

Abstract
Purpose:
The first report from the open-label substudy of the phase III iNNOVATE study (PCYC-1127; NCT02165397) demonstrated that single-agent ibrutinib was efficacious and well tolerated in patients with heavily pretreated, rituximab-refractory Waldenström macroglobulinemia. Results from the final analysis are now reported.
Patients and Methods:
Ibrutinib 420 mg was administered once daily to patients (N = 31) who failed to achieve at least a minor response (MR) or who relapsed <12 months after their last rituximab-containing therapy. Endpoints included progression-free survival (PFS) and overall response rate (ORR; MR or better) per independent review committee, hemoglobin improvement, overall survival (OS), and safety; serum IgM was also assessed.
Results:
After a median follow-up of 58 months (range: 9–61), median PFS was 39 months [95% confidence interval (CI): 25–not evaluable]; 60-month PFS rate was 40%. In MYD88L265P/CXCR4WHIM and MYD88L265P/CXCR4WT subtypes, median PFS was 18 months and not reached, respectively. In all patients, ORR was 87%; responses deepened over time with major response (≥ partial response) rates increasing from 61% at 6 months to 77% at 60 months. Median OS was not reached. Seventeen of 21 patients (81%) with baseline hemoglobin ≤11.0 g/dL had sustained hemoglobin improvement. Improvements in serum IgM levels were sustained, reaching a maximum median change of −37 g/L at 54 months. Ibrutinib maintained a manageable safety profile, with no new safety signals identified. There were no events of major hemorrhage or atrial fibrillation.
Conclusions:
In the final analysis from iNNOVATE, single-agent ibrutinib continued to show sustained efficacy in patients with heavily pretreated, rituximab-refractory Waldenström macroglobulinemia.
Translational Relevance.
Rituximab resistance is commonly observed in patients with Waldenström macroglobulinemia due to its widespread and repeated use as either monotherapy or in a combination regimen. Results from the first report of the phase III iNNOVATE substudy showed that single-agent ibrutinib was effective and well tolerated in patients with previously treated, rituximab-refractory Waldenström macroglobulinemia. Here we present results from the final analysis after up to 5 years of follow-up (median: 58 months; range: 9–61). Patients were heavily pretreated, receiving a median of 4 prior lines of therapy. Continuous treatment with single-agent ibrutinib led to sustained response, durable progression-free survival and overall survival, and an acceptable safety profile. Importantly, sustained improvements in hemoglobin and serum IgM and clinically meaningful improvements in patient-reported outcomes were also demonstrated. Our findings support single-agent ibrutinib as an effective chemotherapy-free treatment option for patients with rituximab-refractory Waldenström macroglobulinemia, a population with limited treatment options.
Introduction
Waldenström macroglobulinemia is a rare lymphoproliferative disorder classified as an indolent non–Hodgkin B-cell lymphoma (1). For patients with Waldenström macroglobulinemia, current treatments include alkylating agents, anti-CD20 mAbs, proteosome inhibitors, nucleoside analogues in combination with rituximab, as well as ibrutinib (2). While rituximab-based regimens are most commonly used for the treatment of symptomatic Waldenström macroglobulinemia, most patients will eventually develop resistance to rituximab (3). For patients with rituximab-refractory Waldenström macroglobulinemia, effective treatment options are limited (2). Experience with ibrutinib in this patient population remains limited.
Ibrutinib is the only once-daily Bruton's tyrosine kinase (BTK) inhibitor approved as either a single agent or in combination with rituximab for patients with Waldenström macroglobulinemia across all lines of therapy and is the only BTK inhibitor approved for the treatment of Waldenström macroglobulinemia (4). Ibrutinib was originally identified as a potential treatment for Waldenström macroglobulinemia based on the high prevalence of MYD88 mutations in patients with Waldenström macroglobulinemia (5). More than 90% of patients carry the MYD88L265P point mutation, and nearly one third of these patients also have a frameshift or nonsense mutation in CXCR4 (6). Mutations in MYD88 and CXCR4 are associated with important differences in disease presentation, including serum IgM levels and symptoms at diagnosis (5). Mutations in both CXCR4 and MYD88 are associated with constitutive activation of the BTK pathway (7), making BTK inhibitors an attractive therapeutic option (5). Results from a pivotal phase II study demonstrated high response rates with single-agent ibrutinib in patients with relapsed Waldenström macroglobulinemia (8).
