

Low-dose radiotherapy triggered T-cell infiltration into poorly inflamed tumors and reprogrammed both the adaptive and innate immune landscape to allow responsiveness to combinatorial immunotherapy.
Abstract
Developing strategies to inflame tumors is critical for increasing response to immunotherapy. Here, we report that low-dose radiotherapy (LDRT) of murine tumors promotes T-cell infiltration and enables responsiveness to combinatorial immunotherapy in an IFN-dependent manner. Treatment efficacy relied upon mobilizing both adaptive and innate immunity and depended on both cytotoxic CD4+ and CD8+ T cells. LDRT elicited predominantly CD4+ cells with features of exhausted effector cytotoxic cells, with a subset expressing NKG2D and exhibiting proliferative capacity, as well as a unique subset of activated dendritic cells expressing the NKG2D ligand RAE1. We translated these findings to a phase I clinical trial administering LDRT, low-dose cyclophosphamide, and immune checkpoint blockade to patients with immune-desert tumors. In responsive patients, the combinatorial treatment triggered T-cell infiltration, predominantly of CD4+ cells with Th1 signatures. Our data support the rational combination of LDRT with immunotherapy for effectively treating low T cell–infiltrated tumors.
Significance:
Low-dose radiation reprogrammed the tumor microenvironment of tumors with scarce immune infiltration and together with immunotherapy induced simultaneous mobilization of innate and adaptive immunity, predominantly CD4+ effector T cells, to achieve tumor control dependent on NKG2D. The combination induced important responses in patients with metastatic immune-cold tumors.
This article is highlighted in the In This Issue feature, p. 1
Introduction
Response to immune checkpoint blockade (ICB) is robust and durable in a proportion of patients. Patients with so-called cold or immune-desert tumors are, however, less likely to respond to ICB (1, 2). Important efforts are thus under way to identify effective and feasible approaches for inflaming these tumors (3). Moreover, the inherent plasticity of tumors and their microenvironment enables dynamic upregulation of a diverse range of inhibitory mechanisms, hence necessitating combinatorial treatment approaches to sustain tumor control by T cells.
Along with its direct tumoricidal effects, hypofractionated (high-dose) radiotherapy can mediate important immunomodulatory effects, including (i) in situ vaccination through release of tumor-associated antigens (4); (ii) the activation of dendritic cells (DC; ref. 5); (iii) the release of danger signals and the upregulation of cytokines and chemokines (6); and (iv) normalization of the tumor vasculature (7). In addition, local radiotherapy can activate DNA-sensing pathways in host (5) and tumor cells (8), triggering production of type I IFN (9) and mobilizing innate and adaptive immunity. Numerous studies have convincingly shown that radiotherapy can promote T-cell generation, migration into the tumor bed, tumor cell recognition, and effector function (3). Moreover, hypofractionated radiotherapy may trigger systemic antitumor immunity enabling control of distal metastases, the so-called abscopal effect (3), and synergizes with ICB in preclinical studies (5, 10–13) and in patients (4, 14).
The effects of low-dose radiotherapy (LDRT; i.e., up to 2 Gy per fraction) remain largely unexplored in the context of cancer immunotherapy. Early evidence in a mouse model of localized neuroendocrine pancreatic tumors suggested that low-dose irradiation (i.e., 0.5–2 Gy) can reprogram the tumor microenvironment (TME), inducing macrophage M1 polarization. In turn, iNOS-positive M1 macrophages produce relevant chemokines to recruit effector T cells, whereas they induce tumor vasculature normalization and inflammation, allowing T-cell infiltration (15). More recently, Barsoumian and colleagues (16) have shown that high-dose irradiation to primary murine lung tumors combined with low-dose irradiation to secondary metastases and ICB was effective in controlling metastatic tumors through the engagement of innate and adaptive immunity, and downregulation of immunosuppressive TGFβ (16).
We sought to investigate treatment options for ovarian cancer, a disease that has so far eluded ICB combinations. Although high-dose radiotherapy has been previously used together with ICB against other tumor types, the diffuse spread of ovarian cancer throughout the peritoneal cavity puts the abdominal viscera at risk from conventional radiotherapy administered to large abdominal volumes (17, 18), which has thus been abandoned due to toxicity. Inspired by previous clinical evidence that weekly LDRT can, however, be administered safely to the entire abdominal cavity (19, 20), we tested LDRT for its ability to safely reprogram the TME (21) and facilitate response to immunotherapy in advanced ovarian cancer. Here we present novel evidence that LDRT transiently inflames tumors, rendering them vulnerable to immunotherapy. Because LDRT elicited upregulation of adaptive immune resistance mechanisms in newly inflamed tumors, a rational orthogonal combinatorial immunotherapy approach was pursued to address simultaneously immune checkpoints on effector T cells, regulatory T cells (Treg), and antigen-presenting cells (APC) to control tumors. The effect of the combination was predicated on simultaneous mobilization of both innate and adaptive immunity. Importantly, high-dimensional interrogation of tumors with single-cell RNA sequencing (scRNA-seq) revealed that effective LDRT profoundly reprogrammed the TME, newly enlisting DCs and activated effector CD4+ and CD8+ T cells executing a cytolytic transcriptional program that was in part hinged on NKG2D expression. These results were translated in a phase I clinical trial, where LDRT resulted in de novo inflammation, and regression of metastatic solid tumors when combined with orthogonal immunotherapy.
Results
Low-Dose Whole Abdominal Radiotherapy Induces Immune-Cell Infiltration in Advanced Orthotopic Ovarian Cancer
To evaluate the impact of low-dose whole abdominal radiotherapy (LD-WART) in ovarian cancer, we chose the orthotopic intraperitoneal (i.p.) murine ID8 model (22), characterized as being propense to losing intraepithelial T cells with progression in vivo (23). In order to gauge the schedule of LDRT delivery, we performed experiments looking at the time course of cell response (Fig. 1A). We ascertained that LDRT induced acute stress in ID8 cells (evidenced by calreticulin exposure), without significant effect on survival in vitro (Supplementary Fig. S1A and S1B). In vivo, it induced tumor cell DNA damage (revealed by γH2AX foci) without affecting tumor growth (Supplementary Fig. S1C and S1D). However, 1 Gy irradiation of tumors was sufficient to induce important transcriptional changes in vivo, notably a significant upregulation of inflammation, including IFNα and IFNγ responses, complement activation, IL6/JAK/STAT3 signaling (Fig. 1B), expression of key chemokines known to attract T and natural killer (NK) cells, as well as cross-presenting DCs (refs. 23–25; Fig. 1C and D) and other inflammatory markers (Supplementary Fig. S1E–S1I).
Figure 1.

LDRT induces immune-cell infiltration in orthotopic ID8 tumors. A, Treatment schema of mice engrafted with intraperitoneal ID8 ovarian tumors. Arrow represents administration of LD-WART (1 Gy). B and C, NanoString analysis of LD-WART treated versus control tumors. MSigDB pathways (B) and intratumoral levels of cytokines and chemokines (C) are displayed as heat maps. Red, upregulated; blue, downregulated. D, mRNA levels of intratumoral cytokines and chemokines. E, Heat map of cell density changes in tumors based on NanoString analysis. The heat map legend applies to B, C, and E. F, Flow cytometry quantification of TILs. G, CD8+:Foxp3+ cell ratio by mIF imaging five days after LD-WART. H, Flow cytometry quantification of CD8+ TILs in control mice and mice subjected to IFNα receptor blockade or IFNγ depletion. Symbols represent individual tumors and bars the mean. Data are representative of three independent experiments and are presented as mean ± SEM. *, P ≤ 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; Student unpaired t test.
Advanced ID8 tumors exhibited minimal inflammation at the steady state, but we detected an important influx of lymphocytes, NK cells, macrophages, and DCs on day 5 after radiotherapy by gene signatures, validated by immune staining on day 7 following 1 Gy radiotherapy (Fig. 1E and F). At the steady state, there were very few intraepithelial CD8+, CD4+, or CD11b+ cells, with most immune cells largely confined to the peritumoral stroma, whereas after radiotherapy CD8+, CD4+, and CD11b+ cells localized in the intraepithelial tumor compartment (Supplementary Fig. S1J). Comparing 0.5, 1, or 2 Gy radiotherapy by IHC, we observed the highest infiltration of CD8+, CD4+, and CD11b+ cells and the highest CD8+:Foxp3+ cell ratio following 1 Gy (Fig. 1G; Supplementary Fig. S1J); this dose was thus chosen for all subsequent experiments. T-cell inflammation tended to subside within a week, but repeat administration of 1 Gy in weekly intervals resulted in the sustained recruitment of immune cells into ID8 tumors (Supplementary Fig. S1K) and was used in all subsequent experiments. T-cell influx was specific to tumor deposits, as we did not observe any changes in T-cell content in retroperitoneal lymph nodes, nor in the spleen, which also received 1 Gy radiotherapy (Supplementary Fig. S1L). Demonstrating its dependence on IFN signaling, infiltration of CD8+ cells was abrogated in vivo by IFNα receptor blockade or IFNγ depletion (Fig. 1H). Thus, cyclical LD-WART reprograms the TME to inflame advanced ID8 tumors in a mechanism involving IFN activation.
Metronomic Radiotherapy Confers Tumor Responsiveness to Combinatorial Immunotherapy
We sought to develop a combinatorial treatment strategy addressing immune targets upregulated by LDRT. Thus, we administered ICB with αPD-1 and αCTLA4-blocking Ab, to activate T cells given the detection of increased Pd1 and Ctla4; agonistic αCD40 Ab, to activate APCs (26) given the increase in Cd40 (Supplementary Fig. S2A); and low-dose cyclophosphamide (CP), which attenuated Tregs (ref. 27; Supplementary Fig. S2B). The combinatorial treatment comprising CP on day 0, and LD-WART, ICB, and αCD40 antibody on day 1 [henceforth dubbed radio-combinatorial immunotherapy (RACIM)], was administered weekly, thrice (Fig. 2A), starting at a time when mice had obvious intraperitoneal ID8 tumors by imaging (luminescence). Strikingly, 83.5% of mice receiving RACIM exhibited tumor response by imaging while on therapy (14% complete response and 11% deep partial response with 97%–98% reduction) by day 20. Most mice with partial response, however, progressed after discontinuation of therapy (Fig. 2B; Supplementary Fig. S2C), with median overall survival of 69 days (Fig. 2C). On day 90, all surviving mice were disease-free by imaging and by pathologic examination, yielding an overall cure rate of 15%. Strikingly, the combination immunotherapy lacking LDRT (i.e., CP + ICB + αCD40, dubbed CIM) exhibited no therapeutic effect (0% response or tumor cure; median survival 50 days; Fig. 2B and C). RACIM therapy was associated with no obvious toxicity; mice experienced no weight loss nor systemic inflammation (Supplementary Fig. S2D and S2E). Importantly, foregoing LDRT at the second and/or third cycle reduced the therapeutic efficacy of the combination (Supplementary Fig. S2F).
Figure 2.

Metronomic radiotherapy enables combinatorial immunotherapy. A, Schema of in vivo study evaluating treatment by LD-WART (1 Gy) versus CIM versus RACIM. B, Left, waterfall plot representing the percentage change in tumor BLI levels at day 20 for mice treated in the different groups (RACIM, n = 36 mice; control, n = 41; CIM, n = 26; LD-WART, n = 24). Complete response (CR), PR (at least 30% decrease in BLI from baseline), SD, PD (at least 20% increase in BLI from baseline). Right, tumor growth curves evaluated by BLI. C, Kaplan–Meier analysis in representative mice treated in six different experiments (RACIM, n = 80 mice; control, n = 92; CIM, n = 30; LD-WART, n = 24). P values were determined by a one-sided log-rank Mantel–Cox test. D, Heat map of cell density changes in tumors based on NanoString analysis. E, mIF imaging reveals immune-cell infiltration in tumors at cycle 2, day 5 (20× magnification; DAPI nuclear counterstaining; images are representative of n = 5 mice/group). Number of cells per HPF plotted as mean ± SEM; P was calculated using unpaired two-tailed Student t tests. F–H, Immune-cell phenotypes evaluated on single-cell suspensions of control, RACIM, or RACIM one component, ID8 tumors (n = 5–7 mice per group). Kaplan–Meier analyses of overall survival following RACIM in the absence of anti–PD-1 (F) or anti-CTLA4 antibody, CP (G), or anti-CD40 agonist antibody (H) for n = 10 mice per group. P values were determined by a one-sided log-rank Mantel–Cox test. I, mRNA levels of Tnfa and Ifng in differently treated ID8 tumors. In vivo data are representative of three independent experiments. *, P ≤ 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
We validated the RACIM treatment in the subcutaneous (s.c.) Lewis lung carcinoma (LLC) model, which is reportedly nonresponsive to ICB (28) and exhibits scarce T-cell infiltration (29). We found that unlike early LLC tumors (∼10 days, ∼100 mm3), advanced tumors (∼15–20 days, 200–400 mm3) are depleted of T cells (Supplementary Fig. S2G). We treated s.c. LLC tumors of 300–350 mm3 with RACIM and observed significant control and survival benefit, similar to the ID8 model (Supplementary Fig. S2H). Thus, orthogonal combinatorial immunotherapy leveraged the immunomodulatory effect of LDRT and led to marked therapeutic response in advanced low T cell–infiltrated tumors.
We used gene-expression analysis (NanoString) to analyze the effects of RACIM 48 hours after cycle 2 (i.e., the peak of therapeutic response) in ID8 tumors. We observed significant upregulation of genes associated with T cells, monocytes, and DCs following RACIM relative to CIM or to untreated control tumors (Fig. 2D). By immune staining, we confirmed a marked influx of CD11b+ myeloid, CD8+, and CD4+ T cells following RACIM or LDRT relative to CIM or control tumors (Fig. 2E), with most pronounced infiltration of CD11b+, CD8+, and CD4+ T cells seen after RACIM.
To unveil the individual contributions of the components of RACIM, we administered take-one-out combinations, that is, RACIM without one component. Importantly, the frequency of CD4+ and CD8+ T cells decreased significantly when ICB was omitted (Fig. 2F). Underscoring the key role of effector T cells, no animal cure was observed when ICB was omitted from RACIM (Fig. 2F). Furthermore, highlighting the importance of attenuating Treg with RACIM, we observed a significant increase in Tregs and a decrease in the CD8/Foxp3 ratio, and no mouse cure when CP was omitted (Fig. 2G). Notably, in the absence of agonistic αCD40 we found a decrease in the frequency of MHC-II–expressing CD11b+CD11c+ cells and an increase in M2 macrophages (F4/80+CD206+, Fig. 2H), confirmed by a significant decrease in Nos2 expression in CD11b+ cells (Supplementary Fig. S2I). Each perturbation of RACIM converged to a common effect, that is, collapse of effector T cells, revealed by a significant decrease in Tnfa and Ifng gene expression in ID8 tumors (Fig. 2I). Hence, leveraging the proinflammatory effect of LDRT, RACIM exhibited a dramatic therapeutic effect on originally low T cell–inflamed tumors, and all components of the combinatorial treatment contributed to mobilizing an effective antitumor immune response.
RACIM Expands Tumor-Rejecting CD4+ and CD8+ Tumor-Infiltrating Lymphocytes with Activation and Exhaustion Features
We sought to understand whether in addition to important quantitative differences in T-cell infiltration, the curative effects of RACIM were further associated with qualitative differences in the tumor-infiltrating lymphocytes (TIL). We analyzed TILs five days after the second LDRT cycle by scRNA-seq and used ProjectTIL (30) to assign T cells to previously described T-cell states. We identified nine distinct T-cell states (Fig. 3A). TILs from tumors treated with RACIM were highly enriched in activated effector T-cell populations, which were assigned largely to exhausted (Tex), progenitor-exhausted (Tpex) and effector memory (TEM) states (Fig. 3A; ref. 31). The Th1/Treg as well as the Tex/Treg cell ratio were significantly increased by RACIM over CIM or control (Supplementary Fig. S3A). Relative to CIM-treated tumors, CD4+ Tex cells were the most differentially expanded following RACIM (Fig. 3B). Importantly, by concomitant T-cell receptor (TCR) sequencing, we determined that CD4+ Tex and Tpex were the most clonally expanded cells, suggesting tumor specificity (Supplementary Fig. S3B; refs. 31–34). Within the top expanded CD4+ T-cell clonotypes, we found the same clonotype largely in a Tex or Tpex state, but less frequent cells from the same clonotypes were also TEM, early activated, Th1 or follicular helper (Tfh) cells (Supplementary Fig. S3B), suggesting that CD4+ Tex cells may evolve from all these precursor states. Indeed, among all expanded clonotypes, CD4+ Tex and Tpex compartments shared numerous T-cell clones, and both Tex and Tpex shared clones with the TEM, Th1, or Tfh compartments (Fig. 3C). To the best of our knowledge, precursor states for exhausted CD4+ TILs have not been described to date.
