

Abstract
Transthyretin (TTR) is a homotetrameric protein mainly synthesised by the liver and the choroid plexus whose function is to carry the thyroid hormone thyroxine and the retinol-binding protein bound to retinol in plasma and cerebrospinal fluid. When the stability of the tetrameric structure is lost, it breaks down, paving the way for the aggregation of TTR monomers into insoluble fibrils leading to transthyretin (ATTR) amyloidosis, a progressive disorder mainly affecting the heart and nervous system. Several TTR gene mutations have been characterised as destabilisers of TTR structure and are associated with hereditary forms of ATTR amyloidosis. The reason why also the wild-type TTR is intrinsically amyloidogenic in some subjects is largely unknown. The aim of the review is to give an overview of the TTR biological life cycle which is largely unknown. For this purpose, the current knowledge on TTR physiological metabolism, from its synthesis to its catabolism, is described. Furthermore, a large section of the review is dedicated to examining in depth the role of mutations and physiological ligands on the stability of TTR tetramers.
Keywords: transthyretin, TTR amyloidosis, retinol-binding protein, thyroxine, retinol, TTR clearance, ER-associated degradation pathway
1. Introduction
Transthyretin (TTR) is a homotetrameric protein found in the plasma or serum and cerebrospinal fluid (CSF), mainly synthesised by the liver and the choroid plexus (CP). TTR was previously known as prealbumin because it migrated in front of albumin in serum protein electrophoresis [1]. Following the discovery of its role as a transporter of thyroxine (T4), the name was converted into “thyroxine-binding prealbumin”. It was only in 1981 that The International Union of Biochemistry converted the name into “transthyretin” (transports thyroxine and retinol) [2], a name suggested when it was discovered that the protein was also able to bind both thyroxine and retinol complexed with retinol-binding protein 4 (RBP) [3]. Indeed, only a small fraction of circulating TTR (less than 20%) is involved in the transport of T4, which is mainly carried by thyroxine-binding globulin (TBG) and albumin [4], while most TTR tetramers are involved in the transport of holo-RBP. The association between TTR and RBP assumes considerable importance as it is necessary to avoid glomerular filtration and renal catabolism of RBP [5] due to the small size of holo-RBP (21kDa). Based on holo-RBP affinity for TTR and on their plasma concentrations (2 µM and 3.6–4.5 µM, respectively), it has been estimated that about 95% of circulating RBP is associated with TTR [6,7,8].
Plasma TTR levels rise after birth, reaching a concentration of 20–40 mg/dL in adults, and then TTR levels decrease after 50 years of age [9]. In clinical practice, TTR is considered a surrogate marker of diet adequacy since it is rich in the essential amino acid tryptophan and has a relatively short half-life (2.5 days) [10]. Indeed, a plasma TTR concentration below 10 mg/dL has been associated with malnutrition [11]. Nevertheless, plasma TTR is also negatively influenced by the acute-phase response due to inflammation, which is often associated with malnutrition. Thus, TTR levels can be helpful in evaluating and diagnosing a malnutrition status, but it is important to consider the involvement of inflammatory processes to interpret TTR levels correctly [12].
In the last years, TTR has gained considerable importance in the setting of amyloidosis, a protein misfolding disorder caused by the extracellular deposition of a β-sheet-rich protein as insoluble fibrils. Amyloid TTR (ATTR) amyloidosis is among the most common forms of amyloidosis in human pathology, and it can present as a hereditary form caused by TTR mutations (variant ATTR, ATTRv) or acquired ones due to the deposition of TTR wild-type (ATTRwt) [13]. The ATTRwt was previously known as “senile systemic amyloidosis” since the amyloid fibrils are mainly observed in elderly patients (>75 years) [14]. A few clinical studies have measured plasma TTR in patients with ATTRv or ATTRwt amyloidosis. These studies reported lower plasma TTR in patients with ATTRv amyloidosis as compared to reference values (18-45 mg/dL) [15,16] as well as low values, but still in the normal range, in ATTRwt amyloidosis [17,18].
TTR fibril formation has been investigated in many in vitro studies, which all agree that ATTR amyloid fibrils form because of tetramers dissociation into monomers which spontaneously misfold, forming amorphous aggregates, oligomers, and then amyloid fibrils [19,20,21,22]. The in vitro oligomerisation occurs in both ATTRv and ATTRwt amyloidosis; this process is faster in ATTRv amyloidosis, where the stability of TTR is compromised by genetic mutations [23,24]. The reason why TTRwt, despite the absence of destabilising mutations, also is intrinsically amyloidogenic in some subjects remains unknown.
Given the clinical importance of TTR, in this review, we will discuss the current knowledge of TTR structure and metabolism, highlighting the variables that stabilise or destabilise the native protein in all stages of its life cycle, from synthesis to catabolism.
2. Tissue-Specific Regulation of Transthyretin Expression
Human TTR is a 127-amino acid (AAs) protein encoded by a single-copy gene mapped to chromosome 18q11.2-q12.1 [25]. The gene has a size of about 6.9kb and consists of four exons, three introns, a TATA box-like sequence at nucleotides 24–30, and a CAAT box-like sequence at nucleotides 95–101 [26,27]. The first exon contains 95 base pairs (bp) and 26 bp of 5’ untranslated region; it codes for a 20 amino acid signal peptide as well as the first 3 AAs residues of the mature protein. Exon 2 (131 bp) codes for AAs residues 4 – 47, exon 3 (136 bp) for residues 48–92, and exon 4 (253 bp) for residues 93-127 [26,28]. TTR mRNA encodes for the pro-TTR monomers (147 AAs), whose N-terminal region corresponds to a hydrophobic signal peptide of 20 AAs, which is cleaved to produce the native TTR monomer [29].
The cleavage of the signal peptide is necessary to free the first 9 AAs of the mature TTR monomers, which represent a structural “disordered region” [30] essential for monomer-monomer assembly into dimers, the first step in tetramer formation [31].
The liver and the CP are the most abundant and well-described sites of TTR synthesis in humans, but TTR expression has also been identified in a minor amount in the placenta, pancreas, yolk sac, and retinal pigment epithelium.
2.1. Liver and Choroid Plexus
The protein synthesised and released by the liver is the main source of TTR in plasma [32], where it circulates associated with T4 and holo-RBP. Thus, the physiological functions of hepatic TTR are related to the distribution of T4 and retinol throughout the body.
The TTR gene expression in the liver is regulated at the transcriptional level and is controlled by hepatocyte nuclear factors (HNF) [33]. During the acute phase response, there is a significant reduction in the binding of HNFs to the TTR promoter, which correlates with a decrease in TTR expression [34]. Indeed, as a typical negative acute-phase plasma protein, the TTR gene in the liver is downregulated after trauma, inflammation, or malnutrition resulting in a marked decrease in circulating protein levels.
In the CP, the TTR gene is not subjected to acute phase negative regulation [35], suggesting it must be regulated independently of the liver TTR gene [36]. The structure of the TTR gene is identical in the two tissues: they have the same starting site for mRNA synthesis and the same enhancer sequences [37]; this suggests that the cell-specific distribution of transcription factors is responsible for tissue-specific expression of the gene [38]. In the liver, HNFs are the transcription factors involved in the activation of TTR gene transcription, but these factors are absent in the CP, where the specific identity of the transcription factors involved is yet to be understood. It is thought that in CP, the TTR gene is regulated by transcription factors that are closely related to the HNFs [38], which have the binding sequences in 3kbp of the 5’ flanking region [37].
Differences in gene regulation could be due to the important role of T4 in the brain. TTR synthesised by epithelial cells [39] of the CP is secreted into the cerebrospinal fluid (CSF) [40], where it is involved in the delivery of T4 to stem cells and progenitor cells within the brain, which requires T4 for the regulation of the cell cycle [41]. Since the adult and developing brain is sensitive to thyroid hormone effects, it is extremely important to maintain adequate levels of thyroid hormones even during trauma or inflammatory conditions when the reduction of plasma TTR and albumin results in a reduction of total circulating thyroid hormones [36]. Furthermore, TTR is the main carrier for T4 in the CSF [42], while in plasma, it carries only 15% of the whole T4 [43,44]; this further highlights the important role of TTR for the nervous system.
2.2. Placenta and Visceral Yolk Sac
The production of TTR by placenta trophoblasts plays a crucial role in foetal development in the first trimester [45]. Since the foetus is not able to produce its own thyroid hormone until 16 weeks of gestation, it must rely on the maternal T4 supply carried by placental TTR for brain development [46,47]. The presence of TTR has also been identified in the visceral yolk sac, supporting the idea of its importance in the active transport of T4 as well as retinol from the maternal circulation to the developing foetus [48]. The TTR gene regulation in these sites is poorly understood, but the transcriptional binding proteins CCAAT/enhancer binding protein (C/EBP) and activator protein-1 (AP-1), implicated in the transcription promotion of important genes during foetal development, could be involved [49,50].
2.3. Pancreas
TTR expression in the pancreas occurs mainly in pancreatic α-cells, whereas β-cells may produce TTR only at a low degree [51]. Gene regulation and function of TTR synthesised by these particular cells are largely unknown, but it seems that TTR promotes glucagon/insulin release and cell survival in both α [52] and β [53] cells. Low levels of plasma TTR have been found in patients affected by diabetes type I [4.4 µmol/L (24 mg/dL) compared to 5.3 µmol/L(29 mg/dL) in normal subjects], reinforcing the hypothesis that TTR may be associated with glucose homeostasis [54].
2.4. Retinal Pigment Epithelium
TTR is synthesised together with RBP by the retinal pigment epithelium of the mammalian eye [55,56]. These proteins may be involved in the delivery of all-trans-retinol to Müller and amacrine cells [57], where it is converted to retinoic acid required for photoreceptor functioning. Little is known about the distribution of TTR in the human eye. A recent study showed that TTR distribution in the vitreous directly correlates with that of retinal tissues, suggesting a local distribution or transport of TTR from the retina to the vitreous [58]. In studies on vitreous amyloidosis, ATTR appeared to result from locally synthesised protein from the retina [59,60].
3. TTR Structure
The X-ray crystal structure of human TTR was determined in the laboratory of Colin Blake in 1971 [61]. These structural studies provided information on both the active tetrameric conformation of TTR and the binding sites of both its ligands.
Human TTR is a homotetrameric, slightly acidic (isoelectric point of 5.3) protein with a molecular mass of 55 kDa composed of four identical monomers, with a molecular weight of approximately 14 kDa [62]. Each monomer consists of eight β-strands structures designated with letters from “A” to “H” and one short α-helix of nine residues included between strands E and F. All strand interactions are antiparallel except for the interaction between strands A and G. These eight β-strands are connected by loops and are arranged in two groups of twisted β-sheets [44,63,64]. Interstrand hydrogen bonds allow the organisation of a tertiary structure of β-strands constituted by an inner (strands DAGH) and an outer (strands CBEF) β-sheet, which are orthogonal to one another [44,63,64] (Figure 1).
Figure 1.
