

Abstract
Early clinical data indicate that some patients with castration-resistant prostate cancer (CRPC) may benefit from program death ligand-1 (PD-L1) inhibition, especially with enzalutamide. IMbassador250 (NCT03016312) enrolled 759 men with metastatic CRPC whose disease progressed on abiraterone. Adding atezolizumab to enzalutamide in an open label randomised trial did not meet the primary endpoint of improved overall survival in unselected patients (stratified HR, 1.12; 95% CI, 0.91-1.37; P=0.28), despite an acceptable safety profile. In archival tumour samples, prostate tumours showed comparatively low expression of key immune biomarkers. DNA damage response alterations, PTEN status, and PD-L1 expression levels were similar between hormone-sensitive and castration-resistant prostate cancers. In planned biomarker analysis, longer progression-free survival (PFS) was seen with atezolizumab in patients with high PD-L1 IC2/3, CD8 expression and established immune gene signatures. Exploratory analysis linked PFS in the atezolizumab arm with immune genes such as CXCL9 and TAP1 together with other potentially relevant biomarkers such as PTEN alterations. Together these data indicate that the expected biology associated with response to immune checkpoint inhibitors is present in prostate cancer, albeit in fewer patients. Careful patient selection may be required for immune checkpoint inhibitors to identify subgroups of patients who may benefit from this treatment approach.
Keywords: metastatic castration-resistant prostate cancer, atezolizumab, PD-L1, enzalutamide, biomarkers, TMB, pre-existing immunity
Metastatic castration-resistant prostate cancer (mCRPC) still has a poor prognosis despite the survival benefits imparted by advancements in therapeutics targeting prostate cancer biology. The majority of men with mCRPC will experience disease progression and ultimately die of their disease.1–6 Men with mCRPC have an approximate median survival of only 3 years.1
Numerous therapies are currently available for mCRPC, including chemotherapy (docetaxel, cabazitaxel), radium 223, sipuleucel-T, abiraterone, and enzalutamide. Enzalutamide, a second-generation oral androgen receptor (AR) antagonist, significantly prolongs survival and improves quality-of-life outcomes in patients with mCRPC before and after chemotherapy.6, 7 Despite these benefits, patients inevitably acquire resistance to enzalutamide over the course of treatment.8
In addition to its effect on the AR pathway, enzalutamide has direct immunomodulatory effects. These effects were evident in a gene expression analysis of peripheral blood mononuclear cells isolated from patients with prostate cancer treated with enzalutamide; the analysis showed increased activation of the interferon (IFN)–ɣ pathway and decreased frequency of immunosuppressive cells such as myeloid-derived suppressor cells.9 Further, enzalutamide-resistant tumour cells have been shown to stimulate the programmed death-ligand 1 (PD-L1)/programmed cell death-1 (PD-1) pathway that downregulates the immune system by modulating the activity of T cells,9–11 leading to the hypothesis that targeting the AR and PD-L1/PD-1 pathways together would have a synergistic effect on the sensitisation of tumour cells to immune-mediated cell killing.
Single-agent immunotherapies have shown mixed results in prostate cancer,12–14 with signs of activity for inhibitors of the PD-L1/PD-1 pathway. Atezolizumab (anti–PD-L1), which improves survival in patients with several types of solid tumours,15, 16 has shown clinical activity in a phase 1 study of heavily pretreated patients with mCRPC.17 Additionally, durable objective responses were observed in a phase 1b study with pembrolizumab (anti–PD-1).18, 19 While encouraging, this activity was less than that observed in other cancers. These mixed results are likely due to prostate cancer having features typical of immunologically ‘cold’ tumours.20, 21 One such feature is lower PD-L1 expression levels compared with other cancers, although up to a third of mCRPC tumours may show some PD-L1 expression on tumour cells, indicating a potential vulnerability to immunotherapy.22
Based on these data, investigations are underway to evaluate immune checkpoint inhibition in combination with other agents in order to overcome the barriers to immunotherapy anti-cancer activity in these patients. One such phase 2 study examined the combination of nivolumab (anti–PD-1) and ipilimumab (anti–cytotoxic T-lymphocyte-associated protein 4 [CTLA-4]), which achieved objective responses in men with mCRPC albeit with safety concerns.23 Further, a phase 2 study of pembrolizumab in combination with enzalutamide showed antitumour activity in men with mCRPC whose tumours had progressed on enzalutamide.24
To extend these data in prostate cancer, the phase 3 IMbassador250 study examined the efficacy and safety of atezolizumab and enzalutamide vs enzalutamide alone in men with mCRPC previously treated with abiraterone. IMbassador250 had a primary endpoint of overall survival (OS) while also providing the opportunity to explore the immunobiology of prostate cancer. Here, we report the primary efficacy and safety results from IMbassador250, as well as an analysis of the immunobiology of prostate cancer.
RESULTS
Efficacy
IMbassador250 (NCT03016312) enroled patients who had previously progressed on an androgen synthesis inhibitor (eg, abiraterone) and had progressed on, were ineligible for, or refused a taxane regimen. Initially, a safety run-in of 12 patients with mCRPC who previously received abiraterone and docetaxel was conducted; these 12 patients were not part of the intention-to-treat (ITT) population or the biomarker-evaluable population. Ten patients had been initially planned to be enroled for the safety analysis, 2 additional patients were added due to rapid screening. Then, between June 2017 and May 2018, an additional 759 patients aged between 40 and 92 years old were enroled from 156 sites.
All randomised patients were included in the ITT population (N=759). Overall, 379 patients were randomised to the atezolizumab + enzalutamide arm and 380 patients to the enzalutamide monotherapy arm (Figure 1). Atezolizumab was administered intravenously at a fixed dose of 1200 mg every 3 weeks, and enzalutamide was administered orally at 160 mg once daily. Patient demographics were comparable across treatment arms (Supplementary Table 1).
Figure 1|.
