

Abstract
ATPases associated with diverse cellular activities (AAA+ proteins) are macromolecular machines that convert the chemical energy contained in ATP molecules into powerful mechanical forces to remodel a vast array of cellular substrates, including protein aggregates, macromolecular complexes and polymers. AAA+ proteins have key functionalities encompassing unfolding and disassembly of such substrates in different subcellular localizations and, hence, power a plethora of fundamental cellular processes, including protein quality control, cytoskeleton remodelling and membrane dynamics. Over the past 35 years, many of the key elements required for AAA+ activity have been identified through genetic, biochemical and structural analyses. However, how ATP powers substrate remodelling and whether a shared mechanism underlies the functional diversity of the AAA+ superfamily were uncertain. Advances in cryo-electron microscopy have enabled high-resolution structure determination of AAA+ proteins trapped in the act of processing substrates, revealing a conserved core mechanism of action. It has also become apparent that this common mechanistic principle is structurally adjusted to carry out a diverse array of biological functions. Here, we review how substrate-bound structures of AAA+ proteins have expanded our understanding of ATP-driven protein remodelling.
ATPases associated with diverse cellular activities (AAA+ proteins) are a superfamily of proteins that harness the energy stored in the γ-phosphate bond of ATP to drive large-scale conformational rearrangements, enabling the remodelling of a plethora of cellular substrates, including nucleic acids and proteins1,2. AAA+ proteins are defined by the presence of a conserved ATPase domain that converts ATP hydrolysis into mechanical force3. Across the AAA+ superfamily, this ATPase domain serves as a versatile engine-like module — hence, AAA+ proteins are often referred to as AAA+ ‘motors’ — that is incorporated into larger assemblies, giving rise to a broad range of structurally variegated, ATP-fuelled machines with diverse functions. For instance, DNA replication, transcription and recombination require AAA+ helicases that unwind nucleic acids via translocation of single strands of nucleic acid4. Viral genome packaging also relies on AAA+ proteins to pump viral DNA or RNA into protein capsids4,5. Meanwhile, AAA+ enzymes that target protein substrates, as opposed to nucleic acids, serve as ubiquitous remodellers that power biological processes as divergent as protein quality control, rearrangement of the cytoskeleton and membrane fusion6 (FIG. 1).
Fig. 1 |. Modular organization of classical AAA+ proteins.
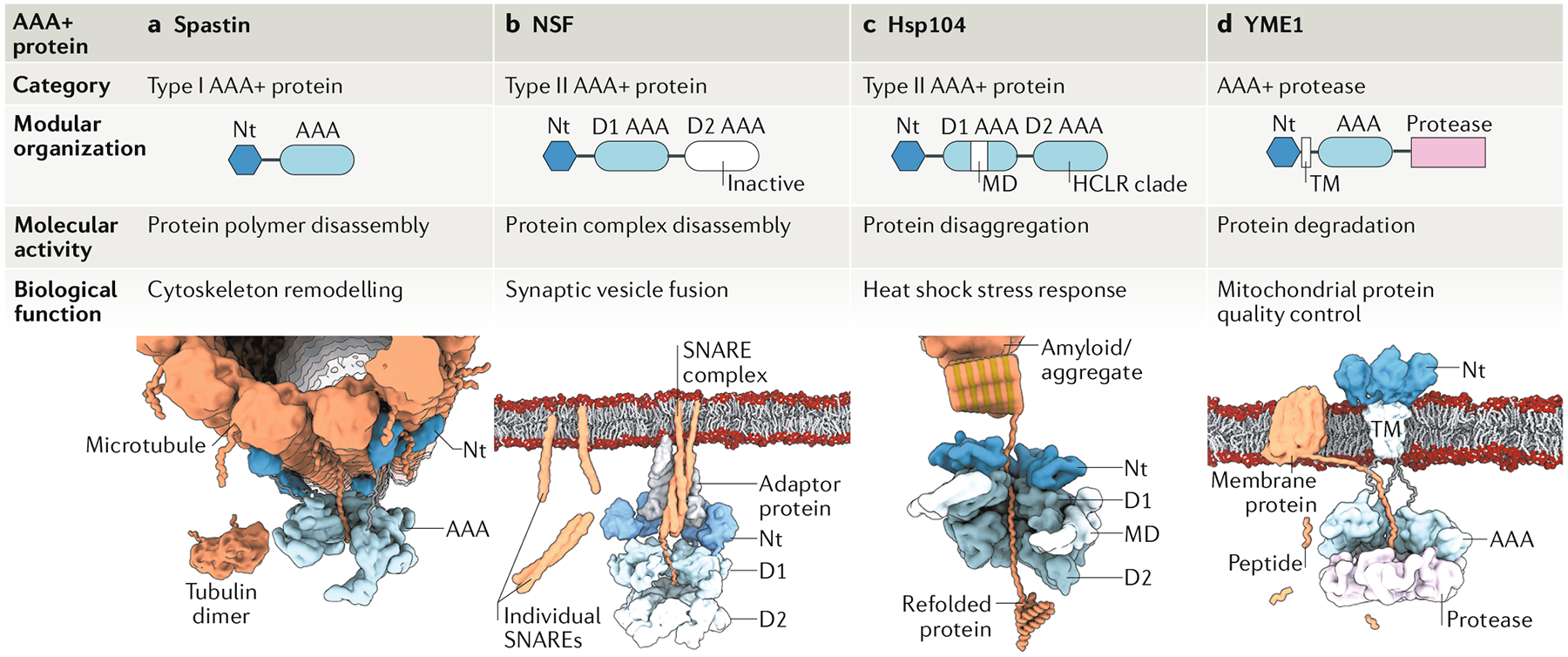
Schematic illustration of representative examples of classical ATPases associated with diverse cellular activities (AAA+ proteins). a | Type I AAA+ protein spastin (pdb:6P07). b | Type II AAA+ protein N-ethylmaleimide sensitive factor (NSF; pdb:6MDO). c | type II AAA+ heat shock protein Hsp104 (pdb:5VYA). d | AAA+ protease YME1 (pdb:6AZ0). In each case, a linear diagram shows the domain composition. The AAA+ protein with its molecular activity and biological function are depicted below. For each AAA+ protein, domains are coloured as indicated in the linear diagram, and the specific protein substrate–tubulin polymer for spastin (part a), SNARE complex for NSF (part b), amyloidic or aggregated protein for Hsp104 (part c) and membrane protein for YME1 (part d) is shown in orange. AAA, ATPase domain; D1 and D2, individual ATPase domains 1 and 2; MD, middle domain; Nt, amino-terminal domain; TM, transmembrane region.
The AAA+ superfamily is subclassified into seven different clades (classical, clamp loader, initiator, superfamily III helicases, HCLR, H2 insert and PS-II insert) based on insertion of distinct, additional elements into the otherwise structurally conserved AAA+ domain5,7 (see next section). Whereas the mechanisms by which AAA+ proteins remodel nucleic acids have been discerned by years of crystallographic studies on DNA or RNA-bound complexes, crystal structures of AAA+ remodellers bound to protein substrates have eluded structural biologists. As a result, how ATP powers protein remodelling remained obscure. Recent advances in cryo-electron microscopy (cryo-EM) have finally enabled the determination of high-resolution structures of AAA+ proteins bound to protein substrates, revealing a core mechanism of action that is conserved across the AAA+ superfamily but uniquely adjusted to the distinct functionality of each AAA+ protein. Thus far, nearly all cryo-EM structures of AAA+ protein translocases bound to a substrate belong to the classical clade (FIG. 1; TABLE 1). In this Review, we focus on the new mechanistic insights provided by these cryo-EM structures.
Table 1 |.
Examples of classical AAA+ proteins recently visualized in a substrate-bound state by cryo-electron microscopy
| AAA+ protein | Type | Subfamily | Primary molecular activity | Main biological process | Refs |
|---|---|---|---|---|---|
| Vps4 | I | Meiotic clade | Disassembly of ESCRT-III polymers | Membrane remodelling | 60,78,79 |
| Spastin and katanin | I | Meiotic clade | Disassembly of tubulin polymers | Cytoskeleton remodelling | 80,81 |
| p97 (higher eukaryotes), Cdc48 (yeast) | II | Cdc48 related | Extraction of polyubiquitylated proteins from membranes and complexes | Endoplasmic reticulum quality control, ubiquitin-dependent pathways | 69,70 |
| VAT | II | Cdc48 related | Dislocation of proteins into the archaeal 20S peptidase | Archaeal protein quality control | 58,59 |
| PEX1-PEX6 | II | Cdc48 related | Dislocation of ubiquitylated PEX5 from the peroxisomal membrane | Peroxisome biogenesis | 76,77 |
| NSF | II | Cdc48 related | Disassembly of SNARE complexes | Synaptic vesicle fusion | 67,68 |
| Hsp104 (yeast), ClpB (bacteria) | II | Hsp100 related | Disaggregase (part of a chaperone complex) | Heat-shock stress response | 61,71–73 |
| Hsp101 | II | Hsp100 related | Translocation of malarial proteins into the host | Malarial infection | 74 |
| YME1 and AFG3L2 | AAA+ protease | FtsH related | Degradation of mitochondrial proteins | Mitochondrial protein quality control | 62,63 |
| Rpt subunits | Proteasomal AAA+ | PAN related | Degradation of cytosolic proteins | Cytosolic protein quality control | 64,65 |
| PAN | Proteasomal AAA+ | PAN related | Degradation of archaeal proteins | Archaeal protein quality control | 66 |
AAA+ protein, ATPase associated with diverse cellular activities; ESCRT-III, endosomal sorting complex required for transport III; Hsp, heat shock protein; NSF, N-ethylmaleimide sensitive factor.
Overview of classical AAA+ proteins
All AAA+ proteins contain a conserved ATPase module. Over the course of evolution, this module has fused with a wide palette of unrelated domains. As a result, we now find the ATPase domain integrated into macromolecular polypeptides containing an array of enzymatic and/or regulatory modules, which has greatly increased the functional diversity of AAA+ proteins3,5. These additional domains confer distinct substrate specificities, modulate ATPase activity, provide additional enzymatic functionalities or mediate interaction with cofactors and accessory proteins. Classical AAA+ proteins can be generally categorized based on the modular organization of the ATPase cassette (FIG. 1).
Type I ATPases.
Type I ATPases, also known as Domain 1 (D1) ring ATPases, contain a single ATPase domain fused to an amino-terminal domain (FIG. 1a). In AAA+ proteins, non-ATPase N-terminal domains serve as the primary substrate recognition sites and are therefore important determinants of substrate preference. Accordingly, the same, conserved ATPase activity can be specifically directed to carry out distinct biological functions through substrate preference and subcellular localization8,9. This is well exemplified by the meiotic clade of type I AAA+ ATPases (named as such owing to the importance for meiosis of two founding members from Caenorhabditis elegans), encompassing katanin, fidgetin, spastin and VPS4 proteins (VPS4A and VPS4B). The N-termini of katanin, spastin and fidgetin recognize tubulin polymers, thereby recruiting these enzymes to sever microtubules for cytoskeleton remodelling9–11.
Meanwhile, the N-termini of VPS4 proteins recognize the carboxy-terminal tails of ESCRT-iii (endosomal sorting complex required for transport III) polymers, leading to ATPase-mediated disassembly of ESCRT-III polymers — a function that is essential for membrane remodelling in various contexts12,13.
ESCRT-III.
(Endosomal sorting complex required for transport III). ESCRT-III proteins are recruited to membrane constriction sites, including nearly all subcellular membrane compartments, where they are activated and assemble into filaments, which in turn recruit VPS4, a type I AAA+ protein. ATP-dependent VPS4 activity remodels and disassembles ESCRT-III polymers, thereby powering ESCRT-dependent membrane fission reactions that are required for diverse biological processes, such as vesicle formation in the secretory system, budding of enveloped viruses from the plasma membrane and membrane repair.
Type II ATPases.
There are numerous AAA+ proteins that, in addition to containing one or more N-terminal domains, contain two fused ATPase domains in tandem, which are commonly referred to as D1 and D2 (FIG. 1b,c). These ATPases, known as type II ATPases, arose through independent fusion events at least three times over the course of evolution. This is demonstrated by the fact that some type II AAA+ proteins contain two AAA+ domains from the classical clade, whereas others combine a classical domain with an AAA+ domain from the HCLR clade14. Type II AAA+ proteins include N-ethylmaleimide sensitive factor (NSF), p97 (also known as VCP; Cdc48 in yeast), PEX1 together with PEX6, and heat shock protein 100 (Hsp100)-related proteins (TABLE 1). NSF mediates membrane fusion, such as the fusion of synaptic vesicles with the presynaptic membrane during neurotransmission, by dissociating SNARE complexes that tether fusing vesicles to a target membrane. This enables the individual SNARE components to be recycled for further fusion events15 (FIG. 1b). p97/Cdc48 dislocates polyubiquitylated proteins from intracellular membranes, including the endoplasmic reticulum and mitochondria, where it functions as a component of the ER-associated protein degradation pathway and mitochondria-associated degradation, respectively. p97/Cdc48 mediates retrotranslocation of misfolded or damaged membrane and secretory proteins (which are selectively ubiquitylated by ubiquitin-conjugating enzymes and ubiquitin ligases) into the cytosol, where they can be captured by the proteasome for degradation16 (FIG. 2). Beyond extraction of proteins from membranes, p97/Cdc48 is also involved in extracting stalled polypeptides from the ribosome17,18 (FIG. 2) as well as in extracting proteins from chromatin to regulate chromatin-dependent processes such as gene expression or DNA repair19. Similarly to p97/Cdc48, the PEX1–PEX6 complex is a central component of extraction machinery — in this case, highly specialized — that removes ubiquitylated peroxisomal protein PEX5 (involved in peroxisomal protein import from the cytosol) from peroxisomal membranes for recycling to the cytosol for new peroxisome biogenesis or for proteasomal degradation (which occurs when PEX5 accumulates on peroxisomal membranes and prevents overloading peroxisomes with imported proteins)16,20. The Hsp100-related type II ATPases, which function as essential chaperones for heat-shock response in yeast (Hsp104) as well as bacteria and eukaryotic mitochondria and plastids (ClpB), use the D1–D2 architecture to disassemble protein aggregates and amyloids21 (FIG. 1c).
Fig. 2 |. Schematic representation of the diverse AAA+ proteins that coordinate eukaryotic protein quality control.
