

Abstract
Background & Aims
The stimulator of interferon genes (STING)/TANK-binding kinase 1 (TBK1) pathway is vital in mediating innate immune and inflammatory responses during oxidative/endoplasmic reticulum (ER) stress. However, it remains unknown whether macrophage thioredoxin-interacting protein (TXNIP) may regulate TBK1 function and cell death pathways during oxidative/ER stress.
Methods
A mouse model of hepatic ischaemia/reperfusion injury (IRI), the primary hepatocytes, and bone marrow-derived macrophages were used in the myeloid-specific TXNIP knockout (TXNIPM-KO) and TXNIP-proficient (TXNIPFL/FL) mice.
Results
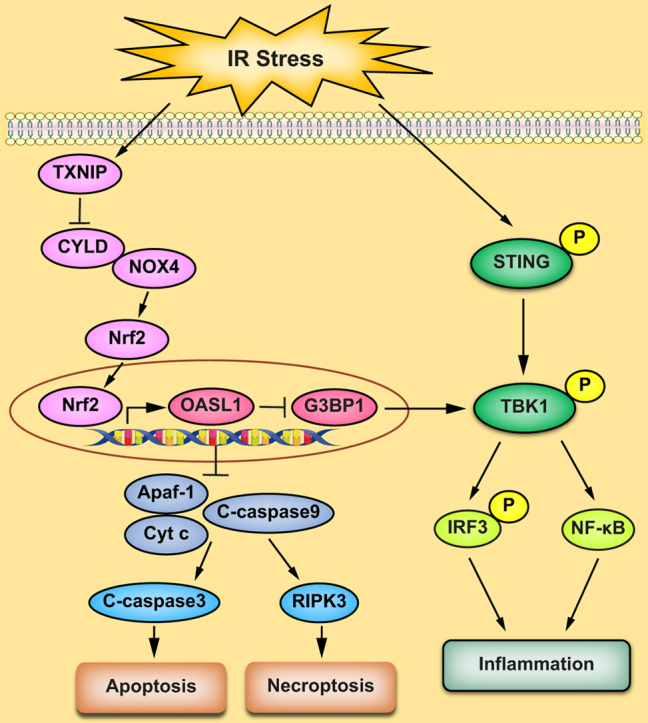
The TXNIPM-KO mice were resistant to ischaemia/reperfusion (IR) stress-induced liver damage with reduced serum alanine aminotransferase (ALT)/aspartate aminotransferase (AST) levels, macrophage/neutrophil infiltration, and pro-inflammatory mediators compared with the TXNIPFL/FL controls. IR stress increased TXNIP, p-STING, and p-TBK1 expression in ischaemic livers. However, TXNIPM-KO inhibited STING, TBK1, interferon regulatory factor 3 (IRF3), and NF-κB activation with interferon-β (IFN-β) expression. Interestingly, TXNIPM-KO augmented nuclear factor (erythroid-derived 2)-like 2 (NRF2) activity, increased antioxidant gene expression, and reduced macrophage reactive oxygen species (ROS) production and hepatic apoptosis/necroptosis in IR-stressed livers. Mechanistically, macrophage TXNIP deficiency promoted cylindromatosis (CYLD), which colocalised and interacted with NADPH oxidase 4 (NOX4) to enhance NRF2 activity by deubiquitinating NOX4. Disruption of macrophage NRF2 or its target gene 2′,5′ oligoadenylate synthetase-like 1 (OASL1) enhanced Ras GTPase-activating protein-binding protein 1 (G3BP1) and TBK1-mediated inflammatory response. Notably, macrophage OASL1 deficiency induced hepatocyte apoptotic peptidase activating factor 1 (APAF1), cytochrome c, and caspase-9 activation, leading to increased caspase-3-initiated apoptosis and receptor-interacting serine/threonine-protein kinase 3 (RIPK3)-mediated necroptosis.
Conclusions
Macrophage TXNIP deficiency enhances CYLD activity and activates the NRF2-OASL1 signalling, controlling IR stress-induced liver injury. The target gene OASL1 regulated by NRF2 is crucial for modulating STING-mediated TBK1 activation and Apaf1/cytochrome c/caspase-9-triggered apoptotic/necroptotic cell death pathway. Our findings underscore a novel role of macrophage TXNIP-mediated CYLD–NRF2–OASL1 axis in stress-induced liver inflammation and cell death, implying the potential therapeutic targets in liver inflammatory diseases.
Lay summary
Liver inflammation and injury induced by ischaemia and reperfusion (the absence of blood flow to the liver tissue followed by the resupply of blood) is a significant cause of hepatic dysfunction and failure following liver transplantation, resection, and haemorrhagic shock. Herein, we uncover an underlying mechanism that contributes to liver inflammation and cell death in this setting and could be a therapeutic target in stress-induced liver inflammatory injury.
Keywords: Innate immunity, STING, G3BP1, IRF3, Apoptosis, Necroptosis, Liver inflammation
Abbreviations: ALT, alanine aminotransferase; APAF1, apoptotic peptidase activating factor 1; ASK1, apoptosis signal-regulating kinase 1; AST, aspartate aminotransferase; BMM, bone marrow-derived macrophage; ChIP, chromatin immunoprecipitation; CXCL-10, C-X-C motif chemokine ligand 10; CYLD, cyclindromatosis; DAMP, damage-associated molecular pattern; DUB, deubiquitinating enzyme; ER, endoplasmic reticulum; ES, embryonic stem; G3BP1, Ras GTPase-activating protein-binding protein 1; GCLC, glutamate-cysteine ligase catalytic subunit; GCLM, glutamate-cysteine ligase regulatory subunit; IHC, immunohistochemistry; INF-β, interferon-β; IR, ischaemia/reperfusion; IRF3, interferon regulatory factor 3; IRF7, IFN-regulating transcription factor 7; IRI, ischaemia/reperfusion injury; KO, knockout; LPS, lipopolysaccharide; Lyz2, Lysozyme 2; MCP-1, monocyte chemoattractant protein 1; NOX2, NADPH oxidase 2; NOX4, NADPH oxidase 4; NQO1, NAD(P)H quinone dehydrogenase 1; NRF2, nuclear factor (erythroid-derived 2)-like 2; NS, non-specific; OASL1, 2′,5′oligoadenylate synthetase-like 1; PAMP, pathogen-derived molecular pattern; RIPK3, receptor-interacting serine/threonine-protein kinase 3; ROS, reactive oxygen species; sALT, serum ALT; sAST, serum AST; siRNA, small interfering RNA; STING, stimulator of interferon genes; TBK1, TANK-binding kinase 1; TLR4, Toll-like receptor 4; TNF-α, tumour necrosis factor-alpha; TRX, thioredoxin; TSS, transcription start sites; TXNIP, thioredoxin-interacting protein; TXNIPFL/FL, floxed TXNIP; TXNIPM-KO, myeloid-specific TXNIP KO; UTR, untranslated region
Graphical abstract
Highlights
-
•
TXNIP is critical in regulating STING-mediated TBK1 activation and inflammatory responses during oxidative/ER stress.
-
•
Macrophage TXNIP deficiency promotes CYLD and activates the NRF2 pathway.
-
•
CYLD interacts with NOX4 and regulates NRF2 activation by deubiquitinating NOX4 in macrophages.
-
•
NRF2 targets OASL1 and inhibits TBK1-mediated inflammatory responses by regulating G3BP1 activation in macrophages.
-
•
OASL1 inhibits stress-induced hepatocyte death via modulating the Apaf1/cytochrome c/caspase-9 pathway.
Introduction
Liver inflammation and injury induced by ischaemia and reperfusion (IR) is a significant cause of hepatic dysfunction and failure following liver transplantation, resection, and haemorrhagic shock.1 IR-induced liver damage involves oxidative stress and endoplasmic reticulum (ER) stress-mediated inflammatory responses.2 Liver macrophages (Kupffer cells), the key components of the hepatic innate immune system, represent the first line of defence in detecting the invading pathogens in the liver.3 IR stress activates macrophages and triggers innate immune responses by recognising exogenous danger signals such as pathogen-derived molecular patterns (PAMPs) or endogenous molecules such as damage-associated molecular patterns (DAMPs) released from damaged or dying cells during inflammatory response.4 Activated macrophages generate reactive oxygen species (ROS) and initiate Toll-like receptor 4 (TLR4) or NLRP3 inflammasome activation, leading to liver inflammation and injury.1,5,6
The stimulator of interferon genes (STING)/TANK-binding kinase 1 (TBK1) pathway has been recognised as a crucial signalling cascade of the innate immune system.7 STING activates the transcription factors NF-κB and interferon (IFN) regulatory factor (IRF) 3 via the TBK1.8 Indeed, STING is a transmembrane protein that predominantly resides in the ER, making STING ideal for response to alarm signals induced by ER stress.9 Activation of STING promoted NLRP3 activation and inflammatory responses,10 whereas disruption of the STING/TBK1 pathway ameliorated liver inflammatory injury.11 Recent studies have demonstrated that STING protein was not detectable in human and murine hepatocytes but was highly expressed in hepatic macrophages.12,13 STING promoted macrophage-mediated inflammatory responses and dampened liver function, whereas disruption of myeloid STING alleviated hepatic inflammation in non-alcoholic fatty liver disease.12,13 These results indicate that the STING/TBK1 pathway is key in mediating innate immune activation during liver inflammatory injury.
