

Abstract
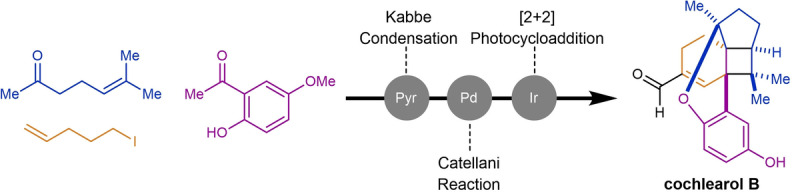
A 14‐step synthesis of (+)‐cochlearol B is reported. This renoprotective meroterpenoid features a unique core structure containing a densely substituted cyclobutane ring with three stereocenters. Our strategy employed an organocatalytic Kabbe condensation in route to the key chromenyl triflate. A subsequent Catellani reaction incorporated the remaining carbon atoms featured in the skeleton of cochlearol B. An ensuing visible‐light‐mediated [2+2] photocycloaddition closed the cyclobutane and formed the central bicyclo[3.2.0]heptane core. Notably, careful design and tuning of the Catellani and photocycloaddition reactions proved crucial in overcoming undesired reactivity, including cyclopropanation reactions and [4+2] cycloadditions.
Keywords: Catellani Reaction, Meroterpenoids, Total Synthesis, Visible Light, [2+2] Photocycloaddition
A 14‐step approach to (+)‐cochlearol B is reported. The strategy involves an organocatalytic Kabbe condensation, Catellani reaction, and visible‐light‐mediated [2+2] cycloaddition to rapidly access the core of this natural product. Careful design and tuning of the Catellani and photocycloaddition reactions proved crucial in overcoming undesired reactivity, including cyclopropanation reactions, and [4+2] cycloadditions.
In 2014, Cheng and co‐workers reported the isolation of cochlearol A (1) together with cochlearol B (2) from Ganoderma cochlear (Scheme 1A). [1] Their studies were initially inspired by the known pharmacological effects of Ganoderma extracts, which are used in traditional Chinese medicine for the prevention and treatment of cancer, hypertension, chronic bronchitis, and asthma. [2] In addition to cochlearol A and B, a number of other structurally diverse meroterpenoids have been isolated from Ganoderma cochlear, including ganocin B (3, Scheme 1A). [3] In comparison to cochlearol B (2), cochlearol A (1) is structurally less complex, incorporating a dioxaspiro[4.5]decane moiety. The structure of cochlearol B (2) was originally deduced based on NMR and HRMS analysis that showed a 4/5/6/6/6‐fused polycyclic ring system with a central hepta‐substituted cyclobutane core, which includes three stereogenic centers and three quaternary carbon atoms. Both cochlearol B (2) and ganocin B (3) contain a common chromane core; however ganocin B possesses a structurally distinct spiro[4,5]decane ring. [3] Notably, both cochlearol A (1) and cochlearol B (2) were isolated as racemates and tested for renoprotective effects on renal fibrosis by inhibiting upregulation of collagen I, fibronectin, and α‐SMA. [1] Interestingly, only (−)‐cochlearol B (2) demonstrated potent antifibrotic efficacy while (−)‐1, (+)‐1, and (+)‐2 were found to be inactive. Furthermore, additional studies suggested that (−)‐2 efficiently inhibits the phosphorylation of Smad2 (Small Mothers Against Decapentaplegic) and Smad3 and consequently disrupts Smad2 and Smad3 activation whereas (+)‐2 does not. While both cochlearol A (1) and ganocin B (3) have been the target of multiple established synthetic strategies[ 4 , 5 ] only one approach to cochlearol B (2) has been reported [6] despite its unique architecture.
Scheme 1.