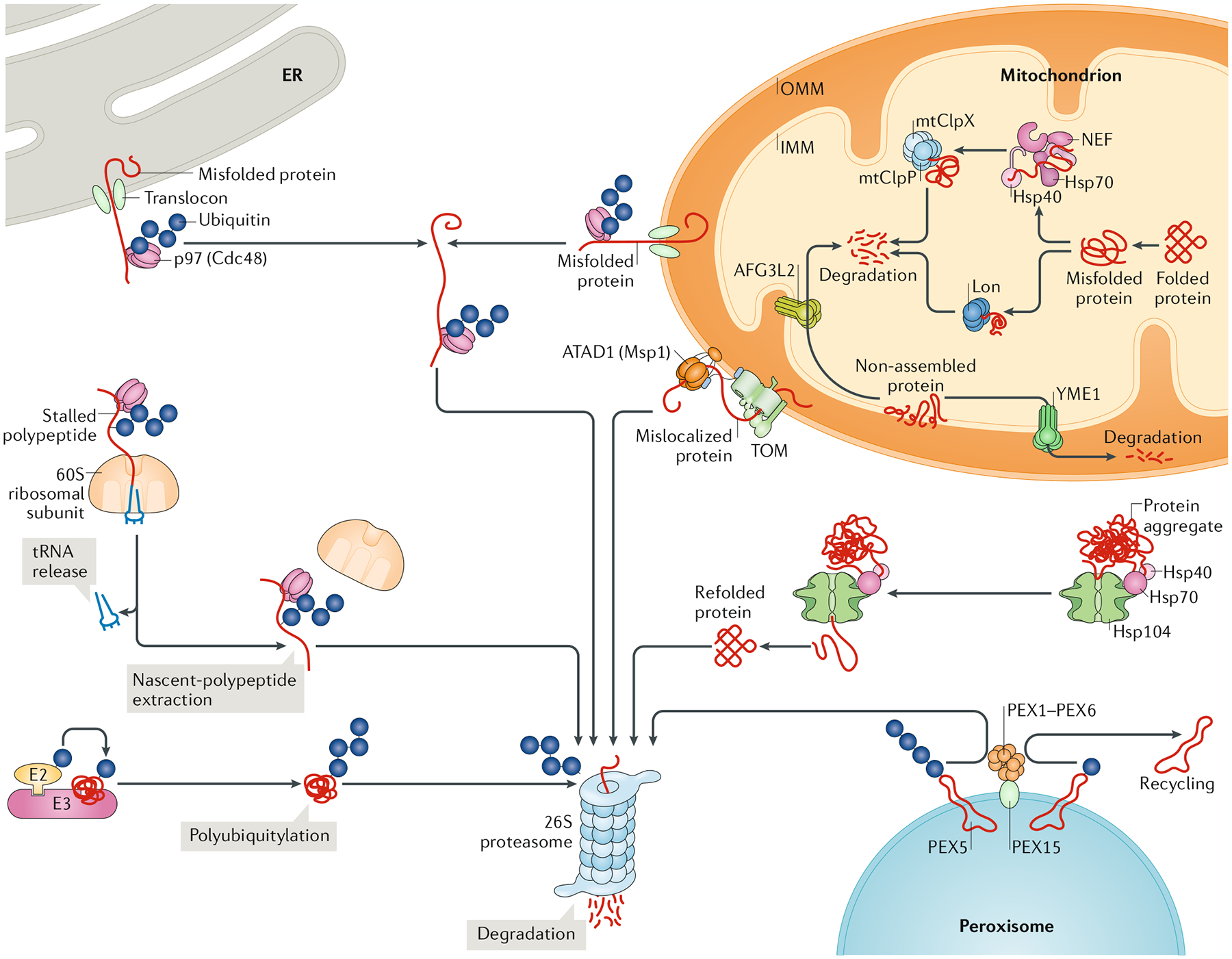
The 26S proteasome is the primary cellular machinery that unfolds and degrades ubiquitylated substrates and constitutes a central component of the protein quality control system, the main function of which is the removal of aberrant (for example, misfolded, truncated or damaged) proteins to maintain protein homeostasis (proteostasis). The 26S proteasome is a complex of numerous adaptor and regulatory components that assemble around a heterohexameric ATPase associated with diverse cellular activities (AAA+) motor, which inserts into a proteolytic barrel. The ubiquitin–proteasome system relies on the activity of E2 ubiquitin-conjugating enzymes and E3 ubiquitin ligases for tightly regulated ubiquitylation of substrates targeted for proteasome-mediated degradation. This pathway operates in the cytoplasm and in eukaryotes also in the nucleus (not shown). Multiple other AAA+ proteins cooperate with the 26S proteasome in protein quality control. Type II AAA+ proteins p97 (Cdc48 in yeast) and hetero-oligomeric PEX1–PEX6 extract polyubiquitylated substrates from the endoplasmic reticulum (ER) and peroxisomal membranes, respectively, for subsequent proteasomal degradation in the cytosol. In the mitochondrial outer membrane (OMM), the type I AAA+ protein ATAD1 (Msp1 in yeast) powers retrotranslocation of mistargeted proteins into the cytosol for degradation. Misfolded OMM proteins are retrotranslocated by p97 in a manner equivalent to ER-associated degradation. p97 is also involved in degrading aberrant polypeptides from stalled ribosomes during ribosome-associated protein quality control. In addition to the activity of the proteasome–ubiquitin system, independent proteolytic systems operate inside mitochondria. Here, AAA+ proteases YME1 and AFG3L2 from the classical clade, as well as ClpXP and Lon from the HCLR clade, degrade aberrant proteins in the inner mitochondrial membrane (IMM) and matrix, respectively. In addition to protein degradation, protein quality control relies on the capacity to refold misfolded proteins. This is mediated by chaperone complexes, including type II AAA+ disaggregases heat shock protein 104 (Hsp104; in yeast) and ClpB (in bacteria, mitochondria and plastids; not shown) that solubilize misfolded proteins and their aggregates, allowing their refolding. mt, mitochondrial; NEF, nucleotide exchange factor; TOM, translocase of the outer membrane.
SNARE complexes.
Protein complexes consisting of syntaxin, synaptobrevin and SNAP25 (synaptosome-associated protein), which assemble into a four-helix bundle that aids in the fusion of membranes.
ER-associated degradation.
A cellular pathway that targets misfolded proteins for selective ubiquitylation by endoplasmic reticulum (ER)-resident ubiquitin ligases. The type II AAA+ protein p97/Cdc48 recognizes and dislocates these polyubiquitylated substrates from the ER membrane and into the cytosol. The resulting unfolded polypeptides are subsequently degraded by the 26S proteasome.
Mitochondria-associated degradation.
The process by which the AAA+ protein p97/Cdc48 recognizes and retrotranslocates polyubiquitylated substrates from the outer mitochondrial membrane for subsequent degradation by the 26S proteasome.
Retrotranslocation.
Following translation in the cytosol, proteins are translocated into the respective cellular subcompartments. When a protein is misfolded, AAA+ proteins extract these proteins from the membrane, dislocating them into the cytosol. This process is known as retrotranslocation (from the subcompartment back into the cytosol).
In some cases, both D1 and D2 retain ATPase activity, such as in the Hsp100-related AAA+ proteins where both ATPase domains actively function to untangle or unfold protein aggregates22–24. In other cases, either D1 or D2 of the AAA+ protein has lost ATP hydrolysis activity (for example, D2 of NSF or D1 of PEX1–PEX6)15 (FIG. 1c). However, these catalytically dead domains typically retain the ability to bind ATP, and the nucleotide state in such inactive domains appears to influence the stability of the AAA+ protein and/or mediate recognition by various adaptor proteins15,25.
Across the AAA+ superfamily, distinct, non-related additional domains foster functional diversity by differentially regulating protein stability and protein–protein interactions with diverse accessory proteins. For instance, the N-terminal domain of NSF specifically binds αSNAP, an adaptor protein that mediates interactions with the SNARE complexes. Meanwhile, Hsp100-related proteins have integrated an additional domain within the D1 ATPase module, termed the ‘middle domain’ (FIG. 1c), that mediates interactions with co-chaperones and other accessory proteins21. The spatial proximity of these accessory proteins to the AAA+ unfoldase promotes cooperative refolding of protein substrates26.
AAA+ proteases.
Numerous AAA+ modules function in tight coordination with proteases, merging ATPase activity with proteolytic cleavage. In this case, the AAA+ module unravels protein substrates and feeds the unfolded polypeptide to adjacent proteases that subsequently degrade the substrate into short peptides27 (FIG. 1d). Cooperativity between these two functionalities establishes a means for processively degrading even tightly folded polypeptides, a functionality that is central to protein quality control across all kingdoms of life28. These AAA+ and protease domains are sometimes brought into close proximity through interlocking protein–protein interactions that position the AAA+ module atop an independent proteolytic oligomer, as in the 26S proteasome27 (and the ClpX family of proteases in the HCLR clade). In other cases, such as in FtsH-related AAA+ proteases, gene fusion events have resulted in tandem ATPase–protease domains incorporated into a single polypeptide29 (FIG. 1d); a similar arrangement is observed in Lon family AAA+ proteases of the HCLR clade. In fact, members of the FtsH-related family of AAA+ proteases (for example, YME1 and AFG3L2) are characterized by a distinct topology that succinctly exemplifies how concatenation of multiple modules facilitates distinct, specialized function (FIG. 1d). FtsH-related proteins are found in bacterial, mitochondrial and chloroplastic membranes, and are required for protein quality control within these membranous environments30 (FIG. 2). These AAA+ proteases are tethered to membranes via an N-terminal transmembrane region that is involved in recognizing membrane-associated or membrane-embedded protein substrates30. Together, N-terminal, ATPase and C-terminal zinc metalloproteinase domains combine distinct functionalities to enable membrane protein degradation (FIG. 1d).
26S proteasome.
A large multisubunit complex located in the cytosol of eukaryotes with numerous ubiquitin receptors that selectively bind polyubiquitylated protein substrates for degradation. Targeted substrates are unfolded by a AAA+ motor within the complex, while another enzyme called a deubiquitinase cleaves the covalently linked ubiquitin chain from the substrate. The AAA+ ATPase directs the unfolded substrate into a barrel-shaped proteolytic chamber which contains six proteolytic active sites that degrade the substrate.
Multicomponent systems that regulate AAA+ motors.
Multiple layers of functionally diverse adaptor and regulatory partner proteins can concomitantly assemble around the AAA+ core. The ubiquitin–proteasome system, a central pathway in cytosolic protein quality control, very well exemplifies a vast and highly regulated protein interaction network with factors that indirectly and directly influence AAA+ motor function. During assembly of the ~2.5-MDa 26S proteasome, six distinct AAA+ proteins (Rpt1–Rpt6 in yeast) attach to one end of a multimeric proteolytic barrel that contains evolutionarily diverse proteolytic domains31. Activity of the AAA+ motor is modulated by over a dozen regulatory and adaptor proteins, which adjust proteasomal activity in response to cellular conditions and requirements31. Substrates are targeted for degradation through a complex regulatory pathway that involves the covalent attachment of specifically linked ubiquitin moieties to protein substrates by ubiquitin ligases32. Even after recruitment for degradation, numerous deubiquitylating enzymes and ubiquitin ligases associate with the 26S proteasome to influence substrate processing32–34, which enables an additional layer of regulation of cytosolic protein quality control.
Importantly, the ubiquitin–proteasome system relies on other AAA+ proteins to liberate certain substrates from different cellular membranes for targeting to the 26S proteasome. Most notably, the retrotranslocation of polyubiquitylated proteins from the endoplasmic reticulum and peroxisomal membranes by type II AAA+ proteins p97/Cdc48 and PEX1–PEX6, respectively, is required for their efficient 26S-mediated degradation16 (FIG. 2). Similarly, ATAD1 (Msp1 in yeast), a type I ATPase, extracts mistargeted proteins from the mitochondrial outer membrane for degradation by the 26S proteasome (FIG. 2). Each of these distantly related AAA+ proteins has evolved to target distinct substrates in different subcellular environments, enabling the ubiquitin–proteasome network to access almost every compartment of the eukaryotic cell. In fact, eukaryotic protein quality control serves as a defining example of how the conserved ATPase domain functions as an energy-providing module upon which a panoply of components can be integrated, giving rise to sophisticated and highly regulated biological functions powered by AAA+ motors (FIG. 2).
Conserved features of the AAA+ module
Despite their functional diversity, AAA+ proteins share a structurally conserved ATPase domain of ~250 amino acids, comprising an N-terminal α–β–α fold35 and a small C-terminal helical bundle, commonly referred to as the large and small subdomains, respectively6 (FIG. 3a,b). Insertion of specific secondary structure elements at distinct locations defines the classification of AAA+ proteins into different clades5,7. The classical AAA+ enzymes we are focusing on in this Review are active as hexamers, where neighbouring subunits assemble into a ring whose central channel (also known as the central pore) binds the substrate (FIG. 3c). This central channel is lined solely by elements of the large subdomain of the ATPase6. The nucleotide-binding pockets are found at the interface between adjacent subunits, where a face of the large and small subdomains of one subunit interact with a face of the large subdomain of the clockwise neighbouring subunit6 (FIG. 3c,d).
Fig. 3 |. The conserved organization of the classical AAA+ domain.