Thioredoxin-interacting protein (TXNIP), the thioredoxin (TRX)-binding protein, has been shown to regulate redox homoeostasis during stress.14 Under normoxic conditions, TXNIP binds to TRX1 in an inactive state. In response to oxidative stress, ROS generation facilitates TXNIP dissociation from TRX1 and activates the TXNIP signal cascade.14 Indeed, activation of TXNIP increased the production of ROS and redox responses involved in multiple inflammatory diseases.15 Disruption of TXNIP reduced oxidative stress and NLRP3-mediated inflammatory responses.16 Moreover, increasing evidence shows that TXNIP is linked to ER stress and tissue inflammation.17 TXNIP deletion inhibited ER stress-induced inflammation and cell injury.18 Although these studies document the critical role of TXNIP in oxidative and ER stress-induced inflammatory responses, it is unknown whether and how macrophage TXNIP regulates STING/TBK1-mediated innate immune response and cell death in IR-triggered liver inflammation.
Here, we identified a novel regulatory mechanism of macrophage TXNIP on TBK1 function and apoptotic/necroptotic cell death pathway in IR stress-mediated liver inflammation. We demonstrated that macrophage TXNIP deficiency controlled STING-mediated TBK1 function and alleviated IR-induced hepatocellular injury by regulating the cylindromatosis (CYLD)–NADPH oxidase 4 (NOX4) axis. Enhancing the CYLD–NOX4 interaction activated nuclear factor (erythroid-derived 2)-like 2 (NRF2) and its target gene 2′,5′ oligoadenylate synthetase-like 1 (OASL1), leading to inhibited Ras-GTPase-activating protein-binding protein 1 (G3BP1) and TBK1-driven inflammatory responses and hepatocyte death in IR stress-induced liver injury.
Materials and methods
Animals
The floxed TXNIP (TXNIPFL/FL) mice (B6;129-Txniptm1Rlee/J) and the mice expressing Cre recombinase under the control of the Lysozyme 2 (Lyz2) promoter (LysM-Cre) were obtained from The Jackson Laboratory (Bar Harbor, ME, USA). A targeting construct incorporating a loxP-exon1-FRT-PGKneobpA-FRT-loxP sequence was introduced to J1 129S4/SvJae-derived embryonic stem (ES) cells. Resultant mice were crossed with 129Sv background animals expressing Gt(ROSA)26Sortm1(FLP1)Dym to excise the FRT-flanked neomycin cassette, leaving exon 1 flanked by loxP sites. This strain was maintained on a mixed 129 and C57BL/6 genetic background. To generate myeloid-specific TXNIP knockout (KO) (TXNIPM-KO) mice, a homozygous loxP-flanked TXNIP mouse was mated with a homozygous Lyz2-Cre mouse to create the F1 mice that were heterozygous for a loxP-flanked TXNIP allele and heterozygous for the Lyz2-Cre. The F1 mice were then backcrossed to the homozygous loxP-flanked TXNIP mice, resulting in the generation of TXNIPM-KO (25% of the offspring), which were homozygous for the loxP-flanked TXNIP allele and heterozygous for the Lyz2-Cre allele (Fig. S1). Mouse genotyping was performed using a standard protocol with primers described in the JAX Genotyping protocols database. Male mice at 6–8 weeks of age were used in all experiments. This study was performed in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health. Animal protocols were approved by the Institutional Animal Care and Use Committee of The University of California at Los Angeles.
Mouse liver IRI model
We used an established mouse model of warm hepatic ischaemia (90 min) followed by reperfusion (6 h).5 Mice were injected with heparin (100 U/kg), and an atraumatic clip was used to interrupt the arterial/portal venous blood supply to the cephalad liver lobes. After 90 min of ischaemia, the clip was removed, and mice were sacrificed at 6 h of reperfusion. Some animals were injected via tail vein with OASL1 small interfering RNAs (siRNAs) or non-specific (NS) control siRNA (2.5 mg/kg) (Santa Cruz Biotechnology, Santa Cruz, CA, USA) mixed with mannose-conjugated polymers (Polyplus transfection™, Illkirch, France) at a ratio according to the manufacturer’s instructions 4 h before ischaemia as described.1,5,6 Some animals were injected via tail vein with TBK1-expressing bone marrow-derived macrophages (BMMs) or control cells (1 × 106 cells in 0.1 ml of PBS/mouse) 24 h before ischaemia.
Statistical analysis
Data are expressed as mean ± SD and analysed using the permutation t-test and Pearson correlation. Per comparison, 2-sided p values <0.05 were considered statistically significant. Multiple-group comparisons were made using 1-way ANOVA followed by Bonferroni’s post hoc test. When groups showed unequal variances, we applied Welch’s ANOVA to make various group comparisons. All analyses were used by SAS/STAT software version 9.4.
For further details regarding the materials and methods used, please refer to the CTAT Table and Supplementary information.
Results
Disruption of myeloid-specific TXNIP ameliorates IR-induced liver injury and diminishes macrophage/neutrophil accumulation and proinflammatory mediators in IR-stressed liver
The myeloid-specific TXNIP-deficient (TXNIPM-KO) and TXNIP-proficient (TXNIPFL/FL) mice were subjected to 90 min of warm ischaemia followed by 6 h of reperfusion. The primary hepatocytes and liver macrophages (Kupffer cells) were isolated from these ischaemic livers. Unlike TXNIPFL/FL livers, TXNIPM-KO lacked TXNIP expression in liver macrophages but not in hepatocytes (Fig. 1A). The liver damage was evaluated using Suzuki’s histological grading of liver ischaemia/reperfusion injury (IRI)19 (Fig. 1B). The TXNIPM-KO livers showed mild to moderate oedema, sinusoidal congestion, and mild necrosis compared to the TXNIPFL/FL livers, which showed severe oedema, sinusoidal congestion, and extensive hepatocellular necrosis (Fig. 1B). Liver injury is measured by the serum ALT (sALT) and sAST levels (IU/L). Myeloid TXNIP deficiency significantly reduced sALT and sAST levels at 6 h post liver reperfusion in the TXNIPM-KO mice compared with the TXNIPFL/FL controls (Fig. 1C). Moreover, the TXNIPM-KO ischaemic livers showed decreased accumulation of CD11b+ macrophages (Fig. 1D) and Ly6G+ neutrophils (Fig. 1E), as well as reduced mRNA levels coding for IL-6, tumour necrosis factor-alpha (TNF-α), C-X-C motif chemokine ligand 10 (CXCL-10), and monocyte chemoattractant protein 1 (MCP-1) in ischaemic livers (Fig. 1F), compared with the TXNIPFL/FL controls. These results suggest that TXNIP plays an important role in IR stress-induced liver inflammation and injury.
Fig. 1.

Disruption of myeloid-specific TXNIP ameliorates IR-induced liver injury and diminishes macrophage/neutrophil accumulation and proinflammatory mediators in IR-stressed liver.
The TXNIPFL/FL and TXNIPM-KO mice were subjected to 90 min of partial liver warm ischaemia, followed by 6 h of reperfusion. (A) The TXNIP expression was detected in hepatocytes and liver macrophages from IR-stressed livers by Western blot assay. Representative of 4 experiments. (B) Representative histological staining (H&E) of ischaemic liver tissue (n = 6 mice/group) and Suzuki’s histological score. Scale bars, 200 and 100 μm. (C) Liver function in serum samples was evaluated by sALT and sAST levels (IU/L) (n = 6 samples/group). (D) Immunofluorescence staining of CD11b+ macrophages in ischaemic livers (n = 6 mice/group). Quantification of CD11b+ macrophages. Scale bars, 100 μm. (E) Immunohistochemistry staining of Ly6G+ neutrophils in ischaemic livers (n = 6 mice/group). Quantification of Ly6G+ neutrophils. Scale bars, 200 and 50 μm. (F) Quantitative RT-PCR analysis of IL-6, TNF-α, CXCL-10, and MCP-1 mRNA levels in ischaemic livers (n = 6 samples/group). All data represent the mean ± SD. Statistical analysis was performed using the Permutation t test. ∗∗p <0.01. ∗∗∗p <0.005, ∗∗∗∗p <0.001. ALT, alanine aminotransferase; AST, aspartate aminotransferase; CXCL-10, chemokine (C-X-C motif) ligand 10; HPF, high power field; IR, ischaemia/reperfusion; MCP-1, monocyte chemoattractant protein 1; sALT, serum ALT; sAST, serum AST; TNF-α, tumour necrosis factor alpha; TXNIP, thioredoxin-interacting protein; TXNIPFL/FL, floxed TXNIP; TXNIPM-KO, myeloid-specific TXNIP knockout.