Figure 3.

Low-dose irradiation and combinatorial immunotherapy expands tumor-rejecting CD4+ and CD8+ TILs exhibiting states of activation and exhaustion. A, UMAP plots of tumor lymphocyte scRNA-seq data (n = 3 tumors/treatment, n = 4 tumors pooled for control, all collected on day 5 of cycle 2). Left, reference map for all groups. Right, contour plots reveal cell density/group. Supervised T-cell state classification by TILPRED identifies functional T-cell subsets: Tpex, Tex, TEM, early activated (EA), Th1, Tfh, Treg, and naïve-like T cells. B, Fold change in T-cell subsets following RACIM versus CIM. C, Cord diagram of the Jaccard similarity coefficient shows the relative number of common TCRs shared between CD4+ T-cell subsets following RACIM. D, Violin plots representing the expression of various activation and cytotoxicity markers in CD4+ T-cell subsets. E, CD4+ T cells expressing indicated genes across subsets and their corresponding average expression (size of dot indicates the percentage of cells in each subset; expression intensity is indicated by color). F, Pseudotime trajectory analysis of CD4_Tpex, CD4_Tex, and Th1 clusters identified by unsupervised single-cell analysis. G, Left, SPICE graphic representing flow-cytometric analysis of GzmB and cytokine production by CD4+ TCF1−PD1+ TILs. Right, bar plots representing cytokine production by CD4+ TCF1−PD-1+ TOX+ cells after PMA/ionomycin or anti-CD3/anti-CD28 TCR stimulation. H, Kaplan–Meier analysis of overall survival of RACIM-treated mice, depleted or not of CD4+ T cells. P values determined by a one-sided log-rank Mantel–Cox test. I, The percentage of CD8+ T cells expressing indicated genes across subsets and their corresponding average expression (as in E). J, Violin plots showing expression of Ifng, Gzmb, and Prf1 in CD8+ T cells following CIM vs. RACIM. K, Bar plots representing the most clonally expanded CD8+ T-cell clonotypes (by TCR-seq) following RACIM treatment (TCRs in all three tumors: #1; in individual tumors: #2–4). L, Cord diagram of the Jaccard similarity coefficient shows the relative number of common TCRs between CD8+ T-cell subsets following RACIM. M, Left, SPICE graphic representing flow cytometric analysis of GzmB and cytokine production by CD8+PD-1+TCF1− TILs. Right: bar plots representing cytokine production by CD8+ TCF1−PD-1+TOX+ cells after PMA/ionomycin or anti-CD3/anti-CD28 TCR stimulation. N, Kaplan–Meier analysis of overall survival of RACIM-treated mice depleted or not of CD8+ T cells. P values were determined by a one-sided log-rank Mantel–Cox test. Data are representative of n = 3 biologically independent experiments (n = 5–10). *, P ≤ 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
CD4+ Tex TILs following RACIM were characterized by significantly higher expression relative to CIM of Ifng, Prf1, and Gzmb, associated with effector function and cytolytic capacity; Pdcd1, Lag3, Havcr2, and Tox, associated with exhaustion; and costimulatory receptors Cd28, Cd27, Icos, and Tnfrsf4; while lacking expression of Tcf7 and Tbet transcription factors, indicating a terminal Tex state (Fig. 3D and E). Pseudotime analysis supported a state evolution model in which preexhausted Th1-like cells differentiate into intratumoral CD4+ Tex cells through an intermediate CD4+ Tpex state (Fig. 3F). Along this trajectory, there was gradual upregulation of exhaustion-associated genes (Tox, Nr4a2, Pdcd1, Havcr2, Lag3, and Tigit), chemokines/chemokine receptors (Ccl2, Ccl3, Ccl5, and Cxcr6), and cytotoxicity-related genes (Prf1, Klrd1, and Nkg7), along with downregulation of progenitor-associated genes (Tcf7 and Il7r). Notably, in the intervening Tpex state, we observed peak gene expression of the chemokine Xcl1, as well as of various activation markers (Tnfsfr9/CD137, Cd81, Cd200, and Crtam; Supplementary Fig. S3C).
We confirmed by flow cytometry a significant enrichment in CD4+PD1+TCF1− cells expressing markers of exhaustion and costimulatory receptors in RACIM-treated tumors (Supplementary Fig. S3D–S3F). We also identified a higher frequency of functional CD4+PD-1+TCF1− TILs expressing IFNγ, with a small fraction of them also expressing IL2, TNFα, or GzmB (Fig. 3G). Finally, we confirmed polyfunctional cytokine production ex vivo upon stimulation in a proportion of CD4+ Tex cells (Fig. 3G).
These findings suggest that RACIM recruits a large amount of oligoclonal CD4+ cells to tumors, which acquire exhaustion but also exhibit important effector functions. We thus asked whether these cells partake in the therapeutic effect of RACIM. Indeed, CD4+ depletion abrogated the therapeutic effect of RACIM, with no cures seen (Fig. 3H).
We also identified CD8+ TIL populations exhibiting Tpex and Tex states with higher exhaustion, costimulation, and effector function following RACIM relative to CIM (Fig. 3I and J). Similar to their CD4+ counterparts, RACIM-treated tumors harbored the highest proportion of expanded CD8+ T-cell clonotypes, with the largest clonal expansion observed among CD8+ Tex cells (Fig. 3K), which shared numerous TCR clones mostly with the CD8+ TEM compartment (Fig. 3L). Expansion of PD-1+TCF1− CD8+ TILs expressing coinhibitory receptors but also CD28, CD27, and CD40L was confirmed by flow cytometry analysis (Supplementary Fig. S3G–S3I). Relative to CIM, CD8+PD-1+TCF1− TILs from RACIM-treated tumors comprised higher frequencies of functional cells secreting Gzmb, IFNγ, and/or TNFα (Fig. 3M), and TOX+ cells were still functional upon ex vivo restimulation (Fig. 3M). We confirmed the critical contribution of CD8+ T cells to the therapeutic efficacy of RACIM, as animals previously depleted of CD8+ T cells with anti-CD8 Ab lost significant survival benefit (Fig. 3N). Thus, LDRT synergizes with combinatorial immunotherapy by enlisting T cells, and specifically by expanding a population of activated, functional effector CD4+ and CD8+ T cells with tumor-rejecting capacity that acquire features of progenitor-exhausted and exhausted cells in the TME, consistent with effective antigen engagement.
RACIM Reprograms Tumor APCs and Enlists NKG2D as a Key Costimulatory Signal
We next examined the effects of therapy on the myeloid compartment. scRNA-seq analysis of CD11b+ cells from untreated, CIM- or RACIM-treated tumors revealed 29 myeloid cell transcriptomic states, which we could assign to three main populations: macrophages, DCs, and monocytes (Supplementary Fig. S4A; ref. 35). Therapy drove important reprogramming of the tumor myeloid compartment (Fig. 4A), with RACIM inducing profound changes in all three myeloid populations relative to CIM alone (Fig. 4B).
Figure 4.

RACIM reprograms tumor APCs and enlists NKG2D as a key signal. A, UMAP plots of intratumoral myeloid cell scRNA-seq data (n = 3 tumors/treatment, n = 4 tumors pooled for control, all collected on day 5 of cycle 2). Left, reference map for all groups. Red, DCs; blue, monocytes; green, macrophages. Right, 29 myeloid states among groups. B, Fold change in myeloid cell subsets for RACIM versus CIM. C, Quantification of DC clusters among groups. D, Rose plot of differentially expressed genes corresponding to DC clusters among groups. E and F, Violin plots showing expression of Batf3 (E) and H2k1 and H2d1 (MHC-I; F) transcripts in cDC1 cells among groups. G and H, Kaplan–Meier analysis of control versus RACIM in Batf3−/− mice (G), and in wild-type (WT) mice (H) in the presence of fingolimod (FTY-720) treatment. P values were determined by a one-sided log-rank Mantel–Cox test. I, Heat map showing expression of the most representative genes for clusters 9, 19, and 21. Gene expression was normalized to median expression value per gene across all clusters shown in the heat map. J, Percentage of cells expressing Rae1, Ulbp1, H60b, and H60c, and average expression in the myeloid compartment by scRNA-seq (size of dot indicates the percentage of cells in each subset; expression level is indicated by color). K, RAE1 expression on intratumoral CD11b+CD11c+MHC-II+ cells determined by flow-cytometric analysis on day 5 of cycle 2. L, Left, mIF imaging reveals RAE1 expression (red) by CD11b+ cells (yellow; 20× magnification; DAPI nuclear counterstaining; representative of n = 5 mice/group). Right, number of CD11b+RAE+ cells per HPF plotted as mean ± SD; P was calculated using unpaired two-tailed Student t tests. M and N, The percentage of CD4+ and CD8+ exhausted T cells expressing NKG2D at the transcriptional (M and N, Klrk1 gene by scRNA-seq analysis) and protein levels (O and P, flow cytometry analysis) on day 5 of cycle 2. Q and R, The percentage of intratumoral Ki-67+proliferating CD4+TCF1+PD-1+ (Q) and CD8+TCF1−PD-1+ (R) cells upon RACIM on day 5 of cycle 2. S–T, NKG2D expression on intratumoral CD4+TCF1−PD-1+ (S) and CD8+TCF1−PD1+ (T) T cells determined by flow cytometry on day 5 of cycle 2 in control or RACIM or RACIM without ICB-treated tumors. U, RACIM survival with NKG2D blockade. P values were determined by a one-sided log-rank Mantel–Cox test. Data are representative of two to three independent experiments (n = 5–10 mice/group). Unless otherwise indicated, statistical analysis was performed using Student unpaired t test; error bars represent mean ± standard deviation. *, P ≤ 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
Canonical macrophages have been classified as M1 and M2, with purported proinflammatory and anti-inflammatory/immunosuppressive roles, respectively (36). We identified 22 monocyte/macrophage clusters according to Immgen signatures (ref. 37; Fig. 4A; Supplementary Fig. S4B). Macrophages at baseline could be assigned collectively to M2 as they expressed Mrc1 (CD206; Supplementary Fig. S4C). Conversely, CIM or RACIM induced a clear shift in macrophage states, among which we could identify M1 macrophages expressing Nos2 (iNOS; Supplementary Fig. S4D). Differential gene-expression analysis showed a further shift in macrophage transcriptional programs by RACIM relative to CIM, with upregulation of genes linked to the inflammasome pathway (Malat1, Tnfrsf1a/TNFα, Cd14, Lrp1, Dusp1, C3ar1, Calr, Itgb2, Nr4a, Il1b, and Nfkia); glycolysis (mt-Nd2); antigen presentation (H2k1, H2d1); type I IFN (Irf8 and Ifrd1); IFNγ sensitivity (ifngr1); chemotaxis and leukocyte migration (Icam1, Cxcl10, Cxcl2, and Ccr2); and wound repair (Socs3, Klf6, and Gnai2), in addition to downregulation of M2 genes (Cd5l), fatty acid metabolism and prostaglandin synthesis (Fabp5, Prdx1, and Tmsb4x), and iron-induced oxidative stress (Ftl1, Fth1, Ftl1-ps1, and Prdx1; Supplementary Fig. S4E). Moreover, RACIM-associated macrophages displayed higher Cd40 and Cd86 (Supplementary Fig. S4F). The above changes were corroborated by flow cytometry, with a substantial reduction of Ly6G−CD11b+F480+CD206+ M2 macrophages observed following RACIM (Supplementary Fig. S4G).
We also found different intratumoral DC states, which could be further annotated as being either conventional (cDC1; Xcr1hi; cluster 25), cDC2 (Il1r2hi; cluster 21), cDC2/mono-like DC (cDC2/MoDC; Clec10ahi; clusters 9 and 19), cDC3 (Ccr7hi; cluster 23), or plasmacytoid (pDC; Cox6a2hi; cluster 28; Fig. 4C and D). These resembled DC states previously reported in other mouse tumor models, indicating that similar to their human counterparts (35, 38, 39), mouse DC states are largely conserved across tumor types (Supplementary Fig. S4H). However, the cDC2/MoDC state (clusters 9 and 19) was specifically associated with RACIM treatment in the ID8 tumor model (Fig. 4C and D).
Although typically found at low frequency in tumors, Batf3-expressing cDC1 efficiently cross-present tumor antigens to CD8+ T cells (40) and are critical mediators of antitumor immunity and response to ICB (41). We found cDC1 (cluster 25) in RACIM-treated tumors, whereas they were almost absent in CIM (Fig. 4C and E). Notably, although cDC1 were also present in control tumors (Fig. 4C and E), upon RACIM treatment they exhibited higher expression of MHC class I (H2K1 and H2D1), suggesting an improved ability to cross-present antigens (Fig. 4F). Batf3 is a key transcription factor driving the development of cDC1 (40, 42, 43). To assess the contribution of Batf3 to tumor control, we administered RACIM to Batf3−/− mice. Loss of Batf3 abrogated the therapeutic benefit of RACIM (Fig. 4G). Because tumor control associated with Batf3-expressing DCs depends on T-cell migration from draining lymph nodes to tumors (44), we administered fingolimod (FTY720), an inhibitor of lymphocyte egress from lymph nodes. This also abrogated the effect of RACIM (Fig. 4H). TCF1−PD-1+CD8+ cells were decreased in the tumors of Batf3−/− mice, indicating that cross-presenting DCs serve to maintain the pool of terminally differentiated effector TCF1− cells (Supplementary Fig. S4I).
Given the dramatic expansion of CD4+ tumor-rejecting TILs upon RACIM, we next interrogated cDC2 (cluster 21) and cDC2/MoDC (clusters 9 and 19), which may interact with CD4+ T cells and support their antitumor activity (45). We found that RACIM induced profound reprogramming of the DC compartment, which explains its impact on adaptive immunity seen above (Fig. 4C). For example, RACIM suppressed DC cluster 21, enriched in anti-inflammatory genes such as Mt, Tgfb1, and Nr4a2, whereas it expanded clusters 9 and 19, enriched in genes involved in MHC class I presentation such as Psmb8/10, Psma1/4/5/7, and Tap1/2; Cd40 and Cxcl9/Cxcl10; and genes related to type I IFN (Gbp2, Isg15, Ifi205, Irf7, and Irf8) associated with antitumor immunity (ref. 46; cluster 9); and class II presentation such as H2Ab1, H2Aa, H2DMa, and Cd74 (cluster 19; Fig. 4I).
We confirmed the important shifts in DCs induced by RACIM via flow cytometry, as we found a significant increase in activated CD11b+CD11c+MHC-II+CCR2+/− DCs (Supplementary Fig. S4G) with increased coexpression of costimulatory ligands CD40, CD70, CD80, and CD86 relative to CIM (Supplementary Fig. S4J). Importantly, we noted that RACIM expanded cDC1 and cDC2 expressing the stress response marker Rae1, a ligand to the NKG2D costimulatory receptor (Fig. 4J). Rae1 upregulation was quite specific, because we did not detect transcripts for other NKG2D ligands in the DCs. This was corroborated by flow cytometry and tissue immune staining, where we identified higher levels of RAE1 on CD11b+CD11c+MHC-II+ DCs (Fig. 4K and L) in RACIM-treated tumors.
We thus asked whether the NKG2D receptor was upregulated in tumor-rejecting lymphocytes mobilized by RACIM. We found by scRNA-seq that both CD4+ and CD8+ Tex cells significantly upregulated Klrk1 (NKG2D) in RACIM-treated tumors (Fig. 4M and N). By flow cytometry, we confirmed that CD4+TCF1−PD-1+ T cells and, to a lesser extent, CD8+TCF1−PD-1+ TILs from RACIM expressed significantly higher levels of NKG2D relative to CIM-treated tumors (Fig. 4O and P).