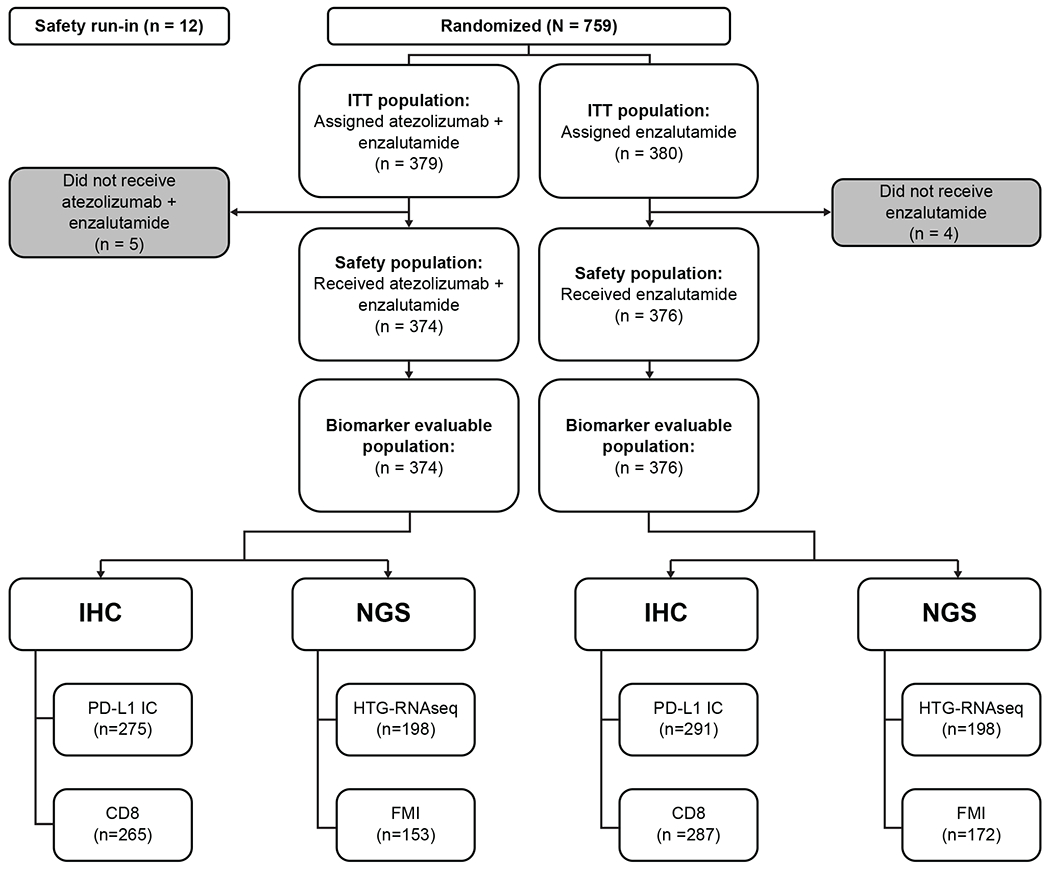
IMbassador250 consort diagram. A total of 759 patients with metastatic castration-resistant prostate cancer who progressed on abiraterone and were ineligible for or declined taxane chemotherapy gave consent and entered the study between June 2017 and May 2018. The number of immunohistochemistry (IHC) and next generation sequencing (NGS) samples of biomarker evaluable population in both arms is shown. FMI: Foundation Medicine.
The study was stopped early after a planned independent data monitoring committee meeting to assess safety. It was deemed it was not in the patients’ best interest to continue as the probability of the trial achieving its primary endpoint was extremely low. Although the tolerability was manageable, patients on study would be at risk of immune mediated adverse events. The median OS follow up was 15.2 months (95% CI, 14.0-17.0) in the atezolizumab + enzalutamide arm and 16.6 months (95% CI, 14.7-18.4) in the enzalutamide arm. No significant difference was observed for OS between arms in the ITT population (stratified HR, 1.12 [95% CI: 0.91, 1.37], P = 0.28; Figure 2a). Forest plot analysis revealed clinical subgroups with similar outcomes to the primary population (Figure 2b and Extended Data Fig. 1).
Figure 2|.
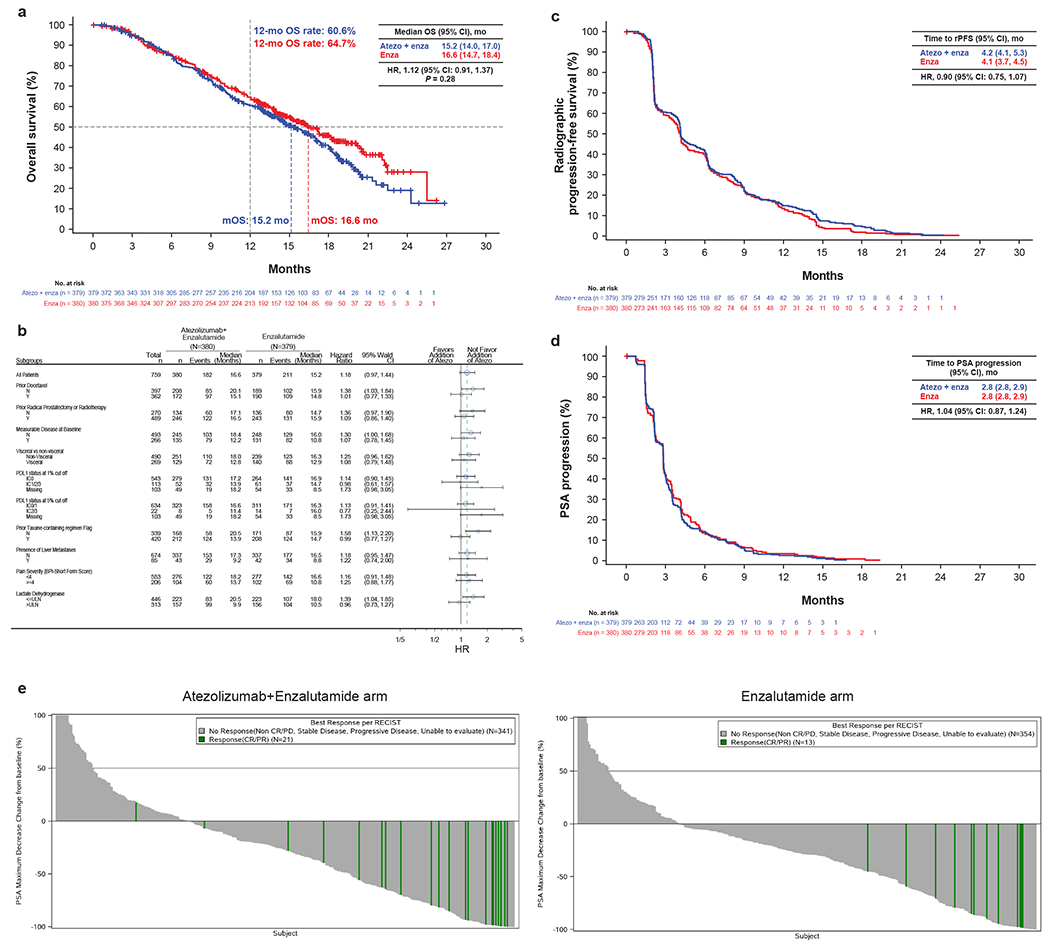
Clinical efficacy in the atezolizumab + enzalutamide vs enzalutamide treatment arms. (a) OS Kaplan-Meier curve. 55.7% of patients (211 of 379) in the atezolizumab + enzalutamide arm and 47.9% of patients (182 of 380) in the enzalutamide arm had an event. The stratified HR interaction P value for OS from the stratified Cox regression model was 0.28. The primary comparison of OS between treatment arms was based on a stratified log-rank test. The HR for death in the experimental arm compared with the control arm was estimated using a stratified Cox regression model, and the 95% CI was provided. (b) Forest plot. P value is from the unstratified Cox regression model. HR is shown for OS in the intention-to-treat (ITT) population. Prior local therapy included prior radical prostatectomy or radiotherapy. PD-L1–positive immune cells (IC) defined as: IC0, <1%; IC1/2/3, ≥1%; IC2/3, ≥5%. Median survival follow-up, 13 months. Minimum follow-up, 12 months. (c) rPFS Kaplan-Meier curve. rPFS was assessed by the investigator and adapted from the Prostate Cancer Working Group 3 (PCWG3) criteria. 74.4% of patients (282 of 379) in the atezolizumab + enzalutamide arm and 72.4% of patients (275 of 380) in the enzalutamide arm had an event. HR is stratified by the Cox proportional hazards model. (d) Time to PSA progression Kaplan-Meier curve for per PCWG3 criteria. 72.3% of patients (274 of 379) in the atezolizumab + enzalutamide arm and 75.0% of patients (285 of 380) in the enzalutamide arm had an event. HR was stratified by Cox proportional hazards model. (e) Waterfall plots showing maximum PSA decline in atezolizumab + enzalutamide and Enzalutamide arm. CR/PR were considered responders. SD, PD, non CR/PD and unable to evaluate were considered non responders. Clinical cutoff for b-e, 24 June 2019.