Disruption of myeloid-specific TXNIP inhibits STING-mediated TBK1 and NF-κB activation in IR-stressed liver
Next, we analysed whether TXNIP may affect the STING/TBK1 pathway in IR-stressed livers. Indeed, IR stress augmented TXNIP expression, STING, and TBK1 phosphorylation in ischaemic livers (Fig. 2A). As previous studies have demonstrated that STING is expressed in nonparenchymal liver cells (mainly macrophages or Kupffer cells) but not in human and murine hepatocytes,12,13 we then detected STING expression in liver macrophages (Kupffer cells). As expected, immunofluorescence staining revealed that IR stress increased p-STING expression in Kupffer cells from ischaemic livers (Fig. 2B). Unlike the TXNIPFL/FL controls, TXNIPM-KO reduced p-TBK1, p-IRF3, p-IκBα, and p-P65 protein expression in ischaemic livers (Fig. 2C). This result was further confirmed by immunohistochemistry (IHC) staining in liver macrophages (Fig. S2). Moreover, immunofluorescence staining revealed that TXNIPM-KO reduced macrophage p-TBK1 expression in IR-stressed livers compared with the TXNIPFL/FL controls (Fig. 2D). Western blot assay showed that TXNIPM-KO diminished p-TBK1, p-IRF3, and p-P65 protein expression (Fig. 2E) accompanied by reduced IFN-β mRNA levels (Fig. 2F) in Kupffer cells from ischaemic livers. These data suggest that macrophage TXNIP is critical in mediating STING-mediated TBK1 activation in IR stress-induced liver injury.
Fig. 2.

Disruption of myeloid-specific TXNIP inhibits STING-mediated TBK1 and IRF3/NF-κB activation in IR-stressed liver.
The WT, TXNIPFL/FL, and TXNIPM-KO mice were subjected to 90 min of partial liver warm ischaemia, followed by 6 h of reperfusion. (A) Western-assisted analysis of TXNIP, p-STING, STING, p-TBK1, and TBK1 in the WT livers after IR stress. (B) Immunofluorescence staining of p-STING and CD68 in ischaemic livers (n = 6 mice/group). Scale bars, 100 μm. (C) Western-assisted analysis and relative density ratio of p-TBK1, TBK1, p-IRF3, IRF3, p-IκBα, IκBα, p-P65, and P65 in the TXNIPFL/FL and TXNIPM-KO livers after IR stress. (D) Immunofluorescence staining of p-TBK1 and CD68 in ischaemic livers (n = 6 mice/group). Scale bars, 50 μm. (E) The Kupffer cells were isolated from the TXNIPFL/FL and TXNIPM-KO livers after IR stress. Western-assisted analysis and relative density ratio of p-TBK1, TBK1, p-IRF3, IRF3, p-P65, and P65 in Kupffer cells from the TXNIPFL/FL and TXNIPM-KO livers after IR stress. Representative of 3 experiments. (F) Quantitative RT-PCR analysis of IFN-β mRNA levels in Kupffer cells from the TXNIPFL/FL and TXNIPM-KO livers after IR stress (n = 6 samples/group). All Western blots represent 4 experiments, and the data represent the mean ± SD. Statistical analysis was performed using the Permutation t test. ∗∗p <0.01. ∗∗∗p <0.005. IFN-β, interferon-β; IR, ischaemia/reperfusion; IRF3, interferon regulatory factor 3; STING, stimulator of interferon genes; TBK1, TANK-binding kinase 1; TXNIP, thioredoxin-interacting protein; TXNIPFL/FL, floxed TXNIP; TXNIPM-KO, myeloid-specific TXNIP knockout; WT, wild type.
Disruption of myeloid-specific TXNIP promotes CYLD and activates the NRF2 pathway in IR-stressed liver
As TXNIP is involved in oxidative and ER stress-induced inflammatory responses, we then examined whether TXNIP influenced the NRF2-antioxidative responses in IR-stressed livers. Indeed, IR stress activated CYLD, NOX4, and NRF2 in ischaemic livers (Fig. 3A) and liver macrophages (Fig. S3). Notably, TXNIPM-KO enhanced CYLD and NRF2 activation in IR-stressed livers (Fig. 3B) and liver macrophages (Fig. S4) with increased antioxidant gene expression coding for NAD(P)H quinone dehydrogenase 1 (NQO1), glutamate-cysteine ligase catalytic subunit (GCLC), and glutamate-cysteine ligase regulatory subunit (GCLM) (Fig. 3C) in ischaemic livers compared with the TXNIPFL/FL controls. As CYLD plays a vital role in inflammation and immune responses,20 we further examined the expression of CYLD by immunofluorescence staining. Indeed, TXNIPM-KO increased macrophage CYLD expression in ischaemic livers compared with the TXNIPFL/FL controls (Fig. 3D). Moreover, TXNIPM-KO augmented CYLD and nuclear NRF2 protein expression (Fig. 3E). Unlike the TXNIPFL/FL controls, the ROS production was significantly reduced in the TXNIPM-KO Kupffer cells from ischaemic livers without or with lipopolysaccharide (LPS) stimulation (Fig. 3F and Fig. S5). Therefore, these data indicate that macrophage TXNIP deficiency promotes CYLD and activates NRF2-mediated antioxidative responses in IR-stressed livers.
Fig. 3.

Disruption of myeloid-specific TXNIP promotes CYLD and activates the NRF2 pathway in IR-stressed liver.
The WT, TXNIPFL/FL, and TXNIPM-KO mice were subjected to 90 min of partial liver warm ischaemia, followed by 6 h of reperfusion. (A) Western-assisted analysis and relative density ratio of CYLD, NOX4, and NRF2 in IR-stressed livers. (B) Western-assisted analysis and relative density ratio of CYLD and NRF2 in the TXNIPFL/FL and TXNIPM-KO livers after IR stress. (C) Quantitative RT-PCR analysis of NQO1, GCLC, and GCLM mRNA levels in ischaemic livers (n = 6 samples/group). (D) Immunofluorescence staining of CYLD and CD68 in ischaemic livers (n = 6 mice/group). Scale bars, 100 μm. (E) The Kupffer cells were isolated from ischaemic livers, and then these cells were cultured for 2 h at 37 °C. Western-assisted analysis and relative density ratio of CYLD and nuclear NRF2 in Kupffer cells from the TXNIPFL/FL and TXNIPM-KO livers after IR stress. (F) The Kupffer cells (2 × 105/each group) were isolated from ischaemic livers and then cultured for 2 h. Detection of ROS production by Carboxy-H2DFFDA in Kupffer cells from the TXNIPFL/FL and TXNIPM-KO livers after IR stress. Quantification of ROS-producing Kupffer cells (green) (n = 6 mice/group). Scale bars, 200 μm. All Western blots represent 4 experiments, and the data represent the mean ± SD. Statistical analysis was performed using the Permutation t test. ∗∗p <0.01, ∗∗∗p <0.005. CYLD, cyclindromatosis; GCLC, glutamate-cysteine ligase catalytic subunit; GCLM, glutamate-cysteine ligase regulatory subunit; H2DFFDA, 2',7'-dihydrofluorescein diacetate; IR, ischaemia/reperfusion; NOX4, NADPH oxidase 4; NQO1, NAD(P)H quinone dehydrogenase 1; NRF2, nuclear factor (erythroid-derived 2)-like 2; ROS, reactive oxygen species; TXNIP, thioredoxin-interacting protein; TXNIPFL/FL, floxed TXNIP; TXNIPM-KO, myeloid-specific TXNIP knockout; WT, wild type.
