

Abstract
Purpose:
There is still no standard nonsurgical regimen for conventional chondrosarcoma (CHS). We aimed to identify whether any CHSs have a favored microenvironment for immunotherapy via multidimensional evaluation of the immunologic characteristics of this tumor.
Experimental Design:
We obtained 98 newly-diagnosed CHS fresh tumors from several institutions and performed comprehensive analysis of data from CyTOF, whole-exome sequencing, and flow cytometry in 22 cases. Clinical data from immunotherapy responders and nonresponders were compared to explore possible biomarkers of immunotherapy response. Mechanism studies were conducted to interpret the biomarker phenotype.
Results:
Based on the integrated data of single-cell CyTOF and flow cytometry, the CHS immune-microenvironment phenotypes were classified into three groups: subtype I, the “granulocytic–myeloid-derived suppressor cell (G-MDSC) dominant” cluster, with high number of HLA-DR− CD14− myeloid cells; subtype II, the “immune exhausted” cluster, with high exhausted T-cell and dendritic-cell infiltration; and subtype III, the “immune desert” cluster, with few immune cells. Immune cell–rich subtypes (subtype I and II) were characterized by IDH mutation, pathologic high grade, and peritumoral edema, while subtype I cases were exclusively featured by myxoid transformation. In clinical practice involving 12 individuals who received PD-1 antibody immunotherapy, all of the 3 cases with controlled diseases were retrospectively classified as subtype II. In mechanism, IDH mutation significantly elevated chemokine levels and immune-cell infiltration in immune-inactivated tumors.
Conclusions:
This study is the first to provide immune characterization of CHS, representing a major step to precise immunotherapy against this malignancy. Immunotherapy is promising for the “immune exhausted” subtype of patients with CHS.
Translational Relevance.
This study provides an immune classification system based on multi-dimensional evaluation in fresh chondrosarcoma (CHS) tumor tissues. Clinical treatment by immunotherapy may be promising in certain immune subtypes of conventional CHS.
Introduction
Conventional chondrosarcoma (CHS), representing about 90% of all chondrosarcomas, is a rare malignancy that is notoriously difficult to treat with nonsurgical regimens (1, 2). It may arise from a benign osteochondroma or enchondroma, although represents the minority of cases. In addition, in recurrent cases, the tumor cells often exhibit transformation to a higher histologic grade (3). The only treatment with efficacy is wide en bloc surgical resection for favorable cases. Although numerous efforts have been made, there are still no effective chemotherapy options for CHS, and tumors are also poorly responsive to radiotherapy. There is a great need for nonsurgical therapeutics to be developed, particular for patients with unresectable, metastatic, or recurrent disease.
Immunotherapy has achieved success in several types of malignancies (4–6). Regarding musculoskeletal sarcomas, immunotherapy is still in its infancy. The approval of an immune stimulator, mifamurtide, by the European Medicine Agency against osteosarcoma and the use of T-cell adoptive transfer against synovial sarcoma (7) represent two early, encouraging attempts at developing immunotherapy against sarcomas. However, there is not yet a sound basis for employing immunotherapy in the treatment of CHS. It remains to be determined whether immunotherapy will be ideally suited to some particular subtypes of this malignancy.
There is an ongoing effort at this time to define predictors and biomarkers for the effectiveness of immunotherapy in cancer. Most recently, these efforts have focused on evaluating the expression of programmed death-ligand 1 (PD-L1; refs. 8, 9), tumor mutational burden (TMB; ref. 10), microsatellite instability (MSI; ref. 11), and Epstein–Barr virus infection (12), etc. To date, information pertaining to the immune-related features of CHS is quite sparse. Although previous studies have provided some evidence on some commonly evaluated immune factors such as PD-L1 expression and TMB in CHS (13, 14), integrated multidimensional analyses involving genetic factors, cytology, pathology, and clinical parameters are lacking. Comprehensive elucidation of the immunologic characteristics of the tumor and microenvironment could improve the identification of patients who would benefit most from immunotherapeutics. Thus, adequate immunotherapy can be developed and applied for this refractory malignancy.
This study aimed to identify immune microenvironment phenotypes of CHS at the single-cell level, and also to determine genetic events that drive the phenotypes. Based on multi-omics data of 98 CHS cases from multiple institutions, we classified the samples into three immune-microenvironment subtypes with different genetic events and clinical features. Response to checkpoint-inhibitor immunotherapy of CHS was observed in the “immune exhausted” subtypes. The potential mechanism was investigated and an association between genetic variation and chemokines was established.
Materials and Methods
Ethics approval
The protocol for use of human subjects was approved by the Institutional Review Board, and written informed consents were obtained from the patients or their legal guardians. The studies were conducted in accordance with the Declaration of Helsinki.
Patients with cartilaginous tumors and fresh tumor tissues processing
Patients diagnosed with cartilaginous tumor by radiological evaluation between February 2017 and June 2019 from three of the partner members of Hangzhou Sarcoma MDT Network were recruited in the study. In total, 98 conventional CHS cases, 4 enchondromas of phalanges with pathologic or incomplete fracture, and 4 individuals with chondroblastoma diagnosed by further pathologic examination were included for further analyses. Final validation of the histologic diagnosis and grading was performed by the pathologist (Y. Fu) of The Second Affiliated Hospital of Zhejiang University School of Medicine, Hangzhou. Regarding the tissue processing, fresh tumor tissues were harvested in 15 minutes from curettage or en bloc resection, washed with saline, and soaked in cold PBS. All surgeries were performed by experienced specialists. Samples were collected from multiple sites of bulk tumor, cut into small pieces by razors, and mixed. To prepare for fresh tissue CyTOF assay, mixed samples were temporally preserved in tissue storage solution (#130–100–008, Miltenyi Biotec) and shipped by cold chain to PLTTECH institution (Hangzhou, China) on the same day for CyTOF assay, to whom the information of the patient was blinded. Some tissues were frozen in liquid nitrogen for further whole-exome sequencing (WES), qRT-PCR, and ELISA. Normal muscle tissue from en bloc–resected tumor was harvested for paired WES evaluation.
MRI image reading
MRI was performed on a 1.5T system. The status of peritumoral edema was recognized on T2-weighted as infiltrating high-signal intensity change in the marrow outside the tumor margins seen on T1-weighted images. One radiologist (X. Zhu) and another experienced orthopaedic surgeon (P. Lin) analyzed the images separately and were blinded to other information. For categorizing cases, the extensive peritumoral edema signal in the images of chondroblastoma was set as the reference for positive cases. Thus, individuals with de facto mild edema signal were also categorized as negative cases.
Conventional multicolor flow cytometry and tumor-infiltrated immune-cell sorting
Mixed disaggregated pieces of tumor were enzymatically digested at 37°C in RPMI 1640 with collagenase IV (2 mg/mL, Sigma), DNase (0.1 mg/mL, Sigma), and hyaluronidase (0.1 mg/mL, Sigma). Cell suspensions were passed through 100-μm filters to remove aggregates. Cells were washed with staining buffer (#420201, Biolegend) and stained with conjugated antibodies. Live cells were identified using LIVE/DEAD staining (#L23105, ThermoFisher). Flow cytometry analyses were performed using BD LSRFortessa. Conjugated antibodies were purchased from Biolegend: CD45 (clone 2D1), CD19 (clone 4G7), CD56 (clone 5.1H11), CD11b (clone ICRF44), CD16 (clone 3G8), CD66b (clone G10F5), HLA-DR (clone L243), CD14 (clone 63D3), and CD15 (clone HI98). Five × 106 total events were collected from each tumor sample to analyze a sufficient number of intracellular lymphocytes. Flow data were analyzed by FlowJo V10. Gating strategies were determined by fluorescence minus one (FMO). Tumor-infiltrated dendritic cells (TIDC) were sorted and cultured in complete 1640 medium without further expansion.
CyTOF sample preparation
Cell suspensions were collected as described above. Antibodies were either purchased preconjugated from Fluidigm (DVS Sciences) or purchased purified and conjugated in-house using MaxPar X8 Polymer Kits (Fluidigm) according to the manufacturer's instructions. Mass cytometry antibodies used are shown in Supplementary Table S2. Three × 106 cells for each sample were washed with protein-free PBS, stained with 1 μmol/L cisplatin for 5 minutes at room temperature, then stained for cell-surface markers in staining media (PBS containing 0.5% BSA and 0.02% NaN3) for 30 minutes at 4°C. Cells were fixed and stained with DNA Intercalator-Ir overnight. Intracellular staining was done using fixation/permeabilization reagents from eBioscience (#00–5521–00).
CyTOF data acquisition and analysis
CyTOF data acquisition and analyses were performed by PLTTECH Institution. In brief, cells were washed twice with deionized water prior to adding EQ normalization beads containing Ce140, Eu151, Eu153, Ho165, and Lu175 (Fluidigm) and acquiring on a Helios instrument. After normalizing and randomizing values near 0 using the Helios software, FCS files were downloaded for further biological information analysis.
Mass cytometry data has been firstly debarcoded using a doublet filtering scheme with mass-tagged barcodes, and then manually gated to retain live, singlet, valid-immune cells. Data generated from different batches have been normalized through bead normalization method. In order to get the accurate immune-subset information, we have run phenograph algorithm to all samples. All cell events in each individual sample have been pooled and included in this analysis.
WES
DNA was extracted from liquid nitrogen–preserved tissues using the extraction Kit (Qiagen) following the manufacturer's instructions. Sequencing and library construction were performed using the Agilent SureSelect Human All Exon Kit (Agilent Technologies). After cluster generation, the DNA libraries were sequenced on an Illumina Hiseq platform (Illumina) by OrigiMed, Shanghai. TMB was calculated based on the number of mutated genes.
