

Abstract
Hereditary spastic paraplegias (HSPs) comprise a large group of inherited neurologic disorders affecting the longest corticospinal axons (SPG1–86 plus others), with shared manifestations of lower extremity spasticity and gait impairment. Common autosomal dominant HSPs are caused by mutations in genes encoding the microtubule-severing ATPase spastin (SPAST; SPG4), the membrane-bound GTPase atlastin-1 (ATL1; SPG3A) and the reticulon-like, microtubule-binding protein REEP1 (REEP1; SPG31). These proteins bind one another and function in shaping the tubular endoplasmic reticulum (ER) network. Typically, mouse models of HSPs have mild, later onset phenotypes, possibly reflecting far shorter lengths of their corticospinal axons relative to humans. Here, we have generated a robust, double mutant mouse model of HSP in which atlastin-1 is genetically modified with a K80A knock-in (KI) missense change that abolishes its GTPase activity, whereas its binding partner Reep1 is knocked out. Atl1KI/KI/Reep1−/− mice exhibit early onset and rapidly progressive declines in several motor function tests. Also, ER in mutant corticospinal axons dramatically expands transversely and periodically in a mutation dosage-dependent manner to create a ladder-like appearance, on the basis of reconstructions of focused ion beam-scanning electron microscopy datasets using machine learning-based auto-segmentation. In lockstep with changes in ER morphology, axonal mitochondria are fragmented and proportions of hypophosphorylated neurofilament H and M subunits are dramatically increased in Atl1KI/KI/Reep1−/− spinal cord. Co-occurrence of these findings links ER morphology changes to alterations in mitochondrial morphology and cytoskeletal organization. Atl1KI/KI/Reep1−/− mice represent an early onset rodent HSP model with robust behavioral and cellular readouts for testing novel therapies.
Introduction
Hereditary spastic paraplegias (HSPs) are a large group of inherited neurologic disorders with a cardinal feature of lower extremity spasticity resulting from a length-dependent axonopathy of corticospinal motor neurons, leading to gait impairment (1–3). They have historically been classified as ‘pure’ or ‘complicated’ on the basis of the absence or presence, respectively, of additional clinical features such as distal amyotrophy, ataxia, neuropathy, central nervous system (CNS) white matter changes, parkinsonism, cognitive changes or visual impairment. HSPs are among the most genetically diverse neurologic disorders, with >80 distinct genetic loci (SPG1–86 plus others) and over 60 identified genes (1,3,4). These gene products coalesce into a relatively small number of common cellular pathogenic themes. Notably, mutations in SPAST (SPG4), ATL1 (SPG3A) and REEP1 (SPG31) comprise about half of all patients with autosomal dominant HSP, and the respective gene products are related via direct protein–protein interactions and shared functions in shaping the morphology of the tubular endoplasmic reticulum (ER) network (5–8).
SPG4 is the most common HSP and is caused by hundreds of different autosomal dominant mutations in SPAST, which encodes the microtubule-severing ATPase spastin that exists as two main isoforms, M1 and M87. These isoforms are generated from the same mRNA transcript via the use of different translation initiation sites (9). Although the larger M1 spastin localizes primarily to tubular ER (8), M87 is cytoplasmic and recruited to various cellular compartments, where it participates in processes such as cytokinesis and endosomal fission (10,11). SPG3A is an autosomal dominant, often early-onset and slowly progressive HSP, resulting mostly from missense mutations in the ATL1 gene encoding the membrane-bound GTPase atlastin-1, which mediates tethering/fusion of ER tubules to form three-way junctions (12,13). The K80A mutant of human atlastin-1 lacks GTPase activity and disrupts ER network morphology in cells (12,14), similar to effects of atlastin-1 depletion as well as many known SPG3A missense mutations. SPG31 results from autosomal dominant mutations of REEP1, which encodes receptor expression-enhancing protein 1 (REEP1); haploinsufficiency is the suspected pathomechanism (15). The CNS-enriched REEP1 protein shapes the tubular ER, binds microtubules and interacts robustly with atlastin-1 (8,16).
Many HSP mouse models have been reported over the years (17,18), but few have early onset or the prominent symptoms exhibited by many HSP patients. SPAST knock out animals (19,20) exhibit a mild phenotype, whereas a transgenic mouse expressing a spastin protein harboring a SPG4 missense mutation is more afflicted (21). Beetz et al. (22) found that Reep1 deletion mice suffer obvious gait abnormalities at 12 months of age. That study as well as our study of another Reep1 null mouse line identified modest motor impairments, and we also identified significant loss of adipose tissue at earlier ages (23). No SPG3A mouse models have been published to date, although the role of atlastin GTPases in ER network formation is widely studied in other systems. Taken together, these relatively mild, later onset phenotypes may reflect the far shorter lengths of corticospinal axons in mice relative to humans.
Here we report an early onset HSP mouse model with severe gait impairment. This line harbors homozygous mutations in genes for both SPG3A and SPG31. These genes were chosen because SPG4 affects the spastin ATPase that has several localizations and functions throughout cells, whereas atlastin-1 and REEP1 appear overwhelmingly localized to ER membranes and bind robustly to one another (8). Furthermore, cases of HSP and related disorders have been described in patients with biallelic, loss-of-function mutations in both ATL1 (24) and REEP1 (25), increasing translational relevance of the model. Employing transmission electron microscopy (EM) as well as focused ion beam-scanning electron microscopy (FIB-SEM), we observe dramatic ER morphological changes in corticospinal axons of double homozygous mutant mice, with less prominent changes in single homozygous mutant mice and heterozygotes/homozygotes of double mutant mice. Coincident with changes in ER morphology, axonal mitochondria are more fragmented, and relative levels of hypophosphorylated neurofilaments H (NFH) and M (NFM) are dramatically increased.
Results
Generation and analysis of mutant mice
Homozygous Atl1KI/KI and heterozygous Atl1WT/KI mice with a GTPase-abolishing K80A missense mutation in the mouse atlastin-1 protein were generated as shown in Fig. 1A. Mutant atlastin-1 K80A protein levels in brain and spinal cord of Atl1KI/KI mice are moderately lower, and the mutant protein migrates slightly faster than wild-type (WT) atlastin-1 in sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gels (Fig. 1B and C). However, Atl1 mRNA levels are similar by quantitative real-time polymerase chain reaction (qPCR) (Fig. 1D and E), prefiguring more rapid degradation of mutant atlastin-1 K80A protein as the primary reason for reduced protein levels. Although mutant atlastin-1 K80A dramatically disrupts the tubular ER network upon overexpression in cultured cells (12–14), homozygous Atl1KI/KI and heterozygous Atl1WT/KI mice (both male and female) show only modest behavioral phenotypes from ages 4–24 weeks, as assessed using body weight measurements, rotarod, ledge test, hindlimb clasping and treadmill test (Supplementary Material, Figs S1 and S2). Similarly, homozygous Atl1−/− null mice (generated by breeding homozygous Atl1KI/KI to a cre-transgenic strain) exhibit mild phenotypes from ages 4–16 weeks, reminiscent of our previously published Reep1−/− mice (23) which are re-assessed here for comparison (Supplementary Material, Fig. S3).
Figure 1.

Generation and characterization of Atl1KI/KI/Reep1−/− mice. (A) Schematic representation of Atl1 gene targeting strategy. (B) Immunoblots of tissue homogenates show atlastin-1 and Reep1 protein levels in brain and spinal cord from WT mice and the indicated mutant genotypes. GAPDH is monitored as a control for protein loading. (C) Quantitative analyses of immunoblot results reveal that mutated atlastin-1 K80A levels are slightly lower than WT, with normalization to GAPDH. Student’s t tests were applied; n = 4. (D, E) qPCR results show no differences in mRNA levels between Atl1 and Atl1KI/KI using two sets of Atl1 primers. (F) Photo comparison of 6-month-old WT and Atl1KI/KI/Reep1−/− female mice. (G, H) Double mutant mice show lower body weights starting at 8 weeks in males (G) and 16 weeks in females (H). Parentheses at the bottom indicate numbers of mice in each group. Two-way ANOVA (Tukey’s test) was applied. *P < 0.05, **P < 0.01, ***P < 0.005, ****P < 0.001.
To intensify the HSP phenotype, we generated Atl1KI/KI/Reep1−/− and Atl1−/−/Reep1−/− double mutant mice by crossing Atl1KI/KI and Atl1−/− mice, respectively, with the Reep1−/− mouse line. Different heterozygous and homozygous combinations of these mutant forms were analyzed and compared using behavioral studies (Supplementary Material, Figs S1–S3). Not surprisingly, Atl1KI/KI/Reep1−/− and Atl1−/−/Reep1−/− mice have by far the earliest onset and strongest motor phenotypes. Atl1KI/KI/Reep1−/− mice were selected for detailed phenotypic and pathologic analyses because the Atl1 K80A missense change is more SPG3A disease mimetic (the vast majority of SPG3A patients harbor missense mutations) than the Alt1 knockout.