CYLD interacts with NOX4 and regulates NRF2 activation by deubiquitinating NOX4 in macrophages
Having demonstrated that macrophage TXNIP deficiency promoted CYLD and activated the NRF2 pathway in ischaemic livers, we next analysed putative crosstalk between CYLD and the NRF2 pathway in macrophages. Indeed, a Western blot assay revealed that TXNIPM-KO augmented the CYLD protein expression in LPS-stimulated BMMs compared with the TXNIPFL/FL controls (Fig. 4A). This result was confirmed by immunofluorescence staining, which showed increased CYLD expression in BMMs after LPS stimulation (Fig. 4B). Strikingly, the co-immunoprecipitation assay revealed CYLD bound to endogenous NOX4 in BMMs after LPS stimulation (Fig. 4C). However, CYLD could not interact with NOX4 in BMMs without LPS stimulation (Fig. S6). Immunofluorescence staining revealed that CYLD co-localised with NOX4 in LPS-stimulated BMMs (Fig. 4D). We then determined whether CYLD influenced NOX4 ubiquitination. Interestingly, CRISPR/Cas9-mediated CYLD activation reduced the ubiquitination of NOX4 in LPS-stimulated BMMs (Fig. 4E). Furthermore, disruption of CYLD diminished NOX4 and NRF2 in TXNIPM-KO BMMs after LPS stimulation compared with the control groups (Fig. 4F). Therefore, these results suggest that the CYLD–NOX4 axis is essential for activating the NRF2 pathway in macrophage TXNIP-mediated immune regulation.
Fig. 4.

CYLD interacts with NOX4 and regulates NRF2 activation by deubiquitinating NOX4 in macrophages.
BMMs (1 × 106) were cultured with LPS (100 ng/ml) for 6 h. (A) Western blot analysis of CYLD in LPS-stimulated macrophages from the TXNIPFL/FL and TXNIPM-KO mice. (B) Immunofluorescence staining for CYLD expression in macrophages after LPS stimulation (n = 3–4 samples/group). DAPI was used to visualise nuclei. Scale bars, 30 μm. (C) Immunoprecipitation analysis of CYLD and NOX4 in LPS-stimulated macrophages. (D) Immunofluorescence staining for macrophage CYLD (green) and NOX4 (red) colocalisation in LPS-stimulated macrophages. DAPI was used to visualise nuclei (blue). Scale bars, 30 μm. (E) BMMs were transfected with CRISPR-mediated CYLD activation or control plasmid for 48 h. An immunoprecipitation assay was performed with anti-NOX4 and Ub antibodies. Representative of 3 experiments. (F) BMMs were transfected with CRISPR/Cas9-mediated CYLD KO or control vector. Western-assisted analysis and relative density ratio of CYLD, NOX4, and nuclear NRF2 in LPS-stimulated macrophages from the TXNIPM-KO mice. All immunoblots represent 4 experiments, and the data represent the mean ± SD. Statistical analysis was performed using the permutation t test. ∗∗p <0.01, ∗∗∗p <0.005. BMM, bone marrow-derived macrophage; Cas9, CRISPR associated protein 9; CRISPR, clustered regularly interspaced short palindromic repeats; CYLD, cyclindromatosis; GCLC, glutamate-cysteine ligase catalytic subunit; GCLM, glutamate-cysteine ligase regulatory subunit; IB, Immunoblotting; IP, Immunoprecipitation; IR, ischaemia/reperfusion; KO, knockout; LPS, lipopolysaccharide; NOX4, NADPH oxidase 4; NQO1, NAD(P)H quinone dehydrogenase 1; NRF2, nuclear factor (erythroid-derived 2)-like 2; ROS, reactive oxygen species; TXNIP, thioredoxin-interacting protein; TXNIPFL/FL, floxed TXNIP; TXNIPM-KO, myeloid-specific TXNIP KO; WT, wild type.
NRF2 targets OASL1 and modulates TBK1-mediated inflammatory response in macrophages
To explore the potential mechanism of the NRF2 in the modulation of TBK1-mediated inflammation in macrophages, we performed NRF2 chromatin immunoprecipitation (ChIP) coupled to massively parallel sequencing (ChIP-Seq). Indeed, the NRF2 ChIP-Seq peaks were identified within the OASL1 gene. One presented the location in the promoter region, and the others were found to locate within the intron/exon of the OASL1 gene (Fig. 5A). To validate the ChIP-seq peak situated in the OASL1 promoter region, ChIP-PCR was performed using NRF2 antibodies in LPS-stimulated BMMs. After ChIP with the NRF2 antibody, the primer was designed to detect the NRF2 DNA-binding site in the OASL1 promoter by PCR analysis, confirming that NRF2 is located at the promoter region of OASL1 (Fig. 5B). This suggests that OASL1 is a target gene regulated by NRF2. Moreover, the immunofluorescence staining assay showed that TXNIPM-KO augmented NRF2 expression (Fig. 5C) with increased OASL1 mRNA and protein expression (Fig. 5D) in LPS-stimulated BMMs compared with the TXNIPFL/FL controls. However, KO of NRF2 diminished OASL1 but augmented p-TBK1 and p-IRF3 expression (Fig. 5E), accompanied by increased ROS production (Fig. 5F) in TXNIPM-KO BMMs after LPS stimulation. Collectively, these data indicate that NRF2 and its target gene OASL1 are crucial for macrophage TXNIP-mediated immune regulation.
Fig. 5.

NRF2 targets OASL1 and modulates TBK1-mediated inflammatory response in macrophages.
BMMs were collected and fixed after incubating LPS (100 ng/ml). Following chromatin shearing and NRF2 antibody selection, the precipitated DNA fragments bound by NRF2-containing protein complexes were used for sequencing. (A) Localisation of NRF2-binding sites on the mouse OASL1 gene. The 15 exons, 14 introns, 3′ UTR, 5′ UTR, and TSS of the mouse OASL1 gene on chromosome 5 are shown. (B) ChIP-PCR analysis of NRF2 binding to the OASL1 promoter. Protein-bound chromatin was prepared from BMMs and immunoprecipitated with NRF2 antibody, and then the immunoprecipitated DNA was analysed by PCR. The normal IgG was used as a negative control. (C) Immunofluorescence staining for nuclear NRF2 expression in LPS-stimulated macrophages from the TXNIPFL/FL and TXNIPM-KO mice (n = 3–4 samples/group). Scale bars, 30 μm. (D) Analysis of OASL1 mRNA levels and protein expression of LPS-stimulated macrophages from the TXNIPFL/FL and TXNIPM-KO mice. (E) BMMs were transfected with CRISPR/Cas9-mediated NRF2 KO or control vector. Western blot analysis of OASL1, p-TBK1, TBK1, p-IRF3, and IRF3 in LPS-stimulated macrophages from the TXNIPM-KO mice. (F) Detection of ROS production by Carboxy-H2DFFDA in LPS-stimulated macrophages from the TXNIPM-KO mice. Quantification of ROS-producing macrophages (green) (n = 4 samples/group). Scale bars, 100 μm. All immunoblots represent 4 experiments, and the data represent the mean ± SD. Statistical analysis was performed using the permutation t test. ∗∗p <0.01. BMM, bone marrow-derived macrophage; Cas9, CRISPR associated protein 9; CRISPR, clustered regularly interspaced short palindromic repeats; IRF3, interferon regulatory factor 3; KO, knockout; LPS, lipopolysaccharide; NRF2, nuclear factor (erythroid-derived 2)-like 2; OASL1, 2′,5′ oligoadenylate synthetase-like 1; ROS, reactive oxygen species; TBK1, TANK-binding kinase 1; TSS, transcription start sites; TXNIP, thioredoxin-interacting protein; TXNIPFL/FL, floxed TXNIP; TXNIPM-KO, myeloid-specific TXNIP KO; UTR, untranslated region.
OASL1 inhibits TBK1-mediated inflammation through regulation of G3BP1 activation in macrophages
To dissect the mechanistic role of OASL1 in the regulation of TBK1 activation in the TXNIP/NRF2-mediated immune regulation, BMMs were isolated from the TXNIPFL/FL and TXNIPM-KO mice and then transfected with CRISPR/Cas9-mediated OASL1 activation or OASL1 KO vector. Indeed, LPS stimulation activated G3BP1, whereas CRISPR/Cas9-mediated OASL1 activation diminished G3BP1 expression in TXNIPFL/FL macrophages (Fig. 6A). This result was confirmed by immunofluorescence staining, which showed that induction of macrophage OASL1 reduced G3BP1 expression (Fig. 6B). Interestingly, TXNIPM-KO increased OASL1 but reduced G3BP1 expression, whereas CRISPR/Cas9-mediated OASL1 KO augmented G3BP1 expression in LPS-stimulated TXNIPM-KO macrophages (Fig. 6C). Immunofluorescence staining further unveiled that disruption of OASL1 increased macrophage G3BP1 expression compared with the control vector-treated groups (Fig. 6D). Moreover, CRISPR/Cas9-mediated G3BP1 KO diminished p-TBK1, p-IRF3, and p-P65 expression (Fig. 6E), accompanied by reduced mRNA levels coding for IL-6, TNF-α, CXCL-10, and IFN-β (Fig. 6F) in TXNIPM-KO macrophages after LPS stimulation. These results indicate that OASL1 controls G3BP1, which is critically involved in activating TBK1-mediated inflammatory response.
Fig. 6.