No difference was observed for radiographic PFS (rPFS; stratified HR, 0.90 [95% CI: 0.75, 1.07]; Figure 2c), and time to PSA progression (stratified HR, 1.04 [95% CI: 0.87, 1.24], Figure 2d). Objective response rates (ORR) were 13.7% (95% CI, 8.4%-20.7%) with atezolizumab + enzalutamide vs 7.4% (95% CI, 3.7%-13.0%) with enzalutamide. Partial responses (PR) were seen in 12.2% of patients (16 of 131) in the atezolizumab + enzalutamide arm vs 6.7% of patients (9 of 135) in the enzalutamide arm. Median duration of response (DOR) was 12.4 months (95% CI, 7.0, not estimable [NE]) in the atezolizumab + enzalutamide arm and NE (95% CI, 5.4, NE) in the enzalutamide arm (Supplementary Table 2). There was a small increase in the number of responders with maximum PSA decline in atezolizumab + enzalutamide shown in Figure 2e, suggesting the addition of atezolizumab may have activity in some patients. At the study cutoff, 38.5% of patients (146 out of 379) receiving atezolizumab + enzalutamide and 42.6% of patients (162 out of 380) receiving enzalutamide remained on treatment.
Safety
Adverse events (AEs) are summarised in Table 1 and Supplementary Tables 3 and 4. Overall, no new AE signals were observed, and no indication that AEs contributed to the trial outcome was noted. Further, AEs were not cited by the IDMC as cause for early termination of the trial.
Table 1|.
Safety summary
| All treated (N=750) | ||
|---|---|---|
| Atezo + Enza (n=374) | Enza (n=376) | |
| Median duration of treatment (range), months | Atezo, 3.5 (0–22.8) Enza, 4.5 (0–23.5) |
4.1 (0–26.2) |
| AEs, n (%) | 361 (96.5) | 345 (91.8) |
| Treatment-related AE | 291 (77.8) | 192 (51.1) |
| Grade 3/4 AEs, n (%) | 203 (54.3) | 131 (34.8) |
| Treatment-related Grade 3/4 AEs | 106 (28.3) | 36 (9.6) |
| Grade 5 AEs, n (%) | 16 (4.3) | 12 (3.2) |
| Treatment-related Grade 5 AE | 7 (1.9) | 1 (0.3) |
| Serious AEs, n (%) | 136 (36.4) | 83 (22.1) |
| Treatment-related serious AEs | 52 (13.9) | 10 (2.7) |
| AEs leading to discontinuation of any treatment component, n (%) |
54 (14.4) | 21 (5.6) |
| Atezo only | 20 (5.3) | 1 (0.3)a |
| Enza only | 6 (1.6) | 20 (5.3) |
AE, adverse event; atezo, atezolizumab; enza, Enzalutamide.
One patient was randomised to the atezo + enza arm. Grade 3 infusion-related reaction occurred during the infusion of first atezolizumab dose, which led to withdrawal of atezolizumab.
Grade 5 AEs were seen in 4.3% of patients (16 of 374) receiving atezolizumab + enzalutamide and 3.2% of patients (12 of 376) receiving enzalutamide; 1.9% (7 of 374) and 0.3% (1 of 376) respectively of these deaths were considered treatment related. Grade 5 adverse events recorded by the investigator as immune related were seen of patients (3 of 374) receiving atezolizumab + enzalutamide (due to myositis, pneumonitis, and myasthenic syndrome; n=1 each) and none in patients receiving enzalutamide (Supplementary Table 4). In the combination arm, 5.3% (20 of 374) of patients discontinued atezolizumab and 1.6% (6 of 374) of patients discontinued enzalutamide. In the enzalutamide arm, 5.3% (20 of 376) of patients discontinued treatment.
Hormone-Sensitive vs Castration-Resistant Prostate Cancer Biomarkers
A total of 750 of 759 patients had tissue available for biomarker analysis (Figure 1). Overall, 280 of 680 biopsies analysed in IMbassador250 were taken prior to progression to mCRPC, and 400 of 680 were taken after progression (Supplementary Table 5), ascertained by comparing the date of sample collection to the date of metastatic diagnosis from the case report form. Overall, most patient biopsies (hormone-sensitive prostate cancer [HSPC] and mCRPC) were archival. mHSPC and mCRPC biopsy samples were from different patients and were not paired samples from the same patients.
We conducted a comparison of DNA damage response (DDR) alterations, phosphatase and tensin homolog (PTEN) status, and PD-L1 immune cell expression levels, T effector gene signatures25 and androgen receptor amplifications between the archived unmatched HSPC and CRPC tumours (Figure 3). The increase in androgen receptor amplifications in CRPC samples was consistent with previous findings with CRPC disease, helping validate the analysis.26 The other three putative immune related biomarkers were selected for three reasons. Firstly, approximately 20% of primary prostate tumours have mutations in DDR genes.27 Secondly, 40% to 50% of patients with prostate cancer can have loss of PTEN function at mCRPC, and PTEN loss results in activation of the phosphoinositide 3-kinase/protein kinase 3 pathway and immune modulatory effects.28–31 Thirdly, higher PD-L1 expression levels (ie, IC2/3, defined as PD-L1 ≥5%) on tumour-infiltrating immune cells (IC) and T effector immune RNA signatures have both been shown to have a positive impact on efficacy outcomes with checkpoint inhibitors across tumour types, and PD-L1 expression can change with therapy.25, 32, 33 While there was evidence of dynamic changes to AR signalling with the development of CRPC, the expression of other putative immune biomarkers appears relatively stable as was reported previously.34 Therefore, although the biomarker work presented consists of a mixture of HSPC and CRPC tissue, this may not have a significant effect on expression of these putative immune biomarkers. This questions one of the original hypotheses of our study that CRPC was associated with dynamic changes to key immune parameters 9–11.