Atl1 KI/KI /Reep1 −/− mice exhibit prominent lower extremity motor dysfunction
Homozygous Atl1KI/KI/Reep1−/− mice generally have short breeding periods (from ages 2–5 months), and about 30–50% of breeding pairs are poor breeders. On visual inspection, Atl1KI/KI/Reep1−/− mice appear noticeably thinner (Fig. 1F), and their body weights are significantly lower than for WT animals (Fig. 1G and H) as well as other mutant lines generated for this study except for Atl1−/−/Reep1−/− mice, which have similarly low body weights (Supplementary Material, Figs S1–S3). This weight reduction likely reflects, at least in part, prominent loss of adipose tissue, as shown previously for Reep1−/− mice (23), as well as muscle wasting (Supplementary Material, Fig. S4). Similar to Atl1KI/KI mice, atlastin-1 protein levels in brain and spinal cord of Atl1KI/KI/Reep1−/− mice are slightly decreased, (Fig. 1B and C), with no differences in Atl1 mRNA levels by qPCR (Fig. 1D and E).
Detailed behavioral assessment results for double-mutant homozygous Atl1KI/KI/Reep1−/− mice are shown in Fig. 2. Notably, Atl1KI/KI/Reep1−/− mice have rapid breathing while on a treadmill as well as marked difficulty with ambulation, exhibiting hindlimb weakness and stiff, slow movements characteristic of spasticity (Fig. 2A; Supplementary Material, Supplementary Video). Furthermore, on the treadmill test, seven out of eight males and all females were unable to run at the lowest treadmill speed of 4 cm/s at 24 weeks of age (Fig. 2B; Supplementary Material, Figs S1 and S2). Both male and female Atl1KI/KI/Reep1−/− mice show impairments in rotarod performance as early as 4 weeks of age (Fig. 2C). Similar but less severe performance deficits are observed in heterozygous/homozygous Atl1WT/KI/Reep1−/− mice (Supplementary Material, Figs S1 and S2). Atl1KI/KI/Reep1−/− mice have compromised ability to walk on a cage ledge starting from 4 weeks and worsening over time (Fig. 2D). Heterozygous/homozygous Atl1WT/KI/Reep1−/− mice also have compromised ability to walk on the cage ledge, though at older ages—beginning at 8 weeks for males and 16 weeks for females (Supplementary Material, Figs S1 and S2). Finally, Atl1KI/KI/Reep1−/− mice show prominent and progressive hindlimb clasping (Fig. 2E). Thus, progressive defects in motor function are detected not only in double homozygous Atl1KI/KI/Reep1−/− mice but also, albeit less severely, in heterozygous/homozygous Atl1WT/KI/Reep1−/− mice.
Figure 2.

Behavioral changes in Atl1KI/KI/Reep1−/− mice. (A) Representative images of treadmill tests. An 8-month old female Atl1KI/KI/Reep1−/− mouse is unable to use its hind paws while ambulating, in contrast to the WT mouse. (B) Atl1KI/KI/Reep1−/− mice progressively lose the ability to run on the treadmill. (C) Atl1KI/KI/Reep1−/− mice fall from the rotarod much earlier than WT mice. (D) Ledge tests show that Atl1KI/KI/Reep1−/− mice have poor ability to walk on cage ledge. (E) Hindlimb clasping is observed in Atl1KI/KI/Reep1−/− mice starting from 8 weeks. n = 8 for WT male, Atl1KI/KI/Reep1−/− male, and Atl1KI/KI/Reep1−/− female; n = 9 for WT female. All data were analyzed by two-way ANOVA (Tukey’s test). ***P < 0.005, ****P < 0.001.
Peripheral nerve changes and skeletal muscle atrophy in Atl1KI/KI/Reep1−/− mice
A general pathologic examination was conducted for WT and Reep1−/−, Atl1KI/KI, Atl1−/−, Atl1KI/KI/Reep1−/− and Atl1−/−/Reep1−/− mice, at 4–4.5 months of age. Full details are outlined for organs and tissues outside of the nervous system in Supplementary Materials, Supplementary Data File. Briefly, in the CNS, all six Atl1KI/KI/Reep1−/− mice examined have rare to occasional spheroids in the brainstem as well as in spinal cord (Supplementary Material, Fig. S5). In the peripheral nervous system, all six Atl1KI/KI/Reep1−/− mice also have multiple swollen myelin sheaths and digestion chambers, as shown in the sciatic nerve. Skeletal muscle atrophy is evident in quadriceps femoris sections of Atl1KI/KI/Reep1−/− mutant mice (Supplementary Material, Fig. S5).
Atl1 KI/KI /Reep1−/− mice have transverse ER expansion in corticospinal axons
Previous studies have shown that atlastin GTPases and REEP proteins function in shaping the morphology of the tubular ER network (8,12,14). To assess changes in ER morphology in corticospinal tracts of mutant mice, we employed traditional transmission EM as well as FIB-SEM. Corticospinal tracts exhibit prominent changes in shape and size of axons in Atl1KI/KI/Reep1−/− mice as compared with WT animals (Fig. 3A). Transmission EM reveals very prominent transverse expansion of ER in myelinated corticospinal axons of Atl1KI/KI/Reep1−/− mice (Fig. 3B and C). This transverse ER manifests as a pattern repeating about every 0.3–1 μm across nearly the full diameter of the axon, resembling the rungs of a ladder in longitudinal sections (Fig. 3C). These transverse ER structures are not observed in WT corticospinal axons.
Figure 3.
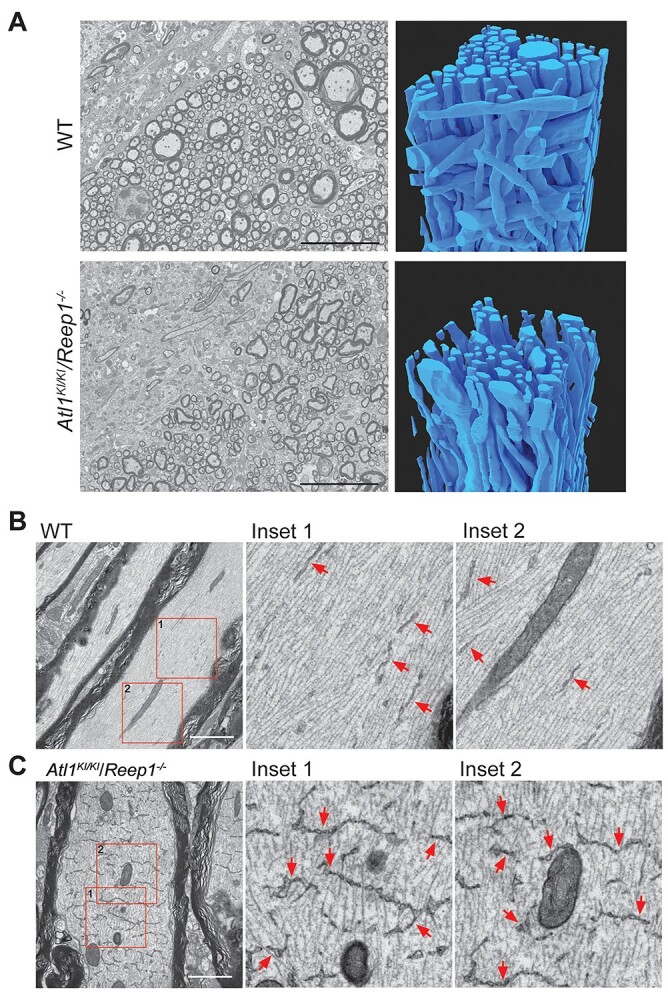
Transmission EM and FIB-SEM images of lumber spinal cord. (A) Transmission EM (left) and FIB-SEM reconstructions (right) of dorsal corticospinal tract area of 6-month-old mice showing degeneration in Atl1KI/KI/Reep1−/− axons relative to WT. Scale bars 10 μm. (B, C) Transmission EM images show dramatically expanded transverse ER in myelinated axons in Atl1KI/KI/Reep1−/− mice (C) as compared with WT mice (B). Boxed areas at the far left are enlarged to the right. Arrows (red) indicate ER. Scale bars 2 μm.
Manual reconstruction of transverse ER structures in Atl1KI/KI/Reep1−/− axons
Single corticospinal axons from FIB-SEM datasets for WT and Atl1KI/KI/Reep1−/− mice (Fig. 3A) were randomly selected for manual reconstruction. A total of 501 consecutive images were manually segmented for each genotype, and ER and mitochondria were identified on the basis of characteristic morphologies within every image. Reconstructions of segmented organelles (ER and mitochondria) are shown in three dimensions (3D) along a total axon length of 3 μm (Fig. 4A and B). Several ER tubules can be seen extending longitudinally along corticospinal axons, with occasional interconnections, in WT mice (Fig. 4A). However, ER shows a radically different morphology in axons of Atl1KI/KI/Reep1−/− mice (Fig. 4B), where there are prominent transverse, sheet-like structures. These periodic structures are connected to one another via a few longitudinal ER tubules and have apertures that allow mitochondria, microtubules and neurofilaments to pass through. Mitochondria also appear more fragmented in corticospinal axons of Atl1KI/KI/Reep1−/− mice as compared with those in WT mice (Figs. 3B and C and4C and D ). In axon cross-sections, the broad extent of these transverse ER structures is evident (compare Fig. 4E and F). Interestingly, transverse ER structures are also seen in Atl1KI/KI mice and Reep1−/− single mutant mice, though rarely (Supplementary Material, Fig. S6), indicating a mutation dosage effect reminiscent of that seen in the behavioral studies (Supplementary Material, Figs S1–S3). We also examined axons of ventral roots at L5 in Atl1KI/KI/Reep1−/− mice but found expanded ER in only a few axons. In contrast, the vast majority of corticospinal axons have expanded transverse ER. Finally, transverse ER structures are also found prominently in Atl1−/−/Reep1−/− double knockout animals, with a similar periodicity to Atl1KI/KI/Reep1−/−mice, but the transverse element does not appear as clearly perpendicular to the long axis (Supplementary Material, Fig. S6).