ELISA
Tumor tissues were homogenized by liquid nitrogen for RIPA protein extraction. Bicinchoninic acid (BCA) assay was utilized to evaluate the concentration of total protein. For tissue cytokine quantification, 60 μg total protein from each sample was added to each well in the 96-well plates. Cell-culture supernatants were also used for cytokine quantification if needed. Cytokine levels were evaluated by Duoset ELISA (#DY206, #DY210, #DY217B, #DY1164, #DY350, #DY350, #DY279, #DY208, #DY266, #DY392, #DY317, #DY278, #DY281, and #DY365; R&D Systems) following the manufacturer's instructions.
D-2-Hydroxyglutarate concentration evaluation
D-2-Hydroxyglutarate Assay Kit (#ab211070, Abcam) was utilized to evaluate the concentration of D-2-Hydroxyglutarate (D-2-HG) in tissue lysates. The levels of D-2-HG were recorded following the manufacturer's instruction.
Hematoxylin and eosin, and IHC staining
Samples were fixed in 10% buffered methanol and embedded in paraffin for hematoxylin and eosin (H&E) staining or IHC. H&E staining was performed following conventional protocols. In IHC assays, DAKO autostainer (DAKO) and DAB chromogen was used for all antigen staining. Antibodies used include anti-CD8a (1:400, Jinqiao Biotech), anti–PD-L1 (clone 28–8, 1:500, #ab205921; Abcam), anti-CD69 (clone EPR21814, 1/250, #ab233396; Abcam), and anti-CD11c (1:200, #ab52632; Abcam).
Cell lines
Mouse liver cancer Hepa 1–6 cells were newly purchased from the Cell Collection of Chinese Academy of Sciences. Cell line was authenticated by the Cell Collection via DNA short tandem repeat (STR) genotyping. Mouse mesothelioma cell line AE17 was a kind gift from Dr. Agnes Kane from Brown University, and cells were authenticated by its unique morphology under microscope as indicated in previous works (15).
Animal studies and in vivo imaging
Immunodeficient nonobese diabetic (NOD)–PrkdcscidIl2rgem1/Smoc M-NSG mice (Shanghai Model Organisms), OT-I transgenic mice (Model Animal Center of Nanjing University), and C57BL/6 mice were handled in a gnotobiotic animal-maintaining facility. Animal studies were approved by the Institutional Animal Care and Use Committee. Mouse homogenous tumors were initiated with 5 × 106 Hepa 1–6 cells or 1 × 106 AE17 cells per mouse administered intraperitoneally. For animals receiving adoptive immune-cell therapy, mice received cyclophosphamide (200 ug) and fludarabine (100 ug) before immune-cell transfusion. OT-I T cells were isolated from splenocytes and delivered intravenously in PBS. Administrations of recombinant IL2 (45,000 IU) were given subcutaneously at the same time as the T cells were injected, during the next 2 consecutive days. In mice receiving CXCL12 or CXCL9/10 blockade, intraperitoneally-implanted osmotic minipumps (Alzet Co.) containing AMD3100 or AMG487 were utilized for continuous blockade as previously described (16). IVIS Spectrum Animal Imaging System (PerkinElmer) was utilized to evaluate the progression of tumor with presence of luciferin (#1–360222, Regis Technologies) for tumor cells transfected with luciferase. For untransfected tumors, the volumes were measured by caliper and calculated using the formula V = a × b2/2, where a is the maximal diameter of the tumor, and b is the minimal diameter.
Statistical analysis and data visualization
GraphPad Prism 7, and R program 3.5.2 with relevant packages were applied for statistical analysis and visualization. R package tidyr 0.8.3, data.table 1.12.0, and plyr 1.8.4 were employed for data cleaning. Oncoplot was depicted using ComplexHeatmap 1.20.0. CyTOF heatmap was visualized using pheatmap 1.0.12.
Unless otherwise specifically indicated in the figure legends, Student t test and Wilcoxon test were applied to compare continuous variables and ordered categorical variables between groups, and the bar plots were created using ggplot2 3.1.0 and ggpubr 0.2 packages. Correlation matrices were constructed with Pearson correlation test, where the clinical status and single-nucleotide variant (SNV)/copy-number variation (CNV) events were converted into binomial distribution, such as SNV/CNV events were classified as 0 for no SNV and CNV, and 1 for SNV or CNV or both, and tumor stage was classified as 0 for stage I, and 1 for stage II or stage III. Data were visualized using corrplot 0.8.4. In the presented study, all the tests were two-sided, and P < 0.05 was regarded as statistical significance.
Results
CyTOF and multicolor flow cytometry revealed three immune subtypes of CHS
A multi-omics scheme was performed to establish the immune subtypes of CHS and to find out possible biomarkers for the subtypes (Fig. 1A). To clarify the phenotypes of immune cells in chondrosarcomas, we performed mass cytometry (i.e., CyTOF) and multicolor flow cytometry analyses of 22 fresh tumor samples from patients with WHO grade I (n = 7), grade II (n = 14), and grade III (n = 1) tumors. One case of grade I tumor was not included for further evaluation because of extremely low number of disaggregated live cells. Eleven CD45+ leukocyte subgroups (Cluster 7–9, 24–31) were identified including T cells, CD11c+ conventional dendritic cells (cDC), and some other myeloid cells (Fig. 1B and C). However, macrophages, which were recognized by CD11b+ CD68hi HLA-DR+ CD11c−/lo phenotype, and monocytes were not detectable, indicating that there were very few macrophages and monocytes in the CHS microenvironment (17, 18).
Figure 1.
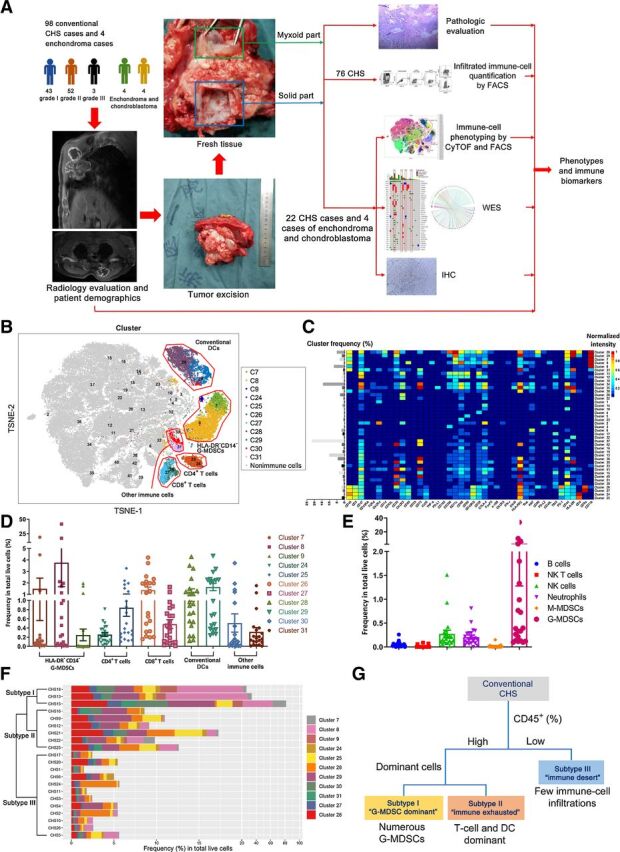
In-depth CyTOF and multicolor flow cytometry identification and quantification of immune-cell subgroups classified CHSs into 3 immunophenotypic subgroups. A, Research scheme. B, t-SNE map illustrating each identified cell subgroup in color by cluster. A total of 42 frequent cell subgroups including both immune cells and nonimmune cells (CD45 negative) were identified. C, Heatmap showing normalized expression of selected markers for the 42 identified cell phenotypes. Clusters were grouped by expression profile. D and E, Column figures showing the frequencies of 11 dominant immune-cell subtypes illustrated by CyTOF (D) and 6 other immune-cell subgroups identified by multicolor flow cytometry (E) in total live cells. Dots represent data from each case (n = 21). F, Hierarchical k-means clustering by the immune-cell signatures of each patient with CHS. G, Scheme showing the classification of CHS according to the immune-cell composition.
A multicolor flow cytometry panel provided additional data on other immune cells, including B cells, natural killer (NK) cells, NK T cells, neutrophils, monocytic myeloid-derived suppressor cells (M-MDSC), and granulocytic MDSCs (G-MDSC; Supplementary Fig. S1B). B cells, NK cells, NK T cells, neutrophils, and M-MDSCs were scarcely found in CHS. However, there were numerous G-MDSCs in 3 samples (Fig. 1E). These cells were also seen in the CyTOF panel as HLA-DR− CD14− myeloid cells (Fig. 1D, cluster 7–9). To be more specific, these 3 samples shared the same clinical patterns (see below).
According to the phenotyping of immune cells, the 21 included cases could be classified into 3 groups by hierarchical k-means clustering algorithm (Fig. 1F). There were 3, 6, and 12 individuals in subtypes I, II, and III, respectively. Further analyses revealed the characteristics of each subtype. Subtype III cases were “cold” tumors. They were poorly infiltrated by immune cells, featured by an “immune desert” status. For immune cell–rich subtypes I and II, a substantial number of G-MDSCs were discovered in subtype I, while dendritic cells and T cells were dominant in subtype II (Fig. 1G). Further analyses in subtype II cases revealed that in the CD8+ T cells, the majority belonged to cluster 26, i.e., CD86+ CD39+ CTLA-4+ TIM-3+ PD-1hi “deeply exhausted” or “post-activated” Tex cells (Fig. 1C and D; refs. 19–22), indicating an exhausted immune microenvironment.