In the phase III randomized iNNOVATE study (PCYC-1127; NCT02165397), ibrutinib demonstrated superior progression-free survival (PFS) in combination with rituximab versus single-agent rituximab in patients with Waldenström macroglobulinemia (9). In the open-label substudy of iNNOVATE for patients with heavily pretreated, rituximab-refractory Waldenström macroglobulinemia, single-agent ibrutinib demonstrated sustained efficacy and was well tolerated (10, 11). With a median follow-up of 18 months, the estimated 18-month PFS rate per investigator assessment was 86%, and the overall survival (OS) rate was 97% (10). Median PFS was still not reached after a median of 39 months of follow-up in a subsequent analysis (11).
Here we present results from the final analysis of the iNNOVATE substudy of single-agent ibrutinib in patients with rituximab-refractory Waldenström macroglobulinemia, representing up to 5 years of follow-up, an additional 40 months since the first report.
Patients and Methods
Study design
The iNNOVATE substudy was an international, multicenter, single-arm, open-label trial designed to assess the efficacy and safety of single-agent ibrutinib in patients with rituximab-refractory Waldenström macroglobulinemia. Study design details have been previously published (10). In brief, eligible patients had centrally-confirmed Waldenström macroglobulinemia defined per criteria from the Second International Workshop on Waldenström macroglobulinemia (IWWM) and had failed to achieve at least a minor response to their last rituximab-containing therapy or relapsed less than 12 months after their last rituximab-containing therapy. Patients received once-daily ibrutinib 420 mg until unacceptable toxicity or progressive disease (PD). iNNOVATE was approved by institutional review boards or independent ethics committees at each institution and was conducted according to the principles of the Declaration of Helsinki and the Good Clinical Practice guidelines from the International Conference on Harmonization. All patients provided written informed consent prior to enrollment.
Endpoints and assessments
The primary endpoint was PFS per independent review committee (IRC). Other endpoints included response per IRC, hematologic improvement as measured by change in hemoglobin, OS, safety, and patient-reported outcomes (10). Change in serum IgM was also assessed.
Efficacy, safety, and response assessments were conducted as described previously (10). Briefly, responses were assessed according to the modified consensus criteria from the sixth IWWM. The proportion of patients with an overall response was defined as the proportion of patients who achieved a minor response (MR; ≥25% but <50% reduction of serum IgM from baseline) or better. Major response was defined as a partial response (PR; ≥50% reduction of serum IgM concentrations from baseline) or better.
Adverse events (AE), regardless of attribution, were collected by investigators throughout the study and graded per National Cancer Institute Common Terminology Criteria for Adverse Events v4.03.
Genotype analysis
Bone marrow aspirates were collected to assess MYD88 and CXCR4 mutational status using the Personalis ACE Extended Cancer Panel with >500X mean coverage depth. Calls of somatic variants for MYD88 and CXCR4 used the Personalis Cancer Panel DNA pipeline operating in the tumor-only mode with no matched normal samples.
Patient-reported outcomes
Patient-reported outcomes and disease-related symptoms were measured according to the Functional Assessment of Cancer Therapy-Anemia (FACT-An) and the EuroQoL 5-Dimension Questionnaire (EQ-5D-5L©; copyright of EuroQol Research Foundation; EQ-5D is a trademark of the EuroQol Research Foundation), as previously described (10). A clinically meaningful improvement in FACT-An was defined as an increase of ≥7 points in the total score or ≥6 points in the anemia-subscale score. A clinically meaningful improvement in 5Q-5D-5L was defined as an increase of ≥7 points in the visual analog scale (VAS) or ≥0.08 points in the utility score.
Statistical analysis
The open-label substudy provided a descriptive analysis of the efficacy, safety, and patient-reported outcomes of single-agent ibrutinib for the treatment of patients with rituximab-refractory Waldenström macroglobulinemia and was not designed to provide statistical comparisons. Analyses included all patients who received at least one dose of ibrutinib.
Kaplan–Meier estimates of PFS, OS, time to next treatment, and duration of response (DOR) were calculated. For overall response rate (ORR; MR or better) and major response rate (PR or better), 95% confidence intervals (CI) were provided. Sustained hematologic improvement was defined as improvement in hemoglobin of ≥2.0 g/dL (intent-to-treat population) or an increase to >11.0 g/dL with improvement of ≥0.5 g/dL (patients with baseline hemoglobin ≤11.0 g/dL) for ≥56 days. DOR was defined as the duration from the date of initial documentation of response to the date of first documented evidence of PD or death for responders.