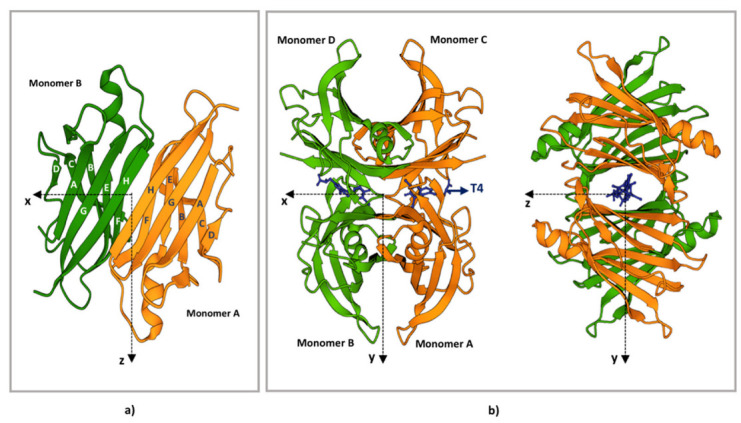
TTR structure. (a) Ribbon diagram of the AB dimer interface (b) Ribbon diagram of the TTR tetramers. The X-axis passes through the T4 binding channels, which are formed at the interface of monomers D-B and C-A. The halogen binding pockets (HBP) are shaped by the following amino acids: HBP1, Lys15-Leu17-Thr106-Val 121; HBP2, Lys15-Leu17-Ala108-Ala109-Leu110; HBP3, Ser117-Leu110-Thr119-Ala108. T4 is represented in blue. The figure has been produced using “www.rcsb.org”web site (protein ID code 1QAB), access date 22 June 2022.
Two TTR monomers are arranged into dimers by hydrogen bonding between two F (F, F’) and two H (H, H’) strands from adjacent monomers. This provides an extensive contact region where the β-sheets are strictly packed in a layer-by-layer manner contributing to the stability of the molecules [44,63,64].
Two dimers then form a tetramer mainly through hydrophobic bonds between loops, including between the β-strands A-B and G-H, finalising the assembly of the globular TTR. Thus, the quaternary structure of TTR occurs in interactions between four identical monomers, but its conformation is maintained by the dimer–dimer interface. Indeed, the dimer is thought to be the basic structural unit of mature TTR. This perspective is strengthened by the observation that the contact region between dimers is smaller than those between monomers and consists of hydrophobic and hydrophilic interactions [44,63,64]. Thus, in physiological conditions, TTR is a globular homo-tetramer organised as a dimer of dimers. Even if the tetrameric structure is considered stable, a monomer exchange process has been shown in in vitro studies on recombinant proteins and consists of a slow disassembly of tetramers into TTR monomers followed by a quick reassembly [65].
4. TTR Structural Stability
In the assembling of the TTR tetramer, two cylindrical hydrophobic channels are generated at the dimer–dimer interface that can accommodate one T4 molecule each (Figure 1). The hydrophobic channel contains two pairs of three symmetrical sets of halogen binding pockets where the four iodine atoms of T4 are placed [61,66]. Even if the two channels are symmetric, there is a 100-fold difference in the binding constants for the first and the second T4 molecule. Indeed, because of negative co-operativity between the two sites, only one site can be occupied by T4 under physiological conditions [67]. Interestingly, each T4 binding site is formed by residues of two monomers belonging to opposite dimers (Figure 1), which become connected by T4 itself, leading to a drastic stabilisation of the native TTR tetramer. One of the current therapeutic strategies against ATTR amyloidosis is based on improving the kinetic stability of the native TTR tetramers by molecules able to bind the T4 pocket, thus preventing the early stage of TTR dissociation [68,69,70]. Guidelines for ATTR amyloidosis treatment [71] suggest the use of tafamidis [72] and diflunisal [73] as stabiliser drugs. A further drug, AG10, is under evaluation in clinical trials [74].
In addition to T4, the central channel of TTR could bind a second endogenous ligand, the triiodothyronine (T3). However, the binding of T3 has a much lower affinity than T4, and circulating TTR binds virtually no T3 [43]. Several natural [75,76] and other chemical ligands [72,74,77] able to bind and stabilise the TTR tetramer have also been found (Figure 2); for reviews on these topics, see for example [78] and [79].
Figure 2.

Chemical structure of some natural (T4, resveratrol, curcumin) and chemical ligands (tafamidis, diflunisal, AG10) able to bind and stabilise TTR tetramer.
Contrary to T4, which is bound to the interior of TTR tetramers, holoRBP binds on its external surface, and each TTR tetramer has four putative binding sites for RBP, two in each dimer.
The three-dimensional X-ray structure of the complex formed by TTR and RBP was first determined in 1995 by analysing crystals containing human TTR and chicken RBP [80]. Then, in 1999, the human TTR–RBP complex was resolved [81]. These studies showed that no more than two RBP molecules could be effectively bound because of their steric hindrance on the other two possible sites [80,82,83]. In humans, the two RBP molecules bind opposite dimers and are arranged in a 2-fold symmetry axis (Figure 3).
Figure 3.
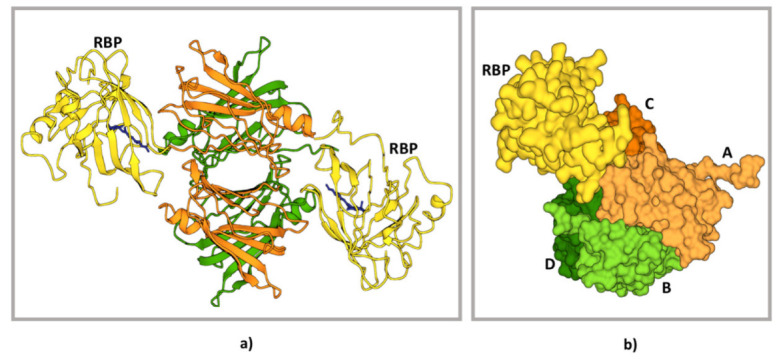
Structure of TTR–RBP complex. (a) Structure representation of the TTR–RBP complex. TTR: monomers A and C, orange; monomers B and D, green. RBP: yellow. Retinol: blue (b) Detail of the contact between the TTR subunits A, C, and D and the RBP molecule [left side, colour codes as in (a)]. Centre and right drawings show the interacting surfaces of RBP (centre) and of the TTR subunits A, C, and D (right). It is possible to appreciate how the RBP surface fits into a site formed by the arrangement of three TTR subunits (A, C, and D). The figure has been produced using “www.rcsb.org” web site, access date 22 June 2022.
Both TTR and RBP contribute 21 amino acids to the protein–protein recognition interface, and most of these residues are in the C-terminal regions of the two proteins [81,84,85]. The RBP–TTR interface can be described with the three-dimensional docking model in which two complementary three-dimensional surfaces constitute the recognition interface. The two surfaces are outlined by amino acids from three TTR monomers and four distinct regions of RBP. The mainly involved amino acids in the interaction between one RBP, and TTR are within the following regions: for RBP 31-38, 63-67, 93-99, and 179-183; for TTR 82-90 and 114-115 for monomer A, 98-102 for B, 82-90, and 19-28 for D [81,85]. Interestingly, retinol is believed to be involved in RBP–TTR interaction since its hydroxyl end group is within hydrogen-bonding distance from the polypeptide chain of TTR [81,85]. Retinol binding induces conformational changes in RBP which strongly increases its affinity for TTR (holo-RBP, Kd 0.2 μM) compared to RBP alone (apo-RBP, Kd 1.2 μM) [7,86]. Accordingly, the stability of the TTR-holo-RBP complexes is reduced when retinol is removed; thus, the remaining TTR-apoRBP complexes are susceptible to degradation [3,87]. From this perspective, holo-RBP and not apo-RBP would exert a stabilisation effect on TTR tetramers. Interactions between TTR and RBP did not cause structural alterations in the proteins involved, but a subtle tertiary structural change was observed. Indeed a slight asymmetry in the extension of recognition surfaces between TTR and one or the other bound RBP was found [81,88]. The dissociation constants of the first and the second RBP molecules were investigated by mass spectrometry resulting in 1.5 × 10–7 M for the first bound RBP molecule and 3.5 × 10–5 M for the second one. It has been hypothesised that the binding of the first RBP induces a negative cooperative effect that decreases the affinity of TTR for the second RBP molecule [8].
The binding of T4 and holoRBP markedly increases the structural stability of TTR tetramers and consequently inhibits amyloid formation [89]. This possibility is supported by in vitro studies on TTR amyloidogenicity, which consider the independent and additive stabilising action of RBP and T4 on TTR tetramer disassembling [87]. The dissociation of TTR tetramers is decreased upon the formation of the TTR-holoRBP complex in a concentration-dependent manner. Indeed, monomers’ exchanges among TTR tetramers were significantly slowed by adding retinol together with RBP [87]. Furthermore, in the presence of holo-RBP and/or T4, the rate of TTR fibril formation was reduced by up to 50% even in the presence of L55P TTR, the most pathogenic TTR variant [87].
TTR stability can be negatively affected also by ageing, metal cations (especially Ca2+), and oxidative modifications [90] at the free cysteine residue in position 10 (Cys10), see later in the text.
TTR proteolysis has recently attracted attention due to its implications for TTR instability and the pathogenesis of amyloidosis. It is challenging to identify the specific structural perturbation caused by proteolysis and how it affects the formation of subsequent dimeric or oligomeric intermediates [91]. Elucidating the detailed structural features of TTR dimers and oligomers is crucial since it may provide critical insights into the aggregation mechanism of TTR and help in identifying novel targets for therapeutic intervention.
5. TTR Variants and Structural Stability
The dissociation of TTR tetramers results in partially unfolded monomers assembling into aggregates with various quaternary structures [90]. TTR aggregates undergo structural rearrangements forming cytotoxic oligomers. These dynamic, heterogeneous species of TTR represent intermediate states of early steps of ATTR amyloidogenesis [92,93].
The pathogenesis of ATTRwt is poorly understood, while it is largely accepted that the hereditary form is caused by autosomal dominant single point mutations in the coding region of the TTR gene that lead to the production of unstable TTR tetramers [94].
ATTRv amyloidosis was initially subdivided into “familial amyloid polyneuropathy (FAP)” and “familial amyloid cardiomyopathy (FAC)” based on amyloid tissue-selective deposition and pathology, but most of the variants were associated with a mixed phenotype, with varying degrees of neurological and cardiac involvement [95]. Recently, the International Society of Amyloidosis (ISA) has updated the nomenclature in favour of more exact definitions: FAC and FAP were renamed to ATTR amyloidosis associated with the specific causative mutation (e.g., ATTRV30M) and the accompanying main symptom (i.e., ATTR with cardiomyopathy) [64].
Among the known TTR variants (to date, more than 140) [96], only a small number have been described as non-amyloidogenic (e.g. G6S, H90N), and only three have been shown to form tetramers more stable than the wild-type one: T119M, R104H, and A108V are considered “trans-suppressor” mutations as they reduce the symptoms of the disease in heterozygous individuals carrying an aggressive mutation in the other allele [97,98,99]. Thr119 and Ala108 are both located in the T4 binding site: the methionine substitution for threonine 119 into methionine produces new hydrophobic contacts between the dimer-dimer interface, which increase the kinetic stability of TTR tetramers and the binding affinity for T4 [98]. The change of alanine 108 into valine induces a similar stabilisation action which determines a higher resistance to tetramer dissociation, although it does not induce an increase in the T4 binding affinity [97]. The arginine 104 is located in loops between strands F and G (F-G loop) at the surface of the TTR monomers [98]. The stabilising effect of R104H mutation seems to depend on thermodynamic stabilisation instead of kinetic stabilisation [98,99]: the final effect is an increase of TTR tetramer form at the expense of the misfolded monomers [99]. The greatest stabilising effect is associated with T119M and A108V variants, while R104H only modestly protects against aggregation [97].
Most of TTR mutations lead to a mature protein more susceptible to tetramer dissociation and monomers aggregation. The most studied TTRv are summarised in Figure 4.
Figure 4.

Positions of amino acids (highlighted in yellow) subjected to mutations described in Table 1. The figure was created through “www.rcsb.org” and “biorender.com” web sites (license agreement number KJ247URGZS), access date 22 June 2022.