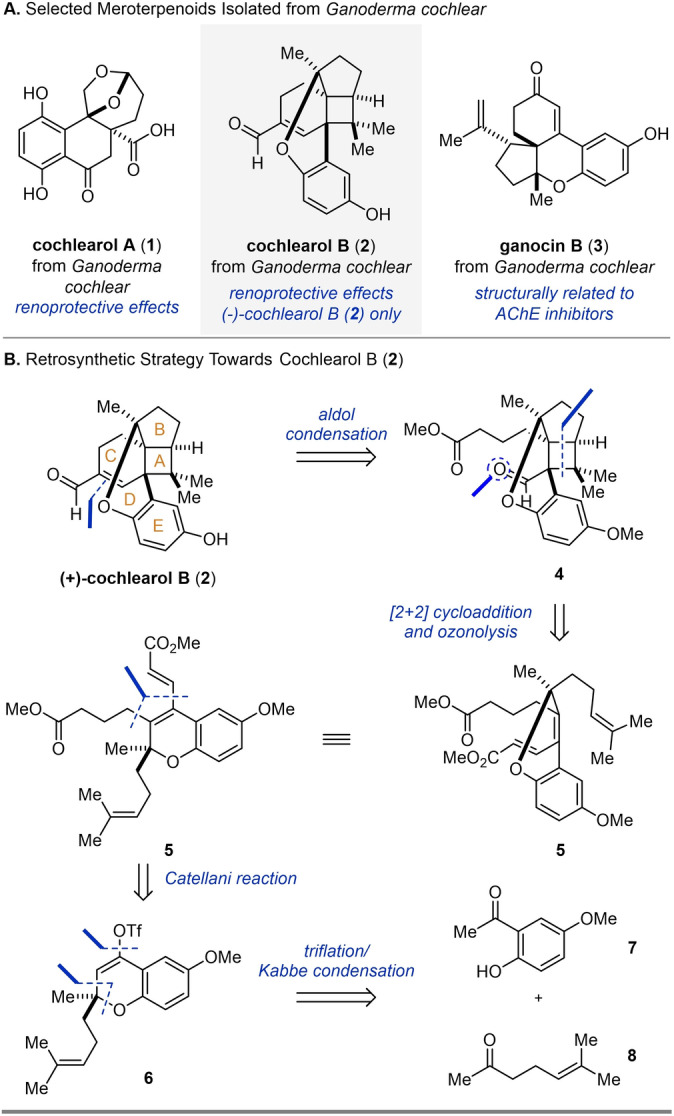
A) Ganoderma meroterpenoids including cochlearol B (2). B) Retrosynthetic strategy towards cochlearol B (2) relying on Catellani and [2+2] cycloaddition reactions. Proceeds through an EDBAC ring formation sequence.
The start of our retrosynthetic analysis of cochlearol B (2) relied on an intramolecular aldol condensation to form the α,β‐unsaturated aldehyde moiety from cyclobutane 4 (Scheme 1B). We envisioned building both the A and B ring systems simultaneously in an intramolecular, visible‐light‐mediated [2+2] cycloaddition of chromene 5. Introduction of the two methyl ester fragments in 5 could proceed concomitantly in a palladium‐ catalyzed Catellani reaction[ 7 , 8 , 9 ] of triflate 6. This represents one of the more complex precursors used in this class of transformations to date.[ 10 , 11 , 12 , 13 , 14 , 15 , 16 , 17 , 18 , 19 , 20 , 21 , 22 , 23 ] Triflate 6 is accessible through a two‐step sequence of triflation preceded by Kabbe condensation [24] of commercially available precursors phenol 7 and sulcatone (8).
In initial studies towards cochlearol B (2), we were able to access 6‐methoxy‐2‐methyl‐4H‐chromen‐4‐one (9) via a one‐pot acylation, Baker–Venkataraman rearrangement, [25] and condensation of 7 using conditions developed by Brown and co‐workers. [26] A subsequent 1,4‐conjugate addition [27] of 9 with homoprenyl magnesium bromide (10) in the presence of catalytic amounts of CuBr⋅(SMe2) initially gave rise to chromanone 11 in 34 % yield. Forming the corresponding pyrilium ion of 9 upon addition of stoichiometric amounts of TMSCl [28] proved beneficial and increased the yield of 11 to 61 % (Scheme 2). The subsequent triflation of chromanone 11 proved more challenging than expected. In addition to isolating 59 % of vinyl triflate 6, cyclopentylchromene 12 was isolated in 35 % yield. The structure of 12 was unambiguously confirmed via X‐ray crystallographic analysis of guanidinium sulfate derivative 13.[ 29 , 30 ] This tricyclic structure is also featured in ganocin B (3), as well as related natural products ganocins A and C. [3] To overcome this undesired reactivity, Comins’ reagent [31] was evaluated as an alternative to phenyltriflimide. This more reactive triflating agent enabled the reaction to proceed at cryogenic temperatures in shorter reaction times, eliminated the formation of 12, and improved the yield of 6 up to 86 % (Scheme 5).