NKG2D serves as an important costimulatory receptor for effector T cells in peripheral tissues (47, 48). We observed significantly increased Ki-67 expression in NKG2D+ TCF1−PD-1+ CD4+ and CD8+ T cells as compared with their NKG2D− counterparts (Fig. 4Q and R), indicating that NKG2D expression defines a subset of canonical CD4+ or CD8+ exhausted TILs that retain proliferative capacity. Remarkably, the increase in NKG2D expression in CD4+PD1+ T cells, and to a lesser extent CD8+PD1+ T cells, was abrogated when anti-CTLA4 or anti–PD-1 blockade was omitted from the treatment cocktail (Fig. 4S and T), revealing the important interdependencies that underpinned the synergies in RACIM. We thus asked whether NKG2D supports the function of tumor-rejecting T cells upon RACIM. Indeed, NKG2D blockade by antibody attenuated NKG2D+TCF1−PD-1+ CD4+ as well as CD8+ Tex cells (Fig. 4O and P) and abrogated the therapeutic efficacy of RACIM (Fig. 4U), without affecting RAE1-expressing DCs (Fig. 4K).
LDRT Combined with ICB Induces Responses in Advanced Immune-Desert Human Tumors
We sought to translate the above advances to the clinic. We thus conducted a phase I clinical study (RACIN, NCT03728179), details available in Supplementary Notes, in which patients with solid tumors exhibiting <5 intraepithelial CD8+ cells per high-power field (HPF), otherwise interpreted as “immune-desert” tumors, were treated with LDRT (at 0.5 or 1 Gy per fraction, every two weeks, total dose 6 Gy or 13 Gy, respectively) delivered to all (target and nontarget) metastatic deposits in combination with ICB, which similar to the mouse comprised low-dose CP (200 mg/m2 every two weeks) to attenuate Tregs (27), combined with anti–PD-1 (nivolumab), 240 mg every two weeks and anti-CTLA4 (ipilimumab), 1 mg/kg every 6 weeks (q6wk), for up to 24 weeks. Doses of ipilimumab and nivolumab were inspired by the previous CheckMate 227 study (49), which demonstrated clinical activity of ipilimumab 1 mg/kg every six weeks and nivolumab 3 mg/kg every two weeks in patients with advanced non–small cell lung cancer. The 240-mg flat dose every two weeks of nivolumab was subsequently approved by the FDA as equivalent to the 3 mg/kg every two weeks dose (50).
In the absence of available drugs to activate myeloid cells, we elected to administer at least aspirin (300 mg orally, daily) to suppress prostaglandin E2 (PGE2), a major immunosuppressive factor elaborated by tumor myeloid cells (51). Following completion of the four cycles with ipilimumab/nivolumab, all eligible patients could receive nivolumab (240 mg every two weeks) with daily aspirin until progression or toxicity. LDRT was applied to all metastatic deposits identified by radiologists as pathologic, sparing the bone marrow as much as possible (52). We treated all patients with the same dose of CIM but varied the dose of radiotherapy: The first three patients received 0.5 Gy, and five additional patients received 1 Gy at each lesion (Fig. 5A). The primary endpoint was dose-limiting toxicity (DLT), defined as grade 4 or worse by Common Terminology Criteria of Adverse Events (version 4.03), during the period from cycle 0/day 1 (C0D1) to C2D1, eight weeks later (Supplementary Table S1).
Figure 5.

Low-dose irradiation plus ICB induces responses in advanced human immune-desert tumors. A, Therapeutic schema of the phase I RACIN study. B, Spider plot depicts the percentage change in the sum of targeted irradiated metastases compared with baseline. C, Swimmer plot depicts patients' response to RACIN over time; each bar, one patient; light orange, time on combination treatment; green, time on maintenance treatment; cohort 1: 0.5 Gy, cohort 2: 1 Gy; orange triangles, completed treatment; asterisk, treatment termination due to toxicity or progression; black circles, death. iRECIST v1.1 was used to indicate PR (iPR, green diamond), SD (iSD, light blue square), confirmed progressive disease (iCPD, maroon circle), or unconfirmed (iUPD, maroon empty circle). D,68Ga-PSMA PET/CT images of irradiated tumors (white arrows) before and after treatment from a patient with metastatic castration-resistant prostate cancer having SD according to Prostate Cancer Clinical Trials Working Group 3 (PCWG3) but an important response on 68Ga-PSMA PET/CT images. Changes in PSA tumor marker. Progression observed outside the irradiated areas 24 weeks after treatment initiation. E,18FDG-PET/CT images of irradiated tumors (white arrows) before and after treatment from a patient with high-grade serous ovarian carcinoma having by iRECIST iPD, but an important response on 18FDG-PET/CT imaging. Changes in the CA125 tumor marker. Progression outside the irradiated areas 24 weeks after treatment initiation. F, CT images of irradiated tumors (white arrows and circles) before and after treatment from a patient with gallbladder cancer having PR by iRECIST and 70% reduction from baseline in targeted irradiated lesions. Changes in the CA 19-9 tumor marker. Progression outside the irradiated areas 17 weeks after treatment initiation. G, Anatomic location of irradiated target and nontargeted lesions in responder patients and the anatomical location of tumor recurrence (D2, second dorsal vertebrae; D12, dorsal 12; R, right; L, left; LN, lymph node; liver segments identified with roman numbers III, IV, and V).
Eight immunotherapy-naïve patients with advanced metastatic prostate (n = 4), high-grade serous ovarian (n = 2), or gastrointestinal tract (n = 2) tumors, with no other therapeutic options after a median of three prior lines of chemotherapy, were recruited between March and August 2019. The median number of intraepithelial CD8+ cells at baseline was 2.4 cells (range, 2–4) per HPF, and seven patients (87.5%) had <1% PD-L1+ tumor cells at baseline. The median number of nonsynonymous somatic mutations per Mb for the tumors was 2.41 Mb (range, 0–6.9). None of the tumors exhibited mismatch repair deficiency or BRCA mutation. At data cutoff on August 17, 2020, the median follow-up was 11.9 months (range, 4.4–17.2 months). Patient and tumor characteristics are summarized in Supplementary Table S2.
Adverse events (AE) of any grade occurred in all eight patients, the most frequent being related to clinical investigations or gastrointestinal events (Supplementary Table S3). None of the AEs were attributed to LDRT. Immune-related serious AEs (SAE) of grade 3 or more occurred in two patients (25%). A grade 3 colitis occurred after two cycles of combination treatment in a patient with prostate cancer. The patient was taken off study and was treated with methylprednisolone (1 mg/kg i.v. bolus once), followed by oral prednisone 25 mg/day for 2 months. The patient progressed based on bone scan one month after treatment discontinuation. Two months after the initial episode, the patient developed off study a second episode of grade 3 colitis, managed with infliximab (5 mg/kg i.v. once), oral steroids, and vedolizumab (300 mg i.v. thrice), with complete resolution. A grade 4 myocarditis occurred during the first cycle of treatment in another prostate cancer patient (the only DLT). The patient (1EEY) was taken off study and was treated with intravenous methylprednisolone (1 g/day for 5 days) followed by oral prednisone 1 mg/kg, in association with mycophenolate mofetil (1 g for 5 days orally, followed by 500 mg for 30 days). A pacemaker was implanted. As troponin levels remained elevated after 20 days of immunosuppressive treatment, three doses of tocilizumab (8 mg/kg) were administered with resolution of the event. The patient died three months after treatment discontinuation. One more patient with gallbladder cancer died during the study, both deaths attributed to disease progression. Two other deaths also occurred due to other reasons.
Ipilimumab alone was discontinued in two patients with prostate cancer (25%) for G2 colitis and G3 hepatitis, respectively, both with slow improvement on oral steroids. In both cases, toxicity occurred after two cycles of treatment and did not recur with the continuation of radiation, CP, and nivolumab. Only one patient was eligible for maintenance with nivolumab and aspirin, which was discontinued after one cycle due to disease progression.
Tumor responses were evaluated by Immune Response Evaluation Criteria in Solid Tumors (iRECIST; ref. 53), and when available, relevant serum tumor markers. We observed tumor size reduction in targeted irradiated lesions in three patients overall (37.5%; Fig. 5B). Four other patients experienced stable disease (SD), with overall disease control rate [partial response (PR) + SD] of 87.5%, whereas one patient (12.5%) had confirmed disease progression (Fig. 5C). The overall response rate by iRECIST was 12.5%, with one of eight patients achieving PR. Notably, a patient with prostate cancer and another with ovarian cancer were regarded as SD and PD, respectively, by iRECIST, but all irradiated lesions demonstrated dramatic metabolic response in fields that received LDRT, based on molecular imaging with 68GaPSMA-PET and 18FDG-PET/CT, respectively. In both cases, disease progression was related to new lesions emerging uniquely outside of the irradiated areas (Fig. 5D and E). Another patient with gallbladder cancer had PR according to iRECIST; this patient subsequently progressed, also outside the irradiated volume (Fig. 5F). Emphasizing the importance of irradiating all lesions, we observed tumor progressions only outside the irradiated volumes in all three patients where responses were documented by imaging (Fig. 5G). Changes in radiographic appearance of tumors over time, biochemical responses, radiation dosimetry, and the location of new metastases are illustrated in Fig. 5D–G.
Immune-Desert Tumors Are Reprogrammed Following LDRT
We sought to assess whether LDRT induced similar biological effects in human tumors as in the mouse tumor model. We analyzed biopsies obtained from the same metastatic deposits at baseline and 7 to 10 days following the first administration of LDRT, prior to initiating ICB. We considered as responding tumor lesions those that exhibited subsequent reduction in size following combined LDRT– immunotherapy and compared responding tumor lesions from three patients who experienced PR or SD (patient 19F7 with cholangiocarcinoma, 1EEY with prostate cancer, and 02F5 with ovarian cancer in Fig. 5B) to nonresponding lesions from four other patients. Similar to the mouse model, we observed a marked influx of T cells, which was mainly composed of CD4+ cells in responding tumors (representative images in Fig. 6A). Differential gene-expression analysis of matched pre- and postirradiation biopsies revealed different patterns of response to LDRT in responding versus nonresponding tumors. For example, in responding tumors, LDRT triggered activation of DNA damage response (POLB, NEIL1, and MLH1), type I IFN response (TBK1, IFIH1, and EIF2AK), immune-cell activation (NFKB, NTF3, and ATF1), antigen presentation and innate immune activation (CD83, C1QBP, and ZIC2), as well as TCR activation and effector memory (CD44, SOS1, and RICTM), whereas we also detected downregulation of genes related to epithelial-to-mesenchymal transition (COL1A2, COL1A1, PLAT, COL6A3, COL5A1, and FN1). Conversely, nonresponding tumors upregulated genes associated with immune suppression (HAVCR2, IL10, LILRB4, and LAIR1) and downregulated genes associated with DNA repair (PRKDC, RAD21, RAD50, and DDIT4) and inflammation (IL6, ISG20, IFNB1, and TNFA; Fig. 6B; Supplementary Fig. S5A). Furthermore, responding tumors exhibited a significant increase in Th1 signatures following LDRT, whereas nonresponding tumors were characterized by increasing M2 macrophage and tolerogenic DC signatures (Fig. 6C). Thus, as in mice, LDRT successfully reset in responding tumors the immune TME, recruiting innate and adaptive immune cells. Indeed, we noted an important overlap in the gene signatures of responding mouse and human tumors, with Th1 signatures reaching statistical significance in both (Fig. 6D).
Figure 6.

Effect of low-dose irradiation on tumor immune landscape. A, TILs before and after LDRT revealed by mIF imaging in two representative responding tumors. Left, representative mIF images (20× magnification; CK, pancytokeratin); right, quantification of CD4+ and CD8+ cells. B, Scatter plot showing differential gene expression between baseline and post-irradiation biopsy in responding (x-axis) versus nonresponding tumors (y-axis). The log2 of the fold change in median gene expression (log2FC) is shown (positive values indicate upregulation post-LDRT). Genes displaying a significant change (unadjusted P < 0.05) are color-coded as shown in the legend. C, Line plots showing the progression of immune gene signature scores from baseline to post-LDRT biopsies in responding versus nonresponding tumors. D, Scatter plot showing differential immune signature score analysis between baseline and post-irradiation biopsy in responding human tumors (x-axis) versus responding mouse (RACIM) ID8 tumors (y-axis; top) and between nonresponding human versus nonresponding (CIM) mouse tumors (bottom). E, NanoString GeoMx analysis of intraepithelial tumor immune infiltrates vs. tumor stroma immune infiltrates in responding versus nonresponding tumors. The log2 of the fold change in the median of the signature score (log2FC) is shown. Immune signature score displaying a significant change (unadjusted P < 0.05) is color-coded as depicted in the legend of B. F, Comparison of TCR CDR3 diversity by clonality, Shannon diversity entropy, Gini coefficient, richness, and shared frequency in three patients with responding tumors.
We used spatial transcriptional profiling (GeoMx) to test whether immune activation in responding tumors involved the intraepithelial tumor compartment. We confirmed that spatial transcriptional profiling accurately captured immune cells by correlating gene-expression data and cell counts of CD3+ and CD8+ cells by multispectral immunofluorescence (mIF; Supplementary Fig. S5B). We acquired topologic transcriptional immune profiles of mIF-guided regions of interest (54) from responding and nonresponding tumors, profiling tumor islets, and stroma at baseline and following LDRT. Using single sample gene set enrichment analysis (ssGSEA) of such signatures (55), we observed distinct immune profiles associated with therapeutic outcome. Responding tumors exhibited an increase in Th1, CD8+, and TEM signatures located mainly in tumor islets following LDRT (Fig. 6E). Conversely, nonresponding tumors displayed an upregulation of M2 macrophage and neutrophil signatures following radiotherapy, which were detected mainly in tumor stroma (Fig. 6E).
Reinvigorated clonal T-cell responses have been reported in the peripheral blood of patients undergoing successful ICB (56). Because in the mouse we observed that mobilization of antitumor T cells from lymph nodes was necessary for response to the combined treatment, we used deep sequencing to evaluate TCR repertoire changes in peripheral blood following LDRT. We observed increased TCR clonality, confirmed by a reduced Shanon entropy index, and a significant increase in inequality of clonal frequencies by Gini coefficient, all revealing the mobilization of expanded peripheral blood T-cell clones following LDRT, specifically in patients with responding tumors (Fig. 6F; Supplementary Fig. S5C). Furthermore, we noted a significant increase in the frequency of the largest dominant TCRs clones between pre- and post-LDRT blood samples in patients with responding tumors. Thus, similar to mouse, RACIN mobilized systemic immunity successfully in patients who benefitted from the combination.
Discussion
T-cell exclusion from the TME represents a major mechanism of intrinsic resistance to ICB (1, 2). Here we show that LDRT drives T-cell inflammation and creates immune vulnerabilities, which can then be successfully exploited with rational combinatorial immunotherapy. Although high-dose radiation (>5 Gy per fraction delivered to small tumors as stereotactic treatment) increases antigen release and presentation, and primes immune cells (3), we chose to deliver LDRT (i.e., doses below the threshold to directly kill cancer cells) to enable administration of large volumes in order to irradiate all metastatic deposits and promote immune-cell infiltration into them without causing toxicity.
Two prior preclinical studies have explored the use of LDRT to enhance immunotherapy, one in combination with adoptive T-cell transfer and one with high-dose radiotherapy plus ICB (15, 16). Here we demonstrate in both mice and humans that a dose of 0.5 to 1 Gy elicits dramatic reprogramming of the TME. In advanced murine ID8 ovarian tumors, this included the triggering of DNA damage and IFN response, the upregulation of numerous cytokines and inflammatory chemokines, as well as of druggable targets including immune checkpoints and CD40, thus offering a window of opportunity to rationally intervene with immune modulation. Based on these TME changes upon 1 Gy irradiation, we devised a combinatorial treatment including αCD40 agonist antibody, ICB with PD-1 and CTLA4 blockade, and depletion of Tregs via CP (57). We demonstrated that all components of the cocktail (referred to as RACIM) and repeat LDRT were required for therapeutic efficacy. Moreover, deconvolution experiments, along with comprehensive characterization of the immune TME, revealed a dynamic interdependence of innate and adaptive immunity activation, a fundamental requisite for eradicating cancers (58).
