

Abstract
Differences by sex in lung cancer incidence and mortality have been reported which cannot be fully explained by sex differences in smoking behavior, implying existence of genetic and molecular basis for sex disparity in lung cancer development. However, the information about sex dimorphism in lung cancer risk is quite limited despite the great success in lung cancer association studies. By adopting a stringent two-stage analysis strategy, we performed a genome-wide gene–sex interaction analysis using genotypes from a lung cancer cohort including ~ 47 000 individuals with European ancestry. Three low-frequency variants (minor allele frequency < 0.05), rs17662871 [odds ratio (OR) = 0.71, P = 4.29×10−8); rs79942605 (OR = 2.17, P = 2.81×10−8) and rs208908 (OR = 0.70, P = 4.54×10−8) were identified with different risk effect of lung cancer between men and women. Further expression quantitative trait loci and functional annotation analysis suggested rs208908 affects lung cancer risk through differential regulation of Coxsackie virus and adenovirus receptor gene expression in lung tissues between men and women. Our study is one of the first studies to provide novel insights about the genetic and molecular basis for sex disparity in lung cancer development.
Introduction
Lung cancer is the leading cause of cancer death for both men and women worldwide with a complex genetic and molecular mechanism. Differences by sex in lung cancer incidence and mortality have been reported (1,2). Historically, the sex difference in lung cancer risk development was explained by sex-differences in smoking behavior as women were less likely to smoke cigarettes, initiated smoking at older ages and smoked fewer cigarettes per day (3,4). In 2018, a study on incidence rate of lung cancer between men and women, on the basis of data including 300 343 cases of lung cancer in non-Hispanic Whites, showed that the female-to-male incidence rate ratio had increased over the past two decades, whereas the prevalence of smoking among women had been approaching to the prevalence among men since 1965 (5). And the ratio exceeded 1 in the age groups of 30–34, 35–39, 40–44 and 45–49 years in people with European ancestry. For example, the female-to-male incidence rate ratio among individuals between 40 and 44 years of age had increased from 0.88 [95% CI (confidence interval): (0.84, 0.92)] during the 1995–1999 period to 1.17 [95% CI: (1.11, 1.23)] during the 2010–2014 period and from 0.81 [95% CI: (0.78, 0.83) to 1.13 [95% CI: (1.09, 1.16)]. A similar trend was also identified in Asian or pacific islander and Hispanic population. As smoking behavior has become increasingly similar between men and women, there is growing evidence that sex differences in lung cancer risk cannot be fully explained by differences in smoking behavior, implying sex-based variations in the genetic and molecular basis for lung cancer (5,6). However, the information for sex differences in lung cancer risk remains poorly understood despite the extensive efforts spent in lung cancer research and great success in identifying genetic factors through lung cancer association studies.
Genome-wide association studies (GWAS) have been used to identify sexual dimorphism in genetic susceptibility to lung cancer. Sex-specific GWAS identified VTI1A, ACVR1B and FOXP4-AS1 genes in women that influence lung cancer development (7,8). However, prior female sex-specific GWAS could not test for statistical significance in males or for gene–sex interactions. Gene–sex interaction analysis, on the other hand, will evaluate the information from both male and female group systematically and can identify the variants with significant difference between men and women, although those variants may not achieve genome-wide significance in stratified analysis. But genome-wide genetic interaction (GWGI) studies still remain challenging as most GWAS were designed for main effect detection and have had limited power for interaction analysis. Analytical studies have shown that a sample size that is at least 4-fold larger is required for detecting significant effects in interaction analysis using a standard case–control design compared with detecting significant main effects. An even larger sample size is required when the effect size is modest or the risk allele has a lower frequency (9). A case-only approach has been shown to be much more powerful in detection of an interaction effect than a standard case–control design in the absence of gene–environment correlation (10–14). However, the test validity is destroyed if the gene–environment independence assumption is violated. Researchers proposed a combined case-only and case–control approach in GWGI: step 1, a case-only analysis to test the association between Single nucleotide polymorphism (SNPs) and environmental/or biological factors; step 2, candidate SNPs from step 1 are further evaluated using standard case–control logistic interaction analysis (15–17). This two-step study design benefits from both increased power from case-only analysis and robustness to gene–environment independence assumption. Researchers have applied this approach in gene–environment interaction analysis and identified several novel variants in various human disorders including lung cancer (17–19). In this report, we applied this two-step approach on genome-wide gene–sex interaction analysis in lung cancer, aiming to identify novel susceptibility loci with different or inverse effects between male and female population that are not significant in main-effect association analysis.
Functional inference of genetic variants is important for gaining insights about the molecular mechanism of the disease and clinical application of GWAS findings. Over 90% of GWAS variants are located within non-coding regions and they may affect disease risk through regulating expression of nearby genes (20,21). Expression quantitative trait loci (eQTL) analysis, an allelic association analysis with gene expression, provides a straightforward method to identify susceptibility genes associated with the GWAS hits and it has been widely used in GWAS to investigate the regulatory effect of variants (22–24). However, there are few or no reports about the application of eQTL in gene–sex interaction analysis in human diseases because of the limited studies in sexual dimorphism in disease risk. Another important approach for assessment of genetic variants is functional annotation analysis. Researchers developed various tools to infer the functional roles for the variants such as combined annotation dependent depletion (CADD) and RegulomeDB (25–27). These tools provide insightful information about the functional inference and are very helpful for fine mapping and pinning down the true causal allele.
In this report, we performed a genome-wide gene–sex interaction analysis using genotype from a large lung cancer cohort including ~ 47 000 individuals with European ancestry. Adopting the two-step analysis strategy, we identified novel variants with different risk effects between men and women that were missed by main-effect association analysis. eQTL and functional annotation analysis provided further information about the functional role for the identified genetic variants and supplied multiple lines of evidence for the sexual disparity in lung cancer development. Our study is one of the first studies for sexual difference in risk of cancer development between men and women. It is also the largest scanning for gene–sex interaction in lung cancer and explored the genetic and molecular basis for sexual differences in risk of this deadly disease.
Results
Novel signals from genome-wide gene–sex interaction analysis
In the case-only analysis stage, a total of 19 943 415 and 10 359 674 SNPs were tested in the discovery and replication studies, respectively. Figure 2a displays the Manhattan plot of joint case-only analysis of gene–sex interactions in lung cancer. No inflated type I error rate was detected (lambda = 0.98). There were eight SNPs with a case-only joint P-value <5×10−8 and joint P-values were more significant than those from both discovery and replication studies. Those SNPs were submitted to further case–control interaction analysis using pooled discovery and replication data including 24 223 lung cancer patients and 22 560 controls. Three of these had interaction P-values < 0.05 in case–control analysis (Supplementary Material, Table S2). All three candidate variants had minor allele frequency (MAF) < 0.05 and thus were further evaluated using Firth logistic regression method. The three variants remained significant in Firth analysis and the results are reported in Table 2.
Figure 2.