Scheme 2.
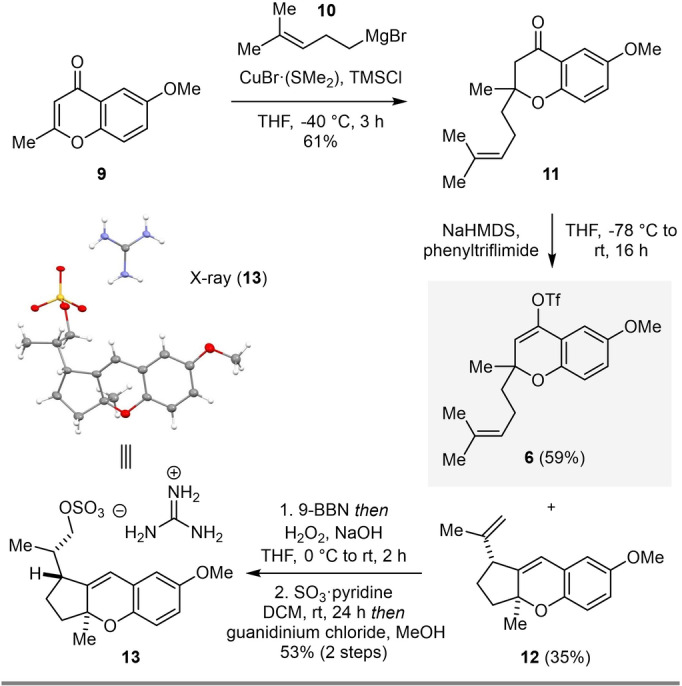
Triflation of chromanone 11 yields unexpected cyclopentyl‐chromene 12, which is also found in ganocin B (3). TMSCl=trimethylsilyl chloride, NaHMDS=sodium bis(trimethylsilyl)amide, 9‐BBN=9‐borabicyclo[3.3.1]nonane.
Scheme 5.

Development of an efficient strategy towards cochlearol B (2) relying on a Catellani reaction and visible‐light‐mediated [2+2] cycloaddition. Comins’ reagent=N‐(5‐chloro‐2‐pyridyl)bis(trifluoromethanesulfonimide), NMMO=N‐methylmorpholine N‐oxide, DMF‐DMA=N,N‐dimethylformaide dimethyl acetal.
Vinyl triflate 6 was subsequently subjected to Catellani conditions [9] to enable concomitant ortho and ipso functionalization to provide tetrasubstituted alkene 5 in 31 % yield (Scheme 3A). However, efforts to optimize this transformation could not overcome the formation of undesired byproduct 15, which forms in up to 11 % yield, likely through a thermal [4+2] cycloaddition. Importantly, further studies confirmed that 15 forms exclusively upon heating of 16 to 100 °C in dioxane, which is consistent with the [4+2] cycloaddition hypothesis. Compound 16 likely forms in situ via a direct Heck reaction [32] of vinyl triflate 6 and methyl acrylate that competes with the desired Catellani reaction. Unfortunately, when tetrasubstituted alkene 5 was subjected to visible‐light‐mediated [2+2] cycloaddition conditions using [Ir(dF(CF3)ppy)2(dtbbpy)]‐(PF6)](17) as a photocatalyst,[ 33 , 34 ] none of the desired cyclobutane 18 was formed. The only product isolated was identified as cyclopropane 19 in 85 % yield (Scheme 3B). [35] We hypothesize that this unexpected product arises upon initial photochemical excitation of the styrenyl olefin in 5 to its excited state 20. The resulting biradical subsequently reacts with the homoprenyl subunit to form ring B (21). From there, instead of forming the desired cyclobutane (ring A) by radical recombination, an addition to the electrophilic carbon of the methyl acrylate fragment occurs, resulting in a second five‐membered ring (22) and ultimately providing cyclopropane 19 upon radical recombination (Scheme 3C). Notably, the evaluation of multiple photocatalysts exhibiting distinct triplet energies [36] (e.g. [Ru(bpy)3](PF6)2, [Ir(dF(CF3)ppy)2(dtbbpy)](PF6) (17)), as well as direct excitation with UV light, did not provide cyclobutane 18; rather cyclopropane 19 remained the exclusive product. To gain support for this mechanistic hypothesis, as well as investigate how to overcome this undesired reactivity, we next evaluated the role of the steric and electronic effects of the substituents by subjecting 16 to the conditions for [2+2] cycloadditions (Scheme 4). Although 16 is electronically comparable to 5, no formation of cyclopropane 23 was observed. Instead, a mixture of the [2+2] and [4+2] cycloadducts 24 and 15+25 were isolated in 29 % and 32 % yield, respectively. These results suggest that steric constraints of the methyl acrylate and methyl butyrate chains favor the formation of cyclopropane 19 over cyclobutane 18. This is consistent with 19 being isolated as a single diastereomer with the cyclopropane and methyl butyrate chains on opposite faces. Furthermore, in addition to the steric constraints, we hypothesized that the electrophilic nature of the acrylate moiety in 21, together