

Abstract
Pediatric tumors are uncommon, yet are the leading cause of cancer-related death in childhood. Tumor types, molecular characteristics, and pathogenesis are unique, often originating from a single genetic driver event. The specific diagnostic challenges of childhood tumors led to the development of the first World Health Organization (WHO) Classification of Pediatric Tumors. The classification is rooted in a multilayered approach, incorporating morphology, IHC, and molecular characteristics. The volume is organized according to organ sites and provides a single, state-of-the-art compendium of pediatric tumor types. A special emphasis was placed on “blastomas,” which variably recapitulate the morphologic maturation of organs from which they originate.
Significance:
In this review, we briefly summarize the main features and updates of each chapter of the inaugural WHO Classification of Pediatric Tumors, including its rapid transition from a mostly microscopic into a molecularly driven classification systematically taking recent discoveries in pediatric tumor genomics into account.
Introduction
Why Pediatric Tumors Need a Separate Classification
Childhood tumors are fundamentally different in many ways from those occurring in adults. Despite being extremely heterogeneous, they account for only approximately one percent of all tumor diagnoses, but at the same time represent the most common cause of disease-related death in children (1). In contrast to malignancies in adults, which are mostly of epithelial origin and often caused by an extended exposure to carcinogens, tumors in childhood are often derived from the mesoderm or neuroectoderm, and, with the exception of hereditary cancer predisposition in approximately 10% of patients, their etiology is largely unknown (2). According to data from the Leukemia and Lymphoma Society, the most common groups of cancer in children, adolescents, and young adults (CAYA; i.e., younger than 20 years) are: leukemia (24.7%), tumors of the nervous system (17.2%), non-Hodgkin lymphoma (7.5%), Hodgkin lymphoma (6.5%), and soft-tissue sarcoma (5.9%). In contrast to cells successively acquiring genetic hits over time in adults, pediatric tumors are typically caused by a maturation block occurring in an immature developing cell type (3). Tumors in children typically carry a much lower burden of genetic aberrations, often driven by a single and thus clonal genetic driver event, such as a translocation leading to an oncogenic fusion (4, 5). Tumors in children predominantly show very limited immune cell infiltration and are thus often considered immunologically “cold” tumors (6–8). All of these unique properties of childhood cancers need to be considered when diagnosing and ultimately treating these children, thus fully justifying a separate World Health Organization (WHO) classification specifically focusing on pediatric tumors. Given the relative rarity of pediatric tumors compared with cancer in adults, cooperation across multiple institutions, national and international consortia, are required to gather enough cases to produce statistically significant data. However, these efforts are complex and face many challenges, including difficulties in communication, sharing biological materials, diverse classification systems applied in different regions of the world, etc. With this in mind, this WHO pediatric tumor classification represents a special effort to use a reproducible and evidence-based taxonomic system, striving for a uniform classification that may result in worldwide improvements.
This new WHO classification of pediatric tumors is intended to support the pathologist responsible for diagnosing the tumor and the multidisciplinary team tailoring treatment intensity to disease risk and matching patients to specific therapies. It should also improve access to molecular genetic testing and, consequently, innovative treatments for children with cancer through increased knowledge of molecularly defined disease subtypes and therapeutic target frequencies resulting from their routine assessment at the time of diagnosis.
Tumor Classification According to WHO Criteria
For the first time, pediatric tumors are covered in a separate volume in the new fifth edition of the WHO classification of tumors. In previous editions, pediatric tumors were covered together with adult tumors in the respective organ systems. As outlined before, it is increasingly clear that many aspects of pediatric tumors differ significantly from those of adults. Even tumors that histologically appear to be of the same type often have distinctive etiology and pathogenesis, which is reflected in their diagnosis and clinical behavior. In the fifth edition series, this has been recognized and the opportunity has been taken to describe these tumors in greater detail.
In keeping with other volumes in the fifth edition series, the WHO Classification of Pediatric Tumors follows a hierarchical classification and lists tumors by site, category, family and type. Each tumor type is described with a common, defined set of characteristics, and where information is not available, this is clearly indicated. The classification is also published on a website, which permits the use of whole-slide images and hyperlinks to evidence cited. Improving the quality of evidence is an important facet of the evolution of the classification of tumors, and hence diagnosis. As a result, we have sought to include new information from methylation studies, and other genomic investigations using HUGO Gene Nomenclature Committee (HGNC) and Human Genome Variation Society (HGVS) notation as appropriate. For those tumors where assessment of proliferation is important for diagnosis or prognosis, we recommend that mitoses are now counted per millimeter squared, thus adhering to standardized international (SI) units, as microscope high-power fields can be of variable size (9). We have also encouraged authors to use medians rather than means when skewed data are being considered. This is particularly important when pediatric age distributions are considered.
Novel Diagnostic Technologies
The diagnostic shift from morphology to molecular analyses is driven by both technology and the need for an even more granular and unbiased classification to optimally serve our patients. Foremost, the introduction of global approaches including next-generation sequencing (NGS), methylome analysis, and proteomics are driving this development. Tumor classification in recent years has been greatly influenced by methylation analysis, being the technology at present best suited for addressing lineage and thereby the cell population of origin of tumors (10, 11). The stability of the diagnostic methylation pattern seen in multiple specimens from the same tumor resection and throughout further progression belies the concept of methylation states representing cellular differentiation rather than neoplastic changes (12). In contrast, NGS focuses on tumor-specific alterations which may be pathognomonic in some instances, especially in the context of genetically “simple” pediatric tumors with gene fusions, focal amplifications, or point mutations. NGS employed in common diagnostic settings addresses differently sized gene panels (13) up to whole-exome (or even whole-genome) sequencing with a tendency to shift toward the latter (14–18). The added value of integrating somatic and germline sequencing data is becoming increasingly evident in this context. One diagnostically valuable NGS technology is RNA sequencing (RNA-seq), which reliably detects pathognomonic gene fusions and provides insight in the activity of gene transcription (19). Diagnostic protein analysis, currently almost exclusively facilitated by IHC, will be supplemented by mass spectrometry–based proteomics allowing simultaneous identification and quantification of several thousand proteins in tumor tissues (20, 21). There is an expectation that this will shed light on the activity of cellular signaling pathways. For example, information about phosphoproteins could lead to recommendations for specific inhibitory therapies. Proteome analyses may turn out to be the most direct approach to tumor characterization, as it combines the readout of cellular responses to epigenetic differentiation settings, tumor-specific structural alterations, and optimization-driven cellular regulation.
In conclusion, future tumor diagnostics are likely to rely on several molecular platforms that contribute orthogonal information to address the questions “where does it come from?” (methylome analyses), “how far has it gone?” (NGS), and “how to treat the patient?” (NGS, proteomics). That said, morphologic tumor diagnostics is also progressing. Identifying novel tumor types based on their molecular profile is followed by focused histologic and IHC evaluation that frequently detects diagnostic features, which can be assessed with classic technologies. Examples are the primary intracranial sarcoma, DICER1-mutant (22, 23), the diffuse glioneuronal tumor with oligodendroglioma-like features and nuclear clusters (24, 25), or the recently described neuroepithelial tumor with PATZ1 fusion (26). Rapid progress is being made with artificial intelligence–based analysis of morphologic images, and this may contribute greatly to tumor evaluation (27, 28). An important task to solve is how to merge the different diagnostic molecular and imaging platforms, including preoperative data, to make a combined evaluation.
Integrated and Layered Diagnoses
A pathologic diagnosis serves the purpose of communicating information relevant for tailored management, including on prognosis and therapeutic options, in the most condensed manner. The diagnosis should be standardized and suitable for local, national, and international communication. Among several approaches, the WHO Classification system emerged as most widely accepted. For a long time, the WHO Classification has provided recommendations on how to reach diagnoses in ways which could be successfully performed in most parts of the world. However, the enormous progress, mainly in molecular diagnosis, which has far-reaching impact on classification, grading, and therapy, is not compatible anymore with such a highly condensed diagnosis or with very different local requirements. When possible, a large body of relevant information needs to be communicated, but the amount of additional information gathered varies from institution to institution and country to country. To overcome this problem, a multilayered diagnosis resulting in an integrated diagnosis has been devised (29, 30). Key to this approach are four of the major characteristics consisting of (i) a compilation of the data from ii–iv to an integrated diagnosis, (ii) a (classic) morphologic evaluation, (iii) a tumor grade, and (iv) a level providing the most salient molecular information. The minimum requirements for reaching the predefined integrated diagnoses are provided by the WHO classification. While many tumors do fit such a matrix, two problems may occur due to either a lack of information or nonmatching information. Where essential molecular tests are not available, the solution is the addition of “not otherwise specified” (NOS) to the morphologic evaluation. In cases where molecular information does not match a WHO tumor type, the addition of “not elsewhere classified” (NEC) to the diagnosis highlights this problem (30, 31). In summary, the integrated diagnosis serves to communicate diagnoses based on different levels of analyses while still maintaining a universal terminology.
Entity-Specific Developments
Leukemias and Lymphomas
Hematolymphoid neoplasms are the most prevalent group of cancers (38.7%) in CAYA. With increasing knowledge of the genetics of hematolymphoid neoplasms, a molecularly oriented classification has a significant impact on the accuracy of diagnosis, treatment, and prognosis. In recent years, the availability of conjugated or unconjugated mAbs against specific targets (such as CD20, CD19, CD22), small molecules interfering with activated molecular pathways (such as tyrosine kinase inhibitors, γ-secretase inhibitors, FLT3 inhibitors), and genetically engineered chimeric antigen receptor T cells (CAR T) as immunotherapy has broadened the opportunities to target the key genetic aberrations in patients with various leukemias and lymphomas (32–36).
Hematopathology has been at the forefront in the adoption of newly available molecular techniques, and indeed the current WHO classification of hematolymphoid neoplasms has long since evolved from a morphologic classification to a classification that integrates clinical, morphologic, immunophenotypical, and molecular features in the definition of entities (37, 38).
The classification of hematolymphoid neoplasms in the WHO Classification of Pediatric Tumors focuses on the landscape of these neoplasms in CAYA and is essentially an adaptation of the revised fourth edition of WHO Classification of Tumors of Hematopoietic and Lymphoid Tissues (Table 1; refs. 32, 39–41). Consequently, adult-type entities that are rare or practically nonexistent in the CAYA age group, such as chronic neutrophilic leukemia, polycythemia vera, essential thrombocythemia, chronic eosinophilic leukemia, and chronic lymphocytic leukemia (CLL), are not part of the pediatric classification, while they are described in detail in the WHO Classification of Hematopoietic and Lymphoid Tissues. Biological and genetic abnormalities are a defining criterion in some entities, such as chronic myeloid leukemia (CML), acute myeloid leukemia (AML) with recurrent genetic abnormalities, large B-cell lymphoma with IRF4 rearrangement, and ALK-positive anaplastic large cell lymphoma. In other entities, genetic aberrations contribute to the diagnosis, identify prognostic categories, or represent targets potentially amenable to therapy.
Table 1.
Classification of pediatric leukemias and lymphomas

Leukemias and myeloid neoplasms
Leukemias comprise one fourth to one third of all malignancies in CAYA, with 80% being acute lymphoblastic leukemia (ALL), 15% AML, and 2% CML. Thus, the proportions of the various leukemia types differ markedly from those seen in adults (38% AML, 30% CLL, 15% CML, 11% ALL).
ALL represents the most common type of leukemia in CAYA, with 85% being of B-lineage (B-ALL; B-lymphoblastic leukemia; ref. 42). Most cases show recurrent genetic abnormalities (Table 1 and more detailed in Supplementary Table S1), which have prognostic significance, for example, B-ALL with ETV6::RUNX1, TCF3::PBX1 and high hyperdiploidy is associated with a favorable outcome, whereas B-ALL with hypodiploidy and KMT2A rearrangement is associated with a poor prognosis (34). BCR::ABL-like (Philadelphia-like) B-ALL is a high-risk B-ALL characterized by heterogeneous genetic alterations, unified by a gene expression profile similar to Ph-positive B-ALL while lacking BCR::ABL1 gene fusion (43). Genetic alterations are variable and may include IKZF1 deletion, CRLF2 rearrangement and overexpression, JAK/IL7R mutations, ABL1 class fusions, EPOR rearrangements, tyrosine kinase pathway activation, and other less common genomic alterations. As a result, for a precise diagnosis it is often necessary to apply multiple techniques such as gene expression profiling/RNA-seq, FISH, reverse transcription PCR, flow cytometry, and NGS. The treatment of ALL has been a remarkable success story in pediatric oncology, with a meager 5-year overall survival rate of 31% in 1975 that has improved to 90% nowadays due to the adoption of risk-stratified dose-intensive chemotherapy (34). Disease-risk stratification can be improved further by the incorporation of genomic data, which will also aid in tailoring the treatment to minimize long-term side effects (35).