OASL1 inhibits TBK1-mediated inflammation through regulation of G3BP1 activation in macrophages.
(A) BMMs were isolated from TXNIPFL/FL mice transfected with p-CRISPR-OASL1 activation or control vector followed by 6 h of LPS (100 ng/ml) stimulation. Western-assisted analysis of OASL1 and G3BP1. (B) Immunofluorescence staining for the G3BP1 expression in LPS-stimulated macrophages after transfecting p-CRISPR-OASL1 activation or control vector (n = 3–4 samples/group). DAPI was used to visualise nuclei. Scale bars, 30 μm. (C) BMMs from TXNIPM-KO mice were transfected with the p-CRISPR-OASL1 KO or control vector followed by 6 h of LPS stimulation. Western blot analysis of OASL1 and G3BP1. (D) Immunofluorescence staining for the G3BP1 expression in LPS-stimulated macrophages after transfecting p-CRISPR-OASL1 KO or control vector (n = 3–4 samples/group). DAPI was used to visualise nuclei. Scale bars, 30 μm. (E) BMMs from TXNIPFL/FL mice were transfected with the p-CRISPR-G3BP1 KO or control vector followed by 6 h of LPS stimulation. Western blot analysis and relative density ratio of p-TBK1, TBK1, p-IRF3, IRF3, p-P65, and P65. (F) Quantitative RT-PCR analysis of IL-6, TNF-α, CXCL-10, and IFN-β mRNA levels in LPS-stimulated macrophages after transfecting p-CRISPR-G3BP1 KO or control vector (n = 4 samples/group). All Western blots represent 4 experiments, and the data represent the mean ± SD. Statistical analysis was performed using the permutation t test. ∗p <0.05, ∗∗p <0.01, ∗∗∗p <0.005. BMM, bone marrow-derived macrophage; CRISPR, clustered regularly interspaced short palindromic repeats; CXCL-10, chemokine (C-X-C motif) ligand 10; G3BP1, Ras GTPase-activating protein-binding protein 1; INF-β, interferon-β; IRF3, interferon regulatory factor 3; KO, knockout; LPS, lipopolysaccharide; OASL1, 2′,5′ oligoadenylate synthetase-like 1; TBK1, TANK-binding kinase 1; TNF-α, tumour necrosis factor-alpha; TXNIP, thioredoxin-interacting protein; TXNIPFL/FL, floxed TXNIP; TXNIPM-KO, myeloid-specific TXNIP KO.
OASL1 is essential for inhibiting G3BP1/TBK1 activation and cell death in myeloid TXNIP-deficient livers in response to IR stress
Having demonstrated the importance of NRF2 target gene OASL1 in macrophage TXNIP-mediated immune regulation in macrophages, we then examined whether OASL1 influenced G3BP1/TBK1 activation in IR-stressed livers. We disrupted OASL1 in TXNIPM-KO livers with an in vivo mannose-mediated OASL1 siRNA delivery system that specifically delivers to macrophages by expressing a mannose-specific membrane receptor as previously described.1,5,6 Indeed, mannose-mediated AlexaFluor488-labelled siRNA (green) delivery was efficiently transduced into macrophages (red) in IR-stressed livers (Fig. 7A). Knockdown of OASL1 in the TXNIPM-KO mice with the mannose-mediated siRNA treatment aggravated IR-induced liver damage as evidenced by increased Suzuki’s histological score (Fig. 7B) and sALT and sAST levels (Fig. S7), compared with the NS siRNA-treated controls. Moreover, OASL1 siRNA treatment in the TXNIPM-KO ischaemic livers increased CD11b+ macrophage (Fig. 7C) and Ly6G+ neutrophil (Fig. 7D) accumulation. Unlike NS siRNA-treated controls, OASL1 siRNA treatment augmented G3BP1, p-TBK1, and p-IRF3 expression (Fig. 7E) in the TXNIPM-KO livers. Intriguingly, OASL1 siRNA treatment augmented hepatocyte apoptosis and necroptosis, as evidenced by increased TUNEL+ cells and receptor-interacting serine/threonine-protein kinase 3 (RIPK3) expression in IR-stressed livers compared with the NS siRNA-treated livers (Fig. 7F and Fig. S8). These results demonstrate that OASL1 is a key regulator of TBK1 activation and cell death in IR-stressed livers. Furthermore, adoptive transfer of TBK1-overexpressing BMMs reversed TXNIPM-KO-mediated cytoprotection and augmented IRF3/NF-κB activation and pro-inflammatory mediators in ischaemic livers (Fig. S9), suggesting that the TBK1 is essential in mediating TXNIP-induced immune and inflammatory response during liver IRI.
Fig. 7.

OASL1 is essential for inhibiting G3BP1/TBK1 activation and cell death in myeloid TXNIP-deficient livers in response to IR.
The TXNIPM-KO mice were injected via tail vein with OASL1 siRNA (2.5 mg/kg) or NS control siRNA mixed with mannose-conjugated polymers at 4 h before ischaemia. (A) Immunofluorescence staining of AlexaFluor488-labelled control siRNA and CD68-positive macrophages in ischaemic liver tissue (n = 6 mice/group), Scale bars, 25 μm. (B) Representative histological staining (H&E) of ischaemic liver tissue (n = 6 mice/group) and Suzuki’s histological score. Scale bars, 200 μm. (C) Immunofluorescence staining of CD11b+ macrophages in ischaemic livers (n = 6 mice/group). Quantification of CD11b+ macrophages. Scale bars, 100 μm. (D) Immunohistochemistry staining of Ly6G+ neutrophils in ischaemic livers (n = 6 mice/group). Quantification of Ly6G+ neutrophils. Scale bars, 200 and 50 μm. (E) Western blot analysis and relative density ratio of G3BP1, p-TBK1, and p-IRF3. Representative of 4 experiments. (F) Liver apoptosis by TUNEL staining in ischaemic livers (n = 6 mice/group). Results were scored semiquantitatively by averaging the number of apoptotic cells. Scale bars, 100 μm. All data represent the mean ± SD. Statistical analysis was performed using the permutation t test. ∗∗p <0.01, ∗∗∗p <0.005. G3BP1, Ras GTPase-activating protein-binding protein 1; HPF, high power field; IR, ischaemia/reperfusion; IRF3, interferon regulatory factor 3; NS, non-specific; OASL1, 2′,5′ oligoadenylate synthetase-like 1; siRNA, small interfering RNA; TBK1, TANK-binding kinase 1; TXNIP, thioredoxin-interacting protein; TXNIPM-KO, myeloid-specific TXNIP knockout.
Macrophage TXNIP deficiency-mediated OASL1 inhibits stress-induced hepatocyte death via modulating Apaf1/cytochrome c/caspase-9 activation
We then determined how macrophage OASL1 might regulate the hepatocyte death pathway under inflammatory and stress conditions. As oxidative and ER stress induces TNF-α, which plays a vital role in caspase-dependent cell death,21 we then explored the mechanistic role of OASL1 in regulating the cell apoptotic/necroptotic pathway. We found increased TNF-α release from LPS-stimulated TXNIPM-KO BMMs after transfection with a CRISPR-OASL1 KO vector compared with the control groups (Fig. 8A). Using a coculture system containing LPS-stimulated CRISPR-OASL1 KO BMMs from the TXNIPM-KO mice and primary hepatocytes supplemented with H2O2 (Fig. 8B), we found that macrophage OASL1 KO augmented hepatocyte apoptotic peptidase activating factor 1 (Apaf1), cytochrome c, cleaved caspase-9, cleaved caspase-3, and RIPK3 expression (Fig. 8C). Unlike the control groups, macrophage OASL1 KO markedly increased LDH release from stressed hepatocytes by H2O2 in the coculture supernatant (Fig. 8D). Furthermore, immunofluorescence staining showed increased TUNEL+ hepatocytes in the CRISPR-OASL1 KO BMMs but not in the control cells after coculture (Fig. 8E). Given that caspase-3 acts as an executioner caspase of apoptosis and RIPK3 is a critical mediator of necroptosis,22,23 our findings indicate that macrophage OASL1 inhibits caspase-3-induced apoptosis and RIPK3-mediated necroptosis by regulating Apaf1/cytochrome c/caspase-9 activation in response to cell stress.
Fig. 8.

Macrophage TXNIP deficiency-mediated OASL1 inhibits stress-induced hepatocyte death via modulating Apaf1/Cyt c/caspase-9 activation.
