

Abstract
Purpose:
The aim of the study was to determine safety, antitumor activity, and pharmacodynamic profile of mogamulizumab, an anti-CCR4 monoclonal antibody targeting effector regulatory T cells (Treg) in combination with the checkpoint inhibitor nivolumab in patients with locally advanced or metastatic solid tumors.
Patients and Methods:
This was a multicenter, dose-finding (phase I), and dose expansion (phase II) study (NCT02705105) in patients with locally advanced or metastatic solid tumors. There were no dose-limiting toxicities in phase I with mogamulizumab 1 mg/kg every week for cycle 1 followed by 1 mg/kg every 2 weeks plus nivolumab 240 mg every 2 weeks intravenously, and cohort expansion occurred at this dose level.
Results:
All 114 patients treated with mogamulizumab 1 mg/kg plus nivolumab 240 mg in phases I (n = 4) and II (n = 110) were assessed for safety and efficacy. Mogamulizumab plus nivolumab showed acceptable safety and tolerability. Objective response rate was 10.5% [95% confidence interval (CI), 5.6–17.7; 3 complete and 9 partial responses]. Disease control rate was 36.8%. Median duration of response was 14.4 months. Median progression-free survival was 2.6 (95% CI, 2.3–3.1) months, and median overall survival was 9.5 (95% CI, 5.9–13.5) months.
Conclusions:
Combination of mogamulizumab with nivolumab for treatment of patients with locally advanced or metastatic solid tumors did not result in enhanced efficacy. Tolerability of mogamulizumab 1 mg/kg plus nivolumab 240 mg was acceptable.
Translational Relevance.
Mogamulizumab is a monoclonal antibody directed at C-C chemokine receptor 4 (CCR4), which is highly expressed by effector regulatory T cells (Treg). The current phase I/II study in 114 patients with locally advanced or metastatic solid tumors evaluated whether depletion of effector Tregs with mogamulizumab would be safe and could improve the efficacy of immune checkpoint inhibitor nivolumab targeting programmed cell death-1 (PD-1). Tolerability of the combination proved acceptable. No additional efficacy was observed with the addition of mogamulizumab compared with that expected with single-agent nivolumab, and the study was ended prematurely. Depletion of the Treg population by mogamulizumab in combination with the checkpoint inhibitor nivolumab might be useful in inducing antitumor immunity but does not appear to be the only factor sufficient to induce potent antitumor efficacy.
Introduction
Programmed cell death-1 (PD-1) is a regulatory receptor commonly expressed in malignant tumor cells that contributes to impaired tumor surveillance by downregulation of T- and B-cell activation (1–3). PD-1 inhibitory immunotherapy with agents such as nivolumab and pembrolizumab is now well recognized as a major advance in cancer treatment (2, 3).
Furthermore, regulatory T cells (Treg) play a key role in antitumor activity and T-cell activation via immune checkpoints, and their expression is associated with advanced malignancy and poor prognosis in patients with solid tumors (4). C-C chemokine receptor 4 (CCR4) is expressed by Tregs, which recognizes two chemokines [CC ligands 17 (CCL17) and 22 (CCL22); ref. 5]. CCR4 is associated with Treg trafficking into the tumor microenvironment (6) and is overexpressed on malignant T cells (7). Furthermore, high CCR4+ Treg levels are found in a wide range of murine and human solid tumors and/or have been associated with tumor progression or metastasis (8–17).
Mogamulizumab is a first-in-class humanized antibody targeting CCR4. Mogamulizumab has been shown to deplete Tregs from peripheral blood and exhibited evidence of single-agent clinical activity in patients with solid tumors (18). Combined treatment with mogamulizumab and a PD-1 inhibitor might therefore present a two-pronged approach to combatting immunosuppressive tumor microenvironments both by depleting Tregs and inhibiting downregulation of T and B cells via PD-1 (19–24).
The aim of the current clinical study was to evaluate the extent that CCR4+ Treg depletion by mogamulizumab enhances antitumor response in combination with the PD-1 checkpoint inhibitor nivolumab in patients with locally advanced or metastatic solid tumors. Nivolumab, an anti–PD-1 monoclonal antibody, is approved by the FDA in the United States for the treatment of various cancers, such as melanoma, squamous cell carcinoma of the head and neck (SCCHN), microsatellite instability (MSI)-high colorectal cancer, hepatocellular carcinoma (HCC), non–small cell lung cancer (NSCLC), renal cell carcinoma, and gastric cancer/gastroesophageal junction cancer/esophageal adenocarcinoma (25).
Patients and Methods
Patients
Adult patients (≥18 years) were included with histologically or cytologically confirmed measurable, locally advanced or metastatic solid tumors that had been previously treated and for which no additional standard or approved therapies were available, with the exception of PD-1 blockade in phase I (patients with tumors for which nivolumab has known survival benefit who had not received PD-1 blockade were eligible). Patients had to have Eastern Cooperative Oncology Group (ECOG) performance status 0 or 1 and have adequate organ and bone marrow function. Patients with primary or uncontrolled metastases to the central nervous system were not eligible. In phase I, patients with all tumor types were eligible. In phase II, patients were not eligible if they had received prior anti–PD-1, anti-programmed cell death ligand 1 (PD-L1), anti–programmed cell death ligand 2, anti-CD137, or anti-cytotoxic T-lymphocyte–associated antigen 4 antibody, or any other antibody or drug specifically targeting T-cell costimulation or checkpoint pathways. In phase II, patients were included in seven disease-specific expansion cohorts: squamous NSCLC, PD-L1 nonexpression nonsquamous cell NSCLC, SCCHN, non–MSI-high colorectal cancer, HCC, pancreatic carcinoma, and ovarian cancer (including primary peritoneal cancer and fallopian tube carcinoma). Full details of inclusion and exclusion criteria are given as Supplementary Information.
Study design and objectives
This open-label, multicenter, dose-finding (phase I) and cohort expansion (phase II) study evaluated the combination of mogamulizumab and nivolumab. Phase I used a standard 3 + 3 design to identify the maximum tolerated dose (MTD) or the highest protocol-defined dose as the recommended phase II dose (RP2D) on the basis of dose-limiting toxicity (DLT). Patients who did not receive all infusions in cycle 1 at the assigned doses or did not complete safety follow-up (until 1 week after the end of cycle 1) were replaced. Full details of DLTs are provided as Supplementary Information.
Two dose combinations were planned in phase I: the starting doses were mogamulizumab 1 mg/kg + nivolumab 240 mg, which could be reduced to mogamulizumab 0.3 mg/kg + nivolumab 240 mg. In phase II, dose expansion occurred with the RP2D in the seven disease-specific cohorts.
