality plasma grading by visual grading or optical density model.
5 days; the Streck tube was free of any fatal deviations; and the plasma tube had high-quality plasma grading by visual grading or optical density model.
For each participant, only 2 parent tubes were randomly selected and processed separately in an automated workflow (tubes 1 and 2). The assay result from tube 1 was reported if it was evaluable; if the assay result from tube 1 was not evaluable, the result from tube 2 was reported.
Pretreatment plasma cfDNA samples were subjected to bisulfite sequencing targeting a panel of over 100,000 informative methylation regions. Cancer status and cancer signal origin were predicted using machine learning classifiers as previously reported (19).
The primary analysis set of the second substudy of CCGA included 2,185 participants with cancer (>50 primary cancer types), separated into a training set (n = 1,531) and an independent validation set (n = 654; ref. 19). For survival analysis, we used both sets jointly, with cancer detection determined using either the cross-validated classifier for the training set or the locked classifier for the independent validation set. 2,129 of the 2,185 participants had vital status available and were included in this analysis (Supplementary Table S1). The remaining 56 participants did not have vital status available due to no electronic medical record and/or the participants were not responsive to at least two contact efforts made by study coordinators.
A subset of the CCGA participants had high clinical suspicion (HCS) of cancer at the time of enrollment, but no confirmed diagnosis. HCS was defined as a high suspicion for a cancer diagnosis by clinical and/or radiologic assessment, with planned biopsy or surgical resection to establish a definitive diagnosis. Cancer status (cancer or non-cancer) was confirmed by pathologic evaluation subsequent to blood draw. Participants in the HCS subgroup who had a confirmed diagnosis of cancer within the enrollment window and were diagnosed following clinical presentation were referred to as HCS cancer participants here.
Lung cancer histology categories were grouped using International Classification of Diseases for Oncology, 3rd Edition (ICD-O-3) morphology codes (20). One case with two morphology codes (8041: small cell carcinoma and 8140: adenocarcinoma) was grouped with small cell lung cancer (21).
Tumor fraction in cfDNA was estimated for samples with matched tissue by determining the methylation variant allele fraction (MVAF), or the methylation variants in paired tissue that were not found in cfDNA of non-cancer individuals, for each cfDNA sample. MVAF for a cfDNA sample was then inferred by counting the frequencies of these methylation variants in cfDNA as previously described (22). MVAF was modeled as follows:
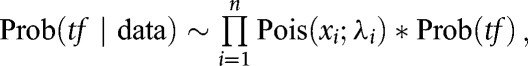 |
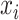 = observed abnormal counts of site i in cfDNA
= observed abnormal counts of site i in cfDNA
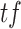 = tumor fraction
= tumor fraction
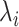 = lambda for site i =
= lambda for site i =
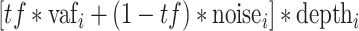
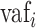 = variant allele fraction for site i in the biopsy
= variant allele fraction for site i in the biopsy
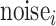 = site-specific noise rate in cfDNA
= site-specific noise rate in cfDNA
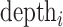 = depth of site i in cfDNA
= depth of site i in cfDNA
Calculation of expected survival
To provide a reference for survival rates accounting for the heterogeneous mix of ages, clinical stages, and cancer types in the CCGA study, we obtained from the National Cancer Institute's Surveillance, Epidemiology, and End Results (SEER) Program and related SEER*Stat program (version 8.38) population-based data encompassing the quarterly OS of patients diagnosed in 18 regions of the United States. These statistics [which included patients with primary cancer diagnosed between 2006 and 2015 stratified by age (20+ to match that of the CCGA participants enrolled; 5-year age group), stage at diagnosis (AJCC 6th edition stage I, II, III, IV, or unknown), and cancer type (SEER site recode)] were adjusted to the CCGA distributions of age, clinical stage, and cancer type, to estimate the expected OS over time in the CCGA MCED detected and not detected populations, for comparisons with observed OS.
Restricted mean survival time (RMST; refs. 23, 24), defined as the area under the survival curve, at 36 months for CCGA was calculated using the survRM2 package. RMST at 36 months for SEER expected survival was calculated using the AUC function of the DescTools package (25).
Statistical analysis
Each participant's vital status during the outcome assessment was recorded by clinical site staff per the CCGA data management plan (19). “Alive” participants refer to the ones whose last confirmed vital status was alive. Overall survival (OS) was defined as the time from the date blood samples were collected to the date of death or the last date the participant was confirmed alive (censored). Survival curves were estimated using the Kaplan–Meier method, and differences were compared using the log-rank test (26, 27). Cox proportional hazards regression model was used for univariate and multivariate analyses (28) to assess the association of potential prognostic factors to OS. For multivariate analysis, we assessed OS association with cancer detection by the MCED test (detected versus not detected), clinical stage (III/IV versus I/II), cancer mortality hazard group (stage II SEER 5-year mortality high versus low), method of clinical diagnosis (clinical presentation vs. screening-diagnosed), sex (male versus female), age (≥ 50 versus < 50), and histologic grade (3 or 2 versus 1). The 95% confidence interval (CI) was calculated using Wilson's score CI (29). A P value of <0.05 was considered significant. All analyses were carried out in R 3.6.0 (30).
