

Abstract
Purpose:
This study reports the safety, tolerability, MTD, recommended phase II dose (RP2D), pharmacokinetic/pharmacodynamic profile, and preliminary antitumor activity of ceralasertib combined with carboplatin in patients with advanced solid tumors. It also examined exploratory predictive and pharmacodynamic biomarkers.
Patients and Methods:
Eligible patients (n = 36) received a fixed dose of carboplatin (AUC5) with escalating doses of ceralasertib (20 mg twice daily to 60 mg once daily) in 21-day cycles. Sequential and concurrent combination dosing schedules were assessed.
Results:
Two ceralasertib MTD dose schedules, 20 mg twice daily on days 4–13 and 40 mg once daily on days 1–2, were tolerated with carboplatin AUC5; the latter was declared the RP2D. The most common treatment-emergent adverse events (Common Terminology Criteria for Adverse Events grade ≥3) were anemia (39%), thrombocytopenia (36%), and neutropenia (25%). Dose-limiting toxicities of grade 4 thrombocytopenia (n = 2; including one grade 4 platelet count decreased) and a combination of grade 4 thrombocytopenia and grade 3 neutropenia occurred in 3 patients. Ceralasertib was quickly absorbed (tmax ∼1 hour), with a terminal plasma half-life of 8–11 hours. Upregulation of pRAD50, indicative of ataxia telangiectasia mutated (ATM) activation, was observed in tumor biopsies during ceralasertib treatment. Two patients with absent or low ATM or SLFN11 protein expression achieved confirmed RECIST v1.1 partial responses. Eighteen of 34 (53%) response-evaluable patients had RECIST v1.1 stable disease.
Conclusions:
The RP2D for ceralasertib plus carboplatin was established as ceralasertib 40 mg once daily on days 1–2 administered with carboplatin AUC5 every 3 weeks, with pharmacokinetic and pharmacodynamic studies confirming pharmacodynamic modulation and preliminary evidence of antitumor activity observed.
Translational Relevance.
Carboplatin is a cytotoxic chemotherapy for treating patients with solid cancers, including those of the ovary, lung, and cervix. Its utility, as with other platinum agents, is limited by its cumulative toxicity and the fact that most patients eventually develop resistance. There is a need, therefore, to develop chemotherapy combinations, which increase efficacy and help defer or overcome resistance. Ceralasertib is a potent and selective orally available inhibitor of ataxia telangiectasia and Rad3-related kinase that is apical in the cellular response to DNA damage, especially replication stress. Preclinical efficacy studies have shown that addition of ceralasertib to carboplatin enhances the antitumor activity of carboplatin in vitro and in vivo. Here, we report the results of a multicenter phase I study of ceralasertib plus carboplatin in patients with advanced solid cancers, providing a recommended phase II dose, pharmacokinetic profile, preliminary evidence of antitumor activity, and an exploratory assessment of novel predictive and pharmacodynamic biomarkers.
Introduction
Ataxia telangiectasia and Rad3-related (ATR) is a serine/threonine kinase involved in DNA repair in response to DNA damage and replication stress (1, 2). During normal replication, ATR is recruited to stalled replication forks and causes cell-cycle arrest, thereby preventing the formation of double-strand breaks (DSB) while DNA is repaired (3). Inhibiting normal ATR function leads to increasing accumulation of DSBs, particularly in genomically unstable tumor cells with preexisting replication stress, resulting in further genomic instability, mitotic catastrophe, and cell death. Targeting ATR is, therefore, a relevant strategy for the development of novel anticancer agents.
Ceralasertib (formerly AZD6738) is an oral potent selective inhibitor of ATR (4, 5) in development as a novel antitumor agent in patients with solid or hematologic malignancies. In nonclinical studies, ceralasertib displayed synergistic activity when combined with cytotoxic treatments such as chemotherapy or novel DNA damage response (DDR) inhibitors because of a dependence on ATR in tumor cells, with a rise in replication stress and genomic instability (6, 7). Putative predictive biomarkers of ceralasertib include ataxia telangiectasia mutated (ATM), for which a synthetic lethal interaction with ATR is thought to exist (8), while phospho-RAD50 is a pharmacodynamic biomarker of ATR inhibition (9). Schlafen 11 (SLFN11) is a recently discovered biomarker that blocks stressed replication forks independently from ATR and whose loss of expression is associated with treatment resistance (10).
Platinum agents, such as cisplatin and carboplatin, are cytotoxic chemotherapies used to treat numerous cancers, including ovarian, lung, cervix, and other solid tumors. Their full potential is, however, limited by the development of drug resistance and cumulative toxicity (11, 12). As such, there is an overarching need to develop novel combination regimens that can increase the activity of platinum agents while overcoming intrinsic or acquired resistance.
Preclinical evidence supporting synergy between platinum agents and ceralasertib was shown in in vitro and in vivo patient-derived xenograft (PDX) combination studies in nude mice, whereby orally dosed ceralasertib showed dose-dependent synergistic potentiation of platinum agents that led to enhanced tumor growth inhibition and regression (ref. 7 and Supplementary Preclinical Data). In vitro, combination cell killing with carboplatin was seen at 0.3 μmol/L ceralasertib but not at 0.1 μmol/L. Greater cell killing with carboplatin was reached at 1 μmol/L ceralasertib, which is close to the 90% inhibitory concentration (IC90) for ATR inhibition in cells of 0.67 μmol/L (4).
Preclinical experiments that investigated distinct dosing schedules guided the clinical schedule of carboplatin and ceralasertib dosing in this clinical trial. For example, in a TP53-mutant triple-negative breast cancer PDX murine model, 3 days of ceralasertib (at 25 mg/kg) with carboplatin administered concurrently on day 1 of ceralasertib yielded optimal tumor control (6). Schedules in which carboplatin was dosed concurrently with ceralasertib on day 2 or 3 resulted in growth inhibition rather than tumor control. In the rat, which is believed to be a better model of bone marrow toxicity than the mouse, dosing of ceralasertib directly after carboplatin was poorly tolerated. To preserve efficacy while ensuring tolerability, it was necessary to introduce gaps between carboplatin and the start of ceralasertib dosing, with a 3-day gap predicted to be tolerable and efficacious in human trials (Supplementary Preclinical Data).
We report here the first results from the phase I clinical trial, which assessed the administration of oral ceralasertib in combination with carboplatin chemotherapy in patients with advanced solid tumors. Data are presented on the clinical experience of this rational combination, including the safety and tolerability, recommended phase II dose (RP2D), pharmacokinetics (PK), pharmacodynamics, and preliminary antitumor activity of ceralasertib and carboplatin chemotherapy and predictive biomarkers of response to this combination.
Patients and Methods
Study design and participants
The phase I study was a two-part, open-label, modular dose-escalation and dose-expansion study (NCT02264678). To date, three modules have enrolled patients, each comprising ceralasertib dose escalation in a rolling-six design in combination with an anticancer agent to determine an RP2D (part 1), as well as optional expansion cohorts at the declared RP2D (part 2). The rolling-six design was employed to improve the efficiency of recruiting into dose cohorts by recruiting at least 3 and up to 6 per cohort to reduce the need for late replacement of patients who became nonevaluable during the dose-limiting toxicity (DLT) assessment period (13). We report here the final results from module 1 (data cutoff April 10, 2018), which evaluated ceralasertib in combination with carboplatin.
The trial was conducted in accordance with the Declaration of Helsinki and the Guidelines for Good Clinical Practice. The trial protocol and the final version of the Informed Consent Form, and any other written information provided to trial participants, were approved by an ethics committee or an Institutional Review Board depending upon the country location of the study center. This trial was registered in ClinicalTrials.gov with the identifier NCT02264678 and EudraCT number 2014-002233-66.
Eligible patients were ≥18 years old with histologic or cytologic confirmation of advanced malignancy considered suitable for study treatment and an Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1. Key exclusion criteria included a diagnosis of ataxia telangiectasia, any unresolved toxicities from prior therapy, and recent treatment with cytotoxic, corticosteroid, or any investigational medicinal product. Full eligibility criteria are listed in Supplementary Table S1.
