

Abstract
Purpose:
Although patients with unresectable or metastatic melanoma can experience long-term survival with BRAF- and MEK-targeted agents or immune checkpoint inhibitors over 5 years, resistance develops in most patients. There is a distinct lack of pretherapeutic biomarkers to identify which patients are likely to benefit from each therapy type. Most research has focused on the predictive role of T cells in antitumor responses as opposed to B cells.
Patients and Methods:
We conducted prespecified exploratory biomarker analysis using gene expression profiling and digital pathology in 146 patients with previously untreated BRAF V600–mutant metastatic melanoma from the randomized, phase III COMBI-v trial and treated with dabrafenib plus trametinib who had available tumor specimens from screening.
Results:
Baseline cell-cycle gene expression signature was associated with progression-free survival (P = 0.007). Patients with high T-cell/low B-cell gene signatures had improved median overall survival (not reached [95% confidence interval (CI), 33.8 months–not reached]) compared with patients with high T-cell/high B-cell signatures (19.1 months; 95% CI, 13.4–38.6 months). Patients with high B-cell signatures had high B-cell infiltration into the tumor compartment, corresponding with decreased MAPK activity and increased expression of immunosuppressive markers.
Conclusions:
B cells may serve as a potential biomarker to predict clinical outcome in patients with advanced melanoma treated with dabrafenib plus trametinib. As separate studies have shown an opposite effect for B-cell levels and response to immunotherapy, B cells may serve as a potential biomarker to facilitate treatment selection. Further validation in a larger patient cohort is needed.
Translational Relevance.
Biomarkers are needed to identify patients who benefit most from targeted or immune checkpoint inhibitor (ICI) therapy in metastatic melanoma. We analyzed biomarkers using pretreatment melanoma samples from the dabrafenib plus trametinib arm of the randomized, phase III COMBI-v trial. To our knowledge, this is the first large-scale analysis of gene expression signatures and tumor cell and T- and B-cell interactions by digital pathology in patients treated with targeted therapy. Distinct T-cell/B-cell signatures at baseline demonstrated prognostic value, as high T-cell/low B-cell signatures associated with prolonged survival versus high T-cell/high B-cell signatures. Patients with high B-cell signatures had high tumor B-cell infiltration associated with a modified tumor cell phenotype. Recent findings show high T-cell/high B-cell signatures at screening correlated with improved response to ICIs versus low B-cell signature. B cells may be potential biomarkers to identify patients who may achieve improved clinical benefit.
Introduction
Pivotal phase III trials, including COMBI-v, evaluating targeted or immune checkpoint inhibitor (ICI) therapy have demonstrated long-term survival in patients with advanced melanoma. However, therapeutic resistance occurs regardless of therapy type, with ≥50% of patients eventually dying of the disease within 5 years (1–3). Although several resistance mechanisms acquired after targeted therapy have been identified, including reactivation of the MAPK pathway, significant heterogeneity exists between patients and within tumors. Furthermore, the association of individual resistance mechanisms with clinical response has yet to be determined (4). Regarding ICIs, features such as insufficient antigen presentation and T-cell activation may contribute to primary resistance; however, the underlying biology and, therefore, effective therapies to overcome these mechanisms remain undefined (5). Predictive clinical and biological characteristics are needed to identify patients who will benefit most from each treatment type.
Previous studies indicated that immune composition of the tumor microenvironment (TME) affects response (6, 7). Although B cells represent up to 33% of infiltrating immune cells, most studies to date have focused on the importance of tumor-infiltrating T cells (8). In addition, findings have been inconsistent regarding the role of B cells due to their contribution during antitumor responses and tumor heterogeneity, growth, and metastasis (8–10). Recent studies in metastatic melanoma show B cells, in conjunction with T-cell signatures are positive prognostic and predictive markers for clinical response to immunotherapies, including ICIs (11–15). However, similar analyses in patients with metastatic melanoma treated with BRAF and MEK inhibitors have not yet been published.
We report findings from gene expression profiling (GEP) and digital pathology studies in pretreatment tumor samples from the dabrafenib (dab) plus trametinib (tram) arm of COMBI-v to evaluate B cells as a potential biomarker to identify patients at screening who may derive improved clinical benefit from targeted therapy.
Patients and Methods
Study design and participants
COMBI-v (ClinicalTrials.gov: NCT01597908) was an open-label, randomized, phase III trial (N = 704) that included previously untreated patients with unresectable stage IIIC or IV BRAF V600E/K–mutant melanoma (per American Joint Committee on Cancer Staging Manual, 7th edition) treated with dab + tram or vemurafenib monotherapy. Study design details were published previously (16). For comparison, treatment-naive tumor samples from 95 patients with BRAF V600 wild-type melanoma were randomly selected at the Essen Department of Dermatology. The study was approved by the ethical committee of the University Hospital Essen (approval No. 16-7190-BO).
COMBI-v was conducted in accordance with the provisions of the Declaration of Helsinki and Good Clinical Practice guidelines. The protocol was approved by the institutional review board or human research ethics committee at each site. All patients provided written informed consent before participation in the study and collection and analyses of tissue samples.
Tissue processing, NanoString CodeSet design, and expression quantification
Tissue samples were submitted for BRAF testing for COMBI-v screening. If patient consent and tissue sample were available, remnant tissue material was used for exploratory biomarker studies. Biomarker analyses were conducted in two sets of patients included in the dab + tram arm. All biomarker analyses were performed using tumor block samples from COMBI-v biomarker set 1 (n = 79; 76 of 79 with gene expression results); biomarker set 2 (n = 67) was an additional cohort with remnant formalin-fixed, paraffin-embedded (FFPE) slides.