To obtain information regarding the structural changes responsible for the destabilisation of TTR tetramers, several researchers tried to solve the crystal structures of amyloidogenic TTR variants. The results derived from these X-ray crystallography studies showed almost identical structures between wild-type and mutated TTRs. Indeed, β-sheet tertiary structures of native TTR are minimally modified by pathogenic mutations [100,101]. Thus, X-ray studies could explain how single amino acid substitutions increase the TTR aggregation propensity only for those mutations highly affecting TTR structure [100,101].
A complementary approach to TTR structural studies has been provided by NMR, which allows the identification of minimal structural changes but is significant for kinetic and/or thermodynamic stability of tetramers and monomers, respectively [94]. One of the most important structural alterations that differentiate the structure of TTRv from TTRwt involves the EF-helix-loop region, positioned within the AB or CD dimer. This region seems to play an important role in TTR structural stability since any change in it affects the dimer–dimer interface and thus the stability of TTR tetramers [100,102,103]. Some information on structural alterations induced by amino acid substitutions is available only for the most known and well-studied pathological mutations, summarised in Table 1. For more details on TTRv, these web sources are recommended: gnomAD (https://gnomad.broadinstitute.org/) or Mutations in Hereditary Amyloidosis (www.amyloidosismutations.com; access date 10 June 2022)).
Table 1.
Main TTR gene mutations and related structural modifications.
| TTRv (Other Name) * |
Mutated Nucleotide (mRNA) | Clinical Phenotype | Involved Secondary Structure [30] | Overall Structural Alteration | Ref. |
|---|---|---|---|---|---|
| Val30Met (p.Val50Met) |
c.148G>A | AN, E, LM, PN |
β-strand B | Destabilisation of B and E strands resulting in a distortion of T4 binding channel. Causes a lower affinity for T4 | [104,105] |
| Ser52Pro (p.Ser72Pro) |
c.214T>C | AN, H, K, PN |
β-bend | Stability alteration of C-D loop in protein monomer | [106] |
| Glu54Lys (p.Glu74Lys) |
c.220G>A | AN, H, PN |
β-strand D | Lys54 destabilises tetramer structure due to increased electrostatic repulsion between Lys15 of two monomers. The T4 binding pocket is markedly narrower in Glu54Lys compared with wtTTR, suggesting a decrease of affinity for T4 | [107] |
| Leu55Pro (p.Leu75Pro) |
c.224T>C | AN, E, H, PN |
β-strand D | Disruption of hydrogen bond interaction between β-strands A and D leads a β-strand D structure highly disordered with different contacts between the subunits as well as a significant variation in the CE region of the monomer | [108,109] |
| Val122Ile (p.Val142Ile) | c.424G>A | H | 122-127 AAs terminal loop | small changes in the region associated with the intra- and inter dimer interactions | [110] |
* The name of TTRv is reported according both to the traditional and to the Human Gene Organization (HUGO) nomenclature which include the 20-AA signal peptide, e.g.: Val30Met (p.Val50Met). AN = autonomic neuropathy; E = eye; H = heart; K = kidney; LM = leptomeningeal; PN = polyneuropathy.
6. TTR Post-Translational Modifications
Post-translational modifications (PTMs) of proteins may be critical for the regular folding of the polypeptide chain, protein stability, and their normal turnover. Altered PTMs mechanisms may cause the structural destabilisation of proteins that form amyloid fibrils [111,112,113,114]. Even in the case of TTR, PTMs seem to participate in protein stabilisation. The most relevant and known PTMs for TTR occur at the free Cys10. Each TTR monomer contains a single Cys, which participates in the thyroid hormone-binding channels within the TTR tetramer; therefore, PTMs of Cys10 may interfere with the binding of thyroid hormones [115], thus indirectly affecting TTR stability. The strong reactivity of Cys10 is due to the fact that it is not involved in any intra- or inter-protein disulphide bond, which makes it susceptible to forming mixed disulphides with several thiol-reactive molecules, such as Cys, CysGly, and glutathione [116]. Other identified modifications on Cys10 are sulfonation and its conversion into organosulfur acids [117,118] (Figure 5). S-sulfonated or S-cysteinylated TTR are the most prevalent circulating forms, while only 10–15% remains unmodified at Cys-10 [117]. The type and grade of Cys10 modifications modulate TTR stability in different ways. S-sulfonation stabilises TTR tetramers [119,120], whereas S-cysteinylation enhances dissociation by 2-fold for the unmodified form [121]. Therefore, it is not surprising that Cys10 modifications are involved in tetramers destabilisation that triggers some forms of TTR familial amyloidosis [122,123]. These studies do not exclude that Cys10 modifications may also destabilise the unmutated protein in ATTRwt amyloidosis [121]. Furthermore, Met and Cys oxidation as well as carbonylation make TTR cytotoxic to human cardiomyocytes cell lines in a dose–responsive manner. Therefore age-related TTR oxidative modifications may play a role in the onset of ATTRwt amyloidosis [124].
Figure 5.

The four Cys 10 are highlighted in green and represented as a spacefill model. The most frequent Cys10 PTMs are indicated on the right. T4 is represented in blue. The figure has been produced by using “www.rcsb.org” web site (protein ID code 1QAB), access date 22 June 2022.
7. TTR–RBP Complex Formation
Only a few studies were conducted to investigate the mechanism of RBP–TTR complex formation. It was initially proposed that the association between RBP and TTR occurred in plasma after the independent secretion of the two proteins [125]. However, the observation that TTR was accumulated within hepatocytes in vitamin A-deficient rats, resulting in decreased plasma TTR [126], led to the belief that RBP–TTR complexes may actually form inside the cells. Data supporting this hypothesis were obtained by Melhus and collaborators [127] using HeLa cells transfected both with RBP and TTR wild-type or modified with the endoplasmic reticulum (ER) retention signal peptide (KDEL). Authors showed that RBP could not be secreted when co-expressed with TTR–KDEL [127].
The importance of TTR in maintaining normal levels in mammals of plasma RBP, retinol, and thyroid hormones was confirmed in vivo in TTR knockout mice: animals were phenotypically normal and viable, but plasma levels of RBP, retinol, and thyroid hormones were significantly decreased compared to controls [128]. Furthermore, RBP–TTR complexes formation and secretion are likely sensitive to retinol supply [129]. Interestingly retinoids that could interact with RBP but, at the same time, prevented its association with TTR also inhibited RBP secretion [130]. These studies suggested that RBP–TTR complexes were formed inside the ER, as proved and confirmed in 1996 by Bellovino and collaborators [131].
8. Endoplasmic Reticulum Quality Control in TTRwt and TTRv Cellular Release
The critical role of ER in TTR synthesis has been confirmed by several studies conducted in the 2000s, concerning the mechanism by which TTR variants can bypass ER quality control systems [132,133,134,135,136,137,138]. This system aims to guarantee that only correctly folded or assembled proteins are translocated from the ER to their final destinations [139]. Misfolded or misassembled proteins are selectively retained in the ER by specific chaperone proteins or retro-translocated across the ER membrane and degraded by the cytosolic proteasome (ER-associated degradation, ERAD, mechanism) [140]. Therefore, ER quality control is a cellular protective mechanism to avoid the production of unstable proteins. However, some amyloidogenic proteins (including TTR) are stable enough to pass through the ER but misfold and aggregate once they reach their final destination [141].
Sekijima et al. tried to explain why almost all TTRv, despite their compromised folding energetics, can bypass the ERAD system showing a secretion efficiency similar to the TTRwt protein [138]. The authors analysed the secretion efficiency of 32 types of TTRv, finding that only the most destabilised variants, D18G [132] and A25T [133], exhibited a very low concentration in blood, suggesting a secretion defect by the liver [138]. Indeed, these TTRv are not associated with severe systemic amyloidosis. On the contrary, D18G and A25T seemed to be normally secreted by CP cells, and cause severe CNS amyloidosis [132,133]. The authors speculated that D18G and A25T are efficiently released by CP thanks to a high local T4 concentration which acts as a chaperone metabolite able to transiently stabilise TTR tetramers which can thus escape the ER quality control and be secreted into CSF. Once TTR is excreted into the CSF, the low local T4 concentration is insufficient to stabilise TTR that undergoes dissociation [132,133]. Therefore, ERAD protects against severe early-onset systemic amyloidosis, decreasing the secretion of most highly destabilised TTR variants. However, the mechanism is not able to prevent the secretion of those TTRv able to form tetramers stable enough to be secreted by the ER-assisted protein folding (ERAF) pathways, which include molecular chaperones and folding enzymes that allow proper folding and subsequent release from the ER of the newly synthetised proteins [142]. The ERAF is an error-prone process whose success depends on the combination of tissue-specific chaperones, metabolite chaperones (such as T4 for TTR tetramers) as well as thermodynamic and kinetic stability of the nascent protein. ER chaperone BiP and the protein disulfide isomerase PDIA4, key components of the ERAF mechanism, are differently involved in the secretory regulation of TTRv in various cellular models (HEK293T, HepG2, HeLa) [135,137].
The complex ER molecular networks, specific and unique for each tissue, with different distributions of protein chaperones, metabolite chaperones and osmolytes, could in part explain the tissue selectivity of TTR secretion and the organ tropism shown by TTR amyloidogenesis [138].
In addition to ER chaperones, some extracellular chaperones are detectable in body fluids, bind misfolded proteins and prevent their inappropriate protein-protein interactions and their aggregation into insoluble deposits [143,144]. Da Costa and collaborators have identified the extracellular chaperones probably involved in countering ATTR: haptoglobin, alpha-1-anti-trypsin, alpha-2-macroglobulin and clusterin, which plasma levels are highly increased in ATTR amyloidosis [136]. Increased levels of some of these extracellular chaperones have been identified for other amyloid diseases [145,146,147], supporting the hypothesis that their increase is necessary to compensate for the larger amount of altered protein prone to forming amyloid fibres [145].
9. TTR Catabolism
9.1. Sites of Degradation
The first data about TTR catabolism were derived from studies conducted by Makover and collaborators in 1988 [148]. To determine the in vivo tissue sites of plasma and CSF TTR degradation, the authors utilised a nonmetabolisable tracer covalently linked to TTR that was administered to rats by intravenous or intraventricular injections. Upon tissue uptake, TTR underwent its normal degradation cycle, while the tracer was retained within cells in amounts proportional to the quantity of catabolised TTR. This tracer allowed the identification of TTR degradation sites and the estimation of the kinetics of TTR catabolism [148]. Tissue sites and quantitative patterns in TTR degradation were almost the same for the protein injected into the cerebrospinal fluid or plasma. No specific degradation of TTR was observed in the nervous system tissues, but, in both cases, the liver was the main organ involved in TTR degradation (36–38% of total body TTR degradation). Hepatocytes were the only liver cells involved in TTR degradation, while RBP was reported to be degraded in comparable amounts by both parenchymal and stellate cells [149]. In addition to the liver, TTR degradation mainly occurred in muscles (12–15%) and skin (8–10%), while kidneys, adipose tissue, testicles, and gastrointestinal tract catalysed about 1–8% of total TTR. Only less than 1% of TTR degradation occurred in other tissues. When the measured catabolic activities were normalised on tissue masses, kidneys, and liver resulted in being the most active organs [148].
On the bases of the mathematical model developed to interpret kinetic data, it was estimated that the entire CSF TTR pool was moved from CSF to plasma in 2.5–3.5 h, a time consistent with the turnover of whole CSF through the subarachnoid villi. The whole body TTR was then turned over in about 24 hours [148].
9.2. TTR Cellular Internalisation
We know little about the TTR cellular uptake and its degradation mechanism. Sousa et al. [150] showed that renal uptake of TTR is mediated by megalin (also known as low-density lipoprotein-related protein 2, LRP2), which is a member of the LDL receptor family [151]. Megalin is a multi-ligand receptor expressed in the epithelium of kidney proximal tubes, where it is involved in the renal reuptake of plasma proteins, including free RBP [152]. Recent studies showed that TTR-internalisation is megalin-mediated in murine sensory neurons as well [153] and in the human placenta [49]. In the latter case, trophoblasts both secrete TTR into the maternal placental circulation and reuptake the protein preferentially as TTR-T4 complex after binding to maternal T4 [154].