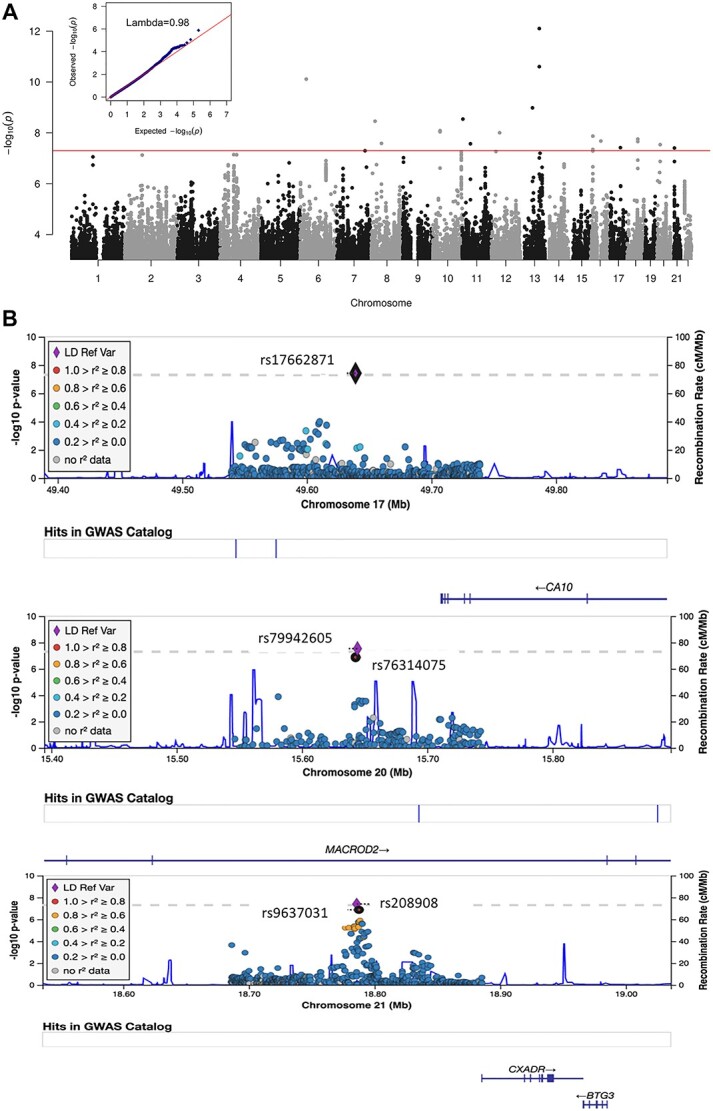
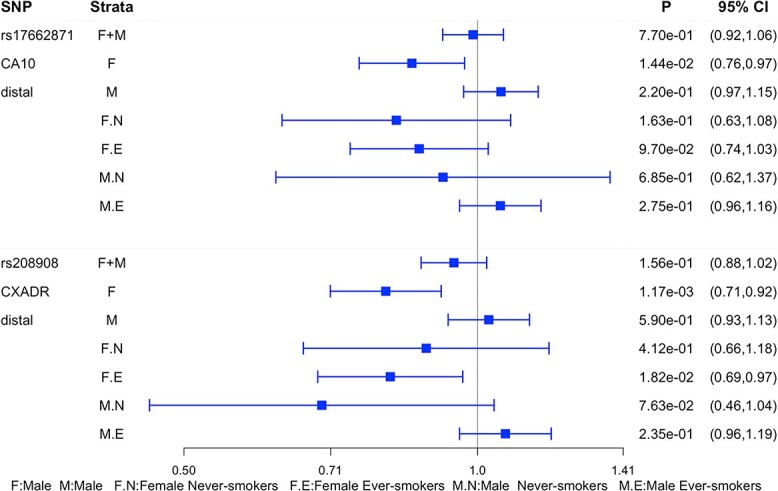
Plot of signals from genome-wide gene–sex interaction analysis in lung cancer. (a) Manhattan plot of P-values from case-only meta-analysis. No inflated type I error was detected in the analysis (lambda = 0.98). (b) Regional plot at three significant regions. (c) Forest plot of stratified lung cancer risk for the most significant SNP from each region.
Table 2.
Significant variants in gene–sex interaction analysis evaluated using Firth regression method
| Case-only | Case–control | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Discovery | Replication | Meta-analysis | Combined dataa | Score | ||||||||||
| SNP | rsID | MAF | GENE | OR | P | OR | P | P | OR | Q | I | OR | P | |
| 17:49639139:G:A | rs17662871 | 0.049 | CA10 distal | 0.69 | 1.21××10−7 | 0.80 | 7.03×10−2 | 4.29×10−8 | 0.71 | 0.27 | 17.10 | 0.78 | 4.06×10−3 | 0.72 |
| 20:15644218:T:C | rs79942605 | 0.012 | MACROD2 | 2.37 | 9.05×10−8 | 1.67 | 6.34×10−2 | 2.81×10−8 | 2.17 | 0.28 | 14.83 | 1.68 | 1.64×10−2 | 0.82 |
| 21:18785818:G:A | rs208908 | 0.039 | CXADR distal | 0.71 | 2.84×10−6 | 0.67 | 4.49×10−3 | 4.54×10−8 | 0.70 | 0.76 | 0.00 | 0.80 | 1.74×10−2 | 0.85 |
The results from analysis using Firth logistic regression method were reported for the three variants. Infor. Q indicated P-values for Cochrane’s Q statistic; I indicated I2 heterogeneity index (0–100); Score indicated the imputation quality score for the variants.
aDiscovery and replication combined data were used for case–control validation analysis.
SNP rs17662871, located near the carbonic anhydrase 10 (CA10) gene, had an OR of 0.69 and case-only P-value of 1.21×10−7 in discovery study, and OR of 0.80 and P-value of 7.03×10−2 in the replication study. Further case–control validation using Firth test detected an interaction OR of 0.78 and P-value of 4.06×10−3. SNP rs208908, located near Coxsackie virus and adenovirus receptor (CXADR) gene, had a P-value of 2.84×10−6 (OR = 0.71) and 4.49×10−3 (OR = 0.67) in case-only discovery and replication studies, respectively. It has a meta-analysis P-value of 4.54×10−8 (OR = 0.70) and P-value of 1.74×10−2 (OR = 0.80) in case–control analysis. rs79942605 had an OR of 2.17 (P = 2.81×10−8) in joint case-only analysis. In addition, case–control analysis identified an interaction OR of 1.68 (P = 1.64×10−2) in lung cancer cohort. We performed the same test in other lung cancer subtypes, such as lung adenocarcinoma and squamous lung cancer, etc. The signals for the top 10 variants in case-only analysis were reported for each lung cancer subtype although no significant variants were identified (Supplementary Material, Table S3).
Regional plot at the significant regions
We examined the regional information around the significant findings. rs17662871, close to CA10 gene, was the single variant achieving genome-wide significance of gene–sex interaction in the region and there were no other variants that were in strong linkage disequilibrium (LD) with this variant. This variant had an imputation quality score of 0.72 (Fig. 2b top). SNP rs79942605, located within mono-ADP ribosylhydrolase 2 (MACROD2) gene, had a P-value of 2.81×10−8 and OR 2.17 in joint case-only analysis. Another variant rs76314075, in high LD with rs79942605 (r2 ≥0.8), had an OR of 2.11 and P-value of 1.33×10−7 in the joint analysis (Fig. 2b middle). For the third novel variant, rs208908 (OR = 0.70, P = 4.54×10−8), located upstream of CXADR gene, we found eight variants with joint case-only P-value <1×10−5 and the strongly associated variant rs9637031 had an OR of 0.70 and P-value of 1.34×10−7 in the joint analysis (Fig. 2b bottom).
Stratified analysis of lung cancer risk at significant SNPs
To explore how sex influenced genetic risk in lung cancer, we conducted stratified lung cancer risk analysis in men and women using discovery and replication combined dataset. We observed very distinct lung cancer risk patterns between men and women at the three identified variants. For example, rs208908 had a protective effect for lung cancer in women with OR of 0.81 (P = 1.17×10−3) but had no significant effect in males (OR = 1.03, P = 0.59) (Fig. 2c). Similarly, SNP rs17662871 had a protective effect for lung cancer in women with OR of 0.86 (P = 1.44×10−2). We further stratified the analysis by smoking status (ever. vs. never smokers) and the risk effect did not vary by smoking status for these two SNPs. rs79942605, on the other hand, had an increased risk for lung cancer in women (OR = 1.52, P-value = 4.76×10−3). And this variant only had risk effect in ever smoking women (OR = 2.03, P = 4.47×10−4), whereas not in never smoking women (OR = 0.60, P = 1.79×10−1).
eQTL analysis
We conducted eQTL analysis to further evaluate the interaction effect associated with nearby gene expression for each of the candidate SNPs. Unfortunately, for rs79942605, in the MARCROD2 gene, no SNP data were available in genotype-tissue expression (GTEx). There were also no data for rs76314075 that is in strong LD with rs79942605 (r2 > 0.8) in GTEx. rs17662871, located upstream to CA10 gene, was an imputed SNP in GTEx with allele frequency of 0.05. We applied a best guess algorithm to assign most likely genotypes to each individual to transform the dosage into genotype. Because the variant is uncommon we did not model an additional group homozygous for the rare variant. After filtering the individuals with very low expression [reads per kilobase million (rpkm) < 0.25], 113 men and 37 women were available in the analysis, and only 5 of them were carriers. The gene expression [log2(rpkm)] between genotype ‘0’ and ‘1’ group was compared and the P-value was 2.21×10−3 in male+female combined group and 4.55×10−3 in male group. However, there was no valid test for women group as there was only one individual in genotype ‘1’ group (Fig. 3a left).