Childhood AML has an overall survival of about 70% despite significant advances in risk classification, chemotherapy intensification, and stem cell transplantation (44, 45). Improved understanding of the tumor biology and molecular pathways of AML provides opportunities to design novel targeted therapies, which will be facilitated by a classification with emphasis on genetic aberrations (45). Subtypes of AML with different recurrent genetic abnormalities are associated with different prognoses (Table 1). Cases of AML that show genetic changes not covered by the defined list can be classified as AML, NOS, appended with the key molecular alterations. The findings of the Children's Oncology Group-National Cancer Institute TARGET AML initiative on the molecular landscape of pediatric AML, based on nearly 1,000 cases, are particularly illuminating, highlighting differences from adult AML (46). Some structural variants, such as new gene fusions and focal deletions of MBNL1, SEB2, and ELF1, are much more prevalent in pediatric compared with adult AML, while some mutations common in adult AML (such as DNMT3A and TP53) virtually never occur in pediatric AML (46). Some new recurrent mutations (such as MYC-ITD, NRAS, KRAS) have also been discovered (46). It is envisaged that the next edition of the WHO Classification of Tumors of Haematopoietic and Lymphoid Tissues will entail more pediatric-specific changes.
Among myeloid neoplasms, juvenile myelomonocytic leukemia (JMML), refractory cytopenia of childhood (RCC), and myeloid proliferations associated with Down syndrome are strictly pediatric diseases.
JMML is characterized by mutations in genes of the RAS signaling pathway (47). PTPN11-, NRAS- or KRAS-mutated JMML shows somatic gain-of-function mutations in nonsyndromic children, while NF1- or CBL-mutated JMML occurs in type 1 neurofibromatosis and CBL mutation–associated syndrome, respectively, characterized by germline mutation and acquired biallelic inactivation of the respective tumor suppressor genes.
RCC is distinct from adult myelodysplastic syndrome, in that the bone marrow is often hypocellular, and somatic alterations commonly seen in the latter, such as mutations in TET2, DNMT3A, TP53, and the spliceosome complex, are usually absent (48–50). In a proportion of cases, monosomy 7 is found (51–54), which was further shown to be associated with germline mutations in GATA2 or SAMD9/9L (55).
Myeloid proliferations associated with Down syndrome encompass transient abnormal myelopoiesis (TAM) and myeloid leukemia of Down syndrome (ML-DS). TAM occurs in newborns, and most cases show spontaneous remission, although ML-DS may supervene in 1 to 3 years in some 25% of cases. The disease is characterized by somatic mutations in GATA1 ML-DS (56–58) and usually occurs before the age of 5 years. Most cases exhibit features of megakaryoblastic leukemia, and harbor GATA1 mutations plus additional mutations. The spectrum of mutations is distinct from other pediatric and adult AML, usually targeting genes encoding cohesin components, signal transducers, and epigenetic regulators (59, 60).
Lymphomas
A subset of pediatric lymphomas is associated with congenital immunodeficiencies or Epstein–Barr virus (EBV) infection, but for the vast majority of children with lymphoma the etiology and predisposing factors are not known (40, 41). Pediatric lymphomas show several features distinct from adult lymphomas. Most are precursor B-cell or T-cell lymphoblastic leukemias/lymphomas, high-grade B-cell lymphomas (particularly Burkitt lymphoma) or, among mature T-cell lymphomas, ALK-positive anaplastic large cell lymphomas (41, 61). Low-grade B-cell lymphomas, such as CLL, follicular lymphoma, lymphoplasmacytic lymphoma, and mantle cell lymphomas, rarely occur in the pediatric age group. High-grade lymphomas (such as Burkitt lymphoma and diffuse large B-cell lymphoma) and Hodgkin lymphomas in the pediatric population have an excellent prognosis (often curable in >90%), superior to that observed in adults (62–64). Several lymphoma types occur almost exclusively in the CAYA age group, including pediatric-type follicular lymphoma, pediatric nodal marginal zone lymphoma, large B-cell lymphoma with IRF4 rearrangement, systemic EBV+ T-cell lymphoma of childhood and hydroa vacciniforme lymphoproliferative disorder.
Although the classification scheme of lymphomas is less molecularly defined compared with the classification of leukemias, most lymphoma types do exhibit distinctive molecular alterations, some of which are defining, such as ALK-positive anaplastic large cell lymphoma and large B-cell lymphoma with IRF4 translocation (as indicated in Table 1).
Soft-Tissue and Bone Tumors
Classifications of soft-tissue and bone tumors have progressively integrated our increasing knowledge regarding recurrent molecular alterations with the traditional diagnostic approach based on morphologic evidence of a lineage differentiation. A token of the limitations of classic morphology for the classification of these tumors was the introduction of a category for “unclassifiable sarcomas” in the 2013 fourth edition of the WHO Classification of Soft Tissue and Bone Tumors. In the current WHO 2020 fifth edition, this has evolved into a growing group of newly characterized tumor types that (at least so far) lack an identifiable lineage of differentiation, but which are now defined by specific recurrent genetic/molecular alterations (ref. 65; Fig. 1A). By and large, the WHO classification of pediatric tumors has been built upon the backbone of the current WHO Classification of Soft Tissue and Bone Tumors. It thoroughly describes entities typical of pediatric age as well as the clinical, pathologic, and molecular features of adult-type tumors frequently occurring in children, including particular pseudotumoral/malformative lesions and hamartomas.
Figure 1.

A, Intertumoral heterogeneity of soft-tissue and bone tumors as assessed by DNA methylation array. Unsupervised, nonlinear t-distributed stochastic neighbor embedding projection of methylation array profiles of 610 soft-tissue and bone tumor samples. Samples have been selected from a large database of sarcoma datasets to serve as reference profiles for training a supervised classification model based on strict criteria. B–G, Undifferentiated small round cell sarcomas of bone and soft tissue. B, Ewing sarcoma with EWSR::FLI 1 fusions. C, Soft-tissue sarcoma with BCOR alteration (BCOR::MAML3 fusion). D, CIC::DUX4 sarcoma. CD99 membranous staining varies from strong and diffuse in ES (E) and BCOR::MAML (F) to focal in CIC::DUX4 (G).
Benign soft-tissue tumors
Benign soft-tissue tumors in children vastly outnumber sarcomas, with benign myofibroblastic and vascular tumors being the most frequently encountered lesions (66). Their accurate characterization requires an expert integration of clinical, histologic, and genetic/molecular findings to define both their potential to progress and their possible role as a sentinel event of more complex syndromes (66, 67). Compared with the 2020 WHO Classification of Soft Tissue and Bone Tumors (65), a special emphasis has been placed on benign vascular lesions, which have been redefined in light of the clinical and pathogenetic orientation of the International Society for the Study of Vascular Anomalies classification (https://www.issva.org/classification). The term “hemangioma” has been dropped and replaced by capillary, venous and arteriovenous malformations, intramuscular vascular anomalies, and lymphatic anomalies, clearly defining their malformative nature and the pathogenetic molecular pathways involved. Furthermore, complex malformations were subdivided into different categories on the basis of molecular alterations and associated syndromes (Table 2).
Table 2.
Classification of pediatric soft-tissue and bone tumors

Soft-tissue sarcomas
Soft-tissue sarcomas in children account for 6% to 7% of all childhood malignancies, with rhabdomyosarcomas (RMS) being the most common, while the others are often referred to by pediatric oncologists as “sarcomas other than rhabdomyosarcomas” (67–70).
In line with the WHO Soft Tissue Tumors Classification, four RMS types can be identified: (i) embryonal (ERMS) including the anaplastic variant; (ii) alveolar with FOXO1 fusions; (iii) spindle/sclerosing RMS including infantile RMS with VGLL2::NCOA2 rearrangements and RMS with MYOD1 mutations (while those lacking fusions are morphologic variants of ERMS); and (iv) pleomorphic RMS, which is extremely rare in children and may represent a diagnostic pitfall when dealing with ERMS with diffuse anaplasia (Supplementary Table S2). RMS is highly aggressive, but remarkably responsive to conventional chemotherapy with an overall 5-year survival rate greater than 70% for localized disease (71, 72).
Soft-tissue sarcomas other than RMS account for about 3% to 4% of all pediatric cancers and can be divided into soft-tissue tumors with intermediate prognosis (locally aggressive and/or rarely metastasizing), such as infantile fibrosarcoma or inflammatory myofibroblastic tumor, and high-grade sarcomas, mostly adult-type sarcomas (refs. 73–77; Table 2).
Infantile fibrosarcoma and inflammatory myofibroblastic tumor are both tyrosine kinase–driven neoplasms and share similar pathogenetic mechanisms with the emerging category of “NTRK-rearranged spindle cell neoplasm” (65, 78). This latter category, currently classified under “tumors with unknown histogenesis” in the 2020 WHO Classification of Soft Tissue Tumors, has been redefined as “pediatric NTRK-rearranged spindle cell neoplasms” and is included in the group of myofibroblastic tumors in the WHO Pediatric Tumor Classification to highlight their clinicopathologic similarities with other pediatric myofibroblastic lesions (e.g., lipofibromatosis, infantile fibrosarcoma, and inflammatory myofibroblastic tumor; ref. 65).
Only the tumor types most frequently occurring in children and adolescents, that is, synovial sarcoma or malignant peripheral nerve sheath tumors, are described in detail. For tumor types only rarely occurring in children, a table with a comprehensive review of reported pediatric cases is provided in the introduction. Adult-type sarcomas in children may differ from their adult counterparts in clinical features, morphology, and/or genetic profile. Examples include (i) myxoid pleomorphic liposarcoma, a liposarcoma type characteristic of the CAYA age group, that can be associated with Li-Fraumeni syndrome, and (ii) synovial sarcoma in children showing minor chromosomal instability (apart from the paradigmatic SYT::SSX1/SSX2 fusions) compared with its adult counterparts (79). In general, despite the overall aggressive clinical behavior and low responsiveness to chemotherapy of most “adult-type sarcomas,” those occurring in children are still associated with a better prognosis (68, 80, 81).
Bone sarcomas
Bone sarcomas represent 4% to 8% of pediatric malignancies, with Ewing sarcomas accounting for about 40% and osteosarcomas for 50% (69). The new section “undifferentiated small round cell sarcomas” introduced in the 2020 WHO Classification of Soft Tissue and Bone Tumors includes four tumor categories (Fig. 1B–G): Ewing sarcoma, round cell sarcomas with EWSR1–non-ETS fusions, CIC-rearranged sarcoma, and sarcomas with BCOR genetic alterations. Sarcomas with BCOR alterations are rare, but increasingly recognized by the use of IHC (BCOR and CCNB3) and molecular tests. While BCOR-internal tandem duplication is typical of infantile undifferentiated sarcomas and primitive myxoid mesenchymal tumor of infancy, BCOR fusions mostly drive the undifferentiated small round cell sarcomas occurring in adolescents and young adults. By contrast, CIC-rearranged sarcomas and EWSR1–non ETS fusion sarcomas are characteristic of adult age.
The differential diagnosis of undifferentiated small round cell sarcomas requires an integrated approach with different techniques, from the faster and less expensive tests, for example, FISH or RT-PCR, when a preliminary diagnosis is suspected based on morphology, to the more sophisticated and traditionally faster and sometimes even less expensive NGS panels or RNA-seq. Methylation profiling, which is widely used in the diagnostic workup of central nervous system (CNS) tumors, also seems to be a promising diagnostic tool for the classification of soft-tissue sarcomas, especially for the group of undifferentiated small round cell sarcomas (Fig. 1A; refs. 11, 82, 83).
Other Solid Tumors
As previously introduced, the WHO Classification of Pediatric Tumors addresses multiple solid tumors by taking a developmental approach as much as possible, because tumors in children differ from those in adults at several levels: Children are developing organisms, undergoing multiple and marked changes at a speed often inversely proportional to the age of the patient. Congenital and neonatal tumors occur in immature tissues, where the histologic similarities between fetal structures and their neoplastic counterparts may not be immediately obvious (Fig. 2A–F). For example, in peripheral neuroblastic tumors, the most common solid tumor in children, their histologic appearance is almost indistinguishable from the fetal adrenal medulla (84), which is formed by migrating neural crest cell precursors that penetrate (one could say “invade”) the fetal adrenal mesoderm–derived cortex. Indeed, congenital adrenal neuroblastoma “in situ” is found in between 0.3% and 1% of neonatal autopsies (85). Details on the classification and molecular makeup of neuroblastoma, including its various clinically relevant molecular subtypes and associated genetic alterations, are provided in Supplementary Table S3.
Figure 2.
