

Abstract
Background and Aims:
Epithelial wound healing is compromised and represents an unleveraged therapeutic target in inflammatory bowel disease (IBD). Intestinal epithelial cells exhibit plasticity that facilitates dedifferentiation and repair during the response to injury. However, it is not known whether epithelial cells of a neighboring organ can be activated to mediate re-epithelialization in acute colitis. Histological findings of a permanent squamous tissue structure in the distal colon in human IBD could suggest diverse cellular origins of repair-associated epithelium. Here, we tested whether skin-like cells from the anus mediate colonic re-epithelialization in murine colitis.
Methods:
We studied dextran sulfate sodium-induced colitis and interleukin 10-deficient colitis in transgenic mice. We performed lineage tracing, three-dimensional imaging, single-cell transcriptomics, and biophysical modeling to map squamous cell fates and to identify squamous cell types involved in colonic repair.
Results:
In acute and chronic colitis, we found a large squamous epithelium, called squamous neo-epithelium of the colon (SNEC), near the anorectal junction. Neighboring squamous cells of the anus rapidly migrate into the ulcerated colon and establish this permanent epithelium of crypt-like morphology. These squamous cells derive from a small unique transition zone, distal to the border of colonic and anal epithelium, that resists colitic injury. The cells of this zone have a pre-loaded program of colonic differentiation and further upregulate key aspects of colonic epithelium during repair.
Conclusion:
Transitional anal cells represent unique reserve cells capable of rebuilding epithelial structures in the colon after colitis. Further study of these cells could reveal novel approaches to direct mucosal healing in inflammation and disease.
Keywords: wound healing, stem cells, regeneration, inflammation
Graphical Abstract
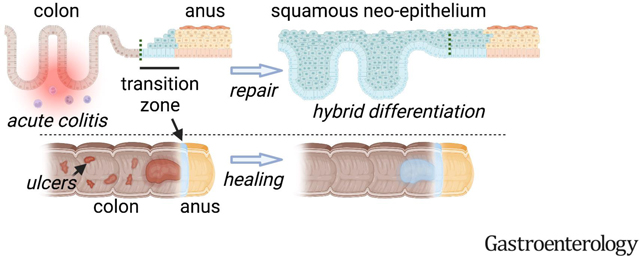
Introduction
Defects in colonic epithelial barrier function and repair have been linked to inflammatory bowel disease (IBD). Direct healing of the epithelial barrier is an attractive therapeutic target1; however, the cellular programs mediating wound repair remain to be elucidated and leveraged. Wound healing in adult mammals is associated with reprogramming or dedifferentiation of surviving epithelium2, but this generally occurs within a lineage-restricted paradigm, e.g.3–5. Whether the wound healing response in colitis could involve the recruitment of non-intestinal cells remains in question. Case reports from human ulcerative colitis, repetitive physical injury, and tumorigenesis have described a squamous epithelium of unknown origin in the distal colon6–12, suggesting a mechanism of lineage plasticity associated with colonic wounding, epithelial ulceration, and IBD.
During the analysis of three-dimensional, whole-mount reconstructions of murine colonic mucosa during repair from acute colitis, we have observed an unusual skin-like epithelial structure in the rectum13–15. We hypothesized that this structure represents part of the regenerative response of the colon during colitis. Here we define its tissue-of-origin: a molecularly unique transition zone (TZ) of squamous cells in the anus, distal to the anorectal junction. We show that squamous cells contribute to overall wound-healing outcomes and establish a long-lived epithelium with hybrid colonic-squamous properties as part of the overall repair process. Thus, colitis engages a specialized set of junctional squamous cells, poised for repair, that contribute to the regenerative response and initiate mucosal healing. Our findings define a distinct mode of colonic epithelial repair that originates from neighboring non-colonic skin-like cells and executes a program of permanent tissue morphogenesis in colitis.
Materials and Methods
Key reagents are listed in Table S1. A full description of Methods is provided as supplementary material.
Mice
All animals were maintained ethically and humanely at Children’s Hospital Los Angeles (CHLA, protocol #288). Genetic recombination for lineage tracing or cell ablation was induced by a single intraperitoneal injection of 0.02–2 mg tamoxifen or enema administration of 25 μg 4-hydroxytamoxifen. Specimens were stained, imaged, and analyzed according to the Deep Mucosal Imaging13, 16 set of procedures.
Single-cell RNA-Seq
The anorectal junction was digested with 0.25% trypsin to a single-cell suspension. Cells were analyzed using the 10X Genomics Chromium platform. Analysis was performed using monocle317.
Modeling
We used a “voter model”18 to quantify cell dynamics within an equipotent progenitor population. The sizes of the tip cell lineages in the rete peg were normalized and fit to statistical scaling forms which enabled quantification of a cellular replacement rate.
Results
Squamous epithelium contributes to wound healing in colitis
To characterize colonic wound healing processes, we orally administered 3% dextran sulfate sodium (DSS) to mice for 6 days (d). This model induces acute injury with epithelial ulceration and inflammation, followed by spontaneous healing in the distal colon over a span of ~3 weeks (wks). In whole-mount total reconstructions of the injured mucosa, obtained with a clearing and imaging protocol we developed previously13, we observed the full extent of a stratified “neo-epithelium” in the distal colon. This neo-epithelium was proximal to the dentate line that, in the uninjured animal, sharply divided KRT8+ colonic columnar epithelium from KRT14+ anal squamous epithelium (Figure 1A,B, Figure S1A). Microscopically the neo-epithelium resembled thick squamous (skin-like) tissue, but lacked a cornified outer layer (Figure 1C,D). We thus refer to this tissue as squamous neo-epithelium of colon (SNEC). The endoscopically visible, KRT14+ SNEC was present from the end of the DSS treatment (experimental day [exp d] 6) and persisted as a proliferative tissue for life (longest timepoint: 15 months post-DSS, Figure 1E, Figure S1B,C). SNEC grew with repeated cycles of DSS (Figure 1F). We have observed SNEC to occupy up to 1 cm of distal mouse colon, equivalent to ~15% of the overall colonic length or ~50% of the rectum. Patches of SNEC were always contiguous with the dentate line; we did not find squamous-like tissue in re-epithelialized regions of the proximal colon. Three-dimensional reconstructions of distal colon revealed a field of filled intestinal crypt-like invaginations (Figure 1G), which we termed rete pegs based on their similarity to structures found in skin and mucous membranes. We did not see other anatomical features of skin, such as sebaceous glands, in SNEC. Rete pegs of SNEC, as well as basal cells of anal squamous epithelium next to the dentate line, stained positively for the progenitor cell markers P63 and SOX2. P63 and SOX2 proteins were not expressed in colonic epithelium before or after DSS treatment (Figure 1H). Thus, acute injury and inflammation of the distal colon are associated with the formation of a permanent squamous tissue structure.