In the liver, the most important site for TTR degradation, megalin is not expressed. Anyway, studies on human and rat hepatoma cell lines highlighted that also, in this case, TTR tetramers were endocytosed by a receptor-mediated process which resulted in being saturable (Kd between ~4 and ~10 nM vs. ~5 µM TTR plasma concentration) [154,155]. Interestingly, the complex TTR-holoRBP showed a 70% decrease in uptake in comparison to the TTR, while the uptake of the T4-saturated TTR was enhanced by 20% [154].
It was observed that the highly amyloidogenic L55P-TTR could not enter the cell line tested, while the mildly (V30M) and the non- amyloidogenic (T119M) TTR present a higher degree of internalisation than the wt proteins [154]. The differential rate of TTR clearance might play a role in the pathogenesis of ATTR amyloidosis.
Suosa and collaborators [156] showed that 1-2% of plasma TTR circulates bound to apolipoprotein AI (apoAI) of HDL lipoproteins. Therefore, the scavenger receptor class B type I (SR-BI) might be involved as TTR-receptor in the liver, but this possibility was then excluded [154]. Alternative candidates for TTR uptake were sought among other components of the LDL receptor family expressed by hepatocytes, i.e., the LDL receptor (LDLr) and the LDL receptor-related protein (LRP): experiments also excluded these two receptors [154]. However, TTR uptake was competitively inhibited by the receptor-associated protein (RAP), a common ligand for all the receptors of the LDLr family, including megalin [154]. These observations confirmed the existence of a receptor-mediated TTR internalisation in the liver and supported the hypothesis of some shared mechanisms between TTR and lipoprotein metabolism [154]. Nevertheless, the hepatocyte TTR receptor has not been identified yet.
9.3. Removal of TTR Aggregates
TTR-related prefibrillar aggregates and amyloid fibrils are detectable in the extracellular matrix (ECM) of ATTR amyloidosis [157]. Several studies conducted on ATTRv and/or ATTRwt amyloidosis patients showed that TTR amyloid deposition manifests organ and tissue tropism, suggesting that the mechanism of TTR fibril deposition is closely related to the surrounding microenvironment [158]. Indeed, the specific composition of the ECM appears to play a key role in amyloidogenesis [159,160].
As previously mentioned, approximately 25% of the systemic TTR degradation occurs diffusely throughout the body (mainly from muscle and skin). Since fibroblasts are widely spread throughout the body and play a key role in maintaining the ECM, Misumi and collaborators [157] hypothesised that they could be the main cellular type involved in the TTR clearance in these sites. Indeed, fibroblast and macrophages can endocyte and degrade TTR aggregates in lysosomes both in vitro and in vivo, thanks to their migratory potential and their proximity to amyloid deposits in ECM [157]. Furthermore, fibroblasts may contribute to the degradation of TTR aggregates and fibrils by secreting matrix metalloproteinases (MMPs) into the ECM [157]. Further studies are needed to clarify the contribution of fibroblasts and macrophages in the clearance of TTR aggregates. Several authors are evaluating the possibility of stimulating phagocytosis by an antibody-related mechanism to promote amyloid reabsorption from tissues [161,162,163].
The follow-up of patients affected by severe forms of systemic ATTRv amyloidosis who underwent liver transplantation as curative therapy provided us with data on human turnover of tissue TTR amyloid. More in detail, liver transplantation in familial amyloidotic polyneuropathy patients was associated with a decrease in the total amount of amyloid deposits in abdominal fat tissues, but, interestingly, a change in amyloid composition was observed. Indeed, the ratio of wt-to-variant TTR shifted towards a greater contribution to the first [164]. This data suggested that remnant amyloid deposits of TTRv could represent a focal point for the deposition of the wild-type TTR (amyloid seeding) [165], furthermore showed that amyloid deposits underwent a turnover likely driven by dynamic processes of amyloid fibril formation and catabolism in the ECM.
10. Conclusions
The journey of the TTR is depicted within this review, from its synthesis to tissue catabolism, with the aim to identify the key points of TTR metabolism likely involved in the onset and progression of organ amyloidosis.
Many studies have been conducted both on cellular and animal models with the aim of clarifying TTR metabolism, but almost all pointed the attention to the TTR tetramer devoid of its physiological ligands, which, however, are essential for TTR stability. Thus, there are many knowledge gaps to be filled to translate experimental data into human pathophysiology. This research effort will provide a better understanding of the pathogenic mechanism of ATTR amyloidosis, especially of ATTRwt amyloidosis.
The main pieces of information available up today about TTR turnover have been summarised in Figure 6. Both in animal and cellular models, it was shown that RBP could be secreted only if complexed with TTR [126], although it is not clear whether in a 1:1 and/or 2:1 ratio. In hepatocyte cellular models, the secretion of TTR tetramers has been described [31,138], and their presence in circulation cannot be excluded. By the way, numerous experiments in animal models, including those for studying the clearance of TTR, were conducted by injecting the tetrameric TTR protein devoid of its ligands. These studies confirmed the possibility that TTR can subsist in plasma by itself. However, TTR tetramers might be cleared much more efficiently than the RBP–TTR complex by hepatocytes [154], so the balance between TTR and RBP–TTR complexes in plasma cannot be foreseen. The clearance of TTR has been studied only for the tetramers [148], so it is not known if the presence of RBP could actually change the rate of clearance. Anyway, it is of interest to observe that the main organs and tissues involved in TTR tetramers clearance (i.e., liver, muscle, skin) are not among those critically affected in ATTR amyloidosis (i.e., heart and nervous system).
Figure 6.

Reconstruction of the possible life cycle of TTR. The figure was created through “biorender.com” web site (license agreement number DL2489YBZI), access date 22 June 2022.
A further point to be evaluated regards the role of retinol in the stability of RBP–TTR complexes; indeed, when absent, the affinity of RBP for TTR decreases; thus, it should be of interest to know if apoRBP–TTR complexes are still stable or if TTR tetramers are released [7,86].
Many in vitro studies investigated the process leading to the metamorphosis of TTR from the globular (physiological) to the fibrils (pathological) structure [165]. All these studies focused on how TTR tetramers are modified, but the role of RBP or T4 in preserving TTR function in vivo was scarcely investigated. The time has come to translate all this knowledge to a more physiological context that would allow us to determine what conditions favour the increase of free TTR tetramers which is the starting point of the amyloid cascade.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
Funding Statement
This research received no external funding.
Footnotes
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Robbins J. Transthyretin from discovery to now. Clin. Chem. Lab. Med. (CCLM) 2002;40:1183–1190. doi: 10.1515/CCLM.2002.208. [DOI] [PubMed] [Google Scholar]
- 2.Nomenclature committee of the International Union of Biochemistry (NC-IUB) Enzyme nomenclature. Recommendations 1978. Supplement 2: Corrections and additions. Eur. J. Biochem. 1981;116:423–435. doi: 10.1111/j.1432-1033.1981.tb05353.x. [DOI] [PubMed] [Google Scholar]
- 3.Noy N., Slosberg E., Scarlata S. Interactions of retinol with binding proteins: Studies with retinol-binding protein and with transthyretin. Biochemistry. 1992;31:11118–11124. doi: 10.1021/bi00160a023. [DOI] [PubMed] [Google Scholar]
- 4.Nilsson S., Rask L., Peterson P. Studies on thyroid hormone-binding proteins. II. Binding of thyroid hormones, retinol-binding protein, and fluorescent probes to prealbumin and effects of thyroxine on prealbumin subunit self association. J. Biol. Chem. 1975;250:8554–8563. doi: 10.1016/S0021-9258(19)40795-3. [DOI] [PubMed] [Google Scholar]
- 5.Wei S., Episkopou V., Piantedosi R., Maeda S., Shimada K., Gottesman M.E., Blaner W.S. Studies on the metabolism of retinol and retinol-binding protein in transthyretin-deficient mice produced by homologous recombination. J. Biol. Chem. 1995;270:866–870. doi: 10.1074/jbc.270.2.866. [DOI] [PubMed] [Google Scholar]
- 6.Hyung S.J., Deroo S., Robinson C.V. Retinol and retinol-binding protein stabilize transthyretin via formation of retinol transport complex. ACS Chem. Biol. 2010;5 doi: 10.1021/cb100144v. [DOI] [PubMed] [Google Scholar]
- 7.Goodman D.S. Plasma retinol-binding protein. Ann. New York Acad. Sci. 1980;348:378–390. doi: 10.1111/j.1749-6632.1980.tb21314.x. [DOI] [PubMed] [Google Scholar]
- 8.Ronne H., Ocklind C., Wiman K., Rask L., Obrink B., A Peterson P. Ligand-dependent regulation of intracellular protein transport: Effect of vitamin a on the secretion of the retinol-binding protein. J. Cell. Biol. 1983;96:907–910. doi: 10.1083/jcb.96.3.907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stabilini R., Vergani C., Agostoni A., Agostoni R.V. Influence of age and sex on prealbumin levels. Clin. Chim. Acta. 1968;20:358–359. doi: 10.1016/0009-8981(68)90173-3. [DOI] [PubMed] [Google Scholar]
- 10.Ingenbleek Y., Bernstein L.H. Plasma Transthyretin as a Biomarker of Lean Body Mass and Catabolic States. Adv. Nutr. Int. Rev. J. 2015;6:572–580. doi: 10.3945/an.115.008508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Beck F.K., Rosenthal T.C. Prealbumin: A marker for nutritional evaluation. Am. Fam. Physician. 2002;65:1575–1578. [PubMed] [Google Scholar]
- 12.Dellière S., Cynober L. Is transthyretin a good marker of nutritional status? Clin. Nutr. 2016;36:364–370. doi: 10.1016/j.clnu.2016.06.004. [DOI] [PubMed] [Google Scholar]
- 13.Picken M.M. The Pathology of Amyloidosis in Classification: A Review. Acta Haematol. 2020;143:322–334. doi: 10.1159/000506696. [DOI] [PubMed] [Google Scholar]
