

Abstract
Hematopoietic stem cell transplantation-associated thrombotic microangiopathy (HSCT-TMA) is a common complication occurring post-HSCT and is associated with substantial morbidity and mortality if not promptly identified and treated. Emerging evidence suggests a central role for the complement system in the pathogenesis of HSCT-TMA. The complement system has also been shown to interact with other pathways and processes including coagulation and inflammation, all of which are activated following HSCT. Three endothelial cell-damaging “hits” are required for HSCT-TMA genesis: a genetic predisposition or existing damage, an endothelial cell-damaging conditioning regimen, and additional damaging insults. Numerous risk factors for the development of HSCT-TMA have been identified (including primary diagnosis, graft type, and conditioning regimen) and validated lists of relatively simple diagnostic signs and symptoms exist, many utilizing routine clinical and laboratory assessments. Despite the relative ease with which HSCT-TMA can be screened for, it is often overlooked or masked by other common post-transplant conditions. Recent evidence that patients with HSCT-TMA may also concurrently present with these differential diagnoses only serve to further confound its identification and treatment. HSCT-TMA may be treated, or even prevented, by removing or ameliorating triggering “hits”, and recent studies have also shown substantial utility of complement-targeted therapies in this patient population. Further investigation into optimal management and treatment strategies is needed. Greater awareness of TMA post-HSCT is urgently needed to improve patient outcomes; the objective of this article is to clarify current understanding, explain underlying complement biology and provide simple tools to aid the early recognition, management, and monitoring of HSCT-TMA.
Supplementary Information
The online version contains supplementary material available at 10.1007/s12325-022-02184-4.
Keywords: Hematopoietic stem cell transplantation, Thrombotic microangiopathy, Complement, HSCT-TMA
Key Summary Points
| Thrombotic microangiopathy (TMA) is a common complication occurring post-hematopoietic stem cell transplantation (HSCT). It causes microvascular thrombosis, leading to thrombocytopenia, ischemic tissue damage, and microangiopathic hemolytic anemia |
| HSCT-TMA is an under-recognized condition associated with substantial morbidity and mortality, with greater awareness of the condition required to improve outcomes |
| The complement system appears to play a key role in the pathogenesis of TMA following HSCT |
| Key panels of risk factors and simple diagnostic and monitoring criteria exist to help identify, diagnose, and monitor patients with HSCT-TMA, and are summarized in this manuscript |
| Prompt management of HSCT-TMA is associated with improved outcomes and complement-targeted therapies also show utility in this population, although further randomized clinical trials and research into long-term outcomes are needed |
Digital Features
This article is published with digital features, including a video and infographic, to facilitate understanding of the article. To view digital features for this article go to https://doi.org/10.6084/m9.figshare.19723117.
Introduction
Thrombotic microangiopathy (TMA) is defined as a histopathologic lesion associated with endothelial damage and dysfunction, which leads to thrombosis of venules and arterioles [1–3]. This type of microvascular thrombosis can result in thrombocytopenia, ischemic tissue/organ damage, and microangiopathic hemolytic anemia (MAHA). The term TMA has been used to describe various conditions presenting as this triad [1–3]. Historically, thrombotic thrombocytopenic purpura (TTP) and the various forms of hemolytic uremic syndrome (HUS)—such as Shiga toxin-producing Escherichia coli-associated HUS (STEC-HUS) and atypical HUS (aHUS)—have perhaps been the best studied and defined forms of TMA, with the clearest etiologies [1, 4].
Historically, many forms of TMA with unknown or unclear etiology or pathophysiology have been classified as forms of aHUS [5]. More recently, the utility of the term “aHUS” has been questioned as other forms of TMA have begun to be recognized clinically. These include chemotherapy-induced TMA, malignancy-induced TMA, and TMA identified following hematopoietic stem cell transplantation (HSCT-TMA) [1, 6–8]. HSCT-TMA appears—at least in part—to belong to the group of complement-mediated TMAs (CM-TMA), as there is emerging evidence of complement involvement in this condition [8–10]. However, the extent of complement involvement in CM-TMAs (direct vs. indirect), importance of triggering events, and involvement of specific pathways are currently unclear.
Recent studies, including a pragmatic, multi-institutional study, have suggested that HSCT-TMA affects around 10–20% of patients following transplantation. This makes it a relatively common post-transplant complication, with some variance seen depending upon factors such as the type of transplantation (e.g., allogenic vs. autologous), type of conditioning regimen (e.g., high-dose vs. non-myeloablative), and underlying primary diagnosis [11, 12]. Potential triggers of complement activation and/or endothelial damage following an HSCT procedure include (among others) the conditioning regimen used, post-transplant complications such as graft versus host disease (GvHD), the type of transplant (e.g., mismatched donor transplants), and viral infections (e.g., BK virus) [8, 13]. Importantly, the mortality rate in patients with HSCT-TMA is extremely high, and patients who do survive are at a great risk of severe organ damage [11, 14–16]. Yet, despite advances in our understanding of the risks posed by HSCT-TMA, questions remain around diagnostic methodologies, risk factors, differences in presentation between pediatric and adult patients, possible treatment choices, and monitoring criteria.
The aims of this review article are to:
provide a background on the complement system, including its physiologic functions/interactions and its pathophysiologic roles, with a focus on HSCT-TMA
explore the clinical presentation of HSCT-TMA
provide clinicians with a practical approach to recognizing and monitoring patients at risk of/with HSCT-TMA
highlight and discuss current misunderstandings and unknowns in the field.
As this is a review article, it is based on previously conducted studies and does not contain any new studies with human participants or animals performed by any of the authors.
The Complement System
What Is the Complement System and What Is Its Physiologic Role?
The complement cascade is a facet of the innate immune system composed of both plasma- and membrane-bound proteins [17, 18]. It is involved in processes including maintenance of homeostasis via opsonization of apoptotic/necrotic cells, clearance of foreign cells through cytolysis, and induction of inflammation [17, 18]. Alongside these key roles, the complement system interacts with many other physiologic pathways and processes, such as the coagulation cascade, the kinin system, the adaptive immune system, and inflammation [17, 19–22]. There are three distinct pathways involved in initiation of the complement system: the classical pathway (CP); the lectin pathway (LP); and the alternative pathway (AP). All initiator pathways culminate in activation of the terminal complement pathway (TP) and formation of C5b-9, also known as the membrane attack complex (MAC) [17, 18]. The video associated with this manuscript provides a detailed overview of the full complement cascade, including activation signals, key regulators, end effectors, and key physiologic and pathologic interactions.
Video: The role of complement in HSCT-TMA: basic science to clinical practice- A Video (MP4 458780 KB)
Importantly, a key feature of the AP is the amplification loop. C3 cleavage via all complement pathways results in increased assembly of the AP C3 convertase, C3bBb, which can further activate nearby C3 molecules, leading to a rapid, exponential amplification of complement activation [17, 18]. Amplification occurs by default on surfaces that cannot prevent complement activation but can also occur in certain pathologi conditions, where excessive activation overwhelms regulatory capacity. The AP amplification loop is reportedly a core process following initiation by both the CP and LP as well as by the AP [23, 24]. Additionally, this exponential amplification leads to rapid generation of the anaphylatoxins C3a and C5a, resulting in induction of inflammation (including inducing inflammatory cytokine production via endothelial cells), immune cell infiltration and activation, and interactions with the coagulation cascade [25].
As well as its role in infection control, there is strong evidence for a role of complement in adaptive immunity alongside its innate immunity functions, with complement components seemingly able to tag and deliver antigens to dendritic cells, macrophages, and B cells for processing and presentation to T cells [26]. Despite key physiologic roles in tissue homeostasis and clearance of unwanted cells/materials, the complement system has also been implicated as the driving force behind numerous pathologic conditions. For example, in aHUS, acquired or inherited dysregulation of the AP results in direct activation and damage of endothelial cells, resulting in the exposure of pro-thrombotic structures and subsequent thrombosis and hemolysis [17, 18, 27, 28].
Complement, Coagulation, and Inflammation
Outside of its direct roles in homeostasis and immunity, key interactions between the complement system and other physiologic systems and processes, such as the coagulation cascade and inflammation, have been demonstrated to occur at many different levels [17, 19, 20]. While the complement and coagulation systems are generally viewed as completely distinct cascades, there is significant evidence to demonstrate cross talk between them, though an in-depth discussion on this is beyond the scope of this publication and the topic has been reviewed elsewhere [29–34]. However, an important point to note is that complement can promote the induction of a procoagulant state in both blood and endothelial cells. C3a and C5b-9 can activate platelets—which in turn can induce complement activation—forming a potent amplification loop [32, 35, 36]. Additionally, C5a has been shown to mediate interactions between complement, coagulation, and immunity, upregulating tissue factor expression on a variety of cells, including monocytes, neutrophils, and endothelial cells [29–31]. Procoagulant enzymes, such as thrombin and factor Xa, have been shown to cleave components of the AP in vitro, while the substrate specificity of MASP1 is reportedly like that of thrombin; MASP1 may also be capable of catalyzing fibrin cross-linking [30, 33, 34]. However, some of the non-canonical enzymatic interactions are reportedly less efficient than the prototypical enzyme–substrate interactions, and the lack of specific animal models of TMA further confounds assessment of their relevance in HSCT-TMA. Further investigations, particularly in the clinical setting, are required to understand their clinical relevance and importance in vivo [19, 29, 30, 37, 38].
