

Abstract
Purpose:
We investigated safety, tolerability, pharmacokinetics, and antitumor activity of the protein tyrosine kinase 7 (PTK7)-targeted, auristatin-based antibody–drug conjugate (ADC) PF-06647020/cofetuzumab pelidotin (NCT02222922).
Patients and Methods:
Patients received PF-06647020 intravenously every 3 weeks at 0.2–3.7 mg/kg or every 2 weeks at 2.1–3.2 mg/kg, in sequential dose escalation, following a modified toxicity probability interval method. In dose expansion, pretreated patients with advanced, platinum-resistant ovarian cancer, non–small cell lung cancer (NSCLC), or triple-negative breast cancer (TNBC) received PF-06647020 2.8 mg/kg every 3 weeks.
Results:
The most common, treatment-related adverse events for PF-06647020 administered every 3 weeks were nausea, alopecia, fatigue, headache, neutropenia, and vomiting (45%–25%); 25% of patients had grade ≥ 3 neutropenia. Two patients experienced dose-limiting toxicities (grade 3 headache and fatigue) at the highest every 3 weeks dose evaluated. The recommended phase II dose was 2.8 mg/kg every 3 weeks. The overall safety profile observed with PF-06647020 administered every 2 weeks was similar to that of the every 3 weeks regimen. Systemic exposure for the ADC and total antibody generally increased in a dose-proportional manner. Antitumor activity was observed in treated patients with overall objective response rates of 27% in ovarian cancer (n = 63), 19% in NSCLC (n = 31), and 21% in TNBC (n = 29). Responders tended to have moderate or high PTK7 tumor expression by IHC.
Conclusions:
This PTK7-targeted ADC demonstrated therapeutic activity in previously treated patients with ovarian cancer, NSCLC, and TNBC at a dose range of 2.1–3.2 mg/kg, supporting further clinical evaluation to refine dose, schedule, and predictive tissue biomarker testing in patients with advanced malignancies.
Translational Relevance.
The transmembrane protein tyrosine kinase 7 (PTK7), involved in Wnt signaling, is overexpressed in multiple tumor types, including advanced non–small cell lung cancer (NSCLC), ovarian cancer, and triple-negative breast cancer (TNBC). Also expressed in tumor-initiating cells (TIC) or cancer stem cells, PTK7 is a tumor-associated target for elimination of cancer cells and TICs.
PF-06647020 (cofetuzumab pelidotin) is a humanized, anti-PTK7 antibody–drug conjugate, designed to deliver an auristatin microtubule inhibitor payload (Aur0101) into target cells. It was shown to induce prolonged tumor regression in patient-derived, tumor xenograft preclinical models. In this first-in-human, dose-finding study, PF-06647020 administered every 2 or 3 weeks demonstrated a tolerable safety profile and preliminary clinical activity in previously treated patients with locally advanced/metastatic, PTK7-positive NSCLC, TNBC, and platinum-resistant ovarian cancer, suggesting the feasibility of this PTK7-targeting approach. Further clinical investigations are ongoing to assess the therapeutic potential of PF-06647020/cofetuzumab pelidotin in advanced, PTK7-positive cancers.
Introduction
Protein tyrosine kinase 7 (PTK7) is a highly conserved, catalytically inactive, transmembrane PTK, involved in Wnt signaling during development of hematopoietic and somatic progenitor cells as well as stem cells (1, 2). It is overexpressed in multiple tumor types, including advanced non–small cell lung cancer (NSCLC), ovarian cancer, triple-negative breast cancer (TNBC), and colon, gastric, and esophageal cancers (3–8).
PTK7 overexpression has been associated with a poor prognosis and worse overall survival in patients with NSCLC, TNBC, and other solid tumor types (3–8). In addition, IHC analysis of tumor tissue from patients with lung adenocarcinoma indicated that PTK7 expression correlated with the presence of lymph node metastases (8). PTK7 has also been shown to be enriched in tumor-initiating cells (TIC) or cancer stem cells in patient-derived tumor tissues (9–11). Thus, PTK7 is a tumor-associated target that may facilitate elimination of cancer cells as well as the TICs responsible for tumor recurrence and progression.
PF-06647020 (cofetuzumab pelidotin) is an antibody–drug conjugate (ADC) comprising a humanized anti-PTK7 mAb (hu6M024, IgG1) joined to a proprietary auristatin microtubule inhibitor payload, auristatin-0101 (Aur0101), by a cleavable valine-citrulline (vc) based linker (drug/mAb ratio: 4; refs. 9, 12–15). Following binding and internalization of the ADC PF-06647020 into PTK7-expressing cells, the cleavable linker is processed by endosomal proteases leading to release of the auristatin payload, microtubule disruption, induction of mitotic arrest (G2–M phase), and apoptotic cell death in target cancer cells (9, 12). Preclinical studies have demonstrated sustained tumor regression in patient-derived xenograft models following administration of PF-06647020, with greater antitumor activity than standard chemotherapy (9). In addition, serial transplantation experiments showed that treatment with PF-06647020 reduced the frequency of TICs (9).
On the basis of these findings, we investigated safety, pharmacokinetics, and preliminary therapeutic activity of PF-06647020 in a first-in-human, phase I study of patients with advanced solid tumors resistant to or with no available standard therapy. Following completion of dose escalation with once every 3 weeks infusion, a range of doses was explored with once every 2 weeks infusion and treatment with single-agent PF-06647020 was further evaluated in expansion cohorts of previously treated patients with advanced NSCLC, TNBC, or platinum-resistant ovarian cancer.
Patients and Methods
Study design and treatment
This was a phase I, open label, multi-center, nonrandomized, dose-escalation and dose-expansion study designed to evaluate PF-06647020, administered every 3 weeks or every 2 weeks, in patients with advanced solid tumors.
Primary objectives of dose escalation were to assess safety and tolerability of treatment with PF-06647020, to determine the MTD, and select the recommended phase II dose (RP2D). Secondary objectives included evaluation of pharmacokinetic profiles (ADC: PF-06647020, total antibody: hu6M024, and unconjugated payload: PF-06380101), immunogenicity, and antitumor activity of PF-06647020 in patients with advanced solid tumors. Dose escalation was guided by the modified toxicity probability interval (mTPI) method, with a target dose-limiting toxicity (DLT) rate of 25% and an acceptable DLT interval of 20%–30%.
Patients received PF-06647020 intravenously every 3 weeks at doses ranging from 0.2 to 3.7 mg/kg or every 2 weeks at 2.1, 2.8, and 3.2 mg/kg, in sequential dose-escalation cohorts (Supplementary Fig. S1). The duration of the intravenous drug infusion was approximately 60 minutes for all dose levels. The starting dose of 0.2 mg/kg given as an intravenous infusion every 3 weeks represented approximately 1/6th of the highest nonseverely toxic dose assessed in monkeys (based on the human equivalent dose, normalized to body surface area). For every 2 weeks dosing, 2.1 mg/kg represented 75% of the most commonly tolerated dose of 2.8 mg/kg for PF-06647020 administered every 3 weeks in the first part of the study. Pharmacokinetic modeling based on the initial data from patients treated every 3 weeks suggested that the cumulative exposure of 6 weeks with 2.1 mg/kg every 2 weeks dosing would be comparable with that achieved with 2.8 mg/kg every 3 weeks dosing with a potentially superior therapeutic index (14).
In dose expansion, patients were treated with intravenous PF-06647020 at the RP2D of 2.8 mg/kg every 3 weeks. Primary objectives of dose expansion were to further evaluate safety and tolerability of treatment specifically in patients with advanced ovarian cancer, NSCLC, and TNBC. Secondary objectives included evaluation of the clinical antitumor activity, pharmacokinetics, and immunogenicity of PF-06647020 in these patient cohorts.