The primary objective was to evaluate the safety and tolerability and determine the RP2D of the combination of mogamulizumab + nivolumab in patients with locally advanced or metastatic solid tumors. The secondary objective was to evaluate the antitumor activity of the combination. Exploratory objectives included evaluation of the pharmacokinetics and immunogenicity of mogamulizumab and nivolumab, the pharmacodynamics of the combination, and whether any biomarkers were correlated with safety or antitumor activity.
Rationale for dose selection
The initial mogamulizumab 1 mg/kg dose was based on the dose administered in a development program in T-cell lymphomas, the marketed dose in Japan, and the RP2D for the combination of mogamulizumab 1 mg/kg weekly + nivolumab 3 mg/kg biweekly in a Japanese study in patients with solid tumors (26).
The nivolumab dose of 240 mg every 2 weeks was selected based on clinical data and modeling and simulation approaches using population pharmacokinetic (PK) and exposure–response analyses of data from studies in multiple tumor types (melanoma, NSCLC, and renal cell carcinoma) where body weight normalized dosing (mg/kg) was used (27). Population PK analyses showed that the PK of nivolumab is linear with proportional exposure over a dose range of 0.1 to 10 mg/kg, and no differences in PK across ethnicities and tumor types were observed. Nivolumab clearance and volume of distribution were found to increase as body weight increases, but less than proportionally with increasing weight, indicating that mg/kg dosing represents an overadjustment for the effect of body weight on nivolumab PK. The population PK model previously developed using data from NSCLC subjects was updated, using data from 1,544 subjects from seven studies investigating nivolumab in the treatment of melanoma, NSCLC, and renal cell carcinoma. In this dataset, the median (range) weight was 77 (35–160) kg and, thus, an approximately equivalent dose of 3 mg/kg for an 80 kg subject, nivolumab 240 mg every 2 weeks was selected for future studies.
To predict relevant summary exposures of nivolumab 240 mg every 2 weeks, the population PK model was used to simulate 100 virtual trials, each consisting of two arms, nivolumab 3 mg/kg every 2 weeks and 240 mg every 2 weeks. In the simulations, the simulated patient populations consisted of 1,000 subjects per treatment arm randomly sampled from the aforementioned pooled database of cancer patients. Because no differences in PK were noted across ethnicities and tumor types, these simulated melanoma and NSCLC data are applicable to patients with other tumor types. The simulated measure of exposure of interest, time-averaged concentrations at steady state for 240 mg every 2 weeks were predicted to be similar for all subjects in reference to 80 kg subjects receiving 3 mg/kg every 2 weeks.
Nivolumab is safe and well tolerated up to a 10 mg/kg every 2 weeks dose level. AEs have been broadly consistent across tumor types following monotherapy and have not demonstrated clear dose–response or exposure–response relationships. In addition, the simulated median and 95th prediction interval of nivolumab summary exposures across body weight range (35–160 kg) are predicted to be maintained below the corresponding observed highest exposure experienced in nivolumab, that is, 95th percentile following nivolumab 10 mg/kg every 2 weeks from a specific clinical study (CA209003). Thus, while subjects in the lower body weight ranges would have greater exposures than 80 kg subjects, the exposures are predicted to be within the range of observed exposures at doses (up to 10 mg/kg every 2 weeks) used in the nivolumab clinical program and are not considered to put subjects at increased risk. For subjects with greater body weights, the simulated ranges of exposures are also not expected to affect efficacy, because the exposures predicted following administration of a 240 mg every 2 weeks are on the flat part of the exposure–response curves for previously investigated tumors, melanoma, and NSCLC. Given the similarity of nivolumab PK across tumor types and the similar exposures predicted following administration of 240 mg flat dose compared with 3 mg/kg, it was expected that the safety and efficacy profile of 240 mg nivolumab would be similar to that of 3 mg/kg nivolumab. Thus, a flat dose of 240 mg every 2 weeks was used this study. Based on population pharmacokinetic modeling, the benefit–risk profile of the flat dose nivolumab 240 mg every 2 weeks is comparable with 3 mg/kg every 2 weeks with respect to exposure–response relationships for efficacy and safety, and clinical safety (27).
Study drug administration
Mogamulizumab was administered by intravenous infusion over ≥1 hour on days 1, 8, 15, and 22 of cycle 1 and on days 1 and 15 of each subsequent 28-day cycle. Nivolumab 240 mg was administered by intravenous infusion on days 1 and 15 of each 28-day cycle. Mogamulizumab infusion was started ≥30 minutes after end of nivolumab infusion when given on the same day. Patients were premedicated with oral acetaminophen 325 to 1,000 mg and intravenous diphenhydramine 50 mg (or equivalent) at least 30 minutes before the first mogamulizumab infusion, and if they had a reaction, this was repeated before subsequent mogamulizumab infusions. Patients were not premedicated before the first infusion of nivolumab, but if they experienced a reaction, they were subsequently premedicated. Mogamulizumab infusion was made using a 0.22- or 0.2-μm inline filter and nivolumab using a 0.22- or 1.2-μm, low-protein-binding polyethersulfone membrane inline filter. Doses could be interrupted, delayed, or stopped depending on tolerability. Treatment was given for up to 24 months. Patients were not allowed to receive concurrent administration of immunosuppressive agents, immunosuppressive doses of systemic corticosteroids [except for <3 weeks as prophylaxis or treatment of study drug hypersensitivity reaction or other adverse event (AE)] related to investigational medicinal product (IMP), or other anticancer therapy.
Assessments
Demographics and medical history were recorded at screening (up to 4 weeks prior to first dose). Vital signs were recorded at every visit. Physical examination was undertaken and ECOG performance status determined at screening, on day 1 of cycles ≥1, and at end of treatment (EOT). Hematology and serum chemistry profile was determined at every visit. Coagulation profile was performed at screening, on day 1 of cycles ≥1, and at EOT. Thyroid function testing was undertaken at screening, on day 1 of cycles ≥2, and at EOT. Standard 12-lead electrocardiogram was performed at screening, on days 1, 8, and 15 of cycle 1, and at EOT. Pulse oximetry was performed on days 1 and 15 of cycles ≥1, and at EOT. Urinalysis was undertaken at screening and EOT. Testing for hepatitis B virus, hepatitis C virus, human immunodeficiency virus, and cytomegalovirus was performed at screening. Serum pregnancy testing was undertaken at screening and EOT, and urinary pregnancy testing on day 1 of cycles ≥1 in women of child-bearing potential.