Unlike the well-established evidence for complement–coagulation interactions, evidence for cross talk between complement and inflammation is still emerging [32, 39]. Studies suggest that complement may play a key role in activating neutrophils, enhancing the release of neutrophil extracellular traps (NETs) in a process known as NETosis [40–42]. NETs may also serve as a scaffold for thrombin generation and AP activation [42]. Further evidence suggests that complement may interact directly with a range of other inflammatory cells such as monocytes and macrophages.
In vitro studies have suggested that stimulation of human endothelial cell lines via tumor necrosis factor α and/or interleukin-1β results in reduced expression of thrombomodulin and increased production of C3 and factor B [43]. In contexts such as pre-existing AP dysregulation or excessive inflammation, these responses may contribute to the development of TMA in vivo. Specifically, in the context of HSCT-TMA, strong inflammatory responses are often seen in response to infections or the development of GvHD, providing an environment conducive to TMA development [8, 44].
Complement and HSCT-TMA Pathogenesis
Following HSCT procedures, patients often experience infections, challenges to the coagulation system, and pro-inflammatory stimuli, all of which may influence the complement system, induce endothelial damage, and—if left unchecked—contribute to the generation of TMA [44–46]. Importantly, in patients with HSCT-TMA, there is a requirement for three different TMA-promoting stimuli to occur to trigger disease; this is known at the “three hits hypothesis” [8, 13]. These three triggering stimuli include the presence of a pre-existing genetic disposition or endothelial injury (hit one), followed by two TMA-promoting factors, an endothelial-damaging conditioning regimen (hit two) and additional endothelial damaging insults (hit three) [8, 13]. While patients with HSCT-TMA may often present with complement dysregulation, akin to patients with other CM-TMAs, such as aHUS, subsequent vascular endothelial damage typically results from insults such as immunosuppressive drugs (i.e., calcineurin or mammalian target of rapamycin [mTOR] inhibitors), viral infections, human leukocyte antigen (HLA) mismatching, and GvHD [8, 11, 13, 14, 47]. While many patients undergoing HSCT will experience hits two and/or three, these only seem able to trigger TMA in patients who also have hit one.
At present, the direct evidence for a central pathogenic role of complement activation in HSCT-TMA remains relatively limited. However, data from some of the largest studies systematically analyzing complement activation to date suggest that approximately 60–80% of patients show evidence of systemic terminal pathway activation (Gantner et al. personal communication, 2022) [48]. In a smaller number of patients with no evidence of systemic complement activation, localized complement activation can be demonstrated by immunohistology in organ biopsies [47]. Further, recent studies assessing the genetic profiles of patients with HSCT-TMA have identified many variants in key components of the complement pathway, particularly in genes coding for AP components [9, 49]. This may be particularly important because of the central role that the AP plays in amplifying complement activation, as discussed above. Some evidence, albeit mostly from preclinical in vitro or animal sources, also exists to suggest that the CP and LP may be involved in the development of HSCT-TMA [50, 51]. The applicability of these findings to HSCT-TMA in humans is currently unclear, however [50, 52–60].
Many patients with HSCT-TMA harbor pathogenic genetic variants (65% of HSCT recipients with TMA vs. 9% of patients without TMA) [9]. However, it is important to note that the genetic fingerprint of susceptibility in patients with HSCT-TMA is extremely complex, with multiple variants often combining to contribute to pathogenesis. Genetic variants in HSCT-TMA also do not appear limited to complement components; indeed, some patients with HSCT-TMA have also presented with variants in genes associated with other forms of TMA, such as ADAMTS13, or in components of the coagulation cascade. Therefore, in the absence of standardized genetic screening panels and interpretation guidance, genetic results should be interpreted cautiously, and the roles of either pre-existing or emerging autoantibodies against individual complement components in HSCT-TMA merit further studies.
Lastly, despite discussions around their relative contributions, the concurrent involvement of all three complement pathways may distinguish HSCT-TMA from other forms of CM-TMA. Other typical causes of CM-TMA mainly appear to involve uncontrolled or misdirected AP attack [9, 51, 61–63]. Further, the magnitude of systemic complement activation and inflammation may be useful for identifying HSCT-TMA. For example, patients presenting with HSCT-TMA have much higher levels of soluble C5b-9 (sC5b-9) compared to patients with other forms of TMA [9, 63]. Coupled with the multi-pathway involvement, this has been suggested as an underlying rationale for the high proportion of multi-system involvement and severe disease observed in patients with HSCT-TMA [9, 63].
HSCT-TMA: Diagnosis
What Are the Risk Factors for the Development of HSCT-TMA?
Patients who present with TMA following HSCT are at a much greater risk of severe organ damage and/or death than patients who have received a transplant but did not develop TMA [11, 64]. Despite this, if patients at risk are identified early enough, and appropriate therapeutic strategies are implemented, the prognosis of patients with HSCT-TMA improves dramatically [14]. Several studies have been conducted to identify potential risk factors that predispose patients to developing HSCT-TMA following transplantation, including a pragmatic, multi-institutional study by Dandoy et al. [11]. An easily referenceable list of key risk factors is summarized in the diagnostic algorithm presented in the infographic of this review (Fig. 1). Further lists of risk factors validated in pediatric patients stratified by inherent/non-modifiable, transplant-associated and post-transplant events can also be found in the review by Dvorak et al. [13]. These risk factors are relatively intuitive, with the majority clearly associated with damage to endothelial cells.
Fig. 1.
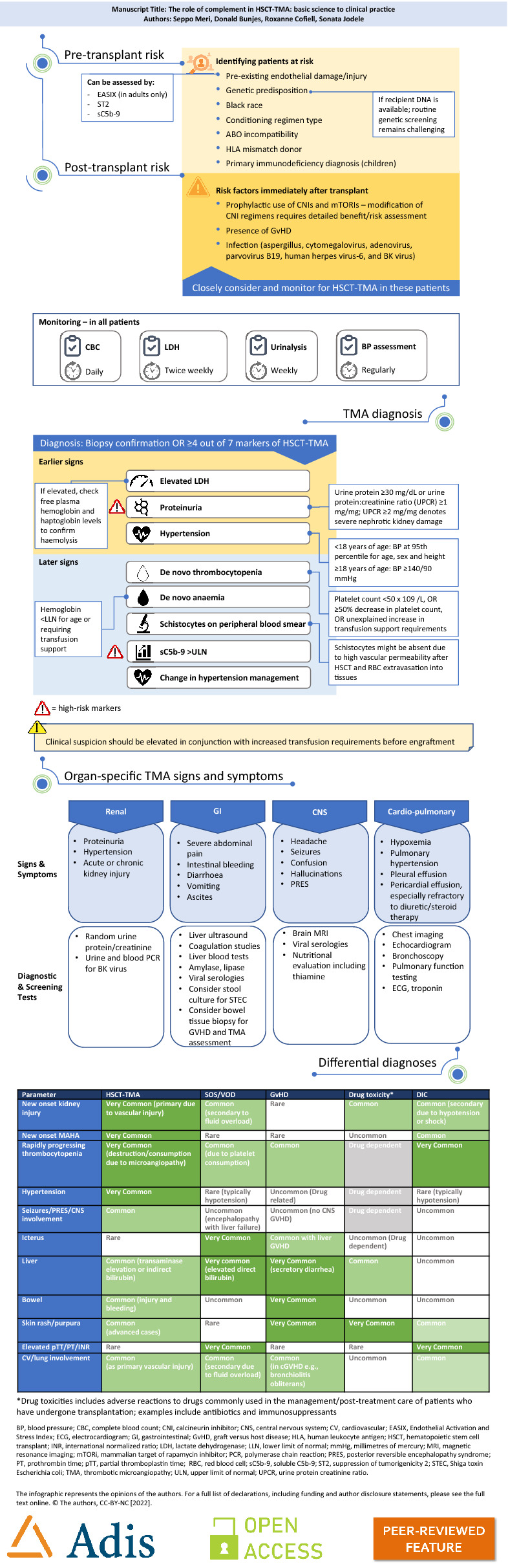
Diagnostic Algorithm Infographic
HSCT-TMA: How Can it Be Identified?
The most important consideration for optimized outcomes in patients with HSCT-TMA is early identification. Since a large proportion of patients undergoing HSCT experience TMA, and given that laboratory testing is inexpensive and widely available, prospective screening at all transplant centers is essential to improving patient outcomes [11]. Further, when HSCT-TMA is suspected, or in patients presenting with risk factors, there are several key signs and symptoms that should be considered and assessed [8, 10]. These are also summarized in the diagnostic algorithm presented in the infographic. This algorithm, generated using published literature and the authors’ clinical experiences, is easy to refer to and contains pre- and post-transplant risk factors for development of TMA, key monitoring tests and timings, a diagnostic panel based on that published by Jodele et al. in 2015, and organ-specific signs and symptoms of HSCT-TMA.