The possible correlations among different immune-cell populations were estimated by gating CD45+ cells. In addition to the 11 dominant phenotypes indicated above, 11 less frequent kinds of leukocytes were further identified (Supplementary Fig. S2A–S2C). Five CD3− CD11b− populations with minimal frequencies (clusters 1, 5, 6, 9, and 10, recognized as neither T-lymphoid nor myeloid cells) were excluded from further analyses. The correlations among populations were tested by linear regression analysis and illustrated (Supplementary Fig. S2E and S2F).
Comparison of immune-cell atlas among cartilaginous tumors revealed unique immune-cell clusters in CHS
To compare and contrast the immune characteristics of CHS with benign cartilaginous tumors, we performed a similar analysis for enchondroma and chondroblastoma (n = 4 of each). Enchondromas, in general, were poorly infiltrated by immune cells, similar to immunophenotypic subtype III CHSs. While all of the immune-cell subtypes in enchondromas were found in CHSs, the latter had more T cells and G-MDSCs (Fig. 2A). The comparison of CHS and chondroblastoma revealed different immune-cell profiles (Fig. 2B). Both tumors were infiltrated by T cells, but there were notable differences in the proportion of various myeloid cells in the 2 tumors.
Figure 2.
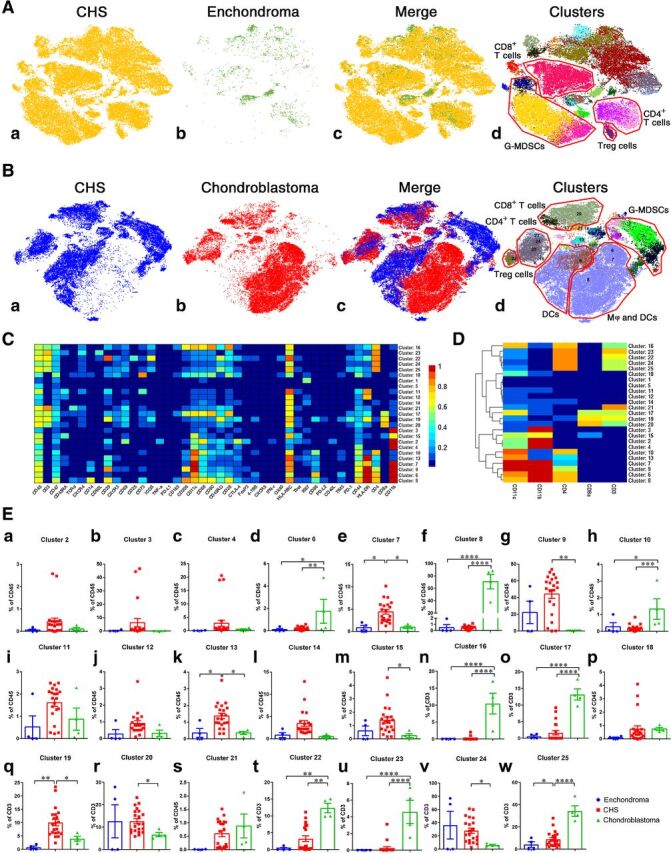
Comparison of intratumoral immune cells among chondrosarcoma, enchondroma, and chondroblastoma. A, t-SNE map showing identified immune-cell subgroups in 21 cases of CHS and 4 cases of enchondromas. The number of intratumoral CD45+ immune cells in enchondromas was very small compared with chondrosarcoma. B, t-SNE map displaying immune-cell phenotypes in CHS and 4 cases of chondroblastomas. Macrophages were almost exclusively seen in chondroblastomas. C and D, Normalized expression of markers for 25 identified immune-cell subgroups from 3 kinds of cartilaginous neoplasms. E, Comparison of clusters among the 3 types of cartilaginous tumors by Bonferroni multiple comparison test. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
A total of 25 kinds of CD45+ immune cells in the 3 cartilaginous tumors were identified (Fig. 2C and D). In HLA-DR+ myeloid cells (clusters 6–10 and 13; Fig. 2D), M1 macrophages (clusters 6 and 10), and M2 macrophages (cluster 8) appeared only in chondroblastomas (Fig. 2B, 2Ed, 2Ef, 2Eh). G-MDSCs (clusters 2–4), however, were only seen in CHSs (Fig. 2B, Ea–c). Regarding CD8+ T-lymphocytes, the activated subtype (cluster 17) were significantly more in chondroblastomas (Fig. 2Eo), while the majority subtype of CD8+ T cells in CHSs were exhausted phenotypes cluster 19 (Fig. 2Ep). Collectively, the “G-MDSC dominant” subtype I and “immune exhausted” subtype II were exclusively seen in CHSs among cartilaginous tumors.
Isocitrate dehydrogenase mutation was associated with immune response and chemokine levels in CHS
To find out possible biomarkers for the immune subtypes of CHS, WES was performed in the 22 CyTOF-evaluated CHS samples. The patient demographics were listed in Supplementary Table S1. The oncoplot of the mutation status of the frequently altered genes in 18 patients is shown in Fig. 3A. The data of 4 patients were not included in the figure as they carried altered genes that only occurred once in the cohort. A mutation in the isocitrate dehydrogenase (IDH) genes IDH1 and IDH2 was detected in 8 and 3 cases, respectively; IDH1/2 mutations had the highest frequency (50.0%) in the series of 22 CHS cases compared with other mutations (Fig. 3A). Reactome enrichment analysis for the top 13 mutated genes and the network of enriched terms in different signaling pathways showed no association between IDH1/2 mutation and signaling pathways (Supplementary Fig. S3).
Figure 3.
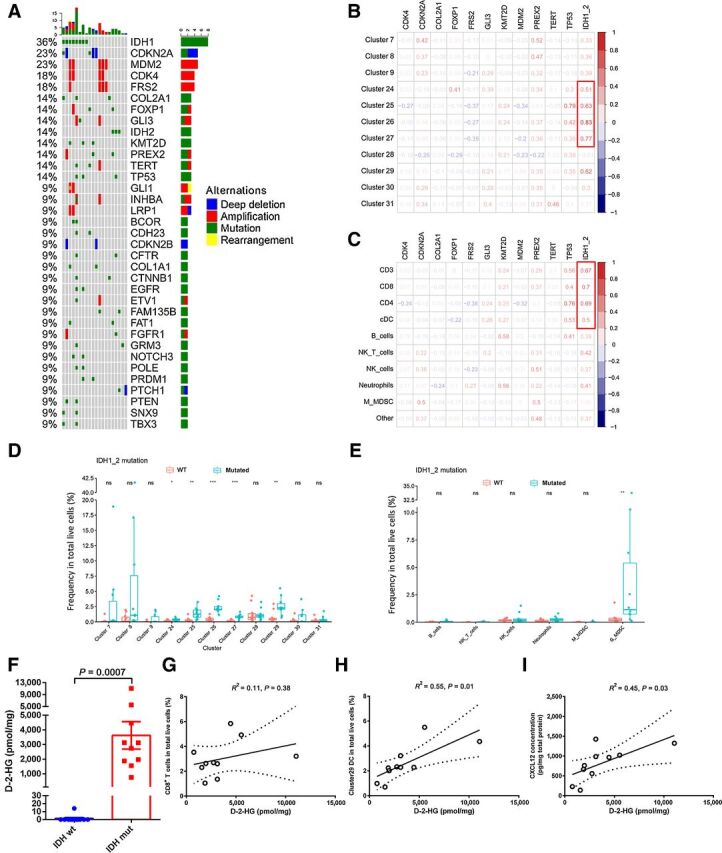
Genetic mutation atlas of CHS and its association with immune cells. t-SNE plots from Fig. 1A were referenced by the data in this figure. A, Oncoplot of mutation status of frequently altered genes in 18 of 22 CHS specimens available for WES. The bar chart on the top indicated the total mutation count per patient; the bar chart to the right indicated the total mutation count per gene. Blue, red, green, and yellow squares represent deletion, amplification, SNV, and rearrangement, respectively. B and C, Pearson correlation evaluation for associations between genetic mutations and immune-cell subgroups. Red figures indicated positive correlation, while blue figures represented negative correlation. Numbers indicated correlation coefficient. D and E, Comparisons of immune-cell proportions between IDH1/2 mutated and WT cases. Immune-cell subgroups were identified by CyTOF (D) and multicolor flow cytometry (E). Wilcoxon signed rank test was used to evaluate potential statistical significance between groups. F, Comparison of D-2-HG concentration in IDH1/2 wildtype (wt) or mutated (mut) CHS tissues. Student t test was utilized to evaluate the statistical difference. G–I, The correlations between the concentration of D-2-HG and immune-cell proportions or CXCL12 were examined by linear regression. R2 was calculated by goodness of fit measurement. P < 0.05 was determined as significantly correlated. Dotted lines indicated 95% confidence interval (CI) of the best-fit line. *, P < 0.05; **, P < 0.01; ***, P < 0.001. ns, no statistical significance.
Since certain genetic aberrations could affect the function of immune cells, we explored the potential association between mutations and the immune-cell subgroups identified by CyTOF for the 21 CHS cases that underwent both CyTOF/multicolor flow cytometry and WES (Supplementary Fig. S4). Cell clusters were defined as shown in Figs. 1B–D. Strong correlations between IDH1/2 mutation and immune-cell subpopulations were discovered. IDH1/2 mutation was associated with a higher number of multiple immune-cell subtypes including clusters 24–27 and 29 (Fig. 3B). Integrated evaluation also revealed positive associations among IDH1/2 mutation, T cells, and antigen-presenting cDCs (Fig. 3C). By comparing the number of intratumoral immune cells between CHS cases carrying mutated or wild-type (WT) IDH1/2, significant differences were found in clusters 24–27 (T cells), cluster 29 [dendritic cells (DC)], and G-MDSCs (Fig. 3D and E). These data indicated that IDH1/2-mutated chondrosarcomas had significantly more immune cells than chondrosarcomas with wild-type IDH1/2.