Procedures
Patients received ceralasertib as oral tablets on various explored doses and schedules in combination with carboplatin administered intravenously at a fixed AUC5 dose on day 1 of each 21-day cycle. The ceralasertib starting dose and schedule, selected on the basis of preclinical modeling, was 20 mg twice daily. Patients received a single dose (cycle 0 day 1), then twice daily dosing for up to 5 days before beginning cycle 1 treatment for pharmacokinetic assessment of ceralasertib monotherapy. For the starting dose and schedule, carboplatin AUC5 was administered on cycle 1 day 1, with ceralasertib re-introduced in cohort 1 at 20 mg twice daily on days 4–20 in 21-day cycles. Patients received combination treatment until disease progression, unacceptable toxicity, withdrawal of consent, or completion of up to six cycles of chemotherapy, at which point ceralasertib was also stopped. The dose of ceralasertib in subsequent cohorts was based on emerging safety and pharmacokinetic data, the daily dose, and the number of days that dosing was escalated or de-escalated accordingly. The MTD was defined as the highest dose at which ≤1 in 6 evaluable patients experienced a DLT (Supplementary Table S2). Dose interruptions or reductions were permitted for patients experiencing clinically significant toxicity. Tumor response was assessed by CT or MRI at baseline and during the combination phase every 6 weeks until disease progression. Venous blood for ceralasertib pharmacokinetic and pharmacodynamic measurements was collected during cycle 0 (on day 1 after a single dose and day 6 after repeat dosing) and the combination phase at cycle 1 (on the first and last dosing days of ceralasertib in cycle 1). Optional tumor biopsies for biomarker analyses were collected from patients with accessible tumors during cycle 0 at any time between days 2 and 6 and up to 24 hours after the last dose of ceralasertib in that cycle.
IHC
All IHC used 4- to 5-μm-thick sections of formalin-fixed paraffin-embedded clinical tissues. ATM IHC was performed as described in Villaruz and colleagues (14). pRAD50 IHC was performed as described in Jones and colleagues (9). SLFN11 IHC was performed using a Leica Bond RX staining platform, the steps of which included dewaxing, pH9 antigen retrieval (ER2) for 25 minutes, protein blocking with SignalStain (Cell Signaling Technology), staining with 2.5 μg/mL anti-SLFN11 ab121731 antibody (Abcam), detection using the Polymer Refine Detection Kit (Leica), and counterstaining with hematoxylin (Leica). Appropriate control tissues and matched isotype controls were used during each staining run. Slides were digitalized using an Aperio AT2 scanner and pathology H-scores generated. H-scores are a product of the intensity of expression (0–3+) and percentage of cells stained. For ATM and SLFN11, which are both expressed in the nucleus, internal tissue control staining (e.g., lymphocytes) must be of 2+ or higher intensity for the sample to be deemed evaluable. A single pathologist performed the scoring at 20× magnification and the entire sample was assessed.
Outcomes
The primary objective was to assess the safety and tolerability of ceralasertib in combination with carboplatin and to establish an MTD and recommend a suitable phase II dose by continual monitoring of adverse events [AE; according to Common Terminology Criteria for Adverse Events (CTCAE) v4.03], laboratory data, vital signs, and ECG parameters. Secondary objectives included an assessment of preliminary antitumor activity by tumor response according to RECIST v1.1 and pharmacokinetic and pharmacodynamic parameters. Pharmacokinetic measurements included area under the plasma concentration–time curve (AUC), maximum concentration (Cmax), time to maximum concentration (tmax), and terminal half-life (t1/2). Exploratory endpoints to determine the clinical or biological activity of ceralasertib alone or in combination with carboplatin included analysis of ATM and SLFN11 (8, 15) in tumors by IHC as potential predictors of response, as well as pRAD50 induction as a pharmacodynamic biomarker of ATR inhibition (9).
Statistical analysis
Data are presented descriptively by cohort; no formal statistical analysis was conducted. To determine an MTD for ceralasertib, cohorts of 3–6 evaluable patients were required. The safety set comprised all patients who received ≥1 dose of ceralasertib, and the evaluable-for-response set comprised all dosed patients with measurable disease at baseline. The pharmacokinetic set comprised all dosed patients with reportable ceralasertib plasma concentrations and no important AEs or protocol deviations that could impact pharmacokinetics. The pharmacodynamic set encompassed all patients who provided biological samples.
Data-sharing statement
Data underlying the findings described in this article may be obtained in accordance with AstraZeneca's data-sharing policy described at: https://astrazenecagrouptrials.pharmacm.com/ST/Submission/Disclosure.
Results
Patient characteristics
Thirty-six patients were enrolled between October 2014 and August 2016 at four study centers in three countries (United Kingdom, France, and United States). All patients received ceralasertib, and 35 patients also received carboplatin chemotherapy; 1 patient in cohort 2 discontinued from the study before the first carboplatin dose. Median patient age was 63 years, and the most common primary tumor site was colorectal (25% of patients). The majority of patients had received prior chemotherapy (89%); the median number of prior regimens was 4. Patient demographics and baseline disease characteristics are shown in Table 1. Eleven patients completed the planned number of cycles of carboplatin and therefore stopped all study treatment (chemotherapy and ceralasertib). The remaining 25 patients discontinued treatment because of disease progression (n = 19, 53%) or an AE (n = 5, 14%), and 1 patient with EGFR-mutant mixed cell–type lung cancer switched to an alternative (EGFR inhibitor) cancer therapy of their choice after cycle 2 because of insufficient response (in the absence of radiologic or clinical progression). AEs leading to discontinuation included grade 3 cerebral venous thrombosis (cohort 2) and grade 2 lower respiratory tract infection (cohort 2), which were believed unrelated to study treatment; grade 3 platelet count decreased (cohort 7); and two cases of grade 4 thrombocytopenia (cohorts 4 and 5), each of which was at least possibly related to carboplatin or ceralasertib. One patient, who was diagnosed with squamous cancer of the cervix and discontinued treatment because of recurrent grade 4 thrombocytopenia, died within the 30-day follow-up period from disease progression. Patient disposition is summarized in Supplementary Table S3.
Table 1.
Patient demographics and baseline disease characteristics (safety analysis set).
| Total (N = 36) | |
|---|---|
| Median age, years (range) | 63 (34–83) |
| Gender, n (%) | |
| Female | 21 (58) |
| Male | 15 (42) |
| Median number of prior chemotherapy regimens (range) | 4 (1–≥6)a |
| Previous treatment | |
| Immunotherapy | 4 (11) |
| Hormonal therapy | 2 (6) |
| Chemotherapy | 32 (89) |
| Systemic therapyb | 7 (19) |
| Other | 11 (31) |
| Radiotherapy | 20 (56) |
| ECOG performance status,c n (%) | |
| 0 | 18 (50) |
| 1 | 18 (50) |
| Primary tumor site, n (%) | |
| Colorectald | 9 (25) |
| Ovary | 7 (19) |
| Lung | 6 (17) |
| Cervix | 4 (11) |
| Mesothelioma | 3 (8) |
| Prostate | 2 (6) |
| Othere | 5 (14) |
aThree patients unknown in total: 1 in cohort 2 and 2 in cohort 7.
bIncludes chemotherapy, anti-VEGF inhibitors, targeted antibodies, and investigational agents.
c0, fully active; 1, restricted in physically strenuous activity.
dIncludes colon, colorectal, colorectal carcinoma, rectal, and sigmoid.
eOther: esophagus, pancreas, parotid gland, small bowel, stomach.
Safety and tolerability
Ceralasertib and carboplatin were investigated across seven dose cohorts (Fig. 1; Supplementary Table S4). All 36 patients experienced ≥1 treatment-emergent AE (TEAE; Table 2), most commonly anemia and thrombocytopenia (each n = 25 patients, 69.4%), nausea (21, 58%), fatigue (17, 47%), and neutropenia (13, 36%). TEAEs of grade ≥3 were observed in 27 (75%) patients, most commonly anemia (14 patients, 39%), thrombocytopenia (13, 36%), and neutropenia (9, 25%).