Some patients with HSCT-TMA may present with mild disease, often limited to hematologic dysfunction. In patients with more serious TMA, severe multiorgan damage and/or organ failure may be observed [10]. Close monitoring of kidney function is important in patients at risk of developing HSCT-TMA, but changes in creatinine clearance may occur too late for optimal treatment initiation, particularly in pediatric patients; monitoring for kidney-related manifestations such as hypertension and proteinuria, alongside more extensive blood panel assessments, should be considered [10]. Seemingly specifically to HSCT-TMA, organ endothelial damage has also been linked to the further development of TMA in the gastrointestinal (GI) tract [8]. While some groups have seen that the Endothelial Activation and Stress Index (EASIX) shows some utility as a prognostic factor for HSCT-TMA in adults—particularly when assessed prior to conditioning—this measure was established and validated for identification of GvHD and VOD, not TMA; EASIX also appears to perform poorly in pediatric patients, meaning more specific tools for measuring endothelial damage/dysfunction are required for patients with HSCT-TMA [65].
Another key measurement in patients with suspected HSCT-TMA is the level of sC5b-9, a key marker of terminal complement activity substantially elevated in most patients with HSCT-TMA [14]. However, measurable plasma sC5b-9 levels are indicative of systemic complement activation, meaning normal levels may not exclude organ-specific TMA or early disease. Further, individualized changes in sC5b-9 levels may be more suitable and informative than absolute levels when assessing systemic complement activation [14]. Unfortunately, while there are a variety of complement function tests available, many have associated technical difficulties, and there appears to be little consistency in the specific tests completed between laboratories. It would be important to have simple and robust tests for measuring complement activation that are standardized between laboratories. As more laboratories begin to adopt the use of biomarkers such as sC5b-9 in HSCT-TMA, testing capability and availability should improve [47, 66, 67]. Indeed, many groups, such as the International Union of Immunological Sciences and International Complement Society (IUIS/ICS) Committee for the Standardization and Quality Assessment in Complement Measurements, have been working in this direction [68]. Others have also suggested that biomarkers, such as the factor B activation product Ba, may show utility in HSCT-TMA [69]. Identification and validation of individual/panels of rapid and simple to identify HSCT-TMA-specific diagnostic and prognostic biomarkers, and associated assays, with acceptable sensitivity and specificity would be of great interest in HSCT-TMA.
Furthermore, while many patients with HSCT-TMA present with genetic variants across a range of pathways—including the complement cascade—the requirement for pre-transplant patient blood samples, insufficient knowledge of variant significance and long turnaround times limit the utility of genetic screening in these patients. Once patients have suspected HSCT-TMA, treatment needs to be initiated rapidly to optimize outcomes and reduce the morbidity/mortality associated with uncontrolled disease; treatment should not be delayed for genetic screening to be completed.
Lastly, in some cases, patients with HSCT-TMA may present atypically, with relatively normal blood counts and parameters but with isolated, unexplained organ dysfunction—again, the kidneys are most commonly affected. In contexts such as this, clinicians should note any recurrent serositis, peripheral neuropathy and muscle weakness, and/or lung dysfunction (hypoxemia), which are key signs suggestive of HSCT-TMA [8, 10]. If there is a strong suspicion of HSCT-TMA despite no evidence of systemic complement activation (e.g., normal sC5b-9), an organ biopsy with staining for C4d and C5b-9 should be considered where possible. If a thorough risk–benefit assessment has been conducted and biopsies are available, evidence of TMA in biopsy samples should be considered as a definitive diagnosis of HSCT-TMA irrespective of the presence of other markers or laboratory assessments. In cases of atypical presentation, differential diagnoses should also be considered carefully because of the similarities of signs and symptoms alongside the non-specific presentation of TMA [13].
Complement in HSCT-TMA: Differential Diagnoses and Comorbid Conditions
As noted, there are numerous post-transplant conditions that share key signs and symptoms with HSCT-TMA and may confound diagnosis. Indeed, misdiagnosis remains relatively common, as it can often be difficult to differentiate patients with HSCT-TMA from patients with other transplant-specific complications. Differential diagnoses and their associated presentations can be found in Table 1. The current body of evidence suggests that patients with HSCT-TMA share more commonalities with patients who have aHUS than with other TMA-associated conditions. For example, some patients present with reduced ADAMTS13 levels, but not to the same extent as in patients with TTP. Further, von Willebrand factor patterns tend to be unaltered in patients with HSCT-TMA, unlike in TTP. A more common feature in HSCT-TMA is complement dysregulation [8, 10, 14]. However, patients with HSCT-TMA typically do not have recurrent disease following successful treatment, as opposed to patients with aHUS where disease recurrence is relatively common; the triggers and underlying pathophysiology of these conditions also tend to differ. Regarding pathogenic genetic variants, different sets of genes appear implicated in the development of HSCT-TMA compared to other forms of CM-TMA [9].
Table 1.
Simple differentiators between HSCT-TMA and other common post-transplant complications
| Parameter | HSCT-TMA | SOS/VOD | GvHD | Drug toxicity* | DIC |
|---|---|---|---|---|---|
| New onset kidney injury | Very common (primary due to vascular injury) | Common (secondary to fluid overload) | Rare | Common | Common (secondary due to hypotension or shock) |
| New onset MAHA | Very common | Rare | Rare | Uncommon | Common |
| Rapidly progressing thrombocytopenia | Very common (destruction/consumption due to microangiopathy) | Common (due to platelet consumption) | Common | Drug dependent | Very common |
| Hypertension | Very common | Rare (typically hypotension) | Uncommon (drug related) | Drug dependent | Rare (typically hypotension) |
| Seizures/PRES/CNS involvement | Common | Uncommon (encephalopathy with liver failure) | Uncommon (no CNS GvHD) | Drug dependent | Uncommon |
| Icterus | Rare | Very common | Common with liver GvHD | Uncommon (drug dependent) | Uncommon |
| Liver | Common (transaminase elevation or indirect bilirubin) | Very common (elevated direct bilirubin) | Very common (secretory diarrhea) | Common | Uncommon |
| Bowel | Common (injury and bleeding) | Uncommon | Very common | Uncommon | Uncommon |
| Skin rash/purpura | Common (advanced cases) | Rare | Very common | Very common | Common |
| Elevated pTT/PT/INR | Rare | Very common | Rare | Rare | Very common |
| CV/lung involvement | Common (as primary vascular injury) | Common (secondary due to fluid overload) | Common (in cGvHD, e.g., bronchiolitis obliterans) | Uncommon | Common |
CNS central nervous system, CV cardiovascular, DIC disseminated intravascular coagulation, GvHD graft versus host disease, INR international normalized ratio, MAHA microangiopathic hemolytic anemia, PRES posterior reversible encephalopathy syndrome, PT prothrombin time, pTT partial thromboplastin time, VOD veno-occlusive disease
*Drug toxicities include adverse reactions to drugs commonly used in the management/post-treatment care of patients who have undergone transplantation, e.g., antibiotics and immunosuppressants [80, 81]
Other transplant-related conditions that may be considered as differential diagnoses when assessing HSCT-TMA are acute GvHD and liver SOS (sometimes also known as veno-occlusive disease [VOD]). The signs and symptoms of GvHD, particularly in the GI tract, may coincide with HSCT-TMA, and as many as one-third of patients presenting with GI dysfunction following HSCT may have both HSCT-TMA and GvHD [1, 8, 70–74]. In these cases, patients should be treated for both conditions concurrently to obtain optimal outcomes. Careful monitoring of TMA worsening is also required if immunosuppressives with known endothelial damaging properties are used to treat GvHD [14, 73, 74]. While TMA and GvHD both affect the GI tract, skin, and the liver, GvHD does not usually involve kidney, lung, or CNS—all common sites of TMA [75]. Further, the presence of vascular damage and bleeding, particularly in the GI tract, appears much more common in patients with TMA. Patients with HSCT-TMA are more likely to present with vascular endothelial damage, whilst patients with GvHD are more likely to have identifiable epithelial injury [48, 76]. There is also some emerging evidence that patients presenting with steroid refractory GvHD may actually have comorbid GvHD and TMA; these patients should be assessed carefully to ensure optimal therapeutic regimens are established [48, 77]. These findings further emphasize the current calls in the literature for endothelial-sparing immunosuppressives in the future [14, 73, 74].
Liver SOS (VOD) is another condition that also presents with some similarities to HSCT-TMA. Both conditions can result in organ injury, although the sites and underlying mechanisms of damage often differ; SOS/VOD rarely affects the GI tract and causes secondary damage to the kidneys and/or lungs due to liver damage-associated fluid overload [13]. SOS/VOD is often associated with rapid weight gain, rising bilirubin levels (direct), and liver damage. HSCT-TMA less commonly results in weight gain or changes in bilirubin [13, 78, 79].
HSCT-TMA: Differences Between Children and Adults
Currently, most HSCT-TMA research has been conducted in pediatric patients, despite the incidence of HSCT-TMA in adults appearing similar to that in pediatric patients; HSCT-TMA may be an underappreciated and under-researched condition in adult transplant patients [14–16]. However, despite very similar underlying mechanisms of TMA, there are some key differences between adult and pediatric patients with HSCT-TMA, which must be considered.