Biomarker (i.e. PTK7 protein expression level) assessments were an eligibility requirement for the every 3 weeks dose expansion and every 2 weeks dose-escalation patients with NSCLC, and a subset of patients with TNBC in the every 3 weeks dose-expansion cohort. Patients continued study treatment until disease progression, unacceptable toxicity, or withdrawal of consent. Administration of G-CSF was allowed after cycle 1 to treat neutropenia; erythropoietin could be used at the investigator's discretion for the supportive treatment of anemia. Additional supportive care medications, such as prophylactic 5-HT3 receptor antagonists or corticosteroids were administered at the treating physician's discretion.
Patients
Eligible, adult patients (18 years or older) had locally advanced or metastatic solid tumors resistant to standard therapy or with no available standard therapy, and Eastern Cooperative Oncology Group (ECOG) performance status 0 or 1. Patients had adequate bone marrow function (absolute neutrophil count ≥1,500/mm3 or ≥1.5 × 109/L, platelets ≥100,000/mm3 or ≥100 × 109/L, and hemoglobin ≥9 g/dL), renal function [serum creatinine ≤1.5 × upper limit of normal (ULN) or estimated creatinine clearance ≥60 mL/minute], liver function [total serum bilirubin ≤1.5 × ULN (unless patient had documented Gilbert syndrome), aspartate and alanine transaminase ≤3.0 × ULN or ≤5.0 × ULN (if liver metastasis at study entry), alkaline phosphatase ≤2.5 × ULN (≤5 × ULN if bone and/or hepatic metastasis at study entry)], and adequate cardiac function. With regards to cardiac function, patients were ineligible if any of the following occurred within 12 months of study entry: myocardial infarction, severe/unstable angina, coronary/peripheral artery bypass graft, symptomatic congestive heart failure, cerebrovascular accident, transient ischemic attack, or symptomatic pulmonary embolism. In addition, eligibility exclusion criteria prohibited patients with any ongoing cardiac dysrhythmias of NCI Common Terminology Criteria for Adverse Event (CTCAE) grade ≥2, any grade of atrial fibrillation, or QTcF interval >470 ms (with exception of right bundle branch block).
Patients with measurable (by RECIST v1.1; ref. 16) and nonmeasurable disease were included in every 3 weeks dose escalation. In the every 3 weeks dose-expansion and every 2 weeks dose-escalation cohorts, measurable disease was required for patients with recurrent NSCLC or TNBC; patients with ovarian cancer had platinum-resistant disease (progression/relapse within 6 months after completion of at least four cycles of the most recently administered platinum-containing therapy and a maximum of three prior lines of chemotherapy) and disease measurable by RECIST v1.1 or assessable by the Gynecologic Cancer Intergroup (GCIG 2011) criteria (17).
Patients with any cancer type in the every 3 weeks cohorts were not preselected at baseline for PTK7 expression in tumors. In the expansion cohorts, patients with ovarian cancer were not preselected for PTK7 baseline expression in tumors; the PTK7 expression rate was assumed to be >80% and confirmed retrospectively by IHC. Patients with NSCLC in the every 3 weeks dose-expansion and every 2 weeks dose-escalation cohorts were selected pretreatment for moderate to high tumor PTK7 expression by IHC. For TNBC, after preliminary assessment in dose expansion of 9 patients initially included without preselection, subsequent patients were preselected for moderately high to high tumor PTK7 expression, as described previously (15).
Patients were not eligible for this study if they had known, symptomatic brain metastases; an active and clinically significant bacterial, fungal, or viral infection; grade ≥ 2 peripheral neuropathy; or known/suspected hypersensitivity to recombinant human or murine proteins. In addition, patients were excluded if they had received major surgery, radiotherapy, or systemic anticancer therapy within 3 weeks of starting study treatment.
The study was approved by the institutional review board or independent ethics committee of the participating institutions and followed the Declaration of Helsinki and the International Conference on Harmonization Good Clinical Practice (ICH GCP) guidelines. The patient informed consent complied with ICH GCP guidelines, local regulatory requirements, and legal requirements. The study was sponsored by Pfizer and registered at ClinicalTrials.gov (NCT02222922).
Assessments
Safety and DLT
The severity of the reported adverse events (AE) was graded using the CTCAE (NCI-CTCAE) v4.03. The monitoring period for DLTs was 28 days.
Any of the following events occurring in the first treatment cycle and deemed related to study treatment was to be considered a DLT: grade 4 neutropenia lasting > 7 days; febrile neutropenia; grade ≥ 3 neutropenic infection; grade 4 anemia or thrombocytopenia; grade ≥ 3 thrombocytopenia with clinically significant bleeding; grade ≥ 3 elevation in serum bilirubin, alanine aminotransferase (ALT), aspartate aminotransferase (AST), or alkaline phosphatase; ALT or AST ≥ 3 × ULN concurrent with elevation in bilirubin ≥ 2 × ULN; grade ≥ 3 nonhematologic, nonhepatic toxicities (excluding alopecia of any grade and grade 3 diarrhea, nausea, and vomiting responding to therapy); or a delay > 2 weeks in receiving the next treatment cycle due to persisting, treatment-related toxicities.
Pharmacokinetics
Blood samples for pharmacokinetic analyses were collected from treated patients at protocol-predefined timepoints: on day 1 in cycle 1 (pre-dose, 1 and 4 hours after start of infusion); day 2 in cycle 1 (24 hours after infusion); days 4, 8, and 15 in cycle 1; day 1 in cycles 2 and 3; days 1, 2, 4, 8, and 15 in cycle 4; then on day 1 of every cycle thereafter, and at the end of treatment. ADC (PF-06647020) and total antibody (conjugated or unconjugated mAb hu6M024) concentrations were quantified by ELISAs; the assays had lower limits of quantitation (LLOQ) of 90 ng/mL and 100 ng/mL for the ADC and total antibody, respectively. Unconjugated payload (PF-06380101) concentrations were determined by a validated LC/MS-MS method (18, 19); the assay had a LLOQ of 15 pg/mL for the unconjugated payload. Samples below the LLOQs were set to zero for analysis. Pharmacokinetic parameters were determined from the respective concentration–time data by standard, noncompartmental analysis, using an internally validated, electronic, noncompartmental analysis software (eNCA) version 2.2.4.
Immunogenicity
Incidence of anti-drug antibodies (ADA) was assessed by a validated electrochemiluminescent (ECL) method (20, 21) in samples collected from treated patients at protocol-predefined timepoints: on day 1 and 15 of cycle 1, day 1 of all subsequent cycles, and at the end of treatment. ADA-positive samples were further evaluated for the presence of neutralizing antibodies (NAb) using a validated, competitive ligand-binding ECL method.
Preliminary therapeutic activity
Objective tumor responses were determined by the investigators every 6 weeks or 8 weeks (every 2 weeks cohorts) using RECIST v1.1 (16) and summarized by calculating objective response rates (ORR). Complete responses (CR) or partial responses (PR) were confirmed in the every 3 weeks and every 2 weeks expansion cohorts by repeat assessment at least 4 weeks after the response criteria were first met.
PTK7 protein expression
Tumor-associated PTK7 protein expression levels were assessed by a Clinical Laboratory Improvement Amendments–validated IHC assay in formalin-fixed, paraffin-embedded tumor tissue samples collected from patients, as described previously (15). Digital tissue analysis was performed using the Flagship Biosciences proprietary Image Analysis platform. At least 90% of cells quantified by the algorithm were tumor cells. H-scores were determined from image analysis mark-up (regions of analysis) and quantified on the basis of percentage of positive cells and staining intensity. If a sample failed digital tissue analysis, a visual assessment was performed for nonquantitative plasma membrane staining. All analytic markups were reviewed and accepted by a board-certified MD pathologist. H-scores were rank ordered lowest to highest and divided into tertiles. Tumor H-scores were divided into relatively low, moderate, and high PTK7 expression categories based on tertiles.
