

Abstract
Purpose:
Lenvatinib has shown efficacy in treating radioiodine-refractory differentiated thyroid cancer (RR-DTC) in the multinational phase III SELECT study; however, it has not been tested in Chinese patients with RR-DTC.
Patients and Methods:
Chinese patients with confirmed RR-DTC (n = 151) were randomly assigned 2:1 to receive lenvatinib 24 mg/day or placebo in 28-day cycles. The primary endpoint was progression-free survival, and key secondary endpoints included objective response rate and safety. Analyses for progression-free survival and objective response rate were conducted using Response Evaluation Criteria in Solid Tumors v1.1 and confirmed by independent imaging review. All adverse events were assessed and monitored.
Results:
Progression-free survival was significantly longer with lenvatinib treatment [n = 103; median 23.9 months; 95% confidence interval (CI), 12.9–not estimable] versus placebo (n = 48; median 3.7 months; 95% CI, 1.9–5.6; hazard ratio = 0.16; 95% CI, 0.10–0.26; P < 0.0001). The objective response rate was 69.9% (95% CI, 61.0–78.8) in the lenvatinib arm and 0% (95% CI, 0–0) in the placebo arm. At data cutoff, 60.2% of patients receiving lenvatinib remained on treatment; treatment-emergent adverse events led to lenvatinib discontinuation in 8.7% of patients. Overall, treatment-emergent adverse events of grade ≥3 occurred in 87.4% of patients in the lenvatinib arm, the most common being hypertension (62.1%) and proteinuria (23.3%).
Conclusions:
Lenvatinib at a starting dose of 24 mg/day significantly improved progression-free survival and objective response rate in Chinese patients with RR-DTC versus placebo. There were no new or unexpected toxicities. Results are consistent with those from SELECT involving patients with RR-DTC.
Translational Relevance.
The tyrosine kinase inhibitor lenvatinib has demonstrated efficacy in the treatment of patients with radioiodine-refractory differentiated thyroid cancer (RR-DTC) in the global phase III SELECT study, but has not been tested in Chinese patients. Rates of DTC are increasing in China, and although DTC can often be cured through surgery with or without radioiodine therapy, there are few options for patients who experience metastasis and are nonresponsive to radioiodine treatment. Current recommendations in China for the treatment of RR-DTC are sorafenib and adriamycin. The aim of this study was to determine if lenvatinib, administered at a starting dose of 24 mg/day to Chinese patients, can improve efficacy outcomes in comparison with placebo, with a tolerable safety profile. It was found that progression-free survival was significantly longer in patients treated with lenvatinib versus placebo, similar to the results from SELECT.
Introduction
Recent data note that approximately 194,000 patients have a diagnosis of thyroid cancer in China (1). A real-world analysis measuring the incidence of differentiated thyroid cancer (DTC) at a hospital in China demonstrated that cases of DTC had increased yearly from 2009 to 2018, and the prevalence of metastasis was nearly 60% (2). Although DTC is often curable through surgery with or without radioactive iodine therapy, approximately two thirds of patients who have recurrence or metastasis develop radioiodine-refractory (RR)-DTC, which typically has a poor prognosis (3). The Chinese Society of Clinical Oncology suggests that systemic therapy, including molecular-targeted therapy, should be considered in patients with progressive and/or symptomatic RR-DTC (4). The level I treatment recommendation for these patients is sorafenib, and the level II treatment recommendation is adriamycin or participation in clinical trials of other agents being assessed to treat this condition (4).
Lenvatinib is an oral, multitargeted, tyrosine kinase inhibitor of vascular endothelial growth factor receptors (VEGFR) 1–3, fibroblast growth factor receptors 1–4, platelet-derived growth factor receptor-α, RET, and KIT (5–8), and is approved as a first-choice systemic therapy for RR-DTC in the United States (9) and Europe (10). Lenvatinib is also approved in multiple countries for the treatment of RR-DTC (11) based on the results of the international, randomized, multicenter, phase III Study of (E7080) LEnvatinib in Differentiated Cancer of the Thyroid (SELECT; ref. 12). In SELECT, lenvatinib treatment resulted in a substantial improvement in progression-free survival (PFS) compared with placebo. Among patients with RR-DTC who received lenvatinib, median PFS duration was 18.3 months [95% confidence interval (CI), 15.1–not estimable (NE)] versus 3.6 months in patients who received placebo (95% CI, 2.2–3.7; hazard ratio (HR) 0.21; 99% CI, 0.14–0.31; P < 0.001). Improved efficacy was also seen in older patients; in a subanalysis of SELECT, overall survival (OS) in patients > 65 years old who received lenvatinib was NE (95% CI, 22.1–NE), compared with OS of 18.4 months (95% CI, 13.3–20.3) in patients > 65 years old who received placebo (13). Subgroup analyses of SELECT also demonstrated that the PFS benefit of lenvatinib for patients with RR-DTC was maintained regardless of BRAF or RAS mutational status (12). However, SELECT did not include patients from China.
To evaluate the efficacy and safety of lenvatinib for the treatment of RR-DTC in the Chinese population, the phase III Study 308 was conducted at 24 sites across China.
Patients and Methods
Study design
Study 308 was a multicenter, randomized, double-blind, placebo-controlled, phase III study of lenvatinib in Chinese patients with RR-DTC (NCT02966093; ref. 14). Patients were randomly assigned 2:1 to receive either daily lenvatinib or placebo in continuous 28-day cycles. Randomization was stratified by tumor subtype (papillary or follicular), number (0 or 1) of prior vascular endothelial growth factor (VEGF)/VEGFR-targeted therapies, and age (≤ 65 years or > 65 years).