Firstly, the underlying diagnoses of adult and pediatric patients requiring transplantation are typically different. Immunodeficiencies are more common reasons for transplantation in pediatric patients, while adult patients typically require transplantation for hematologic malignancies [16, 47]. This is particularly important because of the emerging evidence suggesting that the underlying primary pathology may impact the risk of developing HSCT-TMA [14, 47]. It is important to consider that adults are more likely to present with comorbid conditions, such as hypertension or diabetes, which may contribute to development of HSCT-TMA or further complicate disease progression or treatment decisions [16]. Certain comorbid conditions may also delay consideration or diagnosis of HSCT-TMA if the symptoms of TMA are attributed to a different underlying condition.
Furthermore, the timing of HSCT-TMA appears different between adult and pediatric patients. HSCT-TMA occurs early after transplantation in pediatric settings (typically within the first 28 days post-transplant), while in a study of adult patients, approximately half of HSCT-TMA events occurred “early” (within 100 days of transplant), and half occurred “late” (after 100 days post-transplant) and were often associated with chronic GvHD or late infections [11, 82]. A recent study of adult patients undergoing allogenic HCT showed that HSCT-TMA in the absence of acute GvHD occurs earlier than 100 days, and that comorbid acute GvHD is a key risk factor for late-onset HSCT-TMA in adults [83]. In an unpublished screening study using the Jodele criteria in adult patients, the median time from transplant to the diagnosis of HSCT-TMA was 7 months (Gantner et al. personal communication, 2022).
While it is important to appreciate the differences between adult and pediatric patients, current evidence collected in the pediatric setting suggests that young adults who have received a transplant have an almost identical presentation of TMA to younger children undergoing transplantation [14]. This includes similarities in complement activation, particularly in relation to changes in sC5b-9 levels. Combined with the limited data available in adult transplant recipients, it appears that the incidence and underlying pathophysiology of HSCT-TMA may not be age-dependent, making many of the tools developed for diagnosing and managing HSCT-TMA in pediatric patients potentially applicable to the adult setting [14, 16]. More research would be welcomed to fully validate these tools for use in adult patients.
Treatment and Monitoring
HSCT-TMA: Treatment Decisions
Initially, it may be suitable to implement prevention strategies, such as HLA-matched transplantation, the avoidance of endothelial-toxic medications (i.e., mTOR inhibitors), use of conditioning regimens with reduced toxicity, and strong infection-control. In lower risk patients, adoption of risk-mitigation strategies may result in prevention of HSCT-TMA development entirely [8, 13].
In patients who do develop HSCT-TMA, initial treatment options should include treatment of co-existing conditions such as GvHD (which may exacerbate TMA symptoms), aggressive treatment of co-existing infections, renal replacement therapy, and management of symptoms such as hypertension [8, 10, 13, 14]. In certain clinical situations, modification of endothelial-damaging immunosuppressive regimens may be considered, although there is a substantial risk of triggering GvHD in this context and so careful risk–benefit assessments should be conducted before any modifications. If patients present with several risk factors for severe disease or worse outcomes, or HSCT-TMA progresses despite implementation of supportive therapeutic options, more intensive treatments should be implemented. Therapies including rituximab and defibrotide have been used to treat patients with HSCT-TMA, however neither has been thoroughly investigated in large phase III clinical trials and existing evidence suggests that outcomes are suboptimal [8]. Further, while plasma exchange may be considered in this instance, outcomes are suboptimal compared to patients treated with targeted complement inhibitor therapies [8].
As a result of the involvement of all complement pathways in the pathogenesis of HSCT-TMA, there is a clear and growing body of evidence demonstrating that complement inhibition at the level of C5 appears to be efficacious in a large proportion of patients with HSCT-TMA, improving overall response and survival rates [8, 84–90]. However, not all patients appear to respond appropriately to C5 inhibition, and this may be due to late therapy initiation, the need for increased dosing compared to established aHUS regimens, and the substantial difference in clearance observed in patients with active HSCT-TMA treated with C5 inhibitors [8, 48, 84–89]. A recent study detailing pharmacokinetic (PK)-/pharmacodynamic (PD)-guided eculizumab dosing regimens for pediatric patients with HSCT-TMA demonstrated substantially improved 1-year survival rates when eculizumab dose was guided by baseline sC5b-9 levels [48, 66]. Other studies have proposed that substantial bleeding in patients with TMA may also contribute to eculizumab refractoriness [91–93].
Furthermore, immunosuppressed patients treated with eculizumab are particularly sensitive to meningococcal (Neisseria meningitidis) infections because of an inability to remove the bacteria by the complement membrane attack complexes. Therefore, vaccination against meningococci is mandatory [94]. Interestingly, a recent study has also shown that the use of appropriate antimicrobial prophylaxis is a suitable alternative to vaccination in patients where this is not possible, such as patients in the early post-HSCT period. No significant difference in bacterial and fungal bloodstream infections was seen between patients on antimicrobial prophylaxis treated and not treated with complement C5 inhibitors [95].
While eculizumab is not approved for use in patients with HSCT-TMA, the reported efficacy of complement blockade in this patient population should not be overlooked. It should also be considered that use of standard C5 inhibitor dosing regimens approved and validated in other conditions, such as aHUS, may not be suitable in patients with HSCT-TMA. When using eculizumab, Jodele et al. highlighted the need for ongoing therapeutic drug monitoring (TDM) in patients with HSCT-TMA [91–93]. Unfortunately, TDM may not be universally available, meaning further prospective studies, perhaps utilizing TDM, are required to clarify the optimal regimen of complement inhibition in HSCT-TMA [48, 90, 96]. In the absence of standardized regimens, it is clear that dosing must be tailored and monitored to achieve complete terminal complement blockade and ensure optimal efficacy. Further, a lack of C5 inhibitor responses in severely ill patients seemingly reinforces the need for early diagnosis and intervention in patients with high-risk HSCT-TMA. Since other endothelial injury pathways also play a role, it is hypothesized that combination therapies may be available in the future and enhance response rates in difficult-to-treat patients.
HSCT-TMA: Monitoring
There are a range of parameters that should be monitored to determine treatment efficacy and disease progression as discussed by Jodele et al.; these are presented in Table 2 [10, 47]. These monitoring criteria focus heavily on the key triad of TMA (thrombocytopenia, ischemic organ damage, and MAHA) and on complement activity. They were also applied in the clinical studies conducted by Jodele et al., correspond to the updated diagnostic criteria proposed by the same group, and have been validated in pediatric patients [10, 47]. Notably, routine monitoring of sC5b-9 may be useful, as levels may escalate over time, indicating an overactivated complement system and placing the patient at risk of organ injury. Assessing baseline levels, preferably prior to transplantation if practical, followed by weekly monitoring if a patient develops signs of hematologic TMA may be warranted.
Table 2.
Clinical parameters which should be monitored to determine treatment efficacy and disease progression [10, 47]
| Hematologic measurements | Renal measurements | Other measurements |
|---|---|---|
| Complete blood counts and electrolyte panels should be monitored frequently, even daily | Proteinuria should be assessed via random urine protein and creatinine quantification | Assessment of changes in the need for transfusions (red cell and platelet) should be completed, particularly within first 100 days post-transplant |
| Schistocytes should be assessed routinely via peripheral blood smears | Kidney function monitoring should be assessed to monitor signs of acute kidney injury | Routine monitoring for viremias such as BK virus and adenovirus is required |
| Lactate dehydrogenase levels should be measured twice weekly | Blood pressure measurements should be routinely captured to monitor for hypertension | |
| Serum soluble C5b-9 concentrations should be assessed, if feasible | Cardiac monitoring via echocardiography should be performed in symptomatic patients with suspected/confirmed thrombotic microangiopathy, particularly in hypoxemic patients requiring intensive care | |
| Assessment of oxygen requirements (e.g., unexplained increase in oxygen needs or improvements following treatment) | ||
| If feasible, biopsies can be considered as required, following strict risk–benefit assessments |
HSCT-TMA: Future Treatments Targeting Complement
While most evidence relating to complement-targeted therapy to date has focused on C5 inhibitors, namely eculizumab, much of this has been derived from case studies/series and there remains a need for randomized controlled trials in patients with HSCT-TMA. Indeed, a number of trials are ongoing with various other therapeutics targeting C5, such as ravulizumab and coversin, alongside treatments targeting other components of the complement cascade.
There is some preliminary evidence that inhibitors of specific complement pathways, such as the LP, may show some utility in the management of HSCT-TMA; inhibitors targeting specific complement pathways may preserve the other complement pathways and their function. Further, as a result of possible cross talk between systems, LP inhibitors may be of particular interest in bleeding patients with complement dysregulation. Narsoplimab (OMS721) is a human monoclonal IgG4 inhibitor of MASP2, which has recently completed a phase II study assessing its effectiveness in patients with TMA [97–99]. Data from this phase II study of 28 patients suggest that narsoplimab improves laboratory markers of TMA and organ function and may have a beneficial effect on overall survival in patients with HSCT-TMA [97–99]. However, overall physiologic concentrations of active LP molecules are low, suggesting it may play a minor role in HSCT-TMA pathogenesis, and there may be a risk of reduced efficacy in conditions such as HSCT-TMA because of the apparent involvement of all complement pathways in pathogenesis [97, 98]. Results from phase III studies and greater clinical experience are therefore required to solidify these results. However, complement inhibitors appear to be a current “hot topic,” with a range of novel inhibitors of components such as C5, C3, factor B, and factor D (amongst others) currently being explored for use in treating TMAs. It should also be considered when testing these new therapeutic options that specific PK/PD studies may be required in this patient population, based on the substantially different PK/PD profiles identified by Jodele et al. following eculizumab treatment. A more detailed overview of all therapeutics targeting complement can be found in the review by Gavriilaki et al. [100].