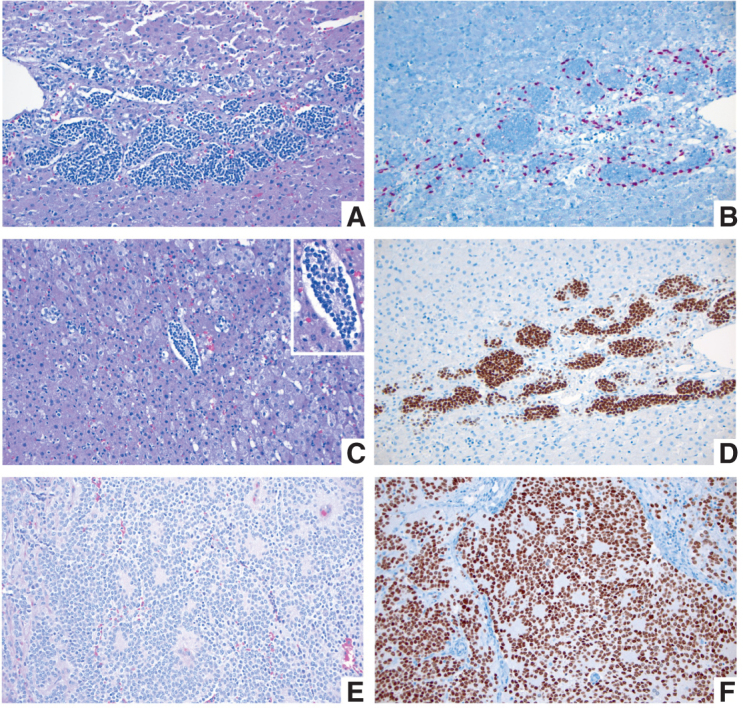
A–D, Fetal adrenal gland at 21–22 weeks of gestation. A, Migrating neural crest cells penetrate through the mesodermally derived fetal adrenal cortex homing into the future adrenal medulla (H&E; original magnification 200×). B, SOX10 IHC stain highlights the nuclei of migrating neural crest cells at the periphery of the migratory clusters, representing future Schwann cell precursors (SOX10 IHC; original magnification 200×). C, Migrating neural crest cells forming a Homer Wright rosette, indistinguishable from a similar structure in a poorly differentiated neuroblastoma (see E and F). The Homer Wright rossete is shown in the center, surrounded by fetal adrenal cortex (H&E; original magnification 200×). Inset shows the nonneoplastic Homer Wright rossete at a higher magnification (400×). Note the fine cytoplasmic prolongations of the future adrenal medullary cells in the center of the rosette. D, PHOX2B IHC stain showing strong nuclear reactivity in the migrating neural crest cells of the future fetal adrenal medulla (PHOX2B IHC; original magnification 200×). E and F, Poorly differentiated neuroblastoma from a 1-year-old patient. E, Several Homer Wright rosettes are seen with their characteristic central area of neuropil (H&E; original magnification 200×). F, PHOX2B IHC stain highlighting the nuclei of the neoplastic neural crest cells (neuroblasts) in multiple Homer Wright rosettes (PHOX2B IHC; original magnification 200×).
A similar situation occurs with other “blastomas,” which variably recapitulate the morphologic maturation of cellular lineages from the organs from which they originate. One of the best examples is nephroblastoma, also known as Wilms tumor, which occurs with a frequency close to that of neuroblastic tumors. This neoplasm reproduces the morphologic steps of renal development to such a high degree that it is challenging to differentiate a nephrogenic rest from small Wilms tumors (86, 87). Approximately 10% of Wilms tumors present histologic changes defined as “anaplasia,” if the following specific criteria are met: nucleomegaly (at least three times the size of nonanaplastic nuclei), nuclear hyperchromatism, and abnormal/atypical mitoses.
Among some of the innovative approaches in this volume, the developmental angle used to present the group of germ cell tumors (GCT) should be highlighted. This group of heterogeneous neoplasms includes entities that may present in multiple body locations, affect both sexes, and are particularly frequent in pediatric patients. From the advantageous position of their shared developmental origins, this chapter (88) moves along a continuum starting with early embryonic cells to gradually maturing germ cells. The extensive migratory pathways followed by primordial germ cells (PGC) during embryonic phases, mostly along the midline of the body, explain their occurrence in seemingly disconnected places such as the brain, mediastinum, gonads, or sacrococcygeal areas (88). Therefore, the grouping of GCTs adopts a rendition that considers their origins and progressive maturation processes responsible for their wide phenotypical varieties (Table 3). The molecular genetic and epigenetic characteristics responsible for the progressive maturation process are taken into consideration for their corresponding classification. The critical mechanism appears to be the reprogramming of nonneoplastic germ cells allowing migrating PGCs to escape apoptosis, and in later developmental stages, their survival within gonadal and extragonadal niches (i.e., mediastinum and brain), from where they can progress to form GCTs in situ and gonadoblastoma-type lesions, early common origins of the germinoma family. Reprogramming of lesions in the germinoma-family line of differentiation may also result in nonseminomatous tumors. Recognizing gonadoblastoma at its incipient stages (89) and discriminating it from its mimics (90) is challenging but important for adequate classification, treatment, and prognostication.
Table 3.
Classification of pediatric solid tumors

Pediatric tumors of the digestive system are another area in which significant progress has been made (91). In this chapter, several blastomas are presented, including: hepatoblastoma, pancreatoblastoma, and gastroblastoma, which are unusual neoplasms that require a high level of experience for their appropriate classification. Molecular pathology information in this chapter has grown exponentially in the last few years, allowing us to better understand the pathogenesis of these rare tumors. Regarding the pathology of hepatoblastoma (92), taxonomic efforts are based on the International Pediatric Tumor Consensus Classification (93), supported by novel molecular pathology information regarding specific genetic events relevant for this tumor, especially the WNT/β-catenin pathway, which is the most important aberrantly activated signaling pathway in hepatoblastoma (94), although other genetic abnormalities, such as those involving NFE2L2, TERT promoter, Notch, Sonic Hedgehog, PI3K/AKT, EGFR, and the Hippo/YAP pathway are also becoming known players in the pathogenesis of hepatoblastoma (93–99).
Pancreatoblastoma, which is extremely rare, occurs predominantly in the first decade of life (100, 101) and is also related to genetic aberrations in the WNT/β-catenin pathway (102–104). Other abnormalities include dysregulation of IGF2 (105, 106). Pancreatoblastoma may be associated with Beckwith–Wiedemann syndrome and familial adenomatous polyposis (107, 108). Gastroblastoma is a recently described tumor (109, 110), arising in the stomach of children and young adults. The tumor shows a recurrent somatic MALAT1::GLI1 fusion gene (111).
Another chapter included in the pediatric tumor classification covers pediatric skin tumors. To understand pediatric melanocytic lesions, such as giant congenital melanocytic naevi (GCMN) and associated disorders, a developmental approach is again necessary. These are neural crest cell–derived lesions (112) in which mutations lead to clonal expansion resulting in congenital melanocytic nevi (CMN). Although most CMNs harbor NRAS mutations (113), up to 8% of them carry BRAF mutations (114). Involvement of the CNS (115), association with neurocutaneous melanocytosis, and malignant transformation to melanoma arising in the context of a GCMN, although infrequent, represent ominous situations. A particularly aggressive form of congenital melanoma associated with amplification of mutated NRAS has been reported (116). Other pediatric skin tumors include hamartomas, epidermal nevi, and additional mosaicism-related abnormalities, which are frequently difficult to classify morphologically and are thus presented together with relevant molecular genetic features (Table 3).
CNS Tumors
The recently published fifth edition of the WHO classification for CNS tumors (summarized in ref. 30) featured a few fundamental paradigm shifts that particularly affected pediatric CNS tumor classification and thus formed the basis for the CNS tumor chapter within the inaugural WHO Classification for Pediatric Tumors. These fundamental changes, among others, included (i) the general concept of integrating histologic patterns with state-of-the art molecular diagnostic readouts to form an integrated diagnosis, (ii) the introduction of designations such as “pediatric-type” and “adult-type” tumor categories for both low- and high-grade gliomas to account for the age-specific biology despite the same histology-related names as well as associated cancer-predisposition syndromes (below), (iii) the inclusion of a multitude of novel tumor entities, many of which are primarily molecularly defined (similar to leukemias and lymphomas and some of the molecularly defined sarcoma types), (iv) the adaptation of tumor grading as a measure for differential aggressiveness of tumors within a tumor type rather than between tumor types, including the suggestion to not report a grade in cases where this could be clinically confusing because the grade would not reflect the expected outcome on current treatment regimens (e.g., WNT-driven medulloblastoma CNS-WHO grade 4), and (v) the widespread introduction of novel molecular diagnostic tools such as DNA methylation analysis for tumor classification, often nominated as an essential diagnostic criterion, particularly for difficult-to-diagnose cases (ref. 30; Fig. 3).
Figure 3.

Molecular groups of pediatric CNS tumors (at the level of superfamilies). Unsupervised, nonlinear t-distributed stochastic neighbor embedding (t-SNE) projection of methylation array profiles from 4,427 tumors. Samples were selected from a large database of >90,000 CNS tumor datasets to serve as reference profiles for training a supervised classification model based on strict criteria: all these samples showed a high calibrated classification score (>0.9) when applying the brain tumor classifier available at https://www.molecularneuropathology.org.
Tumor entities were selected for more detailed discussion in the WHO Classification of Pediatric Tumors if they either mainly occur in children and adolescents, or if a substantial proportion of an “adult-type” CNS tumor class is diagnosed in the pediatric age range (summarized in Table 4). All remaining entities are extensively discussed in the WHO CNS Tumor Classification.
Table 4.
Classification of pediatric CNS tumors

High-grade gliomas
Pediatric-type diffuse high-grade gliomas are now clearly separated from adult-type diffuse high-grade gliomas (the latter typically being IDH–wild-type glioblastomas with EGFR amplification, TERT promoter mutation, and/or combination of gain of chromosome 7 and loss of chromosome 10 or, rarely, high-grade, IDH-mutant astrocytomas or oligodendrogliomas). In the pediatric setting, four different types are distinguished (Table 4). The designation diffuse midline glioma, H3K27-altered was widened to include subtypes with a different mechanism for the loss of H3K27 trimethylation than H3K27 mutations, for example EZHIP overexpression. Diffuse midline glioma, EGFR mutant was newly introduced (Supplementary Table S4).
Infant-type hemispheric glioma was introduced as a new type, which typically occurs in young children and is associated with receptor tyrosine kinase fusions in the NTRK family, ROS1, ALK, or MET (117, 118). Diffuse pediatric-type high-grade glioma, H3-wild-type and IDH-wild-type represents a mixture of quite different molecular subtypes and certainly needs more granularity, including for instance biologically distinct subtypes that can readily be distinguished by DNA methylation analysis (ref. 119; e.g., the methylation classes pedHGG MYCN, pedHGG RTK1, pedHGG RTK2, and HGG_chr6CTX; Fig. 3). They also include tumors with underlying mismatch repair deficiency (120, 121).
Low-grade gliomas
Similar to high-grade pediatric-type gliomas, the designation pediatric-type diffuse low-grade gliomas was introduced to distinguish these latter (mostly MAPK-driven) tumors from their adult-type (typically IDH-driven) counterparts. In contrast to pediatric diffuse low-grade gliomas, in adults these tumors generally progress into high-grade gliomas over the disease course. Several new entities, primarily molecularly defined, were introduced in this group, including diffuse astrocytoma, MYB- or MYBL1-altered (122, 123), polymorphous low-grade neuroepithelial tumor of the young (124), and diffuse low-grade glioma, MAPK pathway–altered (almost as a diagnosis of exclusion), an exemplary family for which a mix-and-match approach can be applied by combining a morphologic diagnosis with a specific genetic alteration, for example diffuse astrocytoma with FGFR1 mutation (Table 4).
Within the category of circumscribed astrocytic gliomas, high-grade astrocytoma with piloid features was newly introduced (125) and astroblastoma, MN1-altered more precisely defined (126, 127). Among glioneuronal and neuronal tumors, diffuse leptomeningeal glioneuronal tumor (128) and diffuse glioneuronal tumor with oligodendroglioma-like features and nuclear clusters (24, 25), as a provisional tumor type, were newly added.
Medulloblastomas
For medulloblastomas, the first layer of classification remained consistent with the fourth edition update of the WHO Classification of CNS Tumors in 2016. However, several aspects have changed: (i) histologic subtypes were condensed into one type (medulloblastoma, histologically defined), underscoring that an integrated molecular classification is preferred over a purely histologic classification, (ii) grading was discouraged for clinical low-risk types such as WNT-driven medulloblastoma to prevent confusion with treating physicians and patients as explained above, and (iii) molecular subtypes were introduced for SHH medulloblastomas (n = 4) and for non-WNT/non-SHH medulloblastomas (n = 8) according to recent publications (129–132). The latter will be of enormous help to prospectively evaluate the predictive and prognostic role of these subtypes in the context of state-of-the-art therapies, for example, allowing for therapy deescalation in the framework of clinical trials for low-risk subtypes other than WNT. Special emphasis was put on the routine assessment of the presence of a cancer predisposition syndrome for all patients with SHH medulloblastoma and CTNNB1–wild-type WNT medulloblastoma (133).