Results
A total of 2,129 out of the 2,185 cancer participants from the second substudy of CCGA had vital status available and were included in these follow-up analyses (Table 1; Supplementary Tables S1 and S2). Of those, 24% (516/2,129) were deceased at the time of analysis. Of the alive participants, 97% had 1-year follow-up, 82% reached 2-year follow-up, and 21% reached 3-year follow-up. Median follow-up duration was 24 months.
Table 1.
Demographic and other baseline characteristics of cancer participants according to vital status.
| Alive | Dead | |
|---|---|---|
| Total | 1,613 | 516 |
| Age, mean ± SD | 61 ± 12 | 66 ± 11 |
| Sex, n (%) | ||
| Female | 857 (81) | 204 (19) |
| Male | 756 (71) | 312 (29) |
| Race/ethnicity, n (%) | ||
| White, non-Hispanic | 1,327 (75) | 440 (25) |
| African American | 115 (77) | 35 (23) |
| Hispanic | 98 (76) | 31 (24) |
| Othera | 73 (88) | 10 (12) |
| Age group,bn (%) | ||
| <50 years | 277 (88) | 37 (12) |
| ≥50 years | 1,336 (74) | 479 (26) |
| Smoking status, n (%) | ||
| Never smoker | 781 (83) | 164 (17) |
| Former smoker | 613 (71) | 253 (29) |
| Current smoker | 184 (65) | 98 (35) |
| Other/missing | 35 (97) | 1 (3) |
| Met the 2013 USPSTF eligibility for lung cancer screening, n (%) | ||
| True | 158 (56) | 124 (44) |
| False | 1,455 (79) | 392 (21) |
| Body mass index, n (%) | ||
| Underweight | 14 (47) | 16 (53) |
| Normal | 384 (72) | 153 (28) |
| Overweight | 527 (74) | 184 (26) |
| Obese | 688 (81) | 163 (19) |
| Clinical stage, n (%) | ||
| I | 544 (94) | 37 (6) |
| II | 479 (88) | 66 (12) |
| III | 310 (71) | 129 (29) |
| IV | 222 (45) | 276 (55) |
| Not expected to be staged | 58 (88) | 8 (12) |
| Method of clinical diagnosis, n (%) | ||
| Screening | 484 (95) | 24 (5) |
| Clinical presentation | 1,129 (70) | 492 (30) |
| Cancer types of > 100 cases or > 20 deaths, n (%) | ||
| Breast | 329 (95) | 17 (5) |
| Colon/rectum | 131 (76) | 41 (24) |
| Esophagus | 38 (54) | 32 (46) |
| Lung | 181 (49) | 186 (51) |
| Lymphoma | 136 (87) | 20 (13) |
| Pancreas | 33 (27) | 88 (73) |
| Prostate | 252 (97) | 7 (3) |
| Uterus | 110 (92) | 9 (8) |
aIncludes Asian, Native American, or Pacific Islander; American Indian, or Alaska Native; Other; Missing.
bAge was calculated from date at enrollment and truncated at 85 years.
Prognosis in cancers detected or not detected by the MCED test
To evaluate the potential prognostic significance of cancer detection using the MCED test, we analyzed OS for participants whose cancers were detected or not detected by the MCED test previously described in Liu et al. (19). In the participants with cancer who died during follow-up, 89% (459/516) had cancer detected by the MCED test. By comparison, in the alive participants, detection was 44% (717/1,613; Supplementary Fig. S1). For most individual cancer types, and across stages I–IV, detection sensitivity was higher in participants who died than alive participants (Supplementary Fig. S1; Supplementary Table S3). This suggested that cfDNA-based cancer detection using the MCED test might be an indicator of prognosis.
Kaplan–Meier survival analysis showed that cancers not detected by the MCED test had significantly better OS than those detected by the MCED test (P < 0.0001; Fig. 1A). Similar patterns were observed in both training and validation sets separately (P < 0.0001; Supplementary Fig. S2A), among a subgroup of participants with HCS of cancer at the time of enrollment and confirmed cancer status subsequent to study blood draw (P < 0.0001; Supplementary Fig. S2B), and in high and low cancer mortality groups (P < 0.0001; Supplementary Fig. S2C).
Figure 1.

Comparison of OS in cancers detected versus not detected by the MCED test. A, Kaplan–Meier curve depicting OS for cancer participants of all stages detected (red) versus not detected (blue) by the MCED test. P value: log-rank test. B, Kaplan–Meier curve depicting OS for cancer participants of stages I–IV, detected (red) versus not detected (blue) by the MCED test. P values: log-rank test. C, Estimated RMST at 36 months for the MCED test detected and not detected cancers. Black diamonds indicate SEER-based expected survival. Error bars, 95% CI. ***, t test, P < 0.0001.