In addition, the involvement and interaction of other molecules and pathways in the pathogenesis of HSCT-TMA—such as interferons and NETosis—may also contribute to refractoriness to current treatment approaches and therefore be key targets for future drug development. Overall, the applicability of these novel treatment modalities, or indeed combinations thereof, in the setting of HSCT-TMA remains a key topic of interest [100].
HSCT-TMA: Myths, Unknowns, and Areas of Discord
While our understanding of HSCT-TMA has advanced substantially, there is still much that remains to be elucidated, and several key topics are discussed below.
Firstly, many of the non-canonical interactions between complement and other pathways/systems discussed in this article have been evaluated in vitro, using purified proteins and non-physiologic conditions, or in animal models. Additionally, some of the non-canonical enzymatic interactions are reportedly less efficient than the prototypical enzyme–substrate interactions, and the lack of specific animal models of TMA further confounds assessment of their relevance in HSCT-TMA. Further investigations, particularly in the clinical setting, are required to understand their importance in vivo and subsequently their relevance to clinical practice [19, 29, 30, 37, 38]. Secondly, although TMA following HSCT is a relatively common occurrence, it is often masked by other common post-transplant complications such as intestinal/GI GvHD and may also present concurrently with these other conditions. Thus, if clinicians are not actively looking for HSCT-TMA, it is unlikely to be diagnosed or treated [11].
Another issue relates to screening tools and differences between adult and pediatric patients. While the tools used in pediatric patients continue to be improved, their utility in adult patients is unclear, and more studies are needed. There is also a need for more prospective studies in adult patients to determine optimal treatment protocols. Further, simple methods and tools to evaluate and characterize the level of endothelial damage in HSCT patients remain elusive, but would be extremely beneficial in this setting, alongside greater understanding of the molecular processes underlying TMA initiation, endothelial cell damage, and C3 deposition in this population. Easier tools for assessing complement pathway function and activity would be extremely helpful and allow for rapid and routine assessment of complement markers such as sC5b-9 levels. This would, in turn, allow for more robust studies of complement activity at different stages of HSCT-TMA progression to be conducted, which is particularly important as there are many difficulties associated with interpreting the plethora of laboratory measurements, commercial assays, and in vitro studies of complement function or activity. For example, the complement system is inherently unstable and prone to activation, meaning that samples must be taken carefully into EDTA tubes and kept on ice to ensure that artificial in vitro activation does not occur. Artificial activation of the complement system in clinical samples may be particularly troublesome, as the samples may not accurately represent the patient’s underlying pathophysiology once analyzed. This can confound interpretation and complicate clinical decision-making. Randomized clinical studies are required to address and alleviate these complications.
Lastly, it is extremely important to determine why some patients are refractory to treatment with existing complement inhibitors: is treatment initiated too late; are the symptoms unrelated to complement; is complement activation too severe; are other pathways such as the coagulation cascade involved, which are bypassing complement blockade; or are there other explanations for the lack of response in all patients [91, 92]? It is also of great interest to explore the efficacy of novel complement-specific therapeutics, or their combination with inhibitors or modulators of other pathways, such as the coagulation cascade, the interferon system, or other inflammatory mechanisms to determine the optimal treatment regimen for this condition [14, 63, 101, 102].
Conclusions
Dysfunction of the complement system can result in the initiation of vascular damage/dysfunction, e.g., TMA. We have highlighted the key roles that all facets of the complement cascade play in the development of HSCT-TMA and have listed a key set of risk factors and diagnostic criteria to aid clinicians in the identification and management of HSCT-TMA in the clinic, including key differences between adult and pediatric patients.
In summary, HSCT-TMA is a common post-transplant complication that, if left untreated, can have serious negative consequences for patients. Despite the simple screening techniques available for HSCT-TMA, it is often mistaken for other common post-transplant conditions. However, when identified correctly and treated promptly, patient outcomes are drastically improved. In situations with clear involvement of the complement system, targeted therapies can be tried. Therefore, it is imperative that both pediatric and adult transplant specialists understand the key signs and symptoms of HSCT-TMA to ensure prompt diagnosis and optimized outcomes for their patients.
Further Reading
Please find below a list of further references of interest which provide more detail on much of the information discussed within this review article.
Merle NS, Church SE, Fremeaux-Bacchi V, et al. Complement system part I—molecular mechanisms of activation and regulation. Frontiers in Immunology 2015;6.
Merle NS, Noe R, Halbwachs-Mecarelli L, et al. Complement system part II: role in immunity. Frontiers in Immunology 2015;6.
Meri S. Complement activation in diseases presenting with thrombotic microangiopathy. Eur J Int Med 2013;24:496–502.
Oikonomopoulou K, Ricklin D, Ward P, et al. Interactions between coagulation and complement—their role in inflammation. Semin Immunupathol 2012;34(1):151–65.
Foley JH. Examining coagulation-complement crosstalk: complement activation and thrombosis. Thromb Res 2016;141(S2):S50–4.
Dandoy CE, Rotz S, Alonso PB, et al. A pragmatic multi-institutional approach to understanding transplant-associated thrombotic microangiopathy after stem cell transplant. Blood Advances 2020;5:1–11.
Young JA, Pallas CR, Knovich MA. Transplant-associated thrombotic microangiopathy: theoretical considerations and a practical approach to an unrefined diagnosis. Bone Marrow Transplantation 2021;56:1805–1817.
Dvorak CC, Higham C, Shimano KA. Transplant-associated thrombotic microangiopathy in pediatric hematopoietic cell transplant recipients: a practical approach to diagnosis and management. Frontiers in Pediatrics 2019;7.
Jodele S, Sabulski A. Transplant-associated thrombotic microangiopathy: elucidating prevention strategies and identifying high-risk patients. Expert Rev Hematol 2021;14:751–763.
Jodele S, Dandoy CE, Lane A, et al. Complement blockade for TA-TMA: lessons learned from a large pediatric cohort treated with eculizumab. Blood. 2020 Mar 26;135(13):1049–1057.
Mizuno K, Dandoy CE, Teusink-Cross A, et al. Eculizumab precision dosing algorithm for thrombotic microangiopathy in children and young adults undergoing HSCT. Blood Adv. 2022 Jan 10:loodadvances.2021006523. https://doi.org/10.1182/bloodadvances.2021006523. Online ahead of print. PMID: 35008105.
de Bont CM, Boelens WC, Pruijn GJM. NETosis, complement, and coagulation: a triangular relationship. Cell Mol Immunol. 2019;16(1):19–27.
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgements
Funding
This manuscript and its publication, including the Journal’s Rapid Service and Open Access Fees, were sponsored and funded by Alexion, AstraZeneca Rare Disease.
Medical Writing, Editorial, and Other Assistance
Editorial assistance in the preparation of this article was provided by Dr Alexander T. Hardy of Bioscript Medical Communications, Macclesfield, UK. Support for this assistance was funded by Alexion, AstraZeneca Rare Disease.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Author Contributions
All authors contributed to the review article conception and design. The first drafts of the manuscript, diagnostic algorithm and complement video storyboard were developed by Dr Alexander T. Hardy of Bioscript Medical Communications, Macclesfield, UK. All authors critically reviewed and provided comments on the first, second and final drafts of all materials. All authors read and approved the final drafts of this manuscript, including the complement video and diagnostic algorithm.
Disclosures
Seppo Meri has received honoraria from Alexion, AstraZeneca Rare Disease, Biogen, Merck, Pfizer and UCB, and research funding from Alexion. Donald Bunjes has received honoraria from Alexion, AstraZeneca Rare Disease and Omeros. Roxanne Cofiell is an employee and shareholder of Alexion, AstraZeneca Rare Disease. Sonata Jodele has received honoraria from Alexion, AstraZeneca Rare Disease, SOBI and Omeros, Medscape Education, Physician Education Resource, LLC (PER).
Compliance with Ethics Guidelines
This article is based on previously conducted studies and does not contain any new studies with human participants or animals performed by any of the authors.