We tried to determine why tumors with mutated IDH1/2 had larger number of intratumoral immune cells. Evaluation of chemokines revealed a more inflamed microenvironment in IDH1/2-mutated cases, with a significantly higher level of CXCL9 and CXCL12, which might account for the attraction of CXCR3+ and CXCR4+ immune cells in these cases (Supplementary Fig. S5). We also tried to find some clues from the function of mutated IDH1/2. Gain-of-function IDH1/2 mutation has been reported to induce the accumulation of an oncometabolite, D-2-HG, which drives malignant transformation via epigenetic mechanisms (23–25). We evaluated the level of tumor D-2-HG in CHS using an enzymatic method and found high D-2-HG concentrations exclusively in IDH1/2-mutated cases (Fig. 3F). We further evaluated the association of the D-2-HG level with immune-cell subgroups identified by CyTOF. The number of CD8+ T cells, including both clusters 26 and 27, was not directly associated with the level of D-2-HG (Fig. 3G). However, cDCs, which play vital roles in antigen presentation for immune recognition, were positively correlated with the level of D-2-HG (Fig. 3H). In addition, a positive correlation between D-2-HG concentration and chemokine CXCL12 level was also revealed (Fig. 3I).
Biomarkers from genetic events and clinical features were established to identify CHS immune phenotypes
Clinical features of cases were also collected to see if any of them could help predict the immune status of CHS. Three characteristics, tumor grade, peritumoral edema in MRI, and myxoid transformation, were statistically associated with higher immune-cell infiltration (Fig. 4A and B).
Figure 4.
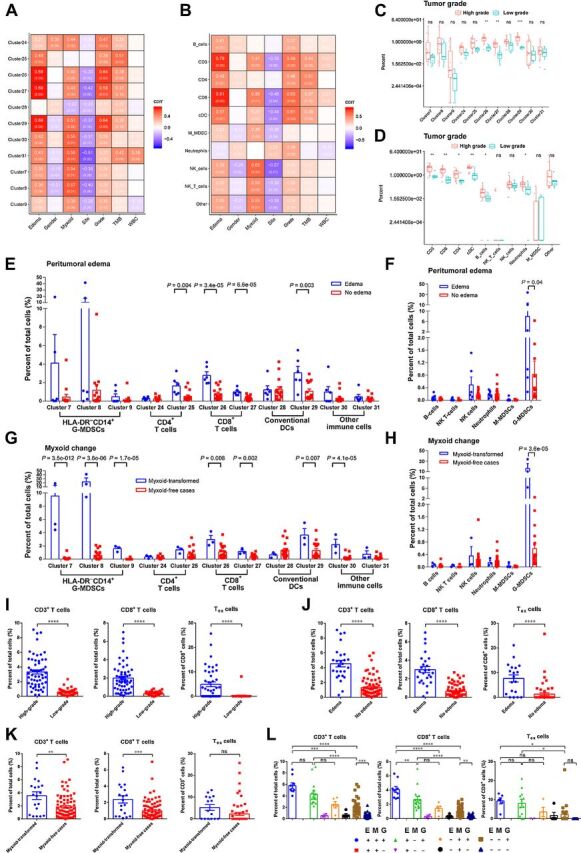
The correlations between clinical features and immune cells. t-SNE plots from Fig. 1B were referenced by the data in this figure. A and B, Pearson correlation assays for associations between clinical features and immune-cell subgroups. Figures in red background indicated positive correlation, while figures in purple background represented negative correlation. Figures in brackets were P values. C and D, Comparisons of immune-cell proportions between high-grade and low-grade cases. Immune-cell subgroups were identified by CyTOF (C) and multicolor flow cytometry (D). Wilcoxon signed rank test was used to evaluate potential statistical difference between 2 groups. E and F, Comparisons of immune-cell proportions between cases with or without peritumoral edema. Wilcoxon signed rank test was used to evaluate potential statistical difference between 2 groups. G and H, Comparisons of immune-cell proportions between cases with or without myxoid change. High proportion of G-MDSCs were only found in myxoid-transformed cases. Wilcoxon signed rank test was utilized to assess potential statistical significance between groups. I–K, Comparison of CD3+ T cells, CD8+ T cells, and Tex cells between groups divided by tumor grade (I), peritumoral edema (J), and myxoid status (K). Tex cells were defined as PD-1+ TIM-3+ T cells in multicolor flow cytometry from 77 cases. The Student t test was used to compare the means of the groups. L, CHS cases were divided into 8 groups according to tumor grade, peritumoral edema, and myxoid status. Comparison of immune-cell proportions between groups was accomplished by one-way ANOVA with Holm–Sidak multiple comparisons test. E, edema condition; G, tumor grade; M, myxoid status; +, positive or high tumor grade; –, negative or low tumor grade. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
High-grade (WHO grade II and III) tumors were associated with a larger number of intratumoral T cells and cDCs compared with low-grade (WHO grade I) tumors (Fig. 4A and B). High-grade CHS had significantly more multiple immune-cell subtypes, especially CD8+ T cells and cDCs (Fig. 4C and D). Similar results were revealed when radiological events were included. In cases with extensive peritumoral edema by MRI evaluation, there were a significantly larger number of CD8+ T cells (clusters 26 and 27), CD4+ T cells (cluster 25), and cDCs (cluster 29; Fig. 4E and F).
Focal myxoid change is not rare in CHS, but what this signifies is unclear. The solid part and myxoid part of a tumor could be quite different in histological examination (Supplementary Fig. S6A). CyTOF revealed that there were barely any immune cells in the myxoid part except for a minor group of myeloid cells, while T cells and most of the other myeloid cells were exclusively located in the solid part of the tumor (Supplementary Fig. S6B and S6C). We further compared the immune status of the solid parts of myxoid-transformed tumors with myxoid-free cases (Fig. 4G and H). A substantial number of HLA-DR− CD14− myeloid cells (clusters 7–9), identified as G-MDSCs, were seen in the 3 cases with myxoid transformation but rarely elsewhere.
Given the preliminary finding that tumor grade, peritumoral edema, and myxoid change may predict immune status in a small CHS cohort, we asked whether these three factors might correctly predict the immune status for a larger cohort. We therefore performed additional multicolor cytometry on 77 CHS cases and assessed whether the three clinical factors correlated with immune-cell infiltration. As shown in Fig. 4I–L, cases with higher grade, peritumoral edema, or myxoid transformation had more T cells and PD-1+ TIM-3+ Tex cells. We divided the 98-case cohort into 8 groups according to their clinical features, and discovered that cases with all three features had the largest number of intratumoral immune cells (Fig. 4L).
A representative clinical case was shown in Fig. 5A. Case number CHS91 was diagnosed with Ollier's disease and suffered from IDH2-mutated CHS with two parts. The acetabular part was identified as WHO grade II (high-grade) CHS with extensive peritumoral edema, while the femoral part was WHO grade I (low-grade) CHS without myxoid transformation or edema (Fig. 5A). Evaluation by IHC (Fig. 5Ac-j) and flow cytometry (Fig. 5B) in both parts indicated that even in the same individual, the high-grade tumor with edema had over 10-fold the number of intratumoral immune cells.
Figure 5.

Biomarkers from genetic events and clinical features were integrated to help immune classification of CHS. A and B, A CHS case with 2 distinct parts of the tumor, one in the pelvis and one in the femur. The green area was high-grade tumor with significant peritumoral edema, while the red area was low-grade tumor without peritumoral edema or myxoid change (A). Pathologic examination showing the cell morphology by H&E staining and immune-cell distribution by IHC. CD69 was used as activation marker. Red arrow indicates positive cells. The number of intratumoral immune cells were quantified by flow cytometry (B). C, Biomarkers were integrated to show their potential associations with the immune subtypes of tumor. D, Flow chart to help determine the immune subtypes of CHS.
We matched the three immune subtypes of CHS cases (Fig. 1F) to the three clinical features above and genetic events (Fig. 5C). In general, all of the immune-cell–infiltrated subtype cases (subtypes I and II) were high-grade tumors. Extensive peritumoral edema was exclusively seen in immune-reactive individuals. The subtype I G-MDSC–dominant cases was featured by myxoid transformation. In addition, in myxoid-free cases, IDH mutation together with edema in high-grade cases indicated possibility of immune-exhausted microenvironment (subtype II). A flow chart to help determine the immune subtypes of CHS based on genetic events and clinical features was shown (Fig. 5D).
Disease control by PD-1 immunotherapy was revealed exclusively in patients with subtype II
To investigate if the immune classification system can help predict response to immunotherapy in patients, we reviewed the medical records of patients with CHS receiving PD-1 immunotherapy in our institutions. Twelve patients suffering from unresectable CHS had received PD-1 antibody-based immunotherapy (Fig. 6A). As there was no standard systemic therapy for inoperable CHS, off-label use of anti–PD-1 antibody with sufficient informed consent was administrated according to the results of recent clinical studies (26, 27). After a thorough collection of pathologic, radiological, and genomic information, the immune subtypes of the included patients were determined according to the flow chart in Fig. 5D. There were 1, 4, and 7 cases in subtype I, subtype II, and subtype III, respectively. All of the enrolled individuals were PD-L1 negative and not MSI-high (MSI-H)/deficient mismatch repair (dMMR). As the diseases were refractory, off-label administration of PD-1 antibodies were performed with informed consents. The patients received at least 4 cycles of PD-1 antibody (pembrolizumab or sintilimab) at 200 mg, for 3 weeks, and underwent at least one response evaluation (Fig. 6B).
Figure 6.