Given the critical need for ICB in the RACIM cocktail for tumor control in conjunction with LDRT, it is not surprising that adaptive immune mechanisms were central in mediating tumor rejection in mice. Both CD4+ and CD8+ cells emerged as necessary, as the elimination of either compartment led to therapeutic collapse. Importantly, we identified CD4+ TILs with cytolytic features following RACIM in mice. Cytolytic CD4+ T cells recognize cognate peptides in the context of class II MHC, normally presented by APCs, and have been implicated in antiviral immunity (59, 60), autoimmune pathology (61), and recently in antitumor responses in the mouse (45, 62, 63) and in patients (64, 65). Transcription factors such as Tbet and Eomes (66, 67), as well as Runx3, ThPOK (68), Hobit (69), and Blimp-1 (70), are involved in the cytolytic differentiation of CD4+ cells. However, cytolytic CD4+ cells with exhausted features have not been described to date. Here we report cytolytic CD4+ cells with polyfunctional effector properties exhibiting features of canonical exhausted cells, including downregulation of Tcf7 and upregulation of Tox and Pdcd1. Importantly, we also detected precursor-exhausted CD4+ cells coexpressing Tcf7 and Tox in these tumors. Interestingly, clonotype analysis revealed that the Tpex (Tcf7+Tox+) and canonical Tex (Tcf7−Tox+) CD4+ compartments shared numerous expanded TCR clones, presently interpreted as tumor specific (71, 72), indicating that exhausted cytolytic CD4+ cells derive from such precursors, similar to what has been reported previously for CD8+ T cells (31, 32, 73). Some of these same clones were also distributed within the TEM, Th1, or Tfh compartments, suggesting that these may also serve as precursors for CD4+ Tex cells. In the CD8+ T-cell compartment, TCF1+PD1+ Tpex cells retain high proliferative potential and undergo long-term self-renewal, while also replenishing the dominant population of TCF1− exhausted effector T cells (34, 74). Response to ICB has been in fact associated with the detection of Tpex CD8+ T cells that can proliferate and give rise to polyfunctional TCF1−PD-1+CD8+ effector cells (31, 75). In this context, Tox ensures stable commitment to the exhausted state (33, 76, 77). Our data suggest a similar evolution of CD4+ precursor–exhausted cells, and a role in the efficacy of the LDRT/immunotherapy combination.
In mouse and human T cells, NKG2D serves as an important costimulatory receptor (47, 48), which enhances CD8+ T-cell cytolytic function (78) and prevents Fas-mediated apoptosis (79). Because its ligands are primarily upregulated in sites of peripheral tissue damage or inflammation, the NKG2D pathway likely plays a key role in regulating effector T-cell responses in the periphery. Interestingly, NKG2D is not expressed by CD4+ T cells at the steady state, but important frequencies of tissue-destructive NKG2D+ CD4+ T cells have been detected in patients with destructive autoimmune disease (80), as well in virally induced cancers (81). The NKG2D pathway likely plays an important role in tumor immune elimination, because tumors develop numerous mechanisms to evade NKG2D (82–85), and neutralization of soluble NKG2D ligands enhances response to ICB (86). Importantly, we identified a subset of CD8+ and especially CD4+ Tex cells expressing NKG2D in tumors treated with RACIM. These Tex cells exhibited higher proliferative capacity relative to Tex cells that did not express NKG2D, suggesting that polyfunctional features are the result partly of in situ NKG2D costimulation. Induction of NKG2D in CD4+ and to a lesser extent CD8+ Tex cells was dependent on activation by CTLA4 or PD-1 blockade, explaining in part how the interdependency of radiotherapy and ICB interventions drove therapeutic synergy in RACIM. Interestingly, NKG2D has been implicated in the acquisition by TILs of the ability to engage tumor target and be retained to radiated tumors upon CTLA4 blockade (87).
The combined treatment also produced profound reprogramming of the myeloid compartment, which explains the effective mobilization of adaptive immunity. DCs underwent important reprogramming upon RACIM, with activation and acquisition of molecular states consistent with competent APCs capable of eliciting T-cell immunity. Importantly, a high frequency of RACIM-induced tumor DCs was found to overexpress the NKG2D ligand RAE1, matching the higher frequency of NKG2D-expressing CD8+ and CD4+ TILs in the same tumors. DCs expressing NKG2D ligands have been described in autoimmune diseases and infection (88), but not in tumors. Our findings support an important functional cross-talk between myeloid cells expressing NKG2D ligands and NKG2D+ T cells driving tumor rejection in the mouse. Indeed, supporting a key role of NKG2D in sustaining immune-rejecting T cells, administration of NKG2D antibody abrogated the efficacy of RACIM.
BATF3-expressing cDC1 have been identified as key APCs for antigen cross-presentation to CD8+ T cells (40) and effective response to ICB (41), whereas cDC2 are purportedly required to drive CD4-mediated antitumor responses (45). We observed profound reprogramming of both compartments. Consistent with the key role of CD8+ T cells in our model, the combination of LDRT and immunotherapy lost its therapeutic efficacy in a Batf3−/− genetic background lacking cDC1, where we also observed an attenuated mobilization of CD8+PD1+TCF1− cells in tumors. Importantly, both DC1 and DC2 upregulated NKG2D following RACIM, and indeed a new state of DC2 (cDC2/MoDC), endowed with features of competent APCs and expressing RAE1, emerged in RACIM-treated tumors. The reprogramming of the DC compartment was also accompanied by marked repolarization of macrophages, with significant activation and acquisition of states that could be collectively assigned to M1, with important implications for T-cell homing (15). CD40L likely contributed to these changes in an important fashion, as the absence of αCD40 agonist antibody in the cocktail was associated with significantly fewer CD11b+CDllc+MHC-II+ DC2s and more F4/80+CD206+ M2 macrophages, resulting in the collapse of T-cell attack (89–91).
We translated the preclinical findings to the clinic with a pilot study in eight patients yielding a response rate of 12.5% by iRECIST, whereas two additional patients achieved a dramatic response by 68GaPSMA-PET and 8FDG-PET/CT, respectively. This is quite remarkable in this patient population (92–94), especially considering that we treated only patients with immune-desert tumors. Our pilot study was not designed to directly compare the two radiotherapy doses tested. However, even with this limitation, paired biopsies confirmed the proinflammatory effect of LDRT at both doses, compatible with simultaneous activation of innate and adaptive immunity, which was associated with tumor response. Although not all immune reprogramming observed in our preclinical study was documented in patient tumors, interestingly, similar to the mouse, the predominant T-cell population infiltrating tumors postradiotherapy were largely CD4+ cells. Although in the mouse we documented the critical role of the NKG2D pathway, unfortunately, tumor biopsies in patients were performed as originally planned 7 to 10 days postradiotherapy, to capture immune infiltration changes, but this fell outside of the short window of upregulation of NKG2D ligands observed in the mouse.
Geospatial resolution localized these responses within epithelial tumor deposits, as required for effective tumor control. Consistent with effective mobilization of immunity and migration of T cells from draining lymph nodes seen in the mouse, responder patients exhibited important mobilization of oligoclonal T-cell response in blood immediately after LDRT.
Importantly, higher than expected persistence of side effects was observed with RACIN. Immune-related SAEs in our trial (25%) were similar to CheckMate 227 (24.5%; ref. 49). However, in CheckMate 227, only 18% of the patients discontinued treatment (49), whereas in our trial all patients with toxicity discontinued treatment after an average of two cycles, and immune toxicity was rather refractory to immune suppression. Low-dose CP combined with anti–PD-1 has been reported as well tolerated (95), suggesting that the addition of ipilimumab to the combination contributed importantly to immune toxicity. Low-dose CP attenuates human Tregs (57, 96, 97), whereas ipilimumab activates systemic effector T cells at the expense of Tregs (98, 99). Therefore, this combination may expose important autoimmune vulnerabilities that remain otherwise compensated in patients. Pavlick and colleagues also found that combining low-dose CP (300 mg/m2) with ipilimumab 10 mg/kg every three weeks resulted in severe toxicity in 10 patients with melanoma, with 40% grade 4 AEs, including steroid-refractory colitis in three patients requiring anti-TNFα therapy, and myasthenia gravis in one patient (100).
In summary, we have demonstrated a novel and important synergy between LDRT and rationally developed combinatorial immunotherapy for the treatment of tumors with poor immune infiltration, hinged on simultaneous activation of multiple innate and adaptive immune pathways revealing interdependencies between LDRT and immune modulation. These led to powerful mobilization of antitumor immunity, with both effector CD4+ and CD8+ T cells implicated, which in the mice led to tumor eradication and in patients to regression of lesions that had been included in the radiation plan. Importantly, our preliminary clinical experience suggests the importance of irradiating all metastatic deposits, because in patients who experienced an objective response, we observed durable responses only in irradiated lesions, whereas lesions that were initially considered nonpathologic (and therefore not radiated) eventually were proven to be metastatic deposits that progressed. Future research should focus on improving the combination strategies to further enhance such synergies and generate important protective memory. For example, in our clinical study, we were unable to use a CD40 agonist. To attenuate macrophage suppression, we used aspirin, which prevents prostaglandin E2 (PGE2)-mediated inhibition of DCs, attenuates Tregs and myeloid-derived suppressor cells (101, 102), and reduces endothelial FasL-mediated killing of homing effector T cells (51). Although delivering systemic CD40 ligands and anti-CTLA4 antibodies may prove intolerable in combinations in humans, strategies focusing on the targeted delivery of these agents in the TME may offer improved approaches to achieve effective and safe immune modulation in combination with enabling LDRT.
Methods
Preclinical Study Experiments
Mouse Strains and Cell Lines
Female C57BL/6 (MGI catalog no. 5658455, RRID:MGI:5658455) mice ages 6 to 8 weeks were purchased from Harlan (Envigo). Female Batf3−/− (IMSR; catalog no. JAX:013755, RRID:IMSR_JAX:013755) mice backcrossed onto a C57BL/6 background, and Foxp3-eGFP mice (IMSR; catalog no. EM:01945, RRID:IMSR_EM:01945), kindly provided by Prof. Pedro Romero (University of Lausanne, Lausanne, Switzerland; UNIL), were bred and housed in pathogen-free conditions in the UNIL animal facility in Epalinges.
All in vivo animal experiments were performed in accordance with relevant guidelines and regulations of the University of Lausanne Ethic Committee for the human care of laboratory animals and were approved by the Service de la Consummation et des Affaires Vétérinaires of the Canton of Vaud (SCAV).
The mouse ovarian epithelial papillary serous adenocarcinoma cell line ID8 was a gift from Dr. K.F. Roby (University of Kansas Medical Center, Kansas City, KS; ref. 22). The Lewis cell carcinoma cell line was purchased from ATCC (catalog no. CRL-1642, RRID:CVCL_4358). Both cell lines tested negative for Mycoplasma contamination (last Mycoplasma test April 2021). Tumor cell lines were authenticated by high polymorphic short tandem repeat loci (Microsynth report; #01230_007070). The median number of passages between thawing and collection was 10 (range, 2–16).
Tumor cell lines were cultured in DMEM w/l-glutamine (DMEM; Gibco, Thermo Fisher Scientific), 4% FBS, and 0.09 mg/mL penicillin–streptomycin. ID8 cells were gene-engineered to express luciferase by transduction with retrovirus prepared with the MSCV-Luciferase-PGK-hygro retroviral plasmid, a gift from Scott Lowe (Memorial Sloan Kettering Cancer Center; RRID:Addgene_18782). Retrovirus was prepared and stored as described previously (103). Transduction was performed with viral supernatant and protamine sulfate (Sigma-Aldrich) when the cells reached a confluence of 30% to 40%. The medium was replaced after 24 hours, and at 48-hour transduction, hygromycin (400 μg/mL final concentration) was added to select for transduced cells. After two weeks, luciferase expression was assessed by bioluminescence imaging (BLI) performed using the Xenogen IVIS Lumina II imaging system, and the photons emitted by the luciferase-expressing cells were quantified using Living Image software v.3.2 (Living Image software, RRID:SCR_014247) following the manufacturer's instructions.
Tumor Engraftment
For the ovarian cancer model, 5 × 106 ID8 tumor cells (luciferase+) were injected intraperitoneally in female C57BL/6 mice, typically ages 6 to 8 weeks on day −21. Successful engraftment of the i.p. tumors was defined as ≥7 × 107 photons/second BLI emission. On day −2, mice were blindly randomized into treatment groups based on BLI emission (average of 7 × 107 photons/second in controls and treated groups). Attrition: mice exhibiting BLI emission of <5 × 107 or higher than 1 × 109 were not included in the experiments. The reason to exclude them was either lack of tumor when the bioluminescence was low or presence of ascites when the bioluminescence was higher than 1 × 109. This strategy was followed for all in vivo work. ID8 tumor–bearing animals were weighed twice a week and euthanized if they exhibited clinical signs of disease or distress (cachexia, anorexia, respiratory problems, ascites, etc.).
For the LLC model, 1 × 106 LL/2 tumor cells were injected subcutaneously in C57BL/6 mice. Once tumors reached an average volume of 350 mm3, mice were randomized, and treatments were initiated. Mice harboring tumors smaller than 300 mm3 were not included due to the presence of T cells in the TME. Tumors were measured with a caliper at randomization and five times a week thereafter. Euthanasia was performed when tumors reached 1,000 mm3. Weight was monitored twice a week as per standard practice.
Tumor Imaging by Bioluminescence
BLI images were taken with Xenogen IVIS using d-luciferin (In Vivo Imaging Solutions), as described by the manufacturer. Images were normalized using Living Image software (PerkinElmer). Maximum luminescent intensity and total flux in photons per second were calculated and reported for the abdominal region of each mouse in photons/second. Significance was determined using one-way ANOVA for BLI. All time points were compared with the earliest time point of day −2.
Clonogenic Survival Assay
Cells were seeded in triplicate into 6-well plates at 100 to 400 cells/well in 4 mL DMEM. Once cells were attached to the wells, a single dose of irradiation (0, 0.5, 1, 2, 3, 4, 6, or 8 Gy) was applied. Cells were then incubated at 37°C in 5% CO2 for 24 hours. Colonies were fixed and stained with crystal violet. All colonies of 50 cells or more were then counted. The survival fraction (SF) was estimated according to the formula: SF = number of colonies formed/number of cells seeded × plating efficiency of the control group.
Immunogenic Cell Stress Assay
5 × 105 cells were seeded in a T25 cm2 flask with 5 mL DMEM. To determine the kinetics of calreticulin upregulation, ID8 cells were treated with 1 Gy radiation or doxorubicin (positive control, 25 μmol/L; Sigma) and harvested 4, 6, 12, and 24 hours after exposure as previously described, washed twice with cold PBS, followed by staining with a calreticulin-specific antibody (Abcam; catalog no. ab83220, RRID:AB_1859755), Annexin V (Thermo Fisher Scientific; catalog no. 88-8007-72, RRID:AB_2575165), which recognizes phosphatidylserine on the surface of apoptotic cells, plus vital dye 4,6-diamidino-2-phenylindole (DAPI), which stains dead cells. Isotype-matched IgG antibody was used as a negative control (Abcam; catalog no. ab91357, RRID:AB_2888649), and the analysis was limited to living (DAPI-negative) tumor cells (104).
Mouse mAb and Chemotherapy Treatment
After successful tumor implantation, C57BL/6 mice were treated with various combinations of 100 μg of agonistic CD40 mAb (Bio X Cell; catalog no. BE0016-2, RRID:AB_1107647), 100 μg of α-mouse PD1 (Bio X Cell; catalog no. BE0146, RRID:AB_10949053), 100 μg of α-CTLA4 (Bio X Cell; catalog no. BE0164, RRID:AB_10949609). Antibodies were administered intraperitoneally in weekly cycles thrice, with and without 1 Gy low-dose irradiation delivered to the whole mouse abdominal cavity (LD-WART). Metronomic CP was used at 100 mg/kg per mouse (27) and was administered once a week for three cycles the day preceding immunotherapy treatment.
For the in vivo depletion or blocking experiments, specific antibodies as well as isotype controls were intraperitoneally injected twice a week starting at day −2. Depletions were confirmed by flow cytometric analysis of peripheral blood. Antibodies and drugs used for in vivo studies are listed in Supplementary Table S4.
LDRT
Radiotherapy was delivered to the whole peritoneal cavity (ID8) or s.c. tumors (LLC). Briefly, mice were anesthetized with isoflurane, and the abdomen or the skin was irradiated with 0.5, 1, and 2 Gy depending on the experiment using the Small Animal Radiation Research Platform (x-Rad-iR-225) using 12 Gy/minute at 225 KV, 13.0 mA, 30 cm SSD. Radiation was focalized using a collimator of 4 × 4 cm or 2 × 2 cm depending on the location. For RACIM, LD-WART was provided with metronomic CP as well as anti–PD-1 and anti-CTLA4 ICB antibodies and anti-CD40 agonist antibody once per week, thrice.