To confirm that the prognostic benefit was not driven only by an increased detection rate at late stages, we investigated whether cancer detection in cfDNA predicted survival beyond clinical stage. As expected, stage was strongly associated with cancer survival (P < 0.0001; Supplementary Fig. S3C). Importantly, cancer detection by the MCED test was prognostic beyond clinical stage alone (P < 0.001; Fig. 1B).
To understand how these patterns compared with those expected in a real-world setting, and to verify that the results were not simply driven by cancer type distribution, we compared the OS of the CCGA cohort analyzed here to the expected OS estimated from SEER (Fig. 1C; Supplementary Fig. S4). Estimated RMST at 36 months for MCED test detected and not detected cancers was compared with SEER expected survival (Fig. 1C).
MCED test not detected cancers, especially stage III and IV cancers, showed notably better survival than that expected from SEER. Conversely, cancers detected by the MCED test showed similar or slightly better survival than that expected from SEER (Fig. 1C; Supplementary Fig. S4).
To further identify clinical and biological factors associated with survival, we performed univariate and multivariate analyses. In univariate analyses, age, sex, clinical stage, and histologic grade were also associated with prognosis (P < 0.0001; Supplementary Fig. S3). In multivariate analyses accounting for other covariates, cancer detection by the MCED remained to be significantly associated with OS (P < 0.0001; Fig. 2). Additionally, detected cancers had better survival if detected at an earlier stage (Supplementary Fig. S5).
Figure 2.
![Figure 2. Multivariate Cox proportional hazards regression model to identify factors associated with OS. Hazard ratios and 95% CIs are indicated by black boxes and gray lines, respectively. P values are indicated. Cancer mortality group is based on SEER 5-year survival of stage II cancers. Cancer mortality hazard-high group includes sarcoma, head and neck, cervix, plasma cell neoplasm, urothelial tract, bladder, myeloid neoplasm, stomach, lung, liver bile duct, esophagus, gallbladder, and pancreas cancer types. Cancer mortality hazard-low group includes thyroid, prostate, breast, kidney, uterus, lymphoid leukemia, lymphoma, ovary, colon/rectum, anus, melanoma, and other [includes brain, mesothelioma, orbit, penis, pleura, skin cancer (not basal cell carcinoma, squamous cell carcinoma, or melanoma), small intestine, testis, thymus, vagina, vulva, and unspecified] cancer types.](https://www.ncbi.nlm.nih.gov/core/lw/2.0/html/tileshop_pmc/tileshop_pmc_inline.html?title=Click%20on%20image%20to%20zoom&p=PMC3&id=9401481_4221fig2.jpg)
Multivariate Cox proportional hazards regression model to identify factors associated with OS. Hazard ratios and 95% CIs are indicated by black boxes and gray lines, respectively. P values are indicated. Cancer mortality group is based on SEER 5-year survival of stage II cancers. Cancer mortality hazard-high group includes sarcoma, head and neck, cervix, plasma cell neoplasm, urothelial tract, bladder, myeloid neoplasm, stomach, lung, liver bile duct, esophagus, gallbladder, and pancreas cancer types. Cancer mortality hazard-low group includes thyroid, prostate, breast, kidney, uterus, lymphoid leukemia, lymphoma, ovary, colon/rectum, anus, melanoma, and other [includes brain, mesothelioma, orbit, penis, pleura, skin cancer (not basal cell carcinoma, squamous cell carcinoma, or melanoma), small intestine, testis, thymus, vagina, vulva, and unspecified] cancer types.
Detection and prognosis in cancer types with current screening options
Next, we explored whether the MCED test provided additional prognostic value within cancer types that have current screening options (i.e., breast, cervical, colorectal, lung, and prostate cancers; ref. 31), and whether prognostic information was imparted on screen-diagnosed cancers. In these cancer types, cancers diagnosed through screening had a more favorable prognosis than cancers with clinical presentation of signs/symptoms (Fig. 3A; Supplementary Fig. S6A). Among participants whose cancers were diagnosed through screening, the MCED test detected more aggressive cancers than less aggressive cancers (Fig. 3B). Statistical significance of the prognostic value of the MCED test detection varied by individual cancer types due to varying sample sizes (Supplementary Fig. S6B and S6C).
Figure 3.

Survival trends in cancers with screening modalities (breast, cervical, colorectal, lung, and prostate cancers). A, OS comparison of cancers diagnosed through standard screening paradigms or clinical presentation. B, OS comparison of cancers detected or not detected by the MCED test in cancers diagnosed through standard screening paradigms.
Specifically, the MCED test preferentially detected the subtypes among lung and breast malignancies that were known to be more aggressive, such as small cell lung cancer and hormone-receptor–negative breast cancer (Fig. 4A and B; refs. 32–34), especially in stage II where sensitivity was moderate (Supplementary Fig. S7A and S7B). Additionally, of the 89% (230/259) of prostate cancers in our analysis population diagnosed through screening [229 by prostate-specific antigen (PSA) and 1 by digital rectal examination], only 6% (14/230) were detected by the MCED test. By contrast, the MCED test detected 41% (12/29) of the prostate cancers diagnosed following clinical presentation; detection was even higher (86%; 6/7) for the participants with prostate cancer who died during follow-up (Supplementary Table S1). In addition, PSA screening detected a large number of prostate cancers with low Gleason scores (35), while the MCED test preferentially detected prostate cancers with high Gleason scores (Fig. 4C) of later stage (Supplementary Fig. S7C).