For GEP, literature-derived genes (N = 780) from typical resistance pathways (Supplementary Materials and Methods) were included in a custom NanoString CodeSet named the phenotypic resistance panel. Probe sets for each gene were designed and synthesized at NanoString Technologies. A commercially available NanoString panel (nCounter PanCancer immune profiling panel) was used to analyze 800 genes covering immune TME pathways, aspects of interferon resistance, and immune escape in melanoma. For both panels, NanoString standard chemistry was used. Per panel, 100-ng RNA from FFPE material was analyzed at the MOLBIZ Molecular Biology Centre (a NanoString core facility) at University Hospital Essen, University of Duisburg-Essen, Essen, Germany. Measurements were performed at maximum resolution; fields of view were adjusted to 555.
Because biomarker set 2 consisted of older FFPE slides containing limited material and did not allow for IHC testing, only GEP was performed using the NanoString nCounter PanCancer immune profiling panel.
Digital pathology
A duplex CD3/CD19 IHC assay was developed. CD3 and CD19 were selected as pan–T-cell and B-cell markers to assess both immature and mature cellular patterns. Duplex CD3/CD19 IHC staining (n = 59) using anti-CD3 (clone LN10) and anti-CD19 (clone BT51E; both of Leica Biosystems) was performed on the Leica Bond Rx autostainer. Slides were scanned at 20× magnification using Aperio AT2, Leica digital whole-slide scanner. Digital image analysis (IA) was performed using the multiplex IHC module, HALO v2.3 software platform (Indica Labs). Analysis enabled clear distinction of melanoma tumor nests (or tumor compartment) and intratumoral and peritumoral stroma (or stroma compartment) and excluded lymphoid tissue (Supplementary Fig. S1); melanoma lesions were annotated by a pathologist in collaboration with imaging scientists. IA was performed blinded to clinical information to avoid unintentional bias. A specific IA algorithm was developed to quantitatively assess the percentage of CD3- and CD19-positive cells within melanoma tumor and stroma compartments. To evaluate the extent of direct T-cell/B-cell interactions, proximity analysis between CD3- and CD19-positive cells was performed with the spatial analysis module using a 10-μm radial distance setting to assess close T-cell/B-cell interactions in tumor and stroma classified regions, respectively.
Statistical analysis
Comparison of groups defined by gene expression was performed using a Cox regression model; HRs and 95% confidence intervals (CIs) are presented. Univariate Cox proportional hazards models were fitted to assess or rank the importance of biomarkers on clinical response. Kaplan–Meier nonparametric survival function estimates were fitted and plotted to visualize the survival characteristics of specific subgroups. For continuous biomarker values, we used optimal cut point determination as implemented in the R Maxstat package v0.52.2. P values derived from this method were corrected using a conditional Monte Carlo–based method. The multivariate Cox proportional hazards model was generated using the R Survminer package v0.4.1. The UpSet plot was generated using the R UpSetR package v1.3.3. All analyses were performed using R v3.4.3. The R package rms v5.1.1 was used to analyze baseline characteristics.
Data sharing
Novartis is committed to sharing with qualified external researchers, access to patient-level data, and supporting clinical documents from eligible studies. Requests are reviewed and approved by an independent review panel on the basis of scientific merit. All data provided are anonymized to respect the privacy of patients who have participated in the trial in line with applicable laws and regulations. This trial data availability is according to the criteria and process described on ClinicalStudyDataRequest.com. The BMS-CA209-038 data set is available in the Gene Expression Omnibus (GSE91061; ref. 17). All TCGA data are available through the NCI Genomics Data Commons Data Portal (https://portal.gdc.cancer.gov/).
Results
Patient characteristics and GEP
Between June 4, 2012, and October 7, 2013, patients were randomized within COMBI-v to receive dab + tram (n = 352) or vemurafenib (n = 352; Supplementary Fig. S2; ref. 16). Baseline characteristics for biomarker cohorts were similar to the overall study population (Supplementary Tables S1 and S2). The techniques performed in this study are summarized in an UpSet plot (Supplementary Fig. S3) used to visualize co-occurrences between data sets.
After quality control and filtering (see Supplementary Materials and Methods), NanoString results were available for 76 patients (biomarker set 1). All genes that had a nominal P < 0.05 in the univariate Cox ranking against progression-free survival (PFS; n = 91) were explored further. Unbiased Cox analysis showed cell-cycle genes were among top pathways associated with poor clinical outcomes with dab + tram. Correlation matrices were derived using the publicly available human cutaneous melanoma data set from The Cancer Genome Atlas (TCGA) to identify clusters of genes that were tightly coregulated across large numbers of samples, which helped in sorting genes from Cox analysis into distinct subgroups on a data-driven basis. The cell-cycle–associated gene expression signature (GES) derived from TCGA (8-gene signature: CENPF, BUB1, CCNA2, KIF2C, BIRC5, CCNB1, UBE2C, E2F1; Supplementary Table S3) was significantly associated with PFS (HR, 1.59; 95% CI, 1.09–2.31; P = 0.007); a Kaplan–Meier curve using an optimized cut point is shown in Fig. 1A. Cell-cycle GES was also associated with overall survival (OS; Fig. 1B), although no statistically significant difference was observed. Patients with durable benefit (PFS ≥ 24 months) had a significantly lower level of cell-cycle–associated GES at baseline than patients with shorter PFS (P = 0.008; Fig. 1C).
Figure 1.

PFS, OS, and durable response for 8-gene cell-cycle signature. Panels show (A) PFS, (B) OS, and (C) durability of responses in patients who received dabrafenib plus trametinib in the COMBI-v trial by expression level of the 8-gene cell-cycle–associated signature (n = 76). Durable response was defined as PFS of ≥24 months. An optimized cutoff was used for Kaplan–Meier PFS analysis and then applied for OS analysis. A box plot shows medians with interquartile range. P values were determined by log-rank test (PFS and OS) and two-sided Wilcoxon signed-rank test (durable response). condMC, conditional Monte Carlo.
Immune markers were among top candidates in Cox analysis associated with improved PFS; predefined immune GES, for example, T-cell–inflamed or IFNγ signatures (18), showed a nonsignificant association with clinical outcome (data not shown). T-cell–inflamed GES did not show strong prognostic or predictive features, indicating other immune markers, for example, those related to the TME and alternative immune-cell populations, may be more important in understanding clinical outcome with BRAF and MEK inhibition.