Results
Patient demographics and characteristics
Thirty-one patients were enrolled; their detailed baseline characteristics were previously reported (10). Patients had received a median 4 prior lines of systemic therapy (range: 1–7), and more than one third of patients (39%) had at least 5 prior treatments (Table 1). Median baseline hemoglobin was 10.3 g/dL (range: 6.4–14.6), and 68% of patients had hemoglobin ≤11.0 g/dL. Mutational data were available for 25 of 31 patients (81%); 6 were missing due to lack of tumor sample or low tumor yield. Of those with available mutational data, 17 patients (68%) had the MYD88L265P/CXCR4WT genotype, 7 (28%) had the MYD88L265P/CXCR4WHIM genotype, and 1 (3%) had the MYD88WT/CXCR4WT genotype.
Table 1.
Patient demographics and baseline characteristics.
| Characteristics | Patients, N = 31 |
|---|---|
| Median age, years (range) | 67 (47–90) |
| IPSSWM, n (%) | |
| Low | 7 (23) |
| Intermediate | 11 (35) |
| High | 13 (42) |
| Median Hgb, g/dL (range) | 10.3 (6.4–14.6) |
| Baseline Hgb ≤11.0 g/dL, n (%) | 21 (68) |
| Median serum IgM, g/L (range) | 39 (9–107) |
| Median bone marrow involvement, % | |
| Cellularity | 75 |
| Intertrabecular space | 53 |
| Median prior therapies, n (range) | 4 (1–7) |
| Number of prior systemic therapies, n (%) | |
| 1–2 | 9 (29) |
| 3–4 | 10 (32) |
| ≥5 | 12 (39) |
| Prior autologous stem-cell transplant, n (%) | 2 (6) |
| Types of prior therapy, n (%) | |
| Rituximab | 31 (100) |
| Alkylating agent | 28 (90) |
| Corticosteroids | 27 (87) |
| Purine analog | 16 (52) |
| Vinca alkaloids | 15 (48) |
| Proteasome inhibitor | 14 (45) |
| Anthracyclines | 12 (39) |
| Immunomodulating agent | 3 (10) |
| Nucleoside analogue (cytarabine) | 2 (6) |
| Other | 8 (26) |
| Genotype, n (%) | |
| MYD88L265P/CXCR4WT | 17 (68) |
| MYD88L265P/CXCR4WHIM | 7 (28) |
| MYD88WT/CXCR4WT | 1 (4) |
| Unknown | 6a |
| Most common concomitant medications, n (%) | |
| Antibacterials | 26 (84) |
| Analgesics | 17 (55) |
| Antivirals | 16 (52) |
| Anti-inflammatory and antirheumatic agents | 14 (45) |
| Medications for acid-related disorders | 13 (42) |
| Antithrombotic agents | 12 (39) |
| Anti-anemic preparations | 12 (39) |
| Vitamins | 11 (36) |
| Ophthalmologicals | 10 (32) |
| Most common reasons for treatment initiation, n (%) | |
| Fatigue | 22 (71) |
| Constitutional symptoms | 13 (42) |
| Hgb ≤10 g/dL | 13 (42) |
| Lymphadenopathy | 7 (23) |
Abbreviations: Hgb, hemoglobin; IPSSWM, International Prognostic Scoring System for Waldenström Macroglobulinemia.
aGenotype data available for 25 patients; percentages were calculated using 25 as the denominator.
Patient disposition
The final analysis was performed at the time of study closure, and the median follow-up was 58 months (range: 9–61). Treatment discontinuations during the study were due to PD (n = 13; 42%), AEs (n = 2; 6%), and withdrawal by patient (n = 2; 6%). Median duration of treatment was 41 months (range: 0.3–61). At study closure, 14 patients (45%) were still receiving ibrutinib: 8 patients opted to enroll in a treatment extension study and 6 continued to receive commercial ibrutinib. Twelve patients (39%) in total received subsequent treatment; subsequent treatments included alkylating agent (n = 7), anti-CD20 antibody therapy (n = 6), corticosteroids (n = 6), proteasome inhibitor (n = 3), vinca alkaloids (n = 2), anthracyclines (n = 1), immunomodulator (n = 1), nucleoside analog (n = 1), and other (n = 4; including 2 patients who received ibrutinib). Median time to next treatment was not reached [95% CI: 42–NE (not evaluable)].
PFS
At the time of the final analysis, median PFS was 39 months (95% CI: 25–NE); 18 patients had PD or died, and the estimated 60-month PFS rate was 40% (Fig. 1). Median PFS was not reached (95% CI: 27–NE) in patients with the MYD88L265P/CXCR4WT genotype (n = 17) and was 18 months (95% CI: 3–28) in patients with the MYD88L265P/CXCR4WHIM genotype (n = 7). The single patient with the MYD88WT/CXCR4WT genotype progressed at 6 months.