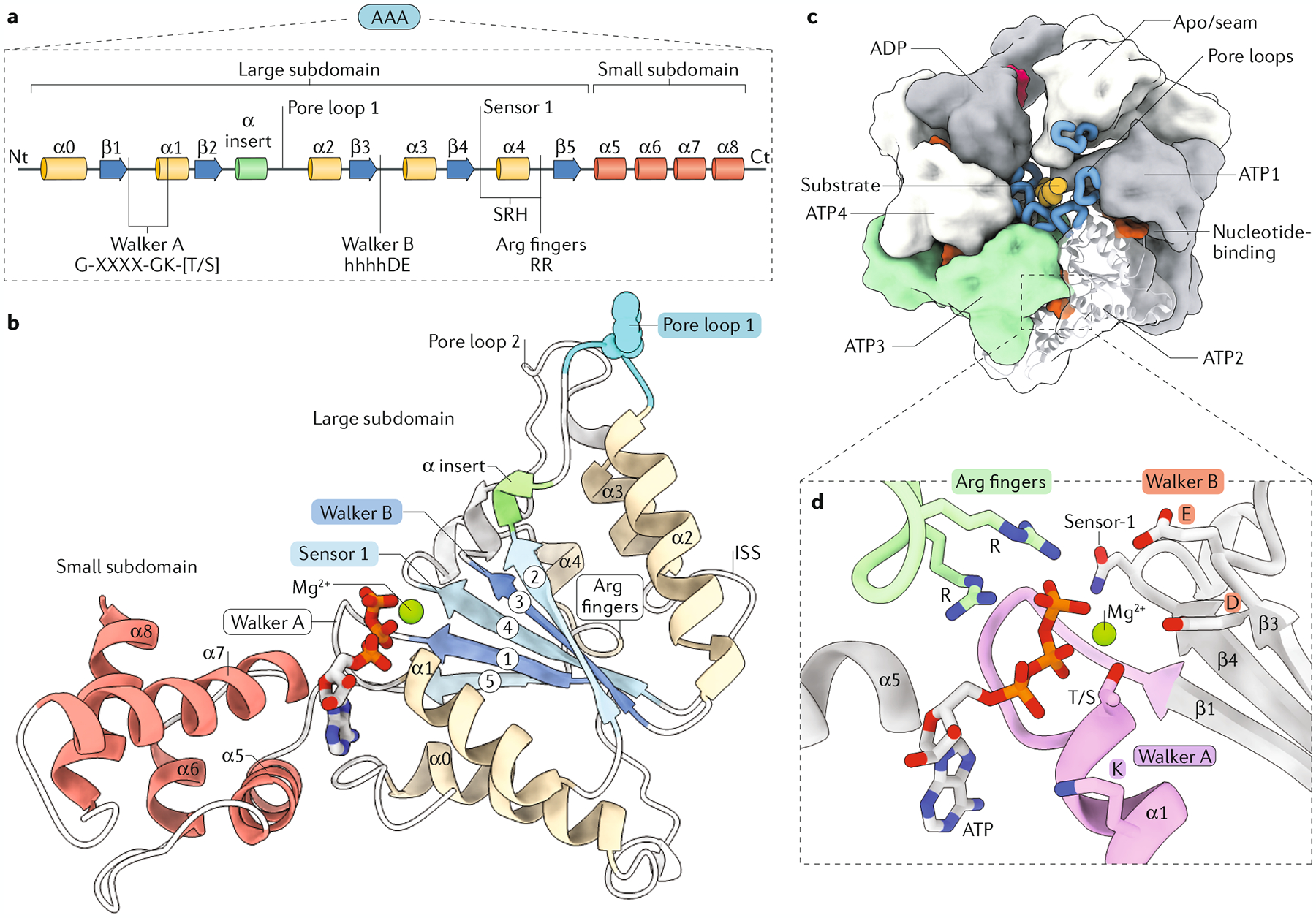
a | Linear diagram of the conserved secondary structure elements of the ATPase associated with diverse cellular activities (AAA+) domain, highlighting the position of the motifs required for activity: Walker A (G-XXXX-GK-[T/S] sequence motif, where X can be any amino acid), pore loop 1, Walker B (comprising conserved aspartic acid (D) and glutamate (E) preceded by several hydrophobic amino acids — hhhhDE), sensor 1 and the arginine (Arg) fingers (RR) found in the second region of homology (SRH). b | Representative atomic model (pdb:6AZ0) of the AAA+ domain labelled as in part a. Inter-subunit signalling (ISS) motif functions in inter-subunit coupling and coordination of the ATPase cycle in some AAA+ proteins (FIG. 7). c | Overview of the hexameric, ring-like organization characteristic of AAA+ proteins, with the pore loops (blue) interacting with the substrate (orange), which is threaded through the centre of the complex. The nucleotide-binding pocket localizes to the inter-subunit interface and is enlarged in part d. The nucleotide state for each subunit is highlighted (bound to ATP (ATP1–4), bound to ADP, nucleotide-free (apo)). d | The ATP-bound nucleotide-binding pocket with the conserved motifs and residues involved in ATP binding and hydrolysis in cis, with the Arg fingers from the adjacent subunit shown in green. Ct, carboxy terminus; Nt, amino terminus.
Phylogenetically, all AAA+ proteins belong to the ‘additional strand, catalytic E’ (ASCE) subclass of ‘P-loop’-type NTPases (nucleotide triphosphate-binding proteins)6. P-loop NTPases are characterized by a conserved α–β Rossman fold that contains the signature nucleotide-binding motifs, Walker A and Walker B36–38 (FIG. 3a,b). The ASCE subgroup is distinguished by a distinct β5–β1–β4–β3–β2 arrangement of the core β-sheet of parallel β-strands and a catalytic glutamate residue within the Walker B motif7,37,39 (FIG. 3a,b). The C-terminal ends of β1, β3 and β4 strands all contain conserved residues that contribute to the nucleotide-binding pocket, including the Walker A motif (found between β1 and the following α1 helix) and the Walker B motif (located on β3)40 (FIG. 3d). ‘Sensor 1’ is a single residue located at the C-terminal end of β4, which is thought to act in concert with the Walker B motif to properly orient a water molecule for nucleophilic attack on the γ-phosphate of ATP6. The sensor 1 residue is typically an asparagine, but other polar residues such as serine, threonine or aspartate are also found in this location.
NTPases.
A generic term that encompasses enzymes capable of binding nucleotide triphosphate (NTP) molecules, such as ATP and GTP. AAA+ proteins are defined as a subclass of P-loop NTPases.
α–β Rossman fold.
A super-secondary structure composed of alternating β-strand–α-helix–β-strand segments. The β-strands form a β-sheet and the α–helices surround both faces of the sheet, producing a three-layered sandwich.
Walker A.
A G-XXXX-GK-[T/S] sequence motif, where X can be any amino acid. This motif (also known as the P-loop) stabilizes the binding of the nucleotide by interacting with the β-phosphate, and is present in many nucleotide-binding proteins.
Walker B.
A consensus sequence (hhhhDE) where h represents any bulky, hydrophobic amino acid. The aspartic acid (D) is important for coordination of a magnesium ion, which in turn helps neutralize the negative charges of the phosphate groups present in the nucleotide. The adjacent glutamate (E) residue serves as a catalytic base, activating water for nucleophilic attack on the γ-phosphate during ATP hydrolysis.
Nucleophilic attack.
A fundamental reaction class in which a partially or fully positively-charged group (electrophile) is attacked by an electron-rich molecule (nucleophile) that substitutes a leaving group.
AAA+ proteins are further characterized by the second region of homology, which contains arginine residues that interact with the γ-phosphate of ATP1,41,42. These residues are referred to as ‘arginine fingers’, as they are located at the inter-subunit interface, where they interact with a nucleotide in trans, extending from the clockwise neighbouring subunit towards the ATP binding pocket (FIG. 3d). Arginine fingers have been shown to be essential for inter-subunit coordination and cooperation within the hexamer, as they can sense and respond to the nucleotide state — the presence of ATP or ADP, or the absence thereof — in the neighbouring subunit43. Classical clade AAA+ proteins are characterized by the presence of two arginines within the second region of homology. This clade is further distinguished by a small insertion between β2 and helix α2 (FIG. 3a,b) that contains a conserved pore region (pore loop 1) (FIG. 3a–c). This pore loop faces the central channel in the hexameric organization, and a conserved aromatic residue within this loop has been repeatedly shown to be required for substrate binding in the central channel44–48 (FIG. 3a–c).
Core mechanism of ATP-driven activity
Numerous biochemical studies of AAA+ proteins indicated that their substrates are threaded through the central pore, which successively imposes a constriction on the substrate polypeptide that eventually forces folded domains to unravel48–54 (FIG. 1). Although the key residues required for this activity have long been established, how conformational changes coupled to ATP binding, hydrolysis and product release might drive peptide substrate translocation has been elusive until recently.
Although X-ray crystallography is an established technique for solving high-resolution structures, it is reliant on crystallization of the protein of interest. The non-physiological conditions often used for crystallization, combined with the structural constraints induced by crystal packing, can sometimes lead to crystallization of a protein conformation that may not represent the predominant or active conformation of a protein in solution. A plethora of X-ray structures of numerous AAA+ protein translocases were solved, but, despite innumerable attempts over many years, high-resolution structures of substrate-engaged protein translocases could not be obtained by crystallography. As a result, the mechanisms underlying protein translocation remained obscure.
Recently, technological and methodological developments in cryo-EM have made it possible to determine the structures of macromolecular complexes to 3-Å resolution or better without the need for crystallization55–57. These advances have profoundly impacted many biological fields, and the impact this ‘resolution revolution’ has had on our understanding of the AAA+ superfamily — especially within the classical clade — is particularly notable. In just under 3 years, high-resolution structures of over a dozen classical AAA+ proteins bound to substrate have been determined58–81 (Table 1). Importantly, and in stark contrast to previously determined X-ray structures, these complexes were trapped in the act of processing substrates. The insights provided by these substrate-bound structures not only explain the precise role of all elements previously shown to be required for activity but also provide strong visual evidence to contextualize decades of analytical studies on AAA+ proteins. Moreover, these structures have revealed the conserved mechanism by which ATP powers substrate translocation.
An ATPase spiral encircles the translocating substrate along the central pore.
Strikingly, substrate-bound structures of AAA+ proteins share a pseudo-helical arrangement of the ATPase domains that resembles a spiral staircase (FIG. 4). A recent study of VAT, an archaeal type II AAA+ protein, combined cryo-EM and nuclear magnetic resonance (NMR) to confirm that the spiralling organization observed in cryo-EM reconstructions is also present in solution58. A spiralling organization for a protein-translocating AAA+ was first observed at subnanometre resolution in the 26S proteasome82–84. The higher resolutions attainable with current cryo-EM methodologies now reveal the molecular relevance of this organization: incoming substrate is threaded through the central pore of the spiralling ATPase ring, with conserved pore loops matching the helical arrangement of the ATPases, forming a staircase that accompanies the substrate along the central channel (FIG. 4). The substrate adopts an extended β-strand conformation, running through the central channel as an unfolded peptide with its side chains radiating outwards towards the spiralling AAA+ pore loops (FIGS 4,5). The conserved aromatic residue within the AAA+ pore loop 1 of each subunit intercalates against the backbone of the substrate (FIGS 4,5,6), whereby one pore loop 1 aromatic residue is inserted every two amino acids (with a distance of approximately 13 Å between individual pore loop 1–substrate interactions). This helical array of pore loop–substrate interactions produces a concomitant grip on the substrate, consistent with previous biochemical studies showing that up to five subunits synergistically engage the translocating substrate85 (FIG. 4a,c). This organization provides a molecular explanation for the essential role of the pore loop 1 aromatic residue across classical AAA+ proteins45–48,86.
Fig. 4 |. Substrate-bound structures of AAA+ proteins reveal a conserved hand-over-hand substrate translocation mechanism.
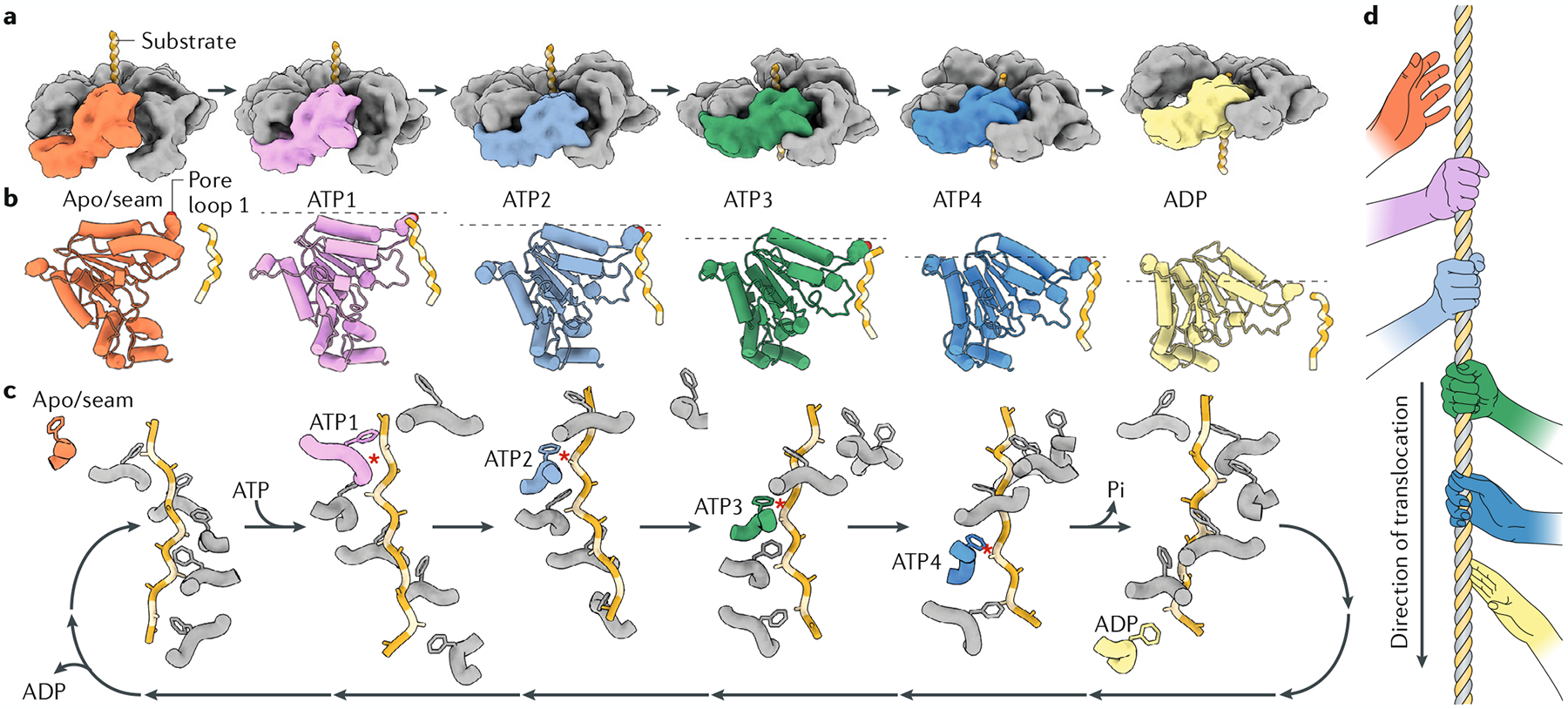
a | Side view of the ATPase associated with diverse cellular activities (AAA+) staircase with a single subunit coloured in each panel to show how it progresses through consecutive positions in the staircase, translocating the substrate with it. Each distinct colour corresponds to the unique position adopted within the hexamer during the translocation cycle. The cycle begins with the subunit that is unbound to nucleotide (apo/seam, coloured orange), and thus displaced from the hexamer and not interacting with the substrate. Upon ATP binding, the subunit progresses through four registers of the spiral staircase within the hexamer (ATP1, pink; ATP2, light blue; ATP3, green; ATP4, dark blue), until hydrolysis in the nucleotide-binding side triggers the subunit to release from the substrate (ADP, coloured yellow). The concerted movement of all six subunits within the hexamer progressing through this cycle sequentially results in hand-over-hand translocation of the substrate. b | The corresponding coloured subunit from part a is shown using a ribbon representation to demonstrate how the ATPase domain progressively tilts downwards through the translocation cycle, giving rise to a downward motion of pore loop 1 (the pore loop 1 aromatic residue is shown using a sphere representation) and the substrate. The dotted grey line emphasizes the downward progression of each step through the cycle. c | Pore loops of the six subunits in an AAA+ hexamer assemble into a spiral staircase that wraps around the translocating substrate. Any given subunit (we follow the cycle of one subunit, coloured according to its register in the ATPase hexamer, as defined in part a) transitions through each position of the staircase hydrolysing ATP at the bottom of the staircase, releasing ADP and detaching from the substrate as it resets to assume a position at the top of the staircase upon rebinding of ATP. Substrate-engaged states are indicated by a red star between the pore loop 1 residue and the substrate. d | Cartoon illustrating the hand-over-hand mechanism of substrate translocation between the subunits of an ATPase associated with diverse cellular activities (AAA+ protein).