Tumor response and safety
Tumor assessment (including MRI or CT, biopsy, and applicable serum tumor markers) was performed by investigators at screening and every 3 cycles using Response Evaluation Criteria in Solid Tumors (RECIST) v1.1 (28) and immune-related RECIST (irRECIST) v1.1 criteria (29). Objective response rate (ORR) was based on the percentage of patients with best overall response of complete response (CR) or partial response (PR) confirmed ≥4 weeks later. Patients who did not meet CR/PR were classified as stable disease (SD) if assessed as SD (or better) ≥9 weeks after first dose of IMP or progressive disease (PD). Time to response (TTR) was defined as days from day 1 of cycle 1 to the first assessment date of confirmed CR or PR. Duration of response (DOR) was defined as days from the first assessment date of confirmed CR or PR to the date of death or PD, whichever was earlier. Progression-free survival (PFS) was defined as days from day 1 of cycle 1 to the date of death or PD, whichever was earlier. Overall survival (OS) was defined as days from day 1 of cycle 1 to the date of death censored for the last date that the patient was known to be alive.
Adverse events (AE) were recorded following observation by the investigator or in response to nonleading questioning during clinic visits, after spontaneous reporting by the patient, or on the basis of clinical or laboratory tests. AEs were graded by National Cancer Institute Common Terminology Criteria for Adverse Events (NCI-CTCAE) v4.03 and classified by the investigator as related or unrelated to study drug administration. Treatment-related AEs included those considered definitely, probably, or possibly related to treatment. Safety was analyzed in the safety analysis set that included all patients who received at least one dose (even a partial dose) of IMP.
In this study, disease progression was not to be reported as an AE. Lymphopenia and leukopenia are expected pharmacologic effects of mogamulizumab and were also not to be reported as AEs for this study. Lymphocytes and white blood cell counts were monitored and evaluated with the laboratory data. The MedDRA preferred term “drug eruption” was used to capture verbatim terms including rash (of any description), pruritus, dermatitis, erythema, redness, urticaria, or itching that were considered related to study treatment. The term “drug eruption rash” is used in this communication to reflect the entire definition when discussing this treatment-related AE.
Antidrug antibody (ADA) testing was undertaken for mogamulizumab using predose blood samples at the start of cycles 1, 2, 3, 4, 8, and 12, and 100 to 110 days after the end of the treatment. ADA testing for nivolumab was planned but not undertaken after the premature termination of the study.
Biomarker assessments
Blood samples were taken for biomarker and pharmacodynamic assessment predose on days 1 and 15 of cycles 1 and 2, predose at the start of cycles 3, 4, and 12, and 100 to 110 days after the end of the treatment. Assessments included circulating CCR4+ Tregs, activated T-cell populations, and other immune cell populations by flow cytometry and immunohistochemistry, which have been previously detailed (30).
Statistical analysis
The sample size for phase I was based on a standard 3 + 3 dose-finding design, and with two dose levels under consideration, a maximum of 12 patients was planned (3–6 patients/cohort). For phase II expansion, a sample size of up to 184 patients (21–36/tumor type) in up to seven tumor-specific cohorts was planned. A Simon's 2-stage optimal design was used to estimate sample size for the expansion cohorts in phase II. A cohort would have been considered for termination due to lack of efficacy if the observed number of tumor responses (either confirmed or unconfirmed) in the first stage was smaller than respective futility boundary. Table 1 presents operating characteristics of the Simon's 2-stage design with 15% false positive rate (FPR) and 10% false negative rate (FNR) and sample size required for each tumor type. A maximum of 188 patients may have participated in the study, including a total of 4 patients who were enrolled in phase I and a total of 184 potential patients (across all tumor types) calculated for the expansion cohort. The decision to terminate a cohort or to allow it to continue was based on the futility boundary for each cohort and after taking into consideration other relevant observations such as duration and depth of response and risk/benefit profile. For some cohorts, number of patients receiving treatment at the time of the stage 1 efficacy evaluation may differ from the specified stage 1 sample size, depending on accrual rate, response lag, and other factors of a clinical nature.
Table 1.
Sample size estimation by tumor type based on Simon's 2-stage optimal design.
| Response assumptions | Simon 2-stage sample sizesa | ||||
|---|---|---|---|---|---|
| Tumor type | Lower boundary historical response rate (%) | Target response rate (%) | Stage 1 target (n1) | Stage 2 target total N | Stage 1 response futility boundary (≤): stop if this many responses or fewer |
| NSCLC SQ | 20 | 40 | 16 | 32 | 3 |
| NSCLC NSQ PD-L1 nonexpressing | 10 | 30 | 10 | 21 | 0 |
| Ovarian | 10 | 30 | 10 | 21 | 0 |
| CRC (non-MSI high) | 5 | 20 | 12 | 29 | 0 |
| SCCHN | 25 | 45 | 14 | 36 | 3 |
| HCC | 16 | 36 | 12 | 28 | 6 |
| Pancreatic | 0 | 15 | 15 | 17 | 0 |
Abbreviations: CRC, colorectal cancer; FNR, false negative rate; FPR, false positive rate; HCC, hepatocellular carcinoma; MSI, microsatellite instability; NSCLC, non–small cell lung cancer; NSQ, nonsquamous; PD-L1, programmed cell death ligand 1; SCCHN, squamous cell carcinoma of the head and neck; SQ, squamous.
aFPR or one-sided alpha = 15% and FNR or 1-power = 10%.
Demographic, baseline characteristics, and efficacy and safety endpoints were summarized descriptively. Frequency and percentages were used for categorical variables and summary statistics [number, mean, standard deviation (SD), median, minimum, and maximum] were calculated for continuous variables.
Efficacy endpoints were analyzed using the efficacy analysis set, which included all patients who had measurable disease and completed the first cycle of combination therapy and who had baseline and at least one post-baseline on-study assessment for response. ORR, PFS, and OS were reported in all patients receiving the same doses in phases I and II combined. For ORR, the percentage of subjects with either CR or PR were calculated, along with two-sided 95% exact CIs using the exact binomial CI method. DOR, PFS, and OS were analyzed by estimating median and respective two-sided 95% CI using the Kaplan–Meier method. TTR was summarized descriptively for responders (i.e., CR or PR).
Ethics
The protocol and its subsequent amendments were approved by the local Institutional Review Boards at the participating centers. All patients provided written informed consent prior to study registration, and the study was performed in accordance with the Declaration of Helsinki and International Conference for Harmonization of Good Clinical Practice Guidelines. The study was registered in ClinicalTrials.gov (NCT02705105).