Figure 1.

PFS as assessed by IRC in all patients and by genetic subtype. The single patient with MYD88WT/CXCR4WT genotype is not shown; this patient progressed at 5.6 months.
Response
At the final analysis, the ORR per IRC was 87%; 29% of patients (n = 9) achieved a best response of very good partial response (VGPR), 48% (n = 15) achieved a best response of PR, and 10% (n = 3) had a best response of MR. Three patients (10%) had stable disease, and 1 patient was not evaluable due to a lack of follow-up assessments after week 1. Over time, a deepening of response was observed with major response rates increasing from 61% at 6 months to 71% at 18 months and 77% at 60 months (Fig. 2A). The median time to overall response (MR or better) was 1 month (range: 1–20), and the median time to major response (PR or better) was 2 months (range 1–52). In patients who achieved a response, the median DOR per IRC was 33 months (range: 2–60+).
Figure 2.
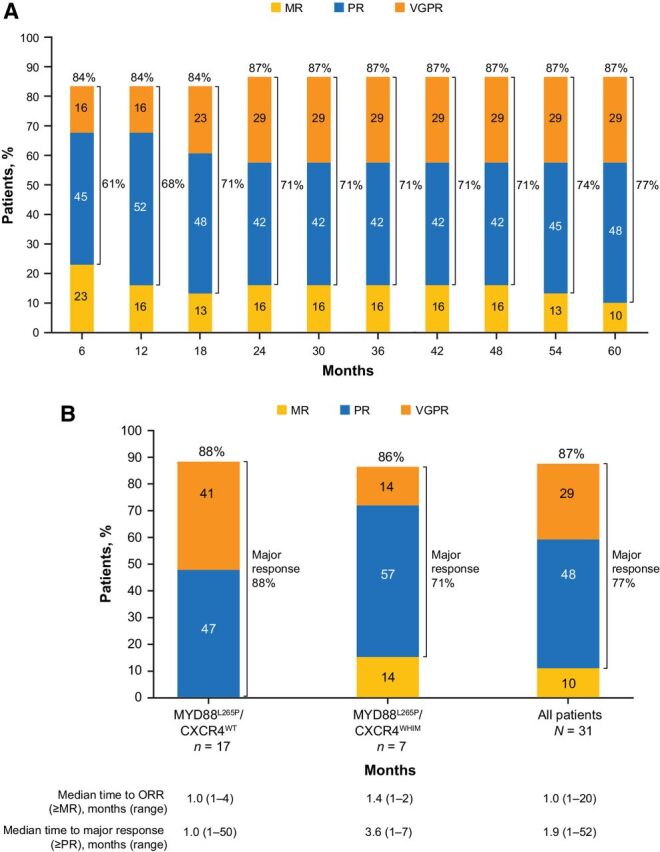
ORR. A, Cumulative best response over time in all patients. ORRs are shown at the top of each bar, and major response rates are shown next to brackets for each bar. B, Best overall response per IRC in all patients and by genetic subtype. The single patient with MYD88WT/CXCR4WT genotype is not shown; this patient had a best response of stable disease. MR, minor response; ORR, overall response rate; PR, partial response; VGPR, very good partial response.
ORRs were similar between the MYD88L265P/CXCR4WT (88%) and MYD88L265P/CXCR4WHIM (86%) genetic subtypes (Fig. 2B). However, a larger proportion of patients with the MYD88L265P/CXCR4WT genotype achieved a VGPR (41% vs. 14%). The 1 patient with MYD88WT/CXCR4WT genotype had a best response of stable disease. Median time to MR or better was 1 month (range: 1–4) in patients with the MYD88 L265P/CXCR4WT genotype and 1 month (range: 1–2) in those with the MYD88L265P/CXCR4WHIM genotype. Median time to major response (PR or better) was 1 month (range: 1–50) and 4 months (range: 1–7) in patients with MYD88L265P/CXCR4WT and MYD88L265P/CXCR4WHIM genotypes, respectively.
Hemoglobin and IgM levels
At baseline, median hemoglobin was 10.3 g/dL (range: 6.4–14.6). Improvements in hemoglobin were generally rapid (median +1.7 g/dL increase from baseline to week 9) and reached a maximum median change of +3.5 g/dL at 35 months (Fig. 3). In patients who had baseline hemoglobin levels ≤11.0 g/dL, the maximum median increase from baseline was +4.3 g/dL (range: 1.2–8.9) at month 50. In total, 22 of 31 patients (71%) exhibited sustained hemoglobin improvement, including 17 of 21 patients (81%) who had baseline hemoglobin levels ≤11.0 g/dL.