with the high stability of the resulting biradical in 22, favors the formation of cyclopropane 19. To enable a synthesis of enantioenriched cochlearol B (2), we revised our synthetic strategy. Specifically, we postulated that a less reactive alkene could mitigate the competing Heck reaction in the Catellani step, while a conformationally restricted diene would be expected to prevent undesired [4+2] cycloadditions. Our final approach to (+)‐cochlearol B (2) takes advantage of these insights and through a revised design for the Catellani reaction to ultimately enable a productive [2+2] cycloaddition by disfavoring competing Heck reaction, [4+2] cycloaddition, and cyclopropanation (Scheme 5). Additionally, a Kabbe condensation [24] was employed to access chromanone 11, which reduced the overall number of steps.
Scheme 3.

Challenges observed in developing a Catellani and subsequent [2+2] cycloaddition approach towards cochlearol B (2).
Scheme 4.
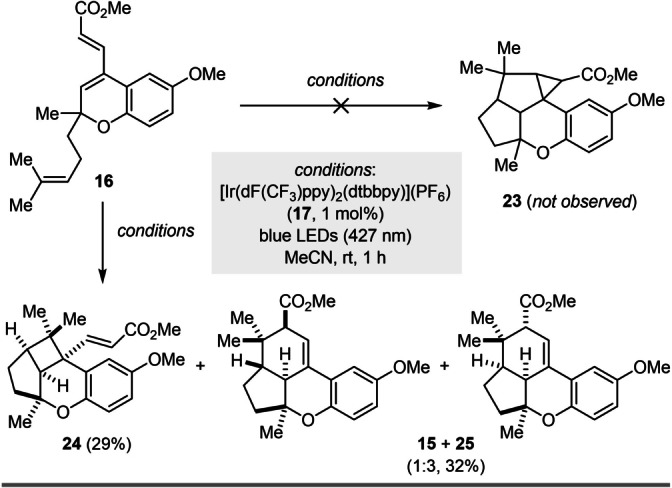
Proof‐of‐principle for a visible‐light‐enabled [2+2] cycloaddition strategy towards cochlearol B (2).
Specifically, a pyrrolidine‐catalyzed condensation between 7 and 8 formed chromanone 11 directly in 76 % yield. Conducting the transformation with chiral pyrrolidine and imidazolinone catalysts [37] provided enantioenriched (−)‐11 albeit with a modest enantioselectivity of 23 %. [38] In comparison, an approach relying on a chiral resolution of chromanone 11 with (R)‐tert‐butanesulfinamide [39] (26) proved superior resulting in the desired product in 95 % ee. Subsequent treatment with Comins’ reagent provided triflate (+)‐6 in 86 % yield. Subjecting (+)‐6 to Catellani conditions with commercially available 5‐iodo‐1‐pentene, [9] which functions as both the nucleophilic and electrophilic coupling partner, gives rise to chromene 28 establishing ring C of (+)‐cochlearol B (2) as well as incorporating an s‐trans diene. This intermediate was expected to exhibit distinct advantages compared to chromene 5. In particular: 1) the locked s‐trans conformation of the diene in 28 prevents the formation of a competing thermal [4+2] cycloadduct under Catellani reaction conditions, while 2) the absence of a methyl acrylate moiety disfavors cyclopropanation, as the alkene is now less electrophilic and the resulting radical is no longer stabilized by an adjacent carbonyl; 3) forming ring C prior to the [2+2] cycloaddition reduces the steric constraints that previously precluded the formation of cyclobutane 18. This also represents a change in our overall ring formation strategy, shifting from an EDBAC sequence to an EDCBA sequence. Importantly, the first supposition was reinforced with the isolation of chromene 28 as the exclusive product. Remarkably, this reaction was amenable to gram scale resulting in the formation of the desired product in 81 % yield. With a viable route to the photocycloaddition precursor established, diene 28 was subjected to visible‐light‐enabled [2+2] cycloaddition conditions giving rise to the pentacyclic cyclobutane 29 as the sole product in 94 % yield. Notably, irradiation of chromene 28 with UV‐light in the absence of a photocatalyst failed to provide the desired product 29. The terminal alkene in 29 was next converted in a two‐step dihydroxylation [40] and oxidative cleavage [41] sequence to ketone 30 in 40 % overall yield. In order to incorporate the desired α,β‐unsaturated aldehyde of cochlearol B (2), ketone 30 was first subjected to a condensation reaction with DMF‐DMA, [42] giving enaminone 31 in 86 % yield. Tandem triflation and hydrolysis, [43] followed by a palladium catalyzed reduction, [44] provided 32 in 76 % yield over two steps. Completion of the synthesis of (+)‐cochlearol B (2) required deprotection of the phenol in 32, which proved challenging due to its instability under Lewis acidic and nucleophilic demethylation conditions. However, following a reduction of the aldehyde with NaBH4, demethylation of the phenol was achieved upon treatment with neat MeMgI at elevated temperatures. [45] A final Swern oxidation completed the total synthesis of (+)‐cochlearol B (2) in 25 % yield over the final 3 steps and in 14 overall steps from commercially available materials.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
We thank the National Science Foundation (NSF CHE‐1654223), the Alfred P. Sloan Foundation (CSS), the David and Lucile Packard Foundation (CSS) the Camille and Henry Dreyfus Foundation (CSS) for financial support. ADR thanks BMS for a predoctoral fellowship and the Rackham Graduate School of the University of Michigan for a dissertation fellowship.
A. D. Richardson, T. R. Vogel, E. F. Traficante, K. J. Glover, C. S. Schindler, Angew. Chem. Int. Ed. 2022, 61, e202201213; Angew. Chem. 2022, 134, e202201213.
A previous version of this manuscript has been deposited on a preprint server (https://doi.org/10.26434/chemrxiv‐2022‐22351).
Data Availability Statement
The data that support the findings of this study are available in the Supporting Information of this article.
References
- 1. Dou M., Di L., Zhou L.-L., Yan Y.-M., Wang X.-L., Zhou F.-J., Yang Z.-L., Li R.-T., Hou F.-F., Cheng Y. X., Org. Lett. 2014, 16, 6064–6067. [DOI] [PubMed] [Google Scholar]
- 2. Russell R., Paterson M., Phytochemistry 2006, 67, 1985–2001. [DOI] [PubMed] [Google Scholar]
- 3. Peng X.-R., Liu J.-Q., Wan L.-S., Li X.-N., Yan Y.-X., Qiu M.-H., Org. Lett. 2014, 16, 5262–5265. [DOI] [PubMed] [Google Scholar]
- 4.For approaches towards cochlearol A, see:
- 4a. Zhang D.-W., Xu W.-D., Fan H.-L., Liu H.-M., Chen D., Liu D.-D., Qin H.-B., Org. Lett. 2019, 21, 6761–6764; [DOI] [PubMed] [Google Scholar]
- 4b. Naruse K., Katsuta R., Yajima A., Nukada T., Watanabe H., Ishigami K., Tetrahedron Lett. 2020, 61, 151845; [Google Scholar]
- 4c. Venkatesh T., Mainkar P. S., Chandrasekhar S., J. Org. Chem. 2021, 86, 5412–5416. [DOI] [PubMed] [Google Scholar]
- 5.For approaches towards ganocin B, see:
- 5a. Liu Y., Zhou C.-J., Li Q., Wang H., Org. Biomol. Chem. 2016, 14, 10362–10365; [DOI] [PubMed] [Google Scholar]
- 5b. Shao H., Gao X., Wang Z.-T., Gao Z., Zhao Y.-M., Angew. Chem. Int. Ed. 2020, 59, 7419–7424; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 7489–7494. [Google Scholar]
- 6. Mashiko T., Shingai Y., Sakai J., Kamo S., Adachi S., Matsuzawa A., Sugita K., Angew. Chem. Int. Ed. 2021, 60, 24484–24487; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 24689–24692. [Google Scholar]
- 7. Catellani M., Frignani F., Rangoni A., Angew. Chem. Int. Ed. Engl. 1997, 36, 119–122; [Google Scholar]; Angew. Chem. 1997, 109, 142–145. [Google Scholar]
- 8.