Results
Patient characteristics
The study was conducted between 18 February, 2016, and 10 October, 2018 (data cutoff), at 13 U.S. centers. A total of 114 patients were enrolled and treated in phase I (n = 4) and phase II (n = 110), all of whom were included in efficacy and safety analyses. The baseline demographic and clinical characteristics of these patients are detailed in Table 2. Disposition of the patients during the study is detailed as Supplementary Fig. S1. The major tumor-specific cohorts for dose expansion in phase II included patients with non–MSI-high colorectal cancer (n = 29), HCC (n = 29), ovarian cancer (n = 24), and pancreatic adenocarcinoma (n = 17). The accrual number of patients in stage 1 of the Simon's 2-stage design was not reached for some cohorts. The number of patients enrolled, compared with the number of patients planned for stage I, was 5 of 16 for squamous cell NSCLC; 4 of 10 for PD-L1 nonexpressing, nonsquamous cell NSCLC; and 10 of 14 for SCCHN cohorts. Aside from accrual rate, response lag and other factors of clinical nature were taken into consideration for enrollment of patients from these cohorts in the first stage.
Table 2.
Baseline patient demographics and clinical characteristics.
| Mogamulizumab + nivolumab | |
|---|---|
| Characteristics | (N = 114) |
| Age (years), median (range) | 62.5 (32–80) |
| ≥65 years, n (%) | 49 (43.0) |
| Gender, n (%) | |
| Male | 61 (53.5) |
| Female | 53 (46.5) |
| Race, n (%) | |
| White | 97 (85.1) |
| Asian | 3 (2.6) |
| Black or African American | 13 (11.4) |
| Not reported | 1 (0.9) |
| ECOG performance status, n (%) | |
| 0 | 45 (39.5) |
| 1 | 69 (60.5) |
| Height (cm), median (range) | 171 (149, 191) |
| Weight (kg), median (range) | 78 (44, 145) |
| Time from diagnosis (months), median (range) | 22.8 (0.8, 103.0) |
| Stage at enrollment, n (%) | |
| Locally advanced | 24 (21.1) |
| Metastatic | 90 (78.9) |
| Primary tumor type, n (%) | |
| Phase I | |
| Bladder | 1 (0.9) |
| Esophageal | 1 (0.9) |
| Pancreatic | 1 (0.9) |
| Sarcoma, alveolar soft part | 1 (0.9) |
| Phase II | |
| Colorectal carcinoma, non-MSI high | 29 (25.4) |
| Hepatocellular carcinoma | 24 (21.1) |
| NSCLC, nonsquamous, PD-L1 nonexpressing | 4 (3.5) |
| NSCLC, squamous cell | 5 (4.4) |
| Ovarian/fallopian tube/primary peritoneal | 21 (18.4) |
| Pancreatic adenocarcinoma | 17 (14.9) |
| SCCHN | 10 (8.8) |
| No. of prior cancer regimens, n (%) | |
| 0 | 4 (3.5) |
| 1 | 35 (30.7) |
| 2 | 26 (22.8) |
| 3 | 20 (17.5) |
| 4 | 8 (7.0) |
| 5 | 8 (7.0) |
| ≥6 | 13 (11.4) |
Abbreviations: ECOG, Eastern Cooperative Oncology Group; MSI, microsatellite instability; NSCLC, non–small cell lung cancer; PD-L1, programmed cell death ligand 1; SCCHN, squamous cell carcinoma of head and neck.
Almost all patients (96.5%) had received prior anticancer regimens. The majority of patients (65.8%) had received ≥2 prior anticancer regimens. Best response to the most recent, previous anticancer regimen was CR (n = 2, 2.3%), PR (n = 4, 4.6%), SD (n = 29, 33.3%), or PD (n = 52, 59.8%) among the 87 patients with available data. None of the patients had previous exposure to checkpoint inhibitor therapies.
The reasons for discontinuation from the study were disease progression (n = 83), AEs not related to cancer progression (n = 17), consent withdrawal (n = 8), and investigator discretion (n = 3); one patient had completed 96 weeks of treatment per protocol (a patient with ovarian cancer who attained CR) and two patients were continuing to receive treatment at cutoff. Drug exposure in phases I and II is detailed in Supplementary Tables S1 and S2. Median relative dose intensity for both mogamulizumab and nivolumab was approximately 94%.
Dose-limiting toxicity and safety
In phase I, four patients (including 1 replacement) received mogamulizumab 1 mg/kg + nivolumab 240 mg: none experienced DLTs. This dose level was therefore used for cohort expansion in phase II.
AEs occurring among the 114 patients treated with mogamulizumab 1 mg/kg + nivolumab 240 mg during phase I/II are summarized in Table 3. All patients experienced at least one AE, and 104 patients (91.2%) had treatment-related AEs. The most common treatment-related grade ≥3 AEs were drug eruption rash (12.3%), increased alanine aminotransferase (7.0%), increased aspartate aminotransferase (5.3%), and anemia (3.5%). Six deaths occurred, none of which were related to treatment. Treatment discontinuation due to AEs (including clinical disease progression) occurred in 45 patients (39.5%). The most common AEs occurring in ≥3 patients leading to treatment discontinuation were fatigue (n = 5, 4.4%), dyspnea (n = 4, 3.5%), and autoimmune colitis, diarrhea, vomiting, and respiratory failure (each n = 3, 2.6%). Serious treatment-related AEs occurred in 24 patients (21.1%). The most common serious treatment-related AEs occurring in ≥2 patients were autoimmune colitis (n = 4, 3.5%), diarrhea, infusion-related reaction, and drug eruption rash (each n = 3, 2.6%), and colitis, erythema multiforme, and thrombocytopenia (each n = 2, 1.8%). Potentially immune-related, drug-related AEs were reported in 18 patients (including 4 patients each with 2 events), including autoimmune colitis (n = 4), pneumonitis (n = 4), hypothyroidism (n = 4), dactylitis (n = 2), and colitis, gastrointestinal inflammation, pancreatitis, alopecia, Stevens–Johnson syndrome, toxic epidermal necrolysis, myositis, and polymyalgia rheumatica (each n = 1; for details, see Supplementary Table S3). AEs of special interest included infusion-related reactions and immune-related AEs. Infusion-related reactions were reported for 36.8% (42/114) of patients, all of which were considered related to treatment. All but one were grade 1 or 2. A total of 22 immune-related AEs occurred in 18 patients. The most frequently reported events were autoimmune colitis, pneumonitis, and hypothyroidism (each n = 4). Details can be found in Supplementary Information. There were no unanticipated laboratory safety signals or any consistent or clinically meaningful changes in vital signs, physical examinations, ECOG performance status, or ECG parameters during treatment compared with baseline apart from one patient who experienced grade 1 treatment-related clinically significant QTc prolongation. Among the 96 patients who were evaluable for ADA testing for mogamulizumab, six (6.3%) were antibody or neutralizing antibody positive at baseline, of whom one was boosted by treatment: none of the remaining 90 patients who were antibody negative had a positive antibody response to mogamulizumab treatment. ADA testing for nivolumab was not undertaken (see Materials and Methods).
Table 3.