Transcriptional B-cell profile
We used an in-house GES (Supplementary Table S3) to evaluate the association of the transcriptional B-cell profile with clinical survival outcomes. High B-cell GES strongly associated with improved OS and contained independent prognostic information compared with T-cell expression in the TCGA melanoma data set (Supplementary Figs. S4A and S4B) and in patients with melanoma treated with nivolumab [BMS-CA209-038 data set (17); Supplementary Figs. S4C and S4D].
When we analyzed the transcriptional immune profiles of COMBI-v biomarker data sets 1 and 2, almost all GES of specific immune-cell subpopulations (e.g., T cells, B cells, macrophages) significantly correlated with each other; therefore, most immune GES were associated with positive clinical outcomes. Importantly, we found B cells to be among top candidate pathways associated with poor OS in univariate Cox analysis using our B-cell signature. To explore this association further, we combined COMBI-v biomarker data sets 1 (n = 76) and 2 (n = 67) after removing the batch effect (see Supplementary Materials and Methods). Multivariate analysis confirmed that batch effects did not impact findings (data not shown). In accordance with previous observations in different cancer types (7, 11, 19), we found a positive correlation between T- and B-cell GES across COMBI-v samples (Fig. 2A) and in a control cohort of BRAF V600 wild-type melanoma samples (Supplementary Tables S4 and S5) comprising NRAS-mutant and NRAS wild-type tumors (Supplementary Fig. S5).
Figure 2.
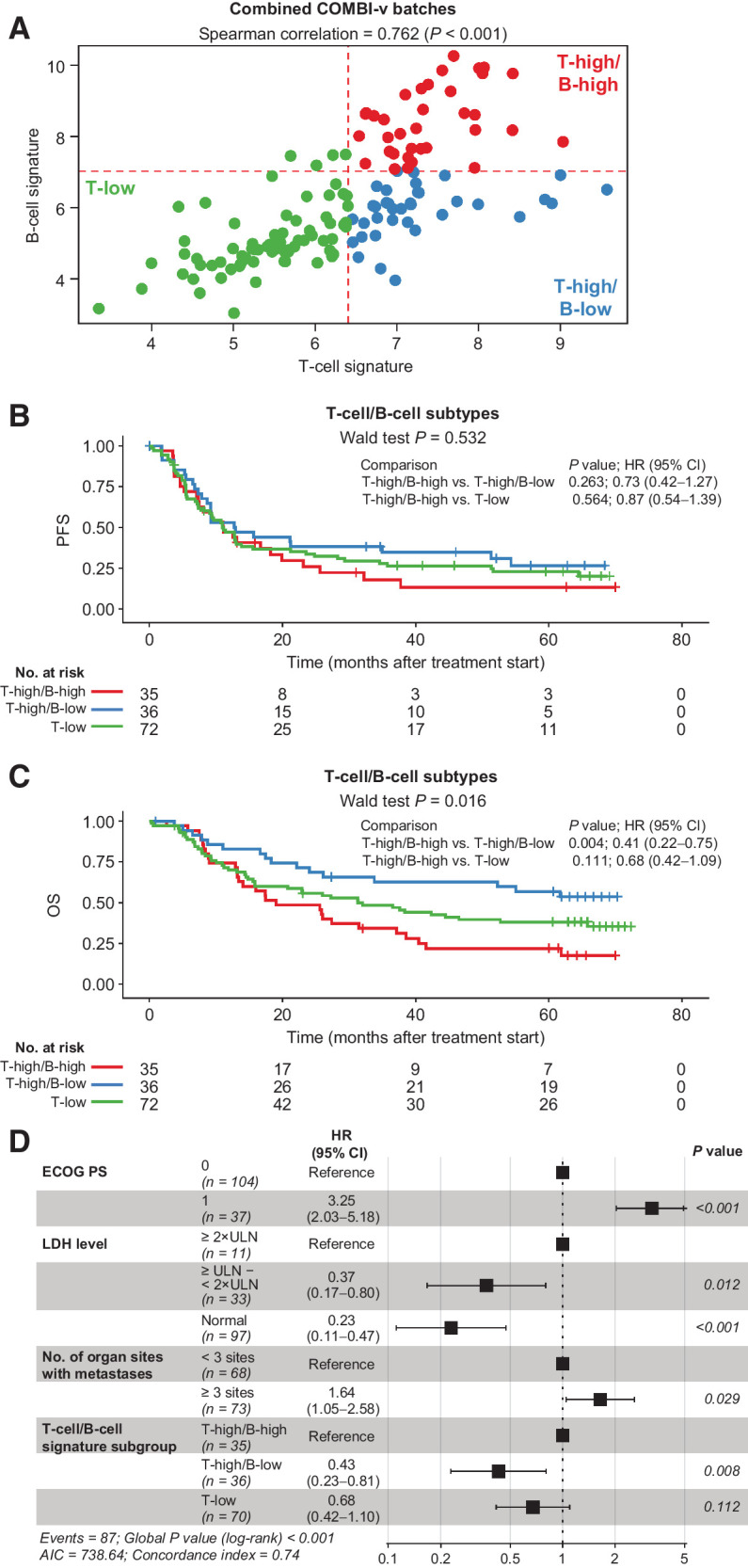
T- and B-cell GESs across COMBI-v tumor samples and survival outcomes. A, Patient tumor samples (n = 143) characterized by level of T-cell/B-cell signature and (B) PFS (P value was determined by Wald test) and (C) OS by T- and B-cell signature subgroup (P value was determined by Wald test). D, Multivariate analysis of baseline factors associated with OS in patients from COMBI-v (n = 141). Data were missing for two patients. P value was determined by log-rank test. AIC, Akaike information criterion; ECOG PS, Eastern Cooperative Oncology Group performance status; LDH, lactate dehydrogenase; ULN, upper limit of normal.