Data Availability
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
References
- 1.Timmermans SAMEG, van Paassen P. The syndromes of thrombotic microangiopathy: a critical appraisal on complement dysregulation. J Clin Med. 2021;10(14):3034. [DOI] [PMC free article] [PubMed]
- 2.El Karoui K, Hill GS, Karras A, et al. A clinicopathologic study of thrombotic microangiopathy in IgA nephropathy. J Am Soc Nephrol. 2012;23(1):137–148. doi: 10.1681/ASN.2010111130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.De Serres SA, Isenring P. Renal thrombotic microangiopathy revisited: when a lesion is not a clinical finding. Saudi J Kidney Dis Transpl. 2010;21(3):411–416. [PubMed] [Google Scholar]
- 4.Meri S. Complement activation in diseases presenting with thrombotic microangiopathy. Eur J Intern Med. 2013;24(6):496–502. doi: 10.1016/j.ejim.2013.05.009. [DOI] [PubMed] [Google Scholar]
- 5.Berger BE. Atypical hemolytic uremic syndrome: a syndrome in need of clarity. Clin Kidney J. 2018;12(3):338–347. doi: 10.1093/ckj/sfy066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Garcia G, Atallah JP. Antineoplastic agents and thrombotic microangiopathy. J Oncol Pharm Pract. 2016;23(2):135–142. doi: 10.1177/1078155216628324. [DOI] [PubMed] [Google Scholar]
- 7.Morton JM, George JN. Microangiopathic hemolytic anemia and thrombocytopenia in patients with cancer. J Oncol Practice. 2016;12(6):523–530. doi: 10.1200/JOP.2016.012096. [DOI] [PubMed] [Google Scholar]
- 8.Young JA, Pallas CR, Knovich MA. Transplant-associated thrombotic microangiopathy: theoretical considerations and a practical approach to an unrefined diagnosis. Bone Marrow Transpl. 2021;56(8):1805–1817. doi: 10.1038/s41409-021-01283-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jodele S, Zhang K, Zou F, et al. The genetic fingerprint of susceptibility for transplant-associated thrombotic microangiopathy. Blood. 2016;127(8):989–996. doi: 10.1182/blood-2015-08-663435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jodele S, Laskin BL, Dandoy CE, et al. A new paradigm: diagnosis and management of HSCT-associated thrombotic microangiopathy as multi-system endothelial injury. Blood Rev. 2015;29(3):191–204. doi: 10.1016/j.blre.2014.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dandoy CE, Rotz S, Alonso PB, et al. A pragmatic multi-institutional approach to understanding transplant-associated thrombotic microangiopathy after stem cell transplant. Blood Adv. 2020;5(1):1–11. doi: 10.1182/bloodadvances.2020003455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Willems E, Baron F, Seidel L, Frère P, Fillet G, Beguin Y. Comparison of thrombotic microangiopathy after allogeneic hematopoietic cell transplantation with high-dose or nonmyeloablative conditioning. Bone Marrow Transpl. 2010;45(4):689–693. doi: 10.1038/bmt.2009.230. [DOI] [PubMed] [Google Scholar]
- 13.Dvorak CC, Higham C, Shimano KA. Transplant-associated thrombotic microangiopathy in pediatric hematopoietic cell transplant tecipients: a practical approach to diagnosis and management. Front Pediatr. 2019;7(133). [DOI] [PMC free article] [PubMed]
- 14.Jodele S, Sabulski A. Transplant-associated thrombotic microangiopathy: elucidating prevention strategies and identifying high-risk patients. Expert Rev Hematol. 2021;14(8):751–763. doi: 10.1080/17474086.2021.1960816. [DOI] [PubMed] [Google Scholar]
- 15.Gavriilaki E, Sakellari I, Batsis I, et al. Transplant-associated thrombotic microangiopathy: incidence, prognostic factors, morbidity, and mortality in allogeneic hematopoietic cell transplantation. Clin Transpl. 2018;32(9):e13371. doi: 10.1111/ctr.13371. [DOI] [PubMed] [Google Scholar]
- 16.Postalcioglu M, Kim HT, Obut F, et al. Impact of thrombotic microangiopathy on renal outcomes and survival after hematopoietic stem cell transplantation. Biol Blood Marrow Transpl. 2018;24(11):2344–2353. doi: 10.1016/j.bbmt.2018.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Merle NS, Church SE, Fremeaux-Bacchi V, Roumenina LT. Complement system part I—molecular mechanisms of activation and regulation. Front Immunol. 2015;6(262). [DOI] [PMC free article] [PubMed]
- 18.Merle NS, Noe R, Halbwachs-Mecarelli L, Fremeaux-Bacchi V, Roumenina LT. Complement system part II: role in immunity. Front Immunol. 2015;6(257). [DOI] [PMC free article] [PubMed]
- 19.Oikonomopoulou K, Ricklin D, Ward PA, Lambris JD. Interactions between coagulation and complement–their role in inflammation. Semin Immunopathol. 2012;34(1):151–165. doi: 10.1007/s00281-011-0280-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Amara U, Rittirsch D, Flierl M, et al. Interaction between the coagulation and complement system. Adv Exp Med Biol. 2008;632:71–79. doi: 10.1007/978-0-387-78952-1_6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Killick J, Morisse G, Sieger D, Astier AL. Complement as a regulator of adaptive immunity. Semin Immunopathol. 2018;40(1):37–48. doi: 10.1007/s00281-017-0644-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bekassy Z, Lopatko Fagerström I, Bader M, Karpman D. Crosstalk between the renin–angiotensin, complement and kallikrein–kinin systems in inflammation. Nat Rev Immunol. 2021. 10.1038/s41577-021-00634-8. [DOI] [PMC free article] [PubMed]
- 23.Harboe M, Ulvund G, Vien L, Fung M, Mollnes TE. The quantitative role of alternative pathway amplification in classical pathway induced terminal complement activation. Clin Exp Immunol. 2004;138(3):439–446. doi: 10.1111/j.1365-2249.2004.02627.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lachmann PJ. The amplification loop of the complement pathways. Adv Immunol. 2009;104:115–149. doi: 10.1016/S0065-2776(08)04004-2. [DOI] [PubMed] [Google Scholar]
- 25.Monsinjon T, Gasque P, Chan P, Ischenko A, Brady JJ, Fontaine MC. Regulation by complement C3a and C5a anaphylatoxins of cytokine production in human umbilical vein endothelial cells. FASEB J. 2003;17(9):1003–1014. doi: 10.1096/fj.02-0737com. [DOI] [PubMed] [Google Scholar]
- 26.Gonzalez SF, Lukacs-Kornek V, Kuligowski MP, et al. Complement-dependent transport of antigen into B cell follicles. J Immunol. 2010;185(5):2659–64. [DOI] [PMC free article] [PubMed]
- 27.Kavanagh D, Goodship TH, Richards A. Atypical hemolytic uremic syndrome. Semin Nephrol. 2013;33(6):508–530. doi: 10.1016/j.semnephrol.2013.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cofiell R, Kukreja A, Bedard K, et al. Eculizumab reduces complement activation, inflammation, endothelial damage, thrombosis, and renal injury markers in aHUS. Blood. 2015;125(21):3253–3262. doi: 10.1182/blood-2014-09-600411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Markiewski MM, Lambris JD. The role of complement in inflammatory diseases from behind the scenes into the spotlight. Am J Pathol. 2007;171(3):715–727. doi: 10.2353/ajpath.2007.070166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Conway EM. Reincarnation of ancient links between coagulation and complement. J Thromb Haemost. 2015;13(Suppl 1):S121–S132. doi: 10.1111/jth.12950. [DOI] [PubMed] [Google Scholar]
- 31.Ritis K, Doumas M, Mastellos D, et al. A novel C5a receptor-tissue factor cross-talk in neutrophils links innate immunity to coagulation pathways. J Immunol. 2006;177(7):4794–802. [DOI] [PubMed]
- 32.Markiewski MM, Nilsson B, Ekdahl KN, Mollnes TE, Lambris JD. Complement and coagulation: strangers or partners in crime? Trends Immunol. 2007;28(4):184–192. doi: 10.1016/j.it.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 33.Amara U, Flierl MA, Rittirsch D, et al. Molecular intercommunication between the complement and coagulation systems. J Immunol. 2010;185(9):5628–36. [DOI] [PMC free article] [PubMed]
- 34.Hess K, Ajjan R, Phoenix F, Dobó J, Gál P, Schroeder V. Effects of MASP-1 of the complement system on activation of coagulation factors and plasma clot formation. PLoS ONE. 2012;7(4):e35690. doi: 10.1371/journal.pone.0035690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Del Conde I, Crúz MA, Zhang H, López JA, Afshar-Kharghan V. Platelet activation leads to activation and propagation of the complement system. J Exp Med. 2005;201(6):871–879. doi: 10.1084/jem.20041497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hamad OA, Ekdahl KN, Nilsson PH, et al. Complement activation triggered by chondroitin sulfate released by thrombin receptor-activated platelets. J Thrombosis Haemostasis. 2008;6(8):1413–1421. doi: 10.1111/j.1538-7836.2008.03034.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Garred P, Genster N, Pilely K, et al. A journey through the lectin pathway of complement-MBL and beyond. Immunol Rev. 2016;274(1):74–97. doi: 10.1111/imr.12468. [DOI] [PubMed] [Google Scholar]