Figure 1: Squamous epithelium contributes to wound healing in colitis.

A-B) Surface representations after 3D imaging of chemically cleared, flattened colonic tissue after DSS injury reveal a distinct epithelium replacing colonic crypts, proximal to the dentate line (orange) but distal to the margin (green) of colonic crypts. C-D) H&E images show that this long-lasting “neo-epithelium” (C) has a broadly squamous structure with rete pegs (*) but lacks a fully differentiated surface (#). The neo-epithelium does not exist in the absence of injury (D). E) Squamous neo-epithelium of colon (SNEC) persists as a KRT14+ tissue structure 15 months after injury (n=3 mice), as shown in whole-mount images. F) Plot of SNEC size (length of incursion from the dentate line) associated with multiple cycles of DSS (n=5–7 mice/condition). G) 3D reconstruction of SNEC surface and abluminal views shows distinct morphology compared to colonic crypts. H) Immunostaining reveals high expression of P63 and SOX2 in squamous cells at the dentate line and in rete pegs, but exclusion from colonic epithelium (n=4 mice). Orange arrows: dentate line. I-K) Apoptotic targeting in Sox2-DTA mice with 4-hydroxytamoxifen (4-OHT) enemas retards body weight recovery (I), leaving ulcers (J,K) in the colon after DSS colitis (n=5 mice/condition, t-test). L) Retrospective analysis of squamous epithelium in colons of Il10−/− mice. H&E photomicrographs show that young (8 wks-old) Il10−/− mice lacking inflammation were devoid of SNEC. Orange arrow marks the dentate line. M) However, older mice (>36 wks-old) with severe distal colitis and buried columnar dysplasia exhibit SNEC. N) Colitis (as defined by histological injury score >0) was a prerequisite for finding SNEC. Each dot represents a mouse. O) Linear regression of animal age and histological colitis score modeled the size of the SNEC tissue. Error bars: SE. See also Figures S1–S3.
The ability of SNEC to rapidly re-epithelialize the colonic surface suggests that it plays a functional role in wound healing. To define the functional contribution of anal-derived cells to outcomes of colonic injury, we targeted apoptotic signals to Sox2+ cells at the anorectal junction. We generated Sox2::CreER;Rosa26::LSL-DTA (Sox2-DTA) mice, which activate in response to topical 4-hydroxytamoxifen (4-OHT) the intracellular expression of the diphtheria toxin A (DTA) polypeptide chain in Sox2-expressing cells. Delivery of 4-OHT during the acute recovery period after DSS exposure prevented the normal rebound in body weight (Figure 1I) and precluded long-term follow-up. Postmortem analysis revealed impaired mucosal healing (Figure 1J) in the form of residual ulcers (Figure 1K), as assessed from 3d reconstructions. Intriguingly, squamous cells resembling SNEC and anal epidermis remained detectable adjacent to the residual ulcers. Although we validated the specificity of apoptotic targeting (Figure S2A–C), surviving anal cells may have undergone compensatory proliferation (Figure S2D) and, after escaping the initial impairment, expanded into a region of exacerbated injury. The submaximal doses of 4-OHT used to achieve anal-specific apoptosis did not change body weights when the compound was administered after healing was complete (Figure S2E). Thus, apoptotic targeting to anorectal squamous cells specifically during acute injury impedes both ulcer repair and clinical recovery from colitis.
To determine if chronic colitis was also associated with formation of SNEC, we studied archived colonic samples19 obtained from Il10−/− mice, which spontaneously develop a progressive colitis. We observed SNEC in animals with severe colitis (Figure 1L,M). We did not find SNEC in uninjured specimens (Figure 1N). SNEC was sometimes observed adjacent to a buried columnar epithelium that may be reflective of a regenerative or dysplastic process (e.g., Figure 1M); however, these two tissue features were easily distinguished by their morphology and were separated by a clear squamocolumnar junction. We next assessed potential study variables associated with SNEC size. We found that a linear mixture of age and histological score was needed to predict the extent of SNEC (Figure 1O). Thus, in a chronic colitis model, the duration and severity of injury both predicted the formation of SNEC. In tissue transcriptomic data from the DSS model, the emergence, proliferation, and gradual intestinal remodeling of SNEC was associated with inflammatory signaling (Figure S3), consistent with the integral function of this tissue within a colitogenic microenvironment.
SNEC derives from cells of the anal transition zone
We next investigated the tissue origins of SNEC. To determine whether SNEC represented a unique epithelium, we screened for keratin expression in our transcriptomic dataset. We found enriched expression of Krt7 in SNEC (Figure 2A). Prior to injury, a thin transition zone (TZ) of squamous Krt7+ cells, representing a narrow (<10 cell-wide) portion of the anal epithelium, was identified next to the dentate line (Figure 2B). These pre-injury TZ cells and their related post-injury Krt7+ SNEC cells were both negative for markers of anal/skin terminal differentiation (LOR, IVL, KRT1), suggesting that these two tissues are related (Figure 2C, Figure S4).
Figure 2: SNEC derives from cells of the anal transition zone.