Statistical analyses
The prior distribution of DLT for the mTPI was set as a beta (0.75, 0.65) and the threshold probability for early termination and dose exclusion was set to 0.975. The algorithm for dose escalation was to be stopped if the maximum, planned sample size had been reached (∼40 patients), at least 9 patients had been treated at a dose level predicted to be at or greater than the MTD, or all doses explored appeared to be too toxic and the MTD could not be determined. The estimated MTD was to be the highest tested dose level with a DLT rate < 0.30 in at least 9 DLT-evaluable patients.
The sample size for the every 3 weeks dose expansion was mainly based on clinical and feasibility considerations. Evaluation of the every 2 weeks regimen in this study ran in parallel with the every 3 weeks expansion cohorts. The every 2 weeks regimen completed dose escalation and did not proceed to dose expansion.
Data sharing statement
Upon request, and subject to certain criteria, conditions, and exceptions (see https://www.pfizer.com/science/clinical-trials/trial-data-and-results for more information), Pfizer will provide access to individual deidentified participant data from Pfizer-sponsored global interventional clinical studies conducted for medicines, vaccines, and medical devices (i) for indications that have been approved in the United States and/or EU or (ii) in programs that have been terminated (i.e., development for all indications has been discontinued). Pfizer will also consider requests for the protocol, data dictionary, and statistical analysis plan. Data may be requested from Pfizer trials 24 months after study completion. The deidentified participant data will be made available to researchers whose proposals meet the research criteria and other conditions, and for which an exception does not apply, via a secure portal. To gain access, data requestors must enter into a data access agreement with Pfizer.
Results
Patients and treatment
A total of 112 patients received PF-06647020 every 3 weeks in the dose-escalation (0.2–3.7 mg/kg) and dose-expansion (2.8 mg/kg) cohorts: 0.2 (n = 2), 0.5 (n = 2), 1.25 (n = 2), 2.1 (n = 4), 2.8 (n = 96), and 3.7 (n = 6) mg/kg (Supplementary Fig. S1). The 2.8 mg/kg every 3 weeks cohort included 15 patients in dose escalation and 11 patients enrolled in a drug–drug interaction substudy with multiple dose fluconazole, the full details of which will be reported separately. In the second part of the study, 25 patients were treated with PF-06647020 every 2 weeks across three dose levels: 2.1 (n = 3), 2.8 (n = 10), and 3.2 mg/kg (n = 12). Demographic and baseline characteristics for all patients are presented in Table 1. The majority of patients (63% and 60% in the every 3 weeks and every 2 weeks cohorts, respectively) had ECOG performance status 1.
Table 1.
Patient demographics and baseline characteristics.
| Every 3 weeks regimen | Every 2 weeks regimen | |
|---|---|---|
| N = 112 | N = 25 | |
| Age, years | 58.4 | 65.4 |
| Mean (range) | (31–80) | (51–79) |
| Male: female, n (%) | 20 (17.9) : 92 (82.1) | 1 (4) : 24 (96) |
| Race, n (%) | ||
| White | 103 (92.0) | 21 (84) |
| Black | 6 (5.4) | 1 (4) |
| Asian | 1 (0.9) | 1 (4) |
| Other | 2 (1.8) | 2 (8) |
| Primary tumor diagnosis, n (%) | ||
| NSCLC | 25 (22.3) | 6 (24) |
| Ovarian cancer | 44 (39.3) | 19 (76) |
| TNBC | 29 (25.9) | 0 |
| Other | 14 (12.5) | 0 |
| ECOG PS, n (%) | ||
| 0 | 40 (35.7) | 10 (40) |
| 1 | 71 (63.4) | 15 (60) |
| 2 | 1 (0.9)a | 0 |
| Prior systemic anticancer therapy, n (%) | 112 (100) | 25 (100) |
| ≥2 regimens | 104 (92.9) | 22 (88) |
| ≥3 regimens | 84 (75.0) | 6 (24) |
| Prior radiotherapy, n (%) | ||
| Yes | 54 (48.2) | 6 (24) |
| No | 58 (51.8) | 19 (76) |
Abbreviations: ECOG PS, Eastern Cooperative Oncology Group performance status; NSCLC, non–small cell lung cancer; TNBC, triple-negative breast cancer.
aProtocol deviation.
The every 3 weeks cohorts included patients with platinum-resistant ovarian cancer (39%), advanced NSCLC (22%), or advanced TNBC (26%); a minority of patients (13%) had other locally advanced/metastatic solid tumor types. Across the every 3 weeks cohorts, 34% of patients with ovarian cancer had high-grade serous carcinoma, 64% of patients with NSCLC had adenocarcinoma, and 79% of patients with TNBC had ductal carcinoma. Patients in the every 2 weeks cohorts had a primary tumor diagnosis of platinum-resistant ovarian cancer (76%) or advanced NSCLC (24%). In these cohorts, 68% of patients with ovarian cancer had high-grade serous carcinoma and 83% of patients with NSCLC had adenocarcinoma.
All patients had received prior systemic, anticancer therapy; 93% of patients in the every 3 weeks cohorts and 88% in the every 2 weeks cohorts had received two or more prior treatment regimens.
DLT and safety
Two DLTs (grade 3 headache and grade 3 fatigue) were reported at the highest dose of PF-06647020 evaluated with the every 3 weeks regimen (3.7 mg/kg, n = 6, 33%). Absent further exploration of doses between 3.7 mg/kg and the previously completed dose cohort of 2.8 mg/kg, the protocol specified that the MTD for every 3 weeks administration of PF-06647020 in patients with solid tumors was estimated to be the last completed, lower dose level, 2.8 mg/kg. Overall, three DLTs were observed with the every 2 weeks dosing regimen: grade 3 neutropenic infection in 1 patient at 2.8 mg/kg (n = 10) and grade 3 abdominal pain in 2 patients at 3.2 mg/kg every 2 weeks (n = 12, 17%). On the basis of the DLT rates observed, the MTD for the every 2 weeks regimen was not reached at the dose levels evaluated in this study.
Treatment-emergent, all-cause AEs were reported in all patients treated with PF-06647020 every 3 weeks or every 2 weeks. Eighty-nine (79.4%) and 18 (72%) patients, respectively, experienced grade ≥ 3 all-cause AEs. Seventeen (15.2%) patients and 4 (16%) patients died on study in the every 3 weeks and every 2 weeks cohorts, respectively, mostly due to disease progression (n = 15 and n = 4, respectively), or respiratory arrest and urosepsis (n = 1 each, every 3 weeks cohorts). None of the patients died because of a treatment-related AE (TRAE).
In the every 3 weeks cohorts, the most common TRAEs were nausea, alopecia, fatigue, headache, neutropenia, and vomiting (45%–25% of patients; Table 2). Treatment-related peripheral sensory neuropathy was observed in 12.5% of patients (all events were grade 1 or 2). Forty-four (39.3%) patients had grade ≥ 3 TRAEs. The most frequent grade ≥ 3 TRAE was neutropenia (25%); 3 (2.7%) patients developed febrile neutropenia. The incidence of TRAEs in the every 3 weeks cohorts was highest in patients dosed at 2.8 mg/kg (Table 3).
Table 2.