Figure 3.

Median IgM and Hgb levels over time.
Likewise, improvements in IgM were generally rapid and sustained. Median serum IgM was 39 g/L (range: 9–107) at baseline. The median change reached −20 g/L at week 9, and reached the maximum −37 g/L at 54 months (Fig. 3).
OS
Median OS was not reached (95% CI: NE–NE), and the 60-month OS rate was 73% (Fig. 4). Median OS was not reached for all subgroups evaluated by number of prior lines of therapy (Fig. 4). When analyzed by genotype, median OS was not reached (95% CI: NE–NE) in patients with the MYD88L265P/CXCR4WT genotype (n = 17) and was 50 months (95% CI: 11–NE) in patients with the MYD88L265P/CXCR4WHIM genotype (n = 7). The single patient with the MYD88WT/CXCR4WT genotype died at 9 months.
Figure 4.

OS in all patients and by lines of prior therapy.
Safety
Patients had received single-agent ibrutinib for a median of 41 months (range: 0.3–61). Overall, 30 patients (97%) experienced a treatment-emergent AE (TEAE), and 25 patients (81%) had a grade ≥3 TEAE. The prevalence of TEAEs of clinical interest of any grade (occurring in >2 patients) during the last 2 years of treatment were infections (years 3–4: 47%, 8/17; years 4–5: 50%, 7/14), hypertension (years 3–4: 29%, 5/17; years 4–5: 36%, 5/14), and diarrhea (years 3–4: 24%, 4/17; years 4–5: 7%, 1/14). The prevalence of grade ≥3 AEs of clinical interest generally declined over time with ibrutinib treatment (Fig. 5). Notably, no patients experienced major hemorrhage or atrial fibrillation. Of 3 patients with grade ≥3 hypertension during the study, all had hypertension at baseline. Grade ≥3 neutropenia and grade ≥3 thrombocytopenia primarily occurred during the first year of treatment, with 1 patient each experiencing these AEs in subsequent years. Five patients experienced a total of 7 AEs requiring dose reduction (diarrhea, n = 2; arthralgia, n = 1; maculopapular rash, n = 1; nausea, n = 1; constipation, n = 1; and fecalith, n = 1); all 7 AEs resolved following dose reduction, and 1 patient was able to resume treatment at the full ibrutinib dose. Two patients experienced a TEAE leading to discontinuation of ibrutinib (grade 2 diarrhea and grade 3 constipation; n = 1 each). No deaths occurred due to TEAEs.
Figure 5.

Prevalence of grade ≥3 AEs of clinical interest by yearly interval. aCombined terms.
Patient-reported outcomes
Patients reported clinically meaningful improvements in quality-of-life measures. FACT-An score, anemia-subscale score, and EQ-5D-5L VAS improved from baseline. At the time of final analysis, most patients had a clinically meaningful improvement in total FACT-An score (77%; n = 24) and anemia-subscale score (84%; n = 26; Supplementary Fig. S1). Clinically meaningful improvements in EQ-5D-5L VAS and utility score were also reported by 71% of patients (n = 22) each.
Discussion
In this final analysis of the open-label substudy of the phase III iNNOVATE trial, once-daily, single-agent ibrutinib demonstrated sustained efficacy in patients with heavily pretreated, rituximab-refractory Waldenström macroglobulinemia. With a median follow-up of 58 months, representing approximately 40 months of additional follow-up since the initial analysis, single-agent ibrutinib continued to demonstrate durable clinical benefit. Response deepened over time, driven by an increase in VGPR from 16% at month 6 to 23% at month 18 and 29% at month 60. Additionally, responses were durable with a median DOR of 33 months. After a median 58 months of follow-up and regardless of the number of prior therapies, median OS was not reached. Median hemoglobin increased and serum IgM levels decreased rapidly and were sustained with treatment. Correspondingly, clinically meaningful improvements in patient-reported outcomes were reported by most patients and were sustained over time.