A) Screening of keratin expression in RNA-Seq data from SNEC identifies enrichment of Krt7 (n=3 mice/timepoint). B) In situ hybridization experiments (n=4 mice) demonstrate mRNA expression of Krt7 in SNEC and in a thin transition zone (TZ) of anal squamous cells next to the dentate line (drawn in orange). C) The TZ and SNEC do not express loricrin (LOR), a marker of terminal differentiation of skin (n=5 mice). D) Summary of contributions of various Cre/CreER driver mice to labeling of SNEC; the Vil1+ lineage (n=3 mice) does not contribute whereas Sox2+ (n=2 mice) and Krt7+ (n=4 mice) lineages do. Values represent % of SNEC surface labeled, normalized to the overall baseline efficiency of labeling. E-I) Visual representations of transgenic labeling data in D, shown as maximum intensity projections of the distal colonic surface (en face) and as rotations (ortho) showing the orthogonal sectioning depth. Crypts were fully labeled in Vil1-mTmG mice, but no labeling was observed in SNEC (E). The lineages of Sox2+ cells were found in the anal epidermis and in SNEC (F). Labeling of Sox+ cells was initiated 28 d prior to DSS treatment in order to trace both progenitor and differentiated anal cells. G-I) Outcomes of tracing in Krt7mTmG mice were assessed in both tamoxifen-free/spontaneous labeling (G,H) (n=4 mice) and 2 mg tamoxifen (n=4 mice) injected-conditions (I). Labeling of TZ and SNEC was observed with both treatment paradigms. Error bars: SE. See also Figures S4, S5.
We considered two possibilities for SNEC origins. Colonocytes may have transdifferentiated to squamous cells to produce a TZ-like epithelium. Alternatively, TZ cells migrated into the colon to make SNEC. To distinguish these possibilities, we performed lineage tracing. We utilized “driver” mice that express either a constitutive Cre recombinase or a tamoxifen-inducible Cre recombinase-estrogen receptor (CreER) fusion protein from lineage-specific promoters. These driver mice were mated to “mTmG” reporter mice to generate lineage-tracing mouse lines that, upon activation of the Cre recombinase, induce heritable expression of membrane-tagged GFP (mG) in specific cellular lineages. Before Cre activation, all cells in the body express membrane-tagged tdTomato (mT), which facilitates 3d reconstruction of tissue architecture. We compared the representations of Vil1+ (colonic, Vil1::Cre;mTmG or “VIl1-mTmG” mice), Sox2+ (anal TZ and anal epidermis, Sox2::CreER;mTmG or “Sox2-mTmG”), and Krt7+ (anal TZ, Krt7::CreER;mTmG or “Krt7-mTmG” mice) lineages in homeostasis and in SNEC. In homeostasis, Vil1-mTmG mice exhibited fully mG-labeled colonic crypts, but the anal squamous epithelium was devoid of mG label (Figure S5A). After DSS-induced injury, the lineage of Vil1+ cells was not observed in SNEC (Figure 2D,E). In contrast, Sox2-mTmG mice injected with tamoxifen and traced for 28 d in homeostasis demonstrated mG labeling in the anal TZ and the distal anal epidermis (Figure S5B). After DSS-induced injury, the lineages of Sox2+ cells were abundant within SNEC (Figure 2F). Thus, SNEC is derived from the migration of anal-derived cells.
To resolve whether TZ cells contribute to SNEC, we analyzed lineage tracing patterns in Krt7-mTmG mice. In homeostasis, Krt7-mTmG mice exhibited labeling of anal cells adjacent to the dentate line to a distal distance of 15 ± 1.1 (n=21 mice) basal cell-widths after low-level induction and tracing for >28 d (Figure 2G). In whole-mount images, we observed exclusion of the anal differentiated epidermal marker KRT10 from dentate line-adjacent anal cells; however, the proximal border of the KRT10+ domain partially overlapped with mG+ TZ-derived lineages (Figure S5C). Thus, TZ-derived cells make overall minor contributions to homeostatic regeneration of anal epidermis. In contrast, after DSS-induced injury, Krt7-cell lineages were found to label extended portions of SNEC in conditions of both low (Figure 2H) and maximal (Figure 2I) induction. These results demonstrate that SNEC is derived from anal TZ epithelium.
Anal TZ is a molecularly distinct squamous tissue with colonic expression
We next asked what molecular properties of anal TZ were associated with its capability to form a wound-associated squamous tissue in colon. To characterize TZ-derived cells in relation to neighboring epithelia, we performed single-cell RNA-Seq analysis of ~35,000 cells. We obtained these cells from gross dissections of the anorectal junction at 3 different timepoints (exp d 0, 7, and 42) around a DSS injury stimulus (exp d 0–6). These samples contained epithelial and stromal cells of the uninjured TZ, of acutely forming SNEC, and of long-lived SNEC, respectively, in addition to colonocytes and anal epidermal cells recovered from the dissection margins. Dimensional reduction, clustering, and plotting of individual cell types showed transcriptional changes associated with acute injury in SNEC and in neighboring colonic epithelium and anal skin (Figure 3A, Figure S6A–C).
Figure 3: Anal TZ is a molecularly distinct squamous tissue with colonic expression.

A) Single-cell RNA sequencing (scRNA-Seq) of >35,000 cells from 3 different timepoints (n=5 pooled mice/timepoint) during DSS-induced injury and repair. Discrete clusters of epithelial and stromal cells from the anorectal junction can be identified in the UMAP visualization. B-C) Louvain subclustering (B) of uninjured (exp d 0) keratinocytes reveals putative TZ (Krt7hi) cell clusters (#1, #2, #4) (C). HF = hair follicle, IFE = interfollicular epithelium. D) COL17A1 staining shows that TZ, anal epidermis, and SNEC have a basal layer that is marked by expression of this collagen isoform. E) Heatmap of aggregated gene expression compared between clusters shows specific genes to clusters #1, #2, and #4 representing unique TZ-related transcripts. F) GSEA of markers of cluster #4 (differentiated TZ) and #1 (basal TZ) identifies junctional and mucus-producing pathways (#4) and mitotic pathways (#1) (blue text) enriched in the TZ. Normalized enrichment score and adjusted p values (q values) were obtained from GSEA software. G) Specific markers of the TZ cross-compared to other tissues using BioGPS. H,I) Summary (H) of colonic or anal epidermal (skin) marker expression in the TZ. By definition, skin and colonic markers were mutually exclusive. The boxplot (I) shows the distribution of expression of colonic or anal epithelial markers in different colonic or anal epithelial cell types. Each dot represents a gene. Expression is quantified as the mean UMI abundance (including the 0-count cells) among the cells within a cluster. Colon and anus markers were defined by identifying genes that had >4-fold expression difference between colonic and anal epithelium (the TZ was excluded for gene identification). The “differentiated” TZ cells exhibit significantly enriched overall expression of colonic epithelial markers compared to differentiated cells of the anal IFE (paired t-test, Holm-Bonferroni correction). In H, a marker was deemed expressed in the TZ if its mean log TZ abundance was >50% of the abundance in the reference anal or colonic tissue. Abbreviations: Prog = progenitors, Abs = absorptive cells, Sec = secretory cells, EEC = enteroendocrine cells, Diff = differentiated cells. Box plot: center line, median; hinges, 25–75th percentiles; whiskers, 1.5x interquartile range; points, outliers. J-L) Krt20 expression in the uninjured anal TZ was confirmed using Krt20-mTmG mice (n=3 mice). Tamoxifen administration induces lineage labeling of Krt20+ cells in colon and in the TZ (J). TZ-labeled cells distal to the dentate line (orange) are KRT14+ in the zoomed image (K) and are found only in the suprabasal layer of cells (L). See also Figures S6, S7.