Figure 3.
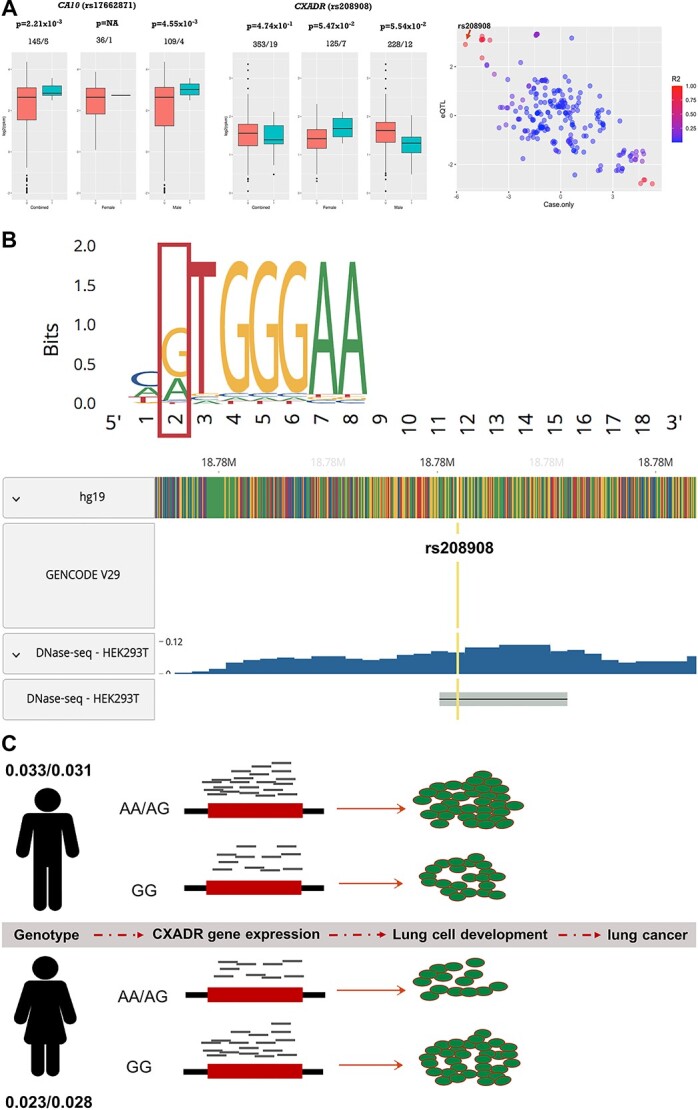
Functional analysis of identified variants. (a) eQTL analysis at candidate variants. Left and middle, box plot of gene expression from lung between individuals with none (0) and at least on risk allele (1) in female and male combined, female and male groups for CA10 and CXADR, respectively. The data from samples with Caucasian ancestry in GTEx were used in the analysis. Individuals with rpkm < 0.25 were removed from the analysis. P-values and number of samples were labeled above each plot. rs208908 was genotyped in GTEx; rs17662871 was imputed in SNP and SNP dosage ≤0.4 was coded as 0; SNP dosage ≥ 0.6 was coded as 1. Linear regression analysis of gene–sex interaction in predicting CXADR gene expression displayed P = 3.85×10−3. Right, scatter plot of z-scores from 229 SNPs, ranging from 18 745 702 to 18 806 105 (hg19) on chromosome 21 was displayed on the plot. X-axis represented gene–sex interaction analysis in lung cancer risk; and y-axis represented the z-scores from gene–sex interaction analysis in CXADR gene expression prediction. LD r2 was computed for each of the SNPs with rs208908 as reference. (b) Functional inference of rs208908. The predicted binding motif at rs208908 (highlighted in red color) for TF using PROMO (Upper). DNase-seq analysis in HEK293T (Human embryonic kidney) cells detected a peak covering rs208908 suggesting it was located within a cis-regulatory DNA sequence element (RegulomeDB bottom). (c) Presumptive disease risk model at rs208908. This variant, located upstream of CXADR, may affect lung cancer risk through gene expression regulation. The numbers indicate MAF for rs208908 in cases/controls between male and female population.
rs208908 was also a low-frequency variant with MAF of 0.04. It was a genotyped SNP in GTEx and there were 240 men and 132 women for gene expression association analysis after gene expression filtering. In the male+female combined group, the CXADR gene expression level was not significantly different between carrier and non-carrier group (P = 0.47). However, this gene had a lower expression in the carrier group compared with the non-carrier group at marginal level in men (P = 5.54×10−2) and had higher expression in carrier group compared with non-carrier group in women (P = 5.47×10−2) (Fig. 3a middle). A generalized linear model was applied to test the gene–sex interaction in CXADR gene expression prediction and there was a significant interaction effect with P-value of 3.85×10−3 (Supplementary Material, Table S4). We further expand the eQTL analysis to 229 SNPs located between 18 745 702 and 18 806 105 bp within the candidate region (hg19). The z-score from joint case-only was plotted against the z-score from eQTL analysis for those SNPs (Fig. 3a right). The variants with significant genetic interactions in lung cancer risk prediction also had significant interactions in gene expression prediction, and those variants were in strong LD with candidate SNP rs208908 (r2 > 0.8). These integrated results from genetic association and eQTL analysis suggested rs208908 had different risk effect in lung cancer between men and women through CXADR gene expression regulation in lung tissues.
Functional annotation analysis
We retrieved the SNPs with case-only meta-analysis P-value <1×10−5 from the three significant regions and queried the functional inference for each of the variants. Twelve SNPs were submitted to the analysis. rs79942605, the significant variant located in MACROD2, had the largest scaled-CADD (PHRED) score of 8.2 indicating top 15% of all reference genome of it being a functional allele. Among the nine SNPs from upstream of CXADR gene, rs208908 was predicted to be a transcription factor binding site (TBFS) of DNase peak with probability of 0.92 by RegulomeDB program (Table 3). Considering the strong probability of rs208908 being a regulatory SNP in TBFS, we used PROMO, a program for the identification of gene expression regulatory motif such as putative TFBS in DNA sequence, to search for motif located upstream of CXADR gene. We identified an 8-bp TBFS motif including six highly conserved variants in the human genome (Fig. 3b upper). The minor allele of rs208908 ‘A’ in the motif is predicted to increase the binding of transcription factor (TF) GR-beta and YY1 at this site. Both of the TFs have been reported involved in tumorigenesis (28,29). RegulomeDB searched the database of annotated SNPs with known and predicted regulatory elements in the intergenic regions of the human genome and found rs208908 being involved in chromatin state with strong transcription activity in human brain, pancreas, embryo tissues, etc. The DNase-seq analysis in HEK293T (Human embryonic kidney) cells also detected a peak covering rs208908 suggesting it was located within a cis-regulatory DNA sequence element (RegulomeDB, Fig. 3b bottom).
Table 3.
Functional annotation of candidate SNPs from significant regions
| SNP | ID | case-only | CADD | RegulomeDB | |
|---|---|---|---|---|---|
| POS | SNP | P | PHREDa | Function annotation | Probabilityb |
| 17:49639139:G:A | rs17662871 | 4.29×10−8 | 2.534 | TF binding or DNase peak | 0.49 |
| 20:15642704:G:A | rs76314075 | 1.33×10−7 | 1.445 | Other | 0.18 |
| 20:15644218:T:C | rs79942605 | 2.81×10−8 | 8.228 | TF binding or DNase peak | 0.04 |
| 21:18776825:C:T | rs208900 | 5.87×10−6 | 0.668 | TF binding or DNase peak | 0.13 |
| 21:18779654:G:A | rs184089 | 5.79×10−6 | 1.066 | Motif hit | 0.48 |
| 21:18783833:G:A | rs11088636 | 4.24×10−6 | 0.039 | Other | 0.18 |
| 21:18784296:T:C | rs423598 | 4.78×10−6 | 2.243 | TF binding or DNase peak | 0.13 |
| 21:18785818:G:A | rs208908 | 4.54×10 −8 | 5.773 | TF binding or DNase peak | 0.92 |
| 21:18787462:T:C | rs208914 | 5.01×10−6 | 2.944 | TF binding + DNase peak | 0.61 |
| 21:18787572:T:C | rs9637031 | 1.34×10−7 | 4.018 | TF binding + DNase peak | 0.61 |
| 21:18787948:A:G | rs6517771 | 1.76×10−6 | 5.095 | Motif hit | 0.34 |
| 21:18788445:A:C | rs1389157 | 1.30×10−6 | 6.257 | TF binding or DNase peak | 0.03 |
SNPs with case-only joint P-value <1×10−5 were selected for annotation analysis. eQTL displayed the information from GTEx Portal. The annotation information for rs208908 was bolded.
aScaled CADD score by expressing the rank in order of magnitude terms.
bRegulomeDB probability score is ranging from 0 to 1, with 1 being most likely to be a regulatory variant.