- 38.Ekdahl KN, Huang S, Nilsson B, Teramura Y. Complement inhibition in biomaterial- and biosurface-induced thromboinflammation. Semin Immunol. 2016;28(3):268–277. doi: 10.1016/j.smim.2016.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Conway EM. Complement-coagulation connections. Blood Coagul Fibrinolysis. 2018;29(3):243–251. doi: 10.1097/MBC.0000000000000720. [DOI] [PubMed] [Google Scholar]
- 40.de Bont CM, Boelens WC, Pruijn GJM. NETosis, complement, and coagulation: a triangular relationship. Cell Mol Immunol. 2019;16(1):19–27. doi: 10.1038/s41423-018-0024-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huang H, Zhang H, Onuma AE, Tsung A. Neutrophil elastase and neutrophil extracellular traps in the tumor microenvironment. Adv Exp Med Biol. 2020;1263:13–23. doi: 10.1007/978-3-030-44518-8_2. [DOI] [PubMed] [Google Scholar]
- 42.Wang H, Wang C, Zhao MH, Chen M. Neutrophil extracellular traps can activate alternative complement pathways. Clin Exp Immunol. 2015;181(3):518–527. doi: 10.1111/cei.12654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sartain SE, Turner NA, Moake JL. TNF regulates essential alternative complement pathway components and impairs activation of protein C in human glomerular endothelial cells. J Immunol. 2016;196(2):832–45. [DOI] [PubMed]
- 44.Sjøqvist C, Snarski E. Inflammatory markers in patients after hematopoietic stem cell transplantation. Arch Immunol Ther Exp. 2013;61(4):301–307. doi: 10.1007/s00005-013-0228-z. [DOI] [PubMed] [Google Scholar]
- 45.Li Z, Rubinstein SM, Thota R, et al. Immune-mediated complications after hematopoietic stem cell transplantation. Biol Blood Marrow Transpl. 2016;22(8):1368–1375. doi: 10.1016/j.bbmt.2016.04.005. [DOI] [PubMed] [Google Scholar]
- 46.Chaturvedi S, Neff A, Nagler A, Savani U, Mohty M, Savani BN. Venous thromboembolism in hematopoietic stem cell transplant recipients. Bone Marrow Transpl. 2016;51(4):473–478. doi: 10.1038/bmt.2015.308. [DOI] [PubMed] [Google Scholar]
- 47.Jodele S, Davies SM, Lane A, et al. Diagnostic and risk criteria for HSCT-associated thrombotic microangiopathy: a study in children and young adults. Blood. 2014;124(4):645–653. doi: 10.1182/blood-2014-03-564997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jodele S, Dandoy CE, Lane A, et al. Complement blockade for TA-TMA: lessons learned from a large pediatric cohort treated with eculizumab. Blood. 2020;135(13):1049–1057. doi: 10.1182/blood.2019004218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jodele S, Licht C, Goebel J, et al. Abnormalities in the alternative pathway of complement in children with hematopoietic stem cell transplant-associated thrombotic microangiopathy. Blood. 2013;122(12):2003–2007. doi: 10.1182/blood-2013-05-501445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gavriilaki E, Ho VT, Schwaeble W, et al. Role of the lectin pathway of complement in hematopoietic stem cell transplantation-associated endothelial injury and thrombotic microangiopathy. Exp Hematol Oncol. 2021;10(1):57. doi: 10.1186/s40164-021-00249-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Khosla J, Yeh AC, Spitzer TR, Dey BR. Hematopoietic stem cell transplant-associated thrombotic microangiopathy: current paradigm and novel therapies. Bone Marrow Transpl. 2018;53(2):129–137. doi: 10.1038/bmt.2017.207. [DOI] [PubMed] [Google Scholar]
- 52.Farrar C, Asgari E, Schwaeble W, Sacks S. Which pathways trigger the role of complement in ischaemia/reperfusion injury? Front Immunol. 2012;3. [DOI] [PMC free article] [PubMed]
- 53.Gorsuch WB, Chrysanthou E, Schwaeble WJ, Stahl GL. The complement system in ischemia-reperfusion injuries. Immunobiology. 2012;217(11):1026–1033. doi: 10.1016/j.imbio.2012.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hart ML, Ceonzo KA, Shaffer LA, et al. Gastrointestinal ischemia-reperfusion injury is lectin complement pathway dependent without involving C1q. J Immunol. 2005;174(10):6373–80. [DOI] [PubMed]
- 55.Collard CD, Väkevä A, Morrissey MA, et al. Complement activation after oxidative stress: role of the lectin complement pathway. Am J Pathol. 2000;156(5):1549–1556. doi: 10.1016/S0002-9440(10)65026-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Buerke M, Prüfer D, Dahm M, Oelert H, Meyer J, Darius H. Blocking of classical complement pathway inhibits endothelial adhesion molecule expression and preserves ischemic myocardium from reperfusion injury. J Pharmacol Exp Ther. 1998;286(1):429–438. [PubMed] [Google Scholar]
- 57.Weiser MR, Williams JP, Moore FD, Jr, et al. Reperfusion injury of ischemic skeletal muscle is mediated by natural antibody and complement. J Exp Med. 1996;183(5):2343–2348. doi: 10.1084/jem.183.5.2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liu H, Jensen L, Hansen S, et al. Characterization and quantification of mouse mannan-binding lectins (MBL-A and MBL-C) and study of acute phase responses. Scand J Immunol. 2001;53(5):489–497. doi: 10.1046/j.1365-3083.2001.00908.x. [DOI] [PubMed] [Google Scholar]
- 59.Schafranski MD, Stier A, Nisihara R, Messias-Reason IJT. Significantly increased levels of mannose-binding lectin (MBL) in rheumatic heart disease: a beneficial role for MBL deficiency. Clin Exp Immunol. 2004;138(3):521–525. doi: 10.1111/j.1365-2249.2004.02645.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jacobson S, Larsson P, Åberg A-M, Johansson G, Winsö O, Söderberg S. Levels of mannose-binding lectin (MBL) associates with sepsis-related in-hospital mortality in women. J Inflamm. 2020;17(1):28. doi: 10.1186/s12950-020-00257-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Elhadad S, Chapin J, Copertino D, Van Besien K, Ahamed J, Laurence J. MASP2 levels are elevated in thrombotic microangiopathies: association with microvascular endothelial cell injury and suppression by anti-MASP2 antibody narsoplimab. Clin Exp Immunol. 2021;203(1):96–104. doi: 10.1111/cei.13497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chua JS, Baelde HJ, Zandbergen M, et al. Complement factor C4d is a common denominator in thrombotic microangiopathy. J Am Soc Nephrol. 2015;26(9):2239–2247. doi: 10.1681/ASN.2014050429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jodele S, Medvedovic M, Luebbering N, et al. Interferon-complement loop in transplant-associated thrombotic microangiopathy. Blood Adv. 2020;4(6):1166–1177. doi: 10.1182/bloodadvances.2020001515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Choi CM, Schmaier AH, Snell MR, Lazarus HM. Thrombotic microangiopathy in haematopoietic stem cell transplantation. Drugs. 2009;69(2):183–198. doi: 10.2165/00003495-200969020-00004. [DOI] [PubMed] [Google Scholar]
- 65.Luft T, Benner A, Terzer T, et al. EASIX and mortality after allogeneic stem cell transplantation. Bone Marrow Transpl. 2020;55(3):553–561. doi: 10.1038/s41409-019-0703-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jodele S, Fukuda T, Mizuno K, et al. Variable eculizumab clearance requires pharmacodynamic monitoring to optimize therapy for thrombotic microangiopathy after hematopoietic stem cell transplantation. Biol Blood Marrow. 2016;22(2):307–315. doi: 10.1016/j.bbmt.2015.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ekdahl KN, Persson B, Mohlin C, Sandholm K, Skattum L, Nilsson B. Interpretation of serological complement biomarkers in disease. Front Immunol. 2018;9(2237). [DOI] [PMC free article] [PubMed]
- 68.Frazer-Abel A, Kirschfink M, Prohászka Z. Expanding horizons in complement analysis and quality control. Front Immunol. 2021;12. [DOI] [PMC free article] [PubMed]
- 69.Sartain S, Shubert S, Wu M-F, Wang T, Martinez C. The alternative complement pathway activation product Ba as a marker for transplant-associated thrombotic microangiopathy. Pediatr Blood Cancer. 2020;67(3):e28070. doi: 10.1002/pbc.28070. [DOI] [PubMed] [Google Scholar]
- 70.Yamada R, Nemoto T, Ohashi K, et al. Distribution of transplantation-associated thrombotic microangiopathy (TA-TMA) and comparison between renal TA-TMA and intestinal TA-TMA: autopsy study. Biol Blood Marrow Transplant. 2020;26(1):178–88. [DOI] [PubMed]
- 71.Cho BS, Min CK, Eom KS, et al. Clinical impact of thrombotic microangiopathy on the outcome of patients with acute graft-versus-host disease after allogeneic hematopoietic stem cell transplantation. Bone Marrow Transpl. 2008;41(9):813–820. doi: 10.1038/sj.bmt.1705976. [DOI] [PubMed] [Google Scholar]