Figure 4.
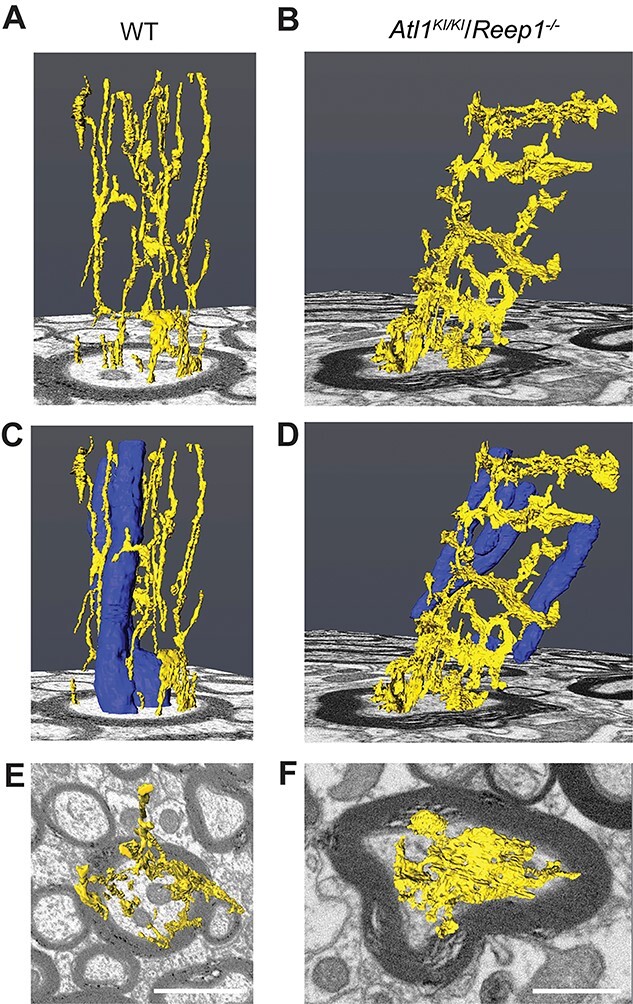
Manual 3D axonal reconstructions of FIB-SEM images. (A–F) ER is shown in yellow and mitochondria in blue. A, C and F show a randomly selected corticospinal axon from a WT mouse, whereas B, D and F show an axon from a Atl1KI/KI/Reep1−/− mouse. E and F are views looking down from the top of A and B, respectively. Scale bar 1 mM.
Machine learning-based reconstruction of transverse axonal ER
To facilitate robust quantitative assessments of ER and mitochondria, plus their contacts with one another, we developed a machine-learning based auto-segmentation algorithm, followed by 3D reconstruction. WT lumbar corticospinal tracts axons show longitudinal ER tubules with some interconnections and interspersed mitochondria, plus occasional ER-mitochondrial contacts (Fig. 5A). In lumbar corticospinal axons of Atl1KI/KI/Reep1−/− mice, dramatic transverse ER expansions are seen periodically along the full extent of the FIB-SEM image dataset, with noticeably more fragmented mitochondria (Fig. 5B). Quantitative analyses reveal an expansion of ER surface area and volume as well as a more transverse orientation of ER in the mutant axons (Fig. 5C and D), consistent with our visual assessment. Mitochondrial volume is slightly increased in the mutant axons, with far more dramatic decreases in mitochondrial length (Fig. 5D). Despite significant changes in ER and mitochondrial morphologies, ER-mitochondrial contact surface areas are surprisingly similar in the WT and Atl1KI/KI/Reep1−/− axons (Supplementary Material, Fig. S7).
Figure 5.

Machine-learning-based reconstructions and analyses of ER and mitochondria in corticospinal axons. (A, B) Orthographically projected 3D renderings of WT (A) and Atl1KI/KI/Reep1−/− (B) myelinated axons (translucent light blue), mitochondria (darker blue) and ER (orange) are shown alongside the FIB-SEM datasets. A representative axon is shown in the right three panels (merged image, ER and mitochondria from left to right). (C) ER volume (top) and surface area (SA; bottom) are quantified as a fraction of axon volume for each axon within the FIB-SEM volume. (D) Quantifications are shown for: orientation of ER relative to local axon orientation; ER length; mitochondrial volume; mitochondrial length and mitochondrial surface area. All measurements are normalized on a per axon basis to axonal volume. Mann–Whitney test was applied for statistical analyses. n = 265 (WT axons) and 268 (Atl1KI/KI/Reep1−/− axons). *P < 0.05, ****P < 0.001, ns—not significant. Scale bars 1 μm.
Transverse ER expansion correlates with behavioral changes
Transverse ER expansions are present in lumbar (L5) and cervical (C4) spinal cord of Atl1KI/KI/Reep1−/− mice at the age of 1 month but are larger and more numerous at 6 months (Supplementary Material, Fig. S8). These increases in transverse ER expansion thus positively correlate with the severity of motor deficits (Fig. 2 and Supplementary Material, Figs S1–S3). Although expanded ER is found in both lumbar and cervical spinal cord regions in Atl1KI/KI/Reep1−/− mice, transverse ER is consistently more prominent in lumbar regions (Supplementary Material, Fig. S8), also in lockstep with motor function studies showing impairment in hindlimbs. Even so, transverse ER is not limited to corticospinal axons; it is also present in spinothalamic axons in spinal cord as well as peripherally in sciatic nerve (Supplementary Material, Fig. S9A). These transverse axonal ER expansions may sometimes be associated with, or give rise to, less-organized ER tubule aggregations that are observed in both cervical and lumbar spinal cord axons of Atl1KI/KI/Reep1−/− mice at 6 months of age (Supplementary Material, Fig. S9B), but not in WT axons of the same age.
Various degrees of motor dysfunction are observed in single mutant homozygous Reep1−/−, double mutant homozygous/heterozygous Atl1WI/KI/Reep1−/− and Atl1KI/KI/Reep1+/− and double mutant homozygous Atl1KI/KI/Reep1−/− and Atl1−/−/Reep1−/− mice (Supplementary Material, Figs S1–S3). Among these, double mutant homozygous mice show both the most severe behavioral phenotypes as well as the greatest degree of transverse ER expansion, followed by double mutant homozygotes/heterozygotes. Of the double mutant homozygotes/heterozygotes, the Atl1WT/KI/Reep1−/− group is most severely afflicted (Supplementary Material, Figs S1 and S2). Reep1−/− mice have much milder motor function defects as compared with double mutant mice, and we observe infrequent transverse ER expansions in a few axons in spinal cord of Reep1−/− mice as well (Supplementary Material, Fig. S6). Noticeably, the degree of transverse ER expansion is much less in Reep1−/− than in Atl1KI/KI/Reep1−/− mice (Supplementary Material, Fig. S6). We detect very few transverse ER expansions (Supplementary Material, Fig. S6) and no significant behavioral changes (Supplementary Material, Figs S1 and S2) in Atl1KI/KI mice.
ER stress plays a role in motor neuronal dysfunction
Although axonal ER of Atl1KI/KI/Reep1−/− mice is dramatically expanded transversely, expression levels of most ER morphology proteins are unchanged in brain and spinal cord (Fig. 6). As described earlier, atlastin-1 protein levels are slightly lower, reflecting greater instability of mutant atlastin-1 K80A (Fig. 1C). Modest increases in levels of two atlastin-1 paralogs—atlastin-2 and atlastin-3—may reflect cellular upregulation in an attempt to compensate for impaired function of atlastin-1 (Fig. 6). Reep5 and reticulon-1C (Rtn1C) levels are slightly increased in Atl1KI/KI/Reep1−/− animals, and reticulon-4C (Rtn4C) levels are modestly decreased in brain. Total actin and β-tubulin levels are unchanged, and tubulin polyglutamylation and acetylation modifications also show no differences by immunoblotting. Levels of the ER stress protein GRP78/immunoglobulin-binding protein (BiP) are significantly increased, suggesting the presence of ER stress in the spinal cord of Atl1KI/KI/Reep1−/− mice; p180 levels show dramatic decreases in spinal cord, for unclear reasons.
Figure 6.

ER protein levels in Atl1KI/KI/Reep1−/− CNS. (A, B) Lysates of brain and spinal cord from 6-month-old mice (WT and the indicated mutant genotypes) were immunoblotted as shown in A, with quantifications in B. One-way ANOVA (Tukey’s test) was applied for statistical analyses. n = 4. *P < 0.05, **P < 0.01, ****P < 0.001.
Neurofilament hypophosphorylation in Atl1KI/KI/Reep1−/− mice
Despite the dramatic changes in ER morphology observed in corticospinal axons, we do not detect obvious changes in levels of many cytoskeletal actin or microtubule proteins, including various post-translationally modified forms. We next investigated another prominent axonal cytoskeletal component—neurofilaments—which are subdivided into heavy (NFH), medium (NFM) and light (NFL) chains. NFH and NFM often exist in CNS neurons in highly phosphorylated forms; phosphorylation levels of NFL are less well understood because of a lack of selective antibodies. We detect lower levels of hyperphosphorylated NFH and NFM and concomitantly higher levels of hypophosphorylated NFH and NFM in brain and spinal cord of Atl1KI/KI/Reep1−/− mice as compared with WT. The changes in Atl1KI/KI/Reep1−/− spinal cord represent an  3-fold increase in hypophosphorylated NFH and 2-fold increase in hypophosphorylated NFM relative to WT spinal cord, whereas overall protein levels of NFH and NFM remain unchanged (Fig. 7A–C).