Adverse events.
| No. of patients (%) | |
|---|---|
| AE | 114 (100) |
| Treatment-related AE | 104 (91.2) |
| Grade ≥3 AE | 91 (79.8) |
| Treatment-related AE grade ≥3 | 49 (43.0) |
| Serious AE | 70 (61.4) |
| Treatment-related serious AE | 24 (21.1) |
| Discontinuation for AE | 45 (39.5) |
| Death | 6 (5.3) |
| Death from treatment-related AE | 0 |
| Most commona treatment-related AEs by preferred termb | |
| Drug eruption | 62 (54.4) |
| Infusion-related reaction | 42 (36.8) |
| Fatigue | 29 (25.4) |
| Diarrhea | 20 (17.5) |
| ALT increased | 16 (14.0) |
| AST increased | 16 (14.0) |
| Nausea | 13 (11.4) |
| Vomiting | 13 (11.4) |
| Pyrexia | 13 (11.4) |
| Anemia | 12 (10.5) |
| Arthralgia | 10 (8.8) |
| Lipase increased | 8 (7.0) |
| Alkaline phosphatase increased | 7 (6.1) |
| Most commonc treatment-related grade ≥3 AEs by preferred termb | |
| Drug eruption | 14 (12.3) |
| AST increased | 8 (7.0) |
| ALT increased | 6 (5.3) |
| Anemia | 4 (3.5) |
| Erythema multiforme | 3 (2.6) |
| Alkaline phosphatase increased | 3 (2.6) |
| Bilirubin increased | 3 (2.6) |
| Thombocytopenia | 3 (2.6) |
| Diarrhea | 3 (2.6) |
| Fatigue | 3 (2.6) |
| Bilirubin increased | 2 (1.8) |
| Diabetes mellitus | 2 (1.8) |
| Hyponatremia | 2 (1.8) |
Abbreviations: AE, adverse event; ALT, alanine aminotransferase; AST, aspartate aminotransferase; MedDRA, Medical Dictionary for Regulatory Activities.
aOccurring in ≥5% of patients overall.
bCoded by MedDRA v18.1.
cOccurring in ≥1 patient overall.
Antitumor activity
Tumor response as assessed by ORR (determined using RECIST criteria) is summarized in Table 4A. Among all of the 114 patients who received mogamulizumab 1 mg/kg + nivolumab 240 mg in phases I/II, ORR was 10.5% (95% CI, 5.6–17.7), with three CRs (two in ovarian cancer and one in hepatocellular carcinoma) and nine PRs. Thirty-three patients (28.9%) had SD.
Table 4.
Tumor response assessments.
| A. ORR | |||||
|---|---|---|---|---|---|
| ORR | CR | PR | SD | PD | |
| Study phase/tumor type | n (%; 95% CI)a | n (%) | n (%) | n (%) | n (%) |
| Phase I/II (N = 114) | 12 (10.5; 5.6–17.7) | 3 (2.6) | 9 (7.9) | 33 (28.9) | 69 (61.5) |
| Phase I (n = 4) | 1 (25.0; 0.6–80.6) | 0 | 1 (25.0)b | 0 | 3 (75.0) |
| Phase II | |||||
| SCCHN (n = 10) | 1 (10.0; 0.3–44.5) | 0 | 1 (10.0) | 4 (40.0) | 5 (50.0) |
| NSCLC, nonsquamous cell, PD-L1 nonexpressing (n = 4) | 1 (25.0; 0.6–80.6) | 0 | 1 (25.0) | 1 (25.0) | 2 (50.0) |
| NSCLC, squamous cell (n = 5) | 1 (20.0; 0.5–71.6) | 0 | 1 (20.0) | 2 (40.0) | 2 (40.0) |
| Ovarian/fallopian tube/primary peritoneal cancer (n = 21) | 3 (14.3; 3.0–36.3) | 2 (9.5) | 1 (4.8) | 7 (33.3) | 11 (52.4) |
| Hepatocellular carcinoma (n = 24) | 4 (16.7; 4.7–37.4) | 1 (4.2) | 3 (12.5) | 11 (45.8) | 9 (37.5) |
| Colorectal cancer, non-MSI high (n = 29) | 1 (3.4; 0.1–17.8) | 0 | 1 (3.4) | 6 (20.7) | 22 (75.9) |
| Pancreatic adenocarcinoma (n = 17) | 0 (0; 0.0–19.5) | 0 | 0 | 2 (11.8) | 15 (88.2) |
| B. TTR, DOR, PFS, and OS | |
|---|---|
| Mogamulizumab 1 mg/kg + Nivolumab 240 mg | |
| Endpoint | (N = 114) |
| TTR, months, median (range) | 3.26 (2.2–13.4) |
| DOR, months, medianc (95% CI) | - (5.1, –)d |
| PFS, months, medianc (95% CI) | 2.6 (2.3–3.1) |
| OS, months, medianc (95% CI) | 9.5 (5.9–13.5) |
Abbreviations: CI, confidence interval; CR, complete response; DOR, duration of response; MSI, microsatellite instability; NSCLC, non–small cell lung cancer; ORR, overall response rate; OS, overall survival; PD, progressive disease; PD-L1, programmed cell death ligand 1; PFS, progression-free survival; PR, partial response; SCCHN, squamous cell carcinoma of the head and neck; SD, stable disease; TTR, time to response.
aExact two-sided 95% CIs using Clopper–Pearson method.
bPatient with alveolar soft part sarcoma.
cKaplan–Meier estimate.
dMedian DOR for subjects with CR/PR only was 14.4 months while the Kaplan–Meier estimate of median DOR was not reached.
Among the most frequently enrolled tumor types, ORR was 16.7% (4/24) for HCC, 14.3% (3/21) for ovarian cancer, 3.4% (1/29) for non-MSI high colorectal cancer, 10.0% (1/10) for SCCHN, and 0% (0/17) for pancreatic cancer. ORRs by irRECIST were the same as those by RECIST except for ovarian cancer (19%, 4/21) and HCC (12.5%, 3/24): ORR for all 114 patients did not therefore differ comparing RECIST and irRECIST.
Tumor response as assessed by other efficacy endpoints is presented in Table 4B. For subjects with CR/PR only, median TTR was 3.26 (range, 2.2–13.4) months, and median DOR was 14.4 months, while the KM estimate of median DOR was not reached. Median PFS was 2.6 (95% CI, 2.3–3.1) months and median OS was 9.5 (95% CI, 5.9–13.5) months. Change in tumor burden over time (spider plots) for patients in phase I and each tumor-specific cohort in phase II are shown in Supplementary Fig. S2A–S2H. Kaplan–Meier curves for PFS and OS are shown in Supplementary Fig. S3.