- 8a. Catellani M., Motti E., Della Ca’ N., Acc. Chem. Res. 2008, 41, 1512–1522; [DOI] [PubMed] [Google Scholar]
- 8b. Della Ca’ N., Fontana M., Motti E., Catellani M., Acc. Chem. Res. 2016, 49, 1389–1400. [DOI] [PubMed] [Google Scholar]
- 9. Wang J., Dong Z., Yang C., Dong G., Nat. Chem. 2019, 11, 1106–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lautens M., Piguel S., Angew. Chem. Int. Ed. 2000, 39, 1045–1046; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2000, 112, 1087–1088. [Google Scholar]
- 11. Catellani M., Motti E., Baratta S., Org. Lett. 2001, 3, 3611–3614. [DOI] [PubMed] [Google Scholar]
- 12. Faccini F., Motti E., Catellani M., J. Am. Chem. Soc. 2004, 126, 78–79. [DOI] [PubMed] [Google Scholar]
- 13. Blaszykowski C., Aktoudianakis E., Bressy C., Alberico D., Lautens M., Org. Lett. 2006, 8, 2043–2045. [DOI] [PubMed] [Google Scholar]
- 14. Gericke K. M., Chai D. I., Bieler N., Lautens M., Angew. Chem. Int. Ed. 2009, 48, 1447–1451; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 1475–1479. [Google Scholar]
- 15. Martins A., Mariampillai B., Lautens M. in C-H Activation Topics in Current Chemistry, Vol. 292 (Eds.: Yu J. Q., Shi Z.), Springer, Berlin, 2010, pp. 1–33. [DOI] [PubMed] [Google Scholar]
- 16. Khanna A., Premachandra I. D. U. A., Sung P. D., Van Vranken D. L., Org. Lett. 2013, 15, 3158–3161. [DOI] [PubMed] [Google Scholar]
- 17. Zhang H., Chen P., Liu G., Angew. Chem. Int. Ed. 2014, 53, 10174–10178; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 10338–10342. [Google Scholar]
- 18. Ye J., Lautens M., Nat. Chem. 2015, 7, 863–870. [DOI] [PubMed] [Google Scholar]
- 19. Qureshi Z., Schlundt W., Lautens M., Synthesis 2015, 47, 2446–2456. [Google Scholar]
- 20. Dong Z., Wang J., Ren Z., Dong G., Angew. Chem. Int. Ed. 2015, 54, 12664–12668; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 12855–12859. [Google Scholar]
- 21. Liu Z.-S., Gao Q., Cheng H.-G., Zhou Q., Chem. Eur. J. 2018, 24, 15461–15476. [DOI] [PubMed] [Google Scholar]
- 22. Yamamoto Y., Murayama T., Jiang J., Yasui T., Shibuya M., Chem. Sci. 2018, 9, 1191–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wang J., Dong G., Chem. Rev. 2019, 119, 7478–7528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kapuriya N. P., Bhalodia J. J., Ambasana M. A., Patel R. B., Bapodra A. H., J. Heterocycl. Chem. 2020, 57, 3369–3374. [Google Scholar]
- 25.
- 25a. Baker W., J. Chem. Soc. 1933, 1381–1389; [Google Scholar]
- 25b. Mahal H. S., Venkataraman K., J. Chem. Soc. 1934, 1767–1769; [Google Scholar]
- 25c. Ameen D., Snape T. J., Synthesis 2015, 47, 141–158. [Google Scholar]
- 26. Abdel Ghani S. B., Mugisha P. J., Wilcox J. C., Gado E. A. M., Medu E. O., Lamb A. J., Brown R. C. D., Synth. Commun. 2013, 43, 1549–1556. [Google Scholar]
- 27. Gallen M. J., Williams C. M., Org. Lett. 2008, 10, 713–715. [DOI] [PubMed] [Google Scholar]
- 28. Jeong Y., Moon Y., Hong S., Org. Lett. 2015, 17, 3252–3255. [DOI] [PubMed] [Google Scholar]
- 29. Brummel B. R., Lee K. G., McMillen C. D., Kolis J. W., Whitehead D. C., Org. Lett. 2019, 21, 9622–9627. [DOI] [PubMed] [Google Scholar]
- 30.Deposition Number 2142345 contains the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service.