3-fold increase in hypophosphorylated NFH and 2-fold increase in hypophosphorylated NFM relative to WT spinal cord, whereas overall protein levels of NFH and NFM remain unchanged (Fig. 7A–C).
Figure 7.

Hypophosphorylation of neurofilaments in Atl1KI/KI/Reep1−/− mice. (A, B) Lysates of brain and spinal cord from 6-month-old WT mice and the indicated mutant genotypes were immunoblotted as shown for total neurofilaments (A) and those with different phosphorylation states (B). GAPDH levels were monitored as a control for protein loading. (C) Quantifications of immunoblots from A and B. One-way ANOVA (Tukey’s test) was applied for statistical analyses (n = 4). *P < 0.05, **P < 0.01, ****P < 0.001. (D, E) Changes of phosphorylation levels were further analyzed using Phos-tag SDS-PAGE (D) and by adding CIP to sample lysates to dephosphorylate neurofilaments (E).
Mouse NFH harbors dozens of phosphorylation sites within the C-terminal domain (26). On the basis of immunoblot intensities of a ~250 kDa smear band from a Phos-tag gel, we observe that hyperphosphorylated NFH is significantly decreased in Atl1KI/KI/Reep1−/− mice (Fig. 7D). NFH from spinal cord lysates of Atl1KI/KI/Reep1−/− mice treated with calf intestinal alkaline phosphatase (CIP) migrates faster on SDS-PAGE than NFH in untreated lysates, indicating that most of the immunoreactive signal detected by antibody (SMI32) represents hypophosphorylated forms of NFH (Fig. 7E); the antibody against non-phosphorylated NFH (SMI32) detects not only the NFH non-phosphorylated form, but also hypo-phosphorylated forms. Hypophosphorylated NFH can be detected as early as 2 weeks of age and reaches peak levels at age 2 months in Atl1KI/KI/Reep1−/− mice (Fig. 8). Lastly, we prepared embryonic stem (ES) cells from Atl1KI/KI/Reep1−/− mice. Motor neurons differentiated from these ES cells show similar results as observed in vivo; hypophosphorylated NFH is significantly increased in Atl1KI/KI/Reep1−/− neurons (Supplementary Material, Fig. S10).
Figure 8.

Age-dependent increases in hypophosphorylated neurofilaments in Atl1KI/KI/Reep1−/− mice. (A) Lysates of spinal cord from WT and Atl1KI/KI/Reep1−/− mice of the indicated ages were immunoblotted for total and hypophosphorylated NFH. GAPDH levels were monitored as a control for protein loading. (B-D) Quantifications of immunoblots as in A for NFH/GAPDH (B), hypophosphorylated-NFH/GAPDH (C), and hypophosphorylated NFH/NFH (D). Two-way ANOVA (Tukey’s test) was applied. n = 4. *P < 0.05, **P < 0.01, ***P < 0.005, ****P < 0.001.
Discussion
HSPs are a large group of genetic disorders characterized by a length-dependent, distal axonopathy of corticospinal tract fibers, resulting in lower limb spasticity and gait impairment (4). Typically, mouse models of HSPs have mild, later onset phenotypes, particularly for ‘pure’ forms, which is not surprising given the far shorter lengths of mouse corticospinal axons relative to humans. The gene products mutated in several of the most common forms of HSP bind one another and function in shaping the tubular ER network, for instance the ER-localized, membrane-bound proteins atlastin-1 (SPG3A) and REEP1 (SPG31) (8). We created mutant mouse models of HSP in which Atl1 is either knocked out or genetically modified with a K80A knock-in missense change that abolishes its GTPase activity, and we also investigated a previously reported mouse line where Reep1 is knocked out (23). Mice harboring homozygous mutations in just one of these two genes have relatively mild phenotypes. However, double mutant HSP mice better mimic the symptoms of HSP patients. Therefore, genetic modification of two genes may be needed in other common HSP cellular pathogenic themes to adequately model the phenotype of length-dependent, human upper motor neuron dysfunction in a small rodent.
In concert with these phenotypic changes, we observe a mutation dosage effect on a novel form of ER morphologic abnormality—periodic, axonal transverse ER expansions resembling the rungs of a ladder that are not seen in WT axons—supporting a causative role for changes in ER morphology in HSP pathogenesis. Indeed, within normal axons, ER tubules are longitudinally oriented with occasional three-way junctions leading to branching, and axonal ER tubules can be as narrow as 20–30 nm in diameter (27). Periodic traverse ER expansions in the mutant mice are a radical departure from this typical morphology. Even so, the mechanism underlying the formation of these expansions remains unclear. Atlastin GTPases are required for the formation of three-way junctions in ER tubules (12–14), whereas Reep1 may shape ER tubules through its hydrophobic hairpin domains. Reep1 also binds microtubules (8), but we do not detect a concomitant change in microtubule appearance, protein levels or several common post-translational modifications. Thus, mutations of either or both of these proteins would not be expected to produce these sheet-like transverse expansions. The mechanism underlying the periodicity is also unclear, with the range of 0.3–1.0 μm noticeably greater than the 180–190 nm spacing of periodic actin rings seen in axons (28).
Maintaining proper ER shape and function requires specific structural shaping and motor proteins along with cytoskeletal interactions, plus remodeling processes including ER-phagy, which delivers fragmented ER to lysosomes for degradation (29). For instance, atlastin-2, atlastin-3 and reticulon-3 are involved in remodeling the ER network and ensuring basal membrane turnover, underscoring important roles for these proteins during ER-phagy (30–32). Interestingly, ER expansion has been described in Atg5 (an autophagy related protein)-deficient plasma cells during differentiation (33), emphasizing the importance of autophagy in clearing ER. In our studies, we observe a significant reticulon-3 decrease in double mutant mice comparing to single mutant Atl1KI/KI or Reep1−/− mice. Importantly, reticulon-3 functions as an ER-phagy receptor that binds to LC3/GABARAP and breaks ER tubules for autophagic clearance (30). It remains unclear what causes the reduction of reticulon-3 in double mutant mice, but this reduction may impair the ability of cells to clear misfolded proteins and dysfunctional ER, perhaps leading to the clumps of tangled ER observed in axons of Atl1KI/KI/Reep1−/− mice. Although the prominent axonal ER morphology changes are consistent with a cell-autonomous disease mechanism, we must also consider the possible contributions of cell non-autonomous processes. In fact, this has been suggested in iPSC studies of SPG3A, where altered cholesterol release from neighboring glia may affect lipid composition of axons and contribute to axonopathy (34).
ER Ca2+ homeostasis is important in maintaining ER shape, and a similar transverse ER expansion has been detected in fish Mauthner cell dendrites after prolonged vestibular stimulation, accompanied by increased Ca2+ within the expanded ER (35). Thus, the periodic transverse ER in the double mutant mice may represent a protective response to ER stress. In fact, ER stress participates in the pathogenesis of neurodegenerative diseases where abnormal BiP activities have been detected (36). For instance, in Huntington disease models, BiP binds to huntingtin aggregates and limits aggregation at high concentrations (37), and elevated expression of BiP has been reported in cellular and animal models of Parkinson disease (38). Lastly, an enhanced BiP chaperone function has been implicated in Alzheimer disease pathogenesis (39). Similarly, we detected significantly increased BiP levels in spinal cord of Atl1KI/KI/Reep1−/− but not Atl1KI/KI or Reep1−/− mice (Fig. 7), indicating ER stress in the more severely affected mice. Yeast cells reduce ER stress by expanding sheet ER (40), possibly linking mechanistically increased BiP levels to the transverse ER expansions observed in double mutant mice.
One of the most prominent changes in protein levels in Atl1KI/KI/Reep1−/− spinal cord is a reduction in p180. Although the reason for this reduction remains unclear, a recent study has highlighted the importance of p180 (in conjunction with several other membrane-bound ER proteins) in distributing ER throughout cells via selective interactions with microtubules on the basis of different levels of microtubule glutamylation (41). Although there are no obvious changes in microtubule polyglutamylation or acetylation in Atl1KI/KI/Reep1−/− spinal cord by immunoblotting, a more comprehensive analysis of other microtubule-binding ER proteins and microtubule modifications may be warranted in the future. Even so, it is not clear how any of these alterations would contribute to the formation of the periodic ladder-like ER expansions.
Accompanying the prominent alterations in ER shape, we observe more fragmented mitochondria in axons of Atl1KI/KI/Reep1−/− mice, and NFH and NFM are hypophosphorylated. Neurofilaments are abundant in myelinated axons, functioning in radial axon growth and nerve conduction (42). In mature axons, NFM and NFH are heavily serine-phosphorylated in their tail domains, which harbor multiple Lys-Ser-Pro (KSP) repeats (43). Tail phosphorylation does not affect filament assembly, but rather contributes to lateral extension and formation of the cytoskeletal lattice (26). Head domain phosphorylation, however, inhibits tail domain phosphorylation in NFM (44). As neurofilament levels reflect nonspecific damage to axons, they have been examined across many neurological disorders, including frontotemporal dementia, amyotrophic lateral sclerosis, Parkinson disease and Alzheimer disease. Our results indicate that monitoring NFH and NFM phosphorylation states in addition to NF protein levels may be necessary. Finally, Reep1 and atlastin-1 play important roles in lipid droplet maintenance (16,23,45,46). We observe weight loss in Reep1−/− mice, consistent with previous findings identifying loss of adipose tissue (23). Atl1KI/KI/Reep1−/− mice have the lowest weights among all groups (Supplementary Material, Fig. S1), in agreement with previous studies demonstrating that atlastin-1 interacts with Reep1 and contributes to lipid droplet formation (8,46).