Discussion
Sex is an important biological factor in human disease development and extensive studies have been conducted to demonstrate sex differences in incidence, prognosis and treatment of various diseases including cancer (1,2). In 2016, a comprehensive characterization of molecular differences between male and female patients in 13 cancer types using multidimensional genomic data in the Cancer Genome atlas(TCGA) categorized lung cancer into the strong sex effect group with more extensive sex-biased genetic and molecular signatures (30). However, differences in risk on the basis of sex are one of the least studied factors in cancer susceptibility. And study of sex disparity in lung cancer development is quite limited despite the success of GWAS in lung cancer research during the past decade. A comprehensive gene–sex interaction scanning combined with functional annotation analysis provided exploratory insights about molecular mechanism underlying difference in lung cancer risk between men and women.
Leveraging the rich resource from integrative analysis of lung cancer etiology and risk (INTEGRAL)-international lung cancer consortium (ILCCO) lung cancer consortium, we conducted one of the largest gene–sex interaction analysis in overall lung cancer as well as lung cancer subtypes using imputed genotype from ~ 47 000 individuals with European ancestry. By adopting a robust and stringent two-stage analysis strategy, we identified three significant gene–sex interactions in overall lung cancer and all the three variants had small MAF between 0.01 and 0.05. Considering all the three variants had MAF < 0.05 and the standard regression method may not preserve the type I error rate for variants with low allele frequency, we further applied the Firth test and validated the signals at the three SNPs. We even retrieved ~ 200 SNPs from the three candidate regions and compared the results between regular logistic regression method and Firth logistic regression method. The beta estimation and P-values between these two methods were highly concordant with each other implying that our results from case-only analysis were reliable (Supplementary Material, Figure S5). Among the three significant variants, rs17662871 and rs208908 displayed a protective effect for lung cancer in women and no effect in men. And the protective effect did not vary much by smoking status (ever vs. never) in women. rs79942605, from MACROD2 gene, displayed strong risk effect in only ever smoking women (OR = 1.52, P = 4.76×10−3), whereas no effect in men or never smoking women was observed. We also conducted the analysis in lung adenocarcinoma and squamous lung cancer but did not identify any significant interactions, possibly because of decreased sample sizes in these subgroups. This suggests that genome-wide scanning for interactions between genes and environmental/biological factors still remains a challenge requiring a more powerful analysis strategy.
All three novel variants identified in this study have significant risk effects in only one gender and non-significant effects in the other, and the non-significant effects tend to be in a reverse direction (Fig. 2C). Those sex-specific effects did not achieve genome-wide significance in regular or sex-specific GWAS analysis thus missed in main-effects analysis. Our findings suggest that gene–sex interaction is a good complement to GWAS in detection of loci with effect in only one gender or with inverse effects between genders.
It is possible that there are some SNPs not significant in case-only analysis but with differential magnitude of associations with lung cancer risk between sexes. We extracted the sex-specific association effects from 22 reported significant variants in lung cancer from European population (Supplementary Material, Table S6) (31,32). All of them have risk effects in both genders although some of them do not achieve genome-wide significance in one gender. Seven SNPs have varied risk effects between male and female groups (|ORmale-ORfemale| > 0.05). The largest variation was from rs9865715 with OR of 1.77 (P = 5.42×10–8) in women and OR of 1.49 (P = 2.38×10–4) in men. None of the seven variants had significant results in case-only analysis. These results suggested that the variants with differential magnitude of association in same direction between genders are usually identified in main-effect analysis using regular GWAS or sex-specific GWAS if their main-effect is very significant in one sex. And the gene–sex interactions may impose very negligible variations in risk effects in lung cancer development for those variants.
eQTL analysis has been very useful in GWAS to provide additional functional evidence for the identified susceptibility loci. However, very few studies were reported about its application in interaction analysis between genes and environmental/biological factors probably because of the limited studies in interaction scanning, restricting the contribution of eQTL in genetic association studies. We explored the application of eQTL in gene–sex interaction study. Take variant rs208908 and CXADR as an example, we did not identify significant association between rs208908 and CXADR gene in main-effect eQTL analysis (P = 0.47, Fig. 3a middle). However, we identified distinct gene expression patterns between men and women and we detected significant gene–sex interaction in CXADR gene expression prediction (P = 3.85×10−3). In addition, the signals from gene–sex interactions between lung cancer risk prediction and CXADR gene expression prediction are highly correlated among the SNPs in high LD with rs208908 (r2 > 0.8) (Fig. 3a right). The eQTL analysis provided strong evidence suggesting rs208908 is a functional variant with difference in lung cancer risk by sex and revealed great potential for application of eQTL in large-scale scanning for interactions between genes and environmental/biological factors.
Further functional annotation analysis inferred rs208908 as a TBFS with probability of 0.92. On the basis of information from existing TFBS, an 8-bp TBFS motif containing rs208908 was predicted and the minor allele ‘A’ at rs208908 is predicted to increase the binding of TF of Glucocorticoid Receptor-β and YY1 (Fig. 3b). Both of these TFs have been reported to be involved in cancer development and YY1 is a dual function TF and has been implicated as a major driver of many cancers including lung cancer (28,29). DNase-seq analysis also identified a cis-regulatory region including rs208908 variant in human cells. These results, combined with eQTL analysis, suggested rs208908 regulated CXADR gene expression by interacting with cellular factors such as TFs that function through recognition of conserved sequence motif located upstream of gene coding sequence. In 2018, a large-scale GWAS including ~ 370 000 individuals with European ancestry showed that CXADR gene was associated with lung function (Forced Expiratory Volume/Forced Vital Capacity (FEV1/FVCratio)) (33). These multiple lines of evidence, from the results from our integrated study to that from previous reports, suggested a disease model for SNP rs208908 with sexual disparity in lung cancer risk. SNP rs208908 has different MAF between women (0.030) and men (0.025). This difference combined with differentiated gene expression regulation mechanism between men and women, leads to different gene expression patterns between these groups resulting in different risk between male and female groups (Fig. 3c).
A remarkable number of genes have been identified to be differentially expressed between male and females in one or more human tissues including lung (34). In addition, sex-biased regulatory targeting patterns have been found for various TFs in human (35). In our study, rs208908 was found to have differential risk effect for lung cancer between men and women, with different MAF between sexes (0.025 in women and 0.032 in men) (Supplementary Material, Table S7). And MAF was 0.023/0.028 between cases/controls in women and 0.033/0.031 in men, indicating more imbalance of allele frequency between lung cancer cases and controls in female group. Similar findings were also found for rs17662871 and rs79942605. We further compared the MAF for the variants between men and women in controls only, to exclude the selection bias for the cases in the data, and the P-value for rs17662871 was 1.88×10−2 and 0.088 for rs208908 which was around the border line statistical significance level. This sex-specific allelic frequency suggested a sexually antagonistic selection, a selection can occur when both sexes have different fitness optima for a trait, leading to genetic variations between male and female in a population (36,37). For example, the difference in allele frequency for rs208908, as a key variant in a TF binding site, may cause differential regulation of TFs between sexes, and then differential CXADR gene expression and different lung cancer risk between men and women.