Figure 1.

Dose cohorts. BID, twice daily; QD, once daily.
Table 2.
TEAEs occurring in >10% of patients in total (safety analysis set) by CTCAE grade.
| Patient cohort | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cohort 1 (N = 2) | Cohort 2 (N = 7) | Cohort 3 (N = 3) | Cohort 4 (N = 3) | Cohort 5 (N = 8) | Cohort 6 (N = 7) | Cohort 7 (N = 6) | ||||||||||
| 20 mg BID ceralasertib 17 days | 20 mg BID ceralasertib 10 days | 40 mg BID ceralasertib 10 days | 40 mg BID ceralasertib 7 days | 60 mg QD ceralasertib 7 days | 60 mg QD codosing ceralasertib 3 days | 40 mg QD codosing ceralasertib 2 days | ||||||||||
| Not tolerated | Tolerated | Not tolerated | Not tolerated | Not tolerated | Not tolerated | Tolerated | Total (N = 36) | |||||||||
| Preferred term | Grade 1–2 | Grade ≥3 | Grade 1–2 | Grade ≥3 | Grade 1–2 | Grade ≥3 | Grade 1–2 | Grade ≥3 | Grade 1–2 | Grade ≥3 | Grade 1–2 | Grade ≥3 | Grade 1–2 | Grade ≥3 | Grade 1–2 | Grade ≥3 |
| Any AE | 1 (50) | 1 (50) | 2 (29) | 5 (71) | 1 (33) | 2 (67) | 0 | 3 (100) | 4 (50) | 4 (50) | 0 | 7 (100) | 1 (17) | 5 (83) | 9 (25) | 27 (75) |
| Thrombocytopenia | 1 (50) | 1 (50) | 4 (58) | 1 (14) | 1 (33) | 2 (67) | 0 | 3 (100) | 4 (50) | 2 (25) | 2 (29) | 4 (57) | 0 | 0 | 12 (33) | 13 (36) |
| Anemia | 1 (50) | 0 | 2 (29) | 2 (29) | 1 (33) | 0 | 1 (33) | 1 (33) | 3 (38) | 3 (38) | 2 (29) | 4 (57) | 1 (17) | 4 (67) | 11 (31) | 14 (39) |
| Nausea | 1 (50) | 0 | 2 (29) | 0 | 0 | 0 | 2 (67) | 0 | 4 (50) | 0 | 6 (86) | 1 (14) | 5 (83) | 0 | 20 (56) | 1 (3) |
| Fatigue | 2 (100) | 0 | 4 (58) | 1 (14) | 1 (33) | 0 | 1 (33) | 0 | 3 (38) | 0 | 1 (14) | 0 | 4 (67) | 0 | 16 (44) | 1 (3) |
| Neutropenia | 0 | 1 (50) | 1 (14) | 0 | 1 (33) | 2 (67) | 1 (33) | 1 (33) | 0 | 0 | 1 (14) | 5 (71) | 0 | 0 | 4 (11) | 9 (25) |
| Constipation | 1 (50) | 0 | 2 (29) | 0 | 1 (33) | 0 | 1 (33) | 0 | 1 (13) | 0 | 1 (14) | 0 | 3 (50) | 0 | 10 (28) | 0 |
| Pyrexia | 1 (50) | 0 | 3 (43) | 0 | 0 | 0 | 1 (33) | 0 | 2 (25) | 0 | 1 (14) | 0 | 1 (17) | 0 | 9 (25) | 0 |
| Vomiting | 1 (50) | 0 | 2 (29) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 4 (57) | 1 (14) | 0 | 1 (17) | 7 (19) | 2 (6) |
| Decreased appetite | 0 | 0 | 1 (14) | 0 | 1 (33) | 0 | 1 (33) | 0 | 2 (25) | 0 | 0 | 0 | 3 (50) | 0 | 8 (22) | 0 |
| Diarrhea | 1 (50) | 0 | 2 (29) | 0 | 0 | 0 | 0 | 0 | 1 (13) | 0 | 1 (14) | 0 | 1 (17) | 0 | 6 (17) | 0 |
| Platelet count decreased | 0 | 0 | 0 | 1 (14) | 1 (33) | 0 | 0 | 0 | 1 (13) | 0 | 0 | 0 | 2 (33) | 1 (17) | 4 (11) | 2 (6) |
| Abdominal pain | 0 | 1 (50) | 0 | 0 | 0 | 0 | 1 (33) | 0 | 1 (13) | 0 | 1 (14) | 0 | 2 (33) | 0 | 5 (14) | 1 (3) |
| Cough | 0 | 0 | 5 (71) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 5 (14) | 0 |
| Dyspnea | 1 (50) | 0 | 3 (43) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (14) | 0 | 0 | 0 | 5 (14) | 0 |
| Insomnia | 0 | 0 | 2 (29) | 0 | 1 (33) | 0 | 0 | 0 | 0 | 0 | 2 (29) | 0 | 0 | 0 | 5 (14) | 0 |
| Lower respiratory tract infection | 0 | 0 | 2 (29) | 0 | 0 | 0 | 0 | 0 | 1 (13) | 1 (13) | 1 (14) | 0 | 0 | 0 | 4 (11) | 1 (3) |
| White blood cell count decreased | 0 | 1 (50) | 0 | 0 | 0 | 0 | 0 | 1 (33) | 0 | 1 (13) | 0 | 2 (29) | 0 | 0 | 0 | 5 (14) |
| Alanine aminotransferase increased | 0 | 0 | 1 (14) | 0 | 0 | 0 | 1 (33) | 0 | 0 | 1 (13) | 0 | 0 | 1 (17) | 0 | 3 (8) | 1 (3) |
| Arthralgia | 0 | 0 | 1 (14) | 0 | 0 | 0 | 0 | 0 | 2 (25) | 0 | 0 | 0 | 1 (17) | 0 | 4 (11) | 0 |
| Neutrophil count decreased | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (33) | 0 | 1 (13) | 0 | 1 (14) | 0 | 1 (17) | 0 | 4 (11) |
| Aspartate aminotransferase increased | 0 | 0 | 1 (14) | 0 | 0 | 0 | 0 | 0 | 1 (13) | 1 (13) | 0 | 0 | 1 (17) | 0 | 3 (8) | 1 (3) |
| Contusion | 0 | 0 | 1 (14) | 0 | 2 (67) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (17) | 0 | 4 (11) | 0 |
| Dizziness | 0 | 0 | 0 | 0 | 0 | 0 | 1 (33) | 0 | 2 (25) | 0 | 1 (14) | 0 | 0 | 0 | 4 (11) | 0 |
| Hypocalcemia | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 (25) | 0 | 1 (14) | 0 | 1 (17) | 0 | 4 (11) | 0 |
| Upper respiratory tract infection | 0 | 0 | 3 (43) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (17) | 0 | 4 (11) | 0 |
Note: Numbers in each column are “patients, n (%)” with specific preferred term and CTCAE grade. Cohort 1, 20 mg twice daily (BID) ceralasertib 17 days + carboplatin; cohort 2, 20 mg BID ceralasertib 10 days + carboplatin; cohort 3, 40 mg BID ceralasertib 10 days + carboplatin; cohort 4, 40 mg BID ceralasertib 7 days + carboplatin; cohort 5, 60 mg once daily (QD) ceralasertib 7 days + carboplatin; cohort 6, co-dosing 60 mg QD ceralasertib + carboplatin days 1–3; cohort 7, co-dosing 40 mg QD ceralasertib + carboplatin days 1 and 2. Each patient has only been represented with the maximum reported CTCAE grade for each system organ class/preferred term. Includes adverse events with an onset date on or after the date of the first dose (earliest date of first dose of ceralasertib or carboplatin), up to and including 28 days following the date of the last dose (latest date of last dose of ceralasertib or carboplatin).