Figure 3|. Ad hoc analysis:
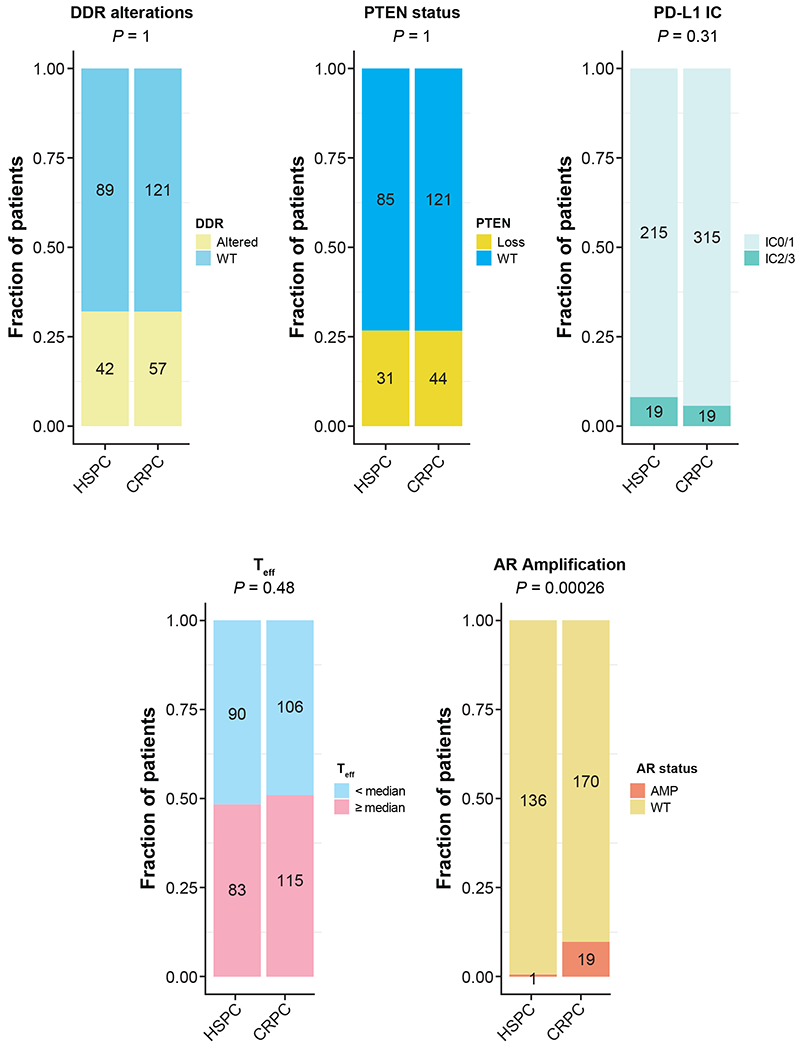
A comparison of DDR alterations, PTEN status, Teff. AR alteration status and PD-L1 expression between hormone-sensitive prostate cancer (HSPC) and castration-resistant prostate cancer (CRPC) biopsy samples. Patient samples were not sequential from the same patient. Each patient contributed only one sample. Numbers in the bars indicate the number of patients being analysed. DDR genes included for analysis were BRCA1, BRCA2, ATM, CDK12, PALB2, ARID1A, ATRX, CHEK1, CHEK2, FANCA, FANCF, FANCG, FANCI, FANCM, MSH2, NBN, RAD52 and WRN. Two-sided Fisher’s exact test with no adjustment of multiplicity was performed in each comparison with P values shown on top.
Biomarkers in Genitourinary Cancers
The negative efficacy results from this trial prompted us to perform a comparative analysis of other genitourinary tumours in which immune checkpoint inhibitors have demonstrated efficacy, in order to put the results in a wider context. These post hoc analyses were performed using identical platforms to facilitate indirect comparisons with available data from atezolizumab in other genitourinary cancers.25, 35–37 Overall, this comparative analysis at the protein, exome, and gene-expression level supported the finding that prostate cancer has low expression of key immune markers, justifying its categorisation as a ‘cold immune phenotype’ tumour (Extended Data Fig. 2).38 This finding was demonstrated by low T effector cell (Teff) and macrophage signatures, as well as reduced major histocompatibility complex class I (MHC I) and immune checkpoint signatures (Figure 4a).
Figure 4|. Ad hoc analysis:
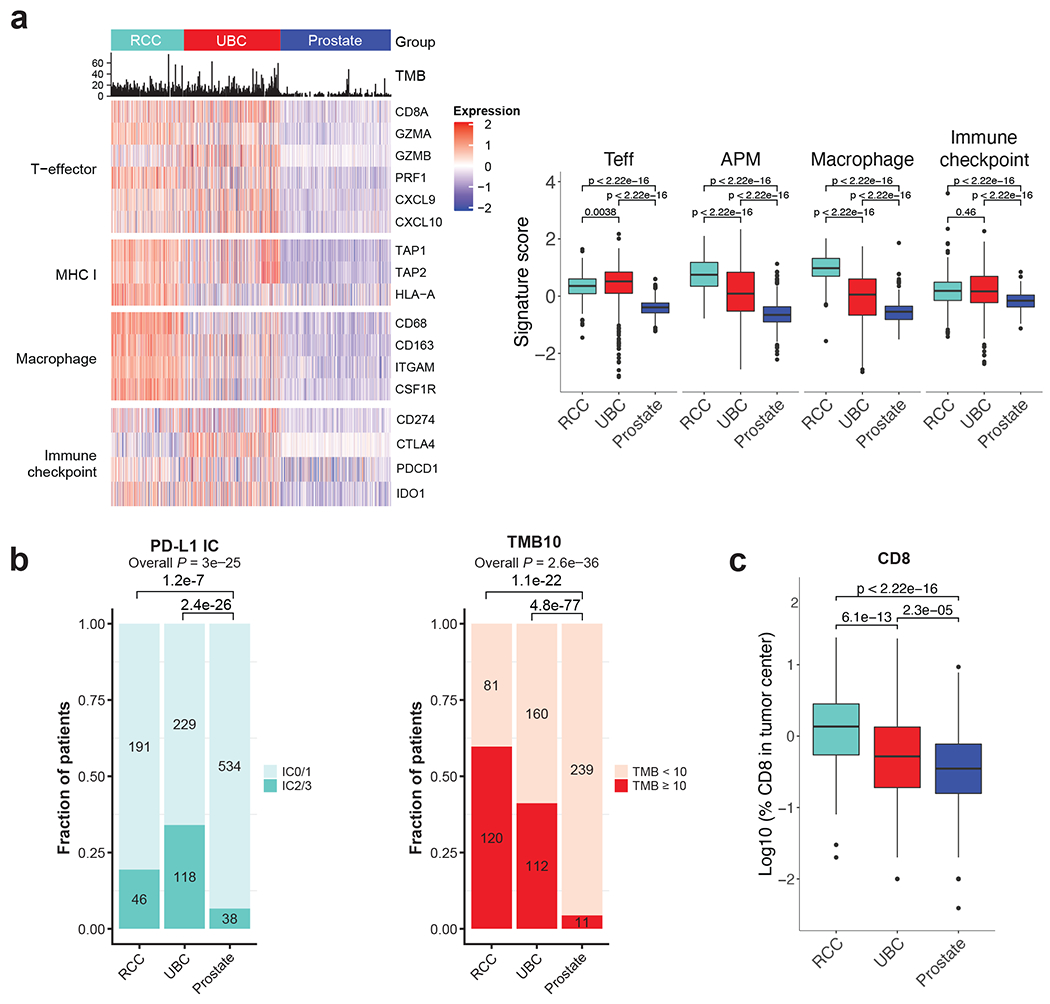
(a) A comparison of Teff, MHC I, macrophage levels and immune checkpoint among 3 genitourinary cancers (renal cell carcinoma, urothelial carcinoma, and prostate cancer). P values were calculated by 2-sided Mann-Whitney u test. ***P < 0.001; **P < 0.01; *P < 0.05. Individual samples analysed: n=263 for RCC; n=348 for UBC and n=400 for prostate. The box plots follow standard Tukey representation by depicting the median at the middle line, with the lower and upper hinges at the first and third quartiles, respectively, the whiskers showing the minima to maxima no greater than 1.5× the interquartile range, and the remaining outlying data points plotted individually. Values for maxima, minima, Q1, Q2, Q3, upper whisker, lower whisker: Teff-RCC: 1.64, −5.53, 0.084, 0.35, 0.60, 1.37, −0.69. UBC: 2.17, −5.48, 0.088, 0.51, 0.84, 1.97, −1.04. Prostate: 0.60, −1.23, −0.58, −0.39, −0.24, 0.28, −1.10. APM- RCC: 2.10, −0.78, 0.35, 0.75, 1.18, 2.43, −0.91. UBC: 2.34, −3.14, −0.53, 0.09, 0.83, 2.88, −2.58. Prostate: 1.13, −2.22, −0.90, −0.66, −0.37, 0.43, −1.70. Macrophage-RCC: 2.02, −1.57, 0.70, 0.98, 1.32, 2.25, −0.24. UBC: 1.74, −2.65, −0.66, 0.05, 0.60, 2.50, −2.56. Prostate: 1.86, −4.62, −0.82, −0.54, −0.35, 0.35, −1.52. Immune checkpoint-RCC: 3.59, −1.42, −0.15, 0.18, 0.49, 1.45, −1.11. UBC: 2.27, −4.44, −0.27, 0.16, 0.68, 2.11, −1.71. Prostate: 0.85, −1.13, −0.38, −0.16, −0.04, 0.66, −1.00. (b) A comparison of PD-L1 IC, TMB and (c) percentage of CD8 in tumours. Individual samples analysed: n=224 for RCC; n=342 for UBC and n=400 for prostate. Numbers on the bars indicate the number of patients. P values of PD-L1 IC, TMB were calculated by Chi square test. P values of CD8 in tumours were calculated by 2-sided Mann-Whitney u test. Values for maxima, minima, Q1, Q2, Q3, upper whisker, lower whisker in (c): RCC: 1.38, −1.70, −0.26, 1.34, 0.45, 1.52, −1.34. UBC: 1.37, −3.10, −0.72, −0.28, 0.13, 1.40, −1.99. Prostate: 0.97, −Inf, −0.82, −0.47, −0.11, 0.95, −1.89.