The long-term results of our study suggest that single-agent ibrutinib offers durable clinical benefit for patients with rituximab-refractory Waldenström macroglobulinemia, a population with few effective treatment options. Other recent trials in previously treated patients with Waldenström macroglobulinemia have reported similar results to those reported here. In a phase II study by Treon and colleagues of 63 patients with relapsed Waldenström macroglobulinemia treated with single-agent ibrutinib, the median 5-year PFS rate was not reached and the 5-year OS rate was 87% (12). Overall and major response rates were 90.5% and 79.4%, respectively, after a median follow-up of 59 months (12); this is highly concordant with the 87% overall and 77% major response rates reported here after a median follow-up of 60 months. Consistent with previous observations that patients with MYD88 wild-type Waldenström macroglobulinemia have shorter survival and a lower probability of response than those with a MYD88 mutation (13) and that mutations in CXCR4 are associated with a lower likelihood of achieving a major response compared with patients with CXCR4 wild-type disease (6), results from Treon and colleagues showed that efficacy responses were genotype dependent. In patients with MYD88 wild-type Waldenström macroglobulinemia, the median PFS was 0.4 years, whereas those with MYD88Mut/CXCR4WT and MYD88Mut/CXCR4Mut genotypes had a median PFS of not reached and 4.5 years, respectively (12).
In addition to studies with ibrutinib, another BTK inhibitor, zanubrutinib, has been evaluated as a treatment for Waldenström macroglobulinemia. In the phase III ASPEN study investigating ibrutinib versus zanubrutinib in both previously untreated and previously treated patients with MYD88L265P Waldenström macroglobulinemia, the primary endpoint of superiority of zanubrutinib was not met; 84% of patients treated with ibrutinib and 85% treated with zanubrutinib were progression-free at 18 months, and major response rates were 78% (ibrutinib) and 77% (zanubrutinib; ref. 14). In the ASPEN single-arm cohort conducted in patients with the MYD88 wild-type genotype, estimated 18-month PFS and OS rates were 68% and 88%, respectively, after treatment with zanubrutinib (median PFS and OS were not reached with a median follow-up of 17.9 months; ref. 15). Interestingly, in an earlier report (median 19 months on treatment) of the above study by Treon and colleagues, the ORR in patients with MYD88 wild-type genotype was 71% (8), which is similar to the rate reported in the ASPEN study (81%) at 17.9 months of follow-up (15), suggesting that longer follow-up may be needed to clearly observe the impact of genotype on survival benefit.
The impact of genotype status in the current analysis should be interpreted with caution due to small sample sizes of the subgroups, particularly those with the MYD88L265P/CXCR4WHIM (n = 7) and MYD88WT/CXCR4WT (n = 1) genotypes. Additionally, indirect comparisons between these studies and our own may be further confounded by differences in patient characteristics and study design. Nonetheless, we did observe lower VGPR rates and shorter median PFS and OS in the subset of patients with the MYD88L265P/CXCR4WHIM genotype compared with those with the MYD88L265P/CXCR4WT genotype. In contrast to these findings, the double-blind randomized portion of this study in which patients with non–rituximab-refractory Waldenström macroglobulinemia received ibrutinib or placebo in combination with rituximab demonstrated sustained clinical benefit with ibrutinib–rituximab among all genotypes; patients treated with ibrutinib–rituximab had a PFS benefit over those treated with placebo–rituximab, regardless of genotype [HR (95% CI): MYD88L265P/CXCR4WT, 0.18 (0.08–0.43); MYD88L265P/CXCR4WHIM, 0.27 (0.12–0.62); MYD88WT/CXCR4WT, 0.29 (0.07–1.19); ref. 16].
Ibrutinib was well tolerated, and no new safety signals emerged over more than 5 years of follow-up. Consistent with observations from other studies, AEs were generally most common during the first year of ibrutinib treatment and decreased over time (17–20). No major hemorrhage or atrial fibrillation events occurred during the entire study. Most AEs were low grade, and few patients (n = 2) discontinued treatment due to an AE. Notably, grade ≥3 neutropenia and thrombocytopenia were observed in a small number of patients, primarily during the first year of treatment. This contrasts with a previous report that neutropenia and thrombocytopenia were more common in heavily pretreated patients (12). All AEs that led to an ibrutinib dose reduction resolved following dose reduction, suggesting that many AEs can be managed effectively with dose modification.
In conclusion, single-agent ibrutinib remains an effective treatment for patients with heavily pretreated, rituximab-refractory Waldenström macroglobulinemia. This regimen provides sustained efficacy, is well tolerated, and provides clinically meaningful improvements in quality of life in a population of patients who have few effective treatment options.
Data sharing statement
Requests for access to individual participant data from clinical studies conducted by Pharmacyclics LLC, an AbbVie Company, can be submitted through Yale Open Data Access (YODA) Project site at http://yoda.yale.edu.