No clinically significant findings were observed in any clinical chemistry laboratory variables, vital signs or ECG parameters, and no AEs with a fatal outcome were reported. TEAEs, as determined by the investigator, are shown in Supplementary Table S5. Overall median (range) total treatment duration for ceralasertib was 2.6 (0.13–5.0) months. Altogether, 11 (31%) patients had treatment interruptions, 9 (25%) had dose reductions, and 17 (47%) had dose modifications in cycle 1 and beyond.
In cohort 1 (N = 2), ceralasertib was dosed at 20 mg twice daily on days 4–20. The 3-day gap between carboplatin and ceralasertib was prospectively scheduled to ameliorate the expected hematologic toxicities predicted from preclinical modeling studies. Both patients experienced grade 1–2 fatigue in cycle 1; one required an interruption in cycle 1 dosing of ceralasertib on day 19 because of grade 3 thrombocytopenia, delaying the start of cycle 2 by 7 days (Supplementary Table S4). The second patient could not start cycle 2 at the scheduled time because of grade 1 thrombocytopenia, grade 2 anemia, and grade 2 fatigue; clinical suspicion of disease progression triggered an early CT scan, which confirmed disease progression. Cohort 1 was declared intolerable based on the toxicities observed in the first 2 patients, and the number of days of ceralasertib dosing was reduced from 17 to 10 in cohort 2. There were no DLTs in 7 evaluable patients in cohort 2, but 1 patient required a dose reduction to 7 days' dosing in cycle 2 and subsequently to 4 days in cycle 3 because of grade 2 thrombocytopenia. Cohort 2 was declared tolerated according to the protocol definition. In cohort 3, the daily dose of ceralasertib was escalated to 40 mg twice daily for 10 days, and 3 patients were recruited. All 3 patients required a reduction to 7 days' dosing (2 patients in cycle 2, 1 in cycle 4) because of grade 2, 3, and 4 thrombocytopenia. While there were no DLTs, cohort 3 was declared intolerable because of the required reduction in the number of dosing days. In cohort 4, ceralasertib was dosed at 40 mg twice daily for 7 days to mirror the dose reductions in cohort 3. There was a DLT of grade 4 thrombocytopenia and grade 3 neutropenia (in the same patient) among 3 patients dosed. All 3 patients required delays to cycle 2 and beyond because of grade 3–4 thrombocytopenia, and all had their dose reduced to 20 mg twice daily ceralasertib for 7 days plus carboplatin AUC4; cohort 4 was therefore declared intolerable. In cohort 5, ceralasertib dose was reduced to 60 mg once daily for 7 days; 8 patients were recruited, but only 6 were evaluable for DLTs because of missed doses in cycle 1 (due to non-drug–related toxicity, n = 1) and cycle 0 (due to dosing error, n = 1). Of these 6 patients, one DLT of grade 4 thrombocytopenia was observed. Of the 8 patients dosed in cohort 5, 5 required dose delays after cycle 1 and 4 had dose reductions. Overall, the safety review committee deemed that cohort 5 was not tolerated because of these chronic toxicities requiring dose delays and reductions.
The mounting evidence for intolerability of the tested sequential schedules led to a change of clinical trial strategy from cohort 6 to concurrent dosing of ceralasertib (60 mg once daily) with carboplatin given on day 1, with further doses of ceralasertib on days 2–3 (allowing more time for bone marrow recovery) and removal of cycle 0 dosing, aside from a single dose for pharmacokinetic sampling on day −7. Seven patients were recruited in cohort 6 and there were no DLTs, but the regimen was not tolerated beyond cycle 1, with dose delays and reductions due to a mixture of grade 2–3 neutropenia and grade 2–4 thrombocytopenia. In cohort 7, ceralasertib was reduced to 40 mg once daily for days 1–2, and 6 patients were recruited. Of these 6, only one DLT of grade 4 platelet count decreased resulted. Overall, this cohort required fewer dose delays, and only 1 patient needed a dose reduction (to 1-day dosing at 40 mg once daily and carboplatin AUC4 because of grade 4 platelet count decreased and grade 3 neutropenia). Cohort 7 was declared safe and well tolerated and established as the MTD and schedule.
Antitumor activity
Thirty-four patients were included in the evaluable-for-response set; 2 were excluded as they had no measurable target lesions at baseline. Overall, 2 patients (both in cohort 6) had confirmed RECIST v1.1 partial responses (PR), and a further 2 (1 in cohort 3, another in cohort 7) had unconfirmed RECIST v1.1 PRs. Eighteen of 34 (53%) patients had a best RECIST v1.1 response of stable disease (SD) for ≥35 days (including the two unconfirmed PRs), and 12 (35%) had a best response of progressive disease (PD). Two patients had no follow-up assessments after baseline. The maximum percentage reduction in sum of target lesions for all patients is shown in Fig. 2.
Figure 2.

Waterfall plot of response-evaluable patients showing best percentage change from baseline. Tumor type and percentage nuclear staining of ATM and SLFN11 are shown in the table. Meso, mesothelioma; N/D, no data available; sm, small.
Of the 2 patients with confirmed RECIST v1.1 PRs from cohort 6, 1 achieved a PR of 55% reduction in the sum of her target lesions and a duration of response of 125 days. This patient had stage IV metastatic squamous cell carcinoma of the cervix with recurrent disease in her lymph nodes. Prior anticancer therapy comprised chemoradiotherapy with four cycles of carboplatin, paclitaxel, and cediranib or placebo administered in a blinded clinical trial approximately 4 years prior to starting this study. The patient achieved a PR to the prior therapy, and disease progression was observed 5 months before commencing study treatment. The second confirmed response was in a patient with stage IIIB colorectal cancer. This patient had received oxaliplatin-based adjuvant chemotherapy approximately 7 years prior to commencing study treatment and six lines of chemotherapy in the metastatic setting. Treatment in the metastatic setting included 5-fluorouracil, bevacizumab, irinotecan, and oxaliplatin-based regimens, with SD as the best response to the most recent oxaliplatin-containing regimen. Study treatment commenced 1 month after completing the sixth line of chemotherapy. A 48% reduction in the sum of target lesions was observed that was maintained for 96 days, with progression as a result of new lesions in the liver, adrenal gland, and bone.
The patient in cohort 3 with an unconfirmed PR completed four cycles of study treatment and achieved a 35% tumor reduction. There was disease progression at the follow-up scan. This patient was diagnosed with locally advanced lung adenocarcinoma and had received four prior lines of chemotherapy, including cisplatin and pemetrexed, erlotinib, docetaxel, and AZD5363 (novel AKT inhibitor). The response to the most recent therapy (AZD5363) was SD, with a best response of PD to the three preceding lines. The second patient with an unconfirmed response had poorly differentiated clear cell ovarian cancer with metastasis to the spleen. Two cycles of combination treatment resulted in a 43% tumor reduction, and the patient discontinued study therapy for surgery.
Pharmacokinetic assessments
Ceralasertib was quickly absorbed when administered as a single dose (median tmax 1.00–1.03 hours) and in combination (1.05–1.07 hours; Supplementary Table S6). Plasma concentrations declined in a generally biphasic manner after reaching Cmax, with a mean half-life of 8.14–10.57 hours when administered alone and 5.14–7.22 hours in combination. No notable difference in between-patient variability was observed when ceralasertib was administered concurrently with carboplatin compared with ceralasertib alone, which is expected given the different routes of metabolism. Overlaying the plasma concentration profiles of ceralasertib with the IC90 for ATR enzyme (4) revealed that ceralasertib exposure only briefly reached IC90 at the RP2D of 40 mg once daily (Fig. 3; Supplementary Fig. S1).
Figure 3.