Fig. 5 |. The universal substrate-interacting pore loop 1 is uniquely adjusted in different AAA+ proteins.
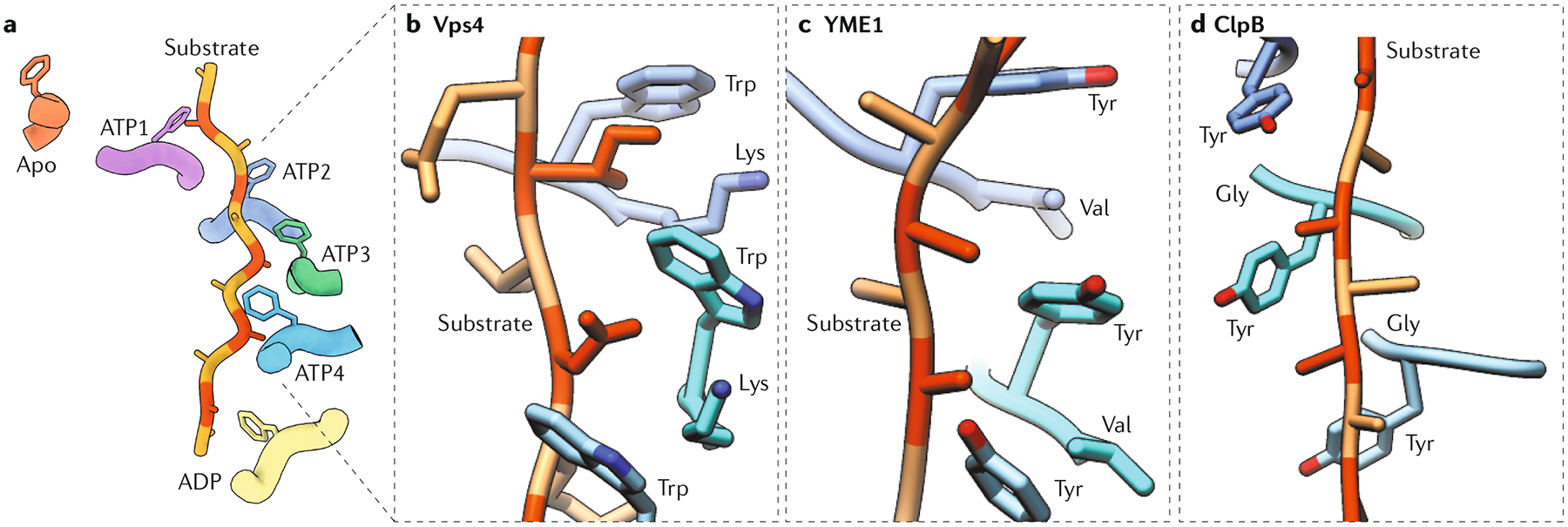
a | The pore loop 1 aromatic residue of the ATP-bound subunits (ATP1–ATP4) intercalates against the substrate every two amino acids, whereas the pore loop of the ADP-bound subunit has limited interactions with the substrate and the apo subunit does not contact the substrate. The pore loop 1–substrate interactions are mediated by the conserved aromatic residue of pore loop 1 of each subunit (see parts b–d where these residues in pore loop 1 of subunits ATP2–ATP4 are shown). b–d | The overall spiralling organization and molecular principles of the pore loop–substrate interface are conserved among classical AAA+ domains (as well as some non-classical domains, part d), but non-conserved residues within pore loop 1 give rise to unique environments around the translocating substrate in different ATPases associated with diverse cellular activities (AAA+ proteins). For example, like in most classical clade AAA+ proteins, the residue preceding this aromatic residue (aromatic-prior position) corresponds to lysine (Lys) in Vps4 (pdb:6BMF, part b). This basic residue engages in cation–π interactions with the conserved aromatic both in cis (within the same subunit) and in trans (with a neighbouring subunit). This configuration likely increases stability of the staircase and strengthens inter-subunit communication. Similar to Vps4, YME1 (pdb:6AZ0, part c) engages the substrate through the pore loop aromatic, which corresponds to tyrosine (Tyr) in this case. However, the aromatic-prior residue corresponds to valine (Val) instead of lysine, which increases the hydrophobicity around the translocating substrate, but at the same time eliminates the stabilizing cation–π interactions within the ATPase staircase. Domain 2 of ClpB (pdb:6OAY, part d) — a domain originating from the HCLR clade — contains glycine (Gly) as an aromatic-prior residue. In consequence, this increases flexibility within the pore loop staircase, which likely weakens the organization of the staircase.
Fig. 6 |. Distinct structural features of different of AAA+ proteins mediate additional interactions with the translocating substrate.
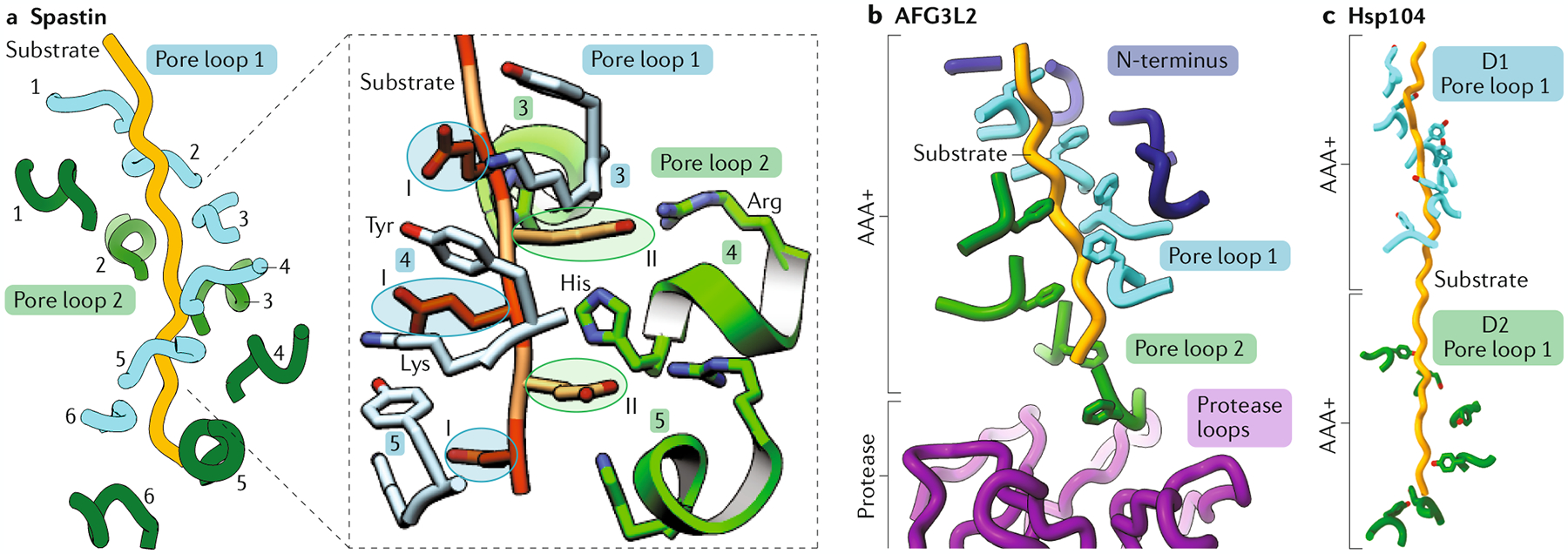
a | Pore loop 2 (green) of type I ATPase associated with diverse cellular activities (AAA+) protein spastin (pdb:6P07) assembles as a staircase parallel to the pore loop 1 (blue) staircase (in both cases, numbers indicate the subunit to which the pore loop belongs). The zoom shows the polyglutamate substrate threaded through the central pore of spastin. The residues of the substrate that insert into the class I pocket formed by pore loop 1 are circled in blue, whereas the residues that face the class II pocket are circled in green. The class I pocket includes the pore loop 1 aromatic (tyrosine, Tyr) and lysine (Lys) residues, both of which are shown in light blue. In the class II pocket, the positively charged residues (arginine (Arg) and histidine (His)) of the non-conserved pore loop 2 are shown in green. b | In AAA+ protease AFG3L2 (pdb:6NYY), pore loop 2 (green) and the amino terminus (N-terminus; dark blue) form spiral staircases additional to the one established by pore loop 1 that wrap around the substrate along the longitudinal axis of the complex. Pore loop 2 of AFG3L2 contains an insertion (Supplementary Fig. 1) that positions the pore loop 2 staircase low within the spiral, such that pore loop 2 of the lowest subunit of AFG3L2 protrudes deep into the proteolytic chamber (purple), contacting the centre of the protease ring. Pore loop 2 escorts the substrate across the degradation chamber, directly transferring it to the protease. c | Pore loop 1 of domain 1 (D1; blue) and domain 2 (D2; green) of the type II AAA+ protein Hsp104 (pdb:5VYA) assemble as individual spiral staircases that interact in tandem with the translocating substrate, thereby providing a cooperative grip on the substrate.
Substrate–pore loop 1 interactions appear to be dominated by non-residue-specific hydrogen-bonding and steric interactions with the substrate backbone, which is compatible with a translocation mechanism that is independent of the substrate sequence and its orientation. These are important requirements for AAA+ activity, given that AAA+ enzymes are able to remodel proteins of diverse sequences and translocate them in either orientation (from the N-terminal or C-terminal end)87,88. However, the intercalating nature of the substrate–pore loop 1 interactions also provides a mechanistic avenue for the substrate to influence these contacts within the central pore. For example, bulky, hydrophobic or aromatic substrate residues intercalate between the pore loop aromatics79, akin to the teeth of two cogs (FIG. 5b), strengthening the grip on the substrate89. By contrast, enrichment of smaller residues in the substrate, such as glycine, which is unable to engage in such interactions, decrease the enzyme’s ability to process such substrates89.
Substrate binding appears to have an important role in the formation of the hydrolysis-competent ATPase spiral staircase. Type I AAA+ enzymes form a single, hexameric ATPase spiral that wraps around the translocating substrate60,79–81 (FIG. 1a). Type II AAA+ enzymes assemble into two stacked hexameric rings, where each ring comprises six equivalent ATPase domains59,61 (FIG. 1b,c). AAA+ proteases similarly form stacked hexameric rings, with one ring consisting of the six protease domains62,63 (FIG. 1d). To date, all ATPase domains with the substrate threaded through the pore have been observed in a spiralling organization, whereas domains within the same complex that do not engage substrate generally form planar symmetric rings. For instance, both the D1 and D2 ATPase domains of Hsp100-related AAA+ proteins are catalytically competent and engage the threaded substrate. Accordingly, in substrate-bound cryo-EM structures of this family of proteins, both D1 and D2 assemble into two stacked homotypic spiralling rings61,71,73,90 (FIG. 1b). Meanwhile, only the D1 of NSF functions as an ATPase and forms a substrate-engaged spiral whereas the catalytically dead D2 assembles into a planar, symmetric ring below the D1 spiral67,68 (FIG. 1b). Similarly, each of the peroxisomal D1–D2 AAA+ proteins PEX1–PEX6 consists of an ATP hydrolysis-incompetent D1 that does not bind the substrate and oligomerizes into a planar symmetric ring, whereas their active, substrate-binding D2s form a spiral76,77. The AAA+ proteases YME1 and AFG3L2 present an analogous organization, wherein the ATPase domains assemble as a spiral atop a planar, six-fold symmetric ring formed by the homotypic protease hexamer62,63 (FIG. 1d).
In some AAA+ proteins, substrate binding itself may play a role in inducing a spiralling organization. For example, cryo-EM structures of D1–D2 AAA+ protein p97 and the yeast homologue Cdc48 in the absence of substrate showed both D1 and D2 oligomerized into planar, symmetric rings70,91, whereas substrate-bound Cdc48 complexes adopted a spiralling organization69,70. The observed correlation between a spiralling quaternary structure of the ATPase domains and the translocase activity strongly suggests a crucial role of this organization in substrate translocation. Together, these findings explain why the presence of substrate promotes hexamerization of the ATPase subunits8,92, stabilizes the hexameric complex81 and increases ATPase activity in AAA+ proteins83.
Sequential ATP hydrolysis cycle powers hand-over-hand substrate translocation.
High-resolution substrate-bound structures of classical clade ATPases have enabled identification of the nucleotide states within the AAA+ binding pocket, revealing that the nucleotide state directly correlates with the pore loop 1 conformation62. The pore loops from subunits that have ATP bound in their pockets intercalate into the substrate, whereas the pore loops of subunits bound to ADP or that do not contain nucleotide have limited, if any, interaction with the substrate62,64,69,70 (FIG. 5a). Consistent with these findings, numerous biochemical studies previously showed that ATP binding is required for substrate engagement by AAA+ proteins. For instance, Walker B mutants, which have increased affinity for ATP but are nearly incapable of hydrolysing it, have increased affinity for substrates6,93. In fact, these Walker B mutations effectively trap the substrate6,94,95, and it is important to note that most substrate-bound cryo-EM structures of AAA+ proteins solved to date are of either Walker B mutants62 or wild-type enzymes bound to non-hydrolysable ATP analogues61.