TRAEs reported in >10% of patients with every 3 weeks dosing of PF-06647020 (all cycles, N = 112).
| Grade 1 | Grade 2 | Grade 3 | Grade 4 | Totala | |
|---|---|---|---|---|---|
| AE | n (%) | ||||
| Nausea | 28 (25.0) | 21 (18.8) | 1 (0.9) | 0 | 50 (44.6) |
| Alopecia | 13 (11.6) | 33 (29.5) | 0 | 0 | 46 (41.1) |
| Fatigue | 19 (17.0) | 19 (17.0) | 3 (2.7) | 0 | 41 (36.6) |
| Headache | 13 (11.6) | 19 (17.0) | 5 (4.5) | 0 | 37 (33.0) |
| Neutropenia | 0 | 4 (3.6) | 17 (15.2) | 11 (9.8) | 32 (28.6) |
| Vomiting | 10 (8.9) | 14 (12.5) | 4 (3.6) | 0 | 28 (25.0) |
| Arthralgia | 16 (14.3) | 2 (1.8) | 1 (0.9) | 0 | 19 (17.0) |
| Decreased appetite | 11 (9.8) | 7 (6.3) | 0 | 0 | 18 (16.1) |
| Diarrhea | 8 (7.1) | 8 (7.1) | 1 (0.9) | 0 | 17 (15.2) |
| Myalgia | 11 (9.8) | 3 (2.7) | 1 (0.9) | 0 | 15 (13.4) |
| Peripheral sensory neuropathy | 7 (6.3) | 7 (6.3) | 0 | 0 | 14 (12.5) |
Abbreviations: AE, adverse event; TRAE, treatment-related AE.
aNo treatment-related grade 5 AEs were observed with the every 3 weeks regimen.
Table 3.
Summary of TRAEs by dose level with every 3 weeks dosing of PF-06647020 (all cycles, N = 112).
| 0.2 mg/kg n (%) | 0.5 mg/kg n (%) | 1.25 mg/kg n (%) | 2.1 mg/kg n (%) | 2.8 mg/kg n (%) | 3.7 mg/kg n (%) | |
|---|---|---|---|---|---|---|
| Evaluable pts | 2 | 2 | 2 | 4 | 96 | 6 |
| Number of AEs | 1 | 1 | 6 | 28 | 475 | 27 |
| Pts with AEs | 1 (50.0) | 1 (50.0) | 1 (50.0) | 4 (100.0) | 84 (87.5) | 5 (83.3) |
| Pts with SAEs | 0 | 0 | 0 | 1 (25.0) | 9 (9.4) | 0 |
| Pts with G3/G4 AEs | 1 (50.0) | 0 | 0 | 0 | 40 (41.7) | 3 (50.0) |
| Pts discontinued due to AEs | 0 | 0 | 0 | 1 (25.0) | 2 (2.1) | 0 |
| Pts with dose reduction due to AEs | 0 | 0 | 0 | 0 | 7 (7.3) | 0 |
| Pts with temporary discontinuation due to AEs | 0 | 0 | 0 | 0 | 16 (16.7) | 0 |
| Pts with dose reduction and temporary discontinuation due to AEs | 0 | 0 | 0 | 0 | 3 (3.1) | 1 (16.7) |
Abbreviations: AE, adverse event; Pts, patients; SAE, serious adverse event.
The most common TRAEs observed with every 2 weeks dosing across dose levels also were alopecia, nausea, fatigue, and headache (Supplementary Table S1). Thirteen (52%) patients had grade ≥ 3 TRAEs. Treatment-related grade 3–4 neutropenia was reported in 24% of patients; 1 patient developed a grade 3 neutropenic infection (DLT). Grade 1–2 peripheral sensory neuropathy occurred in 20% and grade 3 in 8% of the patients on the every 2 weeks regimen.
Treatment with PF-06647020 every 3 weeks was generally tolerable with a manageable safety profile in the majority of patients. Three (2.7%) patients treated with the every 3 weeks regimen and 1 (4%) of the patients on every 2 weeks dosing discontinued because of a TRAE; 7 (6.3%) and 4 (16%) patients, respectively, had a dose reduction of PF-06647020. Temporary discontinuations due to TRAEs occurred less frequently with the every 3 weeks (14.3%) than the every 2 weeks (44%) dosing regimen (Supplementary Table S2). The AE most frequently leading to temporary treatment discontinuation was neutropenia.
Forty-six (41%) and 19 (76%) patients in the every 3 weeks and every 2 weeks cohorts, respectively, received concomitant treatment with ondansetron to manage nausea and vomiting.
Of all the patients enrolled across the every 3 weeks and every 2 weeks cohorts, 54 reported development of headache at some point during the course of the study. Cumulatively, investigators recorded 29 of these patients to have reported a headache after infusion on cycle 1, day 1 or 2. The first such post-infusion event occurred in a patient with breast cancer who was enrolled in the 2.1 mg/kg every 3 weeks dose-escalation cohort. The patient first reported a grade 2 headache, associated with vomiting, on cycle 1 day 1 of treatment, approximately 4 hours after the end of the infusion. The patient was treated with analgesics and antiemetics in the clinic but required hospital admission for the management of persistent symptoms. The symptoms resolved within 48 hours of cycle 1 day 1. She was premedicated for the subsequent cycles with acetaminophen and antiemetics including 5-HT3 antagonists plus dexamethasone. She experienced no recurrent headache or vomiting with further study drug administration. Empirically, as each site managed the care of a few patients, the headaches almost always occurred only after completion of the infusion, and were not associated with fever, chills, or other typical infusion reaction-like symptoms. The collective impression of the investigators was that this headache syndrome (at times accompanied by nausea, photophobia, and/or meningismus-like symptoms) was reminiscent of a transient central nervous system inflammatory syndrome. For all patients, the headaches resolved spontaneously within a maximum of a few days. Analgesics, including opioids, did not seem helpful and anti-inflammatory agents, especially glucocorticoids, seemed most effective. Therefore, although not mandated in the protocol, many investigators ultimately decided to routinely administer pre-infusion steroids to reduce the likelihood of after PF-06647020 infusion headache syndrome. On the basis of these collective observations, the use of steroids warrants further exploration as prophylactic treatment for patients who will receive PF-06647020.
On the basis of overall clinical and statistical assessments, the actual MTD for PF-06647020 is between 2.8 and 3.7 mg/kg every 3 weeks. By protocol definition, the RP2D is 2.8 mg/kg every 3 weeks, but notably there is variance in clearance and tolerability that could lead investigators to revisit higher doses with steroid prophylaxis in the future.
Pharmacokinetics
Pharmacokinetic analyses showed that systemic exposure, based on the observed area under the serum concentration-time curve during the dosing interval (AUCtau) and maximum concentration (Cmax) values for the ADC (PF-06647020), total antibody (hu6M024), and unconjugated payload, generally increased in a dose-related manner across the 0.2 to 3.7 mg/kg every 3 weeks dose levels (Fig. 1). Following single intravenous infusion of PF-06647020 at 2.8 mg/kg every 3 weeks, peak concentrations for the ADC and total antibody were observed at or shortly after the end of the infusion, followed by a multi-phasic decline (Fig. 1), with a mean t1/2 value in cycle 1 of 3.1 days for the ADC (Supplementary Table S3). The AUC ratio between ADC and total antibody was approximately 0.9. Individual Cmax for the payload ranged between 1.2 to 24.5 ng/mL and t1/2 between 0.7 to 5.3 days, across all every 3 weeks dose levels (cycles 1 and 4).
Figure 1.