In summary, we have generated a robust HSP model with mutations in two HSP genes whose products bind one another and function together in shaping the tubular ER. A possible limitation is that this combination is not a physiologic phenotype, and it could instead create new cellular phenotypes that are not HSP relevant. However, this seems very unlikely. Rather, this combination probably optimizes the mouse as a model for HSP from both behavioral and cellular perspectives, providing a crucial model for understanding HSP disease mechanisms and testing treatments.
Materials and Methods
Generation of mutant mice
Animal care and use were carried out in accordance with NIH guidelines and approved by the NINDS/NIDCD Animal Care and Use Committee. Mice were housed in pathogen-free facilities under 12-h light/dark cycles with access to food and water ad libitum. Atl1WT/KI (K80A) mice were generated by Caliper Discovery Alliances & Services. Briefly, the RP23-336 M7 BAC clone was used to generate homology arms and the conditional knockout region for the gene targeting vector, as well as Southern probes for screening targeted events. The 5′ homology arm (4.9 kb), 3′ homology arm (2.9 kb) and conditional knockout region (1.0 kb) were generated by PCR using high-fidelity Taq DNA polymerase. Fragments were cloned in the pCR4.0 vector and confirmed by restriction digestion and end-sequencing. An AA to GC mutation in exon 3 was introduced into the conditional knockout region by PCR-based site-directed mutagenesis with the QuickChange II kit (Stratagene). Aside from homology arms, the final vector also contains LoxP sequences flanking the conditional knockout region (1.0 kb), Frt sequences flanking the Neo expression cassette (for positive selection of ES cells) and a DTA expression cassette (for negative selection of ES cells). The final vector was confirmed by restriction digestion and end sequencing analysis. NotI was used for linearizing the final vector before electroporation. 5′ and 3′ external probes were generated by PCR and were tested by genomic Southern analysis for screening of potentially targeted ES cells. Probes were cloned in the pCR4.0 backbone and confirmed by sequencing (Fig. 1A). The targeting construct was linearized and electroporated into C57BL/6 ES cells. Targeted clones were identified by Southern and PCR analyses, and Flp electroporation was performed. One Neo-deleted clone was identified and injected into a B6/Tyr blastocyst to generate male chimeras. Heterozygous mice were obtained by breeding the chimeras with C57BL/6 WT females. The heterozygous mice were then backcrossed with WT C57BL/6j for at least 10 generations before performing any tests. For PCR genotyping, DNAs were isolated from tail biopsies by DirectPCR lysis reagent (Viagen, 102-T) with protease K and used in a single PCR mix containing the primers: Forward, 5′- CCTCATCGTCAAGGACGACCATT—3′; Reverse, 5′- CACCCAAGTGCTCACTGTCAAATCC -3′. A product size of 520 bp was obtained for the WT allele and a 650 bp amplicon for the knock-in allele.
The generation of Reep1−/− mice was previously reported (23). Atl1KI/KI/Reep1−/− mice were created by breeding Atl1KI/KI mice and Reep1−/− mice. Atl1−/− mice were generated by breeding homozygous Atl1KI/KI to a cre-transgenic strain (B6.FVB-Tg(EIIa-cre)C5379Lmgd/J; Stock No: 003724; Jackson Laboratory). Heterozygous Atl1+/−/hemizygous cre mice were then crossed back to homozygous Atl1KI/KI for production of Atl1−/− mice. PCR genotyping was performed using these primer pairs: Forward, 5′—TGAGTTTGCTCTCCACAACCATAC—3′; Reverse, 5′—CATGGAGTGGTGAATGGCTAGTAG—3′, and a 429-bp product was obtained. Atl1−/−/Reep1−/− mice were generated by crossing the Atl1−/− strain and Reep1−/− strain.
Mouse behavioral tests
WT and mutant mice of ages 4–24 weeks were tested biweekly. Observers were blinded to genotype during all tests. Mouse running ability was tested on treadmill machine (TreadScan, CleverSys). Each run was 10 s at various speeds of 4, 7, 10 and 14 cm/s. Mice ran from low-to-high speeds with a 5-min rest between different runs. The maximum speed at which mice completed the run was recorded. A mouse RotaRod apparatus (Ugo Basile) was set to accelerate from 5 to 40 rpm over 120 s and then to maintain constant speed at 40 rpm; latency to fall was recorded. Mice ran three times on testing days, with a 15-min rest between runs.
The ledge test is a measure of coordination (47). Observations are made as the mouse walks along the cage ledge and lowers itself into the cage. A WT mouse will typically walk along the ledge without losing its balance and then lower itself back into the cage gracefully, using its paws. This represents a score of 0. If the mouse loses its footing while walking along the ledge, but otherwise appears coordinated, the score is 1. If the mouse does not effectively use its hind legs, or lands on its head rather than its paws when descending into the cage, the score is 2. If the mouse falls off the ledge, or nearly so, while walking or attempting to lower itself, or shakes and refuses to move despite encouragement, the score is 3.
Hindlimb clasping is a marker of disease progression (47). The tester grasps the proximal tail and lifts the mouse clear of all surrounding objects, observing the hindlimb position for 10 s. If the hindlimbs are consistently splayed outward, away from the abdomen, the score is 0. If one hindlimb is retracted toward the abdomen for >50% of the time suspended, the score is 1. If both hindlimbs are partially retracted toward the abdomen for >50% of the time suspended, the score is 2. If the hindlimbs are entirely retracted and touching the abdomen for >50% of the time suspended, the score is 3.
Antibodies
Mouse monoclonal antibodies included anti-: GAPDH (ProteinTech, 60 004–1), β-tubulin (Sigma-Aldrich, T8328), tubulin, acetylated antibody (Millipore-Sigma T6793), reticulon2 (Sigma-Aldrich, SAB1406405), actin (Sigma-Aldrich, A4700), NFH phosphorylated SMI31 (BioLegend, 801 602), NFH/M non-phosphorylated SMI33 (BioLegend, 835 403), NFH non-phosphorylated SMI32 (BioLegend, 801 701). Rabbit polyclonal antibodies included anti-: atlastin-1 (custom made, 5409) (48,49) atlastin-2 (Bethyl laboratories, A303-333A), atlastin3 (Bethyl laboratories, A303-312A), polyglutamate chain (polyE), (IN105, AdipoGen, AG-25B-0030-C050), Reep1 (ProteinTech, 17 988), Reep2 (ProteinTech, 15 684), Reep3 (Abcam, ab106463), Reep4 (ProteinTech, 26 650), Reep5 (ProteinTech, 14 643), reticulon1c (ProteinTech, 15 048), reticulon3 (ProteinTech, 12 055), reticulon4 (ProteinTech, 10 740), calreticulin (Abcam, Ab2907), BiP (Cell Signaling, 3183S), p180 (Invitrogen, PA5–21392), NFH (ProteinTech, 18 934), NFH (Abcam, ab8135), NFM (ProteinTech, 20 664) and NFL (ProteinTech, 12 998). Goat polyclonal antibodies included anti-calnexin (Santa Cruz Biotechnology, sc-6465) and anti-choline acetyltransferase (ChAT) (Millipore, AB144P).
Immunoblotting
Mouse brain and spinal cord tissues were dissected and homogenized in RIPA buffer with 1% protease inhibit cocktail (48,49). Then, lysates were centrifuged (21 000g, 20 min). Supernatants (10–30 μg protein) were resolved by SDS-PAGE on 4–15% polyacrylamide gradient gels. Electrophoretic transfer to nitrocellulose membranes was performed after SDS-PAGE. Blots were blocked in 5% non-fat milk in Tris-buffered saline with 0.1% Tween (TBST) for 1 h. Primary antibodies in 5% fetal bovine serum were applied at 4°C overnight. Secondary antibodies in 5% non-fat milk were applied for 1 h at room temperature. Blots were washed with TBST three times between steps. Finally, ECL (Amersham) was applied, and immunoreactivity revealed and quantified using a ChemiDoc XRS+ (Bio-Rad). Samples from each mouse were tested at least two to three times and averaged. Four mice were used for each group. Image Lab software was used for image analyses.
Neurofilament phosphorylation
SuperSep Phos-tag gel (Wako, 198–17 981) was used following the manufacturer’s protocol for detecting phosphorylated neurofilaments. For dephosphorylation studies, mouse spinal cord tissue was homogenized in dephosphorylation buffer [50 mM Tris–HCl (pH 8.2), 135 mM NaCl, 1 mM EDTA] including protease inhibitor cocktail (Thermo Fisher, 87 786–78 430), plus phosphatase inhibitor cocktail (Roche, 04906837001) in control samples. Supernatants (30 mg protein) following centrifugation (21 000 g, 15 min) were incubated with 50 IU CIP (New England Biolabs, M0920L) for 16 h at 37°C. Reactions were stopped by adding phosphatase inhibitor cocktail. Samples were then resolved on SDS-PAGE gels and immunoblotted (50).