Importantly, substrate-bound cryo-EM structures further show that different nucleotide states can coexist within the hexamer, such that ATP-bound subunits form the substrate-interacting spiral, whereas the lowermost subunit typically presents an ADP-like state62,64,70 (FIG. 5a). Nucleotide-free subunits are considerably more flexible and have only been found at the ‘seam’ of the spiral between the lowest and highest subunits of the staircase, transitioning from the lowest to the highest position in the staircase (FIGS 4,5a). This organization strongly suggests that ATP hydrolysis occurs in the lowermost subunit of the staircase, whereupon the subunit detaches from the hexamer and releases nucleotide as it moves towards the uppermost subunit of the staircase (FIG. 4). Next, ATP binding within this subunit at the top of the staircase establishes stabilizing interactions with the neighbouring subunit and its pore loop 1 rearranges to interact with the substrate (FIG. 4c). Thus, if we follow a single subunit through the ATP hydrolysis cycle, we see the pore loop engage and escort the substrate along the central channel, maintaining close interaction with the substrate as the subunit progresses through the successively lower steps of the staircase, and subsequently detaching from the substrate at the bottom (FIG. 4b). The movements of the subunit arise from rotations of the large and small subdomains of the ATPases relative to one another. The positions of these domains are dictated by the nucleotide state in each subunit, and the motions are used to influence interactions with substrate.
The tight allosteric relationships between the nucleotide state of a given subunit and the subunit’s position within the staircase, as well as its mode of interaction with the substrate, establish the mechanistic infrastructure for a sequential ATP hydrolysis cycle. These findings have led several groups to propose that ATP hydrolysis is coordinated within the hexamer such that the subunits fire one at a time in sequential order around the hexameric ring59–62. An immediate consequence of this around-the-ring progression of hydrolysis is that, in the context of the hexamer, each subunit occupies one of the positions within the staircase, and they all progress through each position of the cycle in a coordinated fashion. At any given time during the translocation process, four ATP-bound subunits are engaged with the substrate, escorting the substrate along the central pore, one ADP-bound subunit is disengaging from the substrate at the bottom of the staircase and the last subunit is returning to the topmost position of the hexamer. The resultant cascading cycle of subunits engaging and releasing substrate is analogous to a hand-over-hand sled pull on a rope, where six hands work coordinately to alternatively reach, grab and pull on the rope (FIG. 4d). As the hands take turns to release the rope at the bottom and grab it at the top, the rope remains tightly engaged by four hands at all times, and a pulling force is applied in each cycle. Together, these motions lead to a constant grip on the substrate and stepwise translocation that is powered by a sequential ATP hydrolysis cycle that proceeds anticlockwise around the hexameric ring. As two amino acids are engaged between the pore loop 1 aromatics of adjacent subunits, in the absence of any other forces, every ATP hydrolysis event would lead to a translocation step of exactly two amino acids. Recent structures representing coexisting states of the actively hydrolysing 26S proteasome ATPases, which form a heterohexamer where each ATPase subunit is unique and therefore distinguishable from the rest, further support this rotary model for ATP hydrolysis64,65.
A direct consequence of this around-the-ring model for ATP hydrolysis is the capacity for producing a constant hand-over-hand conveyance of substrate through the central pore (FIG. 4). This unified model and the structural framework provided by substrate-bound structures of diverse AAA+ proteins are particularly relevant for understanding the enzymatic properties of these motors. Single-molecule studies of AAA+ proteins from different clades showed that tight ATP binding is the first irreversible step in the hydrolysis cycle96,97, which can be explained by the spiralling architecture. As a subunit assumes the uppermost position of the staircase, ATP binding within this subunit establishes interactions with the neighbouring subunit. The sub unit immediately below this uppermost subunit is now trapped within the context of the spiral and can only exit the staircase after progressing downwards through the spiral, concluded by ATP hydrolysis and release from the spiral at the bottom of the staircase. Numerous biophysical studies also indicated that phosphate release, which is an irreversible transition, is the primary force-generating step of the cycle. Indeed, the largest domain movements of the classical clade ATPases occur during the transition from an ATP-bound to an ADP-bound (post-phosphate release) state62. However, substrate-bound structures of the actively hydrolysing 26S proteasome challenge the notion that any single step of the ATP hydrolysis cycle can be ascribed as a power stroke64. Instead, the forces that drive translocation likely arise from the concomitant effect of multiple coordinated steps occurring in concert between neighbouring subunits. ATP hydrolysis in the lowermost subunit results in loss of the γ-phosphate, disrupting the ATP-dependent inter-subunit interactions established by the trans-acting arginine fingers (FIG. 3d), as well as additional elements discussed below (see subsection ‘AAA+ proteins utilize different mechanisms for inter-subunit communication‘). As a result, this subunit is released from the rest of the hexamer, and transitions towards the seam position between the lowest and highest subunits of the staircase. The spiralling subunits of the hexamer are then free to undergo a register shift via a downward rigid body movement. The subunit that previously occupied the seam position now completes nucleotide exchange, and rebinds ATP at the top of the staircase, restarting the cycle.
This hand-over-hand mechanistic model is rapidly emerging as the conserved mechanism by which ATP powers protein translocation in classical AAA+ proteins. In fact, the recently solved cryo-EM structures of bacterial AAA+ proteases Lon and ClpXP, both of which belong to the distantly related HCLR clade, demonstrated conservation of this mechanistic principle across protein-remodelling AAA+ proteins98–100. Furthermore, crystallographic studies of substrate-bound DNA and RNA translocases from other AAA+ clades revealed analogous spiralling organizations of the ATPase oligomers, and similar hand-over-hand mechanisms were proposed101–104. This translocation mechanism may thus be conserved across the entire AAA+ superfamily, driving translocation of not just proteins but also nucleic acid substrates.
Functional divergence
Although a conserved mechanism for ATP-driven substrate translocation through the central pore of AAA+ protein assemblies is emerging, a fundamental question remains unanswered: what unique structural features enable each AAA+ protein to perform a distinct biological function? As more cryo-EM structures of distantly related AAA+ proteins with substrate threaded through the central pore are solved, differential features integrated within the core mechanism are beginning to surface.
Distinct residues in pore loop 1 adjust the properties of the central channel.
By stably intercalating into the incoming polypeptide within the central channel, the pore loops in AAA+ proteins transduce the force generated by ATP hydrolysis-driven domain motions within the motor to the substrate, and are therefore responsible for coupling ATP hydrolysis and substrate remodelling46,62,97,105. The pore loops thus have the potential to influence both the chemical and mechanical properties of the motor97. For example, introducing large, bulky amino acids around the conserved pore loop aromatic residue increased grip on the substrate, but lead to an overall decrease in ATP hydrolysis rates, presumably due to slower resetting of the pore loops to rebind the substrate and restart the cycle following ATP hydrolysis97. Whereas the conserved pore loop 1 aromatic residue is observed intercalating into the substrate in all substrate-bound AAA+ protein translocases solved to date (FIG. 5), the overall properties of the central pore appear to be adapted in each AAA+ protein, likely tuning the translocation speed of the motor and its grip on the substrate to specifically suit the needs of its particular biological function.
The substrate-intercalating residue is always aromatic in nature, but phenylalanine, tyrosine and tryptophan vary significantly in size and polarity, offering an evolutionary means of diversifying the environment around the translocating substrate (FIG. 5b–d). This diversification can be furthered through the residues flanking the aromatic residue, which have a critical role in the mechanical unfolding of protein substrates97. For example, single-point substitutions of the residue preceding the conserved aromatic residue (referred to as aromatic prior) in different AAA+ proteins severely reduced enzymatic efficiency or completely abolished activity63,73. In most classical clade AAA+ proteins, the aromatic-prior residue is a lysine61,64,79,80 (Supplementary Fig. 1). Substrate-bound structures consistently show this lysine residue sandwiched between pore loops 1 of neighbouring subunits, engaging in cation–π interactions with the conserved aromatic residues both in cis (within the same subunit) and in trans (with the neighbouring subunit at the next lower position)64,79 (FIG. 5b). Within the context of the spiral, this configuration establishes a connected network of interactions that spans the entire pore loop 1 staircase, which likely increases stability of the staircase and strengthens inter-subunit communication. Meanwhile, AAA+ proteases of the FtsH-related family all contain either a valine or methionine residue in the aromatic-prior position (FIG. 5c; Supplementary Fig. 1). This increases the hydrophobicity around the translocating substrate62,63, but adjacent pore loops within the staircase are stabilized only by van der Waals interactions, which are significantly weaker than the cation–π interactions enabled by the lysine residue found at this position in other AAA+ proteins. When the aromatic prior-residue instead corresponds to a glycine (for example, in D2 of Hsp100-related proteins; domains that originate from the HCRL clade), the spiral staircase is devoid of these stabilizing interactions (FIG. 5d), likely resulting in a weaker grip on the substrate. Furthermore, the flexibility of the polypeptide backbone introduced by this glycine residue results in a switch in the orientation of the aromatic side chain with regards to the pore loop backbone61,72 (FIG. 5d). Type II AAA+ protein p97/Cdc48 constitutes another notable example of how residues adjacent to the conserved aromatic distinctly adjust the central pore and its characteristics. In this case, the conserved aromatic tryptophan is followed by another aromatic residue, tyrosine (Supplementary Fig. 1), and, together, these residues form a pincer-like staircase, maximizing grip on the substrate, as demonstrated in recent cryo-EM reconstructions of Cdc48 (ReFS69,70).
Cation–π interactions.
Non-covalent interactions between an electron-rich π system (for example, aromatic amino acids — phenylalanine, tryptophan and tyrosine) and an adjacent cation (for example, basic residues — arginine, lysine and histidine).
van der Waals interactions.
Distance-dependent interactions between atoms or molecules that are significantly weaker than other kinds of interactions, such as electrostatic ones.
Thus, the non-conserved residues of pore loop 1 give rise to distinct central pore environments and configurations that are likely fine-tuning substrate grip as well as the mechanochemical properties of the motor to the specific requirements for activity of different AAA+ proteins. Strong correlations between pore loop 1 characteristics, distinct substrate preferences and unfoldase power are to be expected, but the precise implications of each molecular organization remain to be established.
Unique structural features within AAA+ proteins finetune substrate processing.
The translocating substrate adopts an extended β-strand conformation, with successive amino acids facing alternate directions within the central pore (FIG. 6a). As a result, alternating substrate residues face two distinct sites, or ‘substrate-binding pockets’, within the AAA+ channel (FIG. 6a). These substrate interaction sites are termed class I and class II pockets79. Pore loop 1 forms class I pockets in the manner described above (FIG. 5). Class II pockets interact with the opposite face of the substrate, and are typically formed by a secondary pore loop (pore loop 2; FIG. 3b; Supplementary Fig. 1), which are poorly conserved across AAA+ proteins (Supplementary Fig. 1). The non-conserved pore loop 2 is an important determinant of function, and substrate-bound structures are now revealing why.
The microtubule-severing AAA+ proteins katanin and spastin, for example, specifically bind the negatively charged C-terminal tails of tubulin, and contain a pore loop 2 that is rich in positively charged residues45,106 (Supplementary Fig. 1). Mutation of these positively charged residues abolishes activity106, and a recently solved structure of spastin bound to a polyglutamate substrate peptide explains the molecular role of pore loop 2 in these enzymes81. Within the central channel of the spastin ATPase, pore loop 2 forms a spiral staircase immediately below the pore loop 1 staircase, such that two positively charged residues face the class II substrate-binding pocket, where they interact with a glutamate residue from the substrate (FIG. 6a). Thus, the distinct pore loop 2 in these microtubule-severing enzymes appears to stabilize and neutralize the negatively charged substrate target for spastin activity. Similarly, the recently solved structures of FtsH-related AAA+ proteases YME1 and AFG3L2 show that pore 2 loops assemble as an additional spiral staircase around the translocating substrate62,63 (FIG. 6b). In these enzymes, however, the pore loop 2 spiral contains an aromatic residue that directly contacts the substrate in the class II pocket and increases hydrophobicity of the central pore (FIG. 6b). This aromatic residue is important for function, and mediates additional hydrophobic interactions with the substrate within the class II pocket62,63. It is thus likely that this is an adaptation for optimal processing of the hydrophobic membrane substrates of this specialized family of AAA+ proteases. Moreover, pore loop 2 of AFG3L2 contains an insertion (Supplementary Fig. 1) that positions the pore loop 2 staircase lower within the spiral than in YME1, such that pore loop 2 of the lowest subunit of AFG3L2 protrudes deep into the proteolytic chamber, contacting the centre of the protease ring63 (FIG. 6b). This organization appears to mediate more effective transfer of the substrate from the central channel to the protease domains63. This adaptation might have evolved to enable interdomain crosstalk, providing a mechanism to coordinate the two enzymatic functions of AFG3L2, as indicated by the fact that ATPase activity is affected by protease activity, and vice versa63.
Inter-domain coordination between the distinct ATPase domains of the type II AAA+ proteins can also serve as a means of establishing robust substrate engagement and enzymatic function. For example, type II AAA+ proteins of the Hsp100-related family, such as disaggregases Hsp104 and ClpB, contain two tandem ATP hydrolysis-competent subunits that both simultaneously interact with the incoming substrate. The two distinct ATPase domains each contribute a unique pore loop 1 that interacts with different sections of the substrate in the central pore, thereby doubling the amount of engaged substrate at any given time61,72,73 (FIG. 6c). This cooperative grip on the substrate of tandem ATPase domains likely explains why type II enzymes are more efficient protein translocators and are more powerful unfoldases90,107.