The study consisted of three phases: the prerandomization phase, the randomization phase, and the extension phase. During the prerandomization phase, patients underwent screening to determine eligibility and baseline assessments. The randomization phase consisted of the study-treatment cycles, during which patients received lenvatinib orally at a starting dose of 24 mg/day or placebo. Upon identifying an intolerable grade 2 or grade 3 treatment-related toxicity, patients underwent a lenvatinib treatment interruption until the toxicity had resolved to grade ≤ 1 or baseline. Lenvatinib treatment was then resumed at a reduced dose, with stepped reduction level options of 20 mg, 14 mg, and then 10 mg/day for each successive toxicity occurrence. At the completion of the primary analysis, patients who were still undergoing treatment moved to the extension phase. Blinded study-drug administration was discontinued following independent imaging review (IIR) confirmation of disease progression.
During the randomization phase, tumor assessments were performed every 8 weeks until documentation of disease progression or another anticancer therapy was initiated. Patients who received placebo and experienced confirmed disease progression were eligible to cross over to receive optional open-label lenvatinib treatment and were followed according to the schedule of the extension phase, which consisted of the optional open-label lenvatinib treatment period and follow-up assessment period. During the follow-up period, all patients underwent survival follow-up, with the recording of all anticancer treatments. Tumor assessments were performed every 12 weeks, or sooner if clinically indicated, until documentation of disease progression. Tumor assessment by IIR was not required during the extension phase. All patients underwent periodic follow-up for survival, and all anticancer treatments were recorded until time of death.
Patients
Eligible patients were ≥ 18 years of age, had an Eastern Cooperative Oncology Group performance status (ECOG PS) of ≤ 2, ≤ 1 prior VEGF/VEGFR-targeted regimen, and a histologically or cytologically confirmed diagnosis of RR-DTC with demonstrated evidence of disease progression within 12 months of providing informed consent, as measured by Response Evaluation Criteria in Solid Tumors version 1.1 (RECIST v1.1) and confirmed by IIR. Radioiodine-refractory/resistant disease was defined as having one or more of the following attributes: ≥ 1 measurable lesions with no iodine uptake on radioiodine scan; ≥ 1 measurable lesion that has progressed per RECIST v1.1 within 12 months of radioiodine therapy despite radioiodine avidity at time of treatment, or having received cumulative activity of radioiodine of > 600 mCi or 22 gigabecquerels with the last dose administered ≥ 6 months prior to study entry. Patients with the following RR-DTC subtypes were eligible for enrollment: papillary thyroid cancer (follicular variant or variants including but not limited to tall cell, columnar cell, cribriform-morular, solid, oxyphil, Warthin's-like, trabecular, tumor with nodular fasciitis-like stroma, Hürthle cell variant of papillary carcinoma, or poorly differentiated) and follicular thyroid cancer (including Hürthle cell, clear cell, or insular). Definitions of RR-DTC and measurable target lesions are included within the full list of inclusion and exclusion criteria, which can be found on this study's listing on clinicaltrials.gov (NCT02966093).
Written informed consent was provided by all patients before undergoing any study-specific procedures. The study protocol was approved by relevant institutional review boards and was conducted in accordance with the standard operating practices of the Contract Research Organization. These practices are designed to ensure adherence to Good Clinical Practice Guidelines and the World Medical Association Declaration of Helsinki principles.
Efficacy and safety outcomes
The primary endpoint was PFS, defined as the time from randomization to the date of first documentation of disease progression as confirmed by IIR per RECIST v1.1, or death. Secondary endpoints included objective response rate (ORR; the proportion of patients with a best overall response of complete or partial response), OS as measured from randomization until the date of death from any cause, and safety and tolerability, including the monitoring and recording of all adverse events (AE) using Common Terminology Criteria for AEs v4.0 grades. IIR per RECIST v1.1 assessed tumor responses. Exploratory endpoints were disease control rate, clinical benefit rate, and durable stable disease (SD). Disease control rate was the proportion of patients who had the best overall response of complete response, partial response, or SD, with SD defined as stable for ≥ 7 weeks after randomization. The clinical benefit rate was the proportion of patients who had the best overall response of complete response, partial response, or durable SD. Durable SD was defined as SD for ≥ 23 weeks. A post hoc analysis was conducted for time to treatment failure, defined as the time from randomization to the date of the last dose.
Statistical analysis
Efficacy analyses were conducted using the full analysis set, which included all randomly assigned patients. Of the analyses reported herein, only the OS analysis included data from the optional open-label phase in which patients from the placebo arm crossed over to receive open-label lenvatinib during the extension phase. The other analyses were conducted using data from the randomization phase. Safety analyses were performed using the safety analysis set, which included all patients randomly assigned and who received ≥ 1 dose of study drug and 1 postbaseline safety evaluation. For patients randomly assigned to receive placebo but who crossed over to lenvatinib during the optional open-label phase, safety data collected before the first dose of lenvatinib were included in the randomization-phase analysis; safety data collected from the first dose of open-label lenvatinib up to 30 days after the final dose were assessed separately. The sample size was estimated based on the primary endpoint, PFS. Approximately 150 patients were planned to be randomly assigned into the lenvatinib versus placebo groups using a 2:1 ratio. For the primary analysis, 104 PFS events (i.e., progression or death) were targeted. This number would provide > 90% power to detect a HR of 0.5 (i.e., estimated median PFS of 19 months in patients who received lenvatinib vs. 9.5 months in patients who received placebo) with a significance level of 0.05 (two-sided). A sequential testing procedure was used to control the overall family-wise error rate with a two-sided α of 0.05. After achieving statistical significance in favor of the lenvatinib arm at the interim analysis (described below) or the final analysis with PFS (the primary endpoint), the secondary endpoint of ORR was to be tested. If ORR was also significant, then OS was to be tested.