Ependymomas
The classification of ependymomas has changed from a mostly morphologic into a primarily molecular classification (Table 4; ref. 12). In the supratentorial compartment, RELA-driven ependymoma was changed into ZFTA-driven ependymoma because it appeared that this latter fusion partner is the most consistent one found in this entity (134–136). YAP1 fusion–driven ependymoma was introduced as a new type (12). In the infratentorial region, molecularly defined posterior fossa group A and B (PFA and PFB) ependymomas were introduced, the first category based on a loss of H3K27 trimethylation in the tumor and/or a methylation profile indicative of PFA ependymoma (137). In the spinal region, the recently described type of MYCN-amplified ependymoma was introduced, a diagnosis associated with particularly unfavorable outcome (138, 139). The difficulty of standardized grading of ependymoma (especially between grade 2 and grade 3) was flagged with a caveat, and assigning a CNS WHO grade is no longer required as part of the diagnosis of ependymomas in children (140).
Other CNS tumors, provisional entities, and emerging entities
Within the category “other embryonal tumors” (Table 4), molecularly defined CNS neuroblastoma, FOXR2-activated, and CNS tumor with BCOR internal tandem duplication were newly introduced. Cribriform neuroepithelial tumor (CRINET), typically associated with SMARCB1 mutations yet biologically distinct from atypical teratoid/rhabdoid tumor (AT/RT), was introduced as a provisional entity (141). The emerging entity PATZ1 fusion–positive tumor, which was not included in the CNS tumor classification yet, was, however, mentioned in the introduction to the CNS tumor chapter of the classification already (142–145). The largest series of these tumors published to date was published only after the editorial meeting of the WHO (26). In addition, embryonal tumor with multilayered rosettes (ETMR) was divided into two subtypes: (i) ETMR with C19MC amplification and (ii) ETMR with DICER1 mutations (often germline; ref. 146). For both pineoblastoma and AT/RT, the molecular consensus subtypes were introduced as recently published (147, 148).
In summary, the CNS tumor chapter of the WHO classification for Pediatric Tumors was mostly adopted from the new CNS tumor classification, which was written at the same time. This classification will certainly be of enormous value not only for diagnosticians, but also for treating physicians, researchers, and patients across the world.
Cancer Predisposition
Genetic predisposition is the major known cause of childhood cancer. Research in this area, including integrated germline and cancer genomic profiling, is highly relevant, as it provides important biological insights into the causes of childhood cancer and represents a unique opportunity to translate this knowledge into improving individualized childhood cancer prevention, surveillance, and treatment in the future.
Definition of the term cancer predisposition syndrome
Cancer predisposition syndromes (CPS) are distinct genetic or epigenetic conditions associated with an increased cancer risk compared with the general population. Causes vary and may include, but are not limited to, constitutional chromosomal anomalies, pathogenic—mainly inactivating but also activating—variants in single cancer predisposition genes, copy number changes, and epigenetic mechanisms (149–152). Several CPSs are characterized by germline mosaicism (151). Patients with CPS need to be distinguished from individuals harboring cancer risk alleles that are not associated with a defined syndrome identified through genome-wide association studies (153, 154). Such low-penetrant cancer risk alleles are likely to contribute to all childhood cancers. It is estimated that at least 10% of children with cancer have an underlying CPS, with the proportion of children with a CPS being substantially higher for selected cancer types (150). Estimates in some more recent studies (15, 16, 155) are even higher; however, this higher incidence might be based on certain selection biases (e.g., enrichment for relapse patients) and the stringency of filtering in terms of causality of the underlying germline mutation. New syndromes continue to be identified (156, 157). The percentage of underlying germline genetic variants in cancer predisposition genes varies between populations and may be characterized by founder mutations, among other factors (158).
Classification of CPSs
For the purpose of this review, CPSs are classified into the following eight different groups (Fig. 4): (i) Li-Fraumeni syndrome (LFS); (ii) constitutional mismatch repair deficiency (CMMRD); (iii) predisposition to neural tumors, including neuroblastoma, glioma, medulloblastoma, retinoblastoma, and rhabdoid tumors; (iv) Wilms tumor predisposition—these are often overgrowth syndromes (159); (v) endocrine tumor predisposition; (vi) predisposition to gastrointestinal tumors; (vii) predisposition to hematologic malignancies including leukemia, lymphoma, and myelodysplastic syndrome (e.g., Fanconi anemia, among others); and (viii) other CPS (e.g., DICER1 syndrome) not classified within one of the other groups. Many of the syndromes listed within one main category predispose to a broader cancer spectrum in and outside the specific CPS category, while others are associated with neoplasms in restricted organ systems. The tumor risks vary substantially between syndromes and genetic subtypes (e.g., Fanconi anemia; ref. 160). LFS and CMMRD are singled out because of the particularly high cancer risk and broad cancer spectrum (161, 162). LFS is also the most commonly diagnosed CPS among children with cancer (4, 150, 163). The list of selected CPSs as listed in the WHO Classification of Pediatric Tumors is provided in Supplementary Table S5.
Figure 4.

Overview on CPSs. For the purpose of this review, syndromes were grouped into eight categories: (1) Li-Fraumeni syndrome; (2) syndromes predisposing to Wilms tumor; (3) syndromes predisposing to endocrine tumors; (4) syndromes predisposing to hematopoietic malignancies; (5) constitutional mismatch repair deficiency; (6) other syndromes predisposing to gastrointestinal tumors; (7) syndromes predisposing to neural tumors; and (8) other cancer-prone syndromes. Cancer predisposition syndromes listed in the WHO Classification of Pediatric Tumors and displayed in Suppementary Table S5 are marked with an asterisk.
Adult-type CPSs
Diagnostic criteria and distinct associated phenotypic characteristics (164) have been established for the most common CPS; however, with the increasing use of high-throughput genetic and genomic profiling technologies in the clinical laboratory, the number of new abnormalities and phenotypic spectra are evolving including previously unrecognized associations, and patients not meeting diagnostic criteria are being identified (163). This is particularly true for adult-type CPS. The increasing use of agnostic germline sequencing has shown that children with cancer not uncommonly harbor pathogenic/likely pathogenic variants in genes mutated in adult-type CPS. Examples include heterozygous pathogenic variants in mismatch repair genes MSH2, MSH6, MLH1, and PMS2 that typically cause Lynch syndrome and heterozygous pathogenic variants in BRCA1/2 associated with hereditary breast and ovarian cancers. While recessive conditions associated with these genes cause CMMRD and Fanconi anemia, respectively, and are well established high-risk CPSs in children, it is currently unclear to what extent heterozygous variants in such genes that also occur at low frequencies in healthy individuals contribute to cancer risk in children and adolescents. Statistically significant associations have been shown for medulloblastoma (BRCA2, PALB2; ref. 133) and non-Hodgkin lymphoma (BRCA2; ref. 165). It has also been shown that childhood cancer survivors who carry variants in DNA repair genes such as BRCA2 have an increased risk of subsequent neoplasms (166). Studies analyzing both germline and tumor genomes (to search for loss of heterozygosity and other characteristic CPS-specific somatic signatures) are crucial to further clarify these associations as well as their predictivity for the potential therapeutic use of PARP inhibitors (16, 18).
Diagnosis of a CPS
Patients with CPS may have clinical features prompting physicians to suspect and evaluate the diagnosis. These clinical features include individual and family cancer history, tumor type, presentation (e.g., multifocal, bilateral) as well as somatic molecular characteristics, and physical features (167, 168). Clinical tools have been developed to identify these signs systematically (169, 170). However, a significant proportion of CPS is not captured by these tools (171). In addition, agnostic gene panel or exome-based germline analyses are increasingly being employed, leading to the identification of patients with a CPS who lack obvious clinical signs or symptoms, as well as to the discovery of previously unknown CPS associations (156, 157, 163, 166).The diagnosis of a CPS may be challenging due to the notion that variants identified in a CPS gene may be of uncertain significance, and variant interpretation challenges should be taken into consideration. Functional tests such as chromosomal breakage analysis in patients with Fanconi anemia (172) can help to establish the diagnosis. Clinical tumor sequencing of pediatric cancers is also becoming increasingly used, and as a result underlying cancer predisposition germline variants are often identified while sequencing the tumor (173, 174). Pathologists and geneticists have an important role in recognizing specific tumor types associated with cancer syndromes and should actively participate in the multidisciplinary teams evaluating these patients (175).
Clinical implications
While it is essential for children with specific cancer types to be diagnosed or to rule out a specific CPS in order to make appropriate clinical decisions, ethical aspects need to be considered and easy access to genetic counseling should be a requirement for specialized centers diagnosing and treating children with cancer. The diagnosis of a CPS may have broad clinical implications including identification of other affected family members through trio or cascade testing, cancer prevention, cancer surveillance, adjusted cancer therapy to account for resistance to conventional therapy and/or increased toxicity, and need for psychosocial support (150). For some patients carrying a pathogenic or likely pathogenic variant in a CPS gene, direct clinical implications for the affected child may be less obvious. For example, a heterozygous variant in BRCA1/2 may have no immediate clinical implications for the affected child with cancer, but it may be relevant for the patient later in life, and for affected family members identified through cascade testing. This is because the cancer risks associated with these variants increase in adults, warranting specific medical recommendations (e.g., breast cancer screening and prophylactic mastectomy).
Summary and outlook
It is increasingly recognized that childhood cancer has a strong genetic component. While germline genetic factors are likely to play a role in all children with cancer, distinct CPSs (Fig. 4) are currently identified in at least 10% of patients. Genomic testing, including family-based trio sequencing, may reveal a new landscape of childhood cancer predisposition. International collaborative studies are needed to improve treatment strategies, prevention, and surveillance programs for children with CPS.
The following areas, among others, will need to be further addressed in the future: (i) discovery of additional germline (epi)genetic mechanisms contributing to childhood cancer and corresponding somatic signatures; (ii) cancer epidemiologic studies to better define cancer risks and environmental as well as (epi)genetic risk modifiers; (iii) improved cancer surveillance through better imaging and biomarker monitoring; (iv) cancer prevention trials with a focus on high-risk CPS; (v) interventional treatment studies for patients with cancer with various CPSs; and (vi) role of digenic, multigenic mutations as well as the emerging utility of polygenic risk scores (171, 176, 177).
Conclusion
Pediatric tumors represent a particular challenge due to their rarity, heterogeneity, different pathogenetic mechanisms compared with adult tumors, strong impact of hereditary cancer predisposition, and need for therapeutic strategies that optimize for survival chances while minimizing risks for long-term sequelae.
Since the 1970s, international clinical trials have spearheaded a multidisciplinary approach that helped change the natural history of pediatric leukemias as well as solid and brain tumors, resulting in a dramatic increase in overall survival and a better quality of life for the majority of patients. Uniformity in diagnosis is critical to these efforts. Unfortunately, mortality rates remain high for advanced diseases and for specific entities, for which survival rates have plateaued for more than two decades. The integration of classic histologic diagnoses with advanced molecular techniques such as methylation profiling, RNA-seq, whole-genome sequencing, or whole-exome sequencing (including tumor and control tissue) represent a step change in the categorization of pediatric cancers and definition of prognostic and/or predictive subgroups or biomarkers to be included in the standard diagnostic process, paving the way toward more personalized therapeutic strategies.
The inaugural edition of the WHO Classification of Pediatric Tumors provides a basis for a multilayered diagnostic process that reflects two important aspects:
1. Meeting the needs of regions with varying level of access to state-of-the art molecular technologies.
2. Acknowledging the current transition from a traditional system of classification focused on “cell type” to an integrated approach, also comprising many newly recognized “molecular entities.”
In line with this, the section “essential and desirable diagnostic criteria” included in the fifth WHO edition represents the first basic morphologic diagnostic level, enriched by a modern, more focused histologic and IHC as well as broader molecular evaluation (including DNA methylation and NGS), often derived from the experience in the correlation between molecular patterns and histology (including the use of artificial intelligence–based approaches). In the future, this may be further complemented with novel technologies that add additional information to the tissue analysis, such as single-cell approaches and proteomics. Noninvasive, NGS-based liquid biopsies to detect circulating tumor DNA seem a promising tool to plan therapeutic strategies and monitor tumor evolution, although technical variability is currently a limiting factor to implementation in routine clinical practice. The integration with information on tumor microenvironment from circulating extracellular vesicles (exosomes) might, in the future, provide additional important diagnostic/prognostic data (178).