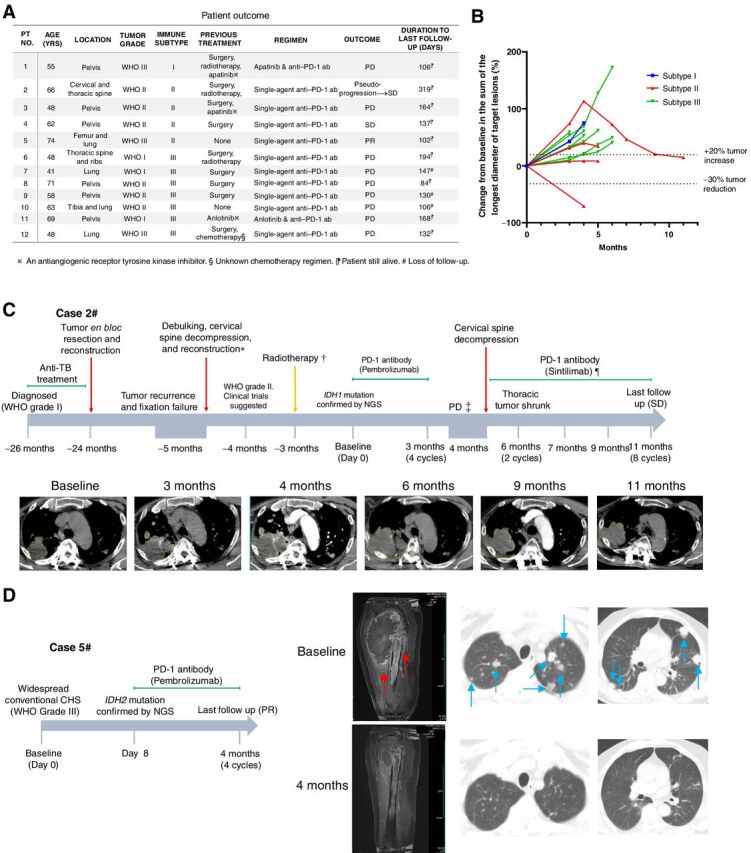
Three conventional CHS cases benefited from immunotherapy. A, Clinical response and outcome of 12 included cases after PD-1 antibody immunotherapy. ab, antibody; PD, progressive disease; PR, partial response; SD, stable disease. B, Spider plot of changes in the sum of unidimensional tumor measurements over time. C and D, Therapeutic information and images of 2 of the cases benefited. Red arrows and blue arrows in D indicate massive peritumoral edema and lung metastases, respectively. C, *, The tumor became refractory after recurrence. The cervical part of tumor was debulked. Clinical trial was suggested but the patient denied. †, The patient received radiotherapy elsewhere, but the detailed information was unclear. The tumor continued progressing afterward. ‡, Although the tumor volume increased and was regarded as PD, it was finally considered to be pseudo-progression. ¶, Although PD-1 inhibition was not recommended from physician's advice, the patient insisted on continuing anti–PD-1 therapy as a compassionate treatment. NGS, next-generation sequencing; TB, tuberculosis.
At the time of cut-off, disease control after immunotherapy was observed in 3 cases. Among them, 1 patient achieved a partial response (Fig. 6D), 1 patient had a pseudo-progression with consequent tumor regression (Fig. 6C), and 1 patient had stable disease (Fig. 6B). In case 5# with partial response, the lung metastases were almost eradicated, and the primary tumor in the femur also dramatically regressed at the first but also the latest follow-up (Fig. 6D). Generalized maculopapular erythema was developed 3 weeks after the first administration of pembrolizumab in case 5#. No significant adverse events were reported in the other patients.
Collectively, all of the 3 cases with controlled disease were subtype II “immune exhausted” cases, while tumor control was observed in none of the subtype I or III cases (P = 0.018, Fisher exact test)
Anti–PD-1–based immunotherapy delayed the progression of immune-resistant tumor with transduced mutated IDH in mice
The unavailability of mouse CHS cell line made it impossible to evaluate anti–PD-1–based immunotherapy in mouse models. We used 2 mouse cancer-cell lines Hepa 1–6 and AE17 to establish homogenous model in immune-competent mice. Hepa 1–6 was mouse liver carcinoma with strong immune-cell infiltration, while AE17 was mouse mesothelioma with few intratumoral immune cells. These 2 cell lines were then transfected with normal or mutated human IDH1. In immune-reactive Hepa 1–6–bearing mice, albeit no statistical difference, a trend in decreased number of CD8+ T cells but elevated number of CD11c+ cells was revealed (Supplementary Fig. S7A). In addition, a significant elevation of CXCL12 concentration in the tumor tissue was illustrated (Supplementary Fig. S7B). In therapeutic assays, we used sorted tumor-infiltrated CD11c+ cells because DCs were frequent in high-grade CHSs. However, treatment by either tumor-infiltrated DC infusion or PD-1 antibody did not delay tumor progression (Supplementary Fig. S7C) or prolong mouse survival (Supplementary Fig. S7D) in mice with WT or mutated IDH Hepa 1–6 tumors (Supplementary Fig. S7E and S7F).
In mice bearing immune-tolerate AE17 mesothelioma, however, there were significantly higher levels of intratumoral T cells, DCs, and chemokines when IDH1 was mutated (Fig. 7A and B). Although treatment did not reveal benefit in mice with WT IDH1 tumor (Fig. 7C and D), delayed tumor progression by PD-1–based therapy was demonstrated in mice with IDH1R132C tumors (Fig. 7E and F). The medium survival was 34.5 days in control group, while it was 38 days in PD-1 solely-treated group and 45 days in combination treatment. CXCL12 was then knocked down by short hairpin RNA (shRNA) to investigate whether it played an important role in mutated IDH-mediated immune response. As was shown in Fig. 7A and G, CXCL12 knockdown weakened the effects of IDH mutation in increasing intratumoral immune cells. In addition, the elevation of levels of T-cell chemotactic chemokines CXCL9 and CXCL10 was also impaired (Fig. 7B and H). Neither PD-1 therapy nor combination treatment with PD-1 antibody and TIDC prolonged mouse survival in CXCL12 knockdown models (Fig. 7I).
Figure 7.
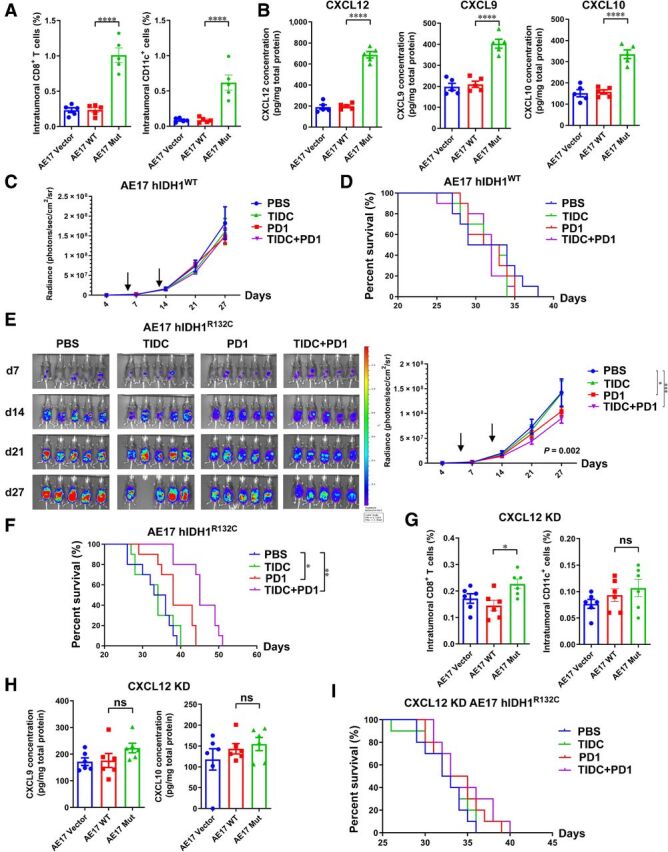
IDH mutation in immune inactive tumor elevated chemokine levels and facilitated response to PD-1 immunotherapy. A and B, Knock-in of hIDH1R132C in AE17 mesothelioma cells significantly elevate the level of CD8+ T cells, DCs (A), and chemokines (B) in vivo. C–F, Immunotherapy by PD-1 antibody or sorted TIDCs showed minimal effects in delaying the progression of tumor with WT IDH (C, D). However, in tumors with mutated IDH, PD-1–based treatment suppressed tumor proliferation and prolonged mouse survival (E, F). G–I, Knockdown of CXCL12 by shRNA impaired the effects of mutated IDH1 in immune-cell number (G) and chemokine levels (H). The antitumor effect of immunotherapy was also significantly weakened when CXCL12 was knocked down (I). KD, knockdown. One-way ANOVA with Bonferroni multiple comparison test was used for column data analyses. Two-way ANOVA was employed to evaluate tumor-signal progression. Data were presented as mean ± SEM. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
IDH mutation–induced immune response was CXCL12 dependent in immune-tolerant tumor in vivo
As indicated above, CXCL12 level was associated with IDH mutation status in patients with CHS (Fig. 3I; Supplementary Fig. S5) and AE17 tumor-bearing mice (Fig. 7). To further investigate the role CXCL12 played in IDH mutation–induced immune response, an adoptive immunotherapy mouse model using ovalbumin (OVA) viral vector and OVA-specific OT-I T cells was established. AE17-OVA hIDH1R132C cells were used for in vivo assays.
In cyclophosphamide and fludarabine–pretreated AE17-OVA hIDH1R132C tumor-bearing mice, transfused OVA-specific OT-I T cells homed to the tumor area, which could be significantly impaired by CXCL12-CXCR4 axis blockade via antagonist AMD3100 (Fig. 8A). Inhibition of CXCL9/10-CXCR3 axis also restricted OT-I T-cell homing. Quantitative evaluation of tumor inflammatory cytokines revealed significantly higher levels of IL6 (Fig. 8B) and IFNγ (Fig. 8C) in OT-I immunotherapy mice, while they were lower in CXCL12-inhibited mice. Furthermore, adoptive-cell immunotherapy using OVA-specific OT-I cells significantly prolonged mouse survival in vivo, but the administration of CXCL12 antagonist partly abrogated the antitumor effects (Fig. 8D).