The statistical significance of the difference in PFS between lenvatinib versus placebo was assessed using a log-rank test. Median PFS, and the PFS rate at each time point, were calculated using Kaplan–Meier product-limit estimates for each treatment arm, presented with two-sided 95% CIs, and the Kaplan–Meier estimates of PFS were plotted over time. The Cox regression model was used to estimate the HR and its 95% CI. The statistical significance of the difference in ORR between treatment arms was evaluated using the chi-square test, and a 95% CI was also presented. OS was analyzed using Kaplan–Meier product-limit estimates and compared between lenvatinib and placebo using a log-rank test. Median OS, and the OS rate at each time point, were analyzed in the same manner as PFS. Time to treatment failure was also analyzed by the Kaplan–Meier method. Patients who discontinued study treatment were considered events; other patients were censored at the last dosing date or the data cutoff date. The interim analysis was planned for the time point when half of the targeted PFS events (i.e., 52) was reached or 6 months after 100 patients were enrolled in the lenvatinib arm, whichever occurred later. A one-sided α of 0.01 (fixed) was allocated to the interim analysis. The interim analysis was conducted by the Data Monitoring Committee and the Statistical Analysis Center, both of which were independent of the sponsor, principal investigators, and coordinating doctors.
Results
Patient disposition and baseline characteristics
Of the 151 patients enrolled in several study centers across China, 103 were randomly assigned to receive lenvatinib and 48 to receive placebo (Table 1). The first patient signed informed consent on January 11, 2017, and the data cutoff date was July 31, 2019. All patients were Chinese. The median age was 60.0 years [range, 22–80 years (lenvatinib arm, median: 61.0 years; placebo arm, median: 60.0 years)]. Most patients (81.5%) had papillary DTC (lenvatinib arm, 80.6%; placebo arm, 83.3%), whereas 18.5% had follicular DTC (lenvatinib arm, 19.4%; placebo arm, 16.7%). Most patients (74.8%) had not received prior VEGF/VEGFR-targeted therapy (lenvatinib arm, 74.8%; placebo arm, 75.0%), and 57.0% of patients had a baseline ECOG PS of 0 (lenvatinib arm, 55.3%; placebo arm, 60.4%). The most common metastatic site for both study arms was the lungs (lenvatinib arm, 88.3%; placebo arm, 79.2%). Histologic subtype details are included in Supplementary Table S1.
Table 1.
Demographic and other baseline characteristics.
| Characteristic | Lenvatinib (n = 103) | Placebo (n = 48) | Total (N = 151) |
|---|---|---|---|
| Sex, n (%) | |||
| Male | 57 (55.3) | 21 (43.8) | 78 (51.7) |
| Female | 46 (44.7) | 27 (56.3) | 73 (48.3) |
| Age, years | |||
| Median | 61.0 | 60.0 | 60.0 |
| Range | 28–80 | 22–80 | 22–80 |
| ≤65, n (%) | 73 (70.9) | 33 (68.8) | 106 (70.2) |
| >65, n (%) | 30 (29.1) | 15 (31.3) | 45 (29.8) |
| Weight (kg) | |||
| Mean (standard deviation) | 67.6 (12.9) | 66.0 (13.0) | 67.1 (12.9) |
| Race, n (%) | |||
| Asian (Chinese) | 103 (100) | 48 (100) | 151 (100) |
| Histology, n (%) | |||
| Papillary | 83 (80.6) | 40 (83.3) | 123 (81.5) |
| Follicular | 20 (19.4) | 8 (16.7) | 28 (18.5) |
| ECOG PS, n (%) | |||
| 0 | 57 (55.3) | 29 (60.4) | 86 (57.0) |
| 1 | 41 (39.8) | 16 (33.3) | 57 (37.7) |
| 2 | 5 (4.9) | 3 (6.5) | 8 (5.3) |
| Prior VEGF/VEGFR therapy, n (%) | |||
| 0 | 77 (74.8) | 36 (75.0) | 113 (74.8) |
| 1 | 26 (25.2) | 12 (25.0) | 38 (25.2) |
| Radioiodine refractory statusa, n (%) | |||
| A | 60 (58.3) | 35 (72.9) | 95 (62.9) |
| B | 43 (41.7) | 13 (27.1) | 56 (37.1) |
| C | 24 (23.3) | 10 (20.8) | 34 (22.5) |
| Metastatic sitesb, n (%) | |||
| Lung | 91 (88.3) | 38 (79.2) | 129 (85.4) |
| Liver | 17 (16.5) | 7 (14.6) | 24 (15.9) |
| Lymph node | 72 (69.9) | 35 (72.9) | 107 (70.9) |
| Bone | 36 (35.0) | 13 (27.1) | 49 (32.5) |
| Other | 43 (41.7) | 23 (47.9) | 66 (43.7) |
Abbreviations: ECOG PS, Eastern Cooperative Oncology Group performance status; RECIST v1.1, Response Evaluation Criteria in Solid Tumors version 1.1; VEGF(R), vascular endothelial growth factor (receptor).
aRadioiodine refractory status categories are: (A) ≥ 1 measurable lesions that did not demonstrate iodine uptake on any radioiodine scan; (B) ≥ 1 measurable lesions that had progressed per RECIST v1.1 within 12 months of radioiodine therapy, despite demonstration of radioiodine avidity at the time of that treatment by pretreatment or posttreatment scanning; or (C) cumulative activity of radioiodine of > 600 mCi or 22 gigabecquerels, with the last dose administered at least 6 months prior to study entry.
bMetastatic sites were identified during baseline tumor assessments.
At the data cutoff date for the interim analysis, 62 patients (60.2%) assigned to receive lenvatinib and 8 patients (16.7%) assigned to receive placebo were still receiving their randomly assigned treatment. Nineteen patients (18.4%) from the lenvatinib arm and 37 patients (77.1%) from the placebo arm discontinued treatment because of radiologic disease progression, which was confirmed by independent review (Fig. 1). The remaining 25 patients (lenvatinib arm, n = 22; placebo arm, n = 3) all discontinued treatment for various reasons: among those receiving lenvatinib, 5 discontinued because of patient choice, 4 because of withdrawal of consent, and 3 for other reasons; additionally, 10 patients (9.7%) discontinued lenvatinib because of an AE (one of these cases was due to dyspnea, but it developed 51 days following the final exposure to lenvatinib and was considered not to be treatment-emergent). The 3 patients (6.3%) in the placebo arm discontinued treatment because of an AE.