Figure 4.

MCED test detection in breast, lung, and prostate cancer subgroups. Sensitivity for MCED test detection and OS in different subgroups of breast (A), lung (B), and prostate (C) cancers are shown. Number of samples, and the proportion of cancers diagnosed through standard screening are indicated.
Tumor fraction as a biological factor for cancer signal detection and prognosis
We previously demonstrated that tumor fraction in cfDNA was an important determinant of cancer signal detection (22). In general, higher tumor fraction was observed in later stages with higher tumor burden and cancer detection increased with stage and tumor fraction (Fig. 5A). Further, cancers with lower tumor fraction had better survival than cancers with higher tumor fraction (Fig. 5B), suggesting that tumor DNA shedding into the bloodstream was a biological factor associated with cfDNA-based cancer signal detection, and was strongly related to prognosis.
Figure 5.

Association of tumor fraction in cfDNA and cancer aggressiveness. A, Box plot depicting clinical stage (x-axis) versus tumor fraction (y-axis). Cancers detected (red) or not detected (blue) by the MCED test are indicated. B, Comparison of OS in tumor fraction high and low cancers. Tumor fraction is divided into four quantiles, with 75%–100% being the highest quantile.
Discussion
These analyses demonstrated that even after accounting for clinical stage and method of diagnosis, an MCED test using machine learning classifiers on a cfDNA-based targeted methylation assay preferentially detected clinically significant cancers. In addition, survival for MCED test–detected cancers was comparable to expected survival based on SEER data, while survival for MCED test not detected cancers was notably better than that expected based on SEER. Moreover, among cancers diagnosed through standard screening, the MCED test detected the subtypes associated with higher mortality. We also observed cancer detection to be associated with tumor fraction across all stages. Together, these data demonstrated that this MCED test provided prognostic information, beyond clinical stage and method of clinical diagnosis.
To implement MCED tests at population scale, it will be important to minimize overdiagnosis of indolent cancers that may not otherwise become symptomatic or cause harm during the patient's lifetime—including conditions that are not progressive, spontaneously regress, or progress too slowly to cause symptoms and harm in a lifetime (36, 37). Existing single-cancer screening tests for early cancer detection improve survival (20, 38), but may still contribute to overdiagnosis. Like many other diseases, cancers have a wide range of severity, rate of progression, and outcomes (39), and overdiagnosis of indolent cancers can lead to overtreatment, which can cause more harm than benefit, in addition to unnecessary medical expense and psychological stress (37, 40). It has been suggested that overdiagnosis occurred in about 25% of breast cancer by mammography, 13%–25% of lung cancer by low-dose computed tomography (LDCT), 50% of lung cancer by chest radiography and/or sputum examination, and 50%–60% prostate cancer by PSA (37, 41). Here, we showed that cancer detection based on cfDNA analysis may provide utility as a surrogate biomarker for cancer aggressiveness across multiple cancer types beyond clinical stage. By preferentially detecting clinically significant cancers, even among the cancers diagnosed through standard screening tests, this MCED test may not significantly increase the risk of overdiagnosis in a general population.
MCED tests should complement, not replace, existing single-cancer screening tests. It has been reported that standard screen–detected cancers generally have more favorable survival than cancers detected when patients present with signs and symptoms, or than interval cancers (42, 43). In both cases, an MCED test could function to complement single-cancer screening tests by identifying cancers missed by screening and cancer for which there are no screening tests. Additionally, LDCT screening was shown to preferentially detect the adenocarcinoma subtype (20) that had relatively better survival than other histologic subtypes, whereas the MCED test referenced herein preferentially detected more aggressive lung cancer subtypes, such as small cell lung cancer. Together, this supports the hypothesis that the MCED test could complement standard single-cancer screening tests, while maintaining a low risk of overdiagnosis.
The comparison of observed survival in CCGA with expected survival based on SEER data (after adjustment to CCGA stage, age, and cancer type composition) led to several observations. First, we observed a difference in the SEER-based expected survival for MCED test detected versus not detected cancers, which could be due to differences among the MCED detected and not detected populations, such as age and cancer type composition. Second, survival in CCGA participants with cancer was generally better than the SEER-based expected survival, suggesting a healthy volunteer effect, which has also been reported in other volunteer screening trials (44–46). Further, survival in participants with cancer detected by the MCED test was comparable to expected survival, suggesting that detected cancers are not more likely to cause death than one would expect given the participants' stage of cancer at diagnosis, age, and cancer type, and are still likely treatable by current standard-of-care treatments (47–49). Last, and most notably, participants with cancer not detected by the MCED test had dramatically better survival than expected for their stage of cancer at diagnosis, age, and cancer type–especially in stages III and IV—which could indicate that false-negatives may represent less aggressive cancers. Together, these observations again support that this test may not contribute significantly to overdiagnosis.