Flow Cytometry Analysis
Analysis of the TME throughout all results and figures was performed exclusively on tumor deposits that were collected, mixed, and analyzed as one sample per mouse, and we did not analyze ascites fluid. Tumor samples were dissociated in DMEM supplemented with 200 μg/mL of Liberase TL (Roche) and 1 mg/mL of DNase I (Sigma-Aldrich) at 37°C for one hour. The same protocol was used for spleen and mesenteric lymph node but with an incubation of 15 minutes. Uniform single-cell suspensions were obtained after smashing digested tissues with a syringe plunger on a 100-μm filter. Subsequently, dead cells were stained with the LIVE/DEAD Fixable Aqua Dead Cell Stain Kit according to the manufacturer's protocol, followed by blocking of Fc receptors during an incubation of 15 minutes at 4°C with purified anti-CD16/CD32 mAb (BD Biosciences; catalog no. 553141, RRID:AB_394656). Cells were then stained for 30 minutes at 4°C with the fluorochrome-conjugated mAbs of interest in 50 μL PBS supplemented with 2% FBS, 2 mmol/L EDTA. The cells were then washed twice and fixed in 1% paraformaldehyde (Sigma) in PBS or in Fix/Perm buffer (eBioscience) for intracellular staining. Cells were prepared for intracellular staining using a permeabilization buffer set (eBioscience) before adding antibodies against intracellular markers according to the manufacturer's instructions (eBioscience). Fluorescence minus one control was stained in parallel using the panel of antibodies with sequential omission of one antibody. For intracellular cytokine secretion detection, cell suspensions were restimulated in vitro in the presence of 500 ng of ionomycin/50 ng of phorbol myristate acetate (PMA; Sigma-Aldrich) or after TCR engagement with 10 μg/mL, immobilized anti-CD3 (BioLegend; catalog no. 100340, RRID:AB_11149115) in association with 2 μg/mL soluble anti-CD28 (BioLegend; catalog no. 102116, RRID:AB_11147170) and GolgiPlug Brefeldin A solution (BD Biosciences; catalog no. 555029, RRID:AB_2869014) for four hours. Analysis of stained cells was performed using an LSR-II cytometer (BD Biosciences) supplied with BD Diva interface and FlowJo software (FlowJo, RRID:SCR_008520). Antibodies used for flow cytometric analysis are listed in Supplementary Table S5.
Cell Sorting and scRNA-seq
Single-cell suspensions for scRNA-seq were prepared and stained as for flow-cytometric analysis (described above). For the control, cells were pooled from four tumors (due to low levels of immune infiltrate), whereas for the treatment groups (CIM and RACIM), three independent single-cell suspensions were evaluated for each (i.e., n = 3 tumors/treatment). DAPI was added at a final concentration of 0.5 μg/mL immediately prior to running samples on the Aria II or Aria III sorter (BD Biosciences). Sorted cells were collected in cold DMEM, 10% FBS. After the sort, cells were assessed for viability by Trypan blue staining and their concentration adjusted to 1,000 live cells/μL to process the single-cell encapsulation using a chromium single-cell instrument and reagents (10X Genomics). A Chromium Next GEM Chip G was loaded with the appropriate number of cells, and the sequencing libraries were prepared with the Chromium Next GEM Single-Cell V(D)J Reagent Kits v1.1 following the manufacturer's recommendations. Briefly, an emulsion encapsulating single cells, reverse transcription reagents, and cell barcoding oligonucleotides was generated. After the actual reverse transcription step, the emulsion was broken, and double-stranded cDNA generated and amplified in a bulk reaction. For 5′ gene-expression library, this cDNA was fragmented, ligated to a sequencing adaptor, and PCR amplified. For V(D)J library preparation, a similar approach was followed except that two steps of PCR based V(D)J target enrichment were performed prior to fragmentation.
Libraries were quantified by a fluorometric method, and their quality was assessed on a Fragment Analyzer (Agilent Technologies). Cluster generation was performed with 140 to 165 pmol/L of an equimolar pool from the resulting libraries using the Illumina HiSeq 3000/4000 PE Cluster Kit reagents. Sequencing was performed on the Illumina HiSeq 4000 using HiSeq 3000/4000 SBS Kit reagents according to 10X Genomics recommendations (26 cycles read 1, 8 cycles i7 index read, and 91 cycles read 2). Sequencing data were demultiplexed using the bcl2fastq2 Conversion Software (bcl2fastq, RRID:SCR_015058), and primary data analysis was performed with Cell Ranger (Cell Ranger, RRID:SCR_017344).
NanoString Analysis
Gene-expression profiling was performed using a custom 770 gene NanoString Cancer Immunology panel comprising immune-related genes and genes pertaining to common cancer signaling pathways. Briefly, for each NanoString assay, 1 μg of RNA was isolated from 30 mg of frozen tissue using the RNeasy Mini Kit (Qiagen) as described by the manufacturer. Samples were first lysed in buffer RLT and disrupted/homogenized using the TissueLyser II system from Qiagen. RNA was quantified using the NanoDrop ND1000 Spectrophotometer (Thermo Fisher Scientific); average RNA was 734.38 ng/mL (range, 298–1,003 ng/mL), and RNA quality was checked with the Fragment Analyzer (Advanced Analytical Technologies Inc). Samples were run by the Centre for Integrative Genomics at the UNIL. Class comparison was performed using nSolver 2.6, PanCancer Analysis module, normalization from the selected set of housekeeping genes using GeNorm algorithm (geNORM, RRID:SCR_006763; the number of housekeeping genes to use is automatically chosen), then normalized by geometric mean of the selected genes.
Gene Expression Analyses
The R software (version 3.6.2) was used for all bioinformatics analyses. Similar methods were used for human and mouse gene and pathway analyses. A database of gene orthologs was built using the R biomaRt package (biomaRt, RRID:SCR_019214) and was used when interrogating human-derived gene signatures on mouse data.
The signatures used in this study include immune gene signatures from Bindea and colleagues (55), cell type signatures from the mouse single-cell atlas Tabula Muris (105), biological “Hallmarks” signatures from MSigDB (https://www.gsea-msigdb.org/gsea/msigdb/index.jsp), and some manually curated pathway (i.e., RAE/NKG2D, cytokine, costimulation, and immune-checkpoint pathways). Gene signature scores were computed using the ssGSEA as implemented in the GSVA R package (with default parameters except mx.diff = FALSE). The absolute composition of the immune component was also assessed using the CIBERSORTx (106) online tool using the LM22 immune signature collection. Differential analyses for gene expression and pathway scores were performed using the lmFit function of the limma R package (LIMMA, RRID:SCR_010943). Differential analyses performed in human patients were carried out by using the patient origin as a covariate (paired analyses). Gene expression, signature/pathway scores, and P values of statistical testing were represented under the form of heat maps using the pheatmap R package (pheatmap, RRID:SCR_016418). In the P value heat map scenario, the P values were −log10 transformed before plotting, and directionality of the comparison was kept and color-coded. Pie charts were plotted using the ggplot2 R package (ggplot2, RRID:SCR_014601).
mIF Imaging
All tumor samples were fixed in 10% neutral buffered formalin (NBF) for a minimum of 16 hours before being dehydrated and formalin-fixed paraffin-embedded (FFPE). The following primary antibodies were used: mouse anti-NK1.1 (Thermo Fisher Scientific; catalog no. MA1-70100, RRID:AB_2296673), rat anti-FoxP3 (Thermo Fisher Scientific; catalog no. 14-5773-82, RRID:AB_467576), rabbit anti-CD4 (Abcam; catalog no. ab183685, RRID:AB_2686917), rabbit anti-CD8 (Bioss; catalog no. bs-0648R, RRID:AB_10857537), rabbit anti-CD11b (Abcam; catalog no. ab133357, RRID:AB_2650514), rabbit polyclonal against FoxP3 (Abcam; catalog no. ab54501, RRID:AB_880110), rabbit polyclonal against human CD8 (Bioss; catalog no. bs-0648R, RRID:AB_10857537), rabbit polyclonal against CD11b (Abcam; catalog no. ab133357, RRID:AB_2650514), rabbit anti-mouse CD4 (Abcam; catalog no. ab183685, RRID:AB_2686917), rabbit polyclonal to yH2AX (Novus Biologicals; catalog no. NB 100-384, RRID:AB_350295), goat polyclonal to Foxp3 (GeneTex; catalog no. GTX89752, RRID:AB_10725476), rabbit polyclonal to Rae-1 (Life-Span; catalog no. LS-B3539-50, RRID:AB_10608707).
Multiplex IF IHC was performed on 5-μm paraffin sections on Ventana Discovery Ultra staining module (Ventana, Roche). The following panels were performed: CD4-Opal 690, CD8-Opal 520, CD11b-Opal 480, DAPI; yH2Ax-Opal 620, DAPI; RAE-Opal 620, DAPI; RAE-Opal 620, CD11b-Opal480, DAPI. Slides were placed on the staining module for deparaffinization, epitope retrieval, and endogenous peroxidase quenching. The multiplex staining method/procedure consists of several/multiple rounds of staining, each round includes nonspecific sites blocking (Ventana, Discovery Inhibitor and Discovery Goat Ig Block), incubation with unlabeled primary antibody, with horseradish peroxidase (HRP)–conjugated secondary antibodies (Discovery OmniMap anti-rabbit, anti-goat, and anti-mouse; Ventana), with Opal (Akoya) reactive fluorophore (Opal 480, 520, 620, 690) detection that covalently labels the primary epitope, followed by an antibody (both primary and secondary) heat denaturation step prior to the next round of antibody staining. Finally, nuclear staining was performed with spectral DAPI (Akoya). mIF images from the stained slides were acquired at 20× and 40× magnification using Akoya Vectra POLARIS multispectral microscope.
RNA Extraction, cDNA Preparation, and Real-Time qPCR
Tissue samples from tumors and purified cells were kept frozen (−80°C) until mRNA extraction. As needed, samples were disrupted with a TissueLyser and homogenized in RLT buffer (Qiagen). RNA extraction was performed using the micro or mini RNeasy kit (Qiagen) using the DNAse treatment step (Qiagen), and cDNA preparation were conducted following standard procedures using the PrimeScript first-strand cDNA Synthesis Kit (Takara). Quantitative PCR was performed using TaqMan Fast Universal PCR Master Mix (2X), no AmpErase UNG (Life Technologies) on the 7500 Real-Time System or QuantStudio 6 (Applied Biosystems) as indicated by the manufacturer. Primers and probes for the quantitative qPCR were analyzed with the following assay: GAPDH (Mm99999915_g1), IFNa4 (Mm00833969_s1_m1), IFNb (Mm00439552_s1), XCL1 (Mm00434772_m1), CXCL1 (Mm04207460_m1), CXCL9 (Mm00434946_m1), CCL5 (Mm01302427_m1), TNFα (Mm00443258_m1), IFNg (Mm01168134_m1), Nos2 (Mm01309897_m1), Perforin 1 (Mm00812512_m1), klrk1 (Mm01183328_m1), Rae (Mm00558293_g1), IL18 (Mm00434226_m1), H60a (Mm01311160_m1), IL12a (Mm00434169_m1), and H2K1 (Mm01612247_mH). All primers were obtained from Life Technologies.
Cytokine and Chemokine Beads Assays
Mice bearing ID8 tumors treated or not with RACIM were bled at cycle 2 day 5. Sera were diluted five times in PBS. Cytokines and chemokine were quantified according to the manufacturer's protocol: IFNγ (BD Biosciences; catalog no. 558296, RRID:AB_2869141), TNFα (BD Biosciences; catalog no. 558299, RRID:AB_2869144), IL1α (BD Biosciences; catalog no. 560157, RRID:AB_2869318), IL1β (BD Biosciences; catalog no. 562278, RRID:AB_2869415), IL2 (BD Biosciences; catalog no. 558297, RRID:AB_2869142), IL6 (BD Biosciences; catalog no. 562236, RRID:AB_2869403), IL10 (BD Biosciences; catalog no. 562263, RRID:AB_2869410), and CXCL9 (BD Biosciences; catalog no. 558341, RRID:AB_2869166).
Statistical Analyses
All statistical analyses were performed using GraphPad Prism program (GraphPad; RRID:SCR_000306). When indicated, we performed statistical analyses using SPICE software (SPICE; RRID:SCR_016603). We used χ2 permutation test for pie chart comparison according to Roederer and colleagues (107). When more than two groups were compared, one-way ANOVA (pairwise comparisons) with Tukey–Kramer post hoc test to compare among all pairs of means was used. Unpaired t tests with Welch correction were performed only in experiments having two groups of animals from the same experiment. Survival curves were estimated by using the Kaplan–Meier method, log-rank test. Time to death was measured from the day tumor cells were injected. Confidence intervals (CI) were calculated from SEs.
The number of animals per experiment (sample size) was calculated using the standardized effect size (SES). SES is the magnitude of the difference between the means of two groups in units of standard deviations. This is the effect of the size/pooled SD. SD of 2.0 was used with an 80%–90% power, a 5% significance level and a one-sided or two-sided t test. Based on this, our experiments had between 5 and 10 animals per group (108).
scRNA-seq Data Analysis
Data Processing of scRNA-seq Libraries
The scRNA-seq reads were aligned to the GRCh38 reference genome and quantified using Cell Ranger count (10X Genomics, version 3.0.2). TCR reads were aligned to the GRCh38 reference genome, and consensus TCR annotation was performed using Cell Ranger vdj (10X Genomics, version 2.1.0; Cell Ranger, RRID:SCR_017344). All additional analyses were performed using R 4.0.2 and Seurat v3.0 (ref. 109; Seurat, RRID:SCR_007322), unless it is indicated otherwise. For analyzing shared clonotypes, the Jaccard index was calculated using the R package scRepertoire (ref. 110; R package version 1.0.0.).
Single-Cell Data Filtering and Normalization
To retain high-quality transcriptomes, total ribosomal and mitochondrial count filters were applied. For the T cells, the maximum percentage counts coming from ribosomal and mitochondrial genes were 60% and 10%, respectively. In addition, the acceptable number of detected genes ranges between 250 and 600. For the myeloid compartment, cells were prefiltered in a different (more permissive) way, with a lower limit of 200 detected genes in the attempt retaining the neutrophils that naturally have less mRNA. After this initial filtering, data were normalized by total counts using the “LogNormalize function” from the “Seurat v3.0” package, which normalizes the feature expression measurements for each cell by the total expression, multiplies this by a scale factor of 10,000 and log-transforms the result.
Unsupervised Clustering
Scaled z-scores for each gene were calculated using the ScaleData function and regressed against the number of UMIs per cell. T cells and myeloid cells were analyzed using different parameters. Concerning T cells, scaled data were used as the input of a principal component analysis (PCA) on the top 1,000 most variable genes. The first 30 principal components were used to generate uniform manifold approximation and projection (UMAP) projections, with a minimum distance of 30 neighbors. Within the UMAP space, neighbors were found using the FindNeighbors function from the “Seurat v3.0” package using two dimensions. Then, the shared nearest-neighbor (SNN) clustering method (111) implemented in Seurat 3 as the FindClusters function was applied, with parameter resolution = 0.05, reduction = “umap,” k.param = 10. Concerning the myeloid cells, the expected vast heterogeneity of this population made us to perform a whole-genome PCA, and the UMAP analysis was based on the first 100 principal components. Within the UMAP space, neighbors were found using the FindNeighbors function using 20 dimensions The SNN clustering method was applied, with parameter resolution = 2.5, reduction = “umap,” k.param = 10.
Cell Annotation
In order to classify T cells, each cell from the data set was projected onto a reference T-cell atlas following the methodology proposed by Andreatta and colleagues (30). For the classification of the myeloid cells into the main subpopulations, namely, macrophages, monocytes, neutrophils, and DCs, we used the R package “SingleR” (112) and the immune cell signatures from the Immgen database (37). In order to further classify DCs into distinct subpopulations, namely, DC1, DC2, DC3, pDC, and MDDC, we used the signatures proposed by Zilionis and colleagues (35). To compare the resulting clusters with DC states across mouse tumor models, a reciprocal similarity score between each tumor-infiltrating DC state comparison pair was calculated using a linear support vector classifier fitted to log2-transformed data from each scRNA-seq study.
Clinical Study and Evaluation of Patient Samples
RACIN Clinical Trial
RACIN (protocol identification NCT03728179) is approved by the Ethical Committee of Canton Vaud, and it is conducted in accordance with the Declaration of Helsinki. All patients signed a written informed consent. RACIN is a single-arm, phase I trial of dose-escalation and safety evaluation, testing the combination of nivolumab plus ipilimumab, aspirin, and low-dose CP, along with escalating doses of low-dose ionizing radiation (0.5–1 Gy), followed by nivolumab maintenance. Inclusion criteria: patients with advanced, TIL-negative solid tumors. Experimental subjects were not randomized into groups because this was deemed irrelevant to this study.