Figure 1.

Patient enrollment, randomization, and treatment.
Efficacy
Based on the encouraging efficacy results of the interim analysis, the Data Monitoring Committee recommended that the randomization phase be stopped early. The interim analysis data cutoff date (July 31, 2019) was used for the primary analysis. Overall PFS was significantly improved with lenvatinib (median 23.9 months; 95% CI, 12.9–NE) versus placebo (median 3.7 months; 95% CI, 1.9–5.6; HR = 0.16; 95% CI, 0.10–0.26; P < 0.0001; Fig. 2). Among patients in the lenvatinib arm, the PFS rate was 82.6% (95% CI, 73.1–89.0) at 6 months, 69.6% (95% CI, 57.6–78.8) at 12 months, and 54.4% (95% CI, 40.4–66.4) at 18 months. In the placebo arm, the PFS rate was 32.7% (95% CI, 19.5–46.6) at 6 months, 8.9% (95% CI, 2.4–21.0) at 12 months, and 3.0% (95% CI, 0.2–13.0) at 18 months. Moreover, PFS with lenvatinib treatment was improved versus placebo irrespective of subgroup at baseline (i.e., tumor subtype, number of prior VEGF/VEGFR therapies, age group, and sex; Supplementary Fig. S1).
Figure 2.

Kaplan–Meier plot of PFS as assessed by IIR using RECIST v1.1. There were 2 patients censored following discontinuation of study drug owing to an AE, both in the lenvatinib arm. Of these, 1 patient experienced dysphagia that was considered by the investigator to be not related to lenvatinib and 1 patient experienced dyspnea that was considered by the investigator to be related to lenvatinib. AE, adverse event; CI, confidence interval; HR, hazard ratio; IIR, independent imaging review; NE, not estimable; PFS, progression-free survival; RECIST v1.1, Response Evaluation Criteria In Solid Tumors version 1.1.
The ORR was 69.9% (95% CI, 61.0–78.8) in the lenvatinib arm versus 0% (95% CI, 0–0) in the placebo arm (69.9% difference, 95% CI 61.0–78.8, P < 0.0001; Table 2). In the lenvatinib arm, the disease control rate was 90.3% (95% CI, 84.6–96.0) versus 56.3% (95% CI, 42.2–70.3) in the placebo arm, and the clinical benefit rate in the lenvatinib arm was 81.6% (95% CI, 74.1–89.0) versus 45.8% (95% CI, 31.7–59.9) in the placebo arm (Table 2). Percentage changes in the sums of diameters of target lesions at postbaseline nadir are shown in Fig. 3. During the extension phase, 30 of 48 patients crossed over from placebo to optional open-label lenvatinib treatment; of these 30, 14 had an objective response (as assessed by investigators) for an ORR of 46.7% (95% CI, 28.8–64.5). At the data cutoff date, median OS had not been reached for either the lenvatinib or the placebo arm (HR 0.84, 95% CI, 0.39–1.83), which included those patients who had crossed over to lenvatinib (Supplementary Fig. S2). The OS rate in the lenvatinib arm was 86.0% (95% CI, 76.3–91.9) at 12 months, 75.6% (95% CI, 63.5–84.2) at 18 months, and 75.6% (95% CI, 63.5–84.2) at 24 months. The OS rate for the placebo arm was 81.7% (95% CI, 66.4–90.5) at 12 months, 78.3% (95% CI, 61.9–88.2) at 18 months, and 71.7% (95% CI, 50.7–85.0) at 24 months. The median duration of survival follow-up was 14.8 months (95% CI, 12.4–16.7) for the lenvatinib arm and 15.6 months (95% CI, 11.6–19.1) for the placebo arm. Median time to treatment failure was 18.5 months (95% CI, 12.5–NE) in the lenvatinib arm and 6.4 months (95% CI, 3.9–7.8) in the placebo arm (HR = 0.27; 95% CI, 0.17–0.42; Supplementary Fig. S3).
Table 2.
Summary of tumor responses in patients who received lenvatinib or placebo.
| Parameter | Lenvatinib (n = 103) | Placebo (n = 48) |
|---|---|---|
| Best overall response, n (%) | ||
| Complete response | 2 (1.9) | 0 |
| Partial response | 70 (68.0) | 0 |
| Stable disease | 21 (20.4) | 27 (56.3) |
| Durable stable diseasea | 12 (11.7) | 22 (45.8) |
| Progressive disease | 6 (5.8) | 16 (33.3) |
| Not evaluable/unknown | 4 (3.9) | 5 (10.4) |
| Objective response rate, n (%) | 72 (69.9) | 0 |
| Difference, % (95% CI) | 69.9 (61.0–78.8) | |
| P value | < 0.0001 | |
| Disease control rateb, n (%) | 93 (90.3) | 27 (56.3) |
| Difference, % (95% CI) | 34.0 (18.9–49.2) | |
| Clinical benefit ratec, n (%) | 84 (81.6) | 22 (45.8) |
| Difference, % (95% CI) | 35.7 (19.8–51.7) | |
Abbreviation: CI, confidence interval.
aDurable stable disease = duration of ≥ 23 weeks.
bDisease control rate = complete response, partial response, and stable disease.
cClinical benefit rate = complete response, partial response, and durable stable disease.
Figure 3.