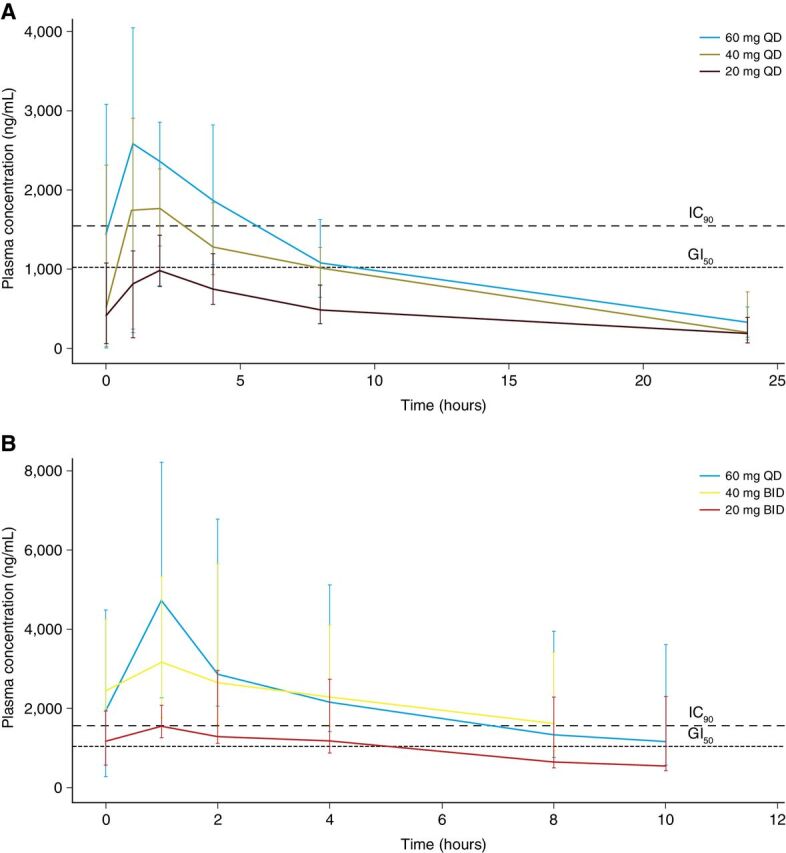
Plasma concentrations of ceralasertib after a single dose (A) and at steady state (B). The bars indicate the 10th and 90th percentiles. The dashed lines indicate the plasma concentrations that result in GI50 for the LoVo cell line and 90% ATR inhibition in vitro. Note that there was no steady-state pharmacokinetic sample collected for 40 mg once-daily ceralasertib in cohort 7. The pharmacokinetics for ceralasertib at each dose in cohort 7 (at 40 mg once daily for days 1–2) will resemble single-dose kinetics for 40 mg.
We compared the exposure of ceralasertib at the RP2D in combination with carboplatin with the exposure achieved by berzosertib (M6620, VX-970, Merck KGaA), an intravenously administered ATR inhibitor, in combination with carboplatin using their respective concentrations for 50% growth inhibition (GI50) for the LoVo cell line (0.44 and 0.09 μmol/L, respectively) as an indication of potency. A total of 1,000 simulations at single-dose 90 mg/m2 berzosertib were performed with a recently published population pharmacokinetic model (16) and superimposed onto the plasma concentration profile for a single dose of 40 mg ceralasertib (Supplementary Fig. S2). For consistent comparison, each pharmacokinetic profile was normalized by molecular weight, unbound fraction, and growth inhibition threshold (GI50). The graphical comparison showed that ceralasertib achieves favorable cover over the GI50 compared with berzosertib at the RP2D.
Pharmacodynamic biomarker analysis
Induction of pRAD50 following ATR inhibition may occur as a result of ATM pathway induction (9). In the analysis of paired tumor biopsies (n = 5 collected; n = 4 evaluable) obtained in this trial, all biopsies were positive for ATM protein expression by IHC. There was variable expression of pRAD50 at baseline, with H-scores ranging from 20 to 90 (Fig. 4A). However, clear increases in pRAD50 expression were observed following treatment with ceralasertib in all four biopsy pairs assessed, indicative of ATR target and pathway engagement. The largest increase in pRAD50 expression was observed in 2 patients with relatively low baseline levels of ATM (20% and 13%) treated in cohort 7, for whom pRAD50 expression increased by 400% (H-scores of 20–80) and 220% (H-scores of 50–110), respectively, although the latter was after combination treatment. Three of the four biopsies were taken from patients who had PD as best response, with the fourth from a patient who completed four cycles of combination treatment and had PD approximately 6 months later.
Figure 4.
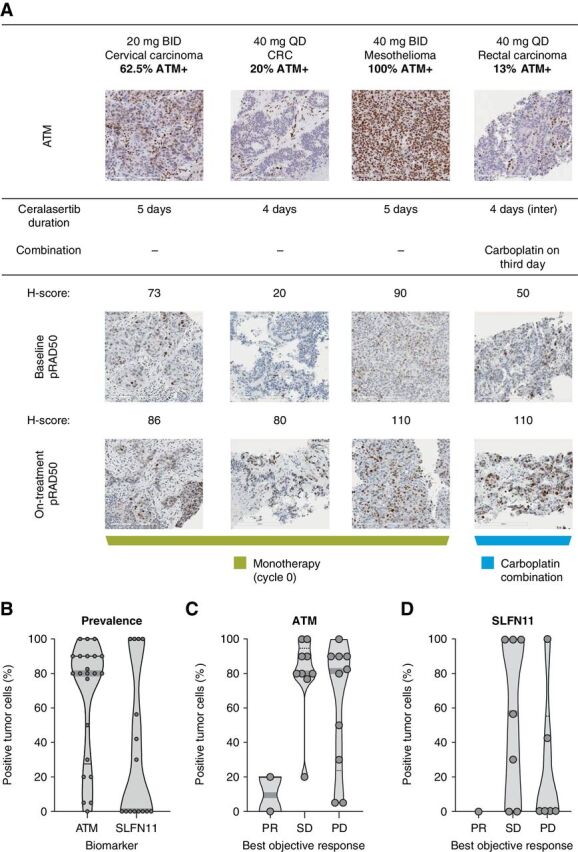
A, Analysis of pRAD50 in paired tumor biopsies. B, Prevalence of ATM and SLFN11 in archival tumor samples. C, Correlation of ATM tumor expression with best objective response. D, Correlation of SLFN11 tumor expression with best objective response. A, top, total ATM expression at baseline. Tumors were scored by percentage of tumor nuclei positive. Previous studies have shown that in ATM-expressing tumors, induction of pRAD50 is a marker of ATR inhibition. Importantly, in each of the tumors for which paired biopsies were collected, ATM expression was documented at baseline. Bottom, Induction of pRAD50 by H-score on treatment compared with baseline. All paired biopsies demonstrated induction of pRAD50 consistent with inhibition of ATR. B, Each point represents the percentage positive tumor cells in an individual tumor. C and D, Each point represents an individual patient.
Predictive biomarker analysis
Archival tumor samples were available for 25 patients, as well as paired fresh biopsies from 4 patients dosed in cohorts 1 and 3 and 2 in cohort 7. This study did not include provisions for DNA sequencing of tumor DNA. Analysis of the archival samples by IHC revealed that 21 (95%) of 22 evaluable tumors had ATM expression, while SLFN11 showed a bimodal distribution, with 8 (53%) of 15 evaluable tumors displaying SLFN11 expression (Fig. 4B). There was no apparent correlation between ATM and SLFN11 expression, although the numbers were too small to draw conclusions. When ATM was correlated with antitumor response (Fig. 4C), it was notable that the 2 patients who had confirmed RECIST v1.1 PRs had among the lowest ATM-expressing tumors, with one having zero detectable nuclear expression. No formal statistical analysis has been conducted given the small sample sizes. For SLFN11, data were available from 14 (41%) patients evaluable for response, including only one of the confirmed responders (Fig. 4D); this patient had an absence of nuclear SLFN11 expression [although they had low (∼20%) nuclear expression of ATM].