For the other two variants, rs17662871 (close to CA10) and rs79942605 (within MACROD2), very few supporting variants were identified nearby, which is not unusual for variants with low allele frequency. The low allele frequency also makes eQTL analysis challenging. rs79942605 was not available in GTEx and there were only a few samples with homogenous minor-allele genotype at rs17662871 in GTEx genotype data, which limited our ability to investigate the gene expression pattern across different genotype groups. Both CA10 and MACROD2 were reported to be associated with smoking behavior and age at menarche in Caucasian (38,39). Controversial results have been reported for association between age at menarche and lung cancer risk (40–42). Our study detected an effect for lung cancer in only women group in these two regions, especially rs79942650 from MACROD2 gene which has a risk effect in only smoking women, suggesting that sex plays an important role in lung cancer susceptibility and may interact with smoking behavior in cancer risk.
In summary, we conducted a large-scale gene–sex interaction scanning in lung cancer and we identified three significant variants with different risk effects on the basis of sex. Our study is one of the first studies to examine sex disparity in lung cancer development and our results provided insights about the genetic and molecular mechanism underlying the differences in lung cancer susceptibility between men and women. Our study is one of the largest scanning for gene–sex interaction in lung cancer in people with European ancestry. The three novel variants identified in our study all have MAF < 0.05. In our previous study to identify cross-ancestry loci contributing to lung cancer using multi-population genome-wide meta-analysis of 61 047cases and 947 237 controls, we identified five novel loci including three rare variants (MAF < 0.05) (31). These results suggest some variants still remain undetected in lung cancer, including those with low allele frequency requiring a larger sample size for more effective methods for detection.
Material and Methods
Materials
The imputed genotypes (reference panel: HRC r1.1) from 46 783 individuals with European ancestry, with lung cancer phenotype, smoking (ever vs. never smokers) and sex information, in the INTEGRAL-ILCCO lung cancer consortium were analyzed in this study (31). The subjects came from nine independent studies: the OncoArray Consortium Lung Study (OncoArray), including 16 845 lung cancer cases and 13 057 controls, was used as the discovery dataset (32,43). Individuals from another eight smaller independent studies: Affymetrix Axiom Array Study, the Genetic Epidemiology of Lung Cancer Consortium, the Environment and Genetics in Lung cancer Etiology study, the International Agency for Research on Cancer, MD Anderson Cancer Center Study, NCI Lung Cancer and Smoking Phenotypes in African-American Cases and Controls (NCI), the Prostate, Lung, Colorectal and Ovarian Cancer Screening Trial and Samuel Lunenfeld Research Institute Study were combined together and used as replication dataset including 7378 lung cancer cases and 9503 controls (Table 1, Supplementary Material, Table S1) (44–47). There were 12 084 women and 17 818 men in discovery dataset; and 6366 women and 10 515 men in replication dataset. About 60.4% of the lung cancer patients were male in the discovery study compared with 64.0% in replication study; 81.6% of individuals in both discovery and validation studies were ever smokers. Stringent quality controls were applied on SNPs and SNPs selected for the analysis had to qualify by two criteria: (1) had imputation information score ≥ 0.7 in discovery data; (2) had information score > 0.2 in each of the eight studies and sample-size weighted information score ≥ 0.7 in replication data. About ~ 20 000 000, out of 39 000 000 imputed SNPs, with information score ≥ 0.7 were analyzed in discovery study and ~ 10 000 000 SNPs were analyzed in replication study (Fig. 1). About 193 050 markers, common to the 9 studies and with linkage disequilibrium r2 value less than or equal to 0.5, were selected to calculate principal components in PLINK. Detailed information about data collection, genotype imputation and quality control procedures can be found from our earlier publication (29).
Table 1.
Characteristics of participants that were studied during the discovery and replication phases
| Discoverya | Replicationb | |||||
|---|---|---|---|---|---|---|
| Male (%) | Female (%) | Total | Male (%) | Female (%) | Total | |
| Controls | 7646 (58.6) | 5411 (41.4) | 13 057 | 5796 (61.0) | 3707 (39.0) | 9503 |
| Cases | 10 172 (60.4) | 6673 (39.6) | 16 845 | 4719 (64.0) | 2659 (36.0) | 7378 |
| Ever smokers | 15 584 (63.9) | 8813 (36.1) | 24 397 | 9230 (67.0) | 4553 (33.0) | 13 783 |
| Never smokers | 2234 (40.6) | 3271 (59.4) | 5505 | 1285 (41.5) | 1813 (58.5) | 3098 |
Samples with European ancestry in INTEGRAL-ILCCO consortium were analyzed in the study. Number of overall lung cancer cases was provided in the table.
aGenotype data from Oncoarray study were used in discovery study.
bGenotype from the other eight studies were used in replication study.
Figure 1.

Flow chart of analysis strategy in genome-wide gene–sex interaction analysis in lung cancer. In genetic association analysis, all the tested SNPs have information score ≥ 0.7 in discovery data; information score > 0.2 in each of the eight studies and sample-size weighted information score ≥ 0.7 in replication data. Smoking status (ever vs. never smokers) and first five principal components were adjusted in case-only, case–control and firth validation analysis.
Statistical methods for GWGI
Following the two-step analysis strategy, case-only analysis was first performed between SNP dosage (additive model) and sex (male and female were coded as 1 and 2, respectively) phenotype using lung cancer patients from discovery study (n = 16 845) and replication study (n = 7378) (case-only model, S denotes smoking status). Fixed-effect meta-analysis was conducted to combine information from both studies. Variants with case-only joint P-value <5×10−8 and joint P-values more significant than those from either study were selected for further validation using case–control analysis. All the samples in discovery and replication study including 24 223 lung cancer patients and 22 560 controls were applied in case–control analysis (Case–control model, D denotes disease status). The SNPs with case–control gene–sex interaction P-value < 0.05 were reported as significant findings. For the significant variants with low allele frequency (MAF < 0.05), we further validated the signals using firth logistic regression, a method designed for rare variants association test to reduce small-sample bias in regular logistic regression (Fig. 1) (48). The statistical analysis was adjusted for smoking status (ever and never smokers) and the first five principal components. The analysis was conducted in overall lung cancer as well as adenocarcinoma (No. Cases = 9630) and squamous lung cancer (No. Cases = 6019).
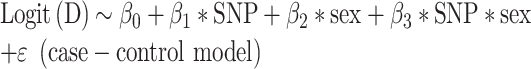 |
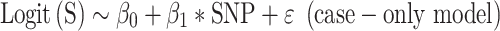 |
eQTL analysis
Genotype dosage and gene expression rpkm data from lung tissue were downloaded from GTEx (phs000424.GTEx.v7.p2). There were 377 individuals with European ancestry available for the analysis, including 244 men and 133 women. Average rpkm for the gene was used if there were duplicated samples. Individuals with rpkm < 0.25 were removed from the analysis. For imputed variants, we applied a best guess algorithm to assign most likely genotypes to each individual to transform the dosage into genotype. For variants with MAF ≥ 0.1, we adopted an additive model: dosage values ≤0.2 were coded as 0; dosage between 0.4 and 0.6 were coded as 1 and dosage ≥ 0.8 was coded as 2. For variants with MAF < 0.1, we adopted a dominant model: dosage values≤0.4 were coded as 0 (non-carrier group with no risk alleles) and dosage ≥ 0.6 was coded as 1 (carrier group with at least 1 risk allele). For each candidate SNP, the boxplot of log2(rpkm) across different genotype was displayed for all the individuals, men and women groups. Student’s test was performed to compare the mean log2(rpkm) across different genotype groups and general linear regression was conducted to test SNP–sex interaction in gene expression prediction.
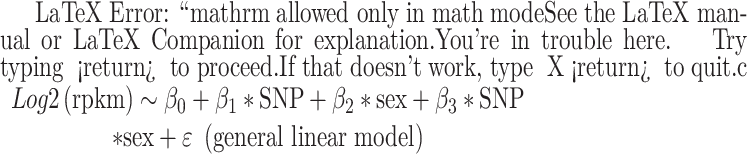 |
Gene–sex interaction and eQTL analyses were conducted using program R-4.0.2. R package logistf 1.23 was applied for Firth logistic regression analysis. PLINK 1.07 was used for meta-analysis.