Using the median for T- and B-cell GES to further dissect the immune-cell composition of our cohort, we identified three subgroups: T-low, T-high/B-low, and T-high/B-high melanomas (Fig. 2A; Supplementary Figs. S6A and S6B). Baseline characteristics are summarized in Supplementary Table S6. Few tumors were observed with low T-cell/high B-cell GES; these samples were included in the T-low group. In the T-high/B-high subgroup, 74% of tumor samples were derived from lymph nodes. In the T-high/B-low and T-low subgroups, tissue source was approximately equally distributed between lymph nodes, skin, or other sites (Supplementary Table S6). Kaplan–Meier analysis showed tumors with high T-cell GES levels stratified into two distinct clinical subgroups based on B-cell gene expression; patients with T-high/B-high tumors had relatively poor clinical outcomes after dab + tram vs patients with T-high/B-low GES, who exhibited improved survival outcomes (Fig. 2B and C). The T-high/B-low subgroup showed fewer PFS events compared with T-low and T-high/B-high subgroups. T-cell/B-cell GES strongly associated with OS (Wald test, P = 0.016); median OS was not reached (95% CI, 33.8 months to not reached) in the T-high/B-low subgroup at the data cutoff versus 31.3 months (95% CI, 15.9–65.8 months) in the T-low subgroup and 19.1 months (95% CI, 13.4–38.6 months) in the T-high/B-high subgroup. Multivariate analysis showed baseline T-cell/B-cell GES was significantly associated with OS alongside previously established prognostic factors, including Eastern Cooperative Oncology Group performance status, lactate dehydrogenase level, and number of organ sites with metastases (Fig. 2D; refs. 1, 20). Tissue source was not associated with OS (Supplementary Fig. S7). The association between T-cell/B-cell subgroups and OS was confirmed when adjusting multivariate analysis for NanoString batch or tissue source (data not shown).
To determine whether increased B-cell infiltration correlated with a specific tumor cell–intrinsic phenotype, we compared gene expression levels from the two NanoString panels among the three subgroups of interest. As expected, in patients with high T-cell infiltration, multiple B-cell–associated GES showed high expression levels in the B-high subgroup compared with tumors with low B-cell infiltration. We also found significant transcriptional upregulation of distinct B-cell chemoattractants and their respective receptors (CXCL13-CXCR5, CCL19/21-CCR7; ref. 21) in the T-high/B-high subgroup (Supplementary Fig. S8).
MAPK activity profile
The MAPK pathway was the top network with significantly decreased MAPK Pathway Activity Score (MPAS)/GES levels in tumors with high B-cell infiltration (Fig. 3A; Supplementary Table S7). MPAS negatively correlated with B-cell GES in all samples (Fig. 3B; Spearman correlation, −0.375; P < 0.001), especially those with high T-cell infiltration (Fig. 3C; Spearman correlation, −0.563; P < 0.001). Patients with T-high/B-high signatures had significantly lower MAPK pathway activity than the other subgroups (P = 0.003; Fig. 3D); a consistent trend was demonstrated with IHC analysis of phosphorylated extracellular signal–regulated kinase (Supplementary Fig. S9). Analysis of MPAS signature in the TCGA data set yielded a result similar to that observed in COMBI-v samples (Supplementary Fig. S10A); MPAS also negatively correlated with B-cell GES in our BRAF wild-type cohort (Supplementary Figs. S10B–S10D), suggesting that the molecular features identified in tumors with high B-cell infiltration were not specific for BRAF-mutant melanoma and could be found in all melanoma subgroups before therapy.
Figure 3.

MPAS signature versus T-cell/B-cell GESs in COMBI-v. A, Upregulated (>0) and downregulated (<0) genes from pathway analysis in COMBI-v tumor samples by T-cell/B-cell subgroup; T-high/B-high (n = 20), T-high/B-low (n = 25). B, Correlation analysis of MPAS and B-cell signature in patients included in the biomarker cohort from COMBI-v (n = 76; P value was determined by Spearman correlation test) and (C) in patients with high T-cell signature (n = 45; P value was determined by Spearman correlation test). D, MPAS signature expression by T-cell/B-cell subgroup. A box plot shows median with interquartile range (P value was determined by two-sided Wilcoxon signed-rank test).
Direct T-cell/B-cell interactions by spatial proximity analysis
Several studies have investigated immune cell and tumor cell density and relative localization using IHC; however, recent technological developments showed digital pathology conducts comprehensive spatial cell analysis (7, 11, 19, 22). We used digital pathology tools to perform blinded analysis of T- and B-cell interactions in tumor (Fig. 4A) versus stroma (Fig. 4B) compartments of T-cell/B-cell subgroups; marked-up tumor and stromal areas defined by the digital IA classifier for this same representative field of view are shown in Supplementary Fig. S1. We evaluated T-cell/B-cell interaction as a sign of direct cell-to-cell co-localization using the number of CD3-positive T cells within a 10-μm distance of CD19-positive B cells based on cell size measurements and morphological shape in classified tumor areas (n = 59). Analysis showed the T-high/B-high (GES) subgroup not only had high B-cell infiltration levels but that each B cell was more likely to exhibit direct cell-to-cell interactions with T cells in stroma and tumor compartments (Fig. 4C–E), and were significantly more likely to interact with T cells compared with those in the T-high/B-low subgroup (Fig. 4F). Because we used the percentage of CD3-positive cells identified within a 10-μm distance in B-high and B-low subgroups, which both had high T-cell infiltration, we can exclude the possibility that significant differences in T-cell/B-cell interaction scores are based only on higher absolute numbers of B cells in one subgroup compared with the other.
Figure 4.