- 31. Comins D. L., Dehghani A., Tetrahedron Lett. 1992, 33, 6299–6302. [Google Scholar]
- 32.
- 32a. Scott W. J., Peña M. R., Swärd K., Stoessel S. J., Stille J. K., J. Org. Chem. 1985, 50, 2302–2308; [Google Scholar]
- 32b. Reddy C. R., Srikanth B., Rao N. N., Shing D.-S., Tetrahedron 2008, 64, 11666–11672. [Google Scholar]
- 33.
- 33a. Lu Z., Yoon T. P., Angew. Chem. Int. Ed. 2012, 51, 10329–10332; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 10475–10478; [Google Scholar]
- 33b. Hurtley A. E., Lu Z., Yoon T. P., Angew. Chem. Int. Ed. 2014, 53, 8991–8994; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 9137–9140. [Google Scholar]
- 34.
- 34a. Poplata S., Tröster A., Zou Y. Q., Bach T., Chem. Rev. 2016, 116, 9748–9815; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34b. Skubi K. L., Blum T. R., Yoon T. P., Chem. Rev. 2016, 116, 10035–10074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Deposition Number 2142344 contains the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service.
- 36. Teegardin K., Day J. I., Chan J., Weaver J., Org. Process Res. Dev. 2016, 20, 1156–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.
- 37a. List B., Chem. Rev. 2007, 107, 5413–5415; [Google Scholar]
- 37b. MacMillan D. W. C., Nature 2008, 455, 304–308; [DOI] [PubMed] [Google Scholar]
- 37c. Xiang S.-H., Nat. Commun. 2020, 11, 3786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.See Supporting Information for details. A selection of 13 different chiral catalysts were evaluated for their ability to promote an asymmetric Kabbe condensation. While enantioenriched product was obtained, the ees did not exceed 23 %. Additionally, an asymmetric copper-catalyzed conjugate addition between 9 and 10 was explored, which failed to provide enantioenriched product likely due to competing background reactivity.
- 39.
- 39a. Liu G., Cogan D. A., Ellman J. A., J. Am. Chem. Soc. 1997, 119, 9913–9914; [Google Scholar]
- 39b. Robak M. T., Herbage M. A., Ellman J. A., Chem. Rev. 2010, 110, 3600–3740. [DOI] [PubMed] [Google Scholar]
- 40.
- 40a. Dupau P., Epple R., Thomas A. A., Fokin V. V., Sharpless K. B., Adv. Synth. Catal. 2002, 344, 421–433; [Google Scholar]
- 40b. Chu H., Smith J. M., Felding J., Baran P. S., ACS Cent. Sci. 2017, 3, 47–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sudalai A., Khenkin A., Neumann R., Org. Biomol. Chem. 2015, 13, 4374–4394. [DOI] [PubMed] [Google Scholar]
- 42. Frolov A. I., Ostapchuk E. N., Pashenko A. E., Chuchvera Y. O., Rusanov E. B., Volochnyuk D. M., Ryabukhin S. V., J. Org. Chem. 2021, 86, 7333–7346. [DOI] [PubMed] [Google Scholar]
- 43.
- 43a. Shiina Y., Tomata Y., Miyashita M., Tanino K., Chem. Lett. 2010, 39, 835–837; [Google Scholar]
- 43b. Kotoku N., Mizushima K., Tamura S., Kobayahsi M., Chem. Pharm. Bull. 2013, 61, 1024–1029. [DOI] [PubMed] [Google Scholar]
- 44.
- 44a. Winkler J. D., Londregan A. T., Hamann M. T., Org. Lett. 2006, 8, 2591–2594; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44b. Hu Y., Bai M., Yang Y., Tian J., Zhou Q., Org. Lett. 2020, 22, 6308–6312. [DOI] [PubMed] [Google Scholar]
- 45. Hoye T. R., Humpal P. E., Moon B., J. Am. Chem. Soc. 2000, 122, 4982–4983. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Data Availability Statement
The data that support the findings of this study are available in the Supporting Information of this article.