We next subclustered the Krt14hi keratinocytes at exp d 0, prior to injury (Figure S6D,E). We identified 3 small clusters (#1, #2, #4) of Krt7hi TZ cells (Figure 3B). Clusters #1 and #2 were non-proliferative and proliferative (Birc5+) squamous basal cells, respectively, expressing high levels of Col17a1 (Figure 3C,D). Cluster #4 was suprabasal TZ cells with a distinct repertoire of expression (Figure 3E). Suprabasal TZ cells demonstrated enrichment of tight junction and mucus-processing pathways not found in other keratinocytes or in basal TZ cells (Figure 3F). In suprabasal TZ cells, we noted specific expression of Muc4, Krt20, and other markers of colonic epithelium (Figure 3G). Quantitative assessment of global similarity of the TZ to colon and anal epidermis validated TZ expression of markers of both colonic and anal epidermal epithelium (Figure 3H). The expression levels of colonic transcripts were significantly higher in the TZ than in the anal epidermis (Figure 3I). We obtained similar results using graph abstraction techniques20 (Figure S7). TZ expression of the colonic marker Krt20 was validated histologically using Krt20-mTmG mice (Figure 3J–L). Thus, the uninjured TZ represents a specialized hybrid anorectal zone.
TZ-derived cells adopt a progenitor state during SNEC formation
In colonic epithelium, wound healing is associated with the adoption of an interferon-induced fetal-like progenitor state marked by expression of Ly6a21, 22. Using the scRNA-Seq data, we tested whether a similar process underpinned squamous cell-mediated colonic re-epithelialization. Krt7hi cells obtained from exp d 7 predominantly segregated as a distinct cluster, #3, which we refer to as wound-associated squamous cells (WASCs) (Figure 4A,B). Compared to other TZ-derived cells, WASCs were enriched for the wound-associated23 keratin Krt17, Ly6a (Sca-1), and the urea-processing enzyme Ass1, among other genes (Figure 4C). Ly6a and Ass1 mRNA expression were found in proximal rete pegs and basal cells at the proximal, “leading” edge of SNEC during wound healing (Figure 4D,E). Gene set enrichment analysis (GSEA) of WASCs showed enriched translational and ribosomal pathways and similarity to stem cell states (Figure 4F), as well as direct upregulation of the fetal intestinal profile24 (Figure 4G), which was not upregulated in anal epidermis (Figure 4H). At exp d 7, WASCs expressed cell proliferation-associated transcripts, unlike inflammatory signaling-enriched colonic epithelium (Figure 4I), demonstrating differential responses of these epithelia to DSS. Thus, WASCs represent leader cells in squamous wound healing and exhibit fetal progenitor-like signaling.
Figure 4: TZ-derived cells adopt a progenitor state during SNEC formation.

A) Clustering of Krt7hi cells combined from all 3 timepoints analyzed via scRNA-Seq; UMAP visualization reveals a distinct cluster (#3) containing wound associated squamous cells (WASCs). B) Plot of cell count per cluster shows that cluster #3 predominantly mapped to Krt7hi cells at exp d 7. C) Enriched genes in WASCs include Ly6a, Krt17, and Ass1. D) Leading edge cells at exp d 6 and proximal rete pegs in SNEC at exp 15 (blue lines) exhibit high expression of Ly6a (Sca-1) mRNA. Ly6a expression is reduced prior to injury and after repair. Orange line denotes the dentate line. E) Leading edge cells (blue line) in SNEC selectively express Ass1 mRNA during injury. F) Gene set enrichment analysis (GSEA) of WASC marker genes demonstrates enrichment of ribosomal, translational, and stem cell pathways. G) Enrichment plots demonstrate upregulation of fetal intestinal genes in WASCs and reprogrammed colonocytes. H) Heatmap of aggregate fetal intestinal gene expression in different types of epithelial cells at exp d 0, 7, or 42 (paired t-tests, Holm-Bonferroni correction to compute adjusted P value as “Q value”). I) Significant MSigDB/Hallmark pathways from GSEA comparing the effect of DSS-induced injury on colonic epithelium vs. TZ-derived cells at exp d 7. The inflammatory response is more pronounced in colonic epithelium.