Transmission EM
Samples for EM were prepared as previously described (51), with minor modifications. Briefly, at least two mice for each genotype and age were anesthetized and transcardially perfused with 1× PBS followed by freshly made EM fixative (2% glutaraldehyde and 2% paraformaldehyde in 0.1 N cacodylate buffer). Spinal cords were removed and post-fixed in EM fixative overnight. The remaining processes were carried out in the NINDS EM Core Facility. Vibratome slices were treated with 0.2% OsO4 in phosphate buffer for 30 min, mordanted en bloc with 0.25% uranyl acetate overnight at 4°C, dehydrated with ethanol and flat embedded in epoxy resin. Thin sections were counterstained with uranyl acetate and lead citrate. After dehydration, embedding and sectioning, images were acquired using a JEOL 1200EX electron microscope.
FIB-SEM sample preparation
Durcupan-embedded spinal cord samples from WT and Atl1KI/KI/Reep1−/− mice were prepared as for EM. They were each first mounted to the top of a 1-mM copper post which was in contact with the metal-stained sample for better charge dissipation, as previously described (52). For each sample, a small vertical sample post was trimmed to the region of interest with a width of 110 μm and depth of 110 μm in the direction of the ion beam. Trimming was guided by X-ray tomography data obtained using a Zeiss Versa XRM-510 microscope as well as optical inspection under a microtome. Thin layers of conductive material of 10-nm gold followed by 100-nm carbon were coated onto the trimmed samples using a Gatan PECS 682 High-Resolution Ion Beam Coater. The coating parameters were 6 keV, 200 nA on both argon gas plasma sources, with 10 rpm sample rotation and 45-degree tilt.
Large volume FIB-SEM 3D imaging
Two FIB-SEM prepared samples, from WT and Atl1KI/KI/Reep1−/− mice, were imaged sequentially using a customized Zeiss NVision40 FIB-SEM system as previously described (52–54). Each sample was biased at 400 V to improve image contrast by filtering out secondary electrons. The block face was imaged using a 1 nA electron beam with 1.5 keV landing energy at a 0.5 MHz scanning rate. The x-y pixel resolution was set at 6 nm. A subsequently applied focused Ga+ beam of 27 nA at 30 keV strafed across the top surface and ablated away 6 nm of the surface. The newly exposed surface was then imaged again. This ablation/imaging cycle continued about once every 2 min for 2 weeks to complete the FIB-SEM imaging for one sample. The sequence of acquired images formed a raw imaged volume, followed by post processing of image registration and alignment using a Scale Invariant Feature Transform-based algorithm. The aligned stack consisted of a final isotropic volume of 40 × 30 × 75 μm3 with 6 × 6 × 6 nm3 voxel size for each sample throughout the entire volume, which can be viewed in any arbitrary orientation.
Reconstruction of EM data
Amira (FEI) was used for 3D manual reconstruction. One myelinated axon from the WT and one from the Atl1KI/KI/Reep1−/− stack were randomly selected. Both WT and Atl1KI/KI/Reep1−/− image stacks were processed in three sets of 167 images (each equal to 1 μm of spinal cord in z-direction) without filtering, for a total of 501 images (3 μm of spinal cord in z-direction) for reconstruction. ER was segmented on the basis of morphology (any ~ 30–60 nm diameter structures that appeared filamentous in y-z and/or x-z planes). All ER was manually selected because of its variable diameter, which would not allow for interpolation between images, whereas cell membrane and mitochondrial segmentations were semi-automated. The 3D surfaces of all manually selected structures were smoothed (200× ats = 0.6) to reduce blockiness. Interpolated structures required minimal, if any, smoothing.
For machine-learning-based segmentation, myelinated axons were first skeletonized by ariadne.ai’s manual annotation service, using the Knossos annotation tool (www.knossostool.org). Mitochondria, ER and myelinated axons were segmented by ariadne.ai’s 3dEMtrace automatic segmentation service. In the case of mitochondria, orientation and length were calculated along the longest axis of each connected component. Axon length was calculated as the path length of the manual skeletons. For the ER, the segmentation was first skeletonized using Lee’s algorithm (55) as implemented by the scikit-image Python library (56). Length and orientation were then individually calculated for every unbranched segment of the skeleton representation.
After segmentation of myelinated axons, mitochondria and ER, we extracted the following morphological features: volume of mitochondria, ER and axons; surface area of mitochondria, ER and axons; and length of mitochondria, ER and axons. Orientations relative to the main axon axis were obtained by calculating the angle to the closest segment in the corresponding manual axon skeleton. When plotting distributions, ER orientation was weighted by the length of the respective segment. Lengths were computed along the longest axis of each object (an arbitrary axis in space, not one of the three cardinal axes). For axons, lengths were calculated along the axon center line, which follows the bending of the axons. Mitochondria, ER and axon surface areas were calculated from surface mesh representations for each segmentation connected component generated by the marching cubes algorithm, as implemented by the zmesh Python library (https://github.com/seung-lab/zmesh).
Contact sites between mitochondria and ER were calculated from surface meshes using the pymesh Python library (https://github.com/PyMesh/PyMesh), by finding all mitochondria mesh facets at a distance of at most 50 nm from ER mesh facets, with the distance understood as the distance between the respective facet centroids. Patches of adjacent ER-proximal facets on the mitochondrial surface meshes were grouped into individual contact sites. The renderings were created by ariadne.ai’s rendering pipeline using the open-source software Blender.
Quantitative PCR of Atl1 expression in mouse brain
Mouse brains were collected at 3 months of age. Tissues were homogenized in TRI Reagent (Sigma-Aldrich; T9424) with 2.38 mM metal beads (OMNI international; 19–620) for 10 s at high speed using a Bead Ruptor 12 (OMNI international). Lysates were spun at 20 000 g for 10 min to remove cell debris. Total RNA was extracted using Direct-zol™ RNA MiniPrep kit (Zymo Research; R2050). Reverse transcription of 5 μg total RNA was performed using the SuperScript™ IV First-Strand Synthesis System (Thermo Fisher; 18 091 050). qPCR was run on a QuantStudio™ 6 Flex System (Thermo Fisher) detecting SYBR-green fluorescence (Thunderbird® SYBR® qPCR Master Mix, Toyobo; QPS-201). Expression of Atl1 was normalized to GAPDH using the 2−ΔΔCT method. Primers used: GAPDH-Forward, 5′-AACTTTGGCATTGTGGAAGG-3′; GAPDH-Reverse, 5′- ACACATTGGGGGTAGGAACA-3′; Atl1-Forward 1, 5′-TTGAGAGATTCGGCCACAGT-3′; Atl1-Reverse 1, 5′-TTCTTCCATTGCCAACCTGC -3′; Atl1-Forward 2, 5′-AGTCCATGTTACAGGCCACA-3′; Atl1-Reverse 2, 5′ACCACCACAAACCTCTTCCA −3′.
Mouse ES cells and motor neuron differentiation
ES cells were isolated from mutant mice (WT, Atl1KI/KI, Reep1−/−, Atl1KI/KI/Reep1−/−). Briefly, E3.5 embryos from naturally mated mice were flushed from oviducts and cultured on primary embryonic mouse fibroblast feeder layers for about a week. ES clones from inner cell mass outgrowths were picked on the basis of morphology and expanded. ES cells were differentiated into motor neurons using a two-step induction protocol (57). ES cells were first primed for neuron induction and embryoid body formation for 2 days with Noggin and fibroblast growth factors. Retinoic acid and smoothened agonist were then added and cultured for 5 days for motor neuron specification. Motor neurons were dissociated and cultured on poly-D/L-ornithine/laminin-coated dishes for an additional 2 weeks when the motor neuron marker choline acetyltransferase (ChAT) was stably expressed.
Pathologic analysis of mice
About 4–4.5 month-old mice were provided to Dr Matthew Starost’s group in the Diagnostic & Research Service Branch, Division of Veterinary Resources, NIH for general pathological analyses. All tissues were fixed in 10% formalin, processed and embedded in paraffin. Sections (5 μm) were cut and stained with hematoxylin and eosin.
Statistical analysis
GraphPad Prism 9 software was used for statistical analyses. One-way ANOVA or two-way ANOVA (Tukey’s test) was used, or as indicated otherwise in each figure legend. All results are expressed as means ± SD. Results are considered significant at P < 0.05. Asterisks indicate significant differences: *P < 0.05, **P < 0.01, ***P < 0.005, ****P < 0.001.
Supplementary Material
Acknowledgements
We thank the animal husbandry staff at NINDS for animal care. We also thank Jung-Hwa Tao-Cheng (NINDS EM Facility) for providing assistance with EM, Huaibin Cai’s lab (NIA) in and the NIMH Rodent Behavioral Core for providing treadmill equipment, James Pickel and the NIMH Transgenic Core Facility for assistance producing ES cells, Jachinta Rooney from Carsten Bonnemann’s lab (NINDS) for providing behavioral testing machines and suggestions, Morgan Musgrove from Jeffrey Diamond’s lab (NINDS) for assistance with Amira software, Chad Douglas Williamson from Juan Bonifacino’s lab (NICHD) for Amira software and technical support, and Claire Le Pichon (NICHD) for helpful comments.
Contributor Information
Peng-Peng Zhu, Neurogenetics Branch, National Institute of Neurological Disorders and Stroke, National Institutes of Health, Bethesda, MD 20892, USA.