Several limitations of this analysis should be considered. First, this subgroup of the CCGA study included 2,129 of the ∼8,000 cancer participants enrolled in the overall study. The current analysis was also limited by the short follow-up time (1–3 years). Annual follow-ups for CCGA participants are ongoing for up to 5 years, and further outcomes analyses will be performed when sufficient data are available. Analyses were also affected by deaths weighted toward lung cancer, which contributed to about one third of all deaths, and thus may limit the generalizability of results. We expect more events from cancer with a longer natural history, such as colorectal cancer, in longer follow-up periods. We also acknowledge limitations regarding the variables that affect survival and death analyses. For example, we drew survival data comparisons from two independent cohorts, which calculated survival differently: SEER survival was calculated from the date of cancer diagnosis to death, whereas CCGA survival was calculated from the date of blood draw (which, in general, could be up to 90 days after or up to 42 days prior to cancer diagnosis). Lead-time bias could be a factor when comparing survival time between screening-diagnosed cancers and cancers with clinical presentations. In addition, we did not have detailed information regarding the cause of death in nearly half of the cases and, therefore, could not assess cancer-specific deaths or survival time. We suspect that most deaths that occurred were in fact related to cancer, given that OS probability at 36 months for participants was 71%, which was lower than expected overall 36-month survival of 94% in a general population of age 20+, based on U.S. mortality data for 2013–2014 through 2017 (50). Furthermore, the fraction of tumor-derived cfDNA in the bloodstream has a large dynamic range (51). Various factors could affect tumor fraction in plasma cfDNA and therefore cancer detection by the MCED test, such as tumor volume, lymph-node involvement, vascularization, tumor cell growth and death rates, and cell morphology (11). In this hypothesis-generating study, we explored a number of potential factors that were collected in the CCGA study and might be associated with cancer aggressiveness and tumor DNA shedding. However, it was likely that we did not account for all confounding factors and that factors selected were biased by data availability and follow-up duration in the study. In addition, different aspects of tumor biology could contribute differently across cancer types. Therefore, analysis of biological and physiologic tumor features would be helpful to assess tumor shedding, circulating tumor DNA detection, and cancer survival on an individual cancer basis. Lastly, CCGA is not an interventional study, and therefore cancer treatments that affected survival were not conditioned on a participant's cancer detection status by the MCED test.
Future work to assess the prognostic value of the MCED test in a larger population will be conducted in prespecified analysis of an independent validation set from the third substudy of CCGA, which contains approximately 3,000 participants with cancer. Ongoing research includes continued analyses of CCGA follow-up data and clinical studies monitoring clinical implementation of this MCED test.
In summary, these data suggest that this MCED test was prognostic beyond clinical stage and method of clinical diagnosis, and that the addition of this MCED test to existing screening paradigms may not add to overdiagnosis.
Authors' Disclosures
X. Chen reports personal fees, non-financial support, and other support from GRAIL, Inc. outside the submitted work. E. Hubbell reports other support from GRAIL outside the submitted work; in addition, E. Hubbell has a patent for cancer detection, sequencing, microarrays (multiple patents) pending and issued. K.N. Kurtzman reports other support from GRAIL and Illumina outside the submitted work. G.R. Oxnard reports personal fees from Foundation Medicine and Roche outside the submitted work. O. Venn reports a patent 2019249422 pending, a patent 3094717 pending, a patent for 201980037495.6 pending, a patent for 20 2019 005 627.0 pending, a patent 2019269742 pending, a patent 3,100,250 pending, a patent for 16/417,336 pending, a patent 2019351130 pending, a patent 3,111,887 pending, a patent for 19866189.4 pending, a patent 281741 pending, a patent for 17/214,105 pending, a patent for 17/214,190 pending, a patent for PCT/US2020/015082 pending, a patent for PCT/US2019/067293 pending, a patent for 16/719,902 pending, a patent for PCT/US2020/016673 pending, a patent for PCT/US2020/016684 pending, a patent for PCT/US2020/032657 pending, a patent 109115936 pending, a patent for 15/931,022 pending, a patent for PCT/US2020/062350 pending, a patent for 17/105,175 pending, a patent for 16/850,634 pending, a patent for PCT/US2020/064577 pending, a patent for 17/119,606 pending, a patent for PCT/US2020/054951 pending, a patent for 63/024,033 pending, a patent for 63/041,699 pending, a patent for 17/066,863 pending, a patent for PCT/US2021/20012 pending, a patent for 17/187,319 pending, a patent for PCT/US2021/019746 pending, a patent for 17/185,885 pending, a patent for 63/041,875 pending, and a patent for 63/171,355 pending and stock ownership in GRAIL Inc. and Illumina Inc, and is an employee of GRAIL Inc. C. Melton reports other support from GRAIL during the conduct of the study; in addition, C. Melton has a patent for identifying methylation patterns that discriminate or indicate a cancer condition pending to GRAIL and a patent for convolutional neural network systems and methods for data classification pending to GRAIL. C.A. Clarke reports other support from GRAIL during the conduct of the study. T. Ma reports other support from GRAIL, Inc outside the submitted work. M.V. Seiden reports other support from GRAIL during the conduct of the study and personal fees from GRAIL outside the submitted work. E.A. Klein reports personal fees from GRAIL during the conduct of the study. E.T. Fung reports personal fees and other support from GRAIL during the conduct of the study; in addition, E.T. Fung has a patent for GRAIL pending. M.C. Liu reports non-financial support and other support from GRAIL during the conduct of the study, as well as other support from Astra Zeneca, Eisai, Genentech, Novartis, Seattle Genetics, Tesaro, and Menarini Silicon Biosystems and non-financial support and other support from Merck and Genomic Health outside the submitted work. No disclosures were reported by the other authors.