Rete pegs progressively form from a proliferative leading edge of WASCs
We next assessed the mechanism of rete peg formation within SNEC. In Sox2-Confetti mice, which enable clonal labeling of cells, initiation of tracing at exp d 6 revealed lateral streams of cells that elongated over time, consistent with the proliferation and inward migration of Sox2+ cells during SNEC formation (Figure S8). To study the link between squamous cell proliferation and rete peg formation, we injected mice with EdU and BrdU at 24 h and 2 h, respectively, prior to euthanasia (Figure 5A). The EdU pulse labels cells in S-phase (i.e., proliferating cells) one day before euthanasia, thereby allowing us to follow their fate over the next 24 h; the BrdU pulse labels any proliferating cells within 2 h of euthanasia. During the DSS injury-repair cycle, colonic epithelium temporarily lost its proliferative activity. In contrast, the anal TZ and SNEC maintained proliferation throughout (Figure 5B). Prior to injury, EdU+ and BrdU+ cells intermingled within the basal layers of the TZ and anal epidermis, consistent with cell division and short-term regeneration along the lateral plane, and within the basal halves of colonic crypts, consistent with vertical regenerative flow (Figure 5C). At exp d 6, colonic crypts were devoid of label (Figure 5D), but a highly proliferative and flat squamous leading edge was visible at the dentate line (Figure 5D’) and expanded proximally (Figure 5E). By exp d 10, SNEC appeared spatially dichotomized, with a highly proliferative and flat leading edge and a rear (distal) end consisting of rete pegs (Figure 5F). At exp d 43, SNEC was structured exclusively as rete pegs, with proliferative cells exclusively located along the periphery of the pegs (Figure 5G). Quantitative analysis (Figure 5H) of 3d images (Figure 5I–K) showed that, during wound healing, proximal rete pegs were filled with BrdU+ cells and that distal rete pegs were filled with exclusively EdU+ cells. These results are consistent with a simple model in which rete pegs are actively formed from highly proliferative cells within the leading edge; after formation and distancing from the wound margin, these rete pegs undergo downregulation of proliferation. Thus, SNEC emerges from the anal TZ as a flat epithelium and gradually remodels to the rete peg morphology through dynamic changes in cell proliferation. Consistent with this idea, lateral clonal streams of Sox2+ cells linking SNEC to the anus were initially observed as flat epithelium, and then later observed to be composed of rete pegs (Figure S9).
Figure 5: Rete pegs progressively form during squamous cell migration.

A) Mice were injected with 2.5 mg EdU 24 h and 2.5 mg BrdU 2 h prior to dissection, at different timepoints during the DSS injury-repair cycle (n=3 mice/timepoint). Orange arrows mark the dentate line in images. B) Quantification of squamous cell BrdU incorporation vs. colonic crypt incorporation (n=3 mice/timepoint) demonstrates a critical time (exp d 6, asterisk) when SNEC proliferates but colonic epithelium does not. C) In thin sections, at exp d 0, proliferative cells (BrdU+) and their short-term descendants (EdU+) are found in colonic crypts and in the anal TZ and skin. D,D’) At exp d 6, crypts (D) are devoid of proliferative signals, whereas the flat leading edge (dotted line) of SNEC (D’) harbors BrdU+ cells close to the ulcer. E) The flat leading edge (dotted line) of SNEC further expands into the ulcerated region at exp d 8. F) Thin-section photomicrograph from exp d 10, demonstrating new BrdU+ rete pegs (asterisk) forming within the leading edge and a population of proliferation-reduced rete pegs (#) near the dentate line. G) Shown is the reduced nucleotide incorporation at exp d 43 in rete pegs, with label restricted to the periphery. H) Quantification of labeled nucleotide incorporation relative to the dentate line, from wholemount 3d images in I-K (n=3 mice/timepoint). The “transition point” between EdU-enriched and BrdU-enriched rete pegs is manually denoted with the dotted green line. I) The homeostatic TZ is a proliferative zone. J) At exp d 10, rete pegs close to the leading edge (left side) are full of BrdU+ cells, indicating rapid proliferation associated with rete peg morphogenesis. However, cells in the interior of rete pegs near the TZ were singly EdU+, suggesting that downregulation of the proliferative rate is involved in maturation of rete pegs. K) Rete pegs at exp d 23 exhibited fewer nucleotide-incorporated cells. See also Figures S8, S9.
SNEC upregulates colonic expression patterns
Because of SNEC’s more-proximal location compared to the TZ, we tested whether SNEC cells would exhibit signs of adaptation to the colonic milieu. In the scRNA-Seq data, we found evidence of altered transcriptional profile in SNEC (exp d 42) versus the uninjured TZ (exp d 0) (Figure 6A) in the suprabasal cell cluster (i.e., cluster #2 as shown in Figure 4A). Through regression analysis, we found that SNEC upregulated a tight junction module and other colonic transcripts, while downregulating epidermal transcripts (Figure 6B). Moreover, the expression of individual claudins and colon-specific keratins was significantly elevated in SNEC vs. TZ (Figure 6C). Globally, SNEC retained some similarity to skin tissue but had significantly elevated expression of colonic genes (Figure 6D). As a hybrid epithelium, SNEC exhibited a distinct differentiated cell profile. Suprabasal cells of SNEC immunoreacted with antibodies targeted against the colonic mucus marker MUC4, the colonic tight junction protein claudin-1 (CLDN1), and the glutathione transferase GSTO1 specifically found in SNEC (Figure 6E, Figure S10). Similar evidence of intestinal remodeling (e.g., Spink4 expression) was found when the analysis was focused on SNEC basal cells (Figure S11A–C); furthermore, some SNEC cells expressed higher mRNA levels of “classic” colonic differentiation markers such as enteroendocrine cell Chga. However, almost no SNEC cells (<5 cells per SNEC per animal) stained positively for CHGA protein (Figure S11D,E).
Figure 6: SNEC upregulates colonic expression patterns.

A) UMAP of differentiated (suprabasal, Col17a1lo) Krt7hi cells coded by experimental day. Datapoints representing SNEC cells at exp d 42 appear to be displaced from datapoints of TZ-derived cells at exp d 0 and exp d 6. B) Comparison of enriched genes at exp d 42 include those associated with tight junction pathways and colonic tissue. Annotations are obtained using enrichr. C) Violin plots of significant increases in junctional and colonic genes at exp d 42; significance was evaluated in monocle3. D) Boxplot similar to that shown in Fig. 3I comparing expression of colonic and anal marker expression at exp d 42. Like uninjured TZ, SNEC has elevated expression of colonic markers compared to anal epidermis. E) Immunostaining for MUC4 shows presence of colon-like “differentiated” cells in SNEC in the suprabasal layer at exp d 36. MUC4+ cells were also found in the uninjured TZ. GSTO1-targeted antibody labels TZ and SNEC suprabasal cells, but not crypt cells. Orange arrows mark the dentate line. F) UMAP of TZ-derived cells at exp d 42 shows 2 distinct clusters. G) The dotted plot indicates a proliferative (Birc5hi) basal (Col17a1hi) cluster (cluster 1) and a nonproliferative suprabasal cluster enriched for Krt19, Krt20, and Muc4. H) Expression of proliferative markers (Birc5) and specific keratins (Krt17, Krt20) in different cells at exp d 42. I) In situ hybridization reveals that Krt17 mRNA expression specifically marks a subset of basal cells of the uninjured TZ and of SNEC. See also Figure S10, S11.