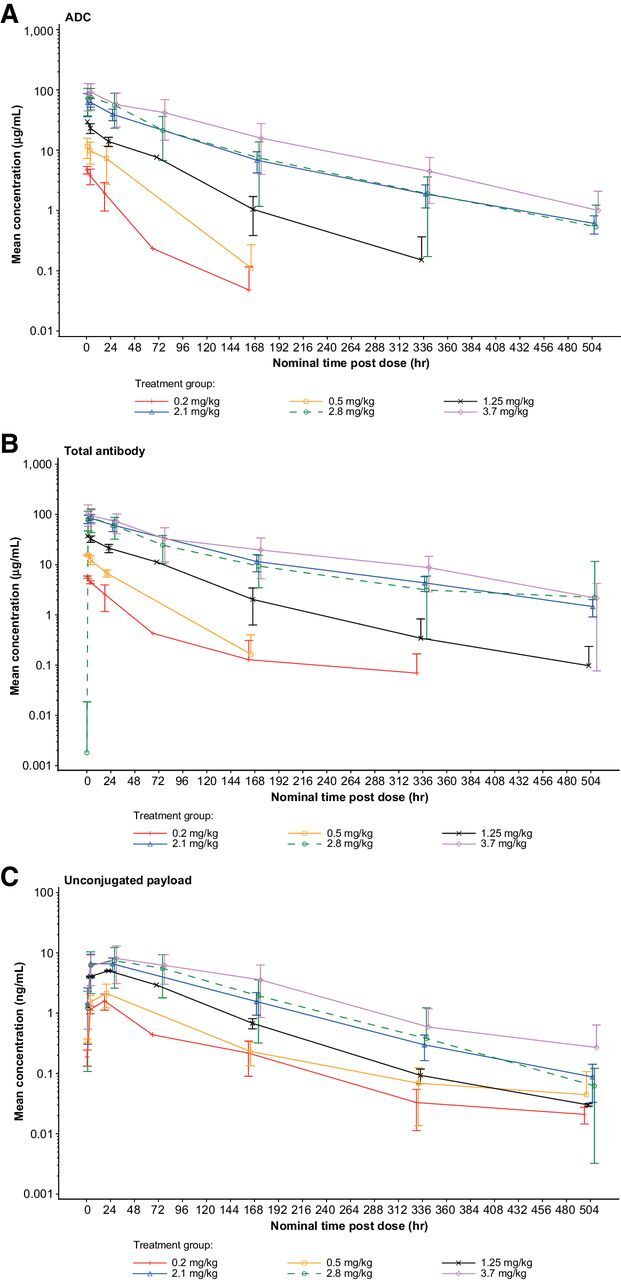
Mean pharmacokinetic profiles for PF-06647020 administered every 3 weeks in patients with solid tumors. Results are shown for the ADC (A), total antibody (B), and unconjugated payload (C). Error bars indicate the SDs. ADC, antibody–drug conjugate.
The pharmacokinetic parameters for every 2 weeks dosing of PF-06647020 are presented in Supplementary Table S4. At the 2.8 mg/kg every 2 weeks dose level, the observed Cmax for the ADC was 99.7 μg/mL in cycle 1 (compared with 79.8 μg/mL with every 3 weeks dosing), with a mean t1/2 value of 2.7 days.
The major cytochrome P450 isoform involved in the metabolism of unconjugated payload (PF-06380101) in humans was predicted to be CYP3A4 based on initial reaction phenotyping experiments. To assess potential CYP3A-mediated drug–drug interaction, the effects of multiple doses of fluconazole, a moderate CYP3A4 inhibitor, on the pharmacokinetic of PF-06380101 (unconjugated payload) following single-dose co-administration of PF-06647020 (ADC) was investigated. For the 11 patients enrolled in this drug–drug interaction substudy, co-administration of PF-06647020 with fluconazole did not have a notable impact on the overall exposure of PF-06380101. Ratios of the adjusted geometric means and [90% confidence interval (CI)] for PF-06380101 exposure based on AUCinf (dn) and Cmax (dn) were 91% (45%, 186%), and 102% (51%, 205%), respectively, when PF-06647020 (ADC) was co-administered with fluconazole in cycle 2 (test) compared with administration alone in cycle 1 (reference). In addition, co-administration of PF-06647020 with fluconazole did not have a notable impact on the overall exposure of PF-06647020 (ADC) and hu6M024 mAb (total antibody).
Immunogenicity
Of the 108 patients treated with PF-06647020 every 3 weeks with at least 1 evaluable, post-dose sample, 11 (10.2%) patients tested positive for treatment-induced ADA. Ten of these ADA-positive patients had NAb. One (4%) of the 25 evaluable patients on the every 2 weeks regimen had treatment-induced ADA and Nab. An additional patient (4%) in the every 2 weeks cohorts had ADA at baseline, but they were not boosted by treatment and had resolved by the second ADA assessment. No hypersensitivity or infusion-related reactions (IRR) were reported in the ADA-positive patients treated every 3 weeks. One ADA-positive patient on the every 2 weeks regimen experienced a grade 2 IRR (day 1, cycle 4), which resolved on the same day.
Antitumor activity and PTK7 biomarker analysis
PF-06647020 demonstrated antitumor activity in the every 3 weeks and every 2 weeks cohorts, with ORRs of 27% and 26%, respectively, in patients with ovarian cancer, 16% and 33% in NSCLC, and 21% in TNBC (every 3 weeks regimen), across the dose levels evaluated (Table 4).
Table 4.
Antitumor activity of PF-06647020 administered every 3 weeks or every 2 weeks in patients with advanced ovarian cancer, NSCLC, or TNBC.
| CR, PR | ORR | mDOR | mPFS | ||
|---|---|---|---|---|---|
| Regimen and tumor | N | N | % (95% CI) | mo (95% CI) | mo (95% CI) |
| Every 3 weeks | |||||
| Ovarian cancer | 44 | 3, 9 | 27 (15–43) | 4.2 (2.8–8.3) | 2.9 (2.3–5.5) |
| NSCLC | 25 | 0, 4 | 16 (5–36) | 5.7 (1.5–9.9) | 2.9 (1.4–6.1) |
| TNBC | 29 | 0, 6 | 21 (8–40) | 4.3 (1.3–10.2) | 1.5 (1.4–4.3) |
| Every 2 weeks | |||||
| Ovarian cancer | 19 | 2, 3 | 26 (9–51) | 6.5 (3.9–8.3) | 3.8 (3.1–7.4) |
| NSCLC | 6 | 0, 2 | 33 (4–78) | NR (3.7–NR) | 2.7 (1.0–NR) |
Abbreviations: CR, complete response; mDOR, median duration of response; mo, months; mPFS, median progression-free survival; NR, not reached; NSCLC, non–small cell lung cancer; ORR, objective response rate; PR, partial response; TNBC, triple-negative breast cancer.
In ovarian cancer, 3 (7%) patients treated with PF-06647020 every 3 weeks achieved a CR and 9 (21%) patients a PR (Fig. 2). All responses, except for 1 CR (2.1 mg/kg), were observed in the 2.8 mg/kg every 3 weeks cohorts. In addition, 2 (11%) patients treated every 2 weeks had a CR and 3 (16%) a PR, in the 2.8 mg/kg (1 CR, 2 PR) or 3.2 mg/kg (1 CR, 1 PR) cohorts. In patients with NSCLC treated with PF-06647020, all responses were observed in the 2.8 mg/kg every 3 weeks (4 PRs) and the 3.2 mg/kg every 2 weeks cohorts (2 PRs). Median duration of response and median PFS (mPFS) results (cut-off data, Dec 19, 2019) are presented in Table 4, according to dosing regimen and tumor type.
Figure 2.

PTK7 protein expression and best overall response to treatment with PF-06647020 administered every 3 weeks (A, B, and C) or every 2 weeks (D and E) in patients with advanced ovarian cancer (A and D), NSCLC (B and E), and TNBC (C). A visual assessment of nonquantitative PTK7 membrane staining was performed for samples that failed digital tissue analysis. CR, complete response; NSCLC, non–small cell lung cancer; OvCa, ovarian cancer; PD, progressive disease; PR, partial response; Q2W, every 2 weeks; Q3W, every 3 weeks; SD, stable disease; TNBC, triple-negative breast cancer.