Tumor and stroma T- and B-cell interactions. Lymphocyte aggregates observed by dual CD3/CD19 IHC staining in a representative pretherapeutic tumor sample from a patient in COMBI-v with a high T-cell/high B-cell signature. This is the same representative field of view shown in Supplementary Fig. S1. Tumor (A) and stroma (B) compartments with CD3-positive T cells (light blue) and CD19-positive B cells (brown) with connected bands representing spatial proximity distribution. Digital (C), spatial (D), and proximity (E) analysis of T- and B-cell interactions that occurred at a distance of ≤10 and >10 μm, respectively. (F) T- and B-cell interactions in tumor and stroma compartments combined in samples from COMBI-v with high T-cell levels based on gene expression subgroups (n = 36). A box plot shows median with interquartile range (P value was determined by two-sided Wilcoxon signed-rank test).
To examine the presence of tertiary lymphoid structures (TLS), we quantitatively evaluated COMBI-v samples using spatial proximity analysis to assess direct (within a 10-μm distance) and distant proximities between T and B cells. Samples (n = 59) were digitally annotated to measure the area of classified melanoma lesions. Although we observed tumor areas ranging from 0.8 to 313.7 mm2 (mean ± SD, 48.0 ± 60.9 mm2), we did not detect TLS that met the previously described morphologic criteria in our cohort of pretreatment samples (13).
T-cell receptor sequencing (TCR-seq; n = 20) revealed that patients with high levels of tumor-infiltrating B cells had an even distribution of T-cell clones, whereas patients with low B-cell infiltration had higher levels of T-cell clonality, suggesting that clonal expansion had already occurred in the T-high/B-low subgroup (Supplementary Fig. S11).
Next, we investigated why the strong presence of direct T-cell/B-cell interaction did not result in improved clinical outcomes, as observed in TCGA data sets and reported in ICI studies (12–15). We found a strong positive correlation (Pearson correlation 0.696; P < 0.001; Supplementary Fig. S12A) between tumor and stroma CD19 levels and between tumor and stroma T- and B-cell interactions (Pearson correlation 0.764; P < 0.001; Supplementary Fig. S12B), suggesting that the T-high/B-high subgroup not only exhibited direct T-cell/B-cell interaction in the stroma but that higher levels of B cells were directly interacting with melanoma cells and T cells in the tumor compartment.
Immunosuppressive features of B-cell-rich tumors
To explore whether immunosuppressive features could further explain our findings in the B-high subgroup, we used a multiplex fluorescence IHC (FIHC) assay by AQUA Technology (Navigate BioPharma, a Novartis subsidiary; refs. 23, 24) to quantify the population of human leukocyte antigen (HLA)-DR/indoleamine 2,3-dioxygenase 1 (IDO-1)–double-positive tumor cells. Melanomas with high B-cell infiltration had higher levels of intrinsic immunosuppressive HLA-DR/IDO-1–double-positive tumor cells than low–B-cell melanomas (Fig. 5A). High levels of HLA-DR/IDO-1–double-positive cells were present in tumors with concomitant high T- and B-cell infiltration and in tumors with low T-cell infiltration but were not present in tumors with high T-cell infiltration and low B-cell levels (Fig. 5B). The presence of HLA-DR/IDO-1–double-positive tumor cells was not associated with PFS but significantly associated with poor OS in univariate Cox analysis (HR, 1.10; 95% CI, 1.01–1.20; P = 0.036; Fig. 5C).
Figure 5.

HLA-DR/IDO-1–positive melanoma cells in B-cell–high and –low gene signature subgroups from COMBI-v. Analysis of HLA-DR/IDO-1 expression in tumor cells using multiplex FIHC in representative samples from (A) patients with a high or low B-cell GES and (B) across all three T-cell/B-cell signature subgroups (n = 54). A box plot shows median with interquartile range (P value was determined by two-sided Wilcoxon signed-rank test). C, Univariate Cox analysis of HLA-DR/IDO-1–positive tumor cells in B-cell–high versus B-cell–low signature subgroups and OS by Kaplan–Meier analysis of patients with high versus low levels of HLA-DR/IDO-1–positive tumors (P value was determined by log-rank test). An optimized cutoff was used for Kaplan–Meier analysis. condMC, conditional Monte Carlo.
Discussion
Pretherapeutic biomarkers are needed to facilitate treatment selection in BRAF V600–mutant melanoma to provide the greatest chance of long-term survival. In this study, GEP of tumor samples from COMBI-v revealed that cell-cycle–related markers were key candidates associated with reduced PFS with dab + tram. A similar pattern was reported with vemurafenib, whereby low cell-cycle GES associated with prolonged survival (25). In our biomarker cohort, although T-cell–inflamed signature did not significantly associate with clinical outcomes, analysis of B-cell signature revealed that high T-cell/low B-cell GES at screening was positively associated with OS with dab + tram, whereas high T-cell/high B-cell GES corresponded with poor survival outcomes.
Our findings complement analyses in metastatic melanoma, renal cell carcinoma, and sarcoma, whereby baseline GES levels of tumor-associated CD8-positive T cells and CD20-positive B cells were enriched in patients with improved survival after ICI treatment, whereas patients with low B-cell tumor infiltration had a significantly increased risk of death (12–15). Helmink and colleagues (14) demonstrated that this T-cell/B-cell profile was independent of BRAF mutation status, consistent with our results. Interestingly, recent B-cell studies identified TLS, characterized as >200-μm aggregates containing germinal centers, in samples with high T-cell/B-cell GES, which positively correlated with ICI responses (13–15). Although the same morphological criteria were applied, TLS were not observed in COMBI-v pretreatment samples. However, these structures can be heterogeneous regarding maturity and applied histomorphologic criteria, such as size of lymphoid aggregates, could be a limiting factor for detecting TLS in small tissue biopsy samples. In addition, lymphocyte aggregation with direct cell-to-cell interaction that was observed with tumor-associated T and B cells in our samples could signify immature TLS that are induced upon treatment, as reported with ICIs (13–15). Analysis of on-treatment samples in future studies would help to address this hypothesis. Because previous studies suggest B-cell–rich aggregates may provide a gateway for naive lymphocytes to enter tumors and sustain T-cell responses (13, 26), we conducted TCR-seq. Our findings support single-cell analysis by Cabrita and colleagues (13), whereby tumor samples with high B-cell levels contained more naive T cells than samples with low B-cell levels. Interestingly, high clonality, as observed in T-high/B-low samples, was described as a favorable parameter for response to BRAF and MEK inhibitors in early and advanced melanoma (27). Together, these findings suggest that the melanoma immune microenvironment is modified by the presence of B cells, and although the composition of lymphocyte aggregates in pretherapeutic high B-cell samples is similar in patients treated with targeted therapy and ICIs, these interactions may be associated with different clinical outcomes based on resistance mechanisms.