None of the 110 patients in phase II had received prior anti–PD-1 or anti–PD-L1 treatment, which is consistent with the enrollment exclusion criteria.
During interim analysis, the threshold for further patient recruitment in phase II was not reached according to the Simon's 2-stage design (futility boundary for each tumor type) and the study was stopped.
Pharmacodynamics
Following treatment with mogamulizumab/nivolumab, four of eight patients evaluated by immunohistochemistry demonstrated a reduction in stromal CCR4+FoxP3+ cells, with most patients retaining similar numbers of FoxP3 single-positive cells (Fig. 1). Of 88 patients evaluated, CCR4+ effector Tregs (CD45RA−, high FoxP3) and CCR4+ nonsuppressive Tregs (CD45RA−, low FoxP3) were depleted from the peripheral blood in the majority of patients irrespective of response. In contrast, CCR4+ naïve Tregs were relatively stable following treatment (Fig. 2A), with few expressing CCR4 at baseline (Fig. 2B). Similarly, CCR4+CD4+ and CCR4+CD8+ cells were depleted irrespective of response (Fig. 3).
Figure 1.
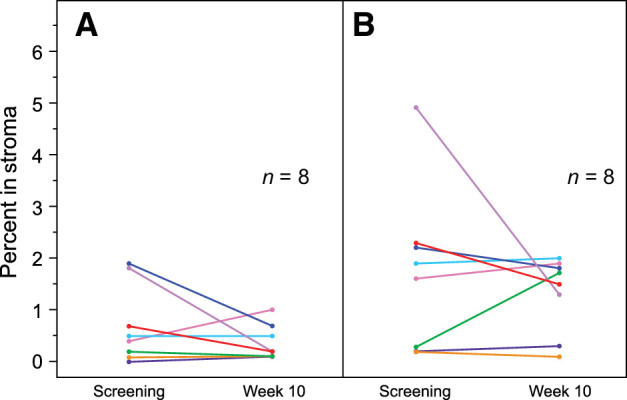
Expression of stromal CCR4+FoxP3+ and FoxP3+ cells in tumor stroma. A and B, The percentage of CCR4+FoxP3+ double-positive (A) and FoxP3+ single-positive stromal cells (B) at screening and at week 10.
Figure 2.

Peripheral blood CCR4+ effector Treg fractions. The percent change from baseline (A) and actual cell count (B) determined from flow cytometric analysis of peripheral blood effector Treg populations were determined prior to administration of drug at cycle 1 day 1 (C1D1), C2D1, C3D1, and C4D1. CR, complete response; PD, progressive disease; PR, partial response; SD, stable disease.
Figure 3.

Peripheral blood CCR4+ CD4 and CD8 cells. The percent change from baseline (A) and actual cell count (B) determined from flow cytometric analysis of peripheral blood CD4 and CD8 cell populations were determined prior to administration of drug at cycle 1 day 1 (C1D1), C2D1, C3D1, and C4D1. CR, complete response; PD, progressive disease; PR, partial response; SD, stable disease.
Discussion
Here, we present results from a multicenter, dose-finding (phase I) and dose expansion (phase II) study (NCT02705105) of mogamulizumab, an anti-CCR4 monoclonal antibody, in combination with nivolumab for patients with locally advanced or metastatic solid tumors. There were no DLTs at the starting dose level of mogamulizumab 1 mg/kg + nivolumab 240 mg in phase I and, therefore, this dose was carried forward for treatment of an additional 110 patients in phase II cohort expansion. The mogamulizumab 1 mg/kg dose was based on the marketed dose in T-cell lymphomas and the previous finding that mogamulizumab 1 mg/kg + nivolumab 3 mg/kg was the RP2D in a Japanese study in patients with advanced or metastatic solid tumors (26). All 114 patients treated in phases I and II were assessed for safety and efficacy.
For the primary endpoint, the combination of mogamulizumab and nivolumab had an acceptable safety profile. The most common treatment-related AEs grade ≥3 were drug eruption rash, transaminase increases, and anemia. The AE profile was similar to that previously reported for the combination of mogamulizumab and nivolumab (26).
With respect to efficacy, ORR was 10.5% (95% CI, 5.6–17.7), which included three CRs (two in ovarian cancer and one in hepatocellular carcinoma) and nine PRs. In the Japanese study of mogamulizumab 1 mg/kg + nivolumab 3 mg/kg, ORR was similar at 12.2% across 90 patients with advanced or metastatic solid tumors in six tumor-specific cohorts, which included NSCLC, SCLC, gastric cancer, esophageal cancer, HCC, and pancreatic cancer (each n = 15). The highest ORRs in the Japanese study were observed in patients with HCC and NSCLC at 27% and 20%, respectively. In the current study, ORRs among tumor subgroups that recruited larger numbers of patients were: HCC 16.7% (n = 24), ovarian cancer 14.3% (n = 21), SCCHN 10.0% (n = 10), non-MSI-high colorectal cancer 3.4% (n = 20), and pancreatic adenocarcinoma 0% (n = 17). Historical ORRs for single-agent nivolumab in patients with heavily pretreated recurrent, locally advanced, or metastatic cancer have generally been similar to those observed in the combination current study. Single-agent nivolumab reported ORRs were 20% for HCC (31, 32), 19.2% for SCCHN (33), 5.9% to 15% for ovarian cancer (34, 35), and 0% for pancreatic cancer or colorectal cancer (34). The combination of mogamulizumab with durvalumab, a PD-L1–blocking antibody did not enhance the efficacy of mogamulizumab (30). While several patients in our study developed durable responses, based on the current and prior studies of mogamulizumab in combination with checkpoint inhibitors, it is not possible to attribute enhanced clinical activity to mogamulizumab.
As reported previously, mogamulizumab effectively depletes cell populations expressing CCR4 (18, 30). Mogamulizumab-mediated depletion of CCR4-expressing immunosuppressive effector Treg cells in the periphery and in the tumor would be expected to augment nivolumab antitumor activity; however, this synergy was not observed. The lack of synergy may be due to the additional depletion of CCR4-expressing antitumor effector cell populations (i.e., CD8 and natural killer cells). Depletion of these populations were observed here and in other studies (18).
The combination of phase I with an extended phase II represents some limitation in terms of resources and costs. We applied full-size cancer type cohorts in order to determine, with a good level of certainty, a significant effect within each cohort, based on prespecified efficacy thresholds. This required a maximum sample size of 188 patients across all cancer types and represents a significant increase from the usual sample size in phase II studies. However, the objective of this study design was to discover the cohort(s) in which the combination could be efficacious. Based on prespecified criteria for stage 1 and other considerations we stopped the study with 114 patients, in this way reducing somewhat the costs of extended phase II of the study.