PTK7 protein expression levels and best overall response to treatment with PF-06647020 administered every 3 weeks or every 2 weeks are shown in Fig. 2. After adjusting for tumor type, patients with high baseline H-score have significantly larger mean percent reduction from baseline in tumor size than that in patients with low baseline H-score (P = 0.027). No significance was observed between H-score high and moderate, or between moderate and low (Supplementary Fig. S2).
Discussion
In this first-in-human dose-finding study, single-agent treatment with a PTK7-targeted ADC, PF-06647020, demonstrated a manageable safety profile and preliminary antitumor activity in patients with advanced ovarian cancer, NSCLC, or TNBC who had received multiple, prior lines of standard-of-care therapy.
The majority of the TRAEs observed with the every 3 weeks regimen were mild or moderate. None of the patients experienced a grade 5 TRAE. Two DLTs (grade 3 headache and grade 3 fatigue) were observed at the highest every 3 weeks dose level evaluated. Although neutropenia was the most frequent grade 3–4 TRAE reported (25%), it resolved in most cases with supportive care; febrile neutropenia occurred only in 2.7% of treated patients. Peripheral sensory neuropathy observed in 12.5% of patients was limited to grade 1–2. A comparable safety profile has been previously observed following treatment with other ADCs containing an auristatin payload, such as the Notch-3 targeted ADC PF-06650808, in patients with advanced solid malignancies (i.e. breast cancer, ovarian cancer, and NSCLC; ref. 22). Treatment with PF-06647020 every 3 weeks was generally tolerable, as only 2.7% of patients discontinued and 6.3% had a dose reduction due to a TRAE. Pharmacokinetic analyses showed that at 2.8 mg/kg every 3 weeks, the mean terminal half-life for PF-06647020 was approximately 3 days.
Further evaluation of single-agent PF-06647020 was undertaken with every 2 weeks dosing in patients with advanced, platinum-resistant ovarian cancer or recurrent NSCLC, based on the hypothesis that therapeutic activity could be augmented by sustaining higher trough concentrations of PF-06647020. Overall safety findings in patients treated with PF-06647020 every 2 weeks were similar to those observed with every 3 weeks dosing. However, 2 patients with ovarian cancer on the every 2 weeks regimen experienced grade 3 abdominal pain (DLT) at the highest dose level evaluated (3.2 mg/kg) and 8% of patients developed grade 3 peripheral sensory neuropathy across dose levels. Grade 2–3 abdominal pain of unknown etiology has also been previously reported with the auristatin-based ADC PF-06650808 (22). Notably, a patient with NSCLC who had no known peritoneal metastases experienced treatment-emergent abdominal pain.
Objective tumor responses were observed following treatment with both PF-06647020 every 3 weeks and every 2 weeks regimens, with ORRs of 26%–27% in patients with ovarian cancer, 16%–33% with NSCLC, and 21% with TNBC, who had progressed on prior standard therapies. Most of the responses associated with either dosing regimen occurred at the 2.8 mg/kg dose level, which was selected as the RP2D for further single-agent every 3 weeks administration of PF-06647020. Biomarker analysis showed that patients with clinical responses tended to have moderate to high H-scores, suggesting the feasibility of patient selection using the IHC assay.
Other ADCs, including mirvetuximab soravtansine (an anti-folate receptor α–DM4 ADC), sacituzumab govitecan-hziy (an anti-human trophoblast cell-surface antigen 2–SN38 ADC), trastuzumab deruxtecan (an anti-HER2–topoisomerase I inhibitor ADC), and lifastuzumab vedotin (an anti-NaPi2b–monomethyl auristatin E ADC) are being developed for the treatment of patients with advanced ovarian cancer or NSCLC and other solid malignancies (23–28). Although our study was not powered to demonstrate antitumor efficacy and despite the small sample size in some tumor types, we observed encouraging preliminary antitumor activity with PF-06647020. Hence, our study results with PF-06647020 suggest that treatment with an ADC directed to PTK7 represents a feasible approach for the management of patients with advanced ovarian cancer, NSCLC, and TNBC.
Further clinical development is in progress for PF-06647020/cofetuzumab pelidotin using the every 3 weeks regimen. Safety and efficacy are being evaluated in a phase Ib study in patients with advanced, recurrent PTK7+ NSCLC, who have received prior treatment with chemotherapy and immune checkpoint inhibitors or targeted agents (NCT04189614, primary study endpoint: ORR). In addition, a combination of PF-06647020 with the investigational PI3K/mTOR inhibitor gedatolisib (29) is being explored in a phase I study in patients with chemotherapy-pretreated, metastatic TNBC (NCT03243331).
Authors' Disclosures
M.L. Maitland reports institutional research funding to Inova Health System from Pfizer. J.C. Sachdev reports personal fees and other support from Pfizer during the conduct of the study, as well as personal fees from Novartis, Puma, AstraZeneca, Tempus, and Immunomedics outside the submitted work. M.R. Sharma reports institutional research funding to START Midwest from Pfizer, as well as ownership of Pfizer stock. V. Moreno reports personal fees from Roche, Janssen, Bayer, BMS, and Basilea outside the submitted work. V. Boni reports institutional funding from Pfizer during the conduct of the study. V. Boni also reports personal fees (consultant/advisory boards) from Oncoart, Ideaya, Loxo Therapeutics, CytomX Therapeutics, Puma Biotechnology, and Guidepoint, as well as institutional funding from AbbVie, ACEO, Adaptimmune, Amcure, Amgen, AstraZeneca, BMS, CytomX, GSK, Genentech/Roche, H3, Incyte, Janssen, Kura, Lilly, Loxo, Nektar, Macrogenics, Menarini, Merck, Merus, Nanobiotix, Novartis, Pfizer, PharmaMar, Principia, Puma, Sanofi, Taiho, Tesaro, BeiGene, Transgene, Takeda, Incyte, Innovio, MSD, PsiOxus, Seattle Genetics, Mersana, Daiichi, Nektar, Astellas, Orca, Boston Therapeutics, Dynavax, DebioPharm, Boehringer Ingelheim, Regeneron, Millennium, Synthon, Spectrum, Rigontec, and Zenith outside the submitted work. S. Kummar reports other support from Boehringer Ingelheim, Springworks Therapeutics, Bayer, Genome & Company, HarbourBiomed, Seattle Genetics, Mundibiopharma, PathomIQ, Cadila Pharmaceuticals, and Arxeon during the conduct of the study. E. Stringer-Reasor reports other support from Pfizer, as well as personal fees and other support from Lilly, Novartis, Immunomedics, and Mylan during the conduct of the study; E. Stringer-Reasor also reports grants from Susan G Komen and VFoundation. N. Lakhani reports other support form Pfizer during the conduct of the