- 14.Westermark P., Bergström J., Solomon A., Murphy C., Sletten K. Transthyretin-derived senile systemic amyloidosis: Clinicopathologic and structural considerations. Amyloid. 2003;10 Suppl 1:48–54. doi: 10.1080/13506129.2003.12088568. [DOI] [PubMed] [Google Scholar]
- 15.Hanson J.L., Arvanitis M., Koch C.M., Berk J.L., Ruberg F.L., Prokaeva T., Connors L.H. Use of Serum Transthyretin as a Prognostic Indicator and Predictor of Outcome in Cardiac Amyloid Disease Associated With Wild-Type Transthyretin. Circ. Hear. Fail. 2018;11:e004000. doi: 10.1161/CIRCHEARTFAILURE.117.004000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Skinner M., Connors L., Rubinow A., Libbey C., Sipe J.D., Cohen A.S. Lowered prealbumin levels in patients with familial amyloid polyneuropathy (FAP) and their non-affected but at risk relatives. Am. J. Med. Sci. 1985;289:17–21. doi: 10.1097/00000441-198501000-00003. [DOI] [PubMed] [Google Scholar]
- 17.Buxbaum J., Koziol J., Connors L.H. Serum transthyretin levels in senile systemic amyloidosis: Effects of age, gender and ethnicity. Amyloid. 2008;15:255–261. doi: 10.1080/13506120802525285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Falk R.H., Haddad M., Walker C.R., Dorbala S., Cuddy S.A. Effect of Tafamidis on Serum Transthyretin Levels in Non-Trial Patients With Transthyretin Amyloid Cardiomyopathy. JACC CardioOncology. 2021;3:580–586. doi: 10.1016/j.jaccao.2021.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Greene M.J., Klimtchuk E.S., Seldin D.C., Berk J.L., Connors L.H. Cooperative stabilization of transthyretin by clusterin and diflunisal. Biochemistry. 2014;54:268–278. doi: 10.1021/bi5011249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miroy G.J., Lai Z., Lashuel H.A., Peterson S.A., Strang C., Kelly J.W. Inhibiting transthyretin amyloid fibril formation via protein stabilization. Proc. Natl. Acad. Sci. 1996;93:15051–15056. doi: 10.1073/pnas.93.26.15051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ueda M., Okada M., Mizuguchi M., Kluve-Beckerman B., Kanenawa K., Isoguchi A., Misumi Y., Tasaki M., Ueda A., Kanai A., et al. A cell-based high-throughput screening method to directly examine transthyretin amyloid fibril formation at neutral pH. J. Biol. Chem. 2019;294:11259–11275. doi: 10.1074/jbc.RA119.007851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hurshman A.R., White J.T., Powers E.T., Kelly J.W. Transthyretin aggregation under partially denaturing conditions is a downhill polymerization. Biochemistry. 2004;43:7365–7381. doi: 10.1021/bi049621l. [DOI] [PubMed] [Google Scholar]
- 23.Dasari A.K.R., Hughes R.M., Wi S., Hung I., Gan Z., Kelly J.W., Lim K.H. Transthyretin Aggregation Pathway toward the Formation of Distinct Cytotoxic Oligomers. Sci. Rep. 2019;9:33. doi: 10.1038/s41598-018-37230-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Quintas A., Vaz D.C., Cardoso I., Saraiva M.J.M., Brito R.M.M. Tetramer dissociation and monomer partial unfolding precedes protofibril formation in amyloidogenic transthyretin variants. J. Biol. Chem. 2001;276:27207–27213. doi: 10.1074/jbc.M101024200. [DOI] [PubMed] [Google Scholar]
- 25.Sparkes R.S., Sasaki H., Mohandas T., Yoshioka K., Klisak I., Sakaki Y., Heinzmann C., Simon M.I. Assignment of the prealbumin (PALB) gene (familial amyloidotic polyneuropathy) to human chromosome region 18q11.2-q12.1. Hum. Genet. 1987;75:151–154. doi: 10.1007/BF00591077. [DOI] [PubMed] [Google Scholar]
- 26.Hiroyuki S., Naoko Y., Yasuyuki T., Yoshiyuki S. Structure of the chromosomal gene for human serum prealbumin. Gene. 1985;37:191–197. doi: 10.1016/0378-1119(85)90272-0. [DOI] [PubMed] [Google Scholar]
- 27.Tsuzuki T., Mita S., Maeda S., Araki S., Shimada K. Structure of the human prealbumin gene. J. Biol. Chem. 1985;260:12224–12227. doi: 10.1016/S0021-9258(17)39013-0. [DOI] [PubMed] [Google Scholar]
- 28.Power D., Elias N., Richardson S., Mendes J., Soares C., Santos C. Evolution of the thyroid hormone-binding protein, transthyretin. Gen. Comp. Endocrinol. 2000;119:241–255. doi: 10.1006/gcen.2000.7520. [DOI] [PubMed] [Google Scholar]
- 29.Ingenbleek A.Y., Young V. Transthyretin (prealbumin) in health and disease: Nutritional implications. Annu. Rev. Nutr. 1994;14:495–533. doi: 10.1146/annurev.nu.14.070194.002431. [DOI] [PubMed] [Google Scholar]
- 30.Blake C., Geisow M., Oatley S., Rérat B., Rérat C. Structure of prealbumin: Secondary, tertiary and quaternary interactions determined by Fourier refinement at 1.8 A. J. Mol. Biol. 1978;121:339–356. doi: 10.1016/0022-2836(78)90368-6. [DOI] [PubMed] [Google Scholar]
- 31.Bellovino D., Morimoto T., Pisaniello A., Gaetani S. In vitro and in vivo studies on transthyretin oligomerization. Exp. Cell Res. 1998;243:101–112. doi: 10.1006/excr.1998.4137. [DOI] [PubMed] [Google Scholar]
- 32.Schreiber G., Richardson S.J. The evolution of gene expression, structure and function of transthyretin. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 1997;116:137–160. doi: 10.1016/S0305-0491(96)00212-X. [DOI] [PubMed] [Google Scholar]
- 33.Costa R.H., Lai E., E Darnell J. Transcriptional control of the mouse prealbumin (transthyretin) gene: Both promoter sequences and a distinct enhancer are cell specific. Mol. Cell. Biol. 1986;6:4697–4708. doi: 10.1128/mcb.6.12.4697-4708.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang Z., Burke P.A. Hepatocyte nuclear factor-4α interacts with other hepatocyte nuclear factors in regulating transthyretin gene expression. FEBS. 2010;277:4066–4075. doi: 10.1111/j.1742-4658.2010.07802.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dickson P.W., Howlett G.J., Schreiber G. Rat transthyretin (prealbumin). Molecular cloning, nucleotide sequence, and gene expression in liver and brain. J. Biol. Chem. 1985;260:8214–8219. doi: 10.1016/S0021-9258(17)39583-2. [DOI] [PubMed] [Google Scholar]
- 36.Dickson P.W., Aldred A.R., Marley P.D., Bannister D., Schreiber G. Rat choroid plexus specializes in the synthesis and the secretion of transthyretin (prealbumin). Regulation of transthyretin synthesis in choroid plexus is independent from that in liver. J. Biol. Chem. 1986;261:3475–3478. doi: 10.1016/S0021-9258(17)35671-5. [DOI] [PubMed] [Google Scholar]
- 37.Yan C., Costa R., Darnell J., Chen J., Van Dyke T. Distinct positive and negative elements control the limited hepatocyte and choroid plexus expression of transthyretin in transgenic mice. EMBO J. 1990;9:869–878. doi: 10.1002/j.1460-2075.1990.tb08184.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Costa R.H., A Van Dyke T., Yan C., Kuo F., E Darnell J. Similarities in transthyretin gene expression and differences in transcription factors: Liver and yolk sac compared to choroid plexus. Proc. Natl. Acad. Sci. USA. 1990;87:6589–6593. doi: 10.1073/pnas.87.17.6589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.SStauder A.J., Dickson P.W., Aldred A.R., Schreiber G., A Mendelsohn F., Hudson P. Synthesis of transthyretin (pre-albumin) mRNA in choroid plexus epithelial cells, localized by in situ hybridization in rat brain. J. Histochem. Cytochem. 1986;34:949–952. doi: 10.1177/34.7.3458812. [DOI] [PubMed] [Google Scholar]
- 40.Schreiber G., Aldred A.R., Jaworowski A., Nilsson C., Achen M.G., Segal M.B. Thyroxine transport from blood to brain via transthyretin synthesis in choroid plexus. Am. J. Physiol. Integr. Comp. Physiol. 1990;258:R338–R345. doi: 10.1152/ajpregu.1990.258.2.R338. [DOI] [PubMed] [Google Scholar]
- 41.Lemkine G.F., Raji A., Alfama G., Turque N., Hassani Z., Alegria-Prévot O., Samarut J., Levi G., Demeneix B.A. Adult neural stem cell cycling in vivo requires thyroid hormone and its alpha receptor. FASEB J. 2005;19:863–865. doi: 10.1096/fj.04-2916fje. [DOI] [PubMed] [Google Scholar]
- 42.Schreiber G., Southwell B.R., Richardson S.J. Hormone delivery systems to the brain - transthyretin. Exp. Clin. Endocrinol. Diabetes. 1995;103:75–80. doi: 10.1055/s-0029-1211332. [DOI] [PubMed] [Google Scholar]
- 43.Palha J.A. Transthyretin as a thyroid hormone carrier: Function revisited. Clin. Chem. Lab. Med. (CCLM) 2002;40:1292–1300. doi: 10.1515/CCLM.2002.223. [DOI] [PubMed] [Google Scholar]
- 44.Hamilton J.A., Benson M.D. Transthyretin: A review from a structural perspective. Cell Mol. Life Sci. 2001;58:1491–1521. doi: 10.1007/PL00000791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McKinnon B., Li H., Richard K., Mortimer R. Synthesis of thyroid hormone binding proteins transthyretin and albumin by human trophoblast. J. Clin. Endocrinol. Metab. 2005;90:6714–6720. doi: 10.1210/jc.2005-0696. [DOI] [PubMed] [Google Scholar]
- 46.Patel J., Landers K., Li H., Mortimer R., Richard K. Ontogenic changes in placental transthyretin. Placenta. 2011;32:817–822. doi: 10.1016/j.placenta.2011.09.007. [DOI] [PubMed] [Google Scholar]
- 47.Patel J., Landers K., Li H., Mortimer R.H., Richard K. Delivery of maternal thyroid hormones to the fetus. Trends Endocrinol. Metab. 2011;22:164–170. doi: 10.1016/j.tem.2011.02.002. [DOI] [PubMed] [Google Scholar]
- 48.Ross C., Boroviak T.E. Origin and function of the yolk sac in primate embryogenesis. Nat. Commun. 2020;11:3760. doi: 10.1038/s41467-020-17575-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Landers K., Mortimer R., Richard K. Transthyretin and the human placenta. Placenta. 2013;34:513–517. doi: 10.1016/j.placenta.2013.04.013. [DOI] [PubMed] [Google Scholar]
- 50.Costa R.H., Grayson D.R. Site-directed mutagenesis of hepatocyte nuclear factor (HNF) binding sites in the mouse transthyretin (TTR) promoter reveal synergistic interactions with its enhancer region. Nucleic Acids Res. 1991;19:4139–4145. doi: 10.1093/nar/19.15.4139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Westermark G.T., Westermark P. Transthyretin and amyloid in the islets of Langerhans in type-2 diabetes. Exp. Diabetes Res. 2008;2008:1–7. doi: 10.1155/2008/429274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Su Y., Jono H., Misumi Y., Senokuchi T., Guo J., Ueda M., Shinriki S., Tasaki M., Shono M., Obayashi K., et al. Novel function of transthyretin in pancreatic alpha cells. FEBS Lett. 2012;586:4215–4222. doi: 10.1016/j.febslet.2012.10.025. [DOI] [PubMed] [Google Scholar]