The current translational paper presents the experience of the three patients who comprised the first cohort, and the first five patients comprising the second cohort.
The interventions used in the study were:
Low-dose ionizing radiation: one fraction of 0.5, 1, 2, or 3 Gy every two weeks on day 2 of each cycle (from cycle 0 to cycle 4).
ICB antibodies: nivolumab 240 mg flat dose every two weeks and ipilimumab 1 mg/kg every 6 weeks, both administered intravenously and sequentially (when administered concurrently), starting on day 2 of each cycle, for four cycles (cycles 1–4).
CP 200 mg/m2 every two weeks administered intravenously one day prior to each combination of radiation and immunotherapy treatment (day 1 of each cycle C0 to C4).
Aspirin (300 mg) administered orally once daily for four cycles, from C1D1. Aspirin is continued during nivolumab maintenance, according to tolerance. An H2 antagonist is used in combination with aspirin to prevent gastric damage.
At the end of the fourth cycle, patients who did not progress by RECIST receive nivolumab maintenance (240 mg flat dose every two weeks), until progression or unacceptable toxicity. Aspirin is maintained according to tolerability.
According to protocol version 4, in the phase Ia (dose escalation) part of the study, patients received low-dose irradiation at escalating doses, according to rules of the classic phase I escalation design avoiding a DLT in more than 17% (1/6) of subjects (algorithm adapted to multidrug combination). The first cohort of thee (or nine) patients received nivolumab, ipilimumab, CP, and aspirin at predefined doses with 0.5 Gy. Two subsequent cohorts of six patients have been planned, with radiation administered at escalating doses (2, 3 Gy), together with nivolumab, ipilimumab, aspirin, and low-dose CP. The expected maximum number of patients to be treated at the phase Ia was 21 and up to 27 patients.
After choosing the recommended phase Ib dose of radiation, an additional group of 19 patients would enter an expansion cohort in the phase Ib part of the study, until a maximum of 40 or 46 patients have been treated.
The primary outcomes of the study include the following:
-
For phase Ia:
Treatment safety and toxicity measured during the DLT period, the backbone limiting toxicity period and all along the trial period using NCI Common Terminology Criteria for Adverse Events (CTCAE v.4.03, refer to Appendix 4: Adverse Event Grading Scale).
MTD or recommended phase Ib dose (RP1bD) of low-dose irradiation for radioimmunotherapy combination.
-
For phase Ib:
Safety and tolerability of LDRT at the RP1bD, in combination with the backbone treatment (measured during the full trial follow-up).
Statistical analyses of the RACIN clinical data were carried out in SAS version 9.4.
Low-Dose Ionizing Irradiation
One fraction of 0.5 or 1 Gy every two weeks was administered to all metastatic deposits (total radiation dose 6 or 13 Gy, respectively). Before starting radiation treatment, patients underwent a CT scan (planning scan) without intravenous contrast medium. They were scanned in a supine position with a head and knee support. For the CT planning scan, standard acquisition parameters were used (tension, 120 kV; tube rotation time 1 second; tube current, 160 mA; helical acquisition with pitch of 0.938; reconstructed image thickness, 2 mm). CT planning scan was automatically fused with the volumetric information of either [18F]FDG-PET/CT or 68GaPSMA-PET/CT and the largest combined tumor volume identified by each imaging method served as the gross tumor volume (GTV). Velocity Advanced Image Software; Velocity Medical Solutions from Varian Medical Systems, Inc., or Raystation 9.2 planning system software from Raysearch Laboratories AB was used for fusion and contouring. GTV with 5-mm expansion constituted the clinical target volume. The planning target volume (PTV) was automatically derived from the CTV with a 5-mm expansion. All the organs at risk were drawn (active bone marrow, bowel, sigmoid, rectum, bladder, femoral heads, heart, lungs, spleen, liver, and duodenum) and protected from the low-dose irradiation. Treatment plans were performed using the Tomotherapy treatment planning system (Accuray Inc.) with a field width of 5 cm and a pitch of 0.287. Image guidance before each fraction was implemented using the MV fan-beam CT of Tomotherapy. The prescribed total dose to the PTV was 6.5 Gy for the cohort 1 (0.5 Gy per fraction) and 13 Gy for the cohort 2 (1 Gy per fraction) administered every 2 weeks for 26 weeks.
Antibodies and Chemotherapy
ICB antibodies, namely, nivolumab (anti–PD-1) and ipilimumab (anti-CTLA4) antibodies, both products of Bristol-Myers Squibb (BMS), and nivolumab 240 mg flat dose every two weeks and ipilimumab 1 mg/kg every six weeks were both administered i.v. and sequentially (when administered concurrently), starting on day 2 of each cycle, for four cycles (C1–C4). CP 200 mg/m2 every two weeks was administered i.v. one day prior to each combination of radiation and immunotherapy treatment (D1 of each cycle C0–C4). Aspirin (300 mg) was administered orally once daily for four cycles, from C1D1. This was continued during nivolumab maintenance, according to tolerance. An H2 antagonist was used in combination with aspirin to prevent gastric damage. At the end of the fourth cycle, patients were started on nivolumab maintenance at 240 mg flat dose every four weeks, until progression or excessive toxicity.
RNA Extraction and Library Preparation
RNA from snap-frozen tissue biopsies was extracted using the RNeasy kit (Qiagen). RNA quality was assessed using the Fragment Analyzer (Advances Analytical Technologies Inc.). RNA-seq libraries were prepared from 250 ng of total RNA. The RNA samples were depleted for ribosomal RNAs with the Illumina TruSeq Stranded Total RNA Gold kit. Ribosomal-RNA–depleted RNA was then converted in cDNA and amplified with the Nugen Ovation RNA-seq System V2 kit. It was used to generate sequencing libraries with the Illumina TruSeq DNA Nano kit. Libraries were quantified by a fluorimetric method and their quality assessed on a Fragment Analyzer (Agilent Technologies). Cluster generation was performed with 2 nmol/L of an equimolar pool from the resulting libraries using the Illumina HiSeq 3000/4000 SR Cluster Kit reagents and sequenced on the Illumina HiSeq 4000 using HiSeq 3000/4000 SBS Kit reagents for 150 cycles. Sequencing data were demultiplexed using the bcl2fastq2 Conversion Software (bcl2fastq, RRID:SCR_015058).
Tumor RNA-seq Processing
Illumina single-end sequencing reads were aligned to the human reference GRCh37.75 genome using STAR aligner (113) and the two-pass method as briefly followed: the reads were aligned in a first round using the –runMode alignReads parameter, then a sample-specific splice-junction index was created using the –runMode genomeGenerate parameter. Finally, the reads were aligned using this newly created index as a reference. The number of counts was summarized at the gene level using htseq-count (114). The number of uniquely mapped, nonmitochondrial and nonribosomal reads averaged 22′532′108 ± 3′732′619 (SD). Read counts were normalized into reads per kilobase per million (RPKM) and log2 transformed after addition of a pseudo count value of 1.
NanoString NanoGeoMX Processing
The NanoString GeoMx platform was used for spatial profiling of pre- and posttreatment tumor biopsies from five patients. Briefly, slide-mounted 5-μm FFPE tissue sections were subjected to antigen retrieval with Tris-EDTA pH 9 under heat and pressure, then incubated with a cocktail of RNA-binding probes targeting approximately 1,400 unique genes with 5 probes/gene and containing UV photocleavable unique molecular identifiers (UMI). In parallel, the same tissue section was coincubated with fluorescent antibodies for pan-cytokeratin, CD3e, and CD68, as well as the DNA dye Syto13 for visualization. Geometric areas of interest (AOI) were selected for spatial molecular profiling, and each of the regions was sequentially illuminated with UV light to release the UMI-containing oligos, which were captured in corresponding microtiter plates. A next-generation sequencing library was then prepared, during which AOI-specific unique dual indices were added to each molecule. Libraries were sequenced using an Illumina NextSeq, which performed on-instrument demultiplexing. Sequencing reads were then mapped back to the corresponding genes based on the UMIs by an internal data processing pipeline. The AOIs were initially annotated as being either tumor, immune (T cell and macrophage), or mixed (tumor and immune) AOIs. The number of genes represented in the NanoGeoMX data equals 1,404.
RNA Expression Analyses
NanoGeoMX and RNA-seq data were merged together in order to gain in statistical power and in the number of postirradiation versus preirradiation comparisons. To do so, NanoGeoMX data were first rebulked using the following methodology: The gene expression of ROIs from a same patient, treatment, and type (immune, tumor, or mixed) were first averaged. Then, we further averaged tumor, mixed, and immune-averaged profiles together per patient and treatment, which resulted in one expression profile per patient and per treatment (as for bulk RNA-seq). RNA-seq data were narrowed down to the genes represented in the NanoGeoMX platform (1,404 genes). We then merged RNA-seq and NanoGeoMX data in a single expression matrix and applied a batch correction algorithm using the ComBat function of the sva R package. Finally, we performed Pearson correlation tests between samples and found that they cluster primarily by patient origin, which indicated that our merging method was appropriate.
The gene-expression analysis and pathway score analyses were either performed on the rebulked data (merge of RNA-seq and NanoGeoMX) or on the NanoGeoMX data alone as indicated in the figures. Differential expression analyses and pathway/signature scoring were performed as described in the methodology for the mouse data. For the differential gene-expression and pathway analysis, a significant result was defined as an unadjusted P value lower than 0.05. Gene Ontology term enrichment and Reactome pathway enrichment analyses of the differentially expressed genes was achieved using the on-line tool (http://geneontology.org/), only the five best pathways are shown in the supplementary figures.
For the Bindea and colleagues (55) immune signature analyses, M1 and M2 macrophage signatures were manually added in order to deconvolute the Bindea macrophage signature. The M1 macrophage signature was defined as LCN2, SAA3, IL1B, CCL5, IRG1, FPR2, CFB, IL1A, CD38, CD274, STAT1, and the M2 macrophage signature as CCND1, MRC1, CKB, GSN, CD300LD, TREM2, CADM1, IGF1, CD36, GM23766, CLEC7A, FLT1, BCAR3, EGR2, and STAT6.
mIF Imaging
All patient tumor samples were fixed in 4% NBF for a minimum of six hours before being processed in VIP5Jr (Vacuum Infiltration Processor Tissue-Tek, SAKURA), for dehydration and paraffinization of the tissue. This process consists of one-hour immersion in 4% formalin, and then tissues were dehydrated in two baths of one hour in alcohol 96°, followed by three baths of one hour in alcohol 100°, then three times one hour bath in Xylene. Finally, the tissue was placed in paraffin for three hours before being embedded (FFPE). Multiplex IF IHC was performed on 3.5-μm paraffin sections on the Ventana Discovery Ultra staining module (Ventana, Roche).
The following primary antibodies were used: rabbit anti-CD4 (Cell Marque; catalog no. 104R-14, RRID:AB_1516770), rabbit anti-FoxP3 (Abcam; cat. #ab99963, RRID:AB_10675258), rabbit anti-CD56 (Cell Marque; catalog no. 156S, RRID:AB_1516783), rabbit anti-CD3 (Agilent; cat. #A0452, RRID:AB_2335677), mouse anti-cytokeratin (LifeSpan; catalog no. LS-C95422-1, RRID:AB_10565578), rabbit anti-CD8 (Abcam; catalog no. 4207-1, RRID:AB_764503). The following panel was performed on patient tumors: CD4-Opal 570, FoxP3-Opal 480, CD3-Opal 520, cytokeratin-Opal 690, CD8-Opal 780, and DAPI.
Slides were placed on the staining module for deparaffinization, epitope retrieval, and endogenous peroxidase quenching. The multiplex staining method/procedure consists of multiple rounds of staining, each round including nonspecific site blocking (Ventana, Discovery Inhibitor and Discovery Goat Ig Block), incubation with unlabeled primary antibody, incubation with HRP-conjugated secondary antibodies (Discovery OmniMap anti-rabbit, anti-goat and anti-mouse, Ventana), with OpalTM (Akoya) reactive fluorophore (Opal 480, 520, 620, 690, 780, and 570) detection that covalently labels the primary epitope, followed by an antibody (both primary and secondary) heat denaturation step prior to the next round of antibody staining. Finally, nuclear staining is performed with spectral DAPI (Akoya). mIF images from the stained slides were acquired at 20× and 40× magnification using Akoya Vectra POLARIS multispectral microscope.
TCRα and TCRβ Sequencing
mRNA was isolated using the Dynabeads mRNA DIRECT Purification Kit (Life Technologies) and was then amplified using the MessageAmp II aRNA Amplification Kit (Ambion) with the following modifications: in vitro transcription was performed at 37°C for 16 hours. First-strand cDNA was synthesized using the Superscript III (Thermo Fisher) and a collection of TRAV/TRBV-specific primers. TCRs were then amplified by PCR (20 cycles with the Phusion from NEB) with a single primer pair binding to the constant region and the adapter linked to the TRAV/TRBV primers added during the reverse transcription. A second round of PCR (25 cycles with the Phusion from NEB) was performed to add the Illumina adapters containing the different indexes. The TCR products were purified with AMPure XP beads (Beckman Coulter), quantified and loaded on the MiniSeq instrument (Illumina) for deep sequencing of the TCRα/TCRβ chain. The TCR sequences were further processed using ad hoc Perl scripts to (i) pool all TCR sequences coding for the same protein sequence; (ii) filter out all out-frame sequences; (iii) determine the abundance of each distinct TCR sequence. TCR with a single read was not considered for the analysis.
To calculate the TCR metrics of a given repertory, we took the frequencies of such repertory and applied different formulas. For a set of TCR sequences, we have a set of frequencies f1 to fn (where n correspond to the maximum number of unique TCR sequences in that set). In the formula, we refer to each of them as fi. The metrics we have calculated are respectively: Shannon entropy (equation 1), Gini index (equation 2), clonality (equation 3), richness, and shared frequency.
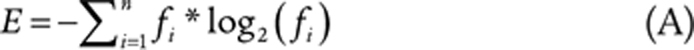 |
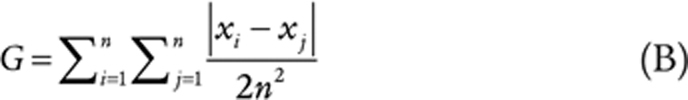 |
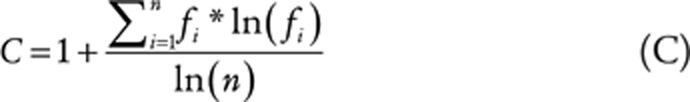 |
The “richness” estimates the number of unique words of a given repertory, i.e., the parameter “n.” Among the various metrics, the “shared frequency” is the only one that needs two (or more) sets. This estimator needs, in fact, to obtain a list of sequences that are present on two (or more) sets. With this list of shared sequences, it is possible to evaluate their size in terms of sum of frequencies, in each set.
Data availability
All data are available from the authors upon reasonable request. For genomic analysis, data can be consulted in GSE169742 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE169742).
Authors' Disclosures
F.G. Herrera reports grants from Bristol-Myers Squibb and Prostate Cancer Foundation Challenge Award during the conduct of the study; grants from Accuray and BioProtect, personal fees from Johnson & Johnson, nonfinancial support from European Organization for Research and Treatment of Cancer, and nonfinancial support from Roche ImFlame cooperative group outside the submitted work. M. Ochoa de Olza reports personal fees from MSD outside the submitted work. R. Genolet reports a patent for Enhanced Immune-Cell Receptor Sequencing Methods pending. B. Navarro Rodrigo reports nonfinancial support from Astellas outside the submitted work. K. Zaman reports other support from Lilly, Novartis, grants and Roche, MSD, Mylan, Daiichi, Pierre Faber, AstraZeneca, Gilead, Pfizer, Seagen, Eisai, and Exact Sciences outside the submitted work. R. Duran reports grants from Society of Interventional Oncology/BTG, grants and personal fees from Guerbet, and Boston Scientific outside the submitted work. S.E. Warren reports other support from NanoString Technologies during the conduct of the study; other support from NanoString Technologies outside the submitted work. M.J. Pittet reports grants from ISREC Foundation during the conduct of the study. S. Zimmermann reports grants from Bristol-Myers Squibb during the conduct of the study; other support from Hoffmann-La Roche, other support from AstraZeneca, MSD, and Astellas outside the submitted work. U. Dafni reports personal fees and nonfinancial support from CHUV/CTE during the conduct of the study; personal fees from Roche outside the submitted work; and visiting professorship at UNIL/CHUV. G. Coukos reports grants from BMS, Prostate Cancer Foundation, and Cancera, Biltema and Paul Matson Foundations during the conduct of the study; other support from AstraZeneca AG, Bristol-Myers Squibb SA, F. Hoffmann-La Roche AG, MSD Merck AG, and Geneos Therapeutics outside the submitted work; and G. Coukos has received grants, research support or is coinvestigator in clinical trials by Bristol-Myers Squibb, Celgene, Boehringer Ingelheim, Roche, Tigen Pharma, Iovance, and Kite. CHUV has received honoraria for advisory services G. Coukos has provided to Roche, BMS, AstraZeneca, MSD Merck, and GeneosTx. G. Coukos has patents in the domain of antibodies and vaccines targeting the tumor vasculature as well as technologies related to T-cell expansion and engineering for T-cell therapy. G. Coukos receives royalties from the University of Pennsylvania. No disclosures were reported by the other authors.