It is difficult to predict whether molecular platform analyses or even liquid biopsies will fully replace histologic diagnosis on tumor tissue biopsies in the future. However, it is increasingly clear that molecular techniques are providing a new, powerful lens to current histologic evaluation, while it will remain of key importance to actually investigate representative tumor material.
Authors' Disclosures
S.M. Pfister reports grants from ITCC-P4 IMI-2 project funded by the EU as well as 10 EFPIA companies (www.itccp4.eu) outside the submitted work; in addition, S.M. Pfister has a patent for EP 16710700 A 20160311 issued. S. Rossi reports grants from Ministry of Health, 5 × 1000 funds (202005_ONCO_ALAGGIO) during the conduct of the study. J.A. Jarzembowski reports grants and non-financial support from Children's Oncology Group outside the submitted work. D. Hill reports other support from ResourcePath outside the submitted work. T.S. Jacques reports grants from Cancer Research UK, The Brain Tumour Charity, GOSH Children's Charity, grants from Children with Cancer UK, Olivia Hodson Cancer Fund; personal fees from Bayer; other support from Repath Ltd, and other support from Neuropath Ltd outside the submitted work. A. von Deimling reports a patent for DNA methylation–based method for classifying tumor species of the brain issued. I.A. Cree reports other support from IARC during the conduct of the study. R. Alaggio reports receiving funding from the European Union's Horizon 2020 research and innovation programme under grant agreement no. 668596 (ChiLTERN). No disclosures were reported by the other authors.
Supplementary Material
Acknowledgments
This work was partially supported by the German Childhood Cancer Foundation (DKS 2015.01 and DKS 2017.01, to S.M. Pfister, A. von Deimling; DKS2019.13, to S.M. Pfister and C.P. Kratz), the BMBF ADDRess grant (01GM1909, to C.P. Kratz and S.M. Pfister), the Everest Centre for Low-grade Paediatric Brain Tumours (The Brain Tumour Charity, UK, GN-000382, to S.M. Pfister, T.S. Jacques), the Low Grade Astrocytoma Fund (PLGA Fund) at the Pediatric Brain Tumor Foundation (PBTF; to S.M. Pfister), the Deutsche Forschungsgemeinschaft (DFG; German Research Foundation) – Project-ID 404521405, SFB 1389 - UNITE Glioblastoma (to S.M. Pfister and A. von Deimling), the European Union's Horizon 2020 research and innovation programme under grant agreement No 668596 (ChiLTERN, to R. Alaggio) and Ministry of Health, 5 × 1000 funds RC2021-IRCCS Ospedale bambino Gesu (to R. Alaggio), the Marjorie K. Harmer Endowment for Research in Padiatric Pathology, University of Pittsburgh School of Medicine (to M. Reyes-Múgica), NIH grant NCI R01CA143167 (to D.A. Hill), funding from Children with Cancer UK, Great Ormond Street Hospital Children's Charity, Olivia Hodson Cancer Fund, Cancer Research UK, and the National Institute of Health Research (to T.S. Jacques). All research at Great Ormond Street Hospital NHS Foundation Trust and UCL Great Ormond Street Institute of Child Health is made possible by the NIHR Great Ormond Street Hospital Biomedical Research Centre. We thank Dr. Robert Bendon, Professor of Pathology at the Perinatal Pathology Center of Excellence, UPMC Magee-Womens Hospital, for his kindness in sharing a block from the fetal adrenal used in Fig. 2. We thank Dr. Martin Sill, Hopp Children's Cancer Center Heidelberg (KiTZ), for generating and providing Figs. 1A and 3. We further thank Dr. David Capper, Professor of Neuropathology at the Charité Hospital in Berlin, Germany, for his contributions to the section on pediatric diffuse gliomas.
The content of this article represents the personal views of the authors and does not represent the views of the authors' employers and associated institutions. Where authors are identified as personnel of the International Agency for Research on Cancer/World Health Organization, the authors alone are responsible for the views expressed in this article and they do not necessarily represent the decisions, policy, or views of the International Agency for Research on Cancer/World Health Organization.
Footnotes
Note: Supplementary data for this article are available at Cancer Discovery Online (http://cancerdiscovery.aacrjournals.org/).
References
- 1. Pui CH, Gajjar AJ, Kane JR, Qaddoumi IA, Pappo AS. Challenging issues in pediatric oncology. Nat Rev Clin Oncol 2011;8:540–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ward E, DeSantis C, Robbins A, Kohler B, Jemal A. Childhood and adolescent cancer statistics, 2014. CA Cancer J Clin 2014;64:83–103. [DOI] [PubMed] [Google Scholar]
- 3. Behjati S, Gilbertson RJ, Pfister SM. Maturation block in childhood cancer. Cancer Discov 2021;11:542–4. [DOI] [PubMed] [Google Scholar]
- 4. Grobner SN, Worst BC, Weischenfeldt J, Buchhalter I, Kleinheinz K, Rudneva VAet al. The landscape of genomic alterations across childhood cancers. Nature 2018;555:321–7. [DOI] [PubMed] [Google Scholar]
- 5. Ma X, Liu Y, Liu Y, Alexandrov LB, Edmonson MN, Gawad Cet al. Pan-cancer genome and transcriptome analyses of 1,699 paediatric leukaemias and solid tumours. Nature 2018;555:371–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Grabovska Y, Mackay A, O'Hare P, Crosier S, Finetti M, Schwalbe ECet al. Pediatric pan-central nervous system tumor analysis of immune-cell infiltration identifies correlates of antitumor immunity. Nat Commun 2020;11:4324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wienke J, Dierselhuis MP, Tytgat GAM, Künkele A, Nierkens S, Molenaar JJ. The immune landscape of neuroblastoma: challenges and opportunities for novel therapeutic strategies in pediatric oncology. Eur J Cancer 2021;144:123–50. [DOI] [PubMed] [Google Scholar]
- 8. Wu CC, Beird HC, Andrew Livingston J, Advani S, Mitra A, Cao Set al. Immuno-genomic landscape of osteosarcoma. Nat Commun 2020;11:1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cree IA, Tan PH, Travis WD, Wesseling P, Yagi Y, White VAet al. Counting mitoses: SI(ze) matters! Mod Pathol 2021;34:1651–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Capper D, Jones DTW, Sill M, Hovestadt V, Schrimpf D, Sturm Det al. DNA methylation-based classification of central nervous system tumours. Nature 2018;555:469–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Koelsche C, Schrimpf D, Stichel D, Sill M, Sahm F, Reuss DEet al. Sarcoma classification by DNA methylation profiling. Nat Commun 2021;12:498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pajtler KW, Witt H, Sill M, Jones DT, Hovestadt V, Kratochwil Fet al. Molecular classification of ependymal tumors across all CNS compartments, histopathological grades, and age groups. Cancer Cell 2015;27:728–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sahm F, Schrimpf D, Jones DT, Meyer J, Kratz A, Reuss Det al. Next-generation sequencing in routine brain tumor diagnostics enables an integrated diagnosis and identifies actionable targets. Acta Neuropathol 2016;131:903–10. [DOI] [PubMed] [Google Scholar]
- 14. Worst BC, van Tilburg CM, Balasubramanian GP, Fiesel P, Witt R, Freitag Aet al. Next-generation personalised medicine for high-risk paediatric cancer patients - The INFORM pilot study. Eur J Cancer 2016;65:91–101. [DOI] [PubMed] [Google Scholar]
- 15. Wong M, Mayoh C, Lau LMS, Khuong-Quang DA, Pinese M, Kumar Aet al. Whole genome, transcriptome and methylome profiling enhances actionable target discovery in high-risk pediatric cancer. Nat Med 2020;26:1742–53. [DOI] [PubMed] [Google Scholar]
- 16. Newman S, Nakitandwe J, Kesserwan CA, Azzato EM, Wheeler DA, Rusch Met al. Genomes for Kids: the scope of pathogenic mutations in pediatric cancer revealed by comprehensive DNA and RNA sequencing. Cancer Discov 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pikman Y, Tasian SK, Sulis ML, Stevenson K, Blonquist TM, Apsel Winger Bet al. Matched targeted therapy for pediatric patients with relapsed, refractory, or high-risk leukemias: a report from the LEAP Consortium. Cancer Discov 2021;11:1424–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. van Tilburg CM, Pfaff E, Pajtler KW, Langenberg KPS, Fiesel P, Jones BCet al. The pediatric precision oncology INFORM registry: clinical outcome and benefit for patients with very high-evidence targets. Cancer Discov 2021;11:2764–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Stichel D, Schrimpf D, Casalini B, Meyer J, Wefers AK, Sievers Pet al. Routine RNA sequencing of formalin-fixed paraffin-embedded specimens in neuropathology diagnostics identifies diagnostically and therapeutically relevant gene fusions. Acta Neuropathol 2019;138:827–35. [DOI] [PubMed] [Google Scholar]
- 20. Petralia F, Tignor N, Reva B, Koptyra M, Chowdhury S, Rykunov Det al. Integrated proteogenomic characterization across major histological types of pediatric brain cancer. Cell 2020;183:1962–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Poulos RC, Hains PG, Shah R, Lucas N, Xavier D, Manda SSet al. Strategies to enable large-scale proteomics for reproducible research. Nat Commun 2020;11:3793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Koelsche C, Mynarek M, Schrimpf D, Bertero L, Serrano J, Sahm Fet al. Primary intracranial spindle cell sarcoma with rhabdomyosarcoma-like features share a highly distinct methylation profile and DICER1 mutations. Acta Neuropathol 2018;136:327–37. [DOI] [PubMed] [Google Scholar]
- 23. Lee JC, Villanueva-Meyer JE, Ferris SP, Sloan EA, Hofmann JW, Hattab EMet al. Primary intracranial sarcomas with DICER1 mutation often contain prominent eosinophilic cytoplasmic globules and can occur in the setting of neurofibromatosis type 1. Acta Neuropathol 2019;137:521–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Deng MY, Sill M, Sturm D, Stichel D, Witt H, Ecker Jet al. Diffuse glioneuronal tumour with oligodendroglioma-like features and nuclear clusters (DGONC) - a molecularly defined glioneuronal CNS tumour class displaying recurrent monosomy 14. Neuropathol Appl Neurobiol 2020;46:422–30. [DOI] [PubMed] [Google Scholar]
- 25. Pickles JC, Mankad K, Aizpurua M, Paine SM, Bridges LR, Carceller Fet al. A case series of diffuse glioneuronal tumours with oligodendroglioma-like features and nuclear clusters (DGONC). Neuropathol Appl Neurobiol 2021;47:464–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Alhalabi KT, Stichel D, Sievers P, Peterziel H, Sommerkamp AC, Sturm Det al. PATZ1 fusions define a novel molecularly distinct neuroepithelial tumor entity with a broad histological spectrum. Acta Neuropathol 2021;142:841–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bera K, Schalper KA, Rimm DL, Velcheti V, Madabhushi A. Artificial intelligence in digital pathology - new tools for diagnosis and precision oncology. Nat Rev Clin Oncol 2019;16:703–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Fu Y, Jung AW, Torne RV, Gonzalez S, Vöhringer H, Shmatko Aet al. Pan-cancer computational histopathology reveals mutations, tumor composition and prognosis. Nat Cancer 2020;1:800–10. [DOI] [PubMed] [Google Scholar]
- 29. Louis DN, Perry A, Burger P, Ellison DW, Reifenberger G, von Deimling Aet al. International Society Of Neuropathology–Haarlem consensus guidelines for nervous system tumor classification and grading. Brain Pathol 2014;24:429–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, Figarella-Branger Det al. The 2021 WHO classification of tumors of the central nervous system: a summary. Neuro Oncol 2021;23:1231–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Louis DN, Wesseling P, Paulus W, Giannini C, Batchelor TT, Cairncross JGet al. cIMPACT-NOW update 1: not otherwise specified (NOS) and not elsewhere classified (NEC). Acta Neuropathol 2018;135:481–4. [DOI] [PubMed] [Google Scholar]