In addition to the importance of sequence and structure variability within the motor domains in modulating biological function, functional diversity among AAA+ proteins is also achieved through non-enzymatic domains. Most AAA+ proteins contain N-terminal domains that serve as essential determinants of substrate preference and ATPase activity8,108. These N-terminal domains are highly variable across AAA+ proteins, and our understanding of the structural relationship between the ATPase spiral and the N-terminal domains remains limited. However, recent studies have provided important clues regarding how distinct N-terminal domains might differentially influence substrate processing in AAA+ proteins. For example, the N-termini of the FtsH-related AAA+ protease AFG3L2 appear to follow the rigid body domain movement of the ATPases, adopting a spiralling organization that mirrors the organization of the pore 1 loop63 (FIG. 6b). Intriguingly, this N-terminal staircase directly contacts the substrate and appears to engage and translocate the substrate concomitantly with pore loop 1. These additional contacts with the substrate likely maximize the substrate remodelling force extracted by the enzyme from each ATP hydrolysis event63.
Given that the N-termini of AAA+ proteins are much more diversified than the motor domain, it is unsurprising that the N-termini are used to recruit and engage substrates in different ways. In fact, the N-termini of several Cdc48-related AAA+ proteins have been shown to undergo major nucleotide-dependent movements along the longitudinal axis of the complex (‘up’ and ‘down’ positions) — owing to an allosteric transmission of conformational rearrangements between the motor and N-terminal domains — which have been directly linked to the ATP-dependent protein remodelling activities of these AAA+ proteins25,67,68,91. For instance, NSF unwinds oligomeric SNARE complexes by pulling the subunits apart, rather than by unfolding each individual monomer109. Recent cryo-EM structures show that, immediately above the D1–D2 ring of NSF, the N-termini form a complex with the adaptor proteins SNAPs that bind SNARE complexes68 (FIG. 1b). Although the D1 ring threads the SNARE polypeptide through the central pore, the D2 domain does not, and NSF does not appear to progressively translocate the full SNARE polypeptide67,68. Rather, the SNARE polypeptide is likely anchored within the staircase so that hydrolysis-induced longitudinal movements of the N-terminal domains along the central channel effectively pull and unwind the SNARE complex67,68,109. Such a mechanism would be more advantageous for enzymes that disassemble protein complexes or polymers without requiring global denaturation of the substrate. Given the unique role of the N-terminal domains in different AAA+ proteins, the relationship between the ATPases, the N-terminal domains and the substrate might be distinct in each case.
SNAPs.
(Soluble N-ethylmaleimide-sensitive factor (NSF) attachment proteins). adaptor proteins that bind both the N-terminal domains of type II AAA+ protein NSF and a SNARE complex, giving rise to the so-called 20S complex.
AAA+ proteins utilize different mechanisms for inter-subunit communication.
Although substrate-bound structures of protein translocases support a sequential ATP hydrolysis cycle as the main driver for protein translocation, the timing of successive ATP hydrolysis events in different AAA+ proteins remains debated. As the timing of ATP hydrolysis will directly depend on the rate at which conformational changes in one subunit are able to allosterically influence the nucleotide-binding pocket of the neighbouring subunit, it is entirely possible that different AAA+ motors exhibit widely different hydrolysis rates. In fact, previous bulk and single-molecule biochemical studies demonstrated that enzymatic properties, such as ATP hydrolysis rates, processivity and mechanochemical coupling, vary dramatically in different AAA+ proteins96. Substrate-bound cryo-EM structures of classical AAA+ proteins are now revealing different modes of inter-subunit coordination and allosteric transmission of ATP-dependent conformational changes, providing a potential molecular explanation for the remarkable operational versatility of AAA+ motors (FIG. 7). However, as mentioned above, the vast majority of the structures of substrate-bound AAA+ proteins have been determined using constructs containing hydrolysis-inactivating mutations or in the presence of non-hydrolysable ATP analogues. We are thus presented with stabilized snapshots of these dynamic enzymes trapped in an energetic minimum. As a result, the actual motions associated with hydrolysis events discussed in this section, as well as how quickly rearrangements within one subunit impact the next, can only be speculated.
Fig. 7 |. The ATP hydrolysis cycle is distinctly regulated in different AAA+ proteins.
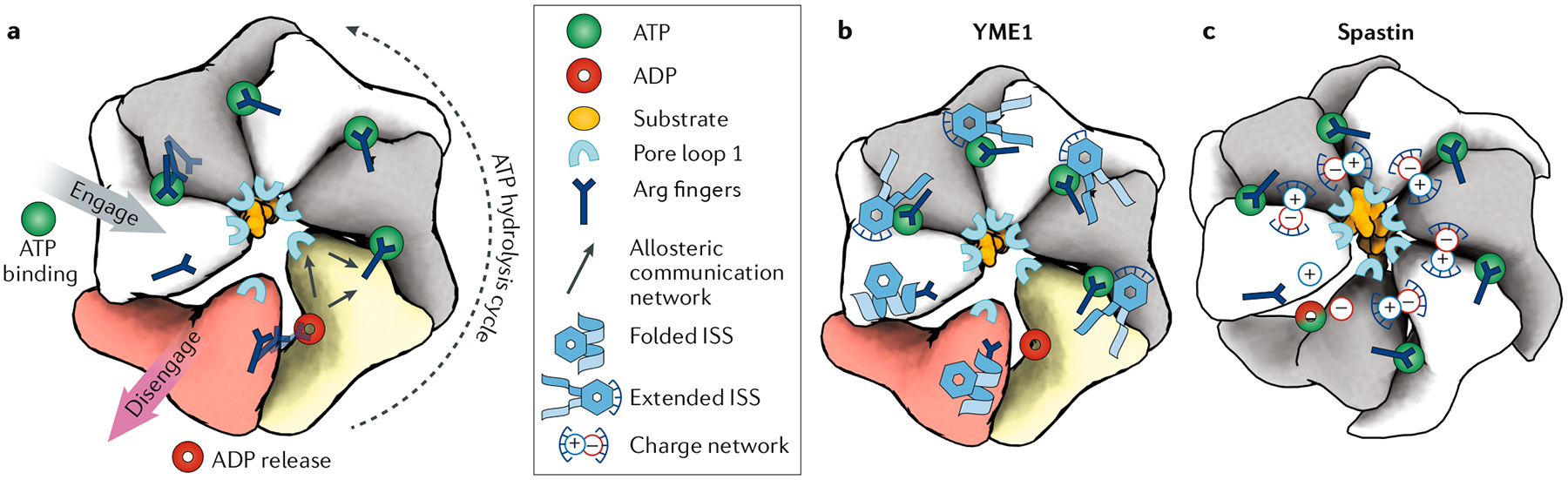
a | Schematic representation of how ATP hydrolysis in one subunit is allosterically transmitted to the pore loops of the adjacent subunit in the anticlockwise position, leading to an around-the-ring ATP hydrolysis cycle that proceeds anticlockwise through the hexamer. Molecular elements involved in this cycle are indicated. ATP-bound subunits are shown in grey and white, the ADP-bound subunit in yellow and the nucleotide-free subunit (apo state) in red. The nucleotide state of the lowermost subunit in spastin (part c) is unclear. Arginine fingers have an essential, conserved role in inter-subunit communication by coordinating the γ-phosphate in trans. Whereas the arginine (Arg) fingers are present in all classical ATPases associated with diverse cellular activities (AAA+ proteins), additional inter-subunit coordination elements give rise to distinct allosteric mechanisms (parts b and c). b | In YME1, inter-subunit coordination is based on the inter-subunit signalling (ISS) motif. This conserved motif (Asp-Gly-Phe) engages in inter-subunit hydrophobic packing and π-stacking interactions with the anticlockwise neighbouring subunit. Upon ATP hydrolysis and loss of the γ-phosphate, the ISS motif disengages from these stabilizing interactions and folds into a helix, thereby abolishing the inter-subunit contact. This conformational switch provides a mechanism for ‘sensing’ and ‘reacting to’ the nucleotide state of the neighbouring subunit. By coupling the ISS to the pore loops, this mechanism also allows the modulation of ATPase–substrate interactions by the nucleotide state. c | In spastin, individual subunits are coupled by a network of positively and negatively charged residues establishing a salt bridge network that connects pore loop 1 of one subunit to the nucleotide-binding pocket of its neighbour. This salt bridge-based allosteric mechanism does not require the reorganization of secondary structure elements. Thus, it could accommodate a fast propagation of conformational changes between subunits, potentially allowing near-simultaneous ATP hydrolysis events in all subunits.
Substrate-bound structures have confirmed that the arginine fingers have an essential, conserved role in inter-subunit communication by coordinating the γ-phosphate in trans43,62,70 (FIGS 3d,7). However, additional, non-conserved inter-subunit coordination elements have now been shown to be essential in different AAA+ proteins (FIG. 7). For instance, protein degradation AAA+ enzymes of the classical clade share a common allosteric mechanism that is based on hydrophobic interactions, mediated by a conserved inter-subunit signalling (ISS) motif62,64–66,110 (FIG. 7b; Supplementary Fig. 1). This conserved motif (Asp-Gly-Phe) forms a loop that extends across the nucleotide-binding pocket of the anticlockwise neighbouring ATP-bound subunit, and engages in inter-subunit hydrophobic packing and π-stacking interactions that lock the two subunits together62,64 (FIG. 7b). Upon ATP hydrolysis and loss of the γ-phosphate, the ISS motif disengages from these stabilizing interactions with the neighbouring subunit and folds into a helix, thereby disrupting all inter-subunit contacts62,64,66 (FIG. 7b). This conformational switch provides a mechanism for ‘sensing’ and ‘reacting to’ the nucleotide state of the neighbouring subunit as a means of propagating ATP hydrolysis sequentially around the ring62 (FIG. 7b). The ISS interacts with β-strands within the ATPase module of the neighbouring subunit, which in turn are directly connected to the substrate-interacting pore loops, so that the nucleotide state also influences ATPase–substrate interactions62. Remarkably, the structures of all processive protein degradation machines solved to date contain at least one subunit that appears to be transitioning between ADP release and ATP binding. Together, these observations support a one-at-time ATP hydrolysis cycle, where ATP hydrolysis in the lowermost subunit of the staircase is timed to coincide with ATP binding in the uppermost subunit. Such a mechanism would be ideally suited to ensure a continuous grip on the translocating substrate for processive unfolding of polypeptides.
π-stacking interactions.
Electrostatic interactions that can occur between two π systems. In proteins, aromatic residues that are in close proximity to each other can engage in such interactions.
Numerous other AAA+ proteins contain ‘vestigial’ ISS motifs (Asp-Gly-h, where h can be Leu, Met or Val; Supplementary Fig. 1). Without an aromatic residue at the turn of the ISS loop, the interactions at the subunit interface are limited to hydrophobic interactions86. These weaker ISS-mediated inter-subunit contacts are expected to decrease the energy barrier for the nucleotide-dependent conformational changes, potentially resulting in faster transmission of the conformational changes to the neighbouring subunit, and therefore a faster ATP hydrolysis cycle. Moreover, some AAA+ proteins with vestigial ISS motifs have evolved additional structural elements that seem to dominate the allosteric mechanism. This is exemplified by the microtubule-severing protein, spastin. The vestigial ISS motif in spastin does not appear to have an important role in inter-subunit communication, and instead a network of positive and negatively charged residues connects the substrate-interacting pore loop 1 of one subunit to the nucleotide-binding pocket of its anticlockwise neighbour81 (FIG. 7c). These residues were found to be required for function, indicative of a salt bridge-based mechanism of allosteric transmission and coordination of ATP hydrolysis81. Unlike the ISS motif, this salt bridge-based allosteric mechanism does not require the reorganization of secondary structure elements, and could therefore accommodate a faster propagation of conformational changes to the neighbouring subunit. A substantially faster sequential ATP hydrolysis cycle would lead to near-synchronous ATP hydrolysis within the hexamer, resulting in near-concerted rounds of ATP hydrolysis. This rapid hydrolysis could concentrate the force exerted by the enzyme into a short time frame to destabilize the microtubule lattice, and this ATP hydrolysis ‘burst’ would then be followed by a dwell period during which all subunits rebind both ATP and substrate for a new round of activity. Such a mechanism would be particularly advantageous for AAA+ proteins that, like spastin and katanin, do not processively translocate and unfold their substrates, and, instead, disassemble protein complexes or polymers, releasing the individual subunits11,92,111 (FIG. 1a).
Nucleotide hydrolysis-induced structural transitions appear to be comparatively slower in Hsp100-related type II AAA+ complexes, where ATP hydrolysis and allosteric propagation of structural events must be coordinated across two tethered ATPase domains112. Substrate-bound cryo-EM structures show that ATP hydrolysis induces a switch in the contacts between D1 and D2, such that D1–D1 contacts are lost at the seam subunit and substituted by D1–D2 contacts61,72. This nucleotide-dependent reorganization likely enables the sequential ATP hydrolysis cycle to be coordinated between both rings. In agreement, these enzymes display both homotypic (D1–D1) and heterotypic (D1–D2) regulation of the ATP hydrolysis cycle113. Notably, these enzymes lack an ISS motif. Instead, the allosteric regulation of ATP hydrolysis in the Hsp100 subfamily of complexes appears to be mediated by a four-helix insertion in D1, referred to as the ‘middle domain’114. The middle domain undergoes major nucleotide-dependent rigid body rotations that seem to be critical for coordination of ATP hydrolysis within these complexes, although the precise mechanism by which these motions influence the nucleotide-binding pockets or mediate inter-subunit interactions to coordinate the mechanochemical cycle remains unclear61,71,73,114. In addition, recent studies indicate that ATP hydrolysis is coordinated between AAA+ D1 and D2 such that the two ATPase rings work synchronously but in alternating cycles with an offset of one subunit. Such an ATP hydrolysis cycle would maximize the number of D1 and D2 domains bound to ATP within the complex, accordingly maximizing substrate interaction, at any given time73,112. Thus, the observed adaptations of inter-subunit coordination of ATP hydrolysis likely provide the molecular basis for the robust, processive unfoldase activity of Hsp100-related enzymes, which have the capacity to unfold even very stable protein aggregates.