- 53.Refai E., Dekki N., Yang S.N., Imreh G., Cabrera O., Yu L., Yang G., Norgren S., Rössner S.M., Inverardi L., et al. Transthyretin constitutes a functional component in pancreatic β-cell stimulus-secretion coupling. Proc. Natl. Acad. Sci. USA. 2005;102:17020–17025. doi: 10.1073/pnas.0503219102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Itoh N., Hanafusa T., Miyagawa J., Tamura S., Inada M., Kawata S., Kono N., Tarui S. Transthyretin (prealbumin) in the pancreas and sera of newly diagnosed type I (insulin-dependent) diabetic patients. J. Clin. Endocrinol. Metab. 1992;74:1372–1377. doi: 10.1210/jcem.74.6.1592883. [DOI] [PubMed] [Google Scholar]
- 55.Cavallaro T., Martone R., Dwork A.J., A Schon E., Herbert J. The retinal pigment epithelium is the unique site of transthyretin synthesis in the rat eye. Investig. Ophthalmol. Vis. Sci. 1990;31:497–501. [PubMed] [Google Scholar]
- 56.Martone R.L., Herbert J., Dwork A., Schon E.A. Transthyretin is synthesized in the mammalian eye. Biochem. Biophys. Res. Commun. 1988;151:905–912. doi: 10.1016/S0006-291X(88)80367-X. [DOI] [PubMed] [Google Scholar]
- 57.Ong D.E., Davis J.T., O’Day W.T., Bok D. Synthesis and secretion of retinol-binding protein and transthyretin by cultured retinal pigment epithelium. Biochemistry. 1994;33:1835–1842. doi: 10.1021/bi00173a029. [DOI] [PubMed] [Google Scholar]
- 58.Eichenbaum J.W., Zheng W. Distribution of lead and transthyretin in human eyes. J. Toxicol. Clin. Toxicol. 2000;38:377–381. doi: 10.1081/CLT-100100946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liepnieks J.J., Wilson D.L., Benson M.D. Biochemical characterization of vitreous and cardiac amyloid in Ile84Ser transthyretin amyloidosis. Amyloid. 2006;13:170–177. doi: 10.1080/13506120600877003. [DOI] [PubMed] [Google Scholar]
- 60.Iakovleva I., Hall M., Oelker M., Sandblad L., Anan I., Sauer-Eriksson A.E. Structural basis for transthyretin amyloid formation in vitreous body of the eye. Nat. Commun. 2021;12:1–10. doi: 10.1038/s41467-021-27481-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Blake C., Swan I., Rerat C., Berthou J., Laurent A., Rerat B. An x-ray study of the subunit structure of prealbumin. J. Mol. Biol. 1971;61:217–224. doi: 10.1016/0022-2836(71)90218-X. [DOI] [PubMed] [Google Scholar]
- 62.Kanda Y., Goodman D.S., Canfield R.E., Morgan F.J. The amino acid sequence of human plasma prealbumin. J. Biol. Chem. 1974;249:6796–6805. doi: 10.1016/S0021-9258(19)42128-5. [DOI] [PubMed] [Google Scholar]
- 63.He S., He X., Liu L., Zhang W., Yu L., Deng Z., Feiyi Z., Mo S., Fan Y., Zhao X., et al. The Structural Understanding of Transthyretin Misfolding and the Inspired Drug Approaches for the Treatment of Heart Failure Associated With Transthyretin Amyloidosis. Front. Pharmacol. 2021;12:628184. doi: 10.3389/fphar.2021.628184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Park G.Y., Jamerlan A., Shim K.H., An S.S.A. Diagnostic and Treatment Approaches Involving Transthyretin in Amyloidogenic Diseases. Int. J. Mol. Sci. 2019;20:2982. doi: 10.3390/ijms20122982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Schneider F., Hammarstrom P., Kelly J.W. Transthyretin slowly exchanges subunits under physiological conditions: A convenient chromatographic method to study subunit exchange in oligomeric proteins. Protein. Sci. 2001;10:1606–1613. doi: 10.1110/ps.8901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tomar D., Khan T., Singh R.R., Mishra S., Gupta S., Surolia A., Salunke D.M. Crystallographic study of novel transthyretin ligands exhibiting negative-cooperativity between two thyroxine binding sites. PLoS ONE. 2012;7:e43522. doi: 10.1371/journal.pone.0043522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ferguson R.N., Edelhoch H., Saroff H.A., Robbins J., Cahnmann H.J. Negative cooperativity in the binding of thyroxine to human serum prealbumin. Biochemistry. 1975;14:282–289. doi: 10.1021/bi00673a014. [DOI] [PubMed] [Google Scholar]
- 68.Johnson S.M., Wiseman L., Sekijima Y., Green N.S., Adamski-Werner S.L., Kelly J.W. Native state kinetic stabilization as a strategy to ameliorate protein misfolding diseases: A focus on the transthyretin amyloidoses. Accounts Chem. Res. 2005;38:911–921. doi: 10.1021/ar020073i. [DOI] [PubMed] [Google Scholar]
- 69.Foss T.R., Kelker M.S., Wiseman R.L., Wilson I.A., Kelly J.W. Kinetic stabilization of the native state by protein engineering: Implications for inhibition of transthyretin amyloidogenesis. J. Mol. Biol. 2005;347:841–854. doi: 10.1016/j.jmb.2005.01.050. [DOI] [PubMed] [Google Scholar]
- 70.Johnson S.M., Connelly S., Fearns C., Powers E.T., Kelly J.W. The transthyretin amyloidoses: From delineating the molecular mechanism of aggregation linked to pathology to a regulatory-agency-approved drug. J. Mol. Biol. 2012;421:185–203. doi: 10.1016/j.jmb.2011.12.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Garcia-Pavia P., Rapezzi C., Adler Y., Arad M., Basso C., Brucato A., Burazor I., Caforio A.L., Damy T., Eriksson U., et al. Diagnosis and treatment of cardiac amyloidosis. A position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur. J. Hear. Fail. 2021;23:512–526. doi: 10.1002/ejhf.2140. [DOI] [PubMed] [Google Scholar]
- 72.Coelho T., Merlini G., Bulawa C.E., Fleming J.A., Judge D., Kelly J.W., Maurer M.S., Planté-Bordeneuve V., Labaudinière R., Mundayat R., et al. Mechanism of Action and Clinical Application of Tafamidis in Hereditary Transthyretin Amyloidosis. Neurol. Ther. 2016;5:1–25. doi: 10.1007/s40120-016-0040-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sekijima Y., Dendle M.A., Kelly J.W. Orally administered diflunisal stabilizes transthyretin against dissociation required for amyloidogenesis. Amyloid. 2006;13:236–249. doi: 10.1080/13506120600960882. [DOI] [PubMed] [Google Scholar]
- 74.Judge D.P., Heitner S.B., Falk R.H., Maurer M.S., Shah S.J., Witteles R.M., Grogan M., Selby V.N., Jacoby D., Hanna M., et al. Transthyretin Stabilization by AG10 in Symptomatic Transthyretin Amyloid Cardiomyopathy. J. Am. Coll. Cardiol. 2019;74:285–295. doi: 10.1016/j.jacc.2019.03.012. [DOI] [PubMed] [Google Scholar]
- 75.Pullakhandam R., Srinivas P., Nair M.K., Reddy G.B. Binding and stabilization of transthyretin by curcumin. Arch. Biochem. Biophys. 2009;485:115–119. doi: 10.1016/j.abb.2009.02.013. [DOI] [PubMed] [Google Scholar]
- 76.Santos L.M., Rodrigues D., Alemi M., Silva S.C., Ribeiro C.A., Cardoso I. Resveratrol administration increases Transthyretin protein levels ameliorating AD features- importance of transthyretin tetrameric stability. Mol. Med. 2016;22:597–607. doi: 10.2119/molmed.2016.00124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yokoyama T., Takaki S., Chosa K., Sato T., Suico M.A., Teranishi Y., Shuto T., Mizuguchi M., Kai H. Structural stabilization of transthyretin by a new compound, 6-benzoyl-2-hydroxy-1H-benzo[de]isoquinoline-1,3(2H)-dione. J. Pharmacol. Sci. 2015;129:240–243. doi: 10.1016/j.jphs.2015.09.006. [DOI] [PubMed] [Google Scholar]
- 78.Ciccone L., Tonali N., Nencetti S., Orlandini E. Natural compounds as inhibitors of transthyretin amyloidosis and neuroprotective agents: Analysis of structural data for future drug design. J. Enzym. Inhib. Med. Chem. 2020;35:1145–1162. doi: 10.1080/14756366.2020.1760262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Almeida M., Gales L., Damas A.M., Cardoso I., Saraiva M.J. Small transthyretin (TTR) ligands as possible therapeutic agents in TTR amyloidoses. Curr. Drug Target.-CNS Neurol. Disord. 2005;4:587–596. doi: 10.2174/156800705774322076. [DOI] [PubMed] [Google Scholar]
- 80.Monaco H.L., Rizzi M., Coda A. Structure of a complex of two plasma proteins: Transthyretin and retinol-binding protein. Science. 1995;268:1039–1041. doi: 10.1126/science.7754382. [DOI] [PubMed] [Google Scholar]
- 81.Naylor H.M., Newcomer M.E. The structure of human retinol-binding protein (RBP) with its carrier protein transthyretin reveals an interaction with the carboxy terminus of RBP. Biochemistry. 1999;38:2647–2653. doi: 10.1021/bi982291i. [DOI] [PubMed] [Google Scholar]
- 82.Berni R., Malpeli G., Folli C., Murrell J., Liepnieks J., Benson M. The Ile-84-->Ser amino acid substitution in transthyretin interferes with the interaction with plasma retinol-binding protein. J. Biol. Chem. 1994;269:23395–23398. doi: 10.1016/S0021-9258(17)31527-2. [DOI] [PubMed] [Google Scholar]
- 83.TTrägårdh L., Anundi H., Rask L., Sege K., A Peterson P. On the stoichiometry of the interaction between prealbumin and retinol-binding protein. J. Biol. Chem. 1980;255:9243–9248. doi: 10.1016/S0021-9258(19)70553-5. [DOI] [PubMed] [Google Scholar]
- 84.Prapunpoj P., Leelawatwattana L. Evolutionary changes to transthyretin: Structure-function relationships. FEBS J. 2009;276:5330–5341. doi: 10.1111/j.1742-4658.2009.07243.x. [DOI] [PubMed] [Google Scholar]
- 85.Zanotti G., Berni R. Plasma retinol-binding protein: Structure and interactions with retinol, retinoids, and transthyretin. Vitam. & Horm. 2004;69:271–295. doi: 10.1016/S0083-6729(04)69010-8. [DOI] [PubMed] [Google Scholar]
- 86.Malpeli G., Folli C., Berni R. Retinoid binding to retinol-binding protein and the interference with the interaction with transthyretin. Biochim. Biophys. Acta. 1996;1294:48–54. doi: 10.1016/0167-4838(95)00264-2. [DOI] [PubMed] [Google Scholar]
- 87.White J.T., Kelly J.W. Support for the multigenic hypothesis of amyloidosis: The binding stoichiometry of retinol-binding protein, vitamin A, and thyroid hormone influences transthyretin amyloidogenicity in vitro. Proc. Natl. Acad. Sci. USA. 2001;98:13019–13024. doi: 10.1073/pnas.241406698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Heller J., Horwitz J. Conformational changes following interaction between retinol isomers and human retinol-binding protein and between the retinol-binding protein and prealbumin. J. Biol. Chem. 1973;248:6308–6316. doi: 10.1016/S0021-9258(19)43449-2. [DOI] [PubMed] [Google Scholar]