Supplementary Material
Acknowledgments
We thank Esther Danenberg, Ekaterina Fortis, Alison Ferguson, Julian Dagher, Bahareh Khormadin, and Stephanie Tissot for IHC technical support; Romain Bedel for supporting with FACS sorting; Corinne Peter, Leonore Wigger, Carolina Bogkowska, and Julian Marquis for NanoString and RNA-sequencing experiments and analysis; Roli Misra for in vivo experiments and qPCR technical support; Petra Baumgarten for support with flow cytometry; Patrick Reichenbach, Bili Seijo, and Evripidis Lanitis for transduction technical support; Wilson Castro, Jennifer Murray, and Nicolas Gestermann for lab organizational support; Marie Catherine Vozenin, Benoit Petit, and Jonathan Olivier for irradiation technical support and help with the clonogenic survival essay; Mauro Delorenzi and Tania Wyss for bioinformatic support; and Camillo Ribi, Denis Comte, and Michel Obeid from the Centre Hospitalier Universitaire Vaudois for their help in patient care. This work was supported by the Ludwig Institute for Cancer Research and grants from Bristol-Myers Squibb, the Prostate Cancer Foundation Challenge Award (18CHAL08), and the Cancera, Biltema and Paul Matson Foundations (all to G. Coukos).
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
Note: Supplementary data for this article are available at Cancer Discovery Online (http://cancerdiscovery.aacrjournals.org/).
Authors' Contributions
F.G. Herrera: Conceptualization, data curation, formal analysis, supervision, funding acquisition, validation, investigation, visualization, methodology, writing–original draft, writing–review and editing. C. Ronet: Data curation, formal analysis, validation, investigation, methodology, writing–review and editing. M. Ochoa de Olza: Data curation, formal analysis, investigation, writing–review and editing. D. Barras: Software, formal analysis, investigation, visualization, methodology, writing–review and editing. I. Crespo: Software, formal analysis, investigation, visualization, methodology, writing–review and editing. M. Andreatta: Software, formal analysis, investigation, visualization, methodology, writing–review and editing. J. Corria-Osorio: Data curation, investigation, methodology, writing–review and editing. A. Spill: Investigation, methodology. F. Benedetti: Software, formal analysis, investigation, visualization, methodology, writing–review and editing. R. Genolet: Software, formal analysis, investigation, methodology, writing–review and editing. A. Orcurto: Investigation, methodology, writing–review and editing. M. Imbimbo: Investigation, methodology, writing–review and editing. E. Ghisoni: Investigation, methodology, writing–review and editing. B. Navarro Rodrigo: Investigation, methodology, writing–review and editing. D.R. Berthold: Investigation, methodology, writing–review and editing. A. Sarivalasis: Investigation, methodology, writing–review and editing. K. Zaman: Investigation, methodology, writing–review and editing. R. Duran: Investigation, methodology, writing–review and editing. C. Dromain: Investigation, methodology, writing–review and editing. J. Prior: Investigation, methodology, writing–review and editing. N. Schaefer: Investigation, methodology, writing–review and editing. J. Bourhis: Investigation, methodology, writing–review and editing. G. Dimopoulou: Data curation, software, formal analysis, validation, investigation, visualization, methodology, writing–review and editing. Z. Tsourti: Data curation, software, formal analysis, validation, investigation, visualization, methodology, writing–review and editing. M. Messemaker: Software, formal analysis, validation, investigation, visualization, methodology, writing–review and editing. T. Smith: Software, formal analysis, investigation, methodology. S.E. Warren: Software, formal analysis, investigation, methodology, writing–review and editing. P. Foukas: Data curation, formal analysis, supervision, investigation, visualization, methodology, writing–review and editing. S. Rusakiewicz: Data curation, software, formal analysis, supervision, validation, investigation, visualization, methodology, writing–review and editing. M.J. Pittet: Data curation, software, formal analysis, supervision, validation, investigation, visualization, methodology, writing–review and editing. S. Zimmermann: Supervision, investigation, methodology, writing–review and editing. C. Sempoux: Data curation, formal analysis, supervision, validation, investigation, visualization, methodology. U. Dafni: Data curation, software, formal analysis, supervision, validation, investigation, visualization, methodology, writing–review and editing. A. Harari: Data curation, software, formal analysis, supervision, validation, investigation, visualization, methodology, writing–review and editing. L.E. Kandalaft: Resources, data curation, supervision, funding acquisition, investigation, methodology, project administration, writing–review and editing. S.J. Carmona: Data curation, software, formal analysis, supervision, validation, investigation, visualization, methodology, writing–review and editing. D. Dangaj Laniti: Data curation, software, formal analysis, supervision, validation, investigation, visualization, methodology, writing–review and editing. M. Irving: Resources, data curation, software, formal analysis, supervision, validation, investigation, visualization, methodology, project administration, writing–review and editing. G. Coukos: Conceptualization, resources, data curation, software, formal analysis, supervision, funding acquisition, validation, investigation, visualization, methodology, writing–original draft, project administration, writing–review and editing.
References
- 1. Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert Let al. PD1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014;515:568–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Herbst RS, Soria JC, Kowanetz M, Fine GD, Hamid O, Gordon MSet al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 2014;515:563–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Herrera FG, Bourhis J, Coukos G. Radiotherapy combination opportunities leveraging immunity for the next oncology practice. CA Cancer J Clin 2017;67:65–85. [DOI] [PubMed] [Google Scholar]
- 4. Formenti SC, Rudqvist NP, Golden E, Cooper B, Wennerberg E, Lhuillier Cet al. Radiotherapy induces responses of lung cancer to CTLA4 blockade. Nat Med 2018;24:1845–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Deng L, Liang H, Xu M, Yang X, Burnette B, Arina Aet al. STING-dependent cytosolic DNA sensing promotes radiation-induced type I interferon-dependent antitumor immunity in immunogenic tumors. Immunity 2014;41:843–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Matsumura S, Wang B, Kawashima N, Braunstein S, Badura M, Cameron TOet al. Radiation-induced CXCL16 release by breast cancer cells attracts effector T cells. J Immunol 2008;181:3099–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lugade AA, Moran JP, Gerber SA, Rose RC, Frelinger JG, Lord EM. Local radiation therapy of B16 melanoma tumors increases the generation of tumor antigen-specific effector cells that traffic to the tumor. J Immunol 2005;174:7516–23. [DOI] [PubMed] [Google Scholar]
- 8. Vanpouille-Box C, Alard A, Aryankalayil MJ, Sarfraz Y, Diamond JM, Schneider RJet al. DNA exonuclease Trex1 regulates radiotherapy-induced tumour immunogenicity. Nat Commun 2017;8:15618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Burnette BC, Liang H, Lee Y, Chlewicki L, Khodarev NN, Weichselbaum RRet al. The efficacy of radiotherapy relies upon induction of type I interferon-dependent innate and adaptive immunity. Cancer Res 2011;71:2488–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Demaria S, Kawashima N, Yang AM, Devitt ML, Babb JS, Allison JPet al. Immune-mediated inhibition of metastases after treatment with local radiation and CTLA4 blockade in a mouse model of breast cancer. Clin Cancer Res 2005;11:728–34. [PubMed] [Google Scholar]
- 11. Deng L, Liang H, Burnette B, Beckett M, Darga T, Weichselbaum RRet al. Irradiation and anti-PD-L1 treatment synergistically promote antitumor immunity in mice. J Clin Invest 2014;124:687–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Twyman-Saint Victor C, Rech AJ, Maity A, Rengan R, Pauken KE, Stelekati Eet al. Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature 2015;520:373–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dovedi SJ, Adlard AL, Lipowska-Bhalla G, McKenna C, Jones S, Cheadle EJet al. Acquired resistance to fractionated radiotherapy can be overcome by concurrent PD-L1 blockade. Cancer Res 2014;74:5458–68. [DOI] [PubMed] [Google Scholar]
- 14. Postow MA, Callahan MK, Barker CA, Yamada Y, Yuan J, Kitano Set al. Immunologic correlates of the abscopal effect in a patient with melanoma. N Engl J Med 2012;366:925–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Klug F, Prakash H, Huber PE, Seibel T, Bender N, Halama Net al. Low-dose irradiation programs macrophage differentiation to an iNOS(+)/M1 phenotype that orchestrates effective T cell immunotherapy. Cancer Cell 2013;24:589–602. [DOI] [PubMed] [Google Scholar]
- 16. Barsoumian HB, Ramapriyan R, Younes AI, Caetano MS, Menon H, Comeaux NIet al. Low-dose radiation treatment enhances systemic antitumor immune responses by overcoming the inhibitory stroma. J Immunother Cancer 2020;8:e000537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fyles AW, Dembo AJ, Bush RS, Levin W, Manchul LA, Pringle JFet al. Analysis of complications in patients treated with abdomino-pelvic radiation therapy for ovarian carcinoma. Int J Radiat Oncol Biol Phys 1992;22:847–51. [DOI] [PubMed] [Google Scholar]
- 18. Herrera FG, Irving M, Kandalaft LE, Coukos G. Rational combinations of immunotherapy with radiotherapy in ovarian cancer. Lancet Oncol 2019;20:e417–e33. [DOI] [PubMed] [Google Scholar]
- 19. Kunos CA, Sill MW, Buekers TE, Walker JL, Schilder JM, Yamada SDet al. Low-dose abdominal radiation as a docetaxel chemosensitizer for recurrent epithelial ovarian cancer: a phase I study of the Gynecologic Oncology Group. Gynecol Oncol 2011;120:224–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ngoi NYL, Heong VYM, Kumarakulasinghe NB, Phyu PS, Peh EY, Lim SEet al. Phase I study of low dose whole abdominal radiation therapy (LDWART) in combination with weekly paclitaxel (wP) for platinum resistant ovarian cancer (PROC). Ann Oncol 2019;30:v422. [DOI] [PubMed] [Google Scholar]
- 21. Yang G, Li W, Jiang H, Liang X, Zhao Y, Yu Det al. Low-dose radiation may be a novel approach to enhance the effectiveness of cancer therapeutics. Int J Cancer 2016;139:2157–68. [DOI] [PubMed] [Google Scholar]
- 22. Roby KF, Taylor CC, Sweetwood JP, Cheng Y, Pace JL, Tawfik Oet al. Development of a syngeneic mouse model for events related to ovarian cancer. Carcinogenesis 2000;21:585–91. [DOI] [PubMed] [Google Scholar]
- 23. Dangaj D, Bruand M, Grimm AJ, Ronet C, Barras D, Duttagupta PAet al. Cooperation between constitutive and inducible chemokines enables T cell engraftment and immune attack in solid tumors. Cancer Cell 2019;35:885–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Crozat K, Guiton R, Contreras V, Feuillet V, Dutertre CA, Ventre Eet al. The XC chemokine receptor 1 is a conserved selective marker of mammalian cells homologous to mouse CD8alpha+ dendritic cells. J Exp Med 2010;207:1283–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dorner BG, Dorner MB, Zhou X, Opitz C, Mora A, Guttler Set al. Selective expression of the chemokine receptor XCR1 on cross-presenting dendritic cells determines cooperation with CD8+ T cells. Immunity 2009;31:823–33. [DOI] [PubMed] [Google Scholar]
- 26. Vonderheide RH, Glennie MJ. Agonistic CD40 antibodies and cancer therapy. Clin Cancer Res 2013;19:1035–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Le DT, Jaffee EM. Regulatory T-cell modulation using cyclophosphamide in vaccine approaches: a current perspective. Cancer Res 2012;72:3439–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Li S, Simoni Y, Zhuang S, Ma S, Chee J, Redwood Aet al. Unsuccessful checkpoint blockade and vaccination therapy despite expansion of tumor neoantigen-specific CD8 T cells in the Lewis lung carcinoma (LLC) model. J Immunol 2020;204:165.13. [Google Scholar]
- 29. Zhong W, Myers JS, Wang F, Wang K, Lucas J, Rosfjord Eet al. Comparison of the molecular and cellular phenotypes of common mouse syngeneic models with human tumors. BMC Genomics 2020;21:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Andreatta M, Corria-Osorio J, Müller S, Cubas R, Coukos G, Carmona SJ. Interpretation of T cell states from single-cell transcriptomics data using reference atlases. Nat Commun 2021;12:2965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Siddiqui I, Schaeuble K, Chennupati V, Fuertes Marraco SA, Calderon-Copete S, Pais Ferreira Det al. Intratumoral Tcf1(+)PD1(+)CD8(+) T cells with stem-like properties promote tumor control in response to vaccination and checkpoint blockade immunotherapy. Immunity 2019;50:195–211. [DOI] [PubMed] [Google Scholar]
- 32. Miller BC, Sen DR, Al Abosy R, Bi K, Virkud YV, LaFleur MWet al. Subsets of exhausted CD8(+) T cells differentially mediate tumor control and respond to checkpoint blockade. Nat Immunol 2019;20:326–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Alfei F, Kanev K, Hofmann M, Wu M, Ghoneim HE, Roelli Pet al. TOX reinforces the phenotype and longevity of exhausted T cells in chronic viral infection. Nature 2019;571:265–9. [DOI] [PubMed] [Google Scholar]
- 34. Utzschneider DT, Charmoy M, Chennupati V, Pousse L, Ferreira DP, Calderon-Copete Set al. T cell factor 1-expressing memory-like CD8(+) T cells sustain the immune response to chronic viral infections. Immunity 2016;45:415–27. [DOI] [PubMed] [Google Scholar]
- 35. Zilionis R, Engblom C, Pfirschke C, Savova V, Zemmour D, Saatcioglu HDet al. Single-cell transcriptomics of human and mouse lung cancers reveals conserved myeloid populations across individuals and species. Immunity 2019;50:1317–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. De Palma M, Lewis CE. Macrophage regulation of tumor responses to anticancer therapies. Cancer Cell 2013;23:277–86. [DOI] [PubMed] [Google Scholar]
- 37. Painter MW, Davis S, Hardy RR, Mathis D, Benoist C, Immunological Genome Project C. Transcriptomes of the B and T lineages compared by multiplatform microarray profiling. J Immunol 2011;186:3047–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Maier B, Leader AM, Chen ST, Tung N, Chang C, LeBerichel Jet al. A conserved dendritic-cell regulatory program limits antitumour immunity. Nature 2020;580:257–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhang L, Li Z, Skrzypczynska KM, Fang Q, Zhang W, O'Brien SAet al. Single-cell analyses inform mechanisms of myeloid-targeted therapies in colon cancer. Cell 2020;181:442–59. [DOI] [PubMed] [Google Scholar]
- 40. Hildner K, Edelson BT, Purtha WE, Diamond M, Matsushita H, Kohyama Met al. Batf3 deficiency reveals a critical role for CD8alpha+ dendritic cells in cytotoxic T cell immunity. Science 2008;322:1097–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sanchez-Paulete AR, Cueto FJ, Martinez-Lopez M, Labiano S, Morales-Kastresana A, Rodriguez-Ruiz MEet al. Cancer immunotherapy with immunomodulatory anti-CD137 and anti-PD-1 monoclonal antibodies requires BATF3-dependent dendritic cells. Cancer Discov 2016;6:71–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Broz ML, Binnewies M, Boldajipour B, Nelson AE, Pollack JL, Erle DJet al. Dissecting the tumor myeloid compartment reveals rare activating antigen-presenting cells critical for T cell immunity. Cancer Cell 2014;26:638–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Grajales-Reyes GE, Iwata A, Albring J, Wu X, Tussiwand R, Kc Wet al. Batf3 maintains autoactivation of Irf8 for commitment of a CD8alpha(+) conventional DC clonogenic progenitor. Nat Immunol 2015;16:708–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature 2015;523:231–5. [DOI] [PubMed] [Google Scholar]