Figure 8.
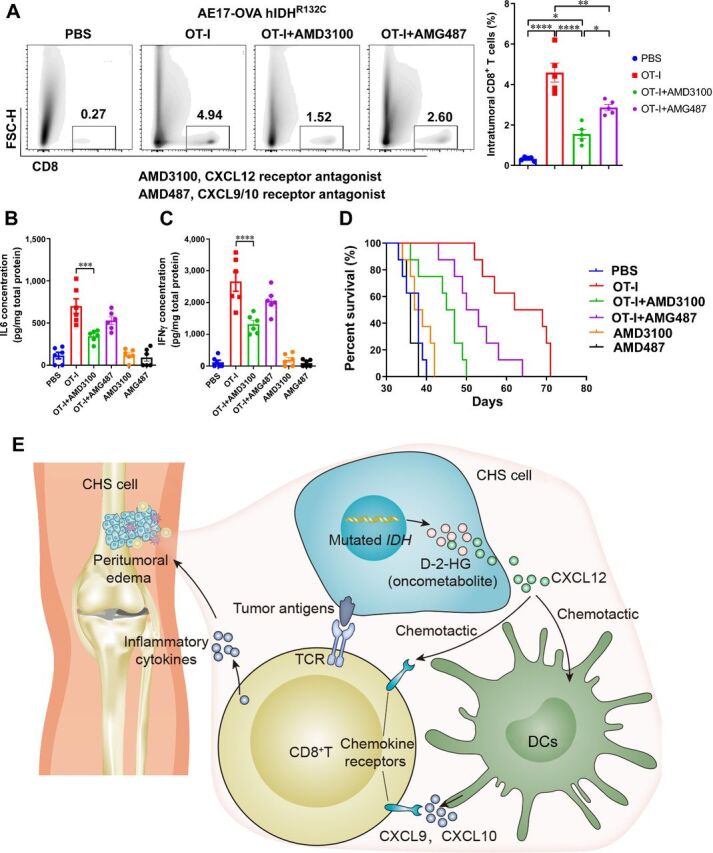
Immune response mediated by IDH mutation was chemokine dependent. A, Transduction of hIDH1R132C and ovalbumin in AE17 cells established optimal targets for tumor-specific OT-I T cells. Adoptively transfused OT-I T cells infiltrated to the tumor, but blockade of CXCL12 or CXCL9/10 chemokine pathway could significantly impair these procedures. B and C, Tumor tissue ELISA assay indicated that homed tumor-specific OT-I T cells secreted substantial amounts of antitumor cytokines including IL6 (B) and IFNγ (C) in a CXCL12-dependent manner. D, Adoptive therapy with tumor-specific cells in IDH-mutated tumor significantly prolonged mouse survival, while blockade of CXCL12 or CXCL9/10 chemokine pathway partly abrogated the effects. E, Proposed mechanical model for IDH mutation-induced immune response. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
The proposed model was shown in Fig. 8E. The mutated IDH induces an epigenetic oncometabolite D-2-HG, driving malignant transformation. However, IDH mutation and D-2-HG level are associated with higher level of CXCL12 and larger number of cDCs. cDCs home to the tumor site driven by CXCL12, and secrete CXCL9/10. Consequently, CXCR3+ or CXCR4+ CD8+ T cells were homed to the tumor following the gradient of CXCL9/10/12. The inflammatory cytokines secreted by CD8+ T cells cause local inflammation, which can be shown in MRI as peritumoral edema.
Discussion
The therapeutic strategy for CHS has remained unchanged for decades. For patients with resectable disease, wide surgical excision is the treatment of choice. Inoperable conventional CHS is fatal because of its resistance to radiotherapy and chemotherapy. However, recent case reports involving PD-1 antibody treatment for metastatic conventional CHS reported a near complete response and tumor regression, highlighting the role that immunotherapy might play against this refractory malignancy (26, 27). A comprehensive immunologic evaluation of the disease seems warranted in order to identify potential candidates for treatment and consequently develop immunotherapeutic regimens for specific profiles.
In this study, based on single-cell mass cytometry, we developed an immune-cell atlas of conventional CHS, dividing the cases into three distinct immune subtypes. The subtype I “G-MDSC dominant” and subtype II “immune exhausted” cases had high immune-cell infiltration, while the former was characteristic of dramatically large number of G-MDSCs in the tumor microenvironment, indicating an immune-suppressive environment mediated by G-MDSCs. This subtype was exclusively seen in samples with focal myxoid transformation. In subtype II, T cells and conventional DCs were more frequent. Among the identified cytotoxic T cells, most of the CD8+ cells were exhausted type Tex cells, expressing high levels of CD39, TIM-3, and PD-1 (19, 28). CD11clo CD68hi macrophages were rarely seen in CHSs. These data indicated that CHS was immunologically different from osteosarcoma and chondroblastoma (29, 30). CHS appeared to be a tumor with few tumor-associated macrophages (TAM), and immunotherapies targeting TAMs might not be the best strategy for this tumor (17). However, the high frequency of cDCs implied that the immune cells were theoretically competent for antigen presenting. Thus, therapeutics targeting exhausted T cells, such as immune checkpoint inhibitors (ICI), may be promising for this tumor, especially subtype II. Our experience in applying PD-1 antibody for patients with subtype II revealed objective response. Further clinical trials are urgently warranted.
In the biomarker study, an association of IDH1/2 mutation with leukocyte infiltration was established. Although IDH mutation seemed to have the potential to impair immune response in immune-reactive tumor Hepa 1–6, which was consistent to previous reports (31, 32), this mutation in immune-inactive tumor AE17 reinforced immune attack. Of note, a notable finding in our study was that most of the CHSs with WT IDH1/2 had a total “immune desert” status, instead of an inflamed environment. In addition, although the number of CD8+ T cells was not directly associated with the concentration of D-2-HG, we did find a positive correlation between D-2-HG concentration and cDCs. The high rate of IDH1/2 mutation in CHSs has been reported previously (33, 34). Interestingly, the only detailed reported conventional CHS case that exhibited a near complete response after using PD-1 antibody also harbored a mutation in IDH and pathologic high grade (27). We provided evidence herein that the mutation of IDH created a favorable microenvironment for immune response by elevating the levels of chemotactic chemokines for immune-cell homing.
It is widely accepted that certain genetic events can affect chemokine levels. For instance, PBRM1, a frequently mutated gene in renal cell carcinoma, is an important marker of ICI response (35, 36). Mechanism study revealed that PBRM1-mutated tumor secreted substantially larger amounts of chemokines, including CXCL9, CXCL10, and CCL5—key chemokines for recruitment of effector T cells (37, 38). In our study, we provided the evidence from both clinical samples and animal studies that in immune-inactive tumors, IDH mutation significantly elevated chemokine levels including CXCL12, CXCL9, and CXCL10, which were critical for immune response. IDH mutation is a potential marker for response to immunotherapy in CHS. Additionally, we provided evidence on the radiological and pathologic features of T-cell–infiltrated CHSs, which may help identify immunotherapy candidates in addition to IDH status.
As far as the authors' knowledge, this is the first report on the identification and quantification of multiple intratumoral immune-cell subtypes in CHS. Unlike renal cell cancer and lung cancer (39, 40), primary bone sarcomas are much more rare neoplasms, which severely hamper fresh tissue-based studies. Recent osteoimmunology discoveries have demonstrated the affect of bone cells on immune regulation, implying that bone tumors may have unique immune-cell subtypes and microenvironments. Examples include multinucleated osteoclast-like giant cells in giant cell tumors and osteosarcomas, and CXCL12-dependent chemotaxis in these 2 tumors (16, 41–43). Similarly, our study also emphasized the role CXCL12 played in the immune response in CHS. Inhibition of CXCL12-mediated pathway significantly impaired the immune-cell homing in vivo. CXCL12 is capable of recruiting multiple CXCR4+ immune cells including DCs and T cells (16). Further study on CXCL12 may unveil the multifaced role it plays in sarcomas.
There are still some limitations to our study. Although we illustrated the immune-cell atlas of CHS by CyTOF, the chosen markers may limit the identification of some unknown immune-cell phenotypes. Further, we recruited 98 cases of fresh tumor tissues, but we were not able to perform multi-omics study in all of the samples. Only 22 cases were comprehensively analyzed, which may have reduced the reliability of our conclusion. Additionally, we proposed our immunologic classification of CHS contained information for clinical decision making, and provided our clinical practice to further prove this system. However, the cohort size was still relatively small. The categorizing system involving MRI image reading of peritumoral edema may be less objective. Although peritumoral soft-tissue edema was used to help differentiate CHS from enchondroma in clinical practice (44–47), clinical features as a means of categorizing were still less precise. Finally, in the current study, we have not fully interpreted the mechanism how IDH mutation affected chemokine levels. We only provided the data of statistical associations and the evidence that chemokine concentrations elevated by IDH mutation were critical for immune response. The use of non-CHS mouse cell line was not an optimal way to investigate IDH mutation in CHS. In solid tumors, IDH mutation can be frequent among glioma, cholangiocarcinoma, and CHS. While IDH mutation was reported to impair immune response in glioma (31, 32), its role in cholangiocarcinoma and CHS was elusive. The lack of homogenous murine-cell line for cholangiocarcinoma or CHS may have restricted the mechanism study in these two deadly diseases. Our data in 2 mouse cell lines revealed different results, indicating that IDH mutation played multifaceted roles in immune response in varied tumors. The establishment of homogenous cartilaginous tumor-cell line for animal study in the future should help clarify the mechanism underlying how IDH mutation affects immune response in CHS.