Markers of pre-existing immunity such as PD-L1 IC expression ≥5% (IC2/3), CD8 T cell infiltration and TMB ≥10mut/mb were uncommon in prostate cancer (Figure 4b). The low expression of immune biomarkers may be responsible for the lack of efficacy of atezolizumab in unselected patients in this setting. To explore this hypothesis further we performed a preplanned efficacy analysis in the biomarker positive population to identify if activity occurred in this subgroup.
Known Biomarkers in Prostate Cancer
Per the protocol (see online Methods), we performed analyses of immunobiological markers (Figure 1). As part of a predefined secondary endpoint, we examined immune gene signatures to identify patients who may have derived a progression-free survival (PFS) benefit from the combination of atezolizumab + enzalutamide (Figure 5a). PFS was used in these planned exploratory analyses because it was considered the most accurate metric of direct efficacy, since several factors may interfere with OS and the interpretability of the biologic efficacy. First, we examined Teff gene signature levels because high levels of this signature indicate an activated immune response.39 Patients with Teff ≥median favored enhanced efficacy in atezolizumab + enzalutamide vs enzalutamide (Teff ≥median: HR, 0.73; 95% CI, 0.52–1.03) (Figure 5a).
Figure 5|.
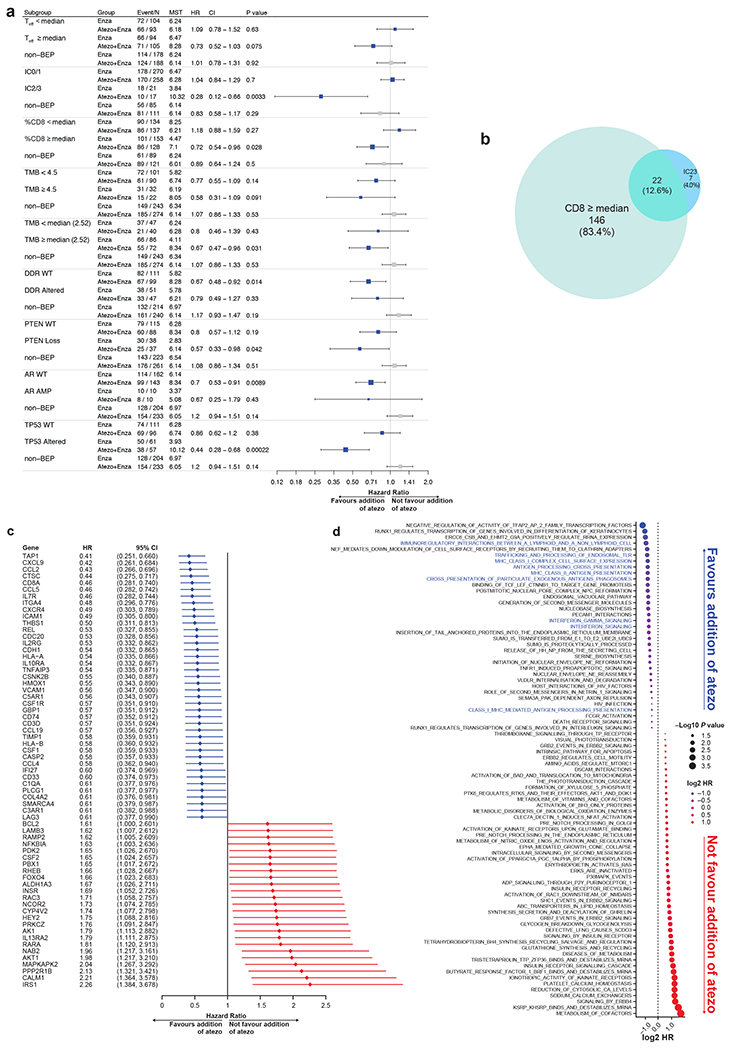
Pre-planned biomarker analysis: PFS in the atezolizumab + enzalutamide vs enzalutamide treatment arms. The numbers with DNA analysis were 325 and RNA analysis were 396. (a) Teff levels (median cutoff), PD-L1 expression, CD8 levels (median cutoff), presence of DDR, and TMB levels. Median was defined by the prostate cancer repository in Foundation Medicine as TMB 4.5 mut/mb. 2.5 mut/mb is the median of TMB of the BEP of this study. The high frequency of the median created imbalance of the two arms. HRs and CIs were calculated using Cox proportional hazards regression model, and P values were two-sided, calculated using unstratified log-rank test without adjustment for multiplicity. MST refers to median survival time (PFS) in months. (b) Venn diagram shows the overlap between patients with ≥median CD8 levels in tumours and high PD-L1 IC (IC2/3). Numbers indicate the number of patients in respective groups. (c) Forest plots summarise the results of PFS for expression of genes, which show an association with outcome. The genes associated with efficacy were ordered based on HR. HRs and CIs were calculated using Cox proportional hazards regression models. (d) Unbiased analysis of PFS modelling based on pathways from Reactome. The pathways that were associated with prognosis were ordered by HRs. Immune signatures that favoured atezo were highlighted in red. Sizes of bubbles represent the two-sided P values calculated using log-rank test with no adjustment for multiplicity. Colours represent the HRs calculated using Cox proportional hazards regression models.
PD-L1 expression levels were then examined. In this study, approximately 2.9% of patients (22 of 759) had tumours that exhibited PD-L1 IC2/3 expression as measured by the VENTANA SP142 IHC assay (immune component) (Roche Diagnostics; Basel, Switzerland). In patients with IC2/3 tumours, fewer PFS events were observed in patients receiving atezolizumab + enzalutamide than in patients receiving enzalutamide alone (HR, 0.28; 95% CI, 0.12–0.66) (Figure 5a). This was inconsistent with the OS results, questioning the clinical utility of this biomarker in this study (Figure 2b).