The rate and synchrony of ATP hydrolysis is likely also directly related to the degree of operational plasticity exhibited by a given AAA+, which varies remarkably across the classical clade. For instance, incorporation of a single defective subunit into a spastin hexamer reduces activity by 50%115, reinforcing the notion that a single, strong tug generated by concerted ATP hydrolysis within all subunits of the hexamer is important for micro tubule severing. By contrast, FtsH-related AAA+ proteases containing three ATP hydrolysis-incompetent subunits retained the ability to process certain substrates, but were incapable of extracting substrates from membranes110. Interestingly, in ClpX, an AAA+ protease from the distantly related HCLR clade that does not contain an ISS motif, it was shown that only two out of six subunits needed to be functional to process less-stable substrates, suggesting that ATPase subunits can function independently116. However, degradation of stable substrates required at least four functional subunits in the ClpX hexamer116. In fact, a near-simultaneous ATP hydrolysis burst of the four functional subunits was required, presumably to trap unfolded substrate intermediate states and prevent their refolding117,118. Intriguingly, type II AAA+ chaperones Hsp104 and ClpB, which also lack the ISS motif, transition between two operational modes: a sequential, hand-over-hand ATP hydrolysis cycle is required for unfolding stable amyloid targets, but the subunits can also function independently of one another to process disordered, less-stable aggregates21,87,114. Such bimodal functionality is supported by recent cryo-EM studies of ClpB and Hsp104, which suggest that distinct interactions between the D1, D2 and middle domains can differentially influence the inter-domain cooperativity to adjust the ATP hydrolysis cycle112,119. It thus appears that AAA+ enzymes are capable of switching between different operational modes in response to different stimuli and conditions90. Nonetheless, how different modes of AAA+ activity are triggered and how these modes may differentially affect the ATP hydrolysis cycle remain poorly understood.
The divergent mechanisms for allosteric communication between the nucleotide state and inter-subunit coordination observed in recent structural studies are beginning to provide a molecular explanation for the remarkable operational versatility and diversity observed in AAA+ proteins. These mechanisms involve structural motifs and additional domains that are generally conserved within subfamilies that share a similar function, but not across AAA+ proteins that carry out different functional roles. These structural differences thus appear to have evolved to adapt the ATP hydrolysis cycle to optimally perform a specific biological activity, but the precise structure–function implications of these observations remain to be established. Importantly, currently available substrate-bound structures mostly represent a single, low-energy snapshot of the translocating state, and the mechanistic characterization of other stages required for substrate processing by AAA+ proteins is still fairly limited. For example, substrate loading must occur prior to translocation of the engaged substrate, but only low-resolution structures of substrate-free pre-engagement states have been solved to date by cryo-EM58,71. Several cycles of binding and release may also be required for complete processing of certain substrates, and different coordination modes of ATP hydrolysis might be common at such stages of AAA+ activity, as suggested by a recent study of the 26S proteasome65. As structure determination methodologies continue to advance, a more complete description of the conformational landscapes associated with substrate engagement, processing and release will continue to emerge. Deciphering the mechanisms underlying the operational plasticity of the AAA+ proteins that target polypeptide substrates will also require further combined biochemical and biophysical analyses, particularly single-molecule studies, which are currently lacking for classical AAA+ proteins.
Concluding remarks
The increasing availability of high-resolution cryo-EM structures of substrate-bound classical AAA+ proteins from archaea to humans has revealed a conserved spiralling organization of ATPase hexamers around the translocating protein substrate. This configuration is reminiscent of the quaternary organization observed for DNA and RNA translocases from distantly related clades within the AAA+ superfamily bound to nucleic acids101,120. Thus, from DNA replication to protein unfolding and degradation, the divergent biological functions of AAA+ proteins appear to converge on a core mechanistic principle: a pseudo-helical oligomeric assembly leads to formation of an ATPase spiral with a central channel through which different biopolymers can be threaded. This organization enables remodelling of protein, DNA and RNA substrates via a conserved hand-over-hand mechanism for substrate translocation, powered by a sequential ATP hydrolysis cycle.
As more substrate-bound structures of different AAA+ proteins have become available, we have begun to appreciate how unique features of each member of this family have been integrated into a core structural motif to enable distinct biological functions. As a result, the ATPase field is quickly moving towards a mechanistic understanding of the molecular principles underlying substrate specific recognition and processing in different AAA+ proteins. Cryo-EM methodologies are now advancing towards in situ structural biology. In the coming years, structures of these enzymes bound to their endogenous substrates in the cell promise to revolutionize our understanding of fundamental ATP-powered biological processes and their regulation, opening up the possibilities for specific manipulation of these molecular motors.
Supplementary Material
Acknowledgements
The authors thank M. Shin for helpful discussions. Preparation of this Review was supported by an American Heart Association predoctoral fellowship to C.P. (17PRE32910005), a National Science Foundation predoctoral fellowship to C.R.S. (2016219351) and a Pew Scholarship in the Biomedical Sciences from the Pew Charitable Trusts and the National Institutes of Health (DP2EB020402 and R21AG06169701) to G.C.L.
Footnotes
Competing interests
The authors declare no competing interests.
Supplementary information
Supplementary information is available for this paper at https://doi.org/10.1038/s41580-019-0183-6.
References
- 1.Neuwald AF, Aravind L, Spouge JL & Koonin EV AAA+: a class of chaperone-like ATPases associated with the assembly, operation, and disassembly of protein complexes. Genome Res. 9, 27–43 (1999). [PubMed] [Google Scholar]
- 2.Ogura T & Wilkinson AJ AAA+ superfamily ATPases: common structure-diverse function. Genes Cells 6, 575–597 (2001). [DOI] [PubMed] [Google Scholar]
- 3.White SR & Lauring B AAA+ ATPases: achieving diversity of function with conserved machinery. Traffic 8, 1657–1667 (2007). [DOI] [PubMed] [Google Scholar]
- 4.Lyubimov AY, Strycharska M & Berger JM The nuts and bolts of ring-translocase structure and mechanism. Curr. Opin. Struct. Biol 21, 240–248 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Erzberger JP & Berger JM Evolutionary relationships and structural mechanisms of AAA+ proteins. Annu. Rev. Biophys. Biomol. Struct 35, 93–114 (2006). [DOI] [PubMed] [Google Scholar]
- 6.Hanson PI & Whiteheart SW AAA+ proteins: have engine, will work. Nat. Rev. Mol. Cell Biol 6, 519–529 (2005). [DOI] [PubMed] [Google Scholar]
- 7.Iyer LM, Leipe DD, Koonin EV & Aravind L Evolutionary history and higher order classification of AAA+ ATPases. J. Struct. Biol 146, 11–31 (2004). [DOI] [PubMed] [Google Scholar]
- 8.Monroe N & Hill CP Meiotic Ccade AAA ATPases: protein polymer disassembly machines. J. Mol. Biol 428, 1897–1911 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roll-Mecak A & McNally FJ Microtubule-severing enzymes. Curr. Opin. Cell Biol 22, 96–103 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hartman JJ et al. Katanin, a microtubule-severing protein, is a novel AAA ATPase that targets to the centrosome using a WD40-containing subunit. Cell 93, 277–287 (1998). [DOI] [PubMed] [Google Scholar]
- 11.Roll-Mecak A & Vale RD Structural basis of microtubule severing by the hereditary spastic paraplegia protein spastin. Nature 451, 363–367 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lata S et al. Helical structures of ESCRT-III are disassembled by VPS4. Science 321, 1354–1357 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Scott A et al. Structural and mechanistic studies of VPS4 proteins. EMBO J. 24, 3658–3669 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Frickey T & Lupas AN Phylogenetic analysis of AAA proteins. J. Struct. Biol 146, 2–10 (2004). [DOI] [PubMed] [Google Scholar]
- 15.Brunger AT & DeLaBarre B NSF and p97/VCP: similar at first, different at last. FEBS Lett. 555, 126–133 (2003). [DOI] [PubMed] [Google Scholar]
- 16.Saffert P, Enenkel C & Wendler P Structure and function of p97 and Pex1/6 type II AAA+ complexes. Front. Mol. Biosci 4, 33 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Verma R, Oania RS, Kolawa NJ & Deshaies RJ Cdc48/p97 promotes degradation of aberrant nascent polypeptides bound to the ribosome. eLife 2, e00308 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vaz B, Halder S & Ramadan K Role of p97/VCP (Cdc48) in genome stability. Front. Genet 4, 60 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Torrecilla I, Oehler J & Ramadan K The role of ubiquitin-dependent segregase p97 (VCP or Cdc48) in chromatin dynamics after DNA double strand breaks. Phil. Trans. R. Soc. B Biol. Sci 372, 20160282 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Platta HW et al. Pex2 and pex12 function as protein–ubiquitin ligases in peroxisomal protein import. Mol. Cell. Biol 29, 5505–5516 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Doyle SM, Genest O & Wickner S Protein rescue from aggregates by powerful molecular chaperone machines. Nat. Rev. Mol. Cell Biol 14, 617–629 (2013). [DOI] [PubMed] [Google Scholar]
- 22.Parsell DA, Kowal AS, Singer MA & Lindquist S Protein disaggregation mediated by heat-shock protein Hsp104. Nature 372, 475–478 (1994). [DOI] [PubMed] [Google Scholar]
- 23.Parsell DA, Sanchez Y, Stitzel JD & Lindquist S Hsp104 is a highly conserved protein with two essential nucleotide-binding sites. Nature 353, 270–273 (1991). [DOI] [PubMed] [Google Scholar]
- 24.Sweeny EA & Shorter J Mechanistic and structural insights into the prion-disaggregase activity of Hsp104. J. Mol. Biol 428, 1870–1885 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bodnar N & Rapoport T Toward an understanding of the Cdc48/p97 ATPase. F1000Res. 6, 1318 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Glover JR & Lindquist S Hsp104, Hsp70, and Hsp40: a novel chaperone system that rescues previously aggregated proteins. Cell 94, 73–82 (1998). [DOI] [PubMed] [Google Scholar]
- 27.Sauer RT & Baker TA AAA+ proteases: ATP-fueled machines of protein destruction. Annu. Rev. Biochem 80, 587–612 (2011). [DOI] [PubMed] [Google Scholar]
- 28.Sauer RT et al. Sculpting the proteome with AAA(+) proteases and disassembly machines. Cell 119, 9–18 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gottesman S Proteolysis in bacterial regulatory circuits. Annu. Rev. Cell Dev. Biol 19, 565–587 (2003). [DOI] [PubMed] [Google Scholar]
- 30.Glynn SE Multifunctional mitochondrial AAA proteases. Front. Mol. Biosci 4, 34 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Livneh I, Cohen-Kaplan V, Cohen-Rosenzweig C, Avni N & Ciechanover A The life cycle of the 26S proteasome: from birth, through regulation and function, and onto its death. Cell Res. 26, 869–885 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Saeki Y Ubiquitin recognition by the proteasome. J. Biochem 161, 113–124 (2017). [DOI] [PubMed] [Google Scholar]
- 33.Bashore C et al. Ubp6 deubiquitinase controls conformational dynamics and substrate degradation of the 26S proteasome. Nat. Struct. Mol. Biol 22, 712–719 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Besche HC et al. Autoubiquitination of the 26S proteasome on Rpn13 regulates breakdown of ubiquitin conjugates. EMBO J. 33, 1159–1176 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rossmann MG, Moras D & Olsen KW Chemical and biological evolution of nucleotide-binding protein. Nature 250, 194–199 (1974). [DOI] [PubMed] [Google Scholar]
- 36.Walker JE, Saraste M, Runswick MJ & Gay NJ Distantly related sequences in the α- and β-subunits of ATP synthase, myosin, kinases and other ATP-requiring enzymes and a common nucleotide binding fold. EMBO J. 1, 945–951 (1982). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Erdmann R et al. PAS1, a yeast gene required for peroxisome biogenesis, encodes a member of a novel family of putative ATPases. Cell 64, 499–510 (1991). [DOI] [PubMed] [Google Scholar]
- 38.Saraste M, Sibbald PR & Wittinghofer A The P-loop-a common motif in ATP- and GTP-binding proteins. Trends Biochem. Sci 15, 430–434 (1990). [DOI] [PubMed] [Google Scholar]
- 39.Leipe DD, Koonin EV & Aravind L Evolution and classification of P-loop kinases and related proteins. J. Mol. Biol 333, 781–815 (2003). [DOI] [PubMed] [Google Scholar]
- 40.Wendler P, Ciniawsky S, Kock M & Kube S Structure and function of the AAA+ nucleotide binding pocket. Biochim. Biophys. Acta 1823, 2–14 (2012). [DOI] [PubMed] [Google Scholar]
- 41.Beyer A Sequence analysis of the AAA protein family. Protein Sci. 6, 2043–2058 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Karata K, Inagawa T, Wilkinson AJ, Tatsuta T & Ogura T Dissecting the role of a conserved motif (the second region of homology) in the AAA family of ATPases. Site-directed mutagenesis of the ATP-dependent protease FtsH. J. Biol. Chem 274, 26225–26232 (1999). [DOI] [PubMed] [Google Scholar]
- 43.Ogura T, Whiteheart SW & Wilkinson AJ Conserved arginine residues implicated in ATP hydrolysis, nucleotide-sensing, and inter-subunit interactions in AAA and AAA+ ATPases. J. Struct. Biol 146, 106–112 (2004). [DOI] [PubMed] [Google Scholar]
- 44.Wang J et al. Crystal structures of the HslVU peptidase–ATPase complex reveal an ATP-dependent proteolysis mechanism. Structure 9, 177–184 (2001). [DOI] [PubMed] [Google Scholar]
- 45.White SR, Evans KJ, Lary J, Cole JL & Lauring B Recognition of C-terminal amino acids in tubulin by pore loops in Spastin is important for microtubule severing. J. Cell Biol 176, 995–1005 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Martin A, Baker TA & Sauer RT Pore loops of the AAA+ ClpX machine grip substrates to drive translocation and unfolding. Nat. Struc. Mol. Biol 15, 1147–1151 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yamada-Inagawa T, Okuno T, Karata K, Yamanaka K & Ogura T Conserved pore residues in the AAA protease FtsH are important for proteolysis and its coupling to ATP hydrolysis. J. Biol. Chem 278, 50182–50187 (2003). [DOI] [PubMed] [Google Scholar]
- 48.Hinnerwisch J, Fenton WA, Furtak KJ, Farr GW & Horwich AL Loops in the central channel of ClpA chaperone mediate protein binding, unfolding, and translocation. Cell 121, 1029–1041 (2005). [DOI] [PubMed] [Google Scholar]
- 49.Weibezahn J et al. Thermotolerance requires refolding of aggregated proteins by substrate translocation through the central pore of ClpB. Cell 119, 653–665 (2004). [DOI] [PubMed] [Google Scholar]
- 50.Zolkiewski M A camel passes through the eye of a needle: protein unfolding activity of Clp ATPases. Mol. Microbiol 61, 1094–1100 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Glynn SE, Martin A, Nager AR, Baker TA & Sauer RT Structures of asymmetric ClpX hexamers reveal nucleotide-dependent motions in a AAA+ protein-unfolding machine. Cell 139, 744–756 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Olivares AO, Baker TA & Sauer RT Mechanistic insights into bacterial AAA+ proteases and protein-remodelling machines. Nat. Rev. Microbiol 14, 33–44 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lum R, Tkach JM, Vierling E & Glover JR Evidence for an unfolding/threading mechanism for protein disaggregation by Saccharomyces cerevisiae Hsp104. J. Biol. Chem 279, 29139–29146 (2004). [DOI] [PubMed] [Google Scholar]
- 54.Lee C, Schwartz MP, Prakash S, Iwakura M & Matouschek A ATP-dependent proteases degrade their substrates by processively unraveling them from the degradation signal. Mol. Cell 7, 627–637 (2001). [DOI] [PubMed] [Google Scholar]
- 55.Bai XC, McMullan G & Scheres SH How cryo-EM is revolutionizing structural biology. Trends Biochem. Sci 40, 49–57 (2015). [DOI] [PubMed] [Google Scholar]
- 56.Kuhlbrandt W Biochemistry. The resolution revolution. Science 343, 1443–1444 (2014). [DOI] [PubMed] [Google Scholar]
- 57.Cheng Y Single-particle cryo-EM at crystallographic resolution. Cell 161, 450–457 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Huang R et al. Unfolding the mechanism of the AAA+ unfoldase VAT by a combined cryo-EM, solution NMR study. Proc. Natl Acad. Sci. USA 113, E4190–E4199 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ripstein ZA, Huang R, Augustyniak R, Kay LE & Rubinstein JL Structure of a AAA+ unfoldase in the process of unfolding substrate. eLife 6, e25754 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper shows that substrate-free and substrate-bound structures of an archaeal type II AAA+ protein, VAT, reveal the conformational rearrangements associated with substrate processing, which suggests hand-over-hand substrate translocation through the central pore.