Authors' Disclosures
J. Trotman reports grants from Roche, Beigene, Janssen, Pharmacyclics LLC, an AbbVie Company, Celgene, and Takeda outside the submitted work. C. Buske reports personal fees from Beigene, Pfizer, Novartis, Gilead, Regeneron, and X4; grants and personal fees from Roche, Janssen, Celltrion, and Bayer; grants from Amgen and MSD; and personal fees from Morphosys outside the submitted work. A. Tedeschi reports other support from Janssen, AstraZeneca, AbbVie, and Beigene outside the submitted work. J.V. Matous reports other support from Pharmacyclics outside the submitted work. D. MacDonald reports personal fees from Roche, AbbVie, Janssen, AstraZeneca, Gilead, and Servier outside the submitted work. C.S. Tam reports personal fees and other support from Janssen during the conduct of the study, as well as personal fees and other support from AbbVie outside the submitted work. O. Tournilhac reports personal fees from Janssen outside the submitted work. S. Ma reports grants and personal fees from AbbVie, AstraZeneca, Beigene, Janssen, and Pharmacyclics LLC, an AbbVie Company; grants from Loxo, Juno, and TG Therapeutics; and personal fees from Genentech during the conduct of the study. S. Ma also reports personal fees from Verastem outside the submitted work. S.P. Treon reports grants, personal fees, and other support from Pharmacyclics LLC, an AbbVie Company; personal fees from Janssen; grants and personal fees from Beigene and BMS; and grants from Eli Lilly during the conduct of the study. A. Oriol reports personal fees from BMS/Celgene, Sanofi, Amgen, GSK, and Oncopeptides outside the submitted work. I. Arango-Hisijara reports other support from Pharmacyclics LLC, an AbbVie Company during the conduct of the study, as well as other support from Bristol Myers Squibb and AbbVie outside the submitted work. M.A. Dimopoulos reports personal fees from Amgen, Takeda, Beigene, BMS, and Janssen outside the submitted work. No disclosures were reported by the other authors.
Supplementary Material
Supplemental Figure S1
Acknowledgments
This study was funded by Pharmacyclics LLC, an AbbVie Company.
We thank all the patients who participated in this trial and their families and Cindi Hoover, PhD, of ApotheCom for medical writing supported by Pharmacyclics LLC, an AbbVie Company. We also thank Bernhard Hauns, MD, PhD, for his contributions to the study and analyses.
Pharmacyclics LLC, an AbbVie Company, sponsored and designed the study. Study investigators and their research teams collected the data. The sponsor confirmed data accuracy and performed analysis of the data.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
Note: Supplementary data for this article are available at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/).
Authors' Contributions
J. Trotman: Conceptualization, data curation, investigation, writing–original draft, writing–review and editing. C. Buske: Data curation, investigation, writing–review and editing. A. Tedeschi: Data curation, investigation, writing–review and editing. J.V. Matous: Data curation, investigation, writing–review and editing. D. MacDonald: Data curation, investigation, writing–review and editing. C.S. Tam: Data curation, investigation, writing–review and editing. O. Tournilhac: Data curation, investigation, writing–review and editing. S. Ma: Data curation, investigation, writing–review and editing. S.P. Treon: Data curation, investigation, writing–review and editing. A. Oriol: Data curation, investigation, writing–review and editing. J. Ping: Data curation, formal analysis, validation, writing–review and editing. E.M. Briso: Data curation, writing–review and editing. I. Arango-Hisijara: Writing-review and editing. M.A. Dimopoulos: Data curation, investigation, writing–review and editing.