CD8+ levels were also examined by IHC. Because CD8+ mediates adaptive immunity, the infiltration of CD8+ T cells into prostate cancer tumours has previously correlated with better prognosis.40, 41 There was a small overlap of 12.6% (22 of 175) between samples of CD8 ≥median and PD-L1 IC2/3 (Figure 5b). For patients with ≥median CD8 T-cell infiltration, fewer PFS events were observed in patients receiving atezolizumab + enzalutamide than in patients receiving enzalutamide alone (HR, 0.72; 95% CI, 0.54–0.96) (Figure 5a).
We then analysed genomic biomarkers using the Foundation Medicine database, including cyclin dependent kinase 12 (CDK12) biallelic deletions, microsatellite instability (MSI) status, TMB levels, and DDR alterations (see methods for details of genes). Biallelic CDK12 loss overall, MSI status, and TMB 10 mut/mb have been associated with improved responses to immunotherapy in solid cancers, potentially due to increased novel neoantigens.42–48 However, our evaluation of the impacts of CDK12 biallelic deletions, MSI, and TMB was limited due to small sample size. Overall, biallelic CDK12 loss is detected in 0% to 1.5% of primary prostate cancer tumours and 5% to 6.3% of metastatic prostate cancer tumours.42,43 However, no CDK12 biallelic deletions were detected in this study population. While numbers were small and data exploratory, CDK12 deleterious mutations (frameshift, nonsense, splice-site) appeared to favour outcome with enzalutamide alone (Extended data Figure 3). Only 9 patients had TMB ≥10 mut/mb, prohibiting outcome analysis. We therefore compared levels of ≥ or < 4.5 mut/mb (the median observed in the Foundation Medicine database) and observed a non-significant trend for longer PFS (HR, 0.58; 95% CI, 0.31–1.09) (Figure 5a). However, TMB ≥2.52 mut/mb (median of this study) favours atezolizumab + enzalutamide (HR, 0.87; 95% CI, 0.47–0.96) (Figure 5a). DDR alterations appeared to have little effect on PFS (Figure 5a). MSI is also uncommon in prostate cancer, with 2.2% of men shown to have MSI-high (MSI-H) tumours.49 In this study, only 2 patients in the biomarker-evaluable population were confirmed to be MSI-H; both were randomised to the enzalutamide monotherapy arm. As such, analyses of outcome by MSI were not performed.
We also examined the IMbassador250 results by PTEN loss status, which is a biomarker of note in prostate cancer due to its frequency and association with poor prognosis.50, 51 PTEN loss is also associated with an immune suppressive microenvironment, potentially making these tumours less susceptible to immune modulation with PD-L1 therapy.31 In this study, patients with PTEN loss had improved PFS with atezolizumab + enzalutamide vs enzalutamide (HR, 0.57; 95% CI, 0.33–0.98; Figure 5a). Despite the exploratory nature of these analyses, it is conceivable that PD-L1 inhibition may overcome the immune suppressive environment generated by PTEN loss improving efficacy. Trials targeting this combination are ongoing (NCT04434040). Indirect immunogenic biomarkers potentially determining outcome with immune therapy.30 We explored the relationship between PTEN, DDR, Teff, AR alterations and PD-L1 IHC expression. DDR alterations were associated with ≥5% PD-L1 expression as described in other cancers (Extended data figure 4).52 There were a number of other DNA alteration biomarkers which were of potential interest but not pre-defined in our SAP (Extended data figure 4). These are presented for descriptive purposes only.
Unbiased Exploratory Biomarkers
Next, an unbiased RNA-Seq based analysis of immune related gene expression was performed to better characterise the immunobiology of prostate cancer. This analysis incorporated the distinct immune-gene signatures previously correlated with mCRPC responses to immunotherapy.27 Overall, the expression of genes related to pre-existing immunity, including antigen presentation (antigen peptide transporter 1 [TAP1]), chemokines mediating T-cell activation and recruitment (C-X-C motif ligand 9 [CXCL9]), and IFN signalling, was associated with longer PFS in patients who responded to atezolizumab + enzalutamide vs patients who responded to enzalutamide alone (Figure 5c and d).
Lastly, an exploratory analysis was performed using gene set enrichment analysis with signatures from the molecular signatures database.53 The increased expression of genes within immune-related pathways, including IFN and PD-1 signalling, was associated with longer PFS in the atezolizumab + enzalutamide arm vs the enzalutamide arm (Figure 5d). These genes/gene signature data underpin the relationship between the addition of atezolizumab and immune active biomarker expression. Low expression of these biomarkers in prostate cancer may account for the lack of activity in unselected patients.
DISCUSSION
IMbassador250 was the first phase 3 trial to investigate a checkpoint immunotherapy combination in patients with mCRPC whose disease progressed on abiraterone. The primary endpoint of OS was not met because the addition of atezolizumab did not increase the efficacy of enzalutamide compared with enzalutamide alone in an unselected population of patients. The clinical trial was therefore negative for overall survival. Secondary endpoints, including rPFS, time to PSA progression, ORR, and DOR, also failed to show a benefit for the treatment combination vs enzalutamide alone. This study was terminated for futility per IDMC recommendation because the likelihood of atezolizumab + enzalutamide demonstrating superior OS over enzalutamide alone was statistically low. No new safety concerns were identified by the IDMC, and the safety profile of the combination was consistent with those of atezolizumab and enzalutamide alone. Patient baseline characteristics were well balanced, and subset analysis unremarkable. Therefore, clinical subgroups or imbalances were not thought to factor into the trial outcomes. This study was launched after promising preclinical data for the combination9–11 and the results of a phase 1 demonstrating some single agent activity of atezolizumab in mCRPC. Performing a phase 2 trial before launching a randomised phase 3 study was considered but not undertaken. Phase 2 trials usually focus on surrogate endpoints for OS such as response or PFS. These endpoints can be challenging in prostate cancer as not all patients have measurable disease, and surrogate endpoints with immune therapy are unproven. As such, a definitive phase 3 trial with a built-in biomarker analysis plan was undertaken. A number of ongoing phase 3 trials investigating immune therapy in CRPC are also addressing efficacy of these agents.
A key aspect of this study was the exploration of potential biomarkers. Given the field’s interest in understanding the low response rate to checkpoint immunotherapy in prostate cancer, we undertook planned and unplanned analyses, beginning with known genitourinary cancer immunobiology biomarkers when there were at least 10 patients with a given biomarker. The results should be interpreted with caution. Our data support the finding that prostate cancer is an immune phenotype ‘cold’ tumour with low expression of immune biomarkers. Nonetheless, data from randomised phase 3 trials demonstrated that sipuleucel-T or ipilimumab with radiotherapy can prolong survival in some men with mCRPC, indicating that a subset of men may benefit from immunotherapy.12, 54
Longer PFS was associated with the addition of atezolizumab with the presence of PD-L1 IC2/3 expression and high levels of CD8+ T cells. The relationship between PD-L1 expression and response to immunotherapy is not clear in prostate cancer.18, 24, 55 In a phase 2 study evaluating pembrolizumab monotherapy in patients with CRPC, in a 3rd cohort evaluating responses in patients with non-measurable disease and bone metastases, investigators found 3 vs 1 PSA responses in patients with PD-L1+ and PD-L1− disease respectively.19 Further work will be necessary to explore the predictiveness of PD-L1 as a biomarker in future studies.