B cells were associated with distinct tumor cell phenotypes, as components linked to B-cell chemoattraction were significantly increased, whereas the MAPK pathway was significantly downregulated in B-cell–rich samples. These suggest tumors with high B-cell chemoattraction could be less dependent on MAPK signaling for growth because B cells may provide additional growth signals. Furthermore, Somasundaram and colleagues (8) found that insulin-like growth factor-1 derived from tumor-activated B cells, among other cytokines, was associated with resistance to BRAF and MEK inhibitors.
Importantly, an immunosuppressive phenotype was identified as positive expression of HLA-DR/IDO-1 in high B-cell samples. In contrast to the effects we saw with targeted therapy, Johnson and colleagues (28) found concomitant high levels of HLA-DR/IDO-1 and programmed death receptor 1 (PD-1)/programmed death ligand 1 (PD-L1) expression in pretreatment tumors, assessed using the same FIHC AQUA assay applied in this study, were associated with positive survival outcomes after anti–PD-1 therapy. Previous studies indicate that IFNy-mediated signaling, often stimulated by T cells, induced both HLA-DR/IDO-1 expression and upregulation of PD-L1 (28). These results further underline differential mechanisms that may dictate response to targeted therapy and ICIs in melanoma.
Potential organ-site bias in our study was addressed by examining baseline characteristics of T-cell/B-cell subgroups and clinical outcomes by tissue source. Our findings are in line with other B-cell analyses in melanoma because, although a higher proportion of B-cell–rich tumors were observed in tissue samples collected from lymph nodes than from skin or other locations, we also identified high B-cell levels in skin samples (13, 14). Additionally, several lymph node samples were not classified as T-high/B-high. T-cell/B-cell subgroups were well balanced for baseline characteristics, including subsequent ICIs, whereas tissue source was not associated with survival. Although we did not find evidence that tissue source affected our findings, tissue heterogeneity in melanoma studies and generally higher tumor-infiltrating B-cell levels in lymph node samples are challenges that need to be addressed if B-cell markers are to be considered for treatment selection in the future.
T-cell/B-cell subgroups showed a stronger effect for OS versus PFS, suggesting a prognostic component rather than a direct treatment effect in the first line. Because of the low number of samples often included in exploratory biomarker studies, we were unable to reliably validate our findings with other studies (29). Future studies with available on-treatment or postprogression samples should assess whether B-cell levels and T-cell/B-cell interactions are modulated by therapy and whether these changes impact response to subsequent therapies and postprogression survival. Somasundaram and colleagues showed an upregulation of B cells and growth factors, such as insulin-like growth factor-1, upon treatment with MAPK inhibitor therapy (8); however, the relationship between baseline B-cell infiltration levels, on-treatment modulation, and clinical outcome is still largely unknown. In our biomarker cohort, few patients were subsequently treated with anti–PD-1 therapy because this treatment was not widely used at that time. Future studies are needed to determine whether patients with disease that progressed with BRAF and MEK inhibitor therapy respond to subsequent anti–PD-1 therapy if B cells are present in the tumor.
We have identified B cells as a potential pretherapeutic biomarker to help select patients with unresectable or metastatic melanoma who may benefit the most from dab + tram, although further validation is needed.
Authors' Disclosures
J.C. Brase reports employment with Novartis and ownership of Novartis stock. R.F.H. Walter reports grants from University Hospital Essen during the conduct of the study. D. Gusenleitner reports other support from Novartis Institutes for Biomedical Research outside the submitted work. D. Castelletti reports employment with NIBR and other support from Novartis stock options outside the submitted work. C. Robert reports personal fees from Bristol Myers Squibb, MSD, Novartis, Pierre Fabre, Sanofi, and Roche outside the submitted work. G.V. Long reports personal fees from Aduro Biotech, Amgen Inc., Array Biopharma Inc., Boehringer Ingelheim International, Bristol Myers Squibb, Hexal AG, Highlight Therapeutics SL, MSD, Novartis Pharma AG, OncoSec, Pierre Fabre, QBiotics Group Ltd., Regeneron Pharmaceuticals Inc., SkylineDX B.V., and Specialised Therapeutics Australia outside the submitted work. P.D. Nathan reports personal fees from Novartis outside the submitted work. A. Ribas reports personal fees from Novartis during the conduct of the study; personal fees from Amgen, Chugai, Genentech, Merck, Novartis, Roche, Sanofi and Vedanta, 4C Biomed, Apricity, Arcus, Highlight, Compugen, ImaginAb, MapKure, Merus, Rgenix, Lutris, PACT Pharma, Tango, Advaxis, CytomX, Five Prime, RAPT, Isoplexis, and Kite-Gilead outside the submitted work. K.T. Flaherty reports personal fees from Clovis Oncology, Strata Oncology, Kinnate BioPharma, Checkmate Pharmaceuticals, Scorpion Therapeutics, PIC Therapeutics, Apricity, Tvardi, xCures, Monopteros, Vibliome, ALX Oncology, Lilly, Takeda, and Boston Biomedical; grants and personal fees from Novartis; and grants from Sanofi during the conduct of the study. J. Schachter reports personal fees from Bristol Myers Squibb, MSD, and Novartis outside the submitted work. L. Zimmer reports personal fees and other support from Novartis during the conduct of the study; personal fees and other support from Bristol Myers Squibb, MSD, Pierre Fabre, Sunpharma, and Sanofi; other support from Amgen; and personal fees from Roche outside the submitted work. E. Livingstone reports personal fees and other support from Bristol Myers Squibb, MSD, Novartis, Amgen, Medac, Pierre Fabre, and Sun Pharma and personal fees from Roche, Actelion, Sanofi, and Janssen outside the submitted work. E. Gasal reports other support from Novartis Pharmaceutical Corporation during the conduct of the study and other support from Novartis Pharmaceutical Corporation outside the submitted work. D. Schadendorf reports grants from Novartis and personal fees and nonfinancial support from Novartis during the conduct of the study; grants from Bristol Myers Squibb and Amgen; personal fees and nonfinancial support from Bristol Myers Squibb, Roche, Pierre-Fabre, MSD, Sanofi, Hexal, and Merck-Serono; and personal fees from Helsinn, Immunocore, Array, Pfizer, Neracare, and Philogen outside the submitted work. A. Roesch reports grants from Novartis during the conduct of the study; nonfinancial support from Amgen, Roche, and TEVA; personal fees and nonfinancial support from Merck/MSD; grants and nonfinancial support from Novartis and Bristol Myers Squibb; and grants from Adtec outside the submitted work. No disclosures were reported by the other authors.