Supplementary Material
Acknowledgments
We thank Sarah A. Prins (GRAIL, Inc., Menlo Park, CA) for medical writing assistance. In addition, Jessica Yecies, Kalyani Chilukuri, Robert Shortt, Hai Liu, Jacqueline D. Brooks, and Tony Wu are recognized for data collection, curation, and analysis, along with Jafi Lipson, Alexander M. Aravanis, and Anne-Renee Hartman for clinical perspective and monitoring, all of whom are or were employees of GRAIL, Inc. at the time of the study. We thank ProEd Communications, Inc. (Beechwood, OH) for graphical support, funded by GRAIL, Inc. This study was funded by GRAIL, Inc.
The publication costs of this article were defrayed in part by the payment of publication fees. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Footnotes
Note: Supplementary data for this article are available at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/).
Authors' Contributions
X. Chen: Conceptualization, formal analysis, visualization, methodology, writing–original draft, writing–review and editing. Z. Dong: Data curation, software, validation, writing–review and editing. E. Hubbell: Conceptualization, methodology, writing–review and editing. K.N. Kurtzman: Conceptualization, supervision, writing–review and editing. G.R. Oxnard: Conceptualization, resources, investigation, writing–original draft, writing–review and editing. O. Venn: Conceptualization, writing–review and editing. C. Melton: Data curation, writing–review and editing. C.A. Clarke: Data curation, methodology, writing–review and editing. R. Shaknovich: Data curation, writing–review and editing. T. Ma: Software, writing–review and editing. G. Meixiong: Software, writing–review and editing. M.V. Seiden: Resources, investigation, writing–original draft, writing–review and editing. E.A. Klein: Resources, investigation, writing–original draft, writing–review and editing. E.T. Fung: Conceptualization, supervision, methodology, writing–review and editing. M.C. Liu: Conceptualization, resources, investigation, writing–original draft, writing–review and editing.
References
- 1. Tan G, Chu C, Gui X, Li J, Chen Q. The prognostic value of circulating cell-free DNA in breast cancer: a meta-analysis. Medicine (Baltimore) 2018;97:e0197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Stover DG, Parsons HA, Ha G, Freeman SS, Barry WT, Guo H, et al. Association of cell-free DNA tumor fraction and somatic copy number alterations with survival in metastatic triple-negative breast cancer. J Clin Oncol 2018;36:543–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chen X, Chang C-W, Spoerke JM, Yoh KE, Kapoor V, Baudo C, et al. Low-pass whole-genome sequencing of circulating cell-free DNA demonstrates dynamic changes in genomic copy number in a squamous lung cancer clinical cohort. Clin Cancer Res 2019;25:2254–63. [DOI] [PubMed] [Google Scholar]
- 4. Chen L, Zhang Y, Cheng Y, Zhang D, Zhu S, Ma X. Prognostic value of circulating cell-free DNA in patients with pancreatic cancer: a systemic review and meta-analysis. Gene 2018;679:328–34. [DOI] [PubMed] [Google Scholar]
- 5. Phallen J, Sausen M, Adleff V, Leal A, Hruban C, White J, et al. Direct detection of early-stage cancers using circulating tumor DNA. Sci Transl Med 2017;9:eaan2415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lee JH, Saw RP, Thompson JF, Lo S, Spillane AJ, Shannon KF, et al. Pre-operative ctDNA predicts survival in high-risk stage III cutaneous melanoma patients. Ann Oncol 2019;30:815–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Balgkouranidou I, Chimonidou M, Milaki G, Tsarouxa EG, Kakolyris S, Welch DR, et al. Breast cancer metastasis suppressor-1 promoter methylation in cell-free DNA provides prognostic information in non-small cell lung cancer. Br J Cancer 2014;110:2054–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Goodall J, Mateo J, Yuan W, Mossop H, Porta N, Miranda S, et al. Circulating cell-free DNA to guide prostate cancer treatment with PARP inhibition. Cancer Discov 2017;7:1006–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Diehl F, Schmidt K, Choti MA, Romans K, Goodman S, Li M, et al. Circulating mutant DNA to assess tumor dynamics. Nat Med 2008;14:985–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Goldberg SB, Narayan A, Kole AJ, Decker RH, Teysir J, Carriero NJ, et al. Early assessment of lung cancer immunotherapy response via circulating tumor DNA. Clin Cancer Res 2018;24:1872–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Abbosh C, Birkbak NJ, Wilson GA, Jamal-Hanjani M, Constantin T, Salari R, et al. Phylogenetic ctDNA analysis depicts early-stage lung cancer evolution. Nature 2017;545:446–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Xu R, Wei W, Krawczyk M, Wang W, Luo H, Flagg K, et al. Circulating tumour DNA methylation markers for diagnosis and prognosis of hepatocellular carcinoma. Nat Mater 2017;16:1155–61. [DOI] [PubMed] [Google Scholar]
- 13. Bettegowda C, Sausen M, Leary RL, Kinde I, Wang Y, et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med 2014;6:224ra24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Powles T, Assaf ZJ, Davarpanah N, Hussain M, Oudard S. Clinical outcomes in post-operative ctDNA-positive muscle-invasive urothelial carcinoma (MIUC) patients after atezolizumab adjuvant therapy [abstract]. Presented at ESMO Immuno-Oncology Virtual Congress 2020; December 9–12, 2020. Abstract 10.