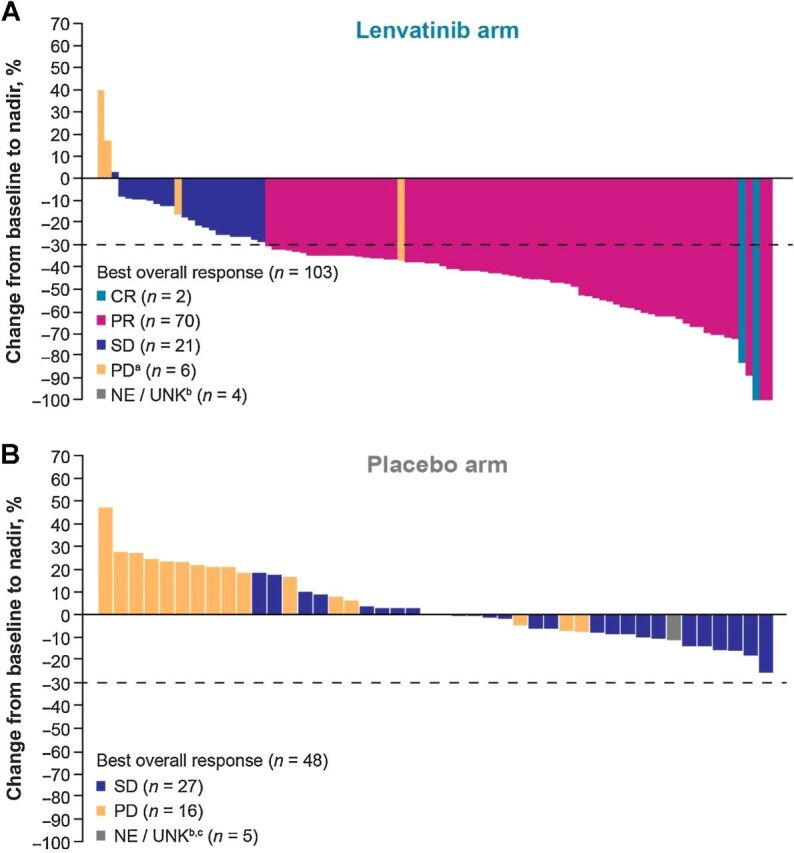
Percentage changes in the sums of diameters of target lesions from baseline to postbaseline nadir for patients treated with (A) lenvatinib or (B) placebo (by IIR using RECIST v1.1). CR, complete response; IIR, independent imaging review; NE/UNK, not evaluable/unknown; PD, progressive disease; PR, partial response; RECIST v1.1, Response Evaluation Criteria In Solid Tumors version 1.1; SD, stable disease. aTwo patients were determined to be PD due to nontarget lesions, but target lesions were not evaluable. These patients are not depicted on the plot. b“Unknown” patients lacked a baseline and/or postbaseline tumor assessment. c1 patient had a single postbaseline assessment at week 5 but this duration was not long enough to be considered SD. This patient is considered “NE” and is shown in gray in this plot.
Safety
The median duration of treatment was 9.26 months for lenvatinib and 6.26 months for placebo. The median dose intensity [dose intensity was defined as the total dose (mg)/duration of treatment (days)] for lenvatinib was 17.49 mg/day.
Among patients who received lenvatinib, the most common grade ≥ 3 treatment-emergent AEs (TEAE; lenvatinib arm vs. placebo arm) recorded were hypertension (62.1% vs. 8.3%), proteinuria (23.3% vs. 0%), and palmar–plantar erythrodysesthesia syndrome (9.7% vs. 0%; Table 3). Serious TEAEs were experienced by 40 patients (38.8%) who received lenvatinib and 16 patients (33.3%) who received placebo (Table 3); serious TEAEs were considered treatment-related in 23 patients (22.3%) in the lenvatinib arm and 6 patients (12.5%) in the placebo arm.
Table 3.
Summary of TEAEs.
| TEAEs, n (%) | Lenvatinib (n = 103) | Placebo (n = 48) | ||
|---|---|---|---|---|
| Any TEAEs | 103 (100) | 47 (97.9) | ||
| Any related TEAEs | 103 (100) | 34 (70.8) | ||
| Grade ≥ 3 TEAEs | 90 (87.4) | 22 (45.8) | ||
| Grade ≥ 3 related TEAEs | 88 (85.4) | 8 (16.7) | ||
| Serious TEAEs | 40 (38.8) | 16 (33.3) | ||
| Fatal TEAEs | 10 (9.7) | 3 (6.3) | ||
| TEAEs leading to study-drug withdrawal | 9 (8.7) | 3 (6.3) | ||
| TEAEs leading to study-drug dose reduction | 83 (80.6) | 3 (6.3) | ||
| TEAEs leading to study-drug interruption | 69 (67.0) | 4 (8.3) | ||
| TEAEs with incidence rates ≥ 20% in the lenvatinib arm | ||||
| Preferred term | Any grade | Grades 3–4 | Any grade | Grades 3–4 |
| Hypertension | 84 (81.6) | 64 (62.1) | 10 (20.8) | 4 (8.3) |
| Proteinuria | 83 (80.6) | 24 (23.3) | 4 (8.3) | 0 |
| PPES | 60 (58.3) | 10 (9.7) | 2 (4.2) | 0 |
| Diarrhea | 51 (49.5) | 7 (6.8) | 2 (4.2) | 0 |
| Weight decreased | 49 (47.6) | 3 (2.9) | 5 (10.4) | 0 |
| Hypocalcemia | 30 (29.1) | 4 (3.9) | 8 (16.7) | 1 (2.1) |
| Platelet count decreased | 27 (26.2) | 7 (6.8) | 0 | 0 |
| Blood lactate dehydrogenase increased | 26 (25.2) | 0 | 2 (4.2) | 0 |
| Cough | 22 (21.4) | 0 | 8 (16.7) | 0 |
| Decreased appetite | 21 (20.4) | 2 (1.9) | 3 (6.3) | 0 |
| Hematuria | 21 (20.4) | 0 | 3 (6.3) | 0 |
Abbreviations: PPES, palmar–plantar erythrodysesthesia syndrome; TEAE, treatment-emergent adverse event.