- 89.Ferreira N., Saraiva M.J., Almeida M.R. Uncovering the Neuroprotective Mechanisms of Curcumin on Transthyretin Amyloidosis. Int. J. Mol. Sci. 2019;20:1287. doi: 10.3390/ijms20061287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wieczorek E., Ożyhar A. Transthyretin: From Structural Stability to Osteoarticular and Cardiovascular Diseases. Cells. 2021;10:1768. doi: 10.3390/cells10071768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Si J.B., Kim B., Kim J.H. Transthyretin Misfolding, A Fatal Structural Pathogenesis Mechanism. Int. J. Mol. Sci. 2021;22:4429. doi: 10.3390/ijms22094429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Pires R.H., Karsai A., Saraiva M.J., Damas A.M., Kellermayer M.S.Z. Distinct annular oligomers captured along the assembly and disassembly pathways of transthyretin amyloid protofibrils. PLoS ONE. 2012;7:e44992. doi: 10.1371/journal.pone.0044992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Koike H., Katsuno M. Transthyretin Amyloidosis: Update on the Clinical Spectrum, Pathogenesis, and Disease-Modifying Therapies. Neurol. Ther. 2020;9:317–333. doi: 10.1007/s40120-020-00210-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Leach B.I., Zhang X., Kelly J.W., Dyson H.J., Wright P.E. NMR Measurements Reveal the Structural Basis of Transthyretin Destabilization by Pathogenic Mutations. Biochemistry. 2018;57:4421–4430. doi: 10.1021/acs.biochem.8b00642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Conceição I., Coelho T., Rapezzi C., Parman Y., Obici L., Galán L., Rousseau A. Assessment of patients with hereditary transthyretin amyloidosis - understanding the impact of management and disease progression. Amyloid. 2019;26:103–111. doi: 10.1080/13506129.2019.1627312. [DOI] [PubMed] [Google Scholar]
- 96.Rowczenio D.M., Noor I., Gillmore J.D., Lachmann H.J., Whelan C., Hawkins P.N., Obici L., Westermark P., Grateau G., Wechalekar A.D. Online registry for mutations in hereditary amyloidosis including nomenclature recommendations. Hum. Mutat. 2014;35:E2403–E2412. doi: 10.1002/humu.22619. [DOI] [PubMed] [Google Scholar]
- 97.Sant’Anna R., Almeida M., Varejao N., Gallego P., Esperante S., Ferreira P., Pereira-Henriques A., Palhano F., De Carvalho M., Foguel D., et al. Cavity filling mutations at the thyroxine-binding site dramatically increase transthyretin stability and prevent its aggregation. Sci. Rep. 2017;7:srep44709. doi: 10.1038/srep44709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Almeida M.R., Alves I.L., Terazaki H., Ando Y., Saraiva M.J. Comparative studies of two transthyretin variants with protective effects on familial amyloidotic polyneuropathy: TTR R104H and TTR T119M. Biochem. Biophys. Res. Commun. 2000;270:1024–1028. doi: 10.1006/bbrc.2000.2554. [DOI] [PubMed] [Google Scholar]
- 99.Sekijima Y., Dendle M.T., Wiseman R.L., White J.T., D’Haeze W., Kelly J.W. R104H may suppress transthyretin amyloidogenesis by thermodynamic stabilization, but not by the kinetic mechanism characterizing T119 interallelic trans-suppression. Amyloid. 2006;13:57–66. doi: 10.1080/13506120600722449. [DOI] [PubMed] [Google Scholar]
- 100.Palaninathan S.K. Nearly 200 X-ray crystal structures of transthyretin: What do they tell us about this protein and the design of drugs for TTR amyloidoses? Curr. Med. Chem. 2012;19:2324–2342. doi: 10.2174/092986712800269335. [DOI] [PubMed] [Google Scholar]
- 101.Hörnberg A., Eneqvist T., Olofsson A., Lundgren E., Sauer-Eriksson E. A comparative analysis of 23 structures of the amyloidogenic protein transthyretin. J. Mol. Biol. 2000;302:649–669. doi: 10.1006/jmbi.2000.4078. [DOI] [PubMed] [Google Scholar]
- 102.Ferguson J.A., Sun X., Dyson H.J., Wright P.E. Thermodynamic Stability and Aggregation Kinetics of EF Helix and EF Loop Variants of Transthyretin. Biochemistry. 2021;60:756–764. doi: 10.1021/acs.biochem.1c00073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Saldaño T.E., Zanotti G., Parisi G., Fernandez-Alberti S. Evaluating the effect of mutations and ligand binding on transthyretin homotetramer dynamics. PLoS ONE. 2017;12:e0181019. doi: 10.1371/journal.pone.0181019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hamilton J., Steinrauf L., Braden B., Liepnieks J., Benson M., Holmgren G., Sandgren O., Steen L. The X-ray crystal structure refinements of normal human transthyretin and the amyloidogenic Val-30-->Met variant to 1.7-A resolution. J. Biol. Chem. 1993;268:2416–2424. doi: 10.1016/S0021-9258(18)53792-3. [DOI] [PubMed] [Google Scholar]
- 105.Terry C., Damas A., Oliveira P., Saraiva M.J., Alves I., Costa P., Matias P., Sakaki Y., Blake C. Structure of Met30 variant of transthyretin and its amyloidogenic implications. EMBO J. 1993;12:735–741. doi: 10.1002/j.1460-2075.1993.tb05707.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Yee A.W., Aldeghi M., Blakeley M.P., Ostermann A., Mas P.J., Moulin M., de Sanctis D., Bowler M.W., Mueller-Dieckmann C., Mitchell E.P., et al. A molecular mechanism for transthyretin amyloidogenesis. Nat. Commun. 2019;10:1–10. doi: 10.1038/s41467-019-08609-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Miyata M., Sato T., Mizuguchi M., Nakamura T., Ikemizu S., Nabeshima Y., Susuki S., Suwa Y., Morioka H., Ando Y., et al. Role of the glutamic acid 54 residue in transthyretin stability and thyroxine binding. Biochemistry. 2009;49:114–123. doi: 10.1021/bi901677z. [DOI] [PubMed] [Google Scholar]
- 108.Sebastião P., Dauter Z., Saraiva M.J., Damas A.M. Crystallization and preliminary X-ray diffraction studies of Leu55Pro variant transthyretin. Acta Crystallogr. Sect. D Biol. Crystallogr. 1996;52:566–568. doi: 10.1107/S0907444995013965. [DOI] [PubMed] [Google Scholar]
- 109.Sebastião M.P., Saraiva M.J., Damas A.M. The crystal structure of amyloidogenic Leu55 --> Pro transthyretin variant reveals a possible pathway for transthyretin polymerization into amyloid fibrils. J. Biol. Chem. 1998;273:24715–24722. doi: 10.1074/jbc.273.38.24715. [DOI] [PubMed] [Google Scholar]
- 110.Damas A.M., Ribeiro S., Lamzin V., Palha J.A., Saraiva M.J. Structure of the Val122Ile variant transthyretin - a cardiomyopathic mutant. Acta Crystallogr. Sect. D Biol. Crystallogr. 1996;52:966–972. doi: 10.1107/S0907444996003307. [DOI] [PubMed] [Google Scholar]
- 111.Vugmeyster L., Au D.F., Ostrovsky D., Kierl B., Fu R., Hu Z.W., Qiang W. Effect of Post-Translational Modifications and Mutations on Amyloid-β Fibrils Dynamics at N Terminus. Biophys. J. 2019;117:1524–1535. doi: 10.1016/j.bpj.2019.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Dorval V., Fraser P.E. Small ubiquitin-like modifier (SUMO) modification of natively unfolded proteins tau and and α-Synuclein. J. Biol. Chem. 2006;281:9919–9924. doi: 10.1074/jbc.M510127200. [DOI] [PubMed] [Google Scholar]
- 113.Barykin E.P., Mitkevich V.A., Kozin S., Makarov A.A. Amyloid β Modification: A Key to the Sporadic Alzheimer’s Disease? Front. Genet. 2017;8:58. doi: 10.3389/fgene.2017.00058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Radamaker L., Karimi-Farsijani S., Andreotti G., Baur J., Neumann M., Schreiner S., Berghaus N., Motika R., Haupt C., Walther P., et al. Role of mutations and post-translational modifications in systemic AL amyloidosis studied by cryo-EM. Nat. Commun. 2021;12:1–11. doi: 10.1038/s41467-021-26553-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Henze A., Homann T., Serteser M., Can O., Sezgin O., Coskun A., Unsal I., Schweigert F., Ozpinar A. Post-translational modifications of transthyretin affect the triiodonine-binding potential. J. Cell. Mol. Med. 2015;19:359–370. doi: 10.1111/jcmm.12446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Pettersson T., Carlstroem A., Joernvall H. Different types of microheterogeneity of human thyroxine-binding prealbumin. Biochemistry. 1987;26:4572–4583. doi: 10.1021/bi00388a061. [DOI] [PubMed] [Google Scholar]
- 117.Poulsen K., Bahl J.M., Tanassi J.T., Simonsen A.H., Heegaard N.H. Characterization and stability of transthyretin isoforms in cerebrospinal fluid examined by immunoprecipitation and high-resolution mass spectrometry of intact protein. Methods. 2012;56:284–292. doi: 10.1016/j.ymeth.2011.12.009. [DOI] [PubMed] [Google Scholar]
- 118.Henze A., Homann T., Rohn I., Aschner M., Link C.D., Kleuser B., Schweigert F., Schwerdtle T., Bornhorst J. Caenorhabditis elegans as a model system to study post-translational modifications of human transthyretin. Sci. Rep. 2016;6:37346. doi: 10.1038/srep37346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Gales L., Saraiva M.J., Damas A.M. Structural basis for the protective role of sulfite against transthyretin amyloid formation. Biochim. et Biophys. Acta (BBA)-Proteins Proteom. 2007;1774:59–64. doi: 10.1016/j.bbapap.2006.10.015. [DOI] [PubMed] [Google Scholar]
- 120.Altland K., Winter P. Potential treatment of transthyretin-type amyloidoses by sulfite. Neurogenetics. 1999;2:183–188. doi: 10.1007/s100480050081. [DOI] [PubMed] [Google Scholar]
- 121.Kingsbury J., Laue T.M., Klimtchuk E.S., Théberge R., Costello C., Connors L.H. The modulation of transthyretin tetramer stability by cysteine 10 adducts and the drug diflunisal: Direct analysis by fluorescence-detected analytical ultracentrifugation. J. Biol. Chem. 2008;283:11887–11896. doi: 10.1074/jbc.M709638200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Takaoka Y., Ohta M., Miyakawa K., Nakamura O., Suzuki M., Takahashi K., Yamamura K., Sakaki Y. Cysteine 10 is a key residue in amyloidogenesis of human transthyretin Val30Met. Am. J. Pathol. 2004;164:337–345. doi: 10.1016/S0002-9440(10)63123-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Zhang Q., Kelly J.W. Cys10 mixed disulfides make transthyretin more amyloidogenic under mildly acidic conditions. Biochemistry. 2003;42:8756–8761. doi: 10.1021/bi030077a. [DOI] [PubMed] [Google Scholar]
- 124.Zhao L., Buxbaum J.N., Reixach N. Age-related oxidative modifications of transthyretin modulate its amyloidogenicity. Biochemistry. 2013;52:1913–1926. doi: 10.1021/bi301313b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Navab M., E Smith J., Goodman D.S. Rat plasma prealbumin. Metabolic studies on effects of vitamin A status and on tissue distribution. J. Biol. Chem. 1977;252:5107–5114. doi: 10.1016/S0021-9258(17)40164-5. [DOI] [PubMed] [Google Scholar]
- 126.Rask L., Valtersson C., Anundi H., Kvist S., Eriksson U., Dallner G., A Peterson P. Subcellular localization in normal and vitamin A-deficient rat liver of vitamin A serum transport proteins, albumin, ceruloplasmin and class I major histocompatibility antigens. Exp. Cell Res. 1983;143:91–102. doi: 10.1016/0014-4827(83)90112-X. [DOI] [PubMed] [Google Scholar]
- 127.Melhus H., Nilsson T., Peterson P.A., Rask L. Retinol-binding protein and transthyretin expressed in HeLa cells form a complex in the endoplasmic reticulum in both the absence and the presence of retinol. Exp. Cell Res. 1991;197:119–124. doi: 10.1016/0014-4827(91)90488-G. [DOI] [PubMed] [Google Scholar]