- 15. Li M, Chen W-D, Papadopoulos N, Goodman SN, Bjerregaard NC, Laurberg S, et al. Sensitive digital quantification of DNA methylation in clinical samples. Nat Biotechnol 2009;27:858–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Siegel R, Miller K, Fuchs HE, Jemal A. Cancer statistics, 2021. CA Cancer J Clin 2021;71:7–33. [DOI] [PubMed] [Google Scholar]
- 17. McPhail S, Johnson S, Greenberg D, Peake M, Rous B. Stage at diagnosis and early mortality from cancer in England. Br J Cancer 2015;112:S108–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ahlquist DA. Universal cancer screening: revolutionary, rational, and realizable. NPJ Precis Oncol 2018;2:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Liu MC, Oxnard GR, Klein EA, Swanton C, Seiden MV, Cummings SR, et al. Sensitive and specific multi-cancer detection and localization using methylation signatures in cell-free DNA. Ann Oncol 2020;31:745–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. de Koning HJ, van der Aalst CM, de Jong PA, Scholten ET, Nackaerts K, Heuvelmans MA, et al. Reduced lung-cancer mortality with volume CT screening in a randomized trial. N Engl J Med 2020;382:503–13. [DOI] [PubMed] [Google Scholar]
- 21. Zhao X, McCutcheon JN, Kallakury B, Chahine JJ, Pratt D, Raffeld M, et al. Combined small cell carcinoma of the lung: Is it a single entity? J Thorac Oncol 2018;13:237–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Melton C, Singh P, Venn O, Hubbell E, Gross S, Saito Y, et al. Tumor methylation patterns to measure tumor fraction in cell-free DNA. JCO 2020;38:3052. [Google Scholar]
- 23. Uno H, Claggett B, Tian L, Inoue E, Gallo P, Miyata T, et al. Moving beyond the hazard ratio in quantifying the between-group difference in survival analysis. JCO 2014;32:2380–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pak K, Uno H, Kim DH, Tian L, Kane RC, Takeuchi M, et al. Interpretability of cancer clinical trial results using restricted mean survival time as an alternative to the hazard ratio. JAMA Oncol 2017;3:1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Signorell A, Aho K, Alfons A, Anderegg N, Aragon T, Arppe A, et al. DescTools: Tools for Descriptive Statistics [Internet]. 2020. Available from: https://github.com/AndriSignorell/DescTools/.
- 26. Kassambara A, Kosinski M, Biecek P, Fabian S. survminer: Drawing Survival Curves using “ggplot2” [Internet]. 2020. Available from: https://CRAN.R-project.org/package=survminer.
- 27. Kassambara A, Kosinski M, Przemyslaw B. survminer: Survival Analysis and Visualization [Internet]. Available from: http://www.sthda.com/english/rpkgs/survminer/.
- 28. Terry M. Therneau. A package for survival analysis in R. [Internet]. Available from: https://CRAN.R-project.org/package=survival.