In conclusion, the efficacy of nivolumab was not enhanced by the addition of mogamulizumab in patients with advanced solid tumors.
Authors' Disclosures
D.S. Hong reports research (Inst)/grant funding (Inst) from AbbVie, Adaptimmune, Aldi-Norte, Amgen, AstraZeneca, Bayer, BMS, Daiichi-Sankyo, Deciphera, Erasca, Fate Therapeutics, Genentech, Genmab, Infinity, Kyowa, Lilly, LOXO, Merck, Medimmune, Mirati, Mologen, Navier, NCI-CTEP, Novartis, Numab, Pfizer, Pyramid Bio, SeaGen, Takeda, Turning Point Therapeutics, Verstatem, and VM Oncology; travel, accommodations, and expenses from Bayer, Genmab, AACR, ASCO, SITC, and Telperian; consulting, speaker, or advisory role with Adaptimmune, Alpha Insights, Acuta, Alkermes, Amgen, Aumbiosciences, Atheneum, Axiom, Barclays, Baxter, Bayer, Boxer Capital, BridgeBio, CDR-life AG, COR2ed, COG, Ecor1, Genentech, Gilead, GLG, Group H, Guidepoint, HCW Precision, Immunogen, Infinity, Janssen, Kymera, Liberium, Medscape, Numab, Oncologia Brasil, Pfizer, Pharma Intelligence, POET Congress, Prime Oncology, Seattle Genetics, ST Cube, Takeda, Tavistock, Trieza Therapeutics, Turning Point, WebMD, and Ziopharm; and other ownership interests from OncoResponse (Founder) and Telperian Inc (Advisor). O. Rixe reports grants from Kyowa Kirin during the conduct of the study. P.M. Forde reports grants from Kyowa during the conduct of the study as well as personal fees from Amgen; grants and personal fees from AstraZeneca and BMS; personal fees from Iteos, Janssen, Daiichi, and Delfi; grants and personal fees from Novartis; personal fees from Sanofi, F Star, Surface Oncology, G1 Therapeutics, and Genentech; grants from Corvus; and other support from Flame and Polaris outside the submitted work. Y. Lou reports personal fees from AstraZeneca, Janssen Pharmaceutical, Lilly, and Turning Point and grants from Merck, MacroGenics, Blueprint Medicines, Kyowa Pharmaceuticals, Tesaro, Bayer HealthCare, Daiichi Sankyo, and Mirati Therapeutics outside the submitted work. R. Leidner reports grants and personal fees from Bristol Myers Squibb; grants from Clinigen and Celldex; and personal fees from Merck, Sanofi, and Oncolys outside the submitted work. A. Collaku reports employment with Kyowa Kirin, Inc. during the time of this research. F.E. Fox reports other support from Kyowa Kirin, Inc. during the conduct of the study as well as other support from Kyowa Kirin, Inc. outside the submitted work and employment with Kyowa Kirin, Inc. M.A. Marshall reports employment from Kyowa Kirin, Inc. during the time of this research. A.J. Olszanski reports other support from Kyowa during the conduct of the study as well as personal fees from Merck, BMS, Pfizer, Array, Takeda, Novartis, Alkermes, Sanofi, and Eisai outside the submitted work, and clinical trial work (Institution compensated for work performed - Recruiting pts, treatment pts): Adaptimmune, Alkermes, Astellas, BMS, Checkmate, EMD Serono, Gan & Lee, GlycoNex, Immunocore, Intensity, Kadmon, Kartos, Kura, Kyowa, Oncoceutics, Sound Biologics, SpringBank, Takeda, and Targovax. No disclosures were reported by the other authors.
Acknowledgments
Medical writing and editorial support were provided by P.A. Todd of Tajut Ltd. and S.E. Johnson of S.E. Johnson Consulting, LLC, which was funded by Kyowa Kirin, Inc. This study was sponsored by Kyowa Kirin, Inc.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
Note: Supplementary data for this article are available at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/).
Authors' Contributions
D.S. Hong: Investigation, writing–original draft, writing–review and editing. O. Rixe: Investigation, writing–original draft, writing–review and editing. V.K. Chiu: Investigation, writing–original draft, writing–review and editing. P.M. Forde: Investigation, writing–original draft, writing–review and editing. T. Dragovich: Investigation, writing–original draft, writing–review and editing. Y. Lou: Investigation, writing–original draft, writing–review and editing. A. Nayak-Kapoor: Investigation, writing–original draft, writing–review and editing. R. Leidner: Investigation, writing–original draft, writing–review and editing. J.N. Atkins: Investigation, writing–original draft, writing–review and editing. A. Collaku: Formal analysis, writing–original draft, writing–review and editing. F.E. Fox: Formal analysis, supervision, investigation, methodology, writing–original draft, writing–review and editing. M.A. Marshall: Conceptualization, supervision, writing–original draft, writing–review and editing. A.J. Olszanski: Investigation, writing–original draft, writing–review and editing.