In conclusion, we provide a comprehensive and integrated immune profile of conventional CHS based on evaluation by CyTOF, multicolor cytometry, WES, radiology, and pathology. The novel immunophenotypic classification can be recapitulated simply by testing the mutation status of IDH and conventional clinical evaluation. This classification may be used to predict the immune status and the response to immunotherapy of CHS in the future. IDH mutation may be the vital role in mediating immune response in CHS by facilitating DC homing through the interaction with CXCL12. This study represents a first step to unlocking the immune environment of CHS, and the results could aid the development of immunotherapies against this disease which has heretofore defied successful medical treatment. The immune-cell profile illustrated herein also provides a valuable resource to further study immune-cell subgroups in other sarcomas and malignancies.
Authors' Disclosures
P.P. Lin reports book royalties from Springer Publishing. No disclosures were reported by the other authors.
Supplementary Material
Supplementary Table 1
Supplementary Table 2
Figure S1
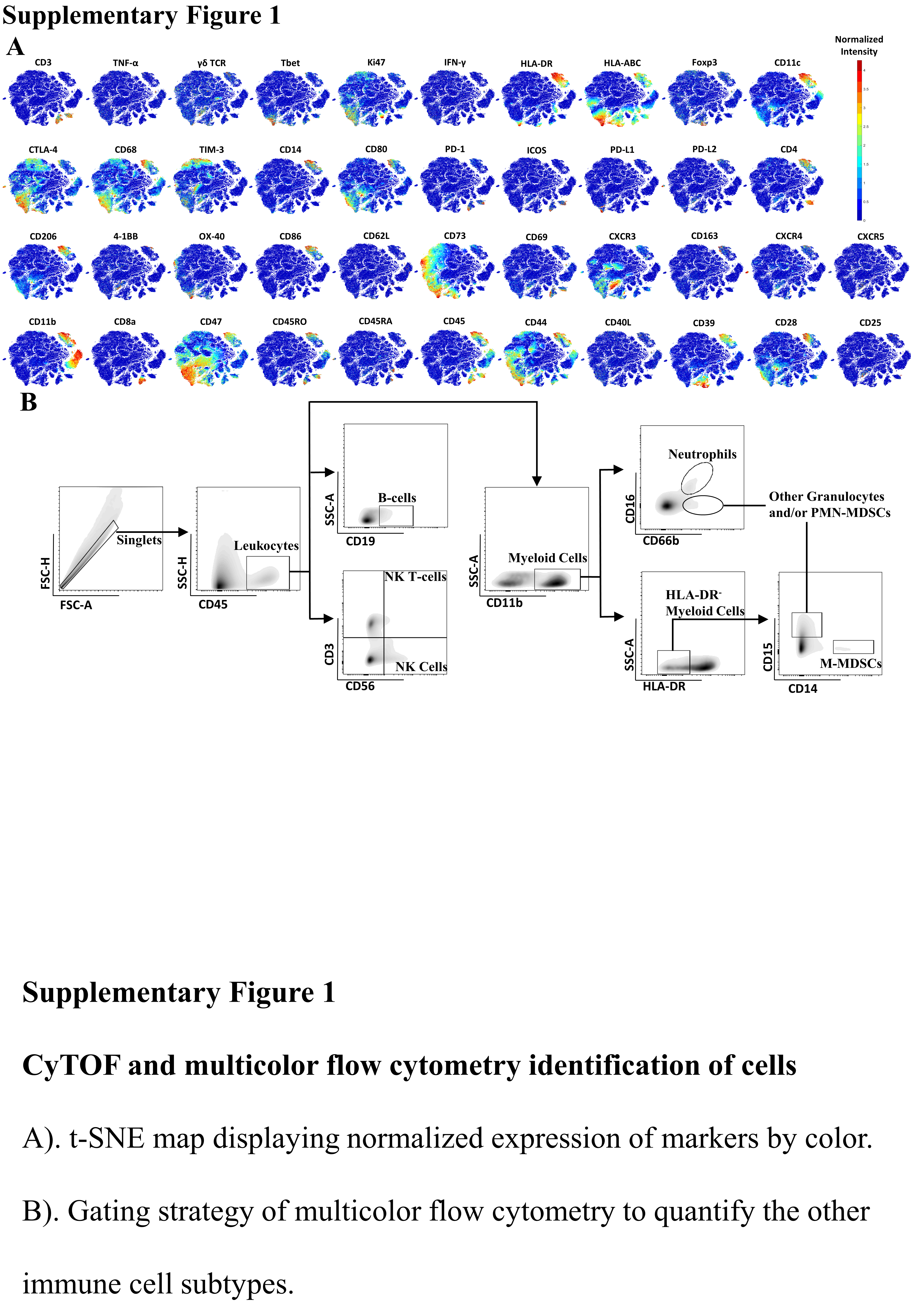{kind=link}
Figure S2
{kind=link}
Figure S3
{kind=link}
Figure S4
{kind=link}
Figure S6
{kind=link}
Figure S5
{kind=link}
Figure S7
{kind=link}
Acknowledgments
We thank Mrs. Li Yuan from Fudan University School of Public Health for her help in statistics.
This work was supported by National Key R&D Program of China (2018YFC1105400, to Z. Ye), National Natural Science Foundation of China (81802684, to B. Li and 81872173, to Z. Ye), and Physician Elite in Training Scholarship of Zhejiang University (171132002, to B. Li).
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
Note: Supplementary data for this article are available at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/).
Authors' Contributions
B. Li: Conceptualization, resources, data curation, formal analysis, funding acquisition, validation, investigation, methodology, writing–original draft, writing–review and editing. G. Li: Data curation, software, visualization, methodology. X. Yan: Data curation. D. Zhu: Data curation, software, visualization, methodology. P.P. Lin: Methodology, writing–review and editing. Z. Wang: Data curation, software. H. Qu: Data curation, investigation. X. He: Resources, methodology, writing–review and editing. Y. Fu: Resources, formal analysis. X. Zhu: Resources, investigation, visualization. P. Lin: Resources. J. Zhang: Resources. X. Li: Resources. H. Dai: Resources. H. Chen: Resources, methodology, writing–review and editing. M.C. Poznansky: Investigation, methodology. N. Lin: Resources, supervision, writing–review and editing. Z. Ye: Conceptualization, resources, supervision, funding acquisition, validation, investigation, methodology, project administration, writing–review and editing.
References
- 1. Giuffrida AY, Burgueno JE, Koniaris LG, Gutierrez JC, Duncan R, Scully SP. Chondrosarcoma in the United States (1973 to 2003): an analysis of 2890 cases from the SEER database. J Bone Joint Surg Am 2009;91:1063–72. [DOI] [PubMed] [Google Scholar]
- 2. van Maldegem A, Conley AP, Rutkowski P, Patel SR, Lugowska I, Desar IME, et al. Outcome of first-line systemic treatment for unresectable conventional, dedifferentiated, mesenchymal, and clear cell chondrosarcoma. Oncologist 2019;24:110–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bovee JV, van Den Broek LJ, Cleton-Jansen AM, Hogendoorn PC. Chondrosarcoma is not characterized by detectable telomerase activity. J Pathol 2001;193:354–60. [DOI] [PubMed] [Google Scholar]
- 4. Horn L, Mansfield AS, Szczesna A, Havel L, Krzakowski M, Hochmair MJ, et al. First-line atezolizumab plus chemotherapy in extensive-stage small-cell lung cancer. N Engl J Med 2018;379:2220–9. [DOI] [PubMed] [Google Scholar]
- 5. June CH, O'Connor RS, Kawalekar OU, Ghassemi S, Milone MC. CAR T cell immunotherapy for human cancer. Science 2018;359:1361–5. [DOI] [PubMed] [Google Scholar]
- 6. Sharma P, Retz M, Siefker-Radtke A, Baron A, Necchi A, Bedke J, et al. Nivolumab in metastatic urothelial carcinoma after platinum therapy (CheckMate 275): a multicentre, single-arm, phase 2 trial. Lancet Oncol 2017;18:312–22. [DOI] [PubMed] [Google Scholar]
- 7. D'Angelo SP, Melchiori L, Merchant MS, Bernstein D, Glod J, Kaplan R, et al. Antitumor activity associated with prolonged persistence of adoptively transferred NY-ESO-1 (c259)T cells in synovial sarcoma. Cancer Discov 2018;8:944–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Reck M, Rodriguez-Abreu D, Robinson AG, Hui R, Csőszi T, Fülöp A, et al. Pembrolizumab versus chemotherapy for PD-L1-positive non-small-cell lung cancer. N Engl J Med 2016;375:1823–33. [DOI] [PubMed] [Google Scholar]
- 9. Fusi A, Festino L, Botti G, Masucci G, Melero I, Lorigan P, et al. PD-L1 expression as a potential predictive biomarker. Lancet Oncol 2015;16:1285–7. [DOI] [PubMed] [Google Scholar]
- 10. Hellmann MD, Ciuleanu TE, Pluzanski A, Lee JS, Otterson GA, Audigier-Valette C, et al. Nivolumab plus ipilimumab in lung cancer with a high tumor mutational burden. N Engl J Med 2018;378:2093–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gibney GT, Weiner LM, Atkins MB. Predictive biomarkers for checkpoint inhibitor-based immunotherapy. Lancet Oncol 2016;17:e542–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kim ST, Cristescu R, Bass AJ, Kim K-M, Odegaard JI, Kim K, et al. Comprehensive molecular characterization of clinical responses to PD-1 inhibition in metastatic gastric cancer. Nat Med 2018;24:1449–58. [DOI] [PubMed] [Google Scholar]
- 13. Simard FA, Richert I, Vandermoeten A, Decouvelaere A-V, Michot J-P, Caux C, et al. Description of the immune microenvironment of chondrosarcoma and contribution to progression. Oncoimmunology 2017;6:e1265716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kostine M, Cleven AH, de Miranda NF, Italiano A, Cleton-Jansen A-M, Bovée JVMG. Analysis of PD-L1, T-cell infiltrate and HLA expression in chondrosarcoma indicates potential for response to immunotherapy specifically in the dedifferentiated subtype. Mod Pathol 2016;29:1028–37. [DOI] [PubMed] [Google Scholar]
- 15. Li B, Zeng Y, Reeves PM, Ran C, Liu Q, Qu X, et al. AMD3100 augments the efficacy of mesothelin-targeted, immune-activating VIC-008 in mesothelioma by modulating intratumoral immunosuppression. Cancer Immunol Res 2018;6:539–51. [DOI] [PubMed] [Google Scholar]
- 16. Li B, Wang Z, Wu H, Xue M, Lin P, Wang S, et al. Epigenetic regulation of CXCL12 plays a critical role in mediating tumor progression and the immune response in osteosarcoma. Cancer Res 2018;78:3938–53. [DOI] [PubMed] [Google Scholar]
- 17. Chou AJ, Kleinerman ES, Krailo MD, Chen Z, Betcher DL, Healey JH, et al. Addition of muramyl tripeptide to chemotherapy for patients with newly diagnosed metastatic osteosarcoma: a report from the Children's Oncology Group. Cancer 2009;115:5339–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kleinerman ES, Jia SF, Griffin J, Seibel NL, Benjamin RS, Jaffe N. Phase II study of liposomal muramyl tripeptide in osteosarcoma: the cytokine cascade and monocyte activation following administration. J Clin Oncol 1992;10:1310–6. [DOI] [PubMed] [Google Scholar]