- 29. Agresti A, Coull BA. Approximate is better than “Exact” for interval estimation of binomial proportions. Am Stat 1998;52:119. [Google Scholar]
- 30. R Team. Core R: a language and environment for statistical computing, version 3.5. 3. Vienna: R Foundation for Statistical Computing [Internet]. 2019. Available from: https://www.R-project.org/. [Google Scholar]
- 31. Smith RA, Andrews KS, Brooks D, Fedewa SA, Manassaram-Baptiste D, Saslow D, et al. Cancer screening in the United States, 2019: a review of current American Cancer Society guidelines and current issues in cancer screening. CA Cancer J Clin 2019;69:184–210. [DOI] [PubMed] [Google Scholar]
- 32. Fallahpour S, Navaneelan T, De P, Borgo A. Breast cancer survival by molecular subtype: a population-based analysis of cancer registry data. CMAJ Open 2017;5:E734–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Győrffy B, Surowiak P, Budczies J, Lánczky A. Online survival analysis software to assess the prognostic value of biomarkers using transcriptomic data in non-small-cell lung cancer. PLoS One 2013;8:e82241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dayen C, Debieuvre D, Molinier O, Raffy O, Paganin F, Virally J, et al. New insights into stage and prognosis in small cell lung cancer: an analysis of 968 cases. J Thorac Dis 2017;9:5101–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Short E, Warren AY, Varma M. Gleason grading of prostate cancer: a pragmatic approach. Diagn Histopathol 2019;25:371–8. [Google Scholar]
- 36. Brodersen J, Schwartz LM, Heneghan C, O'Sullivan JW, Aronson JK, Woloshin S. Overdiagnosis: what it is and what it isn't. BMJ Evidence-Based Medicine 2018;23:1–3. [DOI] [PubMed] [Google Scholar]
- 37. Srivastava S, Koay EJ, Borowsky AD, De Marzo AM, Ghosh S, Wagner PD, et al. Cancer overdiagnosis: a biological challenge and clinical dilemma. Nat Rev Cancer 2019;19:349–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Nishihara R, Wu K, Lochhead P, Morikawa T, Liao X, Qian ZR, et al. Long-term colorectal-cancer incidence and mortality after lower endoscopy. N Engl J Med 2013;369:1095–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Esserman LJ, Thompson IM, Reid B, Nelson P, Ransohoff DF, Welch HG, et al. Addressing overdiagnosis and overtreatment in cancer: a prescription for change. Lancet Oncol 2014;15:e234–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kalager M, Wieszczy P, Lansdorp-Vogelaar I, Corley DA, Bretthauer M, Kaminski MF. Overdiagnosis in colorectal cancer screening: time to acknowledge a blind spot. Gastroenterology 2018;155:592–5. [DOI] [PubMed] [Google Scholar]
- 41. Moynihan R, Doust J, Henry D. Preventing overdiagnosis: how to stop harming the healthy. BMJ 2012;344:e3502. [DOI] [PubMed] [Google Scholar]
- 42. Houssami N, Hunter K. The epidemiology, radiology and biological characteristics of interval breast cancers in population mammography screening. NPJ Breast Cancer 2017;3:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Buskermolen M, Cenin DR, Helsingen LM, Guyatt G, Vandvik PO, Haug U, et al. Colorectal cancer screening with faecal immunochemical testing, sigmoidoscopy or colonoscopy: a microsimulation modelling study. BMJ 2019;367:l5383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Pinsky P, Miller A, Kramer B, Church T, Reding D, Prorok P, et al. Evidence of a healthy volunteer effect in the prostate, lung, colorectal, and ovarian cancer screening trial. Am J Epidemiol 2007;165:874–81. [DOI] [PubMed] [Google Scholar]
- 45. National Lung Screening Trial Research Team, Aberle DR, Adams AM, Berg CD, Black WC, Clapp JD, et al. Reduced lung-cancer mortality with low-dose computed tomographic screening. N Engl J Med 2011;365:395–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Jacobs IJ, Menon U, Ryan A, Gentry-Maharaj A, Burnell M, Kalsi JK, et al. Ovarian cancer screening and mortality in the UK Collaborative Trial of Ovarian Cancer Screening (UKCTOCS): a randomised controlled trial. Lancet 2016;387:945–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Siravegna G, Marsoni S, Siena S, Bardelli A. Integrating liquid biopsies into the management of cancer. Nat Rev Clin Oncol 2017;14:531–48. [DOI] [PubMed] [Google Scholar]
- 48. Kwapisz D. The first liquid biopsy test approved. Is it a new era of mutation testing for non-small cell lung cancer? Ann Transl Med 2017;5:46–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Nakamura Y, Taniguchi H, Ikeda M, Bando H, Kato K, Morizane C, et al. Clinical utility of circulating tumor DNA sequencing in advanced gastrointestinal cancer: SCRUM-Japan GI-SCREEN and GOZILA studies. Nat Med 2020;26:1859–64. [DOI] [PubMed] [Google Scholar]
- 50. Surveillance Research Program, National Cancer Institute. SEER*Stat software (www.seer.cancer.gov/seerstat) version 8.3.8. Data: Surveillance, Epidemiology, and End Results (SEER) Program (www.seer.cancer.gov) SEER*Stat Database: Incidence - SEER Research Data, 9 Registries, Nov 2019 Sub (1975–2017) - Linked To County Attributes - Time Dependent (1990–2017) Income/Rurality, 1969–2018 Counties, National Cancer Institute, DCCPS, Surveillance Research Program, based on the November 2019 submission. 2020.
- 51. Crowley E, Di Nicolantonio F, Loupakis F, Bardelli A. Liquid biopsy: mon- itoring cancer-genetics in the blood. Nat Rev Clin Oncol 2013;10:472–84. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.