- 32. Metzger ML, Mauz-Körholz C. Epidemiology, outcome, targeted agents and immunotherapy in adolescent and young adult non-Hodgkin and Hodgkin lymphoma. Br J Haematol 2019;185:1142–57. [DOI] [PubMed] [Google Scholar]
- 33. Minard-Colin V, Aupérin A, Pillon M, Burke GAA, Barkauskas DA, Wheatley Ket al. Rituximab for high-risk, mature B-cell non-Hodgkin's lymphoma in children. N Engl J Med 2020;382:2207–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Malard F, Mohty M. Acute lymphoblastic leukaemia. Lancet 2020;395:1146–62. [DOI] [PubMed] [Google Scholar]
- 35. Inaba H, Mullighan CG. Pediatric acute lymphoblastic leukemia. Haematologica 2020;105:2524–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lule ZC, Shiferaw EW, Kim J. Thermomechanical properties of SiC-filled polybutylene succinate composite fabricated via melt extrusion. Polymers 2020;12:418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Harris NL, Jaffe ES, Diebold J, Flandrin G, Muller-Hermelink HK, Vardiman Jet al. World Health Organization classification of neoplastic diseases of the hematopoietic and lymphoid tissues: report of the Clinical Advisory Committee meeting-Airlie House, Virginia, November 1997. J Clin Oncol 1999;17:3835–49. [DOI] [PubMed] [Google Scholar]
- 38. Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein Het al. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon, France: IARC; 2017. [Google Scholar]
- 39. Hasle H, Niemeyer CM, Chessells JM, Baumann I, Bennett JM, Kerndrup Get al. A pediatric approach to the WHO classification of myelodysplastic and myeloproliferative diseases. Leukemia 2003;17:277–82. [DOI] [PubMed] [Google Scholar]
- 40. Allen CE, Kelly KM, Bollard CM. Pediatric lymphomas and histiocytic disorders of childhood. Pediatr Clin North Am 2015;62:139–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cairo MS, Beishuizen A. Childhood, adolescent and young adult non-Hodgkin lymphoma: current perspectives. Br J Haematol 2019;185:1021–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Inaba H, Greaves M, Mullighan CG. Acute lymphoblastic leukaemia. Lancet 2013;381:1943–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Shiraz P, Payne KJ, Muffly L. The current genomic and molecular landscape of philadelphia-like acute lymphoblastic leukemia. Int J Mol Sci 2020;21:2193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chen J, Glasser CL. New and emerging targeted therapies for pediatric acute myeloid leukemia (AML). Children 2020;7:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hou HA, Tien HF. Genomic landscape in acute myeloid leukemia and its implications in risk classification and targeted therapies. J Biomed Sci 2020;27:81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bolouri H, Farrar JE, Triche T Jr, Ries RE, Lim EL, Alonzo TAet al. The molecular landscape of pediatric acute myeloid leukemia reveals recurrent structural alterations and age-specific mutational interactions. Nat Med 2018;24:103–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Niemeyer CM, Flotho C. Juvenile myelomonocytic leukemia: who's the driver at the wheel? Blood 2019;133:1060–70. [DOI] [PubMed] [Google Scholar]
- 48. Pastor V, Hirabayashi S, Karow A, Wehrle J, Kozyra EJ, Nienhold Ret al. Mutational landscape in children with myelodysplastic syndromes is distinct from adults: specific somatic drivers and novel germline variants. Leukemia 2017;31:759–62. [DOI] [PubMed] [Google Scholar]
- 49. Hirabayashi S, Flotho C, Moetter J, Heuser M, Hasle H, Gruhn Bet al. Spliceosomal gene aberrations are rare, coexist with oncogenic mutations, and are unlikely to exert a driver effect in childhood MDS and JMML. Blood 2012;119:e96–9. [DOI] [PubMed] [Google Scholar]
- 50. Schwartz JR, Ma J, Lamprecht T, Walsh M, Wang S, Bryant Vet al. The genomic landscape of pediatric myelodysplastic syndromes. Nat Commun 2017;8:1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Göckeritz W, Borchert HH. [The effect of chlorpromazine and carbamazepine on diagnostically relevant liver enzymes]. Pharmazie 1990;45:579–81. [PubMed] [Google Scholar]
- 52. Kardos G, Baumann I, Passmore SJ, Locatelli F, Hasle H, Schultz KRet al. Refractory anemia in childhood: a retrospective analysis of 67 patients with particular reference to monosomy 7. Blood 2003;102:1997–2003. [DOI] [PubMed] [Google Scholar]
- 53. Moriwaki K, Manabe A, Taketani T, Kikuchi A, Nakahata T, Hayashi Y. Cytogenetics and clinical features of pediatric myelodysplastic syndrome in Japan. Int J Hematol 2014;100:478–84. [DOI] [PubMed] [Google Scholar]
- 54. Niemeyer CM, Baumann I. Classification of childhood aplastic anemia and myelodysplastic syndrome. Hematology Am Soc Hematol Educ Program 2011;2011:84–9. [DOI] [PubMed] [Google Scholar]
- 55. Wlodarski MW, Sahoo SS, Niemeyer CM. Monosomy 7 in pediatric myelodysplastic syndromes. Hematol Oncol Clin North Am 2018;32:729–43. [DOI] [PubMed] [Google Scholar]
- 56. Klusmann JH, Creutzig U, Zimmermann M, Dworzak M, Jorch N, Langebrake Cet al. Treatment and prognostic impact of transient leukemia in neonates with Down syndrome. Blood 2008;111:2991–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Wechsler J, Greene M, McDevitt MA, Anastasi J, Karp JE, Le Beau MMet al. Acquired mutations in GATA1 in the megakaryoblastic leukemia of Down syndrome. Nat Genet 2002;32:148–52. [DOI] [PubMed] [Google Scholar]
- 58. Hollanda LM, Lima CS, Cunha AF, Albuquerque DM, Vassallo J, Ozelo MCet al. An inherited mutation leading to production of only the short isoform of GATA-1 is associated with impaired erythropoiesis. Nat Genet 2006;38:807–12. [DOI] [PubMed] [Google Scholar]
- 59. Labuhn M, Perkins K, Matzk S, Varghese L, Garnett C, Papaemmanuil Eet al. Mechanisms of progression of myeloid preleukemia to transformed myeloid leukemia in children with Down syndrome. Cancer Cell 2019;36:123–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Yoshida K, Toki T, Okuno Y, Kanezaki R, Shiraishi Y, Sato-Otsubo Aet al. The landscape of somatic mutations in Down syndrome-related myeloid disorders. Nat Genet 2013;45:1293–9. [DOI] [PubMed] [Google Scholar]
- 61. Burkhardt B, Hermiston ML. Lymphoblastic lymphoma in children and adolescents: review of current challenges and future opportunities. Br J Haematol 2019;185:1158–70. [DOI] [PubMed] [Google Scholar]
- 62. Sandlund JT, Martin MG. Non-Hodgkin lymphoma across the pediatric and adolescent and young adult age spectrum. Hematology Am Soc Hematol Educ Program 2016;2016:589–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Song KJ, Park JH, Im HJ, Ahn SD. Survival and long-term toxicities of pediatric Hodgkin lymphoma after combined modality treatment: a single institute experience. Radiat Oncol J 2020;38:198–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kahn JM, Kelly KM, Pei Q, Bush R, Friedman DL, Keller FGet al. Survival by race and ethnicity in pediatric and adolescent patients with Hodgkin lymphoma: a children's oncology group study. J Clin Oncol 2019;37:3009–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. WHO Classification of Tumours, Soft Tissue and Bone Tumours. Lyon, France: IARC; 2020. [Google Scholar]
- 66. Myhre-Jensen O. A consecutive 7-year series of 1331 benign soft tissue tumours. Clinicopathologic data. Comparison with sarcomas. Acta Orthop Scand 1981;52:287–93. [DOI] [PubMed] [Google Scholar]
- 67. Coffin CM, Alaggio R, Dehner LP. Some general considerations about the clinicopathologic aspects of soft tissue tumors in children and adolescents. Pediatr Dev Pathol 2012;15:11–25. [DOI] [PubMed] [Google Scholar]
- 68. van der Graaf WTA, Orbach D, Judson IR, Ferrari A. Soft tissue sarcomas in adolescents and young adults: a comparison with their paediatric and adult counterparts. Lancet Oncol 2017;18:e166–e75. [DOI] [PubMed] [Google Scholar]
- 69. Kaatsch P. Epidemiology of childhood cancer. Cancer Treat Rev 2010;36:277–85. [DOI] [PubMed] [Google Scholar]
- 70. Ferrari A, Casanova M. New concepts for the treatment of pediatric nonrhabdomyosarcoma soft tissue sarcomas. Expert Rev Anticancer Ther 2005;5:307–18. [DOI] [PubMed] [Google Scholar]
- 71. Arndt CA, Stoner JA, Hawkins DS, Rodeberg DA, Hayes-Jordan AA, Paidas CNet al. Vincristine, actinomycin, and cyclophosphamide compared with vincristine, actinomycin, and cyclophosphamide alternating with vincristine, topotecan, and cyclophosphamide for intermediate-risk rhabdomyosarcoma: children's oncology group study D9803. J Clin Oncol 2009;27:5182–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Oberlin O, Rey A, Sanchez de Toledo J, Martelli H, Jenney ME, Scopinaro Met al. Randomized comparison of intensified six-drug versus standard three-drug chemotherapy for high-risk nonmetastatic rhabdomyosarcoma and other chemotherapy-sensitive childhood soft tissue sarcomas: long-term results from the International Society of Pediatric Oncology MMT95 study. J Clin Oncol 2012;30:2457–65. [DOI] [PubMed] [Google Scholar]
- 73. Pratt CB, Pappo AS, Gieser P, Jenkins JJ, Salzbergdagger A, Neff Jet al. Role of adjuvant chemotherapy in the treatment of surgically resected pediatric nonrhabdomyosarcomatous soft tissue sarcomas: A Pediatric Oncology Group Study. J Clin Oncol 1999;17:1219. [DOI] [PubMed] [Google Scholar]
- 74. Spunt SL, Poquette CA, Hurt YS, Cain AM, Rao BN, Merchant TEet al. Prognostic factors for children and adolescents with surgically resected nonrhabdomyosarcoma soft tissue sarcoma: an analysis of 121 patients treated at St Jude Children's Research Hospital. J Clin Oncol 1999;17:3697–705. [DOI] [PubMed] [Google Scholar]
- 75. Ferrari A, Casanova M, Collini P, Meazza C, Luksch R, Massimino Met al. Adult-type soft tissue sarcomas in pediatric-age patients: experience at the Istituto Nazionale Tumori in Milan. J Clin Oncol 2005;23:4021–30. [DOI] [PubMed] [Google Scholar]
- 76. Pappo AS, Devidas M, Jenkins J, Rao B, Marcus R, Thomas Pet al. Phase II trial of neoadjuvant vincristine, ifosfamide, and doxorubicin with granulocyte colony-stimulating factor support in children and adolescents with advanced-stage nonrhabdomyosarcomatous soft tissue sarcomas: a Pediatric Oncology Group Study. J Clin Oncol 2005;23:4031–8. [DOI] [PubMed] [Google Scholar]
- 77. Ferrari A, Miceli R, Rey A, Oberlin O, Orbach D, Brennan Bet al. Non-metastatic unresected paediatric non-rhabdomyosarcoma soft tissue sarcomas: results of a pooled analysis from United States and European groups. Eur J Cancer 2011;47:724–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Orbach D, Brennan B, De Paoli A, Gallego S, Mudry P, Francotte Net al. Conservative strategy in infantile fibrosarcoma is possible: The European paediatric Soft tissue sarcoma Study Group experience. Eur J Cancer 2016;57:1–9. [DOI] [PubMed] [Google Scholar]
- 79. Lagarde P, Przybyl J, Brulard C, Pérot G, Pierron G, Delattre Oet al. Chromosome instability accounts for reverse metastatic outcomes of pediatric and adult synovial sarcomas. J Clin Oncol 2013;31:608–15. [DOI] [PubMed] [Google Scholar]
- 80. Ferrari A, Chi YY, De Salvo GL, Orbach D, Brennan B, Randall RLet al. Surgery alone is sufficient therapy for children and adolescents with low-risk synovial sarcoma: A joint analysis from the European paediatric soft tissue sarcoma Study Group and the Children's Oncology Group. Eur J Cancer 2017;78:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Brennan B, Zanetti I, Orbach D, Gallego S, Francotte N, Van Noesel Met al. Alveolar soft part sarcoma in children and adolescents: The European Paediatric Soft Tissue Sarcoma study group prospective trial (EpSSG NRSTS 2005). Pediatr Blood Cancer 2018;65. [DOI] [PubMed] [Google Scholar]
- 82. Röhrich M, Koelsche C, Schrimpf D, Capper D, Sahm F, Kratz Aet al. Methylation-based classification of benign and malignant peripheral nerve sheath tumors. Acta Neuropathol 2016;131:877–87. [DOI] [PubMed] [Google Scholar]