- 45. Binnewies M, Mujal AM, Pollack JL, Combes AJ, Hardison EA, Barry KCet al. Unleashing type-2 dendritic cells to drive protective antitumor CD4(+) T cell immunity. Cell 2019;177:556–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Antonczyk A, Krist B, Sajek M, Michalska A, Piaszyk-Borychowska A, Plens-Galaska Met al. Direct inhibition of IRF-dependent transcriptional regulatory mechanisms associated with disease. Front Immunol 2019;10:1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Diefenbach A, Tomasello E, Lucas M, Jamieson AM, Hsia JK, Vivier Eet al. Selective associations with signaling proteins determine stimulatory versus costimulatory activity of NKG2D. Nat Immunol 2002;3:1142–9. [DOI] [PubMed] [Google Scholar]
- 48. Maasho K, Opoku-Anane J, Marusina AI, Coligan JE, Borrego F. NKG2D is a costimulatory receptor for human naive CD8+ T cells. J Immunol 2005;174:4480–4. [DOI] [PubMed] [Google Scholar]
- 49. Hellmann MD, Paz-Ares L, Bernabe Caro R, Zurawski B, Kim SW, Carcereny Costa Eet al. Nivolumab plus ipilimumab in advanced non-small-cell lung cancer. N Engl J Med 2019;381:2020–31. [DOI] [PubMed] [Google Scholar]
- 50. Zhao X, Suryawanshi S, Hruska M, Feng Y, Wang X, Shen Jet al. Assessment of nivolumab benefit-risk profile of a 240-mg flat dose relative to a 3-mg/kg dosing regimen in patients with advanced tumors. Ann Oncol 2017;28:2002–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Motz GT, Santoro SP, Wang LP, Garrabrant T, Lastra RR, Hagemann ISet al. Tumor endothelium FasL establishes a selective immune barrier promoting tolerance in tumors. Nat Med 2014;20:607–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Elicin O, Callaway S, Prior JO, Bourhis J, Ozsahin M, Herrera FG. [(18)F]FDG-PET standard uptake value as a metabolic predictor of bone marrow response to radiation: impact on acute and late hematological toxicity in cervical cancer patients treated with chemoradiation therapy. Int J Radiat Oncol Biol Phys 2014;90:1099–107. [DOI] [PubMed] [Google Scholar]
- 53. Seymour L, Bogaerts J, Perrone A, Ford R, Schwartz LH, Mandrekar Set al. iRECIST: guidelines for response criteria for use in trials testing immunotherapeutics. Lancet Oncol 2017;18:e143–e52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zollinger DR, Lingle SE, Sorg K, Beechem JM, Merritt CR. GeoMx RNA assay: high multiplex, digital, spatial analysis of RNA in FFPE tissue. Methods Mol Biol 2020;2148:331–45. [DOI] [PubMed] [Google Scholar]
- 55. Bindea G, Mlecnik B, Tosolini M, Kirilovsky A, Waldner M, Obenauf ACet al. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity 2013;39:782–95. [DOI] [PubMed] [Google Scholar]
- 56. Fairfax BP, Taylor CA, Watson RA, Nassiri I, Danielli S, Fang Het al. Peripheral CD8(+) T cell characteristics associated with durable responses to immune checkpoint blockade in patients with metastatic melanoma. Nat Med 2020;26:193–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lutsiak ME, Semnani RT, De Pascalis R, Kashmiri SV, Schlom J, Sabzevari H. Inhibition of CD4(+)25+ T regulatory cell function implicated in enhanced immune response by low-dose cyclophosphamide. Blood 2005;105:2862–8. [DOI] [PubMed] [Google Scholar]
- 58. Moynihan KD, Opel CF, Szeto GL, Tzeng A, Zhu EF, Engreitz JMet al. Eradication of large established tumors in mice by combination immunotherapy that engages innate and adaptive immune responses. Nat Med 2016;22:1402–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Brown DM, Lee S, Garcia-Hernandez Mde L, Swain SL. Multifunctional CD4 cells expressing gamma interferon and perforin mediate protection against lethal influenza virus infection. J Virol 2012;86:6792–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Crawford A, Angelosanto JM, Kao C, Doering TA, Odorizzi PM, Barnett BEet al. Molecular and transcriptional basis of CD4(+) T cell dysfunction during chronic infection. Immunity 2014;40:289–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Peeters LM, Vanheusden M, Somers V, Van Wijmeersch B, Stinissen P, Broux Bet al. Cytotoxic CD4+ T cells drive multiple sclerosis progression. Front Immunol 2017;8:1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Quezada SA, Simpson TR, Peggs KS, Merghoub T, Vider J, Fan Xet al. Tumor-reactive CD4(+) T cells develop cytotoxic activity and eradicate large established melanoma after transfer into lymphopenic hosts. J Exp Med 2010;207:637–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Xie Y, Akpinarli A, Maris C, Hipkiss EL, Lane M, Kwon EKet al. Naive tumor-specific CD4(+) T cells differentiated in vivo eradicate established melanoma. J Exp Med 2010;207:651–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kitano S, Tsuji T, Liu C, Hirschhorn-Cymerman D, Kyi C, Mu Zet al. Enhancement of tumor-reactive cytotoxic CD4+ T cell responses after ipilimumab treatment in four advanced melanoma patients. Cancer Immunol Res 2013;1:235–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Oh DY, Kwek SS, Raju SS, Li T, McCarthy E, Chow Eet al. Intratumoral CD4(+) T cells mediate anti-tumor cytotoxicity in human bladder cancer. Cell 2020;181:1612–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Hirschhorn-Cymerman D, Budhu S, Kitano S, Liu C, Zhao F, Zhong Het al. Induction of tumoricidal function in CD4+ T cells is associated with concomitant memory and terminally differentiated phenotype. J Exp Med 2012;209:2113–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Qui HZ, Hagymasi AT, Bandyopadhyay S, St Rose MC, Ramanarasimhaiah R, Menoret Aet al. CD134 plus CD137 dual costimulation induces Eomesodermin in CD4 T cells to program cytotoxic Th1 differentiation. J Immunol 2011;187:3555–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Serroukh Y, Gu-Trantien C, Hooshiar Kashani B, Defrance M, Vu Manh TP, Azouz Aet al. The transcription factors Runx3 and ThPOK cross-regulate acquisition of cytotoxic function by human Th1 lymphocytes. Elife 2018;7:e30496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Oja AE, Vieira Braga FA, Remmerswaal EB, Kragten NA, Hertoghs KM, Zuo Jet al. The transcription factor hobit identifies human cytotoxic CD4(+) T cells. Front Immunol 2017;8:325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Sledzinska A, Vila de Mucha M, Bergerhoff K, Hotblack A, Demane DF, Ghorani Eet al. Regulatory T cells restrain interleukin-2- and Blimp-1-dependent acquisition of cytotoxic function by CD4(+) T cells. Immunity 2020;52:151–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Scheper W, Kelderman S, Fanchi LF, Linnemann C, Bendle G, de Rooij MAJet al. Low and variable tumor reactivity of the intratumoral TCR repertoire in human cancers. Nat Med 2019;25:89–94. [DOI] [PubMed] [Google Scholar]
- 72. van der Leun AM, Thommen DS, Schumacher TN. CD8(+) T cell states in human cancer: insights from single-cell analysis. Nat Rev Cancer 2020;20:218–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Beltra JC, Manne S, Abdel-Hakeem MS, Kurachi M, Giles JR, Chen Zet al. Developmental relationships of four exhausted CD8(+) T cell subsets reveals underlying transcriptional and epigenetic landscape control mechanisms. Immunity 2020;52:825–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Kallies A, Zehn D, Utzschneider DT. Precursor exhausted T cells: key to successful immunotherapy? Nat Rev Immunol 2020;20:128–36. [DOI] [PubMed] [Google Scholar]
- 75. Kurtulus S, Madi A, Escobar G, Klapholz M, Nyman J, Christian Eet al. Checkpoint blockade immunotherapy induces dynamic changes in PD1(−)CD8(+) tumor-infiltrating T cells. Immunity 2019;50:181–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Khan O, Giles JR, McDonald S, Manne S, Ngiow SF, Patel KPet al. TOX transcriptionally and epigenetically programs CD8(+) T cell exhaustion. Nature 2019;571:211–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Scott AC, Dundar F, Zumbo P, Chandran SS, Klebanoff CA, Shakiba Met al. TOX is a critical regulator of tumour-specific T cell differentiation. Nature 2019;571:270–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Markiewicz MA, Carayannopoulos LN, Naidenko OV, Matsui K, Burack WR, Wise ELet al. Costimulation through NKG2D enhances murine CD8+ CTL function: similarities and differences between NKG2D and CD28 costimulation. J Immunol 2005;175:2825–33. [DOI] [PubMed] [Google Scholar]
- 79. Groh V, Smythe K, Dai Z, Spies T. Fas-ligand-mediated paracrine T cell regulation by the receptor NKG2D in tumor immunity. Nat Immunol 2006;7:755–62. [DOI] [PubMed] [Google Scholar]
- 80. Groh V, Bruhl A, El-Gabalawy H, Nelson JL, Spies T. Stimulation of T cell autoreactivity by anomalous expression of NKG2D and its MIC ligands in rheumatoid arthritis. Proc Natl Acad Sci U S A 2003;100:9452–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Garcia-Chagollan M, Jave-Suarez LF, Haramati J, Bueno-Topete MR, Aguilar-Lemarroy A, Estrada-Chavez Cet al. An approach to the immunophenotypic features of circulating CD4(+)NKG2D(+) T cells in invasive cervical carcinoma. J Biomed Sci 2015;22:91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Lazarova M, Steinle A. Impairment of NKG2D-mediated tumor immunity by TGF-beta. Front Immunol 2019;10:2689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Groh V, Wu J, Yee C, Spies T. Tumour-derived soluble MIC ligands impair expression of NKG2D and T-cell activation. Nature 2002;419:734–8. [DOI] [PubMed] [Google Scholar]
- 84. Salih HR, Rammensee HG, Steinle A. Cutting edge: down-regulation of MICA on human tumors by proteolytic shedding. J Immunol 2002;169:4098–102. [DOI] [PubMed] [Google Scholar]
- 85. Paczulla AM, Rothfelder K, Raffel S, Konantz M, Steinbacher J, Wang Het al. Absence of NKG2D ligands defines leukaemia stem cells and mediates their immune evasion. Nature 2019;572:254–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Zhang J, Larrocha PS, Zhang B, Wainwright D, Dhar P, Wu JD. Antibody targeting tumor-derived soluble NKG2D ligand sMIC provides dual co-stimulation of CD8 T cells and enables sMIC(+) tumors respond to PD1/PD-L1 blockade therapy. J Immunother Cancer 2019;7:223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Ruocco MG, Pilones KA, Kawashima N, Cammer M, Huang J, Babb JSet al. Suppressing T cell motility induced by anti-CTLA4 monotherapy improves antitumor effects. J Clin Invest 2012;122:3718–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Stojanovic A, Correia MP, Cerwenka A. The NKG2D/NKG2DL axis in the crosstalk between lymphoid and myeloid cells in health and disease. Front Immunol 2018;9:827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Ma HS, Poudel B, Torres ER, Sidhom JW, Robinson TM, Christmas Bet al. A CD40 agonist and PD1 antagonist antibody reprogram the microenvironment of nonimmunogenic tumors to allow T-cell-mediated anticancer activity. Cancer Immunol Res 2019;7:428–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Beatty GL, Chiorean EG, Fishman MP, Saboury B, Teitelbaum UR, Sun Wet al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science 2011;331:1612–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Vonderheide RH. CD40 agonist antibodies in cancer immunotherapy. Annu Rev Med 2020;71:47–58. [DOI] [PubMed] [Google Scholar]
- 92. Antonarakis ES, Piulats JM, Gross-Goupil M, Goh J, Ojamaa K, Hoimes CJet al. Pembrolizumab for treatment-refractory metastatic castration-resistant prostate cancer: multicohort, open-label phase II KEYNOTE-199 study. J Clin Oncol 2020;38:395–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Kandalaft LE, Odunsi K, Coukos G. Immunotherapy in ovarian cancer: are we there yet? J Clin Oncol 2019;37:2460–71. [DOI] [PubMed] [Google Scholar]
- 94. Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring ADet al. PD1 blockade in tumors with mismatch-repair deficiency. N Engl J Med 2015;372:2509–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Papadopoulos KP, Johnson ML, Lockhart AC, Moore K, Falchook GS, Formenti SCet al. First-in-human study of cemiplimab alone or in combination with radiotherapy and/or low-dose cyclophosphamide in patients with advanced malignancies. Clin Cancer Res 2020;26:1025–33. [DOI] [PubMed] [Google Scholar]
- 96. Heylmann D, Bauer M, Becker H, van Gool S, Bacher N, Steinbrink Ket al. Human CD4+CD25+ regulatory T cells are sensitive to low dose cyclophosphamide: implications for the immune response. PLoS One 2013;8:e83384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Scurr M, Pembroke T, Bloom A, Roberts D, Thomson A, Smart Ket al. Low-dose cyclophosphamide induces antitumor T-cell responses, which associate with survival in metastatic colorectal cancer. Clin Cancer Res 2017;23:6771–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Liakou CI, Kamat A, Tang DN, Chen H, Sun J, Troncoso Pet al. CTLA4 blockade increases IFNgamma-producing CD4+ICOShi cells to shift the ratio of effector to regulatory T cells in cancer patients. Proc Natl Acad Sci U S A 2008;105:14987–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Wei SC, Anang NAS, Sharma R, Andrews MC, Reuben A, Levine JHet al. Combination anti-CTLA4 plus anti-PD-1 checkpoint blockade utilizes cellular mechanisms partially distinct from monotherapies. Proc Natl Acad Sci U S A 2019;116:22699–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Pavlick AC, Sunandana C, Stein C, Madden K, Kannan R, Escano Cet al. Phase II study of low-dose cyclophosphamide and ipilimumab in metastatic melanoma. J Clin Oncol 2014;32:e20025. [Google Scholar]
- 101. Sinha P, Clements VK, Fulton AM, Ostrand-Rosenberg S. Prostaglandin E2 promotes tumor progression by inducing myeloid-derived suppressor cells. Cancer Res 2007;67:4507–13. [DOI] [PubMed] [Google Scholar]
- 102. Baratelli F, Lin Y, Zhu L, Yang SC, Heuze-Vourc'h N, Zeng Get al. Prostaglandin E2 induces FOXP3 gene expression and T regulatory cell function in human CD4+ T cells. J Immunol 2005;175:1483–90. [DOI] [PubMed] [Google Scholar]
- 103. Lanitis E, Rota G, Kosti P, Ronet C, Spill A, Seijo Bet al. Optimized gene engineering of murine CAR-T cells reveals the beneficial effects of IL-15 coexpression. J Exp Med 2021;218:e20192203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini JLet al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med 2007;13:54–61. [DOI] [PubMed] [Google Scholar]
- 105. Tabula Muris Consortium. Overall coordination, Logistical coordination, Organ collection and processing, Library preparation and sequencing, Computational data analysis, et al. Single-cell transcriptomics of 20 mouse organs creates a Tabula Muris. Nature 2018;562:367–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Newman AM, Steen CB, Liu CL, Gentles AJ, Chaudhuri AA, Scherer Fet al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat Biotechnol 2019;37:773–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Roederer M, Nozzi JL, Nason MC. SPICE: exploration and analysis of post-cytometric complex multivariate datasets. Cytometry A 2011;79:167–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Festing MF. On determining sample size in experiments involving laboratory animals. Lab Anim 2018;52:341–50. [DOI] [PubMed] [Google Scholar]
- 109. Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM IIIet al. Comprehensive integration of single-cell data. Cell 2019;177:1888–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Borcherding N, Bormann NL, Kraus G. scRepertoire: an R-based toolkit for single-cell immune receptor analysis. F1000Res 2020;9:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Waltman L, van Eck NJ. A smart local moving algorithm for large-scale modularity-based community detection. Eur Phys J B 2013;86:471. [Google Scholar]
- 112. Aran D, Looney AP, Liu L, Wu E, Fong V, Hsu Aet al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat Immunol 2019;20:163–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha Set al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 2013;29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Anders S, Pyl PT, Huber W. HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics 2015;31:166–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are available from the authors upon reasonable request. For genomic analysis, data can be consulted in GSE169742 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE169742).