Disclaimer
The Editor-in-Chief of Clinical Cancer Research is an author on this article. In keeping with AACR editorial policy, a senior member of the Clinical Cancer Research editorial team managed the consideration process for this submission and independently rendered the final decision concerning acceptability.
Supplementary Material
Acknowledgments
We thank the patients and their families for their participation. We also thank the study site staff; additional investigators; Noelle Hanoteau, Catarina Campbell; LaSalette Charette, Annette Quail, and Nan Fei Jiang from the Novartis Precision Medicine In situ Diagnostics IHC/ISH Team; Navigate BioPharma; and others for their contributions. We also thank Matthew Squires (Novartis Institutes for BioMedical Research) and Maurizio Voi (Novartis Pharmaceuticals Corporation) for guidance and critical review. Medical writing assistance was provided by Zareen Khan, PhD (ArticulateScience LLC), and was funded by Novartis Pharmaceuticals Corporation. The trial was originally sponsored by GlaxoSmithKline; dabrafenib and trametinib were designated as assets of Novartis on March 2, 2015, after which Novartis took over sponsorship of the trial. This work was also partly supported by the Deutsche Forschungsgemeinschaft (German Research Foundation) [grant numbers RO 3577/7-1, SCHA 422/17-1 (KFO 337)].
The publication costs of this article were defrayed in part by the payment of publication fees. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
This article is featured in Highlights of This Issue, p. 4457
Footnotes
Note: Supplementary data for this article are available at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/).
Authors' Contributions
J.C. Brase: Conceptualization, data curation, formal analysis, supervision, validation, methodology, writing–original draft, writing–review and editing. R.F.H. Walter: Data curation, formal analysis, writing–review and editing. A. Savchenko: Conceptualization, data curation, formal analysis, validation, methodology, writing–review and editing. D. Gusenleitner: Formal analysis, writing–review and editing. J. Garrett: Formal analysis, writing–review and editing. T. Schimming: Data curation, writing–review and editing. R. Varaljai: Data curation, project administration, writing–review and editing. D. Castelletti: Formal analysis, writing–review and editing. J. Kim: Formal analysis, writing–review and editing. N. Dakappagari: Data curation, formal analysis, methodology, writing–review and editing. K. Schultz: Data curation, formal analysis, methodology, writing–review and editing. C. Robert: Data curation, formal analysis, writing–review and editing. G.V. Long: Data curation, formal analysis, project administration, writing–review and editing. P.D. Nathan: Data curation, writing–review and editing. A. Ribas: Writing–review and editing. K.T. Flaherty: Formal analysis, writing–review and editing. B. Karaszewska: Data curation, supervision, writing–review and editing. J. Schachter: Data curation, writing–review and editing. A. Sucker: Writing–review and editing. K.W. Schmid: Conceptualization, writing–review and editing. L. Zimmer: Data curation, writing–review and editing. E. Livingstone: Data curation, project administration, writing–review and editing. E. Gasal: Formal analysis, supervision, writing–review and editing. D. Schadendorf: Conceptualization, formal analysis, writing–original draft, writing–review and editing. A. Roesch: Conceptualization, data curation, formal analysis, supervision, validation, methodology, writing–original draft, project administration, writing–review and editing.