- 83. Koelsche C, Hartmann W, Schrimpf D, Stichel D, Jabar S, Ranft Aet al. Array-based DNA-methylation profiling in sarcomas with small blue round cell histology provides valuable diagnostic information. Mod Pathol 2018;31:1246–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Beckwith JB, Perrin PE. In situ neuroblastomas: a contribution to the natural history of neural crest tumors. Am J Pathol 1963;43:1089–104. [PMC free article] [PubMed] [Google Scholar]
- 85. Kastriti ME, Kameneva P, Adameyko I. Stem cells, evolutionary aspects and pathology of the adrenal medulla: a new developmental paradigm. Mol Cell Endocrinol 2020;518:110998. [DOI] [PubMed] [Google Scholar]
- 86. Beckwith JB. Nephrogenic rests and the pathogenesis of Wilms tumor: developmental and clinical considerations. Am J Med Genet 1998;79:268–73. [DOI] [PubMed] [Google Scholar]
- 87. Beckwith JB, Kiviat NB, Bonadio JF. Nephrogenic rests, nephroblastomatosis, and the pathogenesis of Wilms' tumor. Pediatr Pathol 1990;10:1–36. [DOI] [PubMed] [Google Scholar]
- 88. Oosterhuis JW, Looijenga LHJ. Human germ cell tumours from a developmental perspective. Nat Rev Cancer 2019;19:522–37. [DOI] [PubMed] [Google Scholar]
- 89. Berklite L, Witchel SF, Yatsenko SA, Schneck FX, Reyes-Mugica M. Early bilateral gonadoblastoma associated with 45,X/46,XY mosaicism: the spectrum of undifferentiated gonadal tissue and gonadoblastoma in the first months of life. Pediatr Dev Pathol 2019;22:380–5. [DOI] [PubMed] [Google Scholar]
- 90. Nistal M, Rodriguez JI, Garcia-Fernandez E, Cajaiba MM, Reyes-Mugica M. Fetal gonadoblastoid testicular dysplasia: a focal failure of testicular development. Pediatr Dev Pathol 2007;10:274–81. [DOI] [PubMed] [Google Scholar]
- 91. Alaggio R, Hill DA, Jacques TS, Jarzembowski JA, López-Terrada DH, Pfister SMet al. WHO Classification of Tumors: Pediatric Tumors. Lyon, France: IARC; 2021. [Google Scholar]
- 92. Ranganathan S, Lopez-Terrada D, Alaggio R. Hepatoblastoma and pediatric hepatocellular carcinoma: an update. Pediatr Dev Pathol 2020;23:79–95. [DOI] [PubMed] [Google Scholar]
- 93. Lopez-Terrada D, Alaggio R, de Davila MT, Czauderna P, Hiyama E, Katzenstein Het al. Towards an international pediatric liver tumor consensus classification: proceedings of the Los Angeles COG liver tumors symposium. Mod Pathol 2014;27:472–91. [DOI] [PubMed] [Google Scholar]
- 94. Bell D, Ranganathan S, Tao J, Monga SP. Novel advances in understanding of molecular pathogenesis of hepatoblastoma: a Wnt/beta-catenin perspective. Gene Expr 2017;17:141–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Eichenmuller M, Trippel F, Kreuder M, Beck A, Schwarzmayr T, Haberle Bet al. The genomic landscape of hepatoblastoma and their progenies with HCC-like features. J Hepatol 2014;61:1312–20. [DOI] [PubMed] [Google Scholar]
- 96. Lopez-Terrada D, Gunaratne PH, Adesina AM, Pulliam J, Hoang DM, Nguyen Yet al. Histologic subtypes of hepatoblastoma are characterized by differential canonical Wnt and Notch pathway activation in DLK+ precursors. Hum Pathol 2009;40:783–94. [DOI] [PubMed] [Google Scholar]
- 97. Ranganathan S, Ningappa M, Ashokkumar C, Higgs BW, Min J, Sun Qet al. Loss of EGFR-ASAP1 signaling in metastatic and unresectable hepatoblastoma. Sci Rep 2016;6:38347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Sumazin P, Chen Y, Trevino LR, Sarabia SF, Hampton OA, Patel Ket al. Genomic analysis of hepatoblastoma identifies distinct molecular and prognostic subgroups. Hepatology 2017;65:104–21. [DOI] [PubMed] [Google Scholar]
- 99. Tao J, Calvisi DF, Ranganathan S, Cigliano A, Zhou L, Singh Set al. Activation of beta-catenin and Yap1 in human hepatoblastoma and induction of hepatocarcinogenesis in mice. Gastroenterology 2014;147:690–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Mylonas KS, Doulamis IP, Tsilimigras DI, Nasioudis D, Schizas D, Masiakos PTet al. Solid pseudopapillary and malignant pancreatic tumors in childhood: a systematic review and evidence quality assessment. Pediatr Blood Cancer 2018;65:e27114. [DOI] [PubMed] [Google Scholar]
- 101. Shorter NA, Glick RD, Klimstra DS, Brennan MF, Laquaglia MP. Malignant pancreatic tumors in childhood and adolescence: the Memorial Sloan-Kettering experience, 1967 to present. J Pediatr Surg 2002;37:887–92. [DOI] [PubMed] [Google Scholar]
- 102. Abraham SC, Klimstra DS, Wilentz RE, Yeo CJ, Conlon K, Brennan Met al. Solid-pseudopapillary tumors of the pancreas are genetically distinct from pancreatic ductal adenocarcinomas and almost always harbor beta-catenin mutations. Am J Pathol 2002;160:1361–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Isobe T, Seki M, Yoshida K, Sekiguchi M, Shiozawa Y, Shiraishi Yet al. Integrated molecular characterization of the lethal pediatric cancer pancreatoblastoma. Cancer Res 2018;78:865–76. [DOI] [PubMed] [Google Scholar]
- 104. Jiao Y, Yonescu R, Offerhaus GJ, Klimstra DS, Maitra A, Eshleman JRet al. Whole-exome sequencing of pancreatic neoplasms with acinar differentiation. J Pathol 2014;232:428–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Abraham SC, Wu TT, Klimstra DS, Finn LS, Lee JH, Yeo CJet al. Distinctive molecular genetic alterations in sporadic and familial adenomatous polyposis-associated pancreatoblastomas: frequent alterations in the APC/beta-catenin pathway and chromosome 11p. Am J Pathol 2001;159:1619–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Kerr NJ, Chun YH, Yun K, Heathcott RW, Reeve AE, Sullivan MJ. Pancreatoblastoma is associated with chromosome 11p loss of heterozygosity and IGF2 overexpression. Med Pediatr Oncol 2002;39:52–4. [DOI] [PubMed] [Google Scholar]
- 107. Koh TH, Cooper JE, Newman CL, Walker TM, Kiely EM, Hoffmann EB. Pancreatoblastoma in a neonate with Wiedemann-Beckwith syndrome. Eur J Pediatr 1986;145:435–8. [DOI] [PubMed] [Google Scholar]
- 108. Lee CT, Tung YC, Hwu WL, Shih JC, Lin WH, Wu MZet al. Mosaic paternal haploidy in a patient with pancreatoblastoma and Beckwith-Wiedemann spectrum. Am J Med Genet A 2019;179:1878–83. [DOI] [PubMed] [Google Scholar]
- 109. Miettinen M, Dow N, Lasota J, Sobin LH. A distinctive novel epitheliomesenchymal biphasic tumor of the stomach in young adults (“gastroblastoma”): a series of 3 cases. Am J Surg Pathol 2009;33:1370–7. [DOI] [PubMed] [Google Scholar]
- 110. Shin DH, Lee JH, Kang HJ, Choi KU, Kim JY, Park DYet al. Novel epitheliomesenchymal biphasic stomach tumour (gastroblastoma) in a 9-year-old: morphological, ultrastructural and immunohistochemical findings. J Clin Pathol 2010;63:270–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Graham RP, Nair AA, Davila JI, Jin L, Jen J, Sukov WRet al. Gastroblastoma harbors a recurrent somatic MALAT1-GLI1 fusion gene. Mod Pathol 2017;30:1443–52. [DOI] [PubMed] [Google Scholar]
- 112. Adameyko I, Lallemend F, Aquino JB, Pereira JA, Topilko P, Muller Tet al. Schwann cell precursors from nerve innervation are a cellular origin of melanocytes in skin. Cell 2009;139:366–79. [DOI] [PubMed] [Google Scholar]
- 113. Bauer J, Curtin JA, Pinkel D, Bastian BC. Congenital melanocytic nevi frequently harbor NRAS mutations but no BRAF mutations. J Invest Dermatol 2007;127:179–82. [DOI] [PubMed] [Google Scholar]
- 114. Salgado CM, Basu D, Nikiforova M, Bauer BS, Johnson D, Rundell Vet al. BRAF mutations are also associated with neurocutaneous melanocytosis and large/giant congenital melanocytic nevi. Pediatr Dev Pathol 2015;18:1–9. [DOI] [PubMed] [Google Scholar]
- 115. Reyes-Mugica M, Chou P, Byrd S, Ray V, Castelli M, Gattuso Pet al. Nevomelanocytic proliferations in the central nervous system of children. Cancer 1993;72:2277–85. [DOI] [PubMed] [Google Scholar]
- 116. Salgado CM, Basu D, Nikiforova M, Hamilton RL, Gehris R, Jakacki Ret al. Amplification of mutated NRAS leading to congenital melanoma in neurocutaneous melanocytosis. Melanoma Res 2015;25:453–60. [DOI] [PubMed] [Google Scholar]
- 117. Recurrent MET fusion genes represent a drug target in pediatric glioblastoma. Nat Med 2016;22:1314–20. [DOI] [PubMed] [Google Scholar]
- 118. Clarke M, Mackay A, Ismer B, Pickles JC, Tatevossian RG, Newman Set al. Infant high-grade gliomas comprise multiple subgroups characterized by novel targetable gene fusions and favorable outcomes. Cancer Discov 2020;10:942–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Korshunov A, Schrimpf D, Ryzhova M, Sturm D, Chavez L, Hovestadt Vet al. H3-/IDH-wild type pediatric glioblastoma is comprised of molecularly and prognostically distinct subtypes with associated oncogenic drivers. Acta Neuropathol 2017;134:507–16. [DOI] [PubMed] [Google Scholar]
- 120. Guerrini-Rousseau L, Varlet P, Colas C, Andreiuolo F, Bourdeaut F, Dahan Ket al. Constitutional mismatch repair deficiency-associated brain tumors: report from the European C4CMMRD consortium. Neurooncol Adv 2019;1:vdz033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Suwala AK, Stichel D, Schrimpf D, Kloor M, Wefers AK, Reinhardt Aet al. Primary mismatch repair deficient IDH-mutant astrocytoma (PMMRDIA) is a distinct type with a poor prognosis. Acta Neuropathol 2021;141:85–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Ramkissoon LA, Horowitz PM, Craig JM, Ramkissoon SH, Rich BE, Schumacher SEet al. Genomic analysis of diffuse pediatric low-grade gliomas identifies recurrent oncogenic truncating rearrangements in the transcription factor MYBL1. Proc Natl Acad Sci U S A 2013;110:8188–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Zhang J, Wu G, Miller CP, Tatevossian RG, Dalton JD, Tang Bet al. Whole-genome sequencing identifies genetic alterations in pediatric low-grade gliomas. Nat Genet 2013;45:602–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Huse JT, Snuderl M, Jones DT, Brathwaite CD, Altman N, Lavi Eet al. Polymorphous low-grade neuroepithelial tumor of the young (PLNTY): an epileptogenic neoplasm with oligodendroglioma-like components, aberrant CD34 expression, and genetic alterations involving the MAP kinase pathway. Acta Neuropathol 2017;133:417–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Reinhardt A, Stichel D, Schrimpf D, Sahm F, Korshunov A, Reuss DEet al. Anaplastic astrocytoma with piloid features, a novel molecular class of IDH wildtype glioma with recurrent MAPK pathway, CDKN2A/B and ATRX alterations. Acta Neuropathol 2018;136:273–91. [DOI] [PubMed] [Google Scholar]
- 126. Sturm D, Orr BA, Toprak UH, Hovestadt V, Jones DTW, Capper Det al. New brain tumor entities emerge from molecular classification of CNS-PNETs. Cell 2016;164:1060–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Hirose T, Nobusawa S, Sugiyama K, Amatya VJ, Fujimoto N, Sasaki Aet al. Astroblastoma: a distinct tumor entity characterized by alterations of the X chromosome and MN1 rearrangement. Brain Pathol 2018;28:684–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Deng MY, Sill M, Chiang J, Schittenhelm J, Ebinger M, Schuhmann MUet al. Molecularly defined diffuse leptomeningeal glioneuronal tumor (DLGNT) comprises two subgroups with distinct clinical and genetic features. Acta Neuropathol 2018;136:239–53. [DOI] [PubMed] [Google Scholar]
- 129. Robinson GW, Rudneva VA, Buchhalter I, Billups CA, Waszak SM, Smith KSet al. Risk-adapted therapy for young children with medulloblastoma (SJYC07): therapeutic and molecular outcomes from a multicentre, phase 2 trial. Lancet Oncol 2018;19:768–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Northcott PA, Buchhalter I, Morrissy AS, Hovestadt V, Weischenfeldt J, Ehrenberger Tet al. The whole-genome landscape of medulloblastoma subtypes. Nature 2017;547:311–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Cavalli FMG, Remke M, Rampasek L, Peacock J, Shih DJH, Luu Bet al. Intertumoral heterogeneity within medulloblastoma subgroups. Cancer Cell 2017;31:737–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Sharma T, Schwalbe EC, Williamson D, Sill M, Hovestadt V, Mynarek Met al. Second-generation molecular subgrouping of medulloblastoma: an international meta-analysis of Group 3 and Group 4 subtypes. Acta Neuropathol 2019;138:309–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Waszak SM, Northcott PA, Buchhalter I, Robinson GW, Sutter C, Groebner Set al. Spectrum and prevalence of genetic predisposition in medulloblastoma: a retrospective genetic study and prospective validation in a clinical trial cohort. Lancet Oncol 2018;19:785–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Zheng T, Ghasemi DR, Okonechnikov K, Korshunov A, Sill M, Maass KKet al. Cross-species genomics reveals oncogenic dependencies in ZFTA/C11orf95 fusion-positive supratentorial ependymomas. Cancer Discov 2021;11:2230–47. [DOI] [PubMed] [Google Scholar]