References
- 1. Salmaninejad A, Khoramshahi V, Azani A, Soltaninejad E, Aslani S, Zamani MR, et al. PD-1 and cancer: molecular mechanisms and polymorphisms. Immunogenetics 2018;70:73–86. [DOI] [PubMed] [Google Scholar]
- 2. Seliger B. Basis of PD1/PD-L1 therapies. J Clin Med 2019;pii:E2168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Han Y, Liu D, Li L. PD-1/PD-L1 pathway: current researches in cancer. Am J Cancer Res 2020;10:727–42. [PMC free article] [PubMed] [Google Scholar]
- 4. Shang B, Liu Y, Jiang SJ, Liu Y. Prognostic value of tumor-infiltrating FoxP3+ regulatory T cells in cancers: a systematic review and meta-analysis. Sci Rep 2015;5:15179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yoshie O, Matsushima K. CCR4 and its ligands: from bench to bedside. Int Immunol 2015;27:11–20. [DOI] [PubMed] [Google Scholar]
- 6. Ferenczi K, Fuhlbrigge RC, Pinkus J, Pinkus GS, Kupper TS. Increased CCR4 expression in cutaneous T cell lymphoma. J Invest Dermatol 2002;119:1405–10. [DOI] [PubMed] [Google Scholar]
- 7. Ishida T, Utsunomiya A, Iida S, Inagaki H, Takatsuka Y, Kusumoto S, et al. Clinical significance of CCR4 expression in adult T-cell leukemia/lymphoma: its close association with skin involvement and unfavorable outcome. Clin Cancer Res 2003;9:3625–34. [PubMed] [Google Scholar]
- 8. Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med 2004;10:942–9. [DOI] [PubMed] [Google Scholar]
- 9. Gobert M, Treilleux I, Bendriss-Vermare N, Bachelot T, Goddard-Leon S, Arfi V, et al. Regulatory T cells recruited through CCL22/CCR4 are selectively activated in lymphoid infiltrates surrounding primary breast tumors and lead to an adverse clinical outcome. Cancer Res 2009;69:2000–9. [DOI] [PubMed] [Google Scholar]
- 10. Olkhanud PB, Baatar D, Bodogai M, Hakim F, Gress R, Anderson RL, et al. Breast cancer lung metastasis requires expression of chemokine receptor CCR4 and regulatory T cells. Cancer Res 2009;69:5996–6004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Watanabe Y, Katou F, Ohtani H, Nakayama T, Yoshie O, Hashimoto K. Tumor-infiltrating lymphocytes, particularly the balance between CD8+ T cells and CCR4+ regulatory T cells, affect the survival of patients with oral squamous cell carcinoma. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 2010;109:744–52. [DOI] [PubMed] [Google Scholar]
- 12. Svensson H, Olofsson V, Lundin S, Yakkala C, Björck S, Börjesson L, et al. Accumulation of CCR4+CTLA-4hiFOXP3+CD25hi regulatory T cells in colon adenocarcinomas correlate to reduced activation of conventional T cells. PLoS One 2012;7:e30695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Spranger S, Spaapen RM, Zha Y, Williams J, Meng Y, Ha TT, et al. Up-regulation of PD-L1, IDO, and Tregs in the melanoma tumor microenvironment is driven by CD8+ T cells. Sci Transl Med 2013;5:200ra116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Berlato C, Khan MN, Schioppa T, Thompson R, Maniati E, Montfort A, et al. A CCR4 antagonist reverses the tumor-promoting microenvironment of renal cancer. J Clin Invest 2017;127:801–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cheng X, Wu H, Jin ZJ, Ma D, Yuen S, Jing XQ, et al. Up-regulation of chemokine receptor CCR4 is associated with human hepatocellular carcinoma malignant behavior. Sci Rep 2017;7:12362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Maolake A, Izumi K, Shigehara K, Natsagdorj A, Iwamoto H, Kadomoto S, et al. Tumor-associated macrophages promote prostate cancer migration through activation of the CCL22-CCR4 axis. Oncotarget 2017;8:9739–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Karasaki T, Qiang G, Anraku M, Sun Y, Shinozaki-Ushiku A, Sato E, et al. High CCR4 expression in the tumor microenvironment is a poor prognostic indicator in lung adenocarcinoma. J Thorac Dis 2018;10:4741–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kurose K, Ohue Y, Wada H, Iida S, Ishida T, Kojima T, et al. Phase Ia study of FoxP3+ CD4 Treg depletion by infusion of a humanized anti-CCR4 antibody, KW-0761, in cancer patients. Clin Cancer Res 2015;21:4327–36. [DOI] [PubMed] [Google Scholar]
- 19. Cohen J, Sznol M. Therapeutic combinations of immune-modulating antibodies in melanoma and beyond. Semin Oncol 2015;42:488–94. [DOI] [PubMed] [Google Scholar]
- 20. Pennock GK, Chow LQ. The evolving role of immune checkpoint inhibitors in cancer treatment. Oncologist 2015;20:812–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Antonia SJ, López-Martin JA, Bendell J, Ott PA, Taylor M, Eder JP, et al. Nivolumab alone and nivolumab plus ipilimumab in recurrent small-cell lung cancer (CheckMate 032): a multicentre, open-label, phase 1/2 trial. Lancet Oncol 2016;17:883–95. [DOI] [PubMed] [Google Scholar]
- 22. Takeuchi Y, Nishikawa H. Roles of regulatory T cells in cancer immunity. Int Immunol 2016;28:401–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wang H, Franco F, Ho PC. Metabolic regulation of Tregs in cancer: opportunities for immunotherapy. Trends Cancer 2017;3:583–92. [DOI] [PubMed] [Google Scholar]
- 24. Yan S, Zhang Y, Sun B. The function and potential drug targets of tumour-associated Tregs for cancer immunotherapy. Sci China Life Sci 2019;62:179–86. [DOI] [PubMed] [Google Scholar]
- 25. OPDIVO® (nivolumab) injection, for intravenous use. Prescribing information. Princeton, NJ: Bristol-Myers Squibb Company; 2021. [Google Scholar]
- 26. Doi T, Muro K, Ishii H, Kato T, Tsushima T, Takenoyama M, et al. A phase I study of the anti-CC chemokine receptor 4 antibody, mogamulizumab, in combination with nivolumab in patients with advanced or metastatic solid tumors. Clin Cancer Res 2019;25:6614–22. [DOI] [PubMed] [Google Scholar]
- 27. Zhao X, Suryawanshi S, Hruska M, Feng Y, Wang X, Shen J, et al. Assessment of nivolumab benefit-risk profile of a 240-mg flat dose relative to a 3-mg/kg dosing regimen in patients with advanced tumors. Ann Oncol 2017;28:2002–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, et al. New response evaluation criteria in solid tumors: revised RECIST guideline (version 1.1). Eur J Cancer 2009;45:228–47. [DOI] [PubMed] [Google Scholar]
- 29. Bohnsack O, Hoos A, Ludajic K. Adaptation of the immune related response criteria: irRECIST. Ann Oncol 2014;25:iv369.[abstract 1070P]. [Google Scholar]
- 30. Zamarin D, Hamid O, Nayak-Kapoor A, Sahebjam S, Sznol M, Collaku A, et al. Mogamulizumab in combination with durvalumab or tremelimumab in patients with advanced solid tumors: a phase I study. Clin Can Res 2020;26:4531–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. El-Khoueiry AB, Sangro B, Yau T, Crocenzi TS, Kudo M, Hsu C, et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): an open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet 2017;389:2492–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ferris RL, Blumenschein G, Jr FJ, Guigay J, Colevas AD, Licitra L, et al. Nivolumab vs investigator's choice in recurrent or metastatic squamous cell carcinoma of the head and neck: 2-year long-term survival update of CheckMate 141 with analyses by tumor PD-L1 expression. Oral Oncol 2018;81:45–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gillison ML, Blumenschein G Jr, Fayette J, Guigay J, Colevas AD, Licitra L, et al. CheckMate 141: 1-year update and subgroup analysis of nivolumab as first-line therapy in patients with recurrent/metastatic head and neck cancer. Oncologist 2018;23:1079–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med 2012;366:2455–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hamanishi J, Mandai M, Ikeda T, Minami M, Kawaguchi A, Murayama T, et al. Safety and antitumor activity of anti-PD-1 antibody, nivolumab, in patients with platinum-resistant ovarian cancer. J Clin Oncol 2015;33:4015–22. [DOI] [PubMed] [Google Scholar]