Functional annotation analysis
Two public functional annotation tools, CADD and RegulomeDB were applied for functional inference. CADD was designed to predict functional variants by integrating diverse information from wide range of function categories (25). It computed a score inferring the functional ranking of the variants which is helpful for fine mapping. RegulomeDB is a tool designed to predict regulatory DNA elements in the human genome on the basis of information from Gene Expression Omnibus, ENCODE and public literature (26). It adopted score of unified regulatory features model, trained on data from massively parallel reporter assays, to predict the functional variants in enhancer and promoter elements. RegulomeDB provides a probability score for the variant being TFBS, promotor or DNase hypersensitivity, etc. PROMO, an online tool to perform computational-based searches for gene expression regulatory sequence motif on the basis of TFBS database, was used to search for the functional motif with sequence containing the candidate variants as input (27).
Supplementary Material
Acknowledgement
The Genotype-Tissue Expression (GTEx) Project was supported by the Common Fund of the Office of the Director of the National Institutes of Health, and by NCI, NHGRI, NHLBI, NIDA, NIMH and NINDS. The data used for the analyses described in this manuscript were obtained from: dbGaP accession number phs000424.v7.p2.
Conflict of Interest statement. None declared.
Contributor Information
Yafang Li, Institute for Clinical and Translational Research, Baylor College of Medicine, Houston, TX 77030, USA; Section of Epidemiology and Population Sciences, Department of Medicine, Baylor College of Medicine, Houston, TX 77030, USA; Dan L Duncan Comprehensive Cancer Center, Baylor College of Medicine, Houston, TX 77030, USA.
Xiangjun Xiao, Institute for Clinical and Translational Research, Baylor College of Medicine, Houston, TX 77030, USA.
Jianrong Li, Institute for Clinical and Translational Research, Baylor College of Medicine, Houston, TX 77030, USA.
Jinyoung Byun, Institute for Clinical and Translational Research, Baylor College of Medicine, Houston, TX 77030, USA; Section of Epidemiology and Population Sciences, Department of Medicine, Baylor College of Medicine, Houston, TX 77030, USA.
Chao Cheng, Institute for Clinical and Translational Research, Baylor College of Medicine, Houston, TX 77030, USA; Section of Epidemiology and Population Sciences, Department of Medicine, Baylor College of Medicine, Houston, TX 77030, USA; Dan L Duncan Comprehensive Cancer Center, Baylor College of Medicine, Houston, TX 77030, USA.
Yohan Bossé, Institut universitaire de cardiologie et de pneumologie de Québec, Department of Molecular Medicine, Laval University, Quebec City G1V 4G5, Canada.
James McKay, Section of Genetics, International Agency for Research on Cancer, World Health Organization, Lyon 69372, France.
Demetrios Albanes, Division of Cancer Epidemiology and Genetics, National Cancer Institute, National Institutes of Health, Bethesda, MD 20850, USA.
Stephen Lam, Department of Integrative Oncology, University of British Columbia, Vancouver, BC V5Z 1L3, Canada.
Adonina Tardon, Public Health Department, University of Oviedo, ISPA and CIBERESP, Asturias 33003, Spain.
Chu Chen, Program in Epidemiology, Public Health Sciences Division, Fred Hutchinson Cancer Research Center, Seattle, WA 98109, USA.
Stig E Bojesen, Department of Clinical Biochemistry, Copenhagen University Hospital, Copenhagen 2600, Denmark; Faculty of Health and Medical Sciences, University of Copenhagen, Copenhagen 2177, Denmark.
Maria T Landi, Division of Cancer Epidemiology and Genetics, National Cancer Institute, National Institutes of Health, Bethesda, MD 20850, USA.
Mattias Johansson, Section of Genetics, International Agency for Research on Cancer, World Health Organization, Lyon 69372, France.
Angela Risch, Thoraxklinik at University Hospital Heidelberg, Heidelberg 69126, Germany; Translational Lung Research Center Heidelberg (TLRC-H), Heidelberg 69120, Germany; University of Salzburg and Cancer Cluster Salzburg, 5020, Austria.
Heike Bickeböller, Department of Genetic Epidemiology, University Medical Center, Georg-August-University Göttingen, 37099, Germany.
H-Erich Wichmann, Institute of Medical Statistics and Epidemiology, Technical University Munich, 80333, Germany.
David C Christiani, Departments of Environmental Health and Epidemiology, Harvard TH Chan School of Public Health, Boston, MA 02115, USA.
Gad Rennert, Clalit National Cancer Control Center at Carmel Medical Center and Technion Faculty of Medicine, Haifa 3436212, Israel.
Susanne Arnold, University of Kentucky, Markey Cancer Center, Lexington, Kentucky 40536, USA.
Gary Goodman, Swedish Cancer Institute, Seattle, WA 98104, USA.
John K Field, Institute of Translational Medicine, University of Liverpool, Liverpool L69 7BE, United Kingdom.
Michael P A Davies, Institute of Translational Medicine, University of Liverpool, Liverpool L69 7BE, United Kingdom.
Sanjay S Shete, Department of Biostatistics, The University of Texas, M.D. Anderson Cancer Center, Houston, TX 77030, USA; Department of Epidemiology, The University of Texas MD Anderson Cancer Center, Houston, TX 77030, USA.
Loic Le Marchand, Epidemiology Program, University of Hawaii Cancer Center, Honolulu, HI 96813, USA.
Olle Melander, Faculty of Medicine, Lund University, Lund 22184, Sweden.
Hans Brunnström, Faculty of Medicine, Lund University, Lund 22184, Sweden.
Geoffrey Liu, University Health Network- The Princess Margaret Cancer Centre, Toronto, CA ON, M5G 2C1, Canada.
Rayjean J Hung, Luenfeld-Tanenbaum Research Institute, Sinai Health System, Toronto ON, M5G 1X5, Canada; Division of Epidemiology, Dalla Lana School of Public Health, University of Toronto, Toronto ON, M5T 3M7, Canada.
Angeline S Andrew, Departments of Epidemiology and Community and Family Medicine, Dartmouth College, Hanover, NH 03755, USA.
Lambertus A Kiemeney, Radboud University Medical Center, Nijmegen 6525, The Netherlands.
Hongbing Shen, Department of Epidemiology and Biostatistics, Jiangsu Key Lab of Cancer Biomarkers, Prevention and Treatment, Collaborative Innovation Center for Cancer Personalized Medicine, School of Public Health, Nanjing Medical University, Nanjing 211166, P.R. China.
Ryan Sun, Department of Biostatistics, The University of Texas, M.D. Anderson Cancer Center, Houston, TX 77030, USA.
Shan Zienolddiny, National Institute of Occupational Health, Oslo 0304, Norway.
Kjell Grankvist, Department of Medical Biosciences, Umeå University, Umeå 901 87, Sweden.
Mikael Johansson, Department of Radiation Sciences, Umeå University, Umeå 901 87, Sweden.
Neil Caporaso, Division of Cancer Epidemiology and Genetics, National Cancer Institute, National Institutes of Health, Bethesda, MD 20850, USA.
Dawn M Teare, Population Health Sciences Institute, Newcastle University, Newcastle upon Tyne, NE2 4AX, UK.
Yun-Chul Hong, Department of Preventive Medicine, Seoul National University College of Medicine, Seoul 03080, Republic of Korea.
Philip Lazarus, Department of Pharmaceutical Sciences, College of Pharmacy, Washington State University, Spokane, Washington 99202, USA.
Matthew B Schabath, Department of Cancer Epidemiology, H. Lee Moffitt Cancer Center and Research Institute, Tampa, FL 33612, USA.
Melinda C Aldrich, Department of Thoracic Surgery, Division of Epidemiology, Vanderbilt University Medical Center Nashville, TN 37232, USA.
Ann G Schwartz, Department of Oncology, Wayne State University School of Medicine, Detroit, MI 48201, USA; Karmanos Cancer Institute, Detroit, MI 48201, USA.
Ivan Gorlov, Institute for Clinical and Translational Research, Baylor College of Medicine, Houston, TX 77030, USA; Section of Epidemiology and Population Sciences, Department of Medicine, Baylor College of Medicine, Houston, TX 77030, USA; Dan L Duncan Comprehensive Cancer Center, Baylor College of Medicine, Houston, TX 77030, USA.