We interrogated the diversity of cells within SNEC. We analyzed and re-clustered all SNEC (Krt7hi) cells at exp d 42 (Figure 6F). The projection showed a gradient of cell transcriptional identities that were broadly grouped into 2 clusters, a suprabasal Krt20+ cluster and a basal (Col17a1hi) proliferative (Birc5hi) cluster (Figure 6G). These basal cells were unique to the TZ and SNEC through their expression of Krt17 (Figure 6H,I). However, not all basal cells of SNEC expressed Krt17 mRNA, suggesting potential diversification of progenitors after tissue establishment.
SNEC harbors stem cells that mediate tissue homeostasis in a crypt-like pattern
In addition to upregulation of colonic transcripts, SNEC is composed of crypt-like rete peg structures. To assess further evidence of the adoption of colonic-like features, we tested whether rete pegs functioned similarly as crypt-like units of regeneration. To study the patterns of cell turnover and regeneration in SNEC, we turned to long-term clonal lineage tracing using Sox2-Confetti mice. Over a 28-wk time course, we found that a growing proportion of rete pegs became fully monoclonal, comprising cells marked by a single Confetti color (Figure 7A). Thus, once established, rete pegs function as a closed renewing glandular-like compartment, similar to the epithelial organization of the uninjured colonic crypt. More generally, 3 types of clones could be resolved in the tracing data: (1) clones occupying the lateral boundary of the rete peg (termed “casing” clones); (2) clones extending from the abluminal “tip” (i.e., basal vertex) into the inner, non-cycling “core”; and (3) monoclonal rete pegs (Figure 7B,C). Similar results were obtained from the analysis of patches of nGFP expression, which recombines with ~10% of the efficiency of the other Confetti colors25, allowing us to conclude that spatial variability was not the result of clone merger events. The spatial segregation and relative persistence of these clonal types suggested sub-compartmentalization of the epithelium, with a closed lateral boundary domain and an inner core maintained by proliferative cells at the basal tip. We reasoned that an additional transfer of cells from the tip to the casing region would account for the clonal fixation of rete pegs in the long term (Figure 7D). These results suggest that SNEC is renewed in the long-term by the lineages of stem cells located at the rete peg base.
Figure 7: SNEC harbors stem cells that mediate tissue homeostasis in a crypt-like pattern.

A) Long-term clonal tracing (28 wks) in Sox2-Confetti mice demonstrates clonal fixation (single color) in rete pegs (n=3 mice/timepoint). B) Example images of clonal forms (color patches) identified during 1–28 wks of lineage tracing. A reference rotated xy-image of a rete peg is shown to orient the individual xz-planes demonstrating the fate-mapping. “Tip” and “mid” describe lowest and middle height in the rete peg, respectively. C) Quantification of clone stratification between tip, casing, and core domains demonstrates no correlation in clone size between tip and casing compartments, suggesting that progenitor cells in these compartments function independently. Tip and core clone sizes are correlated (coeff = 0.74). The axes denote the fraction of the compartment that is occupied by the analyzed clone. D) Diagram of predicted progenitor cell populations in the rete peg. E) A good fit is obtained by superimposing the theoretical 1d scaling form to an invariant mean-scaled histogram (Pn(t)) of the mean-normalized tip clone sizes (n(t)). Tracing data are stratified by the chase duration (1–4 wks). This demonstrates scaling behavior in the evolution of tip clone sizes, consistent with neutral drift dynamics in tip progenitor population. F) Fit of neutral drift model to the empirical normalized mean tip clone size over time (λ/N2=0.02 /wk), consistent with replacement dynamics of an equipotent progenitor cell population. See also Figures S12–S14.
Maintenance of the intestinal epithelium relies upon the constant loss and replacement of equipotent crypt base columnar cells leading to neutral drift clone dynamics, culminating in clonal loss or fixation of the crypt18. We next assessed whether neutral drift dynamics also characterize turnover of tip progenitor cells. In tracing initiated with low doses of tamoxifen, we found that the clone size distributions obtained from the independently functioning tip and casing compartments conformed to a statistical “scaling” behavior over the short-term (4 and 7 d) (Figure S12). Over 4 wks of tracing, the tip clone size distribution resembled a statistically time-invariant function whose spread was dictated by the mean, a hallmark of scaling behavior (Figure 7E). Moreover, over the full duration (28 wks) of tracing, we found that the empirical mean surviving tip clone size was well fit by a neutral drift model (Figure 7F), with a key parameter (λ/N2 = 0.02 /wk) describing scaling dynamics similar to previously reported values18 in colon and small intestine (see Supplementary Methods, Figure S13). Thus, we reasoned that rete pegs are sustained by an equipotent progenitor cell population at the abluminal tip. Tip cells expressed low levels of the crypt base columnar cell marker Lgr5 (Figure S14A–C), and the progeny of these Lgr5+ rete peg cells was preferentially found in the tip and core regions, consistent with clonal fate-mapping data (Figure S14D–G). SNEC progenitor cells therefore operate from an analogous invaginated location and with similar dynamic features18 as colonic epithelial stem cells.
Discussion
Colonic epithelial damage represents an immediate crisis requiring rapid repair. Columnar epithelial cells can undergo dedifferentiation to promote healing3, 26. Here, using high-resolution imaging, single-cell transcriptomics, and biophysical modeling, we show that severe colitis induces a profound plasticity response: the formation of a permanent squamous epithelium, SNEC. This represents a regenerative process that engages a heterologous cell type. The injury-associated squamous epithelium remodels to be more like the colon through morphological adoption of cryptlike units in the form of rete pegs, upregulation of some differentiated colonocyte markers, and long-term regeneration through an intestinal stem cell-like population. These are key features that make SNEC suitable for colonic wound healing. SNEC represents a colonic extension of an anal transition zone that exhibits a hybrid of colon-epidermis features in homeostasis. This squamous epithelium expands into the colon because it is partially resistant to the severe injury and inflammation that can induce proliferative arrest in colonocytes. Although repetitive or chronic injury enlarged SNEC, we do not know if there is a fundamental limit to the proximal spread of this tissue.