study. N. Lakhani also reports other support from ALX Therapeutics, Ascentage, Asana, BeiGene, Constellation Pharma, Alexion, Cerulean, Forty Seven, Macrogenics, Merck, Shattuck Labs, Regeneron, TaiRx, Apexian, Formation Biologics (forbius), Symphogen, CytomX, InhibRx, Incyte, Jounce, Livzon, Northern Biologics, Innovent Biologics, Ikena, Odonate, Alpine Immune Sciences, Celgene, Seagen, and Mersana, as well as personal fees from Innovent Biologics outside the submitted work. A.R. Moreau is an employee of and owns stock in Pfizer Inc. D. Xuan was an employee of and owned stock in Pfizer Inc. at the time of this study. R. Li is an employee of and owns stock in Pfizer Inc. E.L. Powell is an employee of and owns stock in Pfizer Inc. A. Jackson-Fisher is an employee of and owns stock in Pfizer Inc. M. Bowers is an employee of and owns stock in Pfizer Inc. S. Alekar is an employee and owns stock in Pfizer Inc. X. Xin was an employee of and owned stock in Pfizer Inc. at the time this investigational work was being conducted. A.W. Tolcher reports personal fees and other support from Pfizer during the conduct of the study, as well as personal fees and other support from AbbVie, Agenus Inc, Asana Biosciences, Ascentage, AxImmune, Bayer, Gilde Healthcare Partners, HBM Partners, Immunomet Therapeutics Inc., Karma Oncology B.V., Mekanistic Therapeutics, Menarini Ricerche, Mersana, Nanobiotix, Partner Therapeutics, Pfizer Inc., Pierre Fabre, Ryvu Therapeutics, Seattle Genetics, Sioto Biotechnology Co., Spirea Limited Inc., Transcenta Therapeutics Inc., Trillium Therapeutics Inc., Mirati, Zymeworks Biopharmaceuticals Inc., Pyxis Oncology, Pieris Pharma, Pelican, NBE Therapeutics, Immunome, EMD Serono/Merck KGaA, Elucida, Eleven Bio, Deka Biosciences, Boehringer Ingelheim International GmbH, BioInvent, Aro Biotherapeutics, Adagene Inc., ABL Bio Inc., ADC Therapeutics SA, Aminex Therapeutics Inc., Amphivena Therapeutics Inc., Apros Therapeutics Inc., Arcellx Inc., Armo Biosciences, Arrys Therapeutics Inc., Artios Pharma Limited, Astex Pharmaceuticals, Basilea Pharmaceutica International Ltd, BioInvent International AB, BioNTech RNA Pharmaceuticals GmbH, Birdie Biopharmaceuticals, HK Ltd, BJ Bioscience Inc., Boston Biomedical Inc., Calgent Biotechnology Co. Ltd., Codiak Sciences Inc., CStone Pharmaceuticals (Suzhou) Co. Ltd., Cybrexa Therapeutics Inc., Daiichi Sankyo Inc., Deciphera Pharmaceuticals LLC, eFFECTOR Therapeutics Inc., Eli Lilly and Company, Gilead Sciences Inc., GlaxoSmithKline Research & Development Limited, Haihe Biopharma Co. Ltd., Shanghai Huaota, Heat Biologics, Ideaya Biosciences, ImmuneOncia Therapeutics Inc., IMPACT Therapeutics Inc., Inhibrx Inc., Innate Pharma SA, Janssen Research & Development, K-Group Beta Inc., KeChow Pharma Inc., Kiromic Biopharma Inc, Mabspace Biosciences (Suzhou) Co. Ltd., Merck Sharp & Dohme Corp., a subsidiary of Merck & Co. Inc., NatureWise Biotech & Medicals Corporation, Navire Pharma Inc., Kirovax, MV Pharmaceuticals, 3's Bio, NextCure Inc, Nitto BioPharma Inc., Odonate Therapeutics Inc., Oric Pharmaceuticals, Pelican Therapeutics Inc., Petra Pharma, Pieris Pharmaceuticals Inc., PMV Pharmaceuticals Inc., Qilu Puget Sound Biotherapeutics Corporation, Samumed LLC, Seattle Genetics Inc., Spring Bank Pharmaceuticals Inc., Sunshine Guojian Pharmaceutical (Shanghai) Co. Ltd., Symphogen A/S, Syndax Pharmaceuticals Inc., Synthorx Inc., Takeda, Tizona Therapeutics, and Zymeworks Inc. outside the submitted work. E. Calvo reports non-financial support and other support from Adcendo, Amcure, Janssen, T-knife, Chugai Pharmaceuticals, and PsiOxus Pharmaceuticals; personal fees and other support from Alkermes, Amunix, Anaveon, AstraZeneca, BMS, MSD, Nanobiotix, Novartis, OncoDNA, PharmaMar, Roche/Genentech, Servier, and TargImmune; and grants from Achilles and BeiGene outside the submitted work. E. Calvo is an employee with HM Hospitals Group and START Program of Early Phase Clinical Drug Development in Oncology (Medical Oncologist, Clinical Investigator; Director, Clinical Research), holds ownership in START Corporation and Oncoart Associated: International Cancer Consultants; and is Founder and President of non-profit foundation INTHEOS (Investigational Therapeutics in Oncological Sciences).
Supplementary Material
Figure S1, Figure S2, Table S1, Table S2, Table S3, Table S4
Acknowledgments
The authors thank the patients, their families/caregivers, the investigators, research nurses, study coordinators, and operations staff who contributed to this study; Qionghua (Barbara) Zhao and Yingzhi Chen for their statistical programming support; and Xiaoying Chen and Shibing Deng for their contributions to this study. This study was sponsored by Pfizer. Medical writing was provided by S. Mariani of Engage Scientific Solutions, and funded by Pfizer.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
Note: Supplementary data for this article are available at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/).
Authors' Contributions
M.L. Maitland: Conceptualization, formal analysis, supervision, investigation, methodology, writing–original draft, writing–review and editing. J.C. Sachdev: Conceptualization, formal analysis, investigation, methodology, writing–original draft, writing–review and editing. M.R. Sharma: Formal analysis, investigation, writing–review and editing. V. Moreno: Formal analysis, investigation, writing–review and editing. V. Boni: Formal analysis, investigation, writing–review and editing. S. Kummar: Formal analysis, investigation, writing–review and editing. E. Stringer-Reasor: Formal analysis, investigation, writing–review and editing. N. Lakhani: Formal analysis, investigation, writing–review and editing. A.R. Moreau: Formal analysis, investigation, writing–review and editing. D. Xuan: Formal analysis, investigation, writing–review and editing. R. Li: Data curation, formal analysis, validation, methodology, writing–review and editing. E.L. Powell: Formal analysis, validation, investigation, writing–review and editing. A. Jackson-Fisher: Conceptualization, data curation, formal analysis, validation, investigation, writing–original draft, writing–review and editing. M. Bowers: Formal analysis, investigation, writing–review and editing. S. Alekar: Formal analysis, supervision, investigation, methodology, writing–original draft, writing–review and editing. X. Xin: Conceptualization, formal analysis, supervision, investigation, methodology, writing–original draft, writing–review and editing. A.W. Tolcher: Conceptualization, investigation, methodology, writing–review and editing. E. Calvo: Conceptualization, formal analysis, supervision, investigation, methodology, writing–original draft, writing–review and editing.