- 72.Yamada-Fujiwara M, Miyamura K, Fujiwara T, et al. Diagnosis of intestinal graft-versus-host disease and thrombotic microangiopathy after allogeneic stem cell transplantation. Tohoku J Exp Med. 2012;227(1):31–37. doi: 10.1620/tjem.227.31. [DOI] [PubMed] [Google Scholar]
- 73.El-Bietar J, Warren M, Dandoy C, et al. Histologic features of intestinal thrombotic microangiopathy in pediatric and young adult patients after hematopoietic stem cell transplantation. Biol Blood Marrow Transpl. 2015;21(11):1994–2001. doi: 10.1016/j.bbmt.2015.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Inamoto Y, Ito M, Suzuki R, et al. Clinicopathological manifestations and treatment of intestinal transplant-associated microangiopathy. Bone Marrow Transpl. 2009;44(1):43–49. doi: 10.1038/bmt.2008.419. [DOI] [PubMed] [Google Scholar]
- 75.Saidu NEB, Bonini C, Dickinson A, et al. New approaches for the treatment of chronic graft-versus-host disease: current status and future directions. Front Immunol. 2020;11(2625). [DOI] [PMC free article] [PubMed]
- 76.Warren M, Jodele S, Dandoy C, et al. A complete histologic approach to gastrointestinal biopsy from hematopoietic stem cell transplant patients with evidence of transplant-associated gastrointestinal thrombotic microangiopathy. Arch Pathol Lab Med. 2017;141(11):1558–1566. doi: 10.5858/arpa.2016-0599-RA. [DOI] [PubMed] [Google Scholar]
- 77.Wall SA, Zhao Q, Yearsley M, et al. Complement-mediated thrombotic microangiopathy as a link between endothelial damage and steroid-refractory GVHD. Blood Adv. 2018;2(20):2619–2628. doi: 10.1182/bloodadvances.2018020321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bonifazi F, Barbato F, Ravaioli F, et al. Diagnosis and treatment of VOD/SOS after allogeneic hematopoietic stem cell transplantation. Front Immunol. 2020;11:489. [DOI] [PMC free article] [PubMed]
- 79.Corbacioglu S, Carreras E, Ansari M, et al. Diagnosis and severity criteria for sinusoidal obstruction syndrome/veno-occlusive disease in pediatric patients: a new classification from the European Society for Blood and Marrow Transplantation. Bone Marrow Transpl. 2018;53(2):138–145. doi: 10.1038/bmt.2017.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Horton LE, Haste NM, Taplitz RA. Rethinking antimicrobial prophylaxis in the transplant patient in the world of emerging resistant organisms-where are we today? Curr Hematol Malig Rep. 2018;13(1):59–67. doi: 10.1007/s11899-018-0435-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Noè A, Cappelli B, Biffi A, et al. High incidence of severe cyclosporine neurotoxicity in children affected by haemoglobinopaties undergoing myeloablative haematopoietic stem cell transplantation: early diagnosis and prompt intervention ameliorates neurological outcome. Ital J Pediatr. 2010;36(1):14. doi: 10.1186/1824-7288-36-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Heybeli C, Sridharan M, Alkhateeb HB, et al. Characteristics of late transplant-associated thrombotic microangiopathy in patients who underwent allogeneic hematopoietic stem cell transplantation. Am J Hematol. 2020;95(10):1170–1179. doi: 10.1002/ajh.25922. [DOI] [PubMed] [Google Scholar]
- 83.Vasu S, Bostic M, Zhao Q, et al. Acute GVHD, BK virus hemorrhagic cystitis and age are risk factors for transplant-associated thrombotic microangiopathy in adults. Blood Adv. 2022;6(4):1342–1349. doi: 10.1182/bloodadvances.2021004933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.de Fontbrune FS, Galambrun C, Sirvent A, et al. Use of eculizumab in patients with allogeneic stem cell transplant-associated thrombotic microangiopathy: a study from the SFGM-TC. Transplantation. 2015;99(9):1953–1959. doi: 10.1097/TP.0000000000000601. [DOI] [PubMed] [Google Scholar]
- 85.Vasu S, Wu H, Satoskar A, et al. Eculizumab therapy in adults with allogeneic hematopoietic cell transplant-associated thrombotic microangiopathy. Bone Marrow Transpl. 2016;51(9):1241–1244. doi: 10.1038/bmt.2016.87. [DOI] [PubMed] [Google Scholar]
- 86.Rudoni J, Jan A, Hosing C, Aung F, Yeh J. Eculizumab for transplant-associated thrombotic microangiopathy in adult allogeneic stem cell transplant recipients. Eur J Haematol. 2018;101(3):389–398. doi: 10.1111/ejh.13127. [DOI] [PubMed] [Google Scholar]
- 87.Bohl SR, Kuchenbauer F, von Harsdorf S, et al. Thrombotic microangiopathy after allogeneic stem cell transplantation: a comparison of eculizumab therapy and conventional therapy. Biol Blood Marrow Transpl. 2017;23(12):2172–2177. doi: 10.1016/j.bbmt.2017.08.019. [DOI] [PubMed] [Google Scholar]
- 88.Epperla N, Hemauer K, Hamadani M, Friedman KD, Kreuziger LB. Impact of treatment and outcomes for patients with posttransplant drug-associated thrombotic microangiopathy. Transfusion. 2017;57(11):2775–2781. doi: 10.1111/trf.14263. [DOI] [PubMed] [Google Scholar]
- 89.Martínez-Muñoz ME, Forés R, Lario A, et al. Use of defibrotide to treat adult patients with transplant-associated thrombotic microangiopathy. Bone Marrow Transpl. 2019;54(1):142–145. doi: 10.1038/s41409-018-0256-8. [DOI] [PubMed] [Google Scholar]
- 90.Zhang R, Zhou M, Qi J, et al. Efficacy and safety of eculizumab in the treatment of transplant-associated thrombotic microangiopathy: a systematic review and meta-analysis. Front Immunol. 2021;11(3486). [DOI] [PMC free article] [PubMed]
- 91.Krisinger MJ, Goebeler V, Lu Z, et al. Thrombin generates previously unidentified C5 products that support the terminal complement activation pathway. Blood. 2012;120(8):1717–1725. doi: 10.1182/blood-2012-02-412080. [DOI] [PubMed] [Google Scholar]
- 92.Gavriilaki E, Chrysanthopoulou A, Sakellari I, et al. Linking complement activation, coagulation, and neutrophils in transplant-associated thrombotic microangiopathy. Thromb Haemost. 2019;119(9):1433–1440. doi: 10.1055/s-0039-1692721. [DOI] [PubMed] [Google Scholar]
- 93.Mizuno K, Dandoy CE, Teusink-Cross A, Davies SM, Vinks AA, Jodele S. Eculizumab precision dosing algorithm for thrombotic microangiopathy in children and young adults undergoing HSCT. Blood Adv. 2022;6(5):1454–63. [DOI] [PMC free article] [PubMed]
- 94.FDA. Soliris® (Eculizumab) Highlights of Prescribing Information 2007 https://www.accessdata.fda.gov/drugsatfda_docs/label/2011/125166s172lbl.pdf.
- 95.Jodele S, Dandoy CE, Danziger-Isakov L, et al. Terminal complement blockade after hematopoietic stem cell transplantation is safe without meningococcal vaccination. Biol Blood Marrow Transplant. 2016;22(7):1337–1340. doi: 10.1016/j.bbmt.2016.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Jodele S, Fukuda T, Vinks A, et al. Eculizumab therapy in children with severe hematopoietic stem cell transplantation-associated thrombotic microangiopathy. Biol Blood Marrow Transplant. 2014;20(4):518–525. doi: 10.1016/j.bbmt.2013.12.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Khaled SK, Boelens JJ, Cairo MS, et al. 26 - Narsoplimab (OMS721), a MASP-2 inhibitor, for the treatment of adult hematopoietic stem cell transplant-associated thrombotic microangiopathy (HSCT-TMA). Transplant Cell Ther. 2021;27(3, Supplement):S24–S6.
- 98.Khaled SK, Kwong YL, Smith M, Metjian A, Whitaker S. Early results of phase II study using OMS721 in patients with hematopoietic stem cell transplant-associated thrombotic microangiopathy (HCT-TMA) Biol Blood Marrow Transpl. 2017;23(3):S282–S283. doi: 10.1016/j.bbmt.2016.12.192. [DOI] [Google Scholar]
- 99.Khaled SK, Claes K, Goh YT, et al. Narsoplimab, a mannan-binding lectin-associated serine protease-2 inhibitor, for the treatment of adult hematopoietic stem-cell transplantation-associated thrombotic microangiopathy. J Clin Oncol. 2022:Jco2102389. [DOI] [PMC free article] [PubMed]
- 100.Gavriilaki E, Anagnostopoulos A, Mastellos DC. Complement in thrombotic microangiopathies: unraveling Ariadne's thread into the labyrinth of complement therapeutics. Front Immunol. 2019;10:337. doi: 10.3389/fimmu.2019.00337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Gloude NJ, Dandoy CE, Davies SM, et al. Thinking beyond HLH: clinical features of patients with concurrent presentation of hemophagocytic lymphohistiocytosis and thrombotic microangiopathy. J Clin Immunol. 2020;40(5):699–707. doi: 10.1007/s10875-020-00789-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Gloude NJ, Khandelwal P, Luebbering N, et al. Circulating dsDNA, endothelial injury, and complement activation in thrombotic microangiopathy and GVHD. Blood. 2017;130(10):1259–1266. doi: 10.1182/blood-2017-05-782870. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.