BMMs were isolated from TXNIPM-KO mice and transfected with the p-CRISPR-OASL1 KO or control vector followed by LPS stimulation. (A) ELISA analysis of supernatant TNF-α levels in LPS-stimulated BMMs (n = 4 samples/group). (B) The schematic figure for the macrophage/hepatocyte coculture system. (C) BMMs transfected with the p-CRISPR-OASL1 KO were stimulated with LPS and then cocultured with primary hepatocytes supplemented with or without H2O2 for 24 h. Western-assisted analysis and relative density ratio of Apaf-1, Cyt c, cleaved caspase-9, cleaved caspase-3, and RIPK3 in hepatocytes after coculture. Representative of 4 experiments. (D) LDH release from hepatocytes in cocultures (n = 4 samples/group). (E) Immunofluorescence staining of TUNEL+ hepatocytes after coculture (n = 4 samples/group). Scale bars, 100 μm. All data represent the mean ± SD. Statistical analysis was performed using Permutation t-test. ∗p <0.05, ∗∗p <0.01, ∗∗∗p <0.001. Apaf1, apoptotic peptidase activating factor 1; BMM, bone marrow-derived macrophage; CRISPR, clustered regularly interspaced short palindromic repeats; Cyt c, cytochrome c; KO, knockout; LPS, lipopolysaccharide; OASL1, 2′,5′ oligoadenylate synthetase-like 1; TNF-α, tumour necrosis factor-alpha; TXNIP, thioredoxin-interacting protein; TXNIPM-KO, myeloid-specific TXNIP KO; RIPK3, receptor-interacting serine/threonine-protein kinase 3.
Discussion
In this study, we revealed that disruption of macrophage TXNIP activated the NRF2-OASL1 pathway and regulated TBK1 function and apoptotic/necroptotic cell death pathway in IR stress-induced liver inflammatory injury. Notably, we showed that macrophage TXNIP deficiency promoted CYLD, which interacted with NOX4 and enhanced NRF2 and its target gene OASL1 activity, leading to inhibited STING-mediated TBK1 activation and hepatocyte apoptosis/necroptosis in liver IRI. Our results demonstrate that the macrophage CYLD–NRF2–OASL1 axis by TXNIP is a critical regulator of the STING/TBK1-mediated innate immune responses and apoptotic/necroptotic cell death pathway in IR stress-induced liver inflammatory injury.
TXNIP is a multifunctional protein that modulates oxidative and ER stress and innate immune responses. During stress, TXNIP induces ROS production and causes ER stress, initiating various cellular signalling processes, including cellular redox homoeostasis, inflammation, and immune response.24 STING is predominantly associated with ER, where TBK1 recruitment to STING induces IRF3 and NF-κB activation, mediating innate immune response.25 As a downstream of STING kinase, TBK1 has been shown to modulate macrophage-mediated inflammatory responses.26 Activation of TBK1 augmented lipotoxic-induced liver inflammation, whereas inhibition of TBK1 alleviated liver damage in mouse models of inflammatory injury.27 Indeed, TBK1 is required for the TLR-mediated activation of IRF3 and IFN responses in macrophages.28 Disruption of myeloid TBK1 diminished macrophage activation and pro-inflammatory mediators and reduced tissue inflammation.29 Thus, TBK1 acts as a critical player in controlling inflammatory and immune responses. Indeed, many kinases can be induced by a redox-mediated pathway in response to oxidative stress. IR stress activated Src kinase, NADPH oxidase 2 (NOX2), and apoptosis signal-regulating kinase 1 (ASK1) with increased pro-inflammatory mediators in ischaemic livers and Kupffer cells (Fig. S10). Consistent with previous reports that both NOX2 and ASK1 contribute to tissue inflammation and apoptosis in response to oxidative stress,30,31 these data suggest that Src-mediated NOX2/ASK1 signalling may play a role in liver IRI. Therefore, the mechanism of IR stress-induced liver injury could be involved in multiple signalling pathways. Our current study revealed that IR stress promoted TXNIP activation and the STING/TBK1 pathway in ischaemic livers. Disruption of myeloid TXNIP inhibited TBK1-dependent IRF3 and NF-κB activation and ameliorated IR stress-induced liver injury. Our findings demonstrate the distinct ability of macrophage TXNIP in controlling TBK1-mediated innate immune response and inflammatory cascades in IR-stressed livers.
The mechanisms underlying macrophage TXNIP-mediated immune regulation appear to link multiple signal transduction pathways. We found that IR stress promoted CYLD, a multifunctional deubiquitinating enzyme (DUB) that modulates the inflammatory cascade in immune cells via inhibiting the ubiquitination of key signalling molecules.20 Indeed, the CYLD protein, primarily located in the cytoplasm, is ubiquitously expressed and highly conserved in Kupffer cells of human and murine liver tissue. CYLD recognises and cleaves K63-linked ubiquitin chains to promote protein–protein interactions in the assembly of signalling molecule complex.32 CYLD can inhibit NF-κB upstream signalling molecules and regulate inflammatory response by deconjugating the K63-linked ubiquitin chains.33 Disruption of CYLD induces hepatocyte death and activates Kupffer cells, which promotes inflammation and pro-inflammatory mediators,34 suggesting that CYLD is an essential regulator of immune and inflammatory responses. In line with these findings, we found that myeloid-specific TXNIP deficiency promoted CYLD and NRF2, as evidenced by increased CYLD and nuclear NRF2 protein expression in IR-stressed livers. This indicates the importance of CYLD and NRF2 in macrophage TXNIP-mediated immune regulation during liver IRI.
Nrf2 is a transcription factor that regulates the cellular defence against oxidative stress or ER stress via activating antioxidant genes. Under cell stress conditions, Nrf2 is translocated to the nucleus and activates the transcription of its downstream target genes.35 Increasing evidence shows that NRF2 plays a central role in modulating the innate immune response to oxidative and ER stress.36 Our previous studies demonstrated that disruption of NRF2 exacerbated IR stress-induced liver damage, whereas activation of the NRF2 pathway enhanced its antioxidant responses and attenuated IR-triggered liver inflammation.37 Consistent with previous findings, we found that TXNIPM-KO enhanced nuclear NRF2 activity and increased antioxidant NQO1, GCLC, and GCLM expression in ischaemic livers, indicating that NRF2 could be critical in macrophage TXNIP-mediated immune regulation.
The question arises as to what mechanisms may confer TXNIP to selectively influence the CYLD–NRF2 axis in the modulation of STING-mediated TBK1 activation in IR-stressed liver. Using the in vitro culture system, we found that TXNIPM-KO markedly increased macrophage CYLD expression after LPS stimulation. Interestingly, IR stress activated NOX4, an essential modulator of redox signalling in response to ER stress.38 Under cell stress conditions, NOX4 activated the NRF2-dependent pathway.39 In contrast, disruption of NOX4 resulted in reduced NRF2-mediated antioxidant responses,40 suggesting that NOX4 may be essential for the modulation of the CYLD–NRF2 axis in macrophage TXNIP-mediated immune regulation. Moreover, the in vitro study revealed that CYLD and NOX4 colocalised in LPS-stimulated macrophages. Interestingly, CRISPR-mediated CYLD activation depressed NOX4 ubiquitination. NOX4 was deubiquitinated via direct interaction with CYLD. Disruption of CYLD reduced NOX4 and nuclear NRF2 expression in TXNIPM-KO macrophages after LPS stimulation. These results indicate that NOX4 mediates the CYLD–NRF2 pathway, crucial for the macrophage TXNIP-mediated immune modulation in IR stress-mediated inflammation.
However, how NRF2 may modulate TBK1-mediated inflammatory response in IR-stressed liver remains unclear. In line with our in vivo findings, which showed that TXNIPM-KO increased NRF2 expression in IR-stressed livers, our in vitro data revealed that nuclear NRF2 was upregulated in TXNIPM-KO macrophages after LPS stimulation. Strikingly, using the ChIP and ChIP-sequencing approaches, we found that NRF2 was localised on the promoter of OASL1, suggesting that OASL1 is a target gene of NRF2. Indeed, TXNIPM-KO augmented macrophage OASL1 expression in response to LPS stimulation. Disruption of macrophage NRF2 diminished OASL1 yet enhanced TBK1 activation and ROS production. Our findings revealed the distinct role of NRF2 in modulating OASL1 activity and TBK1 function in IR stress-induced liver injury.