References
- 1. Peradziryi H, Tolwinski NS, Borchers A. The many roles of PTK7: a versatile regulator of cell-cell communication. Arch Biochem Biophys 2012;52471–6. [DOI] [PubMed] [Google Scholar]
- 2. Puppo F, Thome V, Lhoumeau AC, Cibois M, Gangar A, Lembo F, et al. Protein tyrosine kinase 7 has a conserved role in Wnt/beta-catenin canonical signalling. EMBO Rep 2011;12:43–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gärtner S, Gunesch A, Knyazeva T, Wolf P, Högel B, Eiermann W, et al. PTK 7 is a transforming gene and prognostic marker for breast cancer and nodal metastasis involvement. PLoS One 2014;9:e84472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chen R, Khatri P, Mazur PK, Polin M, Zheng Y, Vaka D, et al. A meta-analysis of lung cancer gene expression identifies PTK7 as a survival gene in lung adenocarcinoma. Cancer Res 2014;74:2892–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chen G, Qi S, Yang X, Chen W. Prognostic significance of PTK7 in human malignancies. Histol Histopathol 2018;33:379–88. [DOI] [PubMed] [Google Scholar]
- 6. Lhoumeau AC, Martinez S, Boher JM, Monges G, Castellano R, Goubard A, et al. Overexpression of the promigratory and prometastatic PTK7 receptor is associated with an adverse clinical outcome in colorectal cancer. PLoS One 2015;10:e0123768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Shin WS, Kwon J, Lee HW, Kang MC, Na HW, Lee ST, et al. Oncogenic role of protein tyrosine kinase 7 in esophageal squamous cell carcinoma. Cancer Sci 2013;104:1120–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jiang W, He J, Lv B, Xi X, He G, He J. PTK7 expression is associated with lymph node metastasis, ALK and EGFR mutations in lung adenocarcinomas. Histol Histopathol 2020;35:489–95. [DOI] [PubMed] [Google Scholar]
- 9. Damelin M, Bankovich A, Bernstein J, Lucas J, Chen L, Williams S. A PTK7-targeted antibody-drug conjugate reduces tumor-initiating cells and induces sustained tumor regressions. Sci Transl Med 2017;9:eaag2611. [DOI] [PubMed] [Google Scholar]
- 10. Katoh M. Canonical and non-canonical WNT signaling in cancer stem cells and their niches: cellular heterogeneity, omics reprogramming, targeted therapy and tumor plasticity. Int J Oncol 2017;51:1357–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bie J, Hu X, Yang M, Shi X, Zhang X, Wang Z. PTK7 promotes the malignant properties of cancer stem-like cells in esophageal squamous cell lines. Hum Cell 2020;33:356–65. [DOI] [PubMed] [Google Scholar]
- 12. Maderna A, Doroski M, Subramanyam C, Porte A, Leverett CA, Vetelino BC, et al. Discovery of cytotoxic dolastatin 10 analogues with N-terminal modifications. J Med Chem 2014;57:10527–43. [DOI] [PubMed] [Google Scholar]
- 13. Sachdev JC, Maitland ML, Sharma M, Moreno V, Boni V, Kummar S, et al. PF-06647020 (PF-7020), an antibody-drug conjugate (ADC) targeting protein tyrosine kinase 7 (PTK7), in patients (pts) with advanced solid tumors: results of a phase I dose escalation and expansion study. J Clin Oncol 36:15s, 2018(suppl; abstr 5565). [Google Scholar]
- 14. Xuan D, Xin X, Gibson B, Joh T, Yin H, Garzone P, et al. Clinical pharmacology assessment of PF-06647020, an antibody-drug conjugate (ADC) targeting protein tyrosine kinase 7 (PTK7), in patients with advanced solid tumors. J Clin Oncol 36:15s, 2018(suppl; abstr 2574). [Google Scholar]
- 15. Jackson-Fisher A, Mehra N, Gianani R, Whalen P, Vizcarra P, Deng S, et al. Protein tyrosine kinase 7 (PTK7) biomarker analysis in patients (pts) treated with PF-06647020, a PTK7 antibody-drug conjugate (ADC), in a phase I dose expansion study [abstract]. In: Proceedings of the American Association for Cancer Research Annual Meeting 2019; 2019 Mar 29–Apr 3; Atlanta, GA. Philadelphia (PA): AACR; Cancer Res 2019;79(13 Suppl):Abstract nr 4035. [Google Scholar]
- 16. Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 2009;45228–47. [DOI] [PubMed] [Google Scholar]
- 17. Rustin GJ, Vergote I, Eisenhauer E, Pujade-Lauraine E, Quinn M, Thigpen T, et al. Definitions for response and progression in ovarian cancer clinical trials incorporating RECIST 1.1 and CA 125 agreed by the Gynecological Cancer Intergroup (GCIG). Int J Gynecol Cancer 2011;21:419–23. [DOI] [PubMed] [Google Scholar]
- 18. Kaur S, Xu K, Saad OM, Dere RC, Carrasco-Triguero M. Bioanalytical assay strategies for the development of antibody–drug conjugate biotherapeutics. Bioanalysis 2013;5:201–26. [DOI] [PubMed] [Google Scholar]
- 19. Jenkins R, Duggan JX, Aubry AF, Zeng J, Lee JW, Cojocaru L, et al. Recommendations for validation of LC-MS/MS bioanalytical methods for protein biotherapeutics. AAPS J 2015;17:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jani D, Nowak J, Chen Y, Gorovits B. Assessment of clinical immunogenicity of inotuzumab ozogamicin in patients with non-Hodgkin lymphoma and acute lymphoblastic leukemia. AAPS Open 2018;4:s41120-018-0021-5. [Google Scholar]
- 21. Shankar G, Devanarayan V, Amaravadi L, Barrett YC, Bowsher R, Finco-Kent D, et al. Recommendations for the validation of immunoassays used for detection of host antibodies against biotechnology products. J Pharm Biomed Anal 2008;48:1267–81. [DOI] [PubMed] [Google Scholar]
- 22. Rosen LS, Wesolowski R, Baffa R, Liao KH, Hua SY, Gibson BL, et al. A phase I, dose-escalation study of PF-06650808, an anti-Notch3 antibody-drug conjugate, in patients with breast cancer and other advanced solid tumors. Invest New Drugs 2020;38:120–30. [DOI] [PubMed] [Google Scholar]
- 23. O'Malley DM, Matulonis UA, Birrer MJ, Castro CM, Gilbert L, Vergote I, et al. Phase Ib study of mirvetuximab soravtansine, a folate receptor alpha (FRα)-targeting antibody-drug conjugate (ADC), in combination with bevacizumab in patients with platinum-resistant ovarian cancer. Gynecol Oncol 2020;157:379–85. [DOI] [PubMed] [Google Scholar]
- 24. Bardia A, Mayer IA, Vahdat LT, Tolaney SM, Isakoff SJ, Diamond JR, et al. Sacituzumab govitecan-hziy in refractory metastatic triple-negative breast cancer. N Engl J Med 2019;380:741–51. [DOI] [PubMed] [Google Scholar]
- 25. Lopez S, Perrone E, Bellone S, Bonazzoli E, Zeybek B, Han C, et al. Preclinical activity of sacituzumab govitecan (IMMU-132) in uterine and ovarian carcinosarcomas. Oncotarget 2020;11:560–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Doi T, Shitara K, Naito Y, Shimomura A, Fujiwara Y, Yonemori K, et al. Safety, pharmacokinetics, and antitumour activity of trastuzumab deruxtecan (DS-8201), a HER2-targeting antibody-drug conjugate, in patients with advanced breast and gastric or gastro-oesophageal tumours: a phase 1 dose-escalation study. Lancet Oncol 2017;18:1512–22. [DOI] [PubMed] [Google Scholar]
- 27. Tsurutani J, Iwata H, Krop I, Jänne PA, Doi T, Takahashi S, et al. Targeting HER2 with trastuzumab deruxtecan: a dose-expansion, phase I study in multiple advanced solid tumors. Cancer Discov 2020;10:688–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gerber DE, Infante JR, Gordon MS, Goldberg SB, Martín M, Felip E, et al. Phase Ia study of anti-NaPi2b antibody-drug conjugate lifastuzumab vedotin DNIB0600A in patients with non-small cell lung cancer and platinum-resistant ovarian cancer. Clin Cancer Res 2020;26:364–72. [DOI] [PubMed] [Google Scholar]
- 29. Wainberg ZA, Alsina M, Soares HP, Braña I, Britten CD, Del Conte G, et al. A multi-arm phase I study of the PI3K/mTOR inhibitors PF-04691502 and gedatolisib (PF-05212384) plus irinotecan or the MEK inhibitor PD-0325901 in advanced cancer. Target Oncol 2017;12:775–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1, Figure S2, Table S1, Table S2, Table S3, Table S4