Overall, 13 patients experienced a fatal TEAE (10 patients received lenvatinib and 3 patients received placebo). Of these, two deaths in the lenvatinib arm and one death in the placebo arm were because of disease progression. Fatal serious TEAEs that occurred in 2 or more patients included death of unknown cause (lenvatinib arm, n = 2; placebo arm, n = 2), multiple organ dysfunction syndrome (lenvatinib arm, n = 2), and pleural effusion (lenvatinib arm, n = 2). Five deaths were considered related to lenvatinib treatment (cardiogenic shock, death, sudden death, pleural effusion, and hemorrhage; n = 1 each); one death was considered related to placebo (suicide attempt).
Overall, TEAEs led to study-drug discontinuation in 9 patients (8.7%) who received lenvatinib and 3 patients (6.3%) who received placebo (Table 3). TEAEs leading to dose reduction occurred in 83 patients (80.6%) who received lenvatinib and 3 patients (6.3%) who received placebo. Lastly, TEAEs leading to study-drug interruption were reported in 69 patients (67.0%) who received lenvatinib and in 4 patients (8.3%) who received placebo.
Discussion
The results of this analysis indicate that when compared with placebo, lenvatinib, administered at a starting dose of 24 mg/day, significantly prolonged PFS in Chinese patients with RR-DTC. Median PFS in patients treated with lenvatinib was 23.9 months (95% CI, 12.9–NE) compared with 3.7 months (95% CI, 1.9–5.6) in patients treated with placebo. ORR was 69.9% in patients treated with lenvatinib versus 0% in patients treated with placebo. There were no unexpected toxicities, and most AEs were managed with dose modifications and medications.
Overall, efficacy results from the multinational SELECT (12) were similar to the results in this study of Chinese patients with RR-DTC. However, caution should be used when comparing trials. Aside from region/race, baseline characteristics were generally similar between the two studies. Median PFS in patients who received lenvatinib was 18.3 months in SELECT (95% CI, 15.1–NE; ref. 12) versus 23.9 (95% CI, 12.9–NE) months in this analysis. The HR for progression or death in SELECT [0.21; 99% CI, 0.14–0.31; ref. 12) was comparable to the HR for progression or death in this analysis (0.16; 95% CI, 0.10–0.26). In addition, ORR was similar in patients who received lenvatinib in both studies (SELECT, 64.8%; ref. 12; this analysis, 69.9%).
The safety profile of lenvatinib in this study was also generally consistent with the well-established safety profile of this drug, and no new signals were detected. Specifically, in the current analysis, 10 patients (9.7%) who received lenvatinib experienced a fatal AE, and 5 (4.9%) of those events were considered related to treatment; these results were comparable to those of SELECT, in which 20 patients (7.7%) who received lenvatinib experienced a fatal AE, and 6 (2.3%) of those events were considered related to treatment (12). Notably, among patients who received lenvatinib, 50% of patients in SELECT experienced a serious AE, compared with 38.8% of patients in this analysis. Moreover, the numerically lower incidence of TEAEs leading to study-drug withdrawal in this study (8.7%) compared with patients who received lenvatinib in SELECT (14.2%; ref. 12) indicates that the safety profile of lenvatinib is tolerable in Chinese patients with RR-DTC. The lower rates of study-drug withdrawal could be attributed to differences in the management of Asian patients versus global populations. However, more information would be needed to draw conclusions regarding this difference. A relatively higher rate of dose reductions because of TEAEs was seen in this study (80.6%), and in Japanese patients from SELECT (90%; ref. 15), compared with the multinational SELECT population (67.8%; ref. 12). Of note, the two most common AEs in this study among Chinese patients who received lenvatinib [hypertension (81.6%) and proteinuria (80.6%)] were also found to occur at higher rates in the Asian subpopulation of SELECT compared with the total study population (16). Specifically, grade ≥ 3 hypertension occurred at a rate of 71.7% in the Asian subpopulation of SELECT, and 62.1% in this study, compared with 44.4% in the total SELECT population (12, 16). Grade ≥ 3 proteinuria occurred in 21.7% of patients in the Asian subpopulation of SELECT, and 23.3% in this study, compared with 10.0% of patients in the overall SELECT population (16). It has been suggested that East Asian patients have more severe AEs following tyrosine kinase inhibitor treatment compared with European patients, which is likely due to a lower body mass (17). It is possible that lower body mass in Asian patients contributed to the increased occurrence of grade ≥ 3 AEs observed in this study compared with rates in SELECT.
A limitation of this study is that it was not designed with sufficient statistical power to demonstrate a difference in OS; therefore, OS results should be interpreted carefully. Furthermore, the OS findings are confounded because of crossover from the placebo arm into the active-treatment lenvatinib arm during the open-label phase of the study. In the current study, the median duration of treatment was 9.3 months, and the median PFS was 23.9 months. Although the medians for these parameters may differ due to differences in the calculation methods, the duration of treatment and PFS for individual patients were similar. Inherently, duration of treatment is shorter with shorter follow-up. The study data reported herein are from the preplanned interim analysis. The same data were later used for the primary analysis after the randomization phase was stopped early based on encouraging efficacy data. However, this resulted in a shorter duration of follow-up, which may be a potential limitation of this study. In general, it is possible for censoring of patients in PFS analyses to cause a difference between treatment duration and PFS results. However, only two patients in the lenvatinib arm of this study were censored during the PFS analysis owing to an AE that led to discontinuation of study drug (1 patient with dysphagia that was considered not related to lenvatinib and 1 patient with dyspnea that was considered related to lenvatinib). No patients in the placebo arm were censored owing to an AE. When time to treatment failure was assessed, the results were similar to the PFS results, indicating that the major reason for censoring was ongoing treatment. Therefore, the impact of censoring owing to discontinuation for reasons other than disease progression is likely small.