Indeed, our findings revealed that OASL1 is critical for NRF2-mediated regulation of TBK1 activation in IR-stressed livers. OASL1 is the major protein that regulates IFN signalling and its downstream genes during inflammatory and immune responses.41 Activation of OASL1 inhibits the translation of IFN-regulatory factor 7 (IRF7), the master transcription factor for type I IFN production.42 Upon activation, IRF7 forms a transcriptional complex enhanceosome with IRF3 to activate the IFN-β response.43 Moreover, TBK1 is pivotal for the IRF3 and NF-κB activation in the innate immune response.44 Thus, we speculate that OASL1 is necessary for the modulation of TBK1-driven inflammatory response in IR-induced liver injury. Further evidence was provided by the in vivo study, in which disruption of OASL1 reversed myeloid TXNIP deficiency-mediated cytoprotection, as evidenced by augmented IR-induced liver injury and enhanced TBK1 and IRF3 activity. Interestingly, OASL1 knockdown upregulated G3BP1 activation in IR-stressed livers, suggesting that G3BP1 may be involved in TBK1-mediated inflammatory response during liver IRI. Indeed, G3BP1 promoted cyclic GMP-AMP synthase (cGAS) activation, whereas disruption of G3BP1 dampened cGAS-mediated IFN response.45 In line with these findings, we found that disruption of G3BP1 inhibited TBK1, IRF3, and NF-κB activation, accompanied by reduced IFN-β production in the TXNIPFL/FL macrophages under inflammatory stimulation. Therefore, these results reveal a novel role of OASL1 in controlling dynamic crosstalk with G3BP1/TBK1 in macrophage TXNIP-mediated immune regulation.
Of particular interest, macrophage TXNIP-mediated OASL1 could be involved in regulating IR-induced cell death pathways. Indeed, oxidative and ER stress induces TNF-α and ROS production. ROS are considered potent intrinsic stimuli to trigger cell death (apoptosis and necrosis) by multiple pathways.46 ROS can trigger cytochrome c release, a crucial mediator of the intrinsic apoptotic pathway.47 Cytochrome c binds to Apaf1 to form a procaspase-9-activating heptameric protein complex named apoptosome, which sequentially activates the caspase-9 and the effector caspases-3.48 Thus, cytochrome c and Apaf1 are essential for initiating an apoptotic protease cascade in response to oxidative and ER stress. The current study revealed that CRISPR/Cas9-mediated OASL1 KO increased TNF-α release in TXNIPM-KO macrophages after LPS stimulation. Disruption of macrophage OASL1 augmented hepatocyte cytochrome c, Apaf1, cleaved caspase-9, cleaved caspase-3 expression, LDH release, and cell apoptosis from H2O2-treated hepatocytes after coculture. Indeed, both apoptosis and necrosis can occur in the same cells in TNF-induced cell death.46 IR-induced hepatocellular necrosis is an inflammatory cell death (necroptosis).49 A recent study showed that caspase-9 is a crucial regulator of stress-induced apoptotic and necroptotic cell death by interacting with RIPK3, a central player in necroptosis.50 Consistent with these findings, we revealed that macrophage OASL1 deficiency promoted RIPK3 in IR-stressed livers and in vitro cocultures. Therefore, our results indicate that macrophage TXNIP deficiency-mediated OASL1 inhibits hepatocyte apoptosis and necroptosis by modulating cytochrome c/Apaf1/caspase-9 activation.
In conclusion, we have identified a previously unrecognised role of macrophage TXNIP-induced CYLD–NRF2–OASL1 signalling on TBK1 function and apoptotic/necroptotic cell death in liver IRI. We have also demonstrated that the CYLD–NRF2–OASL1 axis controls TBK1-dependent inflammation and cell death in response to IR stress. The target gene OASL1 regulated by NRF2 is crucial for modulating TBK1 function and hepatocyte death. By identifying the molecular regulatory mechanism of macrophage TXNIP-mediated CYLD–NRF2–OASL1 pathway in IR-stressed livers, our findings provide potential therapeutic targets for stress-induced liver inflammation and injury.
Financial support
This work was supported by the NIH grants R01AI139552, R21AI146742, R21AI112722, and R21AI115133.
Authors’ contributions
Performed in vivo and in vitro experiments and data analysis: YZ, DX, YT, XQ. Performed in vitro experiments: MS, MK, LJ. Generated conditional knockout mice: DX, YT, YL. Participated in scientific discussion: QX, FMK, DGF. Contributed to the study concept, research design, data analysis, and wrote the manuscript: BK.
Data availability statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Conflicts of interest
The authors declare no conflict of interest.
Please refer to the accompanying ICMJE disclosure forms for further details.
Footnotes
Author names in bold designate shared co-first authorship
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jhepr.2022.100532.
Supplementary data
The following are the supplementary data to this article:
References
- 1.Li C., Sheng M., Lin Y., Xu D., Tian Y., Zhan Y., et al. Functional crosstalk between myeloid Foxo1-beta-catenin axis and Hedgehog/Gli1 signaling in oxidative stress response. Cell Death Differ. 2021;28:1705–1719. doi: 10.1038/s41418-020-00695-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dara L., Ji C., Kaplowitz N. The contribution of endoplasmic reticulum stress to liver diseases. Hepatology. 2011;53:1752–1763. doi: 10.1002/hep.24279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Soehnlein O., Lindbom L. Phagocyte partnership during the onset and resolution of inflammation. Nat Rev Immunol. 2010;10:427–439. doi: 10.1038/nri2779. [DOI] [PubMed] [Google Scholar]
- 4.Lotze M.T., Zeh H.J., Rubartelli A., Sparvero L.J., Amoscato A.A., Washburn N.R., et al. The grateful dead: damage-associated molecular pattern molecules and reduction/oxidation regulate immunity. Immunol Rev. 2007;220:60–81. doi: 10.1111/j.1600-065X.2007.00579.x. [DOI] [PubMed] [Google Scholar]
- 5.Yue S., Zhu J., Zhang M., Li C., Zhou X., Zhou M., et al. The myeloid heat shock transcription factor 1/beta-catenin axis regulates NLR family, pyrin domain-containing 3 inflammasome activation in mouse liver ischemia/reperfusion injury. Hepatology. 2016;64:1683–1698. doi: 10.1002/hep.28739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lu L., Yue S., Jiang L., Li C., Zhu Q., Ke M., et al. Myeloid Notch1 deficiency activates the RhoA/ROCK pathway and aggravates hepatocellular damage in mouse ischemic livers. Hepatology. 2018;67:1041–1055. doi: 10.1002/hep.29593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xu D., Tian Y., Xia Q., Ke B. The cGAS–STING pathway: novel perspectives in liver diseases. Front Immunol. 2021;12 doi: 10.3389/fimmu.2021.682736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hopfner K.P., Hornung V. Molecular mechanisms and cellular functions of cGAS–STING signalling. Nat Rev Mol Cell Biol. 2020;21:501–521. doi: 10.1038/s41580-020-0244-x. [DOI] [PubMed] [Google Scholar]
- 9.Ishikawa H., Barber G.N. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature. 2008;455:674–678. doi: 10.1038/nature07317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang W., Hu D., Wu C., Feng Y., Li A., Liu W., et al. STING promotes NLRP3 localization in ER and facilitates NLRP3 deubiquitination to activate the inflammasome upon HSV-1 infection. PLoS Pathog. 2020;16 doi: 10.1371/journal.ppat.1008335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cruz V.H., Arner E.N., Wynne K.W., Scherer P.E., Brekken R.A. Loss of Tbk1 kinase activity protects mice from diet-induced metabolic dysfunction. Mol Metab. 2018;16:139–149. doi: 10.1016/j.molmet.2018.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Luo X., Li H., Ma L., Zhou J., Guo X., Woo S.L., et al. Expression of STING is increased in liver tissues from patients with NAFLD and promotes macrophage-mediated hepatic inflammation and fibrosis in mice. Gastroenterology. 2018;155:1971–1984. doi: 10.1053/j.gastro.2018.09.010. e1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yu Y., Liu Y., An W., Song J., Zhang Y., Zhao X. STING-mediated inflammation in Kupffer cells contributes to progression of nonalcoholic steatohepatitis. J Clin Invest. 2019;129:546–555. doi: 10.1172/JCI121842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lane T., Flam B., Lockey R., Kolliputi N. TXNIP shuttling: missing link between oxidative stress and inflammasome activation. Front Physiol. 2013;4:50. doi: 10.3389/fphys.2013.00050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yoshihara E., Masaki S., Matsuo Y., Chen Z., Tian H., Yodoi J. Thioredoxin/Txnip: redoxisome, as a redox switch for the pathogenesis of diseases. Front Immunol. 2014;4:514. doi: 10.3389/fimmu.2013.00514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhou R., Tardivel A., Thorens B., Choi I., Tschopp J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat Immunol. 2010;11:136–140. doi: 10.1038/ni.1831. [DOI] [PubMed] [Google Scholar]