SNEC bears resemblance to a squamous metaplasia described in case reports of human patients with ulcerative colitis, distal colonic injury, or carcinoma6–12; however, accurate estimates of prevalence in humans are challenged by lack of sampling at the anorectal junction during endoscopic screening, and limited surgical samples of completion proctectomy with removal of the anal canal. The underlying processes giving rise to SNEC may be similar to those of early metaplasia. For example, both SNEC and Barrett’s esophageal metaplasia exhibit elevated expression of Krt727 and share an injurious ontogeny. Whether SNEC can be further directed to an intestinal lineage like Barrett’s metaplasia remains unknown. It may be beneficial to investigate means of extending the relatively limited endogenous intestinalization of SNEC and supporting its longer-term role in the restoration of colonic function. However, retention of a squamous profile may be integral to providing resistance to colonic injury. In addition, future studies are needed to assess the relationship between SNEC and cancer, a sequela of metaplasia. Although SNEC can form in mice in the absence of tumors, the presence of a tumor could amplify SNEC. Additional studies could inform on the physiological tradeoffs between the rapid re-epithelialization of the colon by an injury-resistant squamous tissue and the long-term potential for carcinogenesis.
SNEC is derived from a small but specialized population of Sox2+ Krt14+ Krt7+ squamous cells located within the transitional zone of the anorectal junction. TZ cells may exhibit fundamental plasticity enabling them to contribute to homeostatic maintenance of the narrow proximal edge of anal epidermis. After distal colonic ulceration, TZ cells migrate rapidly to mediate colonic wound healing. The TZ may represent a reserve set of cells that are primed to respond to colonic injury. A similar transition zone has been identified at the gastroesophageal squamocolumnar junction; these cells can be directed to express intestinal markers in vitro28. Thus, by virtue of continuous exposure to two different microenvironments, transitional epithelia in the body may broadly represent important areas of plasticity and wound healing potential. Although a recent study29 has made similar claims about plasticity of the anal TZ, our study, in contrast, does not report squamous cell regeneration of bona fide crypts. In our unique interrogation of colitis models, we find a distinct and robust mode of repair (i.e., SNEC) that was not reported earlier. In addition, further analysis will need to resolve whether SNEC is similar to a squamous tissue associated with mechanical injury in rodents12.
Squamous cell phenotypes appear to be important for optimal outcomes during acute colitis. Restitutive colonic epithelial cells adopt a flattened morphology reminiscent of squamation30. We find transcriptional similarity between the columnar wound healing/fetal intestinal state and squamation (Figure 4H). Furthermore, we complementarily demonstrate the importance of anal-derived squamous Sox2+ cells. Mice with increased apoptosis within Sox2+ cells could not properly recover from acute colitis. We acknowledge some limitations of our experimental approach that preclude a deeper investigation of this phenomenon. As Sox2 is expressed in various organs31, the dose given to achieve specific anal cell apoptosis could not fully ablate the tissue, and the difference in outcomes could have been driven by compensatory regenerative responses including Sox2-negative dedifferentiation, loss of modulatory factors such as epidermal growth factor-like ligands32 (cf. high expression of amphiregulin in SNEC cells, Figure S11C), or altered timing of injury and wound healing in the DSS colitis model. Overall, our results demonstrate that the integrity of anal-derived epithelium is important for optimal outcomes in acute colitis.
A deeper understanding of colonic epithelial repair is therapeutically relevant to IBD. The identification of unique junctional skin-like cells capable of rapid colonic restitution supports that heterologous squamous cell phenotypes might serve as targets for mucosal healing. Our findings illuminate new strategies for the design of heretofore elusive therapies that directly activate regenerative processes in the colon.
Supplementary Material
WHAT YOU NEED TO KNOW
Background and Context
Colonic epithelial wound healing is a key therapeutic endpoint and target in inflammatory bowel disease, but novel mechanistic insights into this biological process are needed to inform and inspire new approaches.
New Findings
Anal cells from a specialized squamous transition zone migrate into the colon to effect wound healing by forming a permanent hybrid intestinal-squamous epithelium after acute colitis and colonic ulceration in mice.
Limitations
The long-term impacts of this type of skin-like colonic wound healing on colonic function and tumorigenesis are not yet known.
Impact
Anal transition zone cells exhibit fundamental plasticity allowing them to serve as potential templates for wound healing therapies in the colon.
LAY SUMMARY
Specialized skin-like cells from the anus migrate into the colon and form a new, hybrid anorectal epithelium to mediate wound healing after acute colitis.
Acknowledgments
Grant support: D.B.P. acknowledges support from the U.S. National Institutes of Health (R01DK108648 and R01DK056008). C.Y.L. acknowledges funding from the Crohn’s and Colitis Foundation (Career Development Award 694110), the California Institute for Regenerative Medicine (postdoctoral fellowship), and the U.S. National Institutes of Health (P30DK042086). B.D.S acknowledges funding from the Wellcome Trust (098357/Z/12/Z) and the Royal Society through an EP Abraham Research Professorship (RP/R1/180165). The study sponsors had no role in the collection, analysis, or interpretation of data.
Footnotes
Disclosures: The authors have no competing interests to disclose.
Transcript profiling: Bulk RNA sequencing data shown in Figure S3 are available on the Gene Expression Omnibus (GEO), with accession number GSE168053: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE168053. The reviewer access code is gzgxewwwltctrqd. Single-cell RNA sequencing data shown in Figs. 3,4,6 are available on GEO, with accession number GSE168033: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE168033. The reviewer access code is ovyjmsegtrwzdef. Analysis scripts for single-cell sequencing are available on GitHub: https://github.com/stalepig/2021-SNEC-single-cell. Image processing routines for images obtained via the Deep Mucosal Imaging (DMI) pipeline are available on GitHub: github.com/stalepig/deep-mucosal-imaging.
Data Transparency Statement: All datasets generated and analyzed during the current study are available from the lead contact on reasonable request.