- 135. Kupp R, Ruff L, Terranova S, Nathan E, Ballereau S, Stark Ret al. ZFTA translocations constitute ependymoma chromatin remodeling and transcription factors. Cancer Discov 2021;11:2216–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Arabzade A, Zhao Y, Varadharajan S, Chen HC, Jessa S, Rivas Bet al. ZFTA-RELA dictates oncogenic transcriptional programs to drive aggressive supratentorial ependymoma. Cancer Discov 2021;11:2200–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Panwalkar P, Clark J, Ramaswamy V, Hawes D, Yang F, Dunham Cet al. Immunohistochemical analysis of H3K27me3 demonstrates global reduction in group-A childhood posterior fossa ependymoma and is a powerful predictor of outcome. Acta Neuropathol 2017;134:705–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Ghasemi DR, Sill M, Okonechnikov K, Korshunov A, Yip S, Schutz PWet al. MYCN amplification drives an aggressive form of spinal ependymoma. Acta Neuropathol 2019;138:1075–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Raffeld M, Abdullaev Z, Pack SD, Xi L, Nagaraj S, Briceno Net al. High level MYCN amplification and distinct methylation signature define an aggressive subtype of spinal cord ependymoma. Acta Neuropathol Commun 2020;8:101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Ellison DW, Kocak M, Figarella-Branger D, Felice G, Catherine G, Pietsch Tet al. Histopathological grading of pediatric ependymoma: reproducibility and clinical relevance in European trial cohorts. J Negat Results Biomed 2011;10:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Johann PD, Hovestadt V, Thomas C, Jeibmann A, Heß K, Bens Set al. Cribriform neuroepithelial tumor: molecular characterization of a SMARCB1-deficient non-rhabdoid tumor with favorable long-term outcome. Brain Pathol 2017;27:411–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Siegfried A, Rousseau A, Maurage CA, Pericart S, Nicaise Y, Escudie Fet al. EWSR1-PATZ1 gene fusion may define a new glioneuronal tumor entity. Brain Pathol 2019;29:53–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Burel-Vandenbos F, Pierron G, Thomas C, Reynaud S, Gregoire V, Duhil de Benaze Get al. A polyphenotypic malignant paediatric brain tumour presenting a MN1-PATZ1 fusion, no epigenetic similarities with CNS high-grade neuroepithelial tumour with MN1 alteration (CNS HGNET-MN1) and related to PATZ1-fused sarcomas. Neuropathol Appl Neurobiol 2020;46:506–9. [DOI] [PubMed] [Google Scholar]
- 144. Lopez-Nunez O, Cafferata B, Santi M, Ranganathan S, Pearce TM, Kulich SMet al. The spectrum of rare central nervous system (CNS) tumors with EWSR1-non-ETS fusions: experience from three pediatric institutions with review of the literature. Brain Pathol 2021;31:70–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Rossi S, Barresi S, Giovannoni I, Alesi V, Ciolfi A, Colafati GSet al. Expanding the spectrum of EWSR1-PATZ1 rearranged CNS tumors: an infantile case with leptomeningeal dissemination. Brain Pathol 2021;31:e12934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Lambo S, Gröbner SN, Rausch T, Waszak SM, Schmidt C, Gorthi Aet al. The molecular landscape of ETMR at diagnosis and relapse. Nature 2019;576:274–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Ho B, Johann PD, Grabovska Y, De Dieu Andrianteranagna MJ, Yao F, Frühwald Met al. Molecular subgrouping of atypical teratoid/rhabdoid tumors-a reinvestigation and current consensus. Neuro Oncol 2020;22:613–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Liu APY, Li BK, Pfaff E, Gudenas B, Vasiljevic A, Orr BAet al. Clinical and molecular heterogeneity of pineal parenchymal tumors: a consensus study. Acta Neuropathol 2021;141:771–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Antonarakis SE. Down syndrome and the complexity of genome dosage imbalance. Nat Rev Genet 2017;18:147–63. [DOI] [PubMed] [Google Scholar]
- 150. Kratz CP, Jongmans MC, Cave H, Wimmer K, Behjati S, Guerrini-Rousseau Let al. Predisposition to cancer in children and adolescents. Lancet Child Adolesc Health 2021;5:142–54. [DOI] [PubMed] [Google Scholar]
- 151. Brioude F, Kalish JM, Mussa A, Foster AC, Bliek J, Ferrero GBet al. Expert consensus document: clinical and molecular diagnosis, screening and management of Beckwith-Wiedemann syndrome: an international consensus statement. Nat Rev Endocrinol 2018;14:229–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Yang XR, Ng D, Alcorta DA, Liebsch NJ, Sheridan E, Li Set al. T (brachyury) gene duplication confers major susceptibility to familial chordoma. Nat Genet 2009;41:1176–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Grunewald TG, Bernard V, Gilardi-Hebenstreit P, Raynal V, Surdez D, Aynaud MMet al. Chimeric EWSR1-FLI1 regulates the Ewing sarcoma susceptibility gene EGR2 via a GGAA microsatellite. Nat Genet 2015;47:1073–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Vijayakrishnan J, Studd J, Broderick P, Kinnersley B, Holroyd A, Law PJet al. Genome-wide association study identifies susceptibility loci for B-cell childhood acute lymphoblastic leukemia. Nat Commun 2018;9:1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Fiala EM, Jayakumaran G, Mauguen A, Kennedy JA, Bouvier N, Kemel Yet al. Prospective pan-cancer germline testing using MSK-IMPACT informs clinical translation in 751 patients with pediatric solid tumors. Nat Cancer 2021;2:357–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Waszak SM, Robinson GW, Gudenas BL, Smith KS, Forget A, Kojic Met al. Germline Elongator mutations in Sonic Hedgehog medulloblastoma. Nature 2020;580:396–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Begemann M, Waszak SM, Robinson GW, Jager N, Sharma T, Knopp Cet al. Germline GPR161 mutations predispose to pediatric medulloblastoma. J Clin Oncol 2020;38:43–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Achatz MI, Hainaut P, Ashton-Prolla P. Highly prevalent TP53 mutation predisposing to many cancers in the Brazilian population: a case for newborn screening? Lancet Oncol 2009;10:920–5. [DOI] [PubMed] [Google Scholar]
- 159. Hol JA, Jewell R, Chowdhury T, Duncan C, Nakata K, Oue Tet al. Wilms tumour surveillance in at-risk children: literature review and recommendations from the SIOP-Europe Host Genome Working Group and SIOP Renal Tumour Study Group. Eur J Cancer 2021;153:51–63. [DOI] [PubMed] [Google Scholar]
- 160. Dutzmann CM, Spix C, Popp I, Kaiser M, Erdmann F, Erlacher Met al. Cancer in children with Fanconi Anemia and Ataxia-Telangiectasia-a nationwide register-based cohort study in Germany. J Clin Oncol 2021:Jco2101495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Villani A, Shore A, Wasserman JD, Stephens D, Kim RH, Druker Het al. Biochemical and imaging surveillance in germline TP53 mutation carriers with Li-Fraumeni syndrome: 11 year follow-up of a prospective observational study. Lancet Oncol 2016;17:1295–305. [DOI] [PubMed] [Google Scholar]
- 162. Wimmer K, Kratz CP. Constitutional mismatch repair-deficiency syndrome. Haematologica 2010;95:699–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Zhang J, Walsh MF, Wu G, Edmonson MN, Gruber TA, Easton Jet al. Germline mutations in predisposition genes in pediatric cancer. N Engl J Med 2015;373:2336–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Wimmer K, Kratz CP, Vasen HF, Caron O, Colas C, Entz-Werle Net al. Diagnostic criteria for constitutional mismatch repair deficiency syndrome: suggestions of the European consortium ‘care for CMMRD' (C4CMMRD). J Med Genet 2014;51:355–65. [DOI] [PubMed] [Google Scholar]
- 165. Wang Z, Wilson CL, Armstrong GT, Hudson MM, Zhang J, Nichols KEet al. Association of germline BRCA2 mutations with the risk of pediatric or adolescent non-Hodgkin lymphoma. JAMA Oncol 2019;5:1362–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Qin N, Wang Z, Liu Q, Song N, Wilson CL, Ehrhardt MJet al. Pathogenic germline mutations in DNA repair genes in combination with cancer treatment exposures and risk of subsequent neoplasms among long-term survivors of childhood cancer. J Clin Oncol 2020;38:2728–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Nguyen TMK, Behnert A, Pietsch T, Vokuhl C, Kratz CP. Proportion of children with cancer that have an indication for genetic counseling and testing based on the cancer type irrespective of other features. Fam Cancer 2021;20:273–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Campbell BB, Light N, Fabrizio D, Zatzman M, Fuligni F, de Borja Ret al. Comprehensive analysis of hypermutation in human cancer. Cell 2017;171:1042–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Goudie C, Cullinan N, Villani A, Mathews N, van Engelen K, Malkin Det al. Retrospective evaluation of a decision-support algorithm (MIPOGG) for genetic referrals for children with neuroblastic tumors. Pediatr Blood Cancer 2018;65:e27390. [DOI] [PubMed] [Google Scholar]
- 170. Schwermer M, Behnert A, Dorgeloh B, Ripperger T, Kratz CP. Effective identification of cancer predisposition syndromes in children with cancer employing a questionnaire. Fam Cancer 2021;20:257–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Byrjalsen A, Hansen TVO, Stoltze UK, Mehrjouy MM, Barnkob NM, Hjalgrim LLet al. Nationwide germline whole genome sequencing of 198 consecutive pediatric cancer patients reveals a high incidence of cancer prone syndromes. PLoS Genet 2020;16:e1009231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Seyschab H, Friedl R, Sun Y, Schindler D, Hoehn H, Hentze Set al. Comparative evaluation of diepoxybutane sensitivity and cell cycle blockage in the diagnosis of Fanconi anemia. Blood 1995;85:2233–7. [PubMed] [Google Scholar]
- 173. Parsons DW, Roy A, Yang Y, Wang T, Scollon S, Bergstrom Ket al. Diagnostic yield of clinical tumor and germline whole-exome sequencing for children with solid tumors. JAMA Oncol 2016;2:616–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Ripperger T, Bielack SS, Borkhardt A, Brecht IB, Burkhardt B, Calaminus Get al. Childhood cancer predisposition syndromes-A concise review and recommendations by the Cancer Predisposition Working Group of the Society for Pediatric Oncology and Hematology. Am J Med Genet A 2017;173:1017–37. [DOI] [PubMed] [Google Scholar]
- 175. Scollon S, Anglin AK, Thomas M, Turner JT, Wolfe Schneider K. A comprehensive review of pediatric tumors and associated cancer predisposition syndromes. J Genet Couns 2017;26:387–434. [DOI] [PubMed] [Google Scholar]
- 176. Campbell BB, Galati MA, Stone SC, Riemenschneider AN, Edwards M, Sudhaman Set al. Mutations in the RAS/MAPK pathway drive replication repair-deficient hypermutated tumors and confer sensitivity to MEK inhibition. Cancer Discov 2021;11:1454–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Wagener R, Taeubner J, Walter C, Yasin L, Alzoubi D, Bartenhagen Cet al. Comprehensive germline-genomic and clinical profiling in 160 unselected children and adolescents with cancer. Eur J Hum Genet 2021;29:1301–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Eisenstein M. Could liquid biopsies help deliver better treatment? Nature 2020;579:S6–S8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.