Kristen Purrington, Karmanos Cancer Institute, Detroit, MI 48201, USA.
Ping Yang, Division of Epidemiology, Department of Health Sciences Research, Mayo Clinics Rochester, MN, 55905, USA.
Yanhong Liu, Section of Epidemiology and Population Sciences, Department of Medicine, Baylor College of Medicine, Houston, TX 77030, USA; Dan L Duncan Comprehensive Cancer Center, Baylor College of Medicine, Houston, TX 77030, USA.
Younghun Han, Institute for Clinical and Translational Research, Baylor College of Medicine, Houston, TX 77030, USA; Section of Epidemiology and Population Sciences, Department of Medicine, Baylor College of Medicine, Houston, TX 77030, USA.
Joan E Bailey-Wilson, National Human Genome Research Institute, NIH, Baltimore, MD 20892, USA.
Susan M Pinney, University of Cincinnati College of Medicine, Cincinnati, OH 45267, USA.
Diptasri Mandal, Louisiana State University Health Sciences Center, New Orleans, LA 70112, USA.
James C Willey, College of Medicine and Life Sciences, University of Toledo, Toledo, OH 43614, USA.
Colette Gaba, The University of Toledo College of Medicine, Toledo, OH 43614, USA.
Paul Brennan, Section of Genetics, International Agency for Research on Cancer, World Health Organization, Lyon 69372, France.
Christopher I Amos, Institute for Clinical and Translational Research, Baylor College of Medicine, Houston, TX 77030, USA; Section of Epidemiology and Population Sciences, Department of Medicine, Baylor College of Medicine, Houston, TX 77030, USA; Dan L Duncan Comprehensive Cancer Center, Baylor College of Medicine, Houston, TX 77030, USA.
Author contribution
Y.L. designed and planned the study, presented the results and wrote the manuscript. X.X. assisted the data preparation and genome-wide gene-sex interaction analysis; J.L. and C.C. assisted the motif analysis in functional annotation; J.B. assisted the data preparation; Y.B. assisted the eQTL analysis. C.I.A. conceived and supervised the study. The other authors contributed to data collection. All authors discussed the results and commented on the manuscript.
Materials & Correspondence
Correspondence should be addressed to Yafang Li. Material requests should be addressed to Christopher I. Amos (chrisa@bcm.edu).
Funding
U19-CA203654 (NIH), RR170048, Cancer Prevention Research Institute of Texas (CPRIT), RR160097T (CPRIT), RR180061 (CPRIT), U01 CA243483-02, Genetic Epidemiology of Lung Cancer Consortium (GELCC), X01HG007491, Department of Health and Human Services contracts HHSN26820100007C, HHSN268201700012C, 75N92020C00001 and 1R21CA235464-01A1 (NIH) for support. CARET is funded by the National Cancer Institute, National Institutes of Health through grants U01-CA063673, UM1-CA167462 and U01-CA167462. The Harvard Lung Cancer Study is funded by NIH (NCI) grant U01CA209414 (Boston Lung Cancer Survival Cohort). The Liverpool Lung Project is supported by MPAD. Roy Castle Lung Foundation (UK). The EAGLE study was supported by the intramural program of the Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH. Joan E. The ReSoLuCENT study is funded by the Weston Park Hospital Cancer Charity. Acknowledgement to the George Isaac Family Fund for Cancer Research for grant support. Bailey-Wilson was supported by the Intramural Program of the National Human Genome Research Institute, NIH. Chris Amos and Chao Cheng are Research Scholars of the Cancer Prevention Institute of Texas.
References
- 1. Zhang, E.A. and Wynder, E.L. (1996) Differences in lung cancer risk between men and women: examination of the evidence. J. Natl. Cancer Inst., 88, 183–192. [DOI] [PubMed] [Google Scholar]
- 2. Kabir, Z., Connolly, G.N. and Clancy, L. (2008) Sex-differences in lung cancer cell-types? An epidemiologic study in Ireland. Ulster Med. J., 77, 31–35. [PMC free article] [PubMed] [Google Scholar]
- 3. Hammond, E.C. (1966) Smoking in relation to the death rates of one million men and women. Natl. Cancer Inst. Monogr., 19, 127–204. [PubMed] [Google Scholar]
- 4. Doll, R., Gray, R., Hafner, B. and Peto, R. (1980) Mortality in relation to smoking: 22 years’ observations on female British doctors. Br. Med. J., 280, 967–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jemal, A., Miller, K.D., Ma, J., Siegel, R.L., Fedewa, S.A., Islami, F., Devesa, S.S. and Thun, M.J. (2018) Higher lung cancer incidence in young women than young men in the United States. N. Engl. J. Med., 378, 1999–2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zang, E.A. and Wynder, E.L. (1996) Differences in lung cancer risk between men and women: examination of the evidence. J. Natl. Cancer Inst., 88, 183–192. [DOI] [PubMed] [Google Scholar]
- 7. Lan, Q., Hsiung, C.A., Matsuo, K., Hong, Y., Seow, A., Wang, Z., Hosgood, H.D., Chen, K., Wang, J., Chatterjee, N.et al. (2012) Genome-wide association analysis identifies new lung cancer susceptibility loci in never-smoking women in Asia. Nat. Genet., 44, 1330–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wang, Z., Seow, W.J., Shiraishi, K., Hsiung, C.A., Matsuo, K., Liu, J., Chen, K., Yamji, T., Yang, Y., Chang, I.et al. (2016) Meta-analysis of genome-wide association studies identifies multiple lung cancer susceptibility loci in never-smoking Asian women. Hum. Mol. Genet., 25, 620–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Smith, P.G. and Day, N.E. (1984) The design of case-control studies: the influence of confounding and interaction effects. Int. J. Epidemiol., 13, 356–365. [DOI] [PubMed] [Google Scholar]
- 10. Piegorsch, W.W., Weinberg, C.R. and Taylor, J.A. (1994) Non-hierarchical logistic models and case-only designs for assessing susceptibility in population-based case-control studies. Stat. Med., 13, 153–162. [DOI] [PubMed] [Google Scholar]
- 11. Khoury, M.J. and Flanders, W.D. (1996) Nontraditional epidemiologic approaches in the analysis of gene-environment interaction: case-control studies with no controls! Am. J. Epidemiol., 144, 207–213. [DOI] [PubMed] [Google Scholar]
- 12. Yang, Q., Khoury, M.J. and Flanders, W.D. (1997) Sample size requirements in case-only designs to detect gene-environment interaction. Am. J. Epidemiol., 146, 713–720. [DOI] [PubMed] [Google Scholar]
- 13. Weinberg, C.R. and Umbach, D.M. (2000) Choosing a retrospective design to assess joint genetic and environmental contributions to risk. Am. J. Epidemiol., 152, 197–203. [DOI] [PubMed] [Google Scholar]
- 14. Umbach, D.M. and Weinberg, C.R. (1997) Designing and analyzing case-control studies to exploit independence of genotype and exposure. Stat. Med., 16, 1731–1743. [DOI] [PubMed] [Google Scholar]
- 15. Li, D. and Conti, D.V. (2009) Detecting gene-environment interactions using a combined case-only and case-control approach. Am. J. Epidemiol., 169, 497–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Murcray, C.E., Lewinger, J.P. and Gauderman, W.J. (2009) Gene-environment interaction in genome-wide association studies. Am. J. Epidemiol., 169, 219–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Li, Y., Xiao, X., Han, Y., Gorlova, O., Qian, D., Leighl, N., Johansen, J.S., Barnett, M., Chen, C., Goodman, G.et al. (2018) Genome-wide interaction study of smoking behavior and non-small cell lung cancer risk in Caucasian population. Carcinogenesis, 39, 336–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gauderman, W.J., Zhang, P., Morrison, J.L. and Lewinger, J.P. (2013) Finding novel genes by testing G×E interactions in a genome-wide association study. Genet. Epidemiol., 37, 603–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Huang, T. and Hu, F.B. (2015) Gene-environment interactions and obesity: recent developments and future directions. BMC Medical Genom., 8, S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Edwards, S.L., Beesley, J., French, J.D. and Dunning, A.M. (2013) Beyond GWASs: illuminating the dark road from association to function. Am. J. Hum. Genet., 93, 779–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hrdlickova, B., deAlmeida, R.C., Borek, Z. and Withoff, S. (2014) Genetic variation in the non-coding genome: involvement of micro-RNAs and long non-coding RNAs in disease. Biochim. Biophys. Acta, 1842, 1910–1922. [DOI] [PubMed] [Google Scholar]