While median TMB levels are low in prostate cancer, we observed a trend that suggests that TMB may help predict better outcomes for atezolizumab + enzalutamide. This is well below median TMB levels observed in other cancers and very few patients in our study had TMB of 10 mut/mb or higher per the pembrolizumab cancer type agnostic approval for the drug.56 Of note, higher TMB was recently shown to be predictive of benefit from combined PD-1 and CTLA-4 inhibition in patients with mCRPC as well as pembrolizumab.23 The trend between TMB ≥2.5 mut/mb and improved PFS and all the results derived from the exome analyses should be interpreted with caution because of the patients with samples submitted for exome analysis in the FMI BEP had a better outcome by chance alone with the addition of atezolizumab, which may introduce a potential bias for the biomarker analyses in favour of the atezolizumab + enzalutamide arm (Supplementary Table 6). Further, there was a lack of correlation between DDR mutations and PFS in our study. Immune checkpoint inhibition has been investigated in cancers with DDR alterations due to the increased genomic instability associated with DDR alterations. However the inconsistency surrounding clinical outcomes in patients bearing cancers with DDR alterations and treated with immune checkpoint inhibitors questions its relevance as a biomarker.37
In conducting an unbiased biomarkers analysis, we sought to broaden the search for prostate cancer biomarkers. Overall, we found that patients benefiting in terms of longer PFS from atezolizumab + enzalutamide vs enzalutamide had evidence of pre-existing immunity despite most prostate cancers being relatively immunologically ‘cold.’ This supports the hypothesis that prostate cancer patients expressing established immune response biomarkers may benefit from atezolizumab, but because of their infrequency trials enroling unselected patients fail to achieve an overall survival advantage.
Overall, a key strength of this phase III randomised controlled trial was the high rate of tumour sample acquisition. Additionally, the unbiased gene sequencing approach allowed for an extensive biomarker analysis. However, the biomarker work is still limited by the small size of some of the resulting biomarker subpopulations. Other limitations include the open-label design of this study, which may have influenced clinical outcomes and because OS results may have been confounded by subsequent therapies. As such, PFS was used in our exploratory biomarker analyses to get a direct measure of atezolizumab + enzalutamide in comparison with enzalutamide and to be consistent with prior work in other genitourinary cancers. Furthermore, some analyses were conducted on a post hoc basis as questions arose due to the clinical results and emerging science during the conduct of the trial. Another potential limitation is that most samples obtained as part of IMbassador250 for biomarker studies were archival. Archival samples have the potential to be degraded because they may have been obtained years before the development of castration resistance. They also may not reflect the current biological status of the tumour. Nonetheless, our results found that the distributions of DDR alterations, PTEN status, and PD-L1 expression levels were similar between HSPC and CRPC tumour samples in patients with metastatic disease, suggesting that these features are stable through the development of mCRPC and may not be a factor in the use of archival samples. The relative stability of DDR pathway genes in patients with metastatic disease including both hormone-sensitive and castration-resistant disease has been previously noted.34 This work indicates there is no role for atezolizumab in combination with enzalutamide in unselected patients with CRPC and it suggests that patient selection may be essential for future drug development. Similar trials to IMbassador250 with other immune checkpoint inhibitors alone are ongoing (NCT04191096). As such, the biomarker data from IMbassador250 provide a valuable baseline of knowledge that will inform future investigations using immune checkpoint inhibition in mCRPC.
ONLINE METHODS
Oversight
This study was approved by local institutional review boards at all study sites and was conducted in accordance with Good Clinical Practice and the Declaration of Helsinki. All patients provided written informed consent. The protocol is available in the Appendix. Protocol approval was obtained from Institutional Review Boards or ethics committees for each site. An independent data monitoring committee reviewed unblinded safety and study conduct data approximately every 6 months. F. Hoffmann-La Roche Ltd/Genentech, Inc., sponsored the study, provided the study drugs, and collaborated with an academic steering committee on study design, data collection, analysis, and interpretation. All drafts of the manuscript were prepared by the authors with editorial assistance from professional medical writers funded by the sponsor. All authors approved the submission.
Patients
Eligible patients were aged ≥18 years and had metastatic castration-resistant prostate cancer (mCRPC) after failure of an androgen synthesis inhibitor (eg, abiraterone acetate) and after failure of, ineligibility for, or refusal of a taxane regimen; patients could not be receiving a hormone receptor inhibitor (eg, enzalutamide). Key eligibility criteria included prostate-specific antigen (PSA) or radiological disease progression in soft tissue or bone prior to enrolment and Eastern Cooperative Oncology Group performance status of 0 to 1.
Patients were excluded if they had a history of or active autoimmune disease or immune deficiency, coinfection with hepatitis B or hepatitis C virus, or prior treatment with immunotherapy, enzalutamide, or any other newer AR antagonist. A complete listing of the inclusion and exclusion criteria is available in the protocol (Appendix).
Study Design and Interventions
This was a phase 3, multicentre, randomised, open-label study consisting of a safety run-in phase followed by a randomised phase. Randomisation (1:1) was stratified according to presence of liver metastasis (yes vs no), use of ≥2 cycles of a taxane-containing regimen (yes vs no), LDH level (≤ upper limit of normal [ULN] vs >ULN), and pain severity (Brief Pain Inventory-Short Form question 3 score <4 vs ≥4). Atezolizumab was administered intravenously at a fixed dose of 1200 mg every 3 weeks, and enzalutamide was administered orally at 160 mg once daily. Atezolizumab and enzalutamide were continued until disease progression, unacceptable toxicity, or symptomatic deterioration.
Tumour assessments (computed tomography with bone scan) were performed every 9 weeks (approximately every 3 cycles) following randomisation for 27 weeks and then every 12 weeks thereafter (or every 6 weeks if treating beyond progression). PSA was assessed every 3 weeks until radiographic disease progression, death, or loss of follow-up. Biopsies were submitted prior to enrolment and collected during treatment per investigator discretion. Samples collected >31 days prior to cycle 1 day 1 were classified as archival.
Endpoints
The primary efficacy endpoint was overall survival (OS; defined as time from randomisation to death from any cause). Key secondary efficacy endpoints included investigator-assessed radiographic progression-free survival (PFS), time to PSA progression, objective response rate, and duration of response, all per Prostate Cancer Working Group 3 (PCWG3) criteria.57 Efficacy endpoints were assessed in the intention-to-treat population, defined as all patients who were randomised, regardless of whether they received any study treatment. Safety was evaluated according to the Common Terminology Criteria for Adverse Events, version 4.0. The safety population included all patients who received any component of study treatments.
A predefined plan was created prior to the trial readout that included RNA (RNAseq), DNA (comprehensive genomic profile panel–based F1CDx; Foundation Medicine, Cambridge, MA) and protein expression (PD-L1 SP124; HistoGeneX; Antwerp, Belgium).