References
- 1. Robert C, Grob JJ, Stroyakovskiy D, Karaszewska B, Hauschild A, Levchenko E, et al. Five-year outcomes with dabrafenib plus trametinib in metastatic melanoma. N Engl J Med 2019;381:626–36. [DOI] [PubMed] [Google Scholar]
- 2. Robert C, Ribas A, Schachter J, Arance A, Grob JJ, Mortier L, et al. Pembrolizumab versus ipilimumab in advanced melanoma (KEYNOTE-006): post-hoc 5-year results from an open-label, multicentre, randomised, controlled, phase 3 study. Lancet Oncol 2019;20:1239–51. [DOI] [PubMed] [Google Scholar]
- 3. McArthur GA, Dréno B, Larkin J, Ribas A, Liszkay G, Maio M, et al. 5-Year survival update of cobimetinib plus vemurafenib in BRAFV600 mutation–positive advanced melanoma: final analysis of the coBRIM study. 2019 Society for Melanoma Research Annual Congress; November 20-23, 2019; Salt Lake City, UT. [DOI] [PMC free article] [PubMed]
- 4. Rizos H, Menzies AM, Pupo GM, Carlino MS, Fung C, Hyman J, et al. BRAF inhibitor resistance mechanisms in metastatic melanoma: spectrum and clinical impact. Clin Cancer Res 2014;20:1965–77. [DOI] [PubMed] [Google Scholar]
- 5. Gide TN, Wilmott JS, Scolyer RA, Long GV.. Primary and acquired resistance to immune checkpoint inhibitors in metastatic melanoma. Clin Cancer Res 2018;24:1260–70. [DOI] [PubMed] [Google Scholar]
- 6. Van Allen EM, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 2015;350:207–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Erdag G, Schaefer JT, Smolkin ME, Deacon DH, Shea SM, Dengel LT, et al. Immunotype and immunohistologic characteristics of tumor-infiltrating immune cells are associated with clinical outcome in metastatic melanoma. Cancer Res 2012;72:1070–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Somasundaram R, Zhang G, Fukunaga-Kalabis M, Perego M, Krepler C, Xu X, et al. Tumor-associated B-cells induce tumor heterogeneity and therapy resistance. Nat Commun 2017;8:607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. DiLillo DJ, Yanaba K, Tedder TF.. B cells are required for optimal CD4+ and CD8+ T cell tumor immunity: therapeutic B cell depletion enhances B16 melanoma growth in mice. J Immunol 2010;184:4006–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ruddell A, Harrell MI, Furuya M, Kirschbaum SB, Iritani BM.. B lymphocytes promote lymphogenous metastasis of lymphoma and melanoma. Neoplasia 2011;13:748–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ladanyi A, Kiss J, Mohos A, Somlai B, Liszkay G, Gilde K, et al. Prognostic impact of B-cell density in cutaneous melanoma. Cancer Immunol Immunother 2011;60:1729–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Griss J, Bauer W, Wagner C, Simon M, Chen M, Grabmeier-Pfistershammer K, et al. B cells sustain inflammation and predict response to immune checkpoint blockade in human melanoma. Nat Commun 2019;10:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cabrita R, Lauss M, Sanna A, Donia M, Skaarup Larsen M, Mitra S, et al. Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature 2020;577:561–5. [DOI] [PubMed] [Google Scholar]
- 14. Helmink BA, Reddy SM, Gao J, Zhang S, Basar R, Thakur R, et al. B cells and tertiary lymphoid structures promote immunotherapy response. Nature 2020;577:549–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Petitprez F, de Reynies A, Keung EZ, Chen TW, Sun CM, Calderaro J, et al. B cells are associated with survival and immunotherapy response in sarcoma. Nature 2020;577:556–60. [DOI] [PubMed] [Google Scholar]
- 16. Robert C, Karaszewska B, Schachter J, Rutkowski P, Mackiewicz A, Stroiakovski D, et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med 2015;372:30–9. [DOI] [PubMed] [Google Scholar]
- 17. Riaz N, Havel JJ, Makarov V, Desrichard A, Urba WJ, Sims JS, et al. Tumor and microenvironment evolution during immunotherapy with nivolumab. Cell 2017;171:934–49.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ayers M, Lunceford J, Nebozhyn M, Murphy E, Loboda A, Kaufman DR, et al. IFN-γ–related mRNA profile predicts clinical response to PD-1 blockade. J Clin Invest 2017;127:2930–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lardone RD, Plaisier SB, Navarrete MS, Shamonki JM, Jalas JR, Sieling PA, et al. Cross-platform comparison of independent datasets identifies an immune signature associated with improved survival in metastatic melanoma. Oncotarget 2016;7:14415–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Long GV, Grob J-J, Nathan P, Ribas A, Robert C, Schadendorf D, et al. Factors predictive of response, disease progression, and overall survival after dabrafenib and trametinib combination treatment: a pooled analysis of individual patient data from randomised trials. Lancet Oncol 2016;17:1743–54. [DOI] [PubMed] [Google Scholar]
- 21. Tokunaga R, Naseem M, Lo JH, Battaglin F, Soni S, Puccini A, et al. B cell and B cell-related pathways for novel cancer treatments. Cancer Treat Rev 2019;73:10–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Heindl A, Nawaz S, Yuan Y.. Mapping spatial heterogeneity in the tumor microenvironment: a new era for digital pathology. Lab Invest 2015;95:377–84. [DOI] [PubMed] [Google Scholar]
- 23. Dolled-Filhart M, Gustavson M, Camp RL, Rimm DL, Tonkinson JL, Christiansen J.. Automated analysis of tissue microarrays. Methods Mol Biol 2010;664:151–62. [DOI] [PubMed] [Google Scholar]
- 24. Schalper KA, Brown J, Carvajal-Hausdorf D, McLaughlin J, Velcheti V, Syrigos KN, et al. Objective measurement and clinical significance of TILs in non-small cell lung cancer. J Natl Cancer Inst 2015;107. doi 10.1093/jnci/dju435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wongchenko MJ, McArthur GA, Dreno B, Larkin J, Ascierto PA, Sosman J, et al. Gene expression profiling in BRAF-mutated melanoma reveals patient subgroups with poor outcomes to vemurafenib that may be overcome by cobimetinib plus vemurafenib. Clin Cancer Res 2017;23:5238–45. [DOI] [PubMed] [Google Scholar]
- 26. Xu H, Li X, Liu D, Li J, Zhang X, Chen X, et al. Follicular T-helper cell recruitment governed by bystander B cells and ICOS-driven motility. Nature 2013;496:523–7. [DOI] [PubMed] [Google Scholar]
- 27. Cooper ZA, Frederick DT, Juneja VR, Sullivan RJ, Lawrence DP, Piris A, et al. BRAF inhibition is associated with increased clonality in tumor-infiltrating lymphocytes. Oncoimmunology 2013;2:e26615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Johnson DB, Bordeaux J, Kim JY, Vaupel C, Rimm DL, Ho TH, et al. Quantitative spatial profiling of PD-1/PD-L1 interaction and HLA-DR/IDO-1 predicts improved outcomes of anti-PD-1 therapies in metastatic melanoma. Clin Cancer Res 2018;24:5250–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yan Y, Wongchenko MJ, Robert C, Larkin J, Ascierto PA, Dréno B, et al. Genomic features of exceptional response in vemurafenib ± cobimetinib-treated patients with BRAF (V600)-mutated metastatic melanoma. Clin Cancer Res 2019;25:3239–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.