- 17.Zhao Q., Che X., Zhang H., Fan P., Tan G., Liu L., et al. Thioredoxin-interacting protein links endoplasmic reticulum stress to inflammatory brain injury and apoptosis after subarachnoid haemorrhage. J Neuroinflammation. 2017;14:104. doi: 10.1186/s12974-017-0878-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Oslowski C.M., Hara T., O'Sullivan-Murphy B., Kanekura K., Lu S., Hara M., et al. Thioredoxin-interacting protein mediates ER stress-induced beta cell death through initiation of the inflammasome. Cell Metab. 2012;16:265–273. doi: 10.1016/j.cmet.2012.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Suzuki S., Toledo-Pereyra L.H., Rodriguez F.J., Cejalvo D. Neutrophil infiltration as an important factor in liver ischemia and reperfusion injury. Modulating effects of FK506 and cyclosporine. Transplantation. 1993;55:1265–1272. doi: 10.1097/00007890-199306000-00011. [DOI] [PubMed] [Google Scholar]
- 20.Mathis B.J., Lai Y., Qu C., Janicki J.S., Cui T. CYLD-mediated signaling and diseases. Curr Drug Targets. 2015;16:284–294. doi: 10.2174/1389450115666141024152421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Micheau O., Tschopp J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell. 2003;114:181–190. doi: 10.1016/s0092-8674(03)00521-x. [DOI] [PubMed] [Google Scholar]
- 22.Porter A.G., Jänicke R.U. Emerging roles of caspase-3 in apoptosis. Cell Death Differ. 1999;6:99–104. doi: 10.1038/sj.cdd.4400476. [DOI] [PubMed] [Google Scholar]
- 23.Orozco S., Oberst A. RIPK3 in cell death and inflammation: the good, the bad, and the ugly. Immunol Rev. 2017;277:102–112. doi: 10.1111/imr.12536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang K., Kaufman R.J. From endoplasmic-reticulum stress to the inflammatory response. Nature. 2008;454:455–462. doi: 10.1038/nature07203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yum S., Li M., Fang Y., Chen Z.J. TBK1 recruitment to STING activates both IRF3 and NF-kappaB that mediate immune defense against tumors and viral infections. Proc Natl Acad Sci USA. 2021;118 doi: 10.1073/pnas.2100225118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yu T., Yi Y.S., Yang Y., Oh J., Jeong D., Cho J.Y. The pivotal role of TBK1 in inflammatory responses mediated by macrophages. Mediators Inflamm. 2012;2012 doi: 10.1155/2012/979105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cho C.S., Park H.W., Ho A., Semple I.A., Kim B., Jang I., et al. Lipotoxicity induces hepatic protein inclusions through TANK binding kinase 1-mediated p62/sequestosome 1 phosphorylation. Hepatology. 2018;68:1331–1346. doi: 10.1002/hep.29742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Perry A.K., Chow E.K., Goodnough J.B., Yeh W.C., Cheng G. Differential requirement for TANK-binding kinase-1 in type I interferon responses to toll-like receptor activation and viral infection. J Exp Med. 2004;199:1651–1658. doi: 10.1084/jem.20040528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hagan R.S., Torres-Castillo J., Doerschuk C.M. Myeloid TBK1 signaling contributes to the immune response to influenza. Am J Respir Cell Mol Biol. 2019;60:335–345. doi: 10.1165/rcmb.2018-0122OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Karim A.S., Reese S.R., Wilson N.A., Jacobson L.M., Zhong W., Djamali A. Nox2 is a mediator of ischemia reperfusion injury. Am J Transpl. 2015;15:2888–2899. doi: 10.1111/ajt.13368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hattori K., Naguro I., Runchel C., Ichijo H. The roles of ASK family proteins in stress responses and diseases. Cell Commun Signal. 2009;7:9. doi: 10.1186/1478-811X-7-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Komander D., Lord C.J., Scheel H., Swift S., Hofmann K., Ashworth A., et al. The structure of the CYLD USP domain explains its specificity for Lys63-linked polyubiquitin and reveals a B box module. Mol Cell. 2008;29:451–464. doi: 10.1016/j.molcel.2007.12.018. [DOI] [PubMed] [Google Scholar]
- 33.Glittenberg M., Ligoxygakis P. CYLD: a multifunctional deubiquitinase. Fly (Austin) 2007;1:330–332. doi: 10.4161/fly.5399. [DOI] [PubMed] [Google Scholar]
- 34.Nikolaou K., Tsagaratou A., Eftychi C., Kollias G., Mosialos G., Talianidis I. Inactivation of the deubiquitinase CYLD in hepatocytes causes apoptosis, inflammation, fibrosis, and cancer. Cancer Cell. 2012;21:738–750. doi: 10.1016/j.ccr.2012.04.026. [DOI] [PubMed] [Google Scholar]
- 35.Tonelli C., Chio I.I.C., Tuveson D.A. Transcriptional regulation by Nrf2. Antioxid Redox Signal. 2018;29:1727–1745. doi: 10.1089/ars.2017.7342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Battino M., Giampieri F., Pistollato F., Sureda A., de Oliveira M.R., Pittalà V., et al. Nrf2 as regulator of innate immunity: a molecular Swiss army knife. Biotechnol Adv. 2018;36:358–370. doi: 10.1016/j.biotechadv.2017.12.012. [DOI] [PubMed] [Google Scholar]
- 37.Ke B., Shen X.D., Zhang Y., Ji H., Gao F., Yue S., et al. KEAP1–NRF2 complex in ischemia-induced hepatocellular damage of mouse liver transplants. J Hepatol. 2013;59:1200–1207. doi: 10.1016/j.jhep.2013.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sciarretta S., Zhai P., Shao D., Zablocki D., Nagarajan N., Terada L.S., et al. Activation of NADPH oxidase 4 in the endoplasmic reticulum promotes cardiomyocyte autophagy and survival during energy stress through the protein kinase RNA-activated-like endoplasmic reticulum kinase/eukaryotic initiation factor 2alpha/activating transcription factor 4 pathway. Circ Res. 2013;113:1253–1264. doi: 10.1161/CIRCRESAHA.113.301787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brewer A.C., Murray T.V., Arno M., Zhang M., Anilkumar N.P., Mann G.E., et al. Nox4 regulates Nrf2 and glutathione redox in cardiomyocytes in vivo. Free Radic Biol Med. 2011;51:205–215. doi: 10.1016/j.freeradbiomed.2011.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nlandu-Khodo S., Dissard R., Hasler U., Schäfer M., Pircher H., Jansen-Durr P., et al. NADPH oxidase 4 deficiency increases tubular cell death during acute ischemic reperfusion injury. Sci Rep. 2016;6 doi: 10.1038/srep38598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Choi U.Y., Kang J.S., Hwang Y.S., Kim Y.J. Oligoadenylate synthase-like (OASL) proteins: dual functions and associations with diseases. Exp Mol Med. 2015;47:e144. doi: 10.1038/emm.2014.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee M.S., Kim B., Oh G.T., Kim Y.J. OASL1 inhibits translation of the type I interferon-regulating transcription factor IRF7. Nat Immunol. 2013;14:346–355. doi: 10.1038/ni.2535. [DOI] [PubMed] [Google Scholar]
- 43.Ning S., Pagano J.S., Barber G.N. IRF7: activation, regulation, modification and function. Genes Immun. 2011;12:399–414. doi: 10.1038/gene.2011.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fitzgerald K.A., McWhirter S.M., Faia K.L., Rowe D.C., Latz E., Golenbock D.T., et al. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat Immunol. 2003;4:491–496. doi: 10.1038/ni921. [DOI] [PubMed] [Google Scholar]
- 45.Liu Z.S., Cai H., Xue W., Wang M., Xia T., Li W.J., et al. G3BP1 promotes DNA binding and activation of cGAS. Nat Immunol. 2019;20:18–28. doi: 10.1038/s41590-018-0262-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fiers W., Beyaert R., Declercq W., Vandenabeele P. More than one way to die: apoptosis, necrosis and reactive oxygen damage. Oncogene. 1999;18:7719–7730. doi: 10.1038/sj.onc.1203249. [DOI] [PubMed] [Google Scholar]
- 47.Redza-Dutordoir M., Averill-Bates D.A. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim Biophys Acta. 2016;1863:2977–2992. doi: 10.1016/j.bbamcr.2016.09.012. [DOI] [PubMed] [Google Scholar]
- 48.Kim H.E., Du F., Fang M., Wang X. Formation of apoptosome is initiated by cytochrome c-induced dATP hydrolysis and subsequent nucleotide exchange on Apaf-1. Proc Natl Acad Sci U S A. 2005;102:17545–17550. doi: 10.1073/pnas.0507900102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Linkermann A., Hackl M.J., Kunzendorf U., Walczak H., Krautwald S., Jevnikar A.M. Necroptosis in immunity and ischemia-reperfusion injury. Am J Transpl. 2013;13:2797–2804. doi: 10.1111/ajt.12448. [DOI] [PubMed] [Google Scholar]
- 50.Molnar T., Pallagi P., Tel B., Kiraly R., Csoma E., Jenei V., et al. Caspase-9 acts as a regulator of necroptotic cell death. FEBS J. 2021;288:6476–6491. doi: 10.1111/febs.15898. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.