Statistical Analyses
The study was designed to enrol approximately 730 patients, 10 in the safety run-in phase and 720 in the randomised phase. To balance treatment assignment across levels of stratification factors, patients were randomly assigned (1:1) using a stratified permuted block method (block size of 4). The primary comparison of OS between treatment arms was based on a stratified log-rank test. The HR for death in the experimental arm compared with the control arm was estimated using a stratified Cox regression model, and the 95% CI was provided. An interim analysis of the primary endpoint of OS was planned to be performed when approximately 432 deaths had occurred, and the final analysis was planned when 540 deaths had occurred. Based on study assumptions, these 540 events would provide 97% power to detect a difference in the duration of OS between treatment arms. For the primary analysis of OS, the stopping boundaries were based on the O’Brien-Fleming α-spending function, and key secondary endpoints were evaluated for statistical significance only if OS was found to be statistically significant. Kaplan-Meier methodology was used to estimate median OS for each treatment arm. The 95% CI for the median OS was estimated using the Brookmeyer-Crowley method. Kaplan-Meier methodology was also used to estimate the median PFS for each treatment arm for exploratory biomarker analyses.
RNAseq gene expression analysis
In an exploratory analysis, patients with enough available tumour samples were assessed for RNA expression levels by HTG EdgeSeq Biomarker Panel (HTG Molecular Diagnostics, Inc.; Tucson, AZ). The HTG EdgeSeq system is an extraction-free specimen preparation and quantitative nuclease protection chemistry, directly from 1 −3 formalin fixed paraffin-embedded (FFPE) sections/slides for downstream NGS applications. RNA was extracted using the High Pure FFPET RNA Isolation Kit (Roche) and assessed by Qubit and Agilent Bioanalyzer for quantity and quality. Following RNA extraction, HTG EdgeSeq Oncology Biomarker Panel analysis was performed to quantitatively measure the expression of 2,559 genes. Detailed methods have been previously described.58 We performed gene signature computation analysis as well as gene expression analysis demultiplexed FASTQ files from 400 samples; the Illumina MiSeq were parsed by the EdgeSeq parser (HTG Molecular Diagnostics; Tucson, AZ) and aligned to the probes of HTG EdgeSeq Oncology Biomarker Panel. For each sample, the parser reported counts on 2568 probes (2559 unique genes plus 4 positive and 5 negative control probes). A statistical process quality-control step filtered out those samples that failed on the basis of the negative control samples. The expression of each gene in a signature was first z score transformed. The Teff gene signature was defined by expression of CD8A, GZMA, GZMB, PRF1, CXCL9, and CXCL10. MHC I, immune checkpoint, and macrophage signatures were obtained from a previous publication.25 For analyses of various biological pathways, a gene set enrichment analysis with signatures from the molecular signatures database (MsigDB; Broad Institute, Cambridge, MA) was used.
DNA isolation and DNA alteration analysis
DNA was extracted using semi-automated genomic DNA isolation from FFPE sections using the Bio-Tek FFPE DNA Extraction Kit (Omega; Norcross, GA) on the KingFisher instrument (ThermoFisher Scientific; Waltham, MA). The FoundationOne® CDx (F1CDx) assay is a next generation sequencing (NGS) comprehensive genomic panel (CGP) in vitro diagnostic device for the detection of DNA alterations including substitutions, insertion and deletion alterations (indels) and copy number alterations in 324 genes and select gene rearrangements. F1CDx also reports select genomic signatures including microsatellite instability (MSI) and tumour mutational burden (TMB). Phosphatase and Tensin Homolog (PTEN) loss was defined as 2 copy deletions (biallelic loss).
Tumour Mutational Burden (TMB)
For analysis of TMB, tumour DNA extraction and preparation were done with HistoGeneX NV. Foundation Medicine performed sequencing library construction, hybridisation capture, DNA sequencing, and genomic alteration detection. In addition to sample processing, Foundation Medicine estimated the mutation burden for each sample using an algorithm that leverages genomic alterations detected by the targeted FoundationOne test (Foundation Medicine) to extrapolate to the whole exome or genome.59 We categorised tumour mutation burden as high (≥4.5) or low (<4.5), which is the median of the Foundation Medicine prostate cancer repository. TMB was defined as the number of nonsynonymous somatic base substitutions and short insertions and deletions identified from coding regions within the FMI test, filtering out known or likely oncogenic driver mutations to reduce bias. All nonsynonymous mutations, including nonsense, missense, frame-shift, splice site, and nonstop changes, were considered. The resultant count was divided by the size of the genomic region of exonic sequence data interrogated to yield a resultant number of mutations per megabase.59
Microsatellite instability (MSI)
MSI was assessed using the FoundationOne CDx sequencing (Foundation Medicine) as previously described.60 An initial pool of 1880 mononucleotide homopolymers of 7 to 39 bp in repeat length, sequenced using the FoundationOne assay was established. A subset of the available loci was selected for inclusion in the PCA training. The selection criteria prioritized coverage and variability in observed allelic lengths at each microsatellite across all the training samples. A minimum of 250× median depth at each locus was enforced to ensure robust detection of variant alleles. In addition, microsatellites that did not show any variability in allelic lengths, compared with the human reference genome and other genomes in the training data set, were removed. All selected loci were also required to be intronic and have the length of 10 to 20 bp of the reference genome because of the limitations of aligning 49-bp reads over a repetitive sequence >20 bp. For each sample at each locus, every NGS read that fully spanned the repeat region was used to determine an allelic length, which allowed a distribution of allelic lengths to be obtained. For a sample that is MSI-High, variability of allelic length is expected to significantly increase while the mean allelic length often decreases due to the higher likelihood that polymerase slippage will result in deletion than insertion. Therefore, for each sample at every locus, the mean and variance of the allelic length were sufficient to predict MSI and were recorded. To combine the separate mean and variance information for all the loci, PCA was used to project the multidimensional data into a corresponding number of PCs. The exemplar training data set was used, and the first principal component (PC1) explained 45% of the data variance, whereas PC2 and onwards were discarded because they explained no more than 5% each of the data variance. The projection vector onto PC1 of the training data set was fixed, and the PC1 value was used as the MSI score. The cutoff of MSI-High versus microsatellite stable (MSS) was established by comparing the MSI score with orthogonal MSI testing data.
DNA Damage Repair (DDR)
The genes associated with the DDR pathway reported in the TOPARP-B trial were considered for analysis.61 The genes included BRCA2, BRCA2, ATM, CDK12, PALB2, ARID1A, ATRX, CHEK1, CHEK2, FANCA, FANCF, FANCG, FANCI, FANCM, MSH2, NBN, RAD52 and WRN. DDR gene alterations were estimated by targeted genomic profiling using Foundation Medicine (Cambridge, MA). Samples with alterations (copy number alterations, fusion/rearrangement and nonsynonymous short variants with known/likely functional impacts) of at least one DDR gene above were considered positive for DDR alterations.
PD-L1
Immunohistochemistry (IHC) was conducted for PD-L1 and centrally evaluated per VENTANA SP142 IHC assay (Roche Diagnostics; Basel, Switzerland). IC0, 1, 2, and 3 refers to <1%, ≥1% to <5%, ≥5% to <10%, and ≥10% PD-L1–expressing IC, respectively.
CD8
Formalin-fixed, paraffin-embedded tumour tissue from biopsies and resections collected before atezolizumab treatment was used for CD8 analysis by IHC. CD8 expression (clone C8/144B; Agilent Technologies; Santa Clara, CA) was assessed in the tumour centre, invasive margin, and periphery in available specimens. In all specimens, total immune infiltrate was assessed in the tumour area based on haematoxylin and eosin staining.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Extended Data
Extended Data Fig. 1. Forest plot of subgroup analysis.