Current guidelines from the Chinese Society of Clinical Oncology recommend sorafenib as the level I option for the treatment of patients with RR-DTC with symptomatic or rapid progression (4). In the pivotal, multinational, phase III study of sorafenib in patients with RR-DTC, median PFS in patients treated with sorafenib was 10.8 months compared with 5.8 months in patients treated with placebo (HR 0.59, 95% CI, 0.45–0.76, P < 0.0001), as confirmed by a central independent review (18). A meta-analysis of sorafenib in RR-DTC revealed that although sorafenib improves PFS, incidences of AEs were high, and 68% of patients required dose reductions because of toxicity (19). Although caution should be used when comparing clinical trials, the results in the current study suggest that the efficacy of lenvatinib in Chinese patients with RR-DTC compares favorably with that of sorafenib in the multinational population (18). Similar results with lenvatinib were also observed in the multinational SELECT study (12).
The results of this study demonstrated similarities in both safety and efficacy between the Chinese population and the multinational population of the randomized, multicenter, phase III SELECT study, and are consistent with the findings of a pharmacokinetic study of lenvatinib in Chinese patients with solid tumors; these data indicate that the pharmacokinetics of lenvatinib in the Chinese population are consistent with those in the multinational population (20). Moreover, prior population pharmacokinetic analyses suggest lenvatinib pharmacokinetics are similar across races/ethnicities (21), which further supports the use of lenvatinib in this patient population. The results of this analysis indicate that lenvatinib shows promising efficacy and has a manageable safety profile in Chinese patients with RR-DTC.
Data Availability Statement
The data will not be available for sharing at this time because the data are commercially confidential. However, Eisai will consider written requests to share the data on a case-by-case basis.
Prior Publication Statement
This manuscript represents an original work, not published elsewhere, except for some limited data excerpts in an abstract presented at the European Society for Medical Oncology Asia Virtual Congress, November 20–22, 2020. Abstract #365.
Authors' Disclosures
X. Zheng reports non-financial support from Eisai Co., Ltd. during the conduct of the study. Q. Ji reports grants and non-financial support from Eisai during the conduct of the study. M. Ge reports non-financial support from Eisai Co., Ltd. during the conduct of the study. G. Chen reports non-financial support and other support from Eisai during the conduct of the study. R. Huang reports other support from Eisai during the conduct of the study. J. Tan reports grants from Eisai during the conduct of the study. T. Huang reports grants from Eisai during the conduct of the study. Z. Lv reports other support from Eisai during the conduct of the study. Y. Lin reports other support from Eisai during the conduct of the study. T. Kubota reports personal fees from Eisai. Co., Ltd. during the conduct of the study, as well as personal fees from Eisai. Co., Ltd. outside the submitted work. T. Suzuki reports personal fees from Eisai Co., Ltd. outside the submitted work. H. Ikezawa reports personal fees from Eisai Co., Ltd. during the conduct of the study. M. Gao reports non-financial support from Eisai Pharmaceutical Group Co., Ltd during the conduct of the study. No disclosures were reported by the other authors.
Supplementary Material
Supplementary Material
Supplementary Figure S1. Forest plot of PFS hazard ratios for lenvatinib versus placebo (by IIR using RECIST v1.1)
Supplementary Figure S2. Kaplan-Meier plot of OS a (full analysis set)
Supplementary Figure S3. Kaplan-Meier plot of time to treatment failure
Acknowledgments
This work was supported by Eisai Inc., Woodcliff Lake, NJ, USA, and also by Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA. Medical writing support was provided by Heather A. Mitchell, PhD, Oxford PharmaGenesis Inc., Newtown, PA, USA, and was funded by Eisai Inc., Woodcliff Lake, NJ, USA, and also by Merck Sharp & Dohme Corp. As several authors are employed by Eisai, there was involvement of this funding body in the study design, data collection, and analysis, and the writing of the manuscript/decision to submit for publication.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
Note: Supplementary data for this article are available at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/).
Authors' Contributions
X. Zheng: Conceptualization, formal analysis, investigation, writing–original draft, writing–review and editing. Z. Xu: Conceptualization, formal analysis, investigation, writing–original draft, writing–review and editing. Q. Ji: Conceptualization, formal analysis, investigation, writing–original draft, writing–review and editing. M. Ge: Conceptualization, formal analysis, investigation, writing–original draft, writing–review and editing. F. Shi: Conceptualization, formal analysis, investigation, writing–original draft, writing–review and editing. J. Qin: Conceptualization, formal analysis, investigation, writing–original draft, writing–review and editing. F. Wang: Conceptualization, formal analysis, investigation, writing–original draft, writing–review and editing. G. Chen: Conceptualization, formal analysis, investigation, writing–original draft, writing–review and editing. Y. Zhang: Conceptualization, formal analysis, investigation, writing–original draft, writing–review and editing. R. Huang: Conceptualization, formal analysis, investigation, writing–original draft, writing–review and editing. J. Tan: Conceptualization, formal analysis, investigation, writing–original draft, writing–review and editing. T. Huang: Conceptualization, formal analysis, investigation, writing–original draft, writing–review and editing. S. Li: Conceptualization, formal analysis, investigation, writing–original draft, writing–review and editing. Z. Lv: Conceptualization, formal analysis, investigation, writing–original draft, writing–review and editing. Y. Lin: Conceptualization, formal analysis, investigation, writing–original draft, writing–review and editing. Z. Guo: Conceptualization, formal analysis, investigation, writing–original draft, writing–review and editing. T. Kubota: Conceptualization, resources, formal analysis, funding acquisition, investigation, writing–original draft, project administration, writing–review and editing. T. Suzuki: Conceptualization, resources, formal analysis, funding acquisition, investigation, writing–original draft, project administration, writing–review and editing. H. Ikezawa: Conceptualization, resources, data curation, software, formal analysis, funding acquisition, validation, investigation, visualization, methodology, writing–original draft, project administration, writing–review and editing. M. Gao: Conceptualization, formal analysis, supervision, investigation, writing–original draft, writing–review and editing.
References
- 1. International Agency for Research on Cancer. GLOBOCAN. [cited 2020 November 5]. Available from: https://gco.iarc.fr/today/home.