References
- 1. Dimopoulos MA, Panayiotidis P, Moulopoulos LA, Sfikakis P, Dalakas M. Waldenström's macroglobulinemia: clinical features, complications, and management. J Clin Oncol 2000;18:214–26. [DOI] [PubMed] [Google Scholar]
- 2. Castillo JJ, Advani RH, Branagan AR, Buske C, Dimopoulos MA, D'Sa S, et al. Consensus treatment recommendations from the tenth International Workshop for Waldenström Macroglobulinaemia. Lancet Haematol 2020;7:e827–e37. [DOI] [PubMed] [Google Scholar]
- 3. Gavriatopoulou M, Ntanasis-Stathopoulos I, Kastritis E, Dimopoulos MA. How I treat rituximab refractory patients with WM. Oncotarget 2018;9:36824–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pharmacyclics LLC, an AbbVie Company. IMBRUVICA (ibrutinib) Prescribing Information. Sunnyvale, CA: Pharmacyclics LLC, an AbbVie Company; 2020. [Google Scholar]
- 5. Treon SP, Cao Y, Xu L, Yang G, Liu X, Hunter ZR. Somatic mutations in MYD88 and CXCR4 are determinants of clinical presentation and overall survival in Waldenstrom macroglobulinemia. Blood 2014;123:2791–6. [DOI] [PubMed] [Google Scholar]
- 6. Castillo JJ, Xu L, Gustine JN, Keezer A, Meid K, Dubeau TE, et al. CXCR4 mutation subtypes impact response and survival outcomes in patients with Waldenström macroglobulinaemia treated with ibrutinib. B J Haematol 2019;187:356–63. [DOI] [PubMed] [Google Scholar]
- 7. Yang G, Zhou Y, Liu X, Xu L, Cao Y, Manning RJ, et al. A mutation in MYD88 (L265P) supports the survival of lymphoplasmacytic cells by activation of Bruton tyrosine kinase in Waldenström macroglobulinemia. Blood 2013;122:1222–32. [DOI] [PubMed] [Google Scholar]
- 8. Treon SP, Tripsas CK, Meid K, Warren D, Varma G, Green R, et al. Ibrutinib in previously treated Waldenstrom's macroglobulinemia. N Engl J Med 2015;372:1430–40. [DOI] [PubMed] [Google Scholar]
- 9. Dimopoulos MA, Tedeschi A, Trotman J, Garcia-Sanz R, Macdonald D, Leblond V, et al. Phase 3 trial of ibrutinib plus rituximab in Waldenstrom's macroglobulinemia. N Engl J Med 2018;378:2399–410. [DOI] [PubMed] [Google Scholar]
- 10. Dimopoulos MA, Trotman J, Tedeschi A, Matous JV, Macdonald D, Tam C, et al. Ibrutinib for patients with rituximab-refractory Waldenstrom's macroglobulinaemia (iNNOVATE): an open-label substudy of an international, multicentre, phase 3 trial. Lancet Oncol 2017;18:241–50. [DOI] [PubMed] [Google Scholar]
- 11. Buske C, Tedeschi A, Trotman J, Garcia-Sanz R, MacDonald D, Leblond V, et al. Ibrutinib treatment in Waldenström's macroglobulinemia: follow-up efficacy and safety from the iNNOVATE study. In: Proceedings of the 60th American Society of Hematology (ASH) Annual Meeting and Exposition; 2018. Dec 1–4; San Diego, CA. [Google Scholar]
- 12. Treon SP, Meid K, Gustine J, Yang G, Xu L, Liu X, et al. Long-term follow-up of ibrutinib monotherapy in symptomatic, previously treated patients with Waldenström macroglobulinemia. J Clin Oncol 2020;39:565–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Treon SP, Xu L, Hunter Z. MYD88 mutations and response to ibrutinib in Waldenström's macroglobulinemia. N Engl J Med 2015;373:584–6. [DOI] [PubMed] [Google Scholar]
- 14. Tam CS, Opat S, D'Sa S, Jurczak W, Lee HP, Cull G, et al. A randomized phase 3 trial of zanubrutinib vs ibrutinib in symptomatic Waldenström macroglobulinemia: the ASPEN study. Blood 2020;136:2038–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dimopoulos M, Sanz RG, Lee HP, Trneny M, Varettoni M, Opat S, et al. Zanubrutinib for the treatment of MYD88 wild-type Waldenström macroglobulinemia: a substudy of the phase 3 ASPEN trial. Blood Adv 2020;4:6009–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Buske C, Tedeschi A, Trotman J, García-Sanz R, MacDonald D, Leblond V, et al. Five-year follow-up of ibrutinib plus rituximab vs placebo plus rituximab for Waldenstrom's macroglobulinemia: final analysis from the randomized phase 3 iNNOVATE™ Study. Blood 2020;136(Suppl 1):S24–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Burger JA, Barr PM, Robak T, Owen C, Ghia P, Tedeschi A, et al. Long-term efficacy and safety of first-line ibrutinib treatment for patients with CLL/SLL: 5 years of follow-up from the phase 3 RESONATE-2 study. Leukemia 2020;34:787–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Coutre SE, Byrd JC, Hillmen P, Barrientos JC, Barr PM, Devereux S, et al. Long-term safety of single-agent ibrutinib in patients with chronic lymphocytic leukemia in 3 pivotal studies. Blood Adv 2019;3:1799–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. O'Brien S, Furman RR, Coutre S, Flinn IW, Burger JA, Blum K, et al. Single-agent ibrutinib in treatment-naive and relapsed/refractory chronic lymphocytic leukemia: a 5-year experience. Blood 2018;131:1910–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gustine JN, Meid K, Dubeau T, Severns P, Hunter ZR, Guang Y, et al. Ibrutinib discontinuation in Waldenstrom macroglobulinemia: etiologies, outcomes, and IgM rebound. Am J Hematol 2018;93:511–7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure S1