- 128.Episkopou V., Maeda S., Nishiguchi S., Shimada K., Gaitanaris G.A., Gottesman M.E., Robertson E.J. Disruption of the transthyretin gene results in mice with depressed levels of plasma retinol and thyroid hormone. Proc. Natl. Acad. Sci. USA. 1993;90:2375–2379. doi: 10.1073/pnas.90.6.2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.E Smith J., Deen D.D., Sklan D., Goodman D.S. Colchicine inhibition of retinol-binding protein secretion by rat liver. J. Lipid Res. 1980;21:229–237. doi: 10.1016/S0022-2275(20)39829-1. [DOI] [PubMed] [Google Scholar]
- 130.Berni R., Clerici M., Malpeli G., Cleris L., Formelli F. Retinoids: In vitro interaction with retinol-binding protein and influence on plasma retinol. FASEB J. 1993;7:1179–1184. doi: 10.1096/fasebj.7.12.8375617. [DOI] [PubMed] [Google Scholar]
- 131.Bellovino D., Morimoto T., Tosetti F., Gaetani S. Retinol binding protein and transthyretin are secreted as a complex formed in the endoplasmic reticulum in HepG2 human hepatocarcinoma cells. Exp. Cell Res. 1996;222:77–83. doi: 10.1006/excr.1996.0010. [DOI] [PubMed] [Google Scholar]
- 132.Hammarström P., Sekijima Y., White J.T., Wiseman R.L., Lim A., Costello C.E., Altland K., Garzuly F., Budka H., Kelly J.W. D18G transthyretin is monomeric, aggregation prone, and not detectable in plasma and cerebrospinal fluid: A prescription for central nervous system amyloidosis? Biochemistry. 2003;42:6656–6663. doi: 10.1021/bi027319b. [DOI] [PubMed] [Google Scholar]
- 133.Sekijima Y., Hammarström P., Matsumura M., Shimizu Y., Iwata M., Tokuda T., Ikeda S., Kelly J.W. Energetic characteristics of the new transthyretin variant A25T may explain its atypical central nervous system pathology. Lab. Investig. 2003;83:409–417. doi: 10.1097/01.LAB.0000059937.11023.1F. [DOI] [PubMed] [Google Scholar]
- 134.Sörgjerd K., Ghafouri B., Jonsson B.H., Kelly J.W., Blond S.Y., Hammarström P. Retention of misfolded mutant transthyretin by the chaperone BiP/GRP78 mitigates amyloidogenesis. J. Mol. Biol. 2006;356:469–482. doi: 10.1016/j.jmb.2005.11.051. [DOI] [PubMed] [Google Scholar]
- 135.Susuki S., Sato T., Miyata M., Momohara M., Suico M.A., Shuto T., Ando Y., Kai H. The Endoplasmic Reticulum-associated Degradation of Transthyretin Variants Is Negatively Regulated by BiP in Mammalian Cells. J. Biol. Chem. 2009;284:8312–8321. doi: 10.1074/jbc.M809354200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.da Costa G., Ribeiro-Silva C., Ribeiro R., Gilberto S., Gomes R.A., Ferreira A., Mateus É., Barroso E., Coelho A.V., Freire A.P., et al. Transthyretin Amyloidosis: Chaperone Concentration Changes and Increased Proteolysis in the Pathway to Disease. PLoS ONE. 2015;10:e0125392. doi: 10.1371/journal.pone.0125392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Mesgarzadeh J.S., Romine I.C., Smith-Cohen E.M., Grandjean J.M.D., Kelly J.W., Genereux J.C., Wiseman R.L. ATF6 Activation Reduces Amyloidogenic Transthyretin Secretion through Increased Interactions with Endoplasmic Reticulum Proteostasis Factors. Cells. 2022;11:1661. doi: 10.3390/cells11101661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Sekijima Y., Wiseman R.L., Matteson J., Hammarström P., Miller S.R., Sawkar A.R., Balch W.E., Kelly J.W. The biological and chemical basis for tissue-selective amyloid disease. Cell. 2005;121:73–85. doi: 10.1016/j.cell.2005.01.018. [DOI] [PubMed] [Google Scholar]
- 139.Sitia R., Braakman I. Quality control in the endoplasmic reticulum protein factory. Nature. 2003;426:891–894. doi: 10.1038/nature02262. [DOI] [PubMed] [Google Scholar]
- 140.Hampton R.Y. ER-associated degradation in protein quality control and cellular regulation. Curr. Opin. Cell Biol. 2002;14:476–482. doi: 10.1016/S0955-0674(02)00358-7. [DOI] [PubMed] [Google Scholar]
- 141.Cohen F.E., Kelly J.W. Therapeutic approaches to protein-misfolding diseases. Nature. 2003;426:905–909. doi: 10.1038/nature02265. [DOI] [PubMed] [Google Scholar]
- 142.Braakman I., Hebert D.N. Protein folding in the endoplasmic reticulum. Cold Spring Harb. Perspect Biol. 2013;5:a013201. doi: 10.1101/cshperspect.a013201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Wyatt A.R., Yerbury J.J., Ecroyd H., Wilson M.R. Extracellular chaperones and proteostasis. Annu. Rev. Biochem. 2013;82:295–322. doi: 10.1146/annurev-biochem-072711-163904. [DOI] [PubMed] [Google Scholar]
- 144.Dabbs R.A., Wyatt A.R., Yerbury J.J., Ecroyd H., Wilson M.R. Extracellular chaperones. Top. Curr. Chem. 2013;328:241–268. doi: 10.1007/128_2011_262. [DOI] [PubMed] [Google Scholar]
- 145.Nilselid A.M., Davidsson P., Nägga K., Andreasen N., Fredman P., Blennow K. Clusterin in cerebrospinal fluid: Analysis of carbohydrates and quantification of native and glycosylated forms. Neurochem. Int. 2006;48:718–728. doi: 10.1016/j.neuint.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 146.Gollin P.A., Kalaria R.N., Eikelenboom P., Rozemuller A., Perry G. Alpha 1-antitrypsin and alpha 1-antichymotrypsin are in the lesions of Alzheimer’s disease. Neuroreport. 1992;3:201–203. doi: 10.1097/00001756-199202000-00020. [DOI] [PubMed] [Google Scholar]
- 147.Yerbury J.J., Poon S., Meehan S., Thompson B., Kumita J.R., Dobson C.M., Wilson M.R. The extracellular chaperone clusterin influences amyloid formation and toxicity by interacting with prefibrillar structures. FASEB J. 2007;21:2312–2322. doi: 10.1096/fj.06-7986com. [DOI] [PubMed] [Google Scholar]
- 148.Makover A., Moriwaki H., Ramakrishnan R., Saraiva M.J., Blaner W.S., Goodman D.S. Plasma transthyretin. Tissue sites of degradation and turnover in the rat. J. Biol. Chem. 1988;263:8598–8603. doi: 10.1016/S0021-9258(18)68346-2. [DOI] [PubMed] [Google Scholar]
- 149.Gjøen T., Bjerkelund T., Blomhoff H.K., Norum K.R., Berg T., Blomhoff R. Liver takes up retinol-binding protein from plasma. J. Biol. Chem. 1987;262:10926–10930. doi: 10.1016/S0021-9258(18)60905-6. [DOI] [PubMed] [Google Scholar]
- 150.Sousa M.M., Norden A.G., Jacobsen C., Willnow T.E., Christensen E.I., Thakker R.V., Verroust P.J., Moestrup S.K., Saraiva M.J. Evidence for the role of megalin in renal uptake of transthyretin. J. Biol. Chem. 2000;275:38176–38181. doi: 10.1074/jbc.M002886200. [DOI] [PubMed] [Google Scholar]
- 151.May P., Woldt E., Matz R.L., Boucher P. The LDL receptor-related protein (LRP) family: An old family of proteins with new physiological functions. Ann. Med. 2007;39:219–228. doi: 10.1080/07853890701214881. [DOI] [PubMed] [Google Scholar]
- 152.Christensen E.I., Moskaug J.O., Vorum H., Jacobsen C., Gundersen T.E., Nykjaer A., Blomhoff R., Willnow T.E., Moestrup S.K. Evidence for an essential role of megalin in transepithelial transport of retinol. J. Am. Soc. Nephrol. 1999;10:685–695. doi: 10.1681/ASN.V104685. [DOI] [PubMed] [Google Scholar]
- 153.Fleming C.E., Mar F.M., Franquinho F., Saraiva M.J., Sousa M.M. Transthyretin internalization by sensory neurons is megalin mediated and necessary for its neuritogenic activity. J. Neurosci. 2009;29:3220–3232. doi: 10.1523/JNEUROSCI.6012-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Sousa M.M., Saraiva M.J. Internalization of transthyretin:. Evidence of a novel yet unidentified receptor-associated protein (RAP)-sensitive receptor. J. Biol. Chem. 2001;276:14420–14425. doi: 10.1074/jbc.M010869200. [DOI] [PubMed] [Google Scholar]
- 155.Divino C.M., Schussler G.C. Receptor-mediated uptake and internalization of transthyretin. J. Biol. Chem. 1990;265:1425–1429. doi: 10.1016/S0021-9258(19)40032-X. [DOI] [PubMed] [Google Scholar]
- 156.Sousa M.M., Berglund L., Saraiva M.J. Transthyretin in high density lipoproteins: Association with apolipoprotein A-I. J. Lipid Res. 2000;41:58–65. doi: 10.1016/S0022-2275(20)32074-5. [DOI] [PubMed] [Google Scholar]
- 157.Misumi Y., Ando Y., Gonçalves N.P., Saraiva M.J. Fibroblasts endocytose and degrade transthyretin aggregates in transthyretin-related amyloidosis. Lab. Investig. 2013;93:911–920. doi: 10.1038/labinvest.2013.83. [DOI] [PubMed] [Google Scholar]
- 158.Misumi Y., Ueda M., Obayashi K., Jono H., Yamashita T., Ando Y. Interaction between amyloid fibril formation and extracellular matrix in the proceedings of VIIIth International Symposium on Familial Amyloidotic Polyneuropathy. Amyloid. 2012;19:8–10. doi: 10.3109/13506129.2012.674987. [DOI] [PubMed] [Google Scholar]
- 159.Bourgault S., Solomon J.P., Reixach N., Kelly J.W. Sulfated glycosaminoglycans accelerate transthyretin amyloidogenesis by quaternary structural conversion. Biochemistry. 2011;50:1001–1015. doi: 10.1021/bi101822y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Madine J., Davies H.A., Hughes E., Middleton D.A. Heparin promotes the rapid fibrillization of a peptide with low intrinsic amyloidogenicity. Biochemistry. 2013;52:8984–8992. doi: 10.1021/bi401231u. [DOI] [PubMed] [Google Scholar]
- 161.Fella E., Sokratous K., Papacharalambous R., Kyriacou K., Phillips J., Sanderson S., Panayiotou E., Kyriakides T. Pharmacological Stimulation of Phagocytosis Enhances Amyloid Plaque Clearance; Evidence from a Transgenic Mouse Model of ATTR Neuropathy. Front. Mol. Neurosci. 2017;10:138. doi: 10.3389/fnmol.2017.00138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Hosoi A., Su Y., Torikai M., Jono H., Ishikawa D., Soejima K., Higuchi H., Guo J., Ueda M., Suenaga G., et al. Novel Antibody for the Treatment of Transthyretin Amyloidosis. J. Biol. Chem. 2016;291:25096–25105. doi: 10.1074/jbc.M116.738138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Michalon A., Hagenbuch A., Huy C., Varela E., Combaluzier B., Damy T., Suhr O.B., Saraiva M.J., Hock C., Nitsch R.M., et al. A human antibody selective for transthyretin amyloid removes cardiac amyloid through phagocytic immune cells. Nat. Commun. 2021;12:3142. doi: 10.1038/s41467-021-23274-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Tsuchiya A., Yazaki M., Kametani F., Takei Y., Ikeda S. Marked regression of abdominal fat amyloid in patients with familial amyloid polyneuropathy during long-term follow-up after liver transplantation. Liver Transpl. 2008;14:563–570. doi: 10.1002/lt.21395. [DOI] [PubMed] [Google Scholar]
- 165.Morfino P., Aimo A., Panichella G., Rapezzi C., Emdin M. Amyloid seeding as a disease mechanism and treatment target in transthyretin cardiac amyloidosis. Hear. Fail. Rev. 2022 doi: 10.1007/s10741-022-10237-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.