- 19. Canale FP, Ramello MC, Nunez N, Araujo Furlan CL, Bossio SN, Gorosito Serrán M, et al. CD39 expression defines cell exhaustion in tumor-infiltrating CD8(+) T cells. Cancer Res 2018;78:115–28. [DOI] [PubMed] [Google Scholar]
- 20. Simoni Y, Becht E, Fehlings M, Loh CY, Koo S-L, Weng Teng KW, et al. Bystander CD8(+) T cells are abundant and phenotypically distinct in human tumour infiltrates. Nature 2018;557:575–9. [DOI] [PubMed] [Google Scholar]
- 21. Zhou Q, Munger ME, Veenstra RG, Weigel BJ, Hirashima M, Munn DH, et al. Coexpression of Tim-3 and PD-1 identifies a CD8+ T-cell exhaustion phenotype in mice with disseminated acute myelogenous leukemia. Blood 2011;117:4501–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Brummelman J, Mazza E, Alvisi G, Colombo FS, Grilli A, Mikulak J, et al. High-dimensional single cell analysis identifies stem-like cytotoxic CD8(+) T cells infiltrating human tumors. J Exp Med 2018;215:2520–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Foote MB, Papadopoulos N, Diaz LJ. Genetic classification of gliomas: refining histopathology. Cancer Cell 2015;28:9–11. [DOI] [PubMed] [Google Scholar]
- 24. Chan SM, Thomas D, Corces-Zimmerman MR, Xavy S, Rastogi S, Hong W-J, et al. Isocitrate dehydrogenase 1 and 2 mutations induce BCL-2 dependence in acute myeloid leukemia. Nat Med 2015;21:178–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gagné LM, Boulay K, Topisirovic I, Huot M-É, Mallette FA. Oncogenic activities of IDH1/2 mutations: from epigenetics to cellular signaling. Trends Cell Biol 2017;27:738–52. [DOI] [PubMed] [Google Scholar]
- 26. Pollack SM, Redman MW, Baker KK, Wagner MJ, Schroeder BA, Loggers ET, et al. Assessment of doxorubicin and pembrolizumab in patients with advanced anthracycline-naive sarcoma: a phase 1/2 nonrandomized clinical trial. JAMA Oncol 2020;6:1778–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wagner MJ, Ricciotti RW, Mantilla J, Loggers ET, Pollack SM, Cranmer LD. Response to PD1 inhibition in conventional chondrosarcoma. J Immunother Cancer 2018;6:94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gupta PK, Godec J, Wolski D, Adland E, Yates K, Pauken KE, et al. CD39 expression identifies terminally exhausted CD8+ T cells. PLoS Pathog 2015;11:e1005177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Buddingh EP, Kuijjer ML, Duim RA, Bürger H, Agelopoulos K, Myklebost O, et al. Tumor-infiltrating macrophages are associated with metastasis suppression in high-grade osteosarcoma: a rationale for treatment with macrophage activating agents. Clin Cancer Res 2011;17:2110–9. [DOI] [PubMed] [Google Scholar]
- 30. Gomez-Brouchet A, Illac C, Gilhodes J, Bouvier C, Aubert S, Guinebretiere J-M, et al. CD163-positive tumor-associated macrophages and CD8-positive cytotoxic lymphocytes are powerful diagnostic markers for the therapeutic stratification of osteosarcoma patients: an immunohistochemical analysis of the biopsies from the French OS2006 phase 3 trial. Oncoimmunology 2017;6:e1331193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kohanbash G, Carrera DA, Shrivastav S, Ahn BJ, Jahan N, Mazor T, et al. Isocitrate dehydrogenase mutations suppress STAT1 and CD8+ T cell accumulation in gliomas. J Clin Invest 2017;127:1425–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bunse L, Pusch S, Bunse T, Sahm F, Sanghvi K, Friedrich M, et al. Suppression of antitumor T cell immunity by the oncometabolite (R)-2-hydroxyglutarate. Nat Med 2018;24:1192–203. [DOI] [PubMed] [Google Scholar]
- 33. Amary MF, Bacsi K, Maggiani F, Damato S, Halai D, Berisha F, et al. IDH1 and IDH2 mutations are frequent events in central chondrosarcoma and central and periosteal chondromas but not in other mesenchymal tumours. J Pathol 2011;224:334–43. [DOI] [PubMed] [Google Scholar]
- 34. Totoki Y, Yoshida A, Hosoda F, Nakamura H, Hama N, Ogura K, et al. Unique mutation portraits and frequent COL2A1 gene alteration in chondrosarcoma. Genome Res 2014;24:1411–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Braun DA, Ishii Y, Walsh AM, Van Allen EM, Wu CJ, Shukla SA, et al. Clinical validation of PBRM1 alterations as a marker of immune checkpoint inhibitor response in renal cell carcinoma. JAMA Oncol 2019;5:1631–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Dizman N, Lyou Y, Salgia N, Bergerot PG, Hsu JA, Enriquez D, et al. Correlates of clinical benefit from immunotherapy and targeted therapy in metastatic renal cell carcinoma: comprehensive genomic and transcriptomic analysis. J Immunother Cancer 2020;8:e000953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pan D, Kobayashi A, Jiang P, de Andrade LF, Tay RE, Luoma AM, et al. A major chromatin regulator determines resistance of tumor cells to T cell-mediated killing. Science 2018;359:770–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Liu T, Xia Q, Zhang H, Wang Z, Yang W, Gu X, et al. CCL5-dependent mast cell infiltration into the tumor microenvironment in clear cell renal cell carcinoma patients. Aging 2020;12:21809–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chevrier S, Levine JH, Zanotelli V, Silina K, Schulz D, Bacac M, et al. An immune atlas of clear cell renal cell carcinoma. Cell 2017;169:736–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lavin Y, Kobayashi S, Leader A, Amir E-AD, Elefant N, Bigenwald C, et al. Innate immune landscape in early lung adenocarcinoma by paired single-cell analyses. Cell 2017;169:750–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Salerno M, Avnet S, Alberghini M, Giunti A, Baldini N. Histogenetic characterization of giant cell tumor of bone. Clin Orthop Relat Res 2008;466:2081–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Galvin RJ, Cullison JW, Avioli LV, Osdoby PA. Influence of osteoclasts and osteoclast-like cells on osteoblast alkaline phosphatase activity and collagen synthesis. J Bone Miner Res 1994;9:1167–78. [DOI] [PubMed] [Google Scholar]
- 43. Liao TS, Yurgelun MB, Chang SS, Zhang HZ, Murakami K, Blaine TA, et al. Recruitment of osteoclast precursors by stromal cell derived factor-1 (SDF-1) in giant cell tumor of bone. J Orthop Res 2005;23:203–9. [DOI] [PubMed] [Google Scholar]
- 44. Afonso PD, Isaac A, Villagran JM. Chondroid tumors as incidental findings and differential diagnosis between enchondromas and low-grade chondrosarcomas. Semin Musculoskelet Radiol 2019;23:3–18. [DOI] [PubMed] [Google Scholar]
- 45. Janzen L, Logan PM, O'Connell JX, Connell DG, Munk PL. Intramedullary chondroid tumors of bone: correlation of abnormal peritumoral marrow and soft-tissue MRI signal with tumor type. Skeletal Radiol 1997;26:100–6. [DOI] [PubMed] [Google Scholar]
- 46. Choi BB, Jee WH, Sunwoo HJ, Cho JH, Kim JY, Chun KA, et al. MR differentiation of low-grade chondrosarcoma from enchondroma. Clin Imaging 2013;37:542–7. [DOI] [PubMed] [Google Scholar]
- 47. Parlier-Cuau C, Bousson V, Ogilvie CM, Lackman RD, Laredo J-D. When should we biopsy a solitary central cartilaginous tumor of long bones? Literature review and management proposal. Eur J Radiol 2011;77:6–12. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1
Supplementary Table 2
Figure S1
Figure S2
Figure S3
Figure S4
Figure S6
Figure S5
Figure S7