- 2. Han L, Wu Z, Li W, Li Y, Ma J, Wu X, et al. The real world and thinking of thyroid cancer in China. Int J Surg Oncol 2019;4:e81. [Google Scholar]
- 3. Jin Y, Van Nostrand D, Cheng L, Liu M, Chen L. Radioiodine refractory differentiated thyroid cancer. Crit Rev Oncol Hematol 2018;125:111–20. [DOI] [PubMed] [Google Scholar]
- 4. Chinese Society of Clinical Oncology (CSCO) diagnosis and treatment guidelines for persistent/recurrent and metastatic differentiated thyroid cancer 2018 (English version). Chin J Cancer Res 2019;31:99–116.30996569 [Google Scholar]
- 5. Matsui J, Funahashi Y, Uenaka T, Watanabe T, Tsuruoka A, Asada M. Multi-kinase inhibitor E7080 suppresses lymph node and lung metastases of human mammary breast tumor MDA-MB-231 via inhibition of vascular endothelial growth factor-receptor (VEGF-R) 2 and VEGF-R3 kinase. Clin Cancer Res 2008;14:5459–65. [DOI] [PubMed] [Google Scholar]
- 6. Matsui J, Yamamoto Y, Funahashi Y, Tsuruoka A, Watanabe T, Wakabayashi T, et al. E7080, a novel inhibitor that targets multiple kinases, has potent antitumor activities against stem cell factor producing human small cell lung cancer H146, based on angiogenesis inhibition. Int J Cancer 2008;122:664–71. [DOI] [PubMed] [Google Scholar]
- 7. Yamamoto Y, Matsui J, Matsushima T, Obaishi H, Miyazaki K, Nakamura K, et al. Lenvatinib, an angiogenesis inhibitor targeting VEGFR/FGFR, shows broad antitumor activity in human tumor xenograft models associated with microvessel density and pericyte coverage. Vasc Cell 2014;6:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tohyama O, Matsui J, Kodama K, Hata-Sugi N, Kimura T, Okamoto K, et al. Antitumor activity of lenvatinib (e7080): an angiogenesis inhibitor that targets multiple receptor tyrosine kinases in preclinical human thyroid cancer models. J Thyroid Res 2014;2014:638747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. National Comprehensive Cancer Network Clinical Practice Guidelines in Oncology (NCCN Guidelines®). Thyroid Carcinoma. Version 2.2020. [cited 2020 November 5]. Available from: https://www.nccn.org/professionals/physician_gls/pdf/thyroid.pdf.
- 10. Filetti S, Durante C, Hartl D, Leboulleux S, Locati LD, Newbold K, et al. Thyroid cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 2019;30:1856–83. [DOI] [PubMed] [Google Scholar]
- 11. Lenvima (lenvatinib) [prescribing information]. Woodcliff Lake, NJ, USA: Eisai Inc.; 2020. [Google Scholar]
- 12. Schlumberger M, Tahara M, Wirth LJ, Robinson B, Brose MS, Elisei R, et al. Lenvatinib versus placebo in radioiodine-refractory thyroid cancer. N Engl J Med 2015;372:621–30. [DOI] [PubMed] [Google Scholar]
- 13. Brose MS, Worden FP, Newbold KL, Guo M, Hurria A. Effect of age on the efficacy and safety of lenvatinib in radioiodine-refractory differentiated thyroid cancer in the phase III SELECT trial. J Clin Oncol 2017;35:2692–9. [DOI] [PubMed] [Google Scholar]
- 14. ClinicalTrials.gov. A Trial of Lenvatinib (E7080) in Radioiodine (131 I)-Refractory Differentiated Thyroid Cancer in China. [cited 2021 January 4]. Available from: https://clinicaltrials.gov/ct2/show/NCT02966093. [Google Scholar]
- 15. Kiyota N, Schlumberger M, Muro K, Ando Y, Takahashi S, Kawai Y, et al. Subgroup analysis of Japanese patients in a phase 3 study of lenvatinib in radioiodine-refractory differentiated thyroid cancer. Cancer Sci 2015;106:1714–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lee EK, Kiyota N, Kim S-B, Muro K, He Y, Baig M, et al. Safety of lenvatinib in an Asian subpopulation of the SELECT trial. Poster presented at: The 12th Asia and Oceania Thyroid Association Congress, Busan, Korea; 16–19 March 2017.
- 17. Touma JA, McLachlan AJ, Gross AS. The role of ethnicity in personalized dosing of small molecule tyrosine kinase inhibitors used in oncology. Transl Cancer Res 2017;6:S1558–91. [Google Scholar]
- 18. Brose MS, Nutting CM, Jarzab B, Elisei R, Siena S, Bastholt L, et al. Sorafenib in radioactive iodine-refractory, locally advanced or metastatic differentiated thyroid cancer: a randomised, double-blind, phase 3 trial. Lancet 2014;384:319–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Feng G, Luo Y, Zhang Q, Zeng F, Xu J, Zhu J. Sorafenib and radioiodine-refractory differentiated thyroid cancer (RR-DTC): a systematic review and meta-analysis. Endocrine 2020;68:56–63. [DOI] [PubMed] [Google Scholar]
- 20. Liu D, Liu L, Shen L, Kubota T, Suzuki T, Ikezawa H, et al. Pharmacokinetic study of lenvatinib in Chinese patients with solid tumors. Future Oncol 2021;17:1855–63. [DOI] [PubMed] [Google Scholar]
- 21. Gupta A, Jarzab B, Capdevila J, Shumaker R, Hussein Z. Population pharmacokinetic analysis of lenvatinib in healthy subjects and patients with cancer. Br J Clin Pharmacol 2016;81:1124–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Supplementary Figure S1. Forest plot of PFS hazard ratios for lenvatinib versus placebo (by IIR using RECIST v1.1)
Supplementary Figure S2. Kaplan-Meier plot of OS a (full analysis set)
Supplementary Figure S3. Kaplan-Meier plot of time to treatment failure
Data Availability Statement
The data will not be available for sharing at this time because the data are commercially confidential. However, Eisai will consider written requests to share the data on a case-by-case basis.