Preprint: A preprint of this manuscript is available with the DOI: doi.org/10.1101/2021.06.02.446836
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Liu CY, Cham CM, Chang EB. Epithelial wound healing in inflammatory bowel diseases: the next therapeutic frontier. Transl Res 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shivdasani RA, Clevers H, de Sauvage FJ. Tissue regeneration: Reserve or reverse? Science 2021;371:784–786. [DOI] [PubMed] [Google Scholar]
- 3.Tetteh PW, Basak O, Farin HF, et al. Replacement of Lost Lgr5-Positive Stem Cells through Plasticity of Their Enterocyte-Lineage Daughters. Cell Stem Cell 2016;18:203–13. [DOI] [PubMed] [Google Scholar]
- 4.van Es JH, Sato T, van de Wetering M, et al. Dll1+ secretory progenitor cells revert to stem cells upon crypt damage. Nat Cell Biol 2012;14:1099–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Donati G, Rognoni E, Hiratsuka T, et al. Wounding induces dedifferentiation of epidermal Gata6(+) cells and acquisition of stem cell properties. Nat Cell Biol 2017;19:603–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Adamsen S, Ostberg G, Norryd C. Squamous-cell metaplasia with severe dysplasia of the colonic mucosa in ulcerative colitis. Report of a case. Dis Colon Rectum 1988;31:558–62. [DOI] [PubMed] [Google Scholar]
- 7.Bujanda L, Iriondo C, Munoz C, et al. Squamous metaplasia of the rectum and sigmoid colon. Gastrointest Endosc 2001;53:255–6. [DOI] [PubMed] [Google Scholar]
- 8.Cheng H, Sitrin MD, Satchidanand SK, et al. Colonic squamous cell carcinoma in ulcerative colitis: Report of a case and review of the literature. Can J Gastroenterol 2007;21:47–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mahesha V, Sehgal K, Saikia UN, et al. Squamous metaplasia of the rectum in a case of Hirschprung’s disease: a coincidence or an association. J Clin Pathol 2006;59:889–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nishi T, Weinstein WM, Makuuchi H. Squamous cell metaplasia in the rectum: a case report and review of the medical literature. Tokai J Exp Clin Med 2004;29:163–6. [PubMed] [Google Scholar]
- 11.Pantanowitz L. Colonic adenoma with squamous metaplasia. Int J Surg Pathol 2009;17:340–2. [DOI] [PubMed] [Google Scholar]
- 12.Sugimoto S, Iwao Y, Shimoda M, et al. Epithelium Replacement Contributes to Field Expansion of Squamous Epithelium and Ulcerative Colitis-Associated Neoplasia. Gastroenterology 2022;162:334–337 e5. [DOI] [PubMed] [Google Scholar]
- 13.Liu CY, Dube PE, Girish N, et al. Optical reconstruction of murine colorectal mucosa at cellular resolution. Am J Physiol Gastrointest Liver Physiol 2015;308:G721–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hale LP, Cianciolo G. Treatment of experimental colitis in mice with LMP-420, an inhibitor of TNF transcription. J Inflamm (Lond) 2008;5:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Seamons A, Treuting PM, Brabb T, et al. Characterization of dextran sodium sulfate-induced inflammation and colonic tumorigenesis in Smad3(−/−) mice with dysregulated TGFbeta. PLoS One 2013;8:e79182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu CY, Polk DB. Cellular maps of gastrointestinal organs: getting the most from tissue clearing. Am J Physiol Gastrointest Liver Physiol 2020;319:G1–G10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cao J, Spielmann M, Qiu X, et al. The single-cell transcriptional landscape of mammalian organogenesis. Nature 2019;566:496–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lopez-Garcia C, Klein AM, Simons BD, et al. Intestinal stem cell replacement follows a pattern of neutral drift. Science 2010;330:822–5. [DOI] [PubMed] [Google Scholar]
- 19.Dube PE, Yan F, Punit S, et al. Epidermal growth factor receptor inhibits colitis-associated cancer in mice. J Clin Invest 2012;122:2780–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wolf FA, Hamey FK, Plass M, et al. PAGA: graph abstraction reconciles clustering with trajectory inference through a topology preserving map of single cells. Genome Biol 2019;20:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nusse YM, Savage AK, Marangoni P, et al. Parasitic helminths induce fetal-like reversion in the intestinal stem cell niche. Nature 2018;559:109–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yui S, Azzolin L, Maimets M, et al. YAP/TAZ-Dependent Reprogramming of Colonic Epithelium Links ECM Remodeling to Tissue Regeneration. Cell Stem Cell 2018;22:35–49 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang X, Yin M, Zhang LJ. Keratin 6, 16 and 17-Critical Barrier Alarmin Molecules in Skin Wounds and Psoriasis. Cells 2019;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mustata RC, Vasile G, Fernandez-Vallone V, et al. Identification of Lgr5-independent spheroid-generating progenitors of the mouse fetal intestinal epithelium. Cell Rep 2013;5:421–32. [DOI] [PubMed] [Google Scholar]
- 25.Snippert HJ, van der Flier LG, Sato T, et al. Intestinal crypt homeostasis results from neutral competition between symmetrically dividing Lgr5 stem cells. Cell 2010;143:134–44. [DOI] [PubMed] [Google Scholar]
- 26.Murata K, Jadhav U, Madha S, et al. Ascl2-Dependent Cell Dedifferentiation Drives Regeneration of Ablated Intestinal Stem Cells. Cell Stem Cell 2020;26:377–390 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cabibi D, Fiorentino E, Pantuso G, et al. Keratin 7 expression as an early marker of reflux-related columnar mucosa without intestinal metaplasia in the esophagus. Med Sci Monit 2009;15:CR203–210. [PubMed] [Google Scholar]
- 28.Jiang M, Li H, Zhang Y, et al. Transitional basal cells at the squamous-columnar junction generate Barrett’s oesophagus. Nature 2017;550:529–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mitoyan L, Chevrier V, Hernandez-Vargas H, et al. A stem cell population at the anorectal junction maintains homeostasis and participates in tissue regeneration. Nat Commun 2021;12:2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Miyoshi H, Ajima R, Luo CT, et al. Wnt5a potentiates TGF-beta signaling to promote colonic crypt regeneration after tissue injury. Science 2012;338:108–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Arnold K, Sarkar A, Yram MA, et al. Sox2(+) adult stem and progenitor cells are important for tissue regeneration and survival of mice. Cell Stem Cell 2011;9:317–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zaiss DMW, Gause WC, Osborne LC, et al. Emerging functions of amphiregulin in orchestrating immunity, inflammation, and tissue repair. Immunity 2015;42:216–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.