- 22. Hazelett, D.J., Rhie, S.K., Gaddis, M., Yan, C., Lakeland, D.L., Coetzee, S.G., Ellipse/GAME-ON consortium, Practical consortium, Henderson, B.E., Noushmehr, H.et al. (2014) Comprehensive functional annotation of 77 prostate cancer risk loci. PLoS Genet., 10, e1004102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pomerantz, M.M., Ahmadiyeh, N., Jia, L., Herman, P., Verzi, M.P., Doddapaneni, H., Beckwith, C.A., Chan, J.A., Hills, A., Davis, M.et al. (2009) The 8q24 cancer risk variant rs6983267 shows long-range interaction with MYC in colorectal cancer. Nat. Genet., 41, 882–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Smemo, S., Tena, S.J., Kim, K., Gamazon, E.R., Sakabe, N.J., Gómez-Marín, C., Aneas, I., Credidio, F.L., Sobreira, D.R., Wasserman, N.F.et al. (2014) Obesity-associated variants within FTO form long-range functional connections with IRX3. Nature, 507, 371–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rentzsch, P., Witten, D., Cooper, G.M., Shendure, J. and Kircher, M. (2019) CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res., 47, D886–D894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Boyle, A.P., Hong, E.L., Hariharan, M., Cheng, Y., Schaub, M.A., Kasowski, M., Karczewski, K.J., Park, J., Hitz, B.C., Weng, S.et al. (2012) Annotation of functional variation in personal genomes using RegulomeDB. Genome Res., 22, 1790–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Messeguer, X., Escudero, R., Farré, D., Nuñez, O., Martínez, J. and Albà, M. (2002) PROMO: detection of known transcription regulatory elements using species-tailored searches. Bioinformatics, 18, 333–334. [DOI] [PubMed] [Google Scholar]
- 28. Sarvagalla, S., Kolapalli, S.P. and Vallabhapurapu, S. (2019) The two sides of YY1 in cancer: a friend and a foe. Front. Oncol., 9, 1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. McBeth, L., Nwaneri, A.C., Grabnar, M., Demeter, J., Nestor-Kalinoski, A. and Hinds, T.D. (2016) Glucocorticoid receptor beta increases migration of human bladder cancer cells. Oncotarget, 7, 27313–27324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yuan, Y. (2016) Comprehensive characterization of molecular differences in cancer between male and female patients. Cancer Cell, 29, 711–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Byun, J., Han, Y., Li, Y., Xia, J., Xiao, X., Sun, R., Walsh, K.M., Gorlov, I., Gorlova, O., Zhou, W.et al. Trans-ethnic genome-wide meta-analysis of 35,732 cases and 34,424 controls identifies novel genomic cross-ancestry loci contributing to lung cancer susceptibility. Accepted by Nature Genetics, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. McKay, J.D., Hung, R.J., Han, Y., Zong, X., Carreras-Torres, R., Christiani, D.C., Caporaso, N.E., Johansson, M., Xiao, X., Li, Y.et al. (2017) Large-scale association analysis identifies new lung cancer susceptibility loci and heterogeneity in genetic susceptibility across histological subtypes. Nat. Genet., 49, 1126–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kichaev, G., Bhatia, G., Loh, P., Gazal, S., Burch, K., Freund, M.K., Schoech, A., Pasaniuc, B. and Price, A.L. (2019) Leveraging polygenic functional enrichment to improve GWAS power. Am. J. Hum. Genet., 104, 65–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gershoni, M. and Pietrokovski, S. (2017) The landscape of sex-differential transcriptome and its consequent selection in human adults. BMC Biol., 15, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lopes-Ramos, C.M., Chen, C.Y., Kuijjer, M.L., Paulson, J.N., Sonawane, A.R., Fagny, M., Platig, J., Glass, K., Quackenbush, J. and DeMeo, D.L. (2020) Sex differences in gene expression and regulatory networks across 29 human tissues. Cell Rep., 31, 107795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lucotte, E.A., Laurent, R., Heyer, E., Ségurel, L. and Toupance, B. (2016) Detection of allelic frequency differences between the sexes in humans: a signature of sexually antagonistic selection. Genome Biol. Evol., 8, 1489–1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hill, M.S., Reuter, M. and Stewart, A.J. (2019) Sexual antagonism drives the displacement of polymorphism across gene regulatory cascades. Proc. R. Soc. B, 286, 20190660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Brazel, D.M., Jiang, Y., Hughey, J.M., Turcot, V., Zhan, X., Gong, J., Batini, C., Weissenkampen, J.D., Liu, M., CHD Exome+ Consortium et al. (2019) Exome chip meta-analysis fine maps causal variants and elucidates the genetic architecture of rare coding variants in smoking and alcohol use. Biol. Psychiatry, 85, 946–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gill, D., Sheehan, N.A., Wielscher, M., Shrine, N., Amaral, A.F.S., Thompson, J.R., Raquel, G.R., Leynaert, B., Real, F.G., Hall, I.P.et al. (2017) Age at menarche and lung function: a Mendelian randomization study. Eur. J. Epidemiol., 32, 701–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Magnus, M.C., Guyatt, A.L., Lawn, R.B., Wyss, A.B., Trajanoska, K., Küpers, L.K., Rivadeneira, F., Martin, D., Tobin, L.S.J., Lawlor, D.A.et al. (2020) Identifying potential causal effects of age at menarche: a Mendelian randomization phenome-wide association study. BMC Med., 18, 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. He, F., Xie, J., Liu, C., Xiong, W., Xu, Q., Liu, Z., Lin, T., Xiao, R., Li, X., Cai, L.et al. (2017) The relationship of lung cancer with menstrual and reproductive factors may be influenced by passive smoking, cooking oil fumes, and tea intake. Medicine, 96, e8816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Khedher, S.B., Neri, M., Papadopoulos, A., Christiani, D.C., Diao, N., Harris, C.C., Susan, O.-M.S., Ann, G., Schwartz, A.G., Cote, M., Koushik, A.et al. (2017) Menstrual and reproductive factors and lung cancer risk: a pooled analysis from the international lung cancer consortium. Int. J. Cancer, 141, 309–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Amos, C.I., Dennis, J., Wang, Z., Byun, J., Schumacher, F.R., Gayther, S.A., Casey, G., Hunter, D.J., Sellers, T.A., Gruber, S.B.et al. (2017) The OncoArray consortium: a network for understanding the genetic architecture of common cancers. Cancer Epidemiol. Biomark. Prev., 26, 126–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kachuri, L., Amos, C.I., McKay, J.D., Johansson, M., Vineis, P., Bueno-de-Mesquita, H.B., Boutron-Ruault, M., Johansson, M., Quirós, J.R., Sieri, S.et al. (2016) Fine mapping of chromosome 5p15.33 based on a targeted deep sequencing and high-density genotyping identifies novel lung cancer susceptibility loci. Carcinogenesis, 37, 96–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Landi, M.T., Consonni, D., Rotunno, M., Bergen, A.W., Goldstein, A.M., Lubin, J.H., Goldin, L., Alavanja, M., Morgan, G., Subar, A.F.et al. (2008) Environment and genetics in lung cancer Etiology (EAGLE) study: an integrative population-based case-control study of lung cancer. BMC Public Health, 8, 203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ji, X., Bossé, Y., Landi, M.T., Gui, J., Xiao, X., Qian, D., Joubert, P., Lamontagne, M., Li, Y., Gorlov, I.et al. (2018) Identification of susceptibility pathways for the role of chromosome 15q25.1 in modifying lung cancer risk. Nat. Commun., 9, 3221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mitchell, K.A., Shah, E., Bowman, E.D., Zingone, A., Nichols, N., Pine, S.R., Kittles, R.A. and Ryan, B.M. (2019) Relationship between west African ancestry with lung cancer risk and survival in African Americans. Cancer Causes Control, 30, 1259–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wang, X. (2014) Firth logistic regression for rare variant association tests. Front. Genet., 5, 187. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.