Hui-Fang Hung, Neurogenetics Branch, National Institute of Neurological Disorders and Stroke, National Institutes of Health, Bethesda, MD 20892, USA; MassGeneral Institute for Neurodegenerative Disease, Charlestown, MA 02129, USA; Department of Neurology, Massachusetts General Hospital and Harvard Medical School, Boston, MA 02114, USA.
Natalia Batchenkova, Neurogenetics Branch, National Institute of Neurological Disorders and Stroke, National Institutes of Health, Bethesda, MD 20892, USA.
Jonathon Nixon-Abell, Neurogenetics Branch, National Institute of Neurological Disorders and Stroke, National Institutes of Health, Bethesda, MD 20892, USA; Howard Hughes Medical Institute, Janelia Research Campus, Ashburn, VA 20147, USA; Cambridge Institute for Medical Research, Cambridge CB2 0XY, UK.
James Henderson, Cambridge Institute for Medical Research, Cambridge CB2 0XY, UK.
Pengli Zheng, Neurogenetics Branch, National Institute of Neurological Disorders and Stroke, National Institutes of Health, Bethesda, MD 20892, USA; MassGeneral Institute for Neurodegenerative Disease, Charlestown, MA 02129, USA; Department of Neurology, Massachusetts General Hospital and Harvard Medical School, Boston, MA 02114, USA.
Benoit Renvoisé, Neurogenetics Branch, National Institute of Neurological Disorders and Stroke, National Institutes of Health, Bethesda, MD 20892, USA.
Song Pang, Howard Hughes Medical Institute, Janelia Research Campus, Ashburn, VA 20147, USA.
C Shan Xu, Howard Hughes Medical Institute, Janelia Research Campus, Ashburn, VA 20147, USA.
Stephan Saalfeld, Howard Hughes Medical Institute, Janelia Research Campus, Ashburn, VA 20147, USA.
Jan Funke, Howard Hughes Medical Institute, Janelia Research Campus, Ashburn, VA 20147, USA.
Yuxiang Xie, Synaptic Function Section, National Institute of Neurological Disorders and Stroke, National Institutes of Health, Bethesda, MD 20892, USA.
Fabian Svara, ariadne.ai ag, CH-6033 Buchrain, Switzerland; Research Center Caesar, D-53175 Bonn, Germany.
Harald F Hess, Howard Hughes Medical Institute, Janelia Research Campus, Ashburn, VA 20147, USA.
Craig Blackstone, Neurogenetics Branch, National Institute of Neurological Disorders and Stroke, National Institutes of Health, Bethesda, MD 20892, USA; MassGeneral Institute for Neurodegenerative Disease, Charlestown, MA 02129, USA; Department of Neurology, Massachusetts General Hospital and Harvard Medical School, Boston, MA 02114, USA.
Conflict of Interest statement
Fabian Svara holds stock in, and is employed by, ariadne.ai ag, provider of image analysis and annotation services. Portions of the technology described herein are covered by U.S. Patent 10 600 615 titled ‘Enhanced FIB-SEM systems for large-volume 3D imaging’, which was issued to C. Shan Xu, Kenneth J. Hayworth and Harald F. Hess and assigned to the Howard Hughes Medical Institute on March 24, 2020.
Data availability
Data supporting findings of this study are available from the corresponding author upon request. FIB-SEM datasets are available at openorganelle.org.
Funding
Intramural Research Program of the NINDS, National Institutes of Health and the Howard Hughes Medical Institute Janelia Research Campus.
References
- 1. Boutry, M., Morais, S. and Stevanin, G. (2019) Update on the genetics of spastic paraplegias. Curr. Neurol. Neurosci. Rep., 19, 18. [DOI] [PubMed] [Google Scholar]
- 2. Fink, J.K. (2014) Hereditary spastic paraplegia: clinical principles and genetic advances. Semin. Neurol., 34, 293–305. [DOI] [PubMed] [Google Scholar]
- 3. Tesson, C., Koht, J. and Stevanin, G. (2015) Delving into the complexity of hereditary spastic paraplegias: how unexpected phenotypes and inheritance modes are revolutionizing their nosology. Hum. Genet., 134, 511–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Blackstone, C. (2018) Hereditary spastic paraplegia. Handb. Clin. Neurol., 148, 633–652. [DOI] [PubMed] [Google Scholar]
- 5. Blackstone, C. (2012) Cellular pathways of hereditary spastic paraplegia. Annu. Rev. Neurosci., 35, 25–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Blackstone, C. (2018) Converging cellular themes for the hereditary spastic paraplegias. Curr. Opin. Neurobiol., 51, 139–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Goyal, U. and Blackstone, C. (2013) Untangling the web: mechanisms underlying ER network formation. Biochim. Biophys. Acta, 1833, 2492–2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Park, S.H., Zhu, P.-P., Parker, R.L. and Blackstone, C. (2010) Hereditary spastic paraplegia proteins REEP1, spastin, and atlastin-1 coordinate microtubule interactions with the tubular ER network. J. Clin. Invest., 120, 1097–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Claudiani, P., Riano, E., Errico, A., Andolfi, G. and Rugarli, E.I. (2005) Spastin subcellular localization is regulated through usage of different translation start sites and active export from the nucleus. Exp. Cell Res., 309, 358–369. [DOI] [PubMed] [Google Scholar]
- 10. Allison, R., Edgar, J.R. and Reid, E. (2019) Spastin MIT disease associated mutations disrupt lysosomal function. Front. Neurosci., 13, 1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yang, D., Rismanchi, N., Renvoisé, B., Lippincott-Schwartz, J., Blackstone, C. and Hurley, J.H. (2008) Structural basis for midbody targeting of spastin by the ESCRT-III protein CHMP1B. Nat. Struct. Mol. Biol., 15, 1278–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hu, J., Shibata, Y., Zhu, P.-P., Voss, C., Rismanchi, N., Prinz, W.A., Rapoport, T.A. and Blackstone, C. (2009) A class of dynamin-like GTPases involved in the generation of the tubular ER network. Cell, 138, 549–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Orso, G., Pendin, D., Liu, S., Tosetto, J., Moss, T.J., Faust, J.E., Micaroni, M., Egorova, A., Martinuzzi, A., McNew, J.A. and Daga, A. (2009) Homotypic fusion of ER membranes requires the dynamin-like GTPase atlastin. Nature, 4, 978–983. [DOI] [PubMed] [Google Scholar]
- 14. Rismanchi, N., Soderblom, C., Stadler, J., Zhu, P.-P. and Blackstone, C. (2008) Atlastin GTPases are required for Golgi apparatus and ER morphogenesis. Hum. Mol. Genet., 17, 1591–1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Beetz, C., Schüle, R., Deconinck, T., Tran-Viet, K.-N., Zhu, H., Kremer, B.P.H., Frints, S.G.M., vanZelst-Stams, W.A.G., Byrne, P., Otto, S.et al. (2008) REEP1 mutation spectrum and genotype/phenotype correlation in hereditary spastic paraplegia type 31. Brain, 131, 1078–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Guglielmi, A. (2020) A complete overview of REEP1: old and new insights on its role in hereditary spastic paraplegia and neurodegeneration. Rev. Neurosci., 31, 351–362. [DOI] [PubMed] [Google Scholar]
- 17. Blackstone, C. (2015) Murine models of autosomal recessive hereditary spastic paraplegia. In LeDoux, M.S. (ed), Movement Disorders, 2nd edn. Academic Press, London, UK, pp. 1087–1093. [Google Scholar]
- 18. Fassier, C., Hazan, J. and Melki, J. (2015) Mouse models of autosomal dominant spastic paraplegia. In LeDoux, M.S. (ed), Movement Disorders, 2nd edn. Academic Press, London, UK, pp. 1073–1086. [Google Scholar]
- 19. Kasher, P.R., De Vos, K.J., Wharton, S.B., Manser, C., Bennett, E.J., Bingley, M., Wood, J.D., Milner, R., McDermott, C.J., Miller, C.C.J.et al. (2009) Direct evidence for axonal transport defects in a novel mouse model of mutant spastin-induced hereditary spastic paraplegia (HSP) and human HSP patients. J. Neurochem., 110, 34–44. [DOI] [PubMed] [Google Scholar]
- 20. Tarrade, A., Fassier, C., Courageot, S., Charvin, D., Vitte, J., Peris, L., Thorel, A., Mouisel, E., Fonknechten, N., Roblot, N.et al. (2006) A mutation of spastin is responsible for swellings and impairment of transport in a region of axon characterized by changes in microtubule composition. Hum. Mol. Genet., 15, 3544–3458. [DOI] [PubMed] [Google Scholar]
- 21. Qiang, L., Piermarini, E. and Baas, P.W. (2019) New hypothesis for the etiology of SPAST-based hereditary spastic paraplegia. Cytoskeleton, 76, 289–297. [DOI] [PubMed] [Google Scholar]
- 22. Beetz, C., Koch, N., Khundadze, M., Zimmer, G., Nietzsche, S., Hertel, N., Huebner, A.K., Mumtaz, R., Schweizer, M., Dirren, E.et al. (2013) A spastic paraplegia mouse model reveals REEP1-dependent ER shaping. J. Clin. Invest., 123, 4273–4282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Renvoisé, B., Malone, B., Falgairolle, M., Munasinghe, J., Stadler, J., Sibilla, C., Park, S.H. and Blackstone, C. (2016) Reep1 null mice reveal a converging role for hereditary spastic paraplegia proteins in lipid droplet regulation. Hum. Mol. Genet., 25, 5111–5125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Willkomm, L., Heredia, R., Hoffmann, K., Wang, H., Voit, T., Hoffman, E.P. and Cirak, S. (2016) Homozygous mutation in Atlastin GTPase 1 causes recessive hereditary spastic paraplegia. J. Hum. Genet., 61, 571–573. [DOI] [PubMed] [Google Scholar]
- 25. Schottmann, G., Seelow, D., Seifert, F., Morales-Gonzalez, S., Gill, E., vonAu, K., vonMoers, A., Stenzel, W. and Schuelke, M. (2015) Recessive REEP1 mutation is associated with congenital axonal neuropathy and diaphragmatic palsy. Neurol. Genet., 1, e32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Perrot, R. and Eyer, J. (2013) Neurofilaments: properties, functions, and gegulation. In Dermietzel, R. (ed), The Cytoskeleton: Imaging, Isolation, and Interaction. Humana Press, Totowa, NJ, pp. 171–236. [Google Scholar]
- 27. Terasaki, M. (2018) Axonal endoplasmic reticulum is very narrow. J. Cell Sci., 131, jcs210450. [DOI] [PubMed] [Google Scholar]
- 28. Xu, K., Zhong, G. and Zhuang, X. (2013) Actin, spectrin and associated proteins form a periodic cytoskeletal structure in axons. Science, 339, 452–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Grumati, P., Dikic, I. and Stolz, A. (2018) ER-phagy at a glance. J. Cell Sci., 131, jcs217364. [DOI] [PubMed] [Google Scholar]
- 30. Chen, Q., Xiao, Y., Chai, P., Zheng, P., Teng, J. and Chen, J. (2019) ATL3 is a tubular ER-phagy receptor for GABARAP-mediated selective autophagy. Curr. Biol., 29, 846–855. [DOI] [PubMed] [Google Scholar]
- 31. Grumati, P., Morozzi, G., Holper, S., Mari, M., Harwardt, M.-L.l., Yan, R., Müller, S., Reggiori, F., Heilemann, M. and Dikic, I. (2017) Full length RTN3 regulates turnover of tubular endoplasmic reticulum via selective autophagy. elife, 6, e25555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Liang, J.R., Lingeman, E., Ahmed, S. and Corn, J.E. (2018) Atlastins remodel the endoplasmic reticulum for selective autophagy. J. Cell Biol., 21, 3354–3367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pengo, N., Scolari, M., Oliva, L., Milan, E., Mainoldi, F., Raimondi, A., Fagioli, C., Merlini, A., Mariani, E., Pasqualetto, E.et al. (2013) Plasma cells require autophagy for sustainable immunoglobulin production. Nat. Immunol., 14, 298–305. [DOI] [PubMed] [Google Scholar]
- 34. Mou, Y., Dong, Y., Chen, Z., Denton, K.R., Duff, M.O., Blackstone, C., Zhang, S.-C. and Li, X.-J. (2020) Impaired lipid metabolism in astrocytes underlies degeneration of cortical projection neurons in hereditary spastic paraplegia. Acta Neuropathol. Commun., 8, 214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Santalova, I.M. and Moshkov, D.A. (1999) Smooth endoplasmic reticulum in fish Mauthner cells at different functional states. Neuroscience, 89, 593–602. [DOI] [PubMed] [Google Scholar]
- 36. Wang, J., Lee, J., Liem, D. and Ping, P. (2017) HSPA5 gene encoding Hsp70 chaperone BiP in the endoplasmic reticulum. Gene, 618, 14–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Muchowski, P.J., Schaffar, G., Sittler, A., Wanker, E.E., Hayer-Hartl, M.K. and Hartl, F.-U. (2000) Hsp70 and hsp40 chaperones can inhibit self-assembly of polyglutamine proteins into amyloid-like fibrils. Proc. Natl. Acad. Sci. U. S. A., 97, 7841–7846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Baek, J.-H., Whitfield, D., Howlett, D., Francis, P., Bereczki, E., Ballard, E.C., Hortobágyi, T., Attems, J. and Aarsland, D. (2016) Unfolded protein response is activated in Lewy body dementias. Neuropathol. Appl. Neurobiol., 42, 352–365. [DOI] [PubMed] [Google Scholar]
- 39. Booth, L., Roberts, J.L. and Dent, P. (2015) HSPA5/DNA K may be a useful target for human disease therapies. DNA Cell Biol., 34, 153–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Schuck, S., Prinz, W.A., Thorn, K.S., Voss, C. and Walter, P. (2009) Membrane expansion alleviates endoplasmic reticulum stress independently of the unfolded protein response. J. Cell Biol., 187, 525–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zheng, P., Obara, C.J., Szczesna, E., Nixon-Abell, J., Mahalingan, K.K., Roll-Mecak, A., Lippincott-Schwartz, J. and Blackstone, C. (2022) ER proteins decipher the tubulin code to regulate organelle distribution. Nature, 601, 132–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yuan, A., Rao, M.V. and Veeranna and Nixon, R.A. (2017) Neurofilaments and neurofilament proteins in health and disease. Cold Spring Harb. Perspect. Biol., 9, a018309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Laser-Azogui, A., Kornreich, M., Malka-Gibor, E. and Beck, R. (2015) Neurofilament assembly and function during neuronal development. Curr. Opin. Cell Biol., 32, 92–101. [DOI] [PubMed] [Google Scholar]
- 44. Zheng, Y.-L., Li, B.-S. and Veeranna and Pant, H.C. (2003) Phosphorylation of the head domain of neurofilament protein (NF-M): a factor regulating topographic phosphorylation of NF-M tail domain KSP sites in neurons. J. Biol. Chem., 278, 24026–24032. [DOI] [PubMed] [Google Scholar]
- 45. Darios, F., Mochel, F. and Stevanin, G. (2020) Lipids in the physiopathology of hereditary spastic paraplegias. Front. Neurosci., 14, 74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Klemm, R.W., Norton, J.P., Cole, R.A., Li, C.S., Park, S.H., Crane, M.M., Li, L., Jin, D., Boye-Doe, A., Liu, T.Y.et al. (2013) A conserved role for atlastin GTPases in regulating lipid droplet size. Cell Rep., 3, 1465–1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Guyenet, S.J., Furrer, S.A., Damian, V.M., Baughan, T.D., La Spada, A.R. and Garden, G.A. (2010) A simple composite phenotype scoring system for evaluating mouse models of cerebellar ataxia. J. Vis. Exp., 39, 1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zhu, P.-P., Patterson, A., Lavoie, B., Stadler, J., Shoeb, M., Patel, R. and Blackstone, C. (2003) Cellular localization, oligomerization, and membrane association of the hereditary spastic paraplegia 3A (SPG3A) protein atlastin. J. Biol. Chem., 278, 49063–49071. [DOI] [PubMed] [Google Scholar]
- 49. Zhu, P.-P., Soderblom, C., Tao-Cheng, J.-H., Stadler, J. and Blackstone, C. (2006) SPG3A protein atlastin-1 is enriched in growth cones and promotes axon elongation during neuronal development. Hum. Mol. Genet., 15, 1343–1353. [DOI] [PubMed] [Google Scholar]
- 50. Porchet, R., Probst, A., Dráberová, E., Dráber, P., Riederer, I.M. and Riederer, B.M. (2003) Differential subcellular localization of phosphorylated neurofilament and tau proteins in degenerating neurons of the human entorhinal cortex. Neuroreport, 14, 929–933. [DOI] [PubMed] [Google Scholar]
- 51. Tao-Cheng, J.-H., Gallant, P.E., Brightman, M.W., Dosemeci, A. and Reese, T.S. (2007) Structural changes at synapses after delayed perfusion fixation in different regions of the mouse brain. J. Comp. Neurol., 501, 731–740. [DOI] [PubMed] [Google Scholar]
- 52. Xu, C.S., Hayworth, K.J., Lu, Z., Grob, P., Hassan, A.M., Garcia-Cerdán, J.G., Niyogi, K.K., Nogales, E., Weinberg, R.J. and Hess, H.F. (2017) Enhanced FIB-SEM systems for large-volume 3D imaging. elife, 6, e25916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Xu, C.S., Hayworth, K.J. and Hess, H.F. (2020) Enhanced FIB-SEM systems for large-volume 3D imaging. US Patent 10,600,615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Xu, C.S., Pang, S., Hayworth, K.J. and Hess, H.F. (2020) Transforming FIB-SEM systems for large-volume connectomics and cell biology. In Wacker, I., Hummel, E., Burgold, S. and Schröder, R. (eds), Volume Microscopy: Neuromethods. Humana Press, Totowa, NJ, Vol. 155, pp. 221–243. [Google Scholar]
- 55. Lee, T.-C. and Kashyap, R.L. (1994) Building skeleton models via 3-D medial surface/axis thinning algorithms. CVGIP: Graph. Models Image Process, 56, 462–478. [Google Scholar]
- 56. van der Walt, S., Schönberger, J.L., Nunez-Iglesias, J., Boulogne, F., Warner, J.D., Yager, N., Gouillart, E., Yu, T. and scikit image contributors. (2014) Scikit-image: image processing in python. Peer J, 2, e453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Wu, C.-Y., Whye, D., Mason, R.W. and Wang, W. (2012) Efficient differentiation of mouse embryonic stem cells into motor neurons. J. Vis. Exp., 64, e3813. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data supporting findings of this study are available from the corresponding author upon request. FIB-SEM datasets are available at openorganelle.org.