- 60.Monroe N, Han H, Shen PS, Sundquist WI & Hill CP Structural basis of protein translocation by the Vps4–Vta1 AAA ATPase. eLife 6, e24487 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gates SN et al. Ratchet-like polypeptide translocation mechanism of the AAA+ disaggregase Hsp104. Science 357, 273–279 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper shows that structures of type II AAA+ protein Hsp104 in two different substrate-bound states support a rotary mechanism for ATP hydrolysis and a hand-over-hand substrate translocation.
- 62.Puchades C et al. Structure of the mitochondrial inner membrane AAA+ protease YME1 gives insight into substrate processing. Science 358, eaao0464 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper shows that coexisting nucleotide states in the substrate-bound structure of AAA+ protease YME1 provide evidence for a sequential ATP hydrolysis cycle powering hand-over-hand substrate translocation. This study establishes the ISS motif as an allosteric modulator of both ATP hydrolysis and pore loop conformation.
- 63.Puchades C et al. Unique structural features of the mitochondrial AAA+ protease AFG3L2 reveal the molecular basis for activity in health and disease. Mol. Cell 75, 1073–1085.e6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.de la Pena AH, Goodall EA, Gates SN, Lander GC & Martin A Substrate-engaged 26S proteasome structures reveal mechanisms for ATP-hydrolysis-driven translocation. Science 362, eaav0725 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper shows that the structures of the actively hydrolysing AAA+ motor of the 26S proteasome stalled during substrate translocation provide a series of snapshots that show the different ATPase subunits proceeding along the staircase.
- 65.Dong Y et al. Cryo-EM structures and dynamics of substrate-engaged human 26S proteasome. Nature 565, 49–55 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Majumder P et al. Cryo-EM structures of the archaeal PAN-proteasome reveal an around-the-ring ATPase cycle. Proc. Natl Acad. Sci. USA 116, 534–539 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhao M et al. Mechanistic insights into the recycling machine of the SNARE complex. Nature 518, 61–67 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.White KI, Zhao M, Choi UB, Pfuetzner RA & Brunger AT Structural principles of SNARE complex recognition by the AAA+ protein NSF. eLife 7, e38888 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper presents the structure of the 20S complex (type II AAA+ protein NSF bound to its endogenous substrate, the SNARE complex, via the adaptor protein αSNAP) and shows that only D1 of NSF engages the substrate.
- 69.Twomey EC et al. Substrate processing by the Cdc48 ATPase complex is initiated by ubiquitin unfolding. Science 365, eaax1033 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper shows the structure of type II AAA+ protein Cdc48 in complex with a polyubiquitylated substrate, trapped in the process of unfolding a ubiquitin molecule.
- 70.Cooney I et al. Structure of the Cdc48 segregase in the act of unfolding an authentic substrate. Science 365, 502–505 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yokom AL et al. Spiral architecture of the Hsp104 disaggregase reveals the basis for polypeptide translocation. Nat. Struct. Mol. Biol 23, 830–837 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rizo AN et al. Structural basis for substrate gripping and translocation by the ClpB AAA+ disaggregase. Nat. Commun 10, 2393 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yu H et al. ATP hydrolysis-coupled peptide translocation mechanism of Mycobacterium tuberculosis ClpB. Proc. Natl Acad. Sci. USA 115, E9560–E9569 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ho CM et al. Malaria parasite translocon structure and mechanism of effector export. Nature 561, 70–75 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Alfieri C, Chang L & Barford D Mechanism for remodelling of the cell cycle checkpoint protein MAD2 by the ATPase TRIP13. Nature 559, 274–278 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gardner BM et al. The peroxisomal AAA-ATPase Pex1/Pex6 unfolds substrates by processive threading. Nat. Commun 9, 135 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Blok NB et al. Unique double-ring structure of the peroxisomal Pex1/Pex6 ATPase complex revealed by cryo-electron microscopy. Proc. Natl Acad. Sci. USA 112, E4017–E4025 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sun S et al. Cryo-EM structures of the ATP-bound Vps4(E233Q) hexamer and its complex with Vta1 at near-atomic resolution. Nat. Commun 8, 16064 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Han H, Monroe N, Sundquist WI, Shen PS & Hill CP The AAA ATPase Vps4 binds ESCRT-III substrates through a repeating array of dipeptide-binding pockets. eLife 6, e31324 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; This article presents an in-depth analysis of the interactions with the substrate along the central pore of the type I AAA+ protein Vps4. This study defines class I and class II substrate binding pockets established by pore loop 1 and pore loop 2, respectively.
- 80.Zehr E et al. Katanin spiral and ring structures shed light on power stroke for microtubule severing. Nat. Struct. Mol. Biol 24, 717–725 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sandate CR, Szyk A, Zehr EA, Lander GC & Roll-Mecak A An allosteric network in spastin couples multiple activities required for microtubule severing. Nat. Struct. Mol. Biol 26, 671–678 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper shows that the substrate-bound structure of type I AAA+ protein spastin reveals a salt bridge-based allosteric mechanism that connects the pore loops of one subunit to the nucleotide-binding pocket of its neighbour.
- 82.Lander GC et al. Complete subunit architecture of the proteasome regulatory particle. Nature 482, 186–191 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Matyskiela ME, Lander GC & Martin A Conformational switching of the 26S proteasome enables substrate degradation. Nat. Struct. Mol. Biol 20, 781–788 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lasker K et al. Molecular architecture of the 26S proteasome holocomplex determined by an integrative approach. Proc. Natl Acad. Sci. USA 109, 1380–1387 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Iosefson O, Nager AR, Baker TA & Sauer RT Coordinated gripping of substrate by subunits of a AAA+ proteolytic machine. Nat. Chem. Biol 11, 201–206 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Han H et al. Binding of substrates to the central pore of the Vps4 ATPase is autoinhibited by the microtubule interacting and trafficking (MIT) domain and activated by MIT interacting motifs (MIMs). J. Biol. Chem 290, 13490–13499 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Doyle SM et al. Asymmetric deceleration of ClpB or Hsp104 ATPase activity unleashes protein-remodeling activity. Nat. Struct. Mol. Biol 14, 114–122 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Barkow SR, Levchenko I, Baker TA & Sauer RT Polypeptide translocation by the AAA+ ClpXP protease machine. Chem. Biol 16, 605–612 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bell TA, Baker TA & Sauer RT Interactions between a subset of substrate side chains and AAA+ motor pore loops determine grip during protein unfolding. eLife 8, e46808 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Shorter J & Southworth DR Spiraling in control: structures and mechanisms of the Hsp104 disaggregase. Cold Spring Harb. Perspect. Biol 11, a034033 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Banerjee S et al. 2.3 A resolution cryo-EM structure of human p97 and mechanism of allosteric inhibition. Science 351, 871–875 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hartman JJ & Vale RD Microtubule disassembly by ATP-dependent oligomerization of the AAA enzyme katanin. Science 286, 782–785 (1999). [DOI] [PubMed] [Google Scholar]
- 93.Burton RE, Baker TA & Sauer RT Nucleotide-dependent substrate recognition by the AAA+ HslUV protease. Nat. Struct. Mol. Biol 12, 245–251 (2005). [DOI] [PubMed] [Google Scholar]
- 94.Weibezahn J, Schlieker C, Bukau B & Mogk A Characterization of a trap mutant of the AAA+ chaperone ClpB. J. Biol. Chem 278, 32608–32617 (2003). [DOI] [PubMed] [Google Scholar]
- 95.Dalal S, Rosser MF, Cyr DM & Hanson PI Distinct roles for the AAA ATPases NSF and p97 in the secretory pathway. Mol. Biol. Cell 15, 637–648 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Liu S, Chistol G & Bustamante C Mechanical operation and intersubunit coordination of ring-shaped molecular motors: insights from single-molecule studies. Biophys. J 106, 1844–1858 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]; This article reviews single-molecule studies of AAA+ proteins from diverse clades, highlighting the operational versatility and different mechanochemical properties of AAA+ motors.
- 97.Rodriguez-Aliaga P, Ramirez L, Kim F, Bustamante C & Martin A Substrate-translocating loops regulate mechanochemical coupling and power production in AAA+ protease ClpXP. Nat. Struct. Mol. Biol 23, 974–981 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper presents a mutational study demonstrating that the bulkiness of pore loop 1 residues influences substrate grip as well as the mechanochemical properties of AAA+ motors.
- 98.Shin M et al. Distinct structural features of the Lon protease drive conserved hand-over-hand substrate translocation. Preprint at bioRxiv 10.1101/617159 (2019). [DOI] [Google Scholar]
- 99.Gatsogiannis C, Balogh D, Merino F, Sieber S & Raunser S Cryo-EM structure of the ClpXP protein degradation machinery. Nat. Struct. Mol. Biol 26, 946–954 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Fei X et al. Structures of the ATP-fueled ClpXP proteolytic machine bound to protein substrate. Preprint at bioRxiv 10.1101/704999 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Enemark EJ & Joshua-Tor L Mechanism of DNA translocation in a replicative hexameric helicase. Nature 442, 270–275 (2006). [DOI] [PubMed] [Google Scholar]
- 102.Thomsen ND & Berger JM Running in reverse: the structural basis for translocation polarity in hexameric helicases. Cell 139, 523–534 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Itsathitphaisarn O, Wing RA, Eliason WK, Wang J & Steitz TA The hexameric helicase DnaB adopts a nonplanar conformation during translocation. Cell 151, 267–277 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Gai D, Zhao R, Li D, Finkielstein CV & Chen XS Mechanisms of conformational change for a replicative hexameric helicase of SV40 large tumor antigen. Cell 119, 47–60 (2004). [DOI] [PubMed] [Google Scholar]
- 105.Maillard RA et al. ClpX(P) generates mechanical force to unfold and translocate its protein substrates. Cell 145, 459–469 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Johjima A et al. Microtubule severing by katanin p60 AAA+ ATPase requires the C-terminal acidic tails of both α- and β-tubulins and basic amino acid residues in the AAA+ ring pore. J. Biol. Chem 290, 11762–11770 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Olivares AO, Nager AR, Iosefson O, Sauer RT & Baker TA Mechanochemical basis of protein degradation by a double-ring AAA+ machine. Nat. Struct. Mol. Biol 21, 871–875 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Djuranovic S et al. Structure and activity of the N-terminal substrate recognition domains in proteasomal ATPases. Mol. Cell 34, 580–590 (2009). [DOI] [PubMed] [Google Scholar]
- 109.Ryu JK et al. Spring-loaded unraveling of a single SNARE complex by NSF in one round of ATP turnover. Science 347, 1485–1489 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Augustin S et al. An intersubunit signaling network coordinates ATP hydrolysis by m-AAA proteases. Mol. Cell 35, 574–585 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]; This article presents the first biochemical study to identify the ISS motif and describe its functional relevance.
- 111.McNally FJ & Vale RD Identification of katanin, an ATPase that severs and disassembles stable microtubules. Cell 75, 419–429 (1993). [DOI] [PubMed] [Google Scholar]
- 112.Deville C, Franke K, Mogk A, Bukau B & Saibil HR Two-step activation mechanism of the ClpB disaggregase for sequential substrate threading by the main ATPase motor. Cell Rep. 27, 3433–3446.e4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hattendorf DA & Lindquist SL Cooperative kinetics of both Hsp104 ATPase domains and interdomain communication revealed by AAA sensor-1 mutants. EMBO J. 21, 12–21 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.DeSantis ME et al. Operational plasticity enables hsp104 to disaggregate diverse amyloid and nonamyloid clients. Cell 151, 778–793 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Eckert T et al. Subunit Interactions and cooperativity in the microtubule-severing AAA ATPase spastin. J. Biol. Chem 287, 26278–26290 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Martin A, Baker TA & Sauer RT Rebuilt AAA+ motors reveal operating principles for ATP-fuelled machines. Nature 437, 1115–1120 (2005). [DOI] [PubMed] [Google Scholar]
- 117.Martin A, Baker TA & Sauer RT Protein unfolding by a AAA+ protease is dependent on ATP-hydrolysis rates and substrate energy landscapes. Nat. Struct. Mol. Biol 15, 139–145 (2008). [DOI] [PubMed] [Google Scholar]
- 118.Sen M et al. The ClpXP protease unfolds substrates using a constant rate of pulling but different gears. Cell 155, 636–646 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Lee S et al. Cryo-EM structures of the Hsp104 protein disaggregase captured in the ATP conformation. Cell Rep. 26, 29–36.e3 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Moffitt JR et al. Intersubunit coordination in a homomeric ring ATPase. Nature 457, 446–450 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.