Discussion
This phase I dose-escalation study in patients with advanced solid tumors determined the safety, tolerability, MTD, RP2D, pharmacokinetics, pharmacodynamics, and preliminary antitumor activity of the ATR inhibitor, ceralasertib, in combination with carboplatin.
While the ceralasertib and carboplatin combination had few protocol-defined DLTs, dose interruptions were needed between cycles at higher ceralasertib exposures to allow bone marrow recovery. Drug-related toxicities, including those of grade ≥3, were consistent with the known safety profiles of single-agent ceralasertib (17) and carboplatin. Synergistic hematologic toxicity was anticipated, especially thrombocytopenia, which did not recover to grade 1 or better by the end of the 21-day cycle, limiting the administration of carboplatin and ceralasertib to the desired schedule. Neutropenia was noticeably more common (together with thrombocytopenia) in cohorts 6 and 7 when ceralasertib was dosed with carboplatin. An RP2D of 40 mg ceralasertib once daily on days 1 and 2 with carboplatin AUC5 on day 1 was established. However, continuation of the combination into an expansion cohort was not supported because of the low doses of ceralasertib achieved in this combination and a strategic decision to focus development on non-chemotherapy combinations. Notably, patients received a median of four lines of chemotherapy before study entry, which may have compromised bone marrow reserve. Additional clinical exploration of ceralasertib combined with less aggressive platinum administration in a platinum-experienced patient population, for example, carboplatin AUC4 versus AUC5 and/or a 28-day versus 21-day administration cycle to reduce bone marrow toxicity and support post-dosing bone marrow recovery, could be considered in future studies, although such a strategy may be suboptimal in compromising the chemotherapy backbone.
Preclinical efficacy studies of ATR inhibitors in combination with DNA-damaging chemotherapy, including ceralasertib with carboplatin, showed synergistic efficacy by scheduling the dosing of both drugs either concurrently with or sequentially after chemotherapy (6). Furthermore, the number of days that ceralasertib was dosed after carboplatin also influenced the degree of antitumor efficacy, with 2 days inducing tumor control and 3 days driving tumor regression in a triple-negative breast cancer PDX model (6). Similar conclusions were reached with the intravenously administered ATR inhibitor, berzosertib (M6620, VX-970; Merck KGaA). Berzosertib was efficacious as a single dose delivered 12–24 hours after chemotherapy and reportedly did not affect the tolerability of the chemotherapy (18). The challenge with oral ceralasertib was delivering a sufficient dose, sustained for several days, without causing intolerable toxicity. Preclinical data suggested that dosing of ceralasertib to reach the IC90 for ATR inhibition on 3 consecutive days was needed to drive efficacy, but this was associated with weight losses in mice during the dosing interval that were regained during the “off period.” Experiments in rats, which may be a more appropriate tolerability model for cytotoxic agents, suggested that a gap of 3 days between carboplatin and the onset of ceralasertib dosing was required for improved tolerability. This drove the clinical investigation of the initially tested sequential schedule in this study, with two clear “ceralasertib-free” days between carboplatin and ceralasertib followed by a prolonged (17-day) period of continuous ceralasertib dosing to maximize efficacy. The number of ceralasertib dosing days was reduced from 17 to 7 to improve tolerability (cohort 2), and the daily dose was escalated (cohort 3) to achieve higher ceralasertib exposure. At ceralasertib 40 mg twice daily (cohorts 3 and 4) and 60 mg once daily (cohort 5), coverage over the IC90 was reached with multiple dosing (≥6 hours), with evidence of ATR inhibition in cohorts 1 and 4 (from on-treatment biopsies). The limited tolerability of this sequential schedule, demonstrated by the need for repeated delays to successive treatment cycles, and the short duration of coverage over the IC90 precluded its further investigation.
The strategy of concurrently administering ceralasertib and carboplatin evolved significantly over the course of this study in response to emerging clinical findings. Limited-duration concurrent dosing of ceralasertib with carboplatin produced better tolerability than sequential schedules of overall longer-duration ceralasertib exposure. Promisingly, we observed two confirmed RECIST v1.1 responses in cohort 6 (with 3-day dosing at 60 mg once daily) and an unconfirmed RECIST v1.1 response in cohort 7 (2-day dosing with 40 mg once daily) using this concurrent dosing strategy. Interestingly, there was anecdotal evidence of pharmacodynamic ATR modulation in tumor biopsies from a patient treated in cohort 7 (demonstrated by an increase in pRAD50 in paired tumor biopsies), suggesting ATR inhibition at this dose level.
In the recently published phase I trial of berzosertib in combination with carboplatin, there were similar challenges associated with schedule-limiting hematologic toxicities, leading to dose delays and reductions (19). Berzosertib was dosed on days 1 and 8 every 21 days, with carboplatin AUC5 on day 1. The starting dose (240 mg/m2) of berzosertib was half the recommended monotherapy dose, but this was not tolerated because of grade 3/4 neutropenia in all 3 patients and the dose of berzosertib was subsequently reduced to 90 mg/m2, which was tolerated. More recently, berzosertib was combined with topotecan in small cell lung cancer at a dose of 210 mg/m2 berzosertib on days 2 and 5 with topotecan given on days 1–5 in a 21-day cycle, with evidence of clinical activity beyond that expected by topotecan alone (20). A comparison of the ceralasertib RP2D achieved with carboplatin with the berzosertib RP2Ds in combination with carboplatin and topotecan demonstrate that we achieved at least as good target inhibition with ceralasertib in combination with carboplatin as that obtained with berzosertib with either of carboplatin or topotecan (Supplementary Fig. S2). This analysis suggests that while the dose of ceralasertib in combination with carboplatin was relatively low compared with doses achieved with ceralasertib monotherapy, it is comparable with berzosertib doses and is meaningful to take into future phase II studies.
Other potential strategies to improve the tolerability of ceralasertib in combination with chemotherapy may be to lengthen the treatment cycle and to consider the choice of cisplatin over carboplatin. Studies of ceralasertib in monotherapy and combination with olaparib (PARP inhibitor) and durvalumab (anti-PD-L1 antibody) all required an intermittent dosing schedule with a treatment break of at least 14 days with every 28-day cycle for optimal bone marrow recovery (17, 21). In this study, we were reluctant to extend the cycle length from 21 to 28 days, as 21 days is the standard of care for carboplatin and we did not want to compromise the efficacy of the approved chemotherapy. Nevertheless, as most treatment delays lasted 7 days, extending the cycle length may have resulted in improved tolerability and may potentially be acceptable in a platinum-experienced patient population. The choice of platinum agent may also influence the outcome as cisplatin has a lower risk of myelosuppression, albeit with higher rates of nausea, vomiting, and nephrotoxicity (22). Ceralasertib has also been combined with the approved weekly dose of paclitaxel with acceptable tolerability at the monotherapy RP2D of ceralasertib (23).
Notably, our experience with ceralasertib is similar to that with olaparib, a PARP inhibitor, in combination with platinum, with which hematologic toxicity was also observed. Here, tolerable combination dose schedules were achieved by starting at lower doses of carboplatin (AUC3 or AUC4) and limiting the number of dosing days or dose of olaparib compared with the monotherapy dose and schedule (24, 25). Our study also highlights the potential challenges of combining platinum-based chemotherapy with other DDR inhibitors that have related but distinct mechanisms of action, such as CHK1 and DNA-PK inhibitors. Synergistic toxicity and efficacy should be expected and consideration of how to schedule the DDR agent to manage toxicity built into phase I studies to deliver optimal schedules for novel DDR–chemotherapy combinations.
This phase I trial also provided evidence of preliminary antitumor activity of the combination of ceralasertib and carboplatin in this population of patients with advanced tumors, with two confirmed and two unconfirmed RECIST v1.1 responses observed. Interestingly, both patients with confirmed RECIST v1.1 responses had low or absent ATM nuclear staining in their tumors, suggesting that the responses may be driven by a synthetic lethal interaction between ATR and ATM. Furthermore, one of these tumors also had low expression of SLFN11, a recently discovered biomarker that blocks stressed replication forks independently from ATR and whose loss of expression is associated with treatment resistance to DNA-damaging chemotherapy, DNA synthesis inhibitors, and PARP inhibitors (10). This tumor occurred in a patient with cervical cancer who had a response to prior platinum therapy approximately 4 years earlier. While this study found no definitive association between ATM and SLFN11 expression and antitumor response to the combination of ceralasertib and carboplatin in our limited series of patients tested, data from this study and others (19) support the further investigation of ATM defects and SLFN11 expression (10) as putative predictive biomarkers of response to ATR inhibitor–based therapies. This study explored ATM expression by IHC but a separate study (NCT04564027) of ceralasertib monotherapy is investigating ATM as a selection biomarker that includes investigation of ATM gene mutation and protein expression to determine which has greatest utility.
Although the combination of ceralasertib and carboplatin was not further investigated in expansion cohorts, the study provided the first insights into the toxicity, pharmacokinetic, and pharmacodynamic profiles for the combination, highlighted the challenges in combining an oral ATR inhibitor with DNA-damaging chemotherapy, and suggested a possible route to a tolerable dose/schedule combination.
Authors' Disclosures
T.A. Yap reports grants from Artios, Bayer, Constellation, Cyteir, Eli Lilly, Forbius, GlaxoSmithKline, Genentech, ImmuneSensor, Ipsen, Jounce, Karyopharm, Kyowa, Merck, Novartis, Pfizer, Ribon Therapeutics, Regeneron, Sanofi, Scholar Rock, Tesaro, and Vertex Pharmaceuticals; grants and personal fees from AstraZeneca, Clovis, EMD Serono, F-Star, Repare, and Seattle Genetics; and personal fees from Almac, Aduro, Atrin, Axiom, Bristol Myers Squibb, Calithera, Cybrexa, Guidepoint, Ignyta, I-Mab, Janssen, Roche, Rubius, Schrodinger, Varian, and Zai Labs outside the submitted work. M.G. Krebs reports personal fees from AstraZeneca during the conduct of the study; M.G. Krebs also reports personal fees from Roche, Bayer, Janssen, Seattle Genetics, BerGenBio, OM Pharma, and Immutep, as well as grants from Roche and BerGenBio outside the submitted work. S. Postel-Vinay reports other support from AstraZeneca during the conduct of the study. S. Postel-Vinay also reports other support from AbbVie, Adaptimmune, Adlai Nortye USA Inc, Aduro Biotech, Agios Pharmaceuticals, Amgen, Argen-X Bvba, Astex Pharmaceuticals, AstraZeneca Ab, Aveo, Basilea Pharmaceutica International Ltd, Bayer Healthcare Ag, Bbb Technologies Bv, Beigene, BicycleTx Ltd, Blueprint Medicines, Boehringer Ingelheim, Boston Pharmaceuticals, Bristol Myers Squibb, Ca, Celgene Corporation, Chugai Pharmaceutical Co, Clovis Oncology, Cullinan-Apollo, Curevac, Daiichi Sankyo, Debiopharm, Eisai, Eisai Limited, Eli Lilly, Exelixis, Faron Pharmaceuticals Ltd, Forma Tharapeutics, Gamamabs, Genentech, GlaxoSmithKline, H3 Biomedicine, F. Hoffmann-La Roche Ag, Imcheck Therapeutics, Innate Pharma, Institut De Recherche Pierre Fabre, Iris Servier, Iteos Belgium SA, Janssen Cilag, Janssen Research Foundation, Kura Oncology, Kyowa Kirin Pharm. Dev, Lilly France, Loxo Oncology, Lytix Biopharma As, Medimmune, Menarini Ricerche, Merck Sharp & Dohme Chibret, Merrimack Pharmaceuticals, Merus, Millennium Pharmaceuticals, Molecular Partners Ag, Nanobiotix, Nektar Therapeutics, Novartis Pharma, Octimet Oncology Nv, Oncoethix, Oncopeptides, Orion Pharma, Ose Pharma, Pfizer, Pharma Mar, Pierre Fabre, Medicament, Roche, Sanofi Aventis, Seattle Genetics, Sotio A.S, Syros Pharmaceuticals, Taiho Pharma, Tesaro, Turning Point Therapeutics, and Xencor; grants from AstraZeneca, BMS, Boehringer Ingelheim, GSK, INCA, Janssen Cilag, Merck, Novartis, Pfizer, Roche, Sanofi, and Merck KgA; and non-financial support from AstraZeneca, Bayer, BMS, Boehringer Ingelheim, GSK, Medimmune, Merck, NH TherAGuiX, Pfizer, and Roche outside the submitted work. A. El-Khouiery reports other support from AstraZeneca during the conduct of the study. A. El-Khouiery also reports personal fees from Agenus, ABL Bio, Bayer, BMS, Exelixis, EISAI, Pieris, Cytomx, Gilead, EMD Serono, Roche/Genentech, and Merck; grants and personal fees from AstraZeneca; and grants from Astex and Fulgent outside the submitted work. J.-C. Soria reports other support from AstraZeneca during the conduct of the study, as well as other support from Hookipa Pharmaceuticals, Gritstone Bio, and Relay Therapeutics outside the submitted work. J. Lopez reports grants from Roche-Genentech and Genmab, as well as grants and personal fees from Basilea outside the submitted work. A. Berges reports other support from AstraZeneca outside the submitted work. S.Y.A. Cheung reports other support from AstraZeneca outside the submitted work. I. Irurzun-Arana reports other support from AstraZeneca outside the submitted work, as well as employment and stock from AstraZeneca. G.N. Jones reports personal fees from AstraZeneca during the conduct of the study, as well as personal fees from AstraZeneca outside the submitted work. A. Lau reports other support from AstraZeneca outside the submitted work. P. Frewer reports other support from AstraZeneca during the conduct of the study, as well as other support from AstraZeneca outside the submitted work; P. Frewer is both an employee and shareholder in AstraZeneca. G. Clack reports other support from AstraZeneca during the conduct of the study, as well as other support from Carrick Therapeutics Ltd., Athenex Inc., Evgen Plc, and Ailse Oncology Ltd outside the submitted work; in addition, G. Clack has a patent for Docket No. 201026-US-PSP[2] pending. C. Stephens is an employee of AstraZeneca and owns shares in AstraZeneca. S.A. Smith reports other support from AstraZeneca outside the submitted work and is an employee and stockholder of AstraZeneca. E. Dean reports other support from AstraZeneca during the conduct of the study, as well as other support from AstraZeneca outside the submitted work; E. Dean is an employee and stockholder at AstraZeneca. S.J. Hollingsworth reports other support from AstraZeneca outside the submitted work. No disclosures were reported by the other authors.
Supplementary Material
Supplementary material
Acknowledgments
The study funder, AstraZeneca, provided organizational support, obtained data, performed the analyses, and had a role in data interpretation and writing of the article. All authors had access to study data, and the corresponding author had final responsibility for the decision to submit the article for publication. M.G. Krebs acknowledges support by the National Institute of Health Research (NIHR) Manchester Biomedical Research Centre, NIHR Manchester Clinical Research Facility at The Christie, and Manchester Experimental Cancer Medicine Centre (Manchester, UK). Medical writing assistance was provided by Martin Goulding, from Mudskipper Business Ltd, funded by AstraZeneca.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
This article is featured in Highlights of This Issue, p. 5151
Footnotes
Note: Supplementary data for this article are available at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/).
Authors' Contributions
T.A. Yap: Formal analysis, investigation, methodology, writing–original draft, writing–review and editing. M.G. Krebs: Investigation, methodology, writing–review and editing. S. Postel-Vinay: Investigation, writing–review and editing. A. El-Khouiery: Investigation, writing–review and editing. J.-C. Soria: Investigation, writing–review and editing. J. Lopez: Investigation, writing–review and editing. A. Berges: Formal analysis, writing–review and editing. S.Y.A. Cheung: Formal analysis, writing–review and editing. I. Irurzun-Arana: Formal analysis, writing–review and editing. A. Goldwin: Project administration, writing–review and editing. B. Felicetti: Project administration, writing–review and editing. G.N. Jones: Formal analysis, investigation, writing–review and editing. A. Lau: Formal analysis, investigation, writing–review and editing. P. Frewer: Formal analysis, methodology, writing–review and editing. A.J. Pierce: Formal analysis, investigation, writing–review and editing. G. Clack: Methodology, writing–review and editing. C. Stephens: Methodology, writing–review and editing. S.A. Smith: Formal analysis, methodology, writing–original draft, writing–review and editing. E. Dean: Formal analysis, writing–original draft, writing–review and editing. S.J. Hollingsworth: Formal analysis, methodology, writing–review and editing.
References
- 1. Yazinski SA, Zou L. Functions, regulation, and therapeutic implications of the ATR checkpoint pathway. Annu Rev Genet 2016;50:155–73. [DOI] [PubMed] [Google Scholar]
- 2. Forment JV, O'Connor MJ. Targeting the replication stress response in cancer. Pharmacol Ther 2018;188:155–67. [DOI] [PubMed] [Google Scholar]
- 3. Cimprich KA, Cortez D. ATR: an essential regulator of genome integrity. Nat Rev Mol Cell Biol 2008;9:616–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Foote KM, Nissink JWM, McGuire T, Turner P, Guichard S, Yates JWT, et al. Discovery and characterization of AZD6738, a potent inhibitor of ataxia telangiectasia mutated and Rad3 related (ATR) kinase with application as an anticancer agent. J Med Chem 2018;61:9889–907. [DOI] [PubMed] [Google Scholar]
- 5. Foote KM, Lau A, Nissink JW. Drugging ATR: progress in the development of specific inhibitors for the treatment of cancer. Future Med Chem 2015;7:873–91. [DOI] [PubMed] [Google Scholar]
- 6. Lau AY, Yates J, Wilson Z, Young LA, Hughes AM, Berges A, et al. ATR inhibitor AZD6738 as monotherapy and in combination with olaparib or chemotherapy: defining pre-clinical dose-schedules and efficacy modelling [abstract]. In:Proceedings of the American Association for Cancer Research Annual Meeting 2017; 2017. Apr 1–5; Washington, DC. Philadelphia (PA): AACR; Cancer Res 2017;77(13 Suppl):Abstract nr 2494. [Google Scholar]
- 7. Vendetti FP, Lau A, Schamus S, Conrads TP, O'Connor MJ, Bakkenist CJ, et al. The orally active and bioavailable ATR kinase inhibitor AZD6738 potentiates the anti-tumor effects of cisplatin to resolve ATM-deficient non-small cell lung cancer in vivo. Oncotarget 2015;6:44289–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Weber AM, Ryan AJ. ATM and ATR as therapeutic targets in cancer. Pharmacol Ther 2015;149:124–38. [DOI] [PubMed] [Google Scholar]
- 9. Jones GN, Rooney C, Griffin N, Roudier M, Young LA, Garcia-Trinidad A, et al. pRAD50: a novel and clinically applicable pharmacodynamic biomarker of both ATM and ATR inhibition identified using mass spectrometry and immunohistochemistry. Br J Cancer 2018;119:1233–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Winkler C, Armenia J, Jones GN, Tobalina L, Sale MJ, Petreus T, et al. SLFN11 informs on standard of care and novel treatments in a wide range of cancer models. Br J Cancer 2021;124:951–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Liu X, Pan CG, Luo ZQ. High expression of NFAT2 contributes to carboplatin resistance in lung cancer. Exp Mol Pathol 2019;110:104290. [DOI] [PubMed] [Google Scholar]
- 12. Griesinger F, Korol EE, Kayaniyil S, Varol N, Ebner T, Goring SM. Efficacy and safety of first-line carboplatin-versus cisplatin-based chemotherapy for non-small cell lung cancer: a meta-analysis. Lung Cancer 2019;135:196–204. [DOI] [PubMed] [Google Scholar]
- 13. Hartford C, Volchenboum SL, Cohn SL. 3 + 3 not equal to (Rolling) 6. J Clin Oncol 2008;26:170–1. [DOI] [PubMed] [Google Scholar]
- 14. Villaruz LC, Jones H, Dacic S, Abberbock S, Kurland BF, Stabile LP, et al. ATM protein is deficient in over 40% of lung adenocarcinomas. Oncotarget 2016;7:57714–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Murai J, Feng Y, Yu GK, Ru Y, Tang SW, Shen Y, et al. Resistance to PARP inhibitors by SLFN11 inactivation can be overcome by ATR inhibition. Oncotarget 2016;7:76534–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Terranova N, Jansen M, Falk M, Hendriks BS. Population pharmacokinetics of ATR inhibitor berzosertib in Phase I studies for different cancer types. Cancer Chemother Pharmacol 2021;87:185–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dillon M, Guevara J, Mohammed K, Smith SA, Dean E, McLellan L, et al. 1915 - A phase I study of ATR inhibitor, AZD6738, as monotherapy in advanced solid tumours (PATRIOT part A, B). Ann Oncol 2019;30:159–93.30596815 [Google Scholar]
- 18. Pollard J, Reaper P, Peek A, Hughes S, Gladwell S, Jones J, et al. Defining optimal dose schedules for ATR inhibitors in combination with DNA damaging drugs: informing clinical studies of VX-970, the first-in-class ATR inhibitor [abstract]. In:Proceedings of the 107th Annual Meeting of the American Association for Cancer Research; 2016. Apr 16–20; New Orleans, LA. Philadelphia (PA): AACR; Cancer Res 2016;76(14 Suppl):Abstract nr 3717. [Google Scholar]
- 19. Yap TA, O'Carrigan B, Penney MS, Lim JS, Brown JS, de Miguel Luken MJ, et al. Phase I trial of first-in-class ATR inhibitor M6620 (VX-970) as monotherapy or in combination with carboplatin in patients with advanced solid tumors. J Clin Oncol 2020;38:3195–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Thomas A, Takahashi N, Rajapakse VN, Zhang X, Sun Y, Ceribelli M, et al. Therapeutic targeting of ATR yields durable regressions in small cell lung cancers with high replication stress. Cancer Cell 2021;39:566–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Krebs MG, Lopez J, El-Khoueiry A, Bang Y-J, Postel-Vinay S, Abida W, et al. Phase I study of AZD6738, an inhibitor of ataxia telangiectasia Rad3-related (ATR), in combination with olaparib or durvalumab in patients (pts) with advanced solid cancers [abstract]. In:Proceedings of the American Association for Cancer Research Annual Meeting 2018; 2018. Apr 14–18; Chicago, IL. Philadelphia (PA): AACR; Cancer Res 2018;78(13 Suppl):Abstract nr CT026. [Google Scholar]
- 22. de Castria TB, da Silva EM, Gois AF, Riera R. Cisplatin versus carboplatin in combination with third-generation drugs for advanced non-small cell lung cancer. Cochrane Database Syst Rev 2013:CD009256. [DOI] [PubMed] [Google Scholar]
- 23. Kim ST, Smith SA, Mortimer P, Loembé AB, Cho H, Kim KM, et al. Phase I study of ceralasertib (AZD6738), a novel DNA damage repair agent, in combination with weekly paclitaxel in refractory cancer. Clin Cancer Res 2021. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lampert EJ, Hays JL, Kohn EC, Annunziata CM, Minasian L, Yu M, et al. Phase I/Ib study of olaparib and carboplatin in heavily pretreated recurrent high-grade serous ovarian cancer at low genetic risk. Oncotarget 2019;10:2855–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. van der Noll R, Jager A, Ang JE, Marchetti S, Mergui-Roelvink MWJ, Lolkema MP, et al. Phase I study of continuous olaparib capsule dosing in combination with carboplatin and/or paclitaxel (part 1). Invest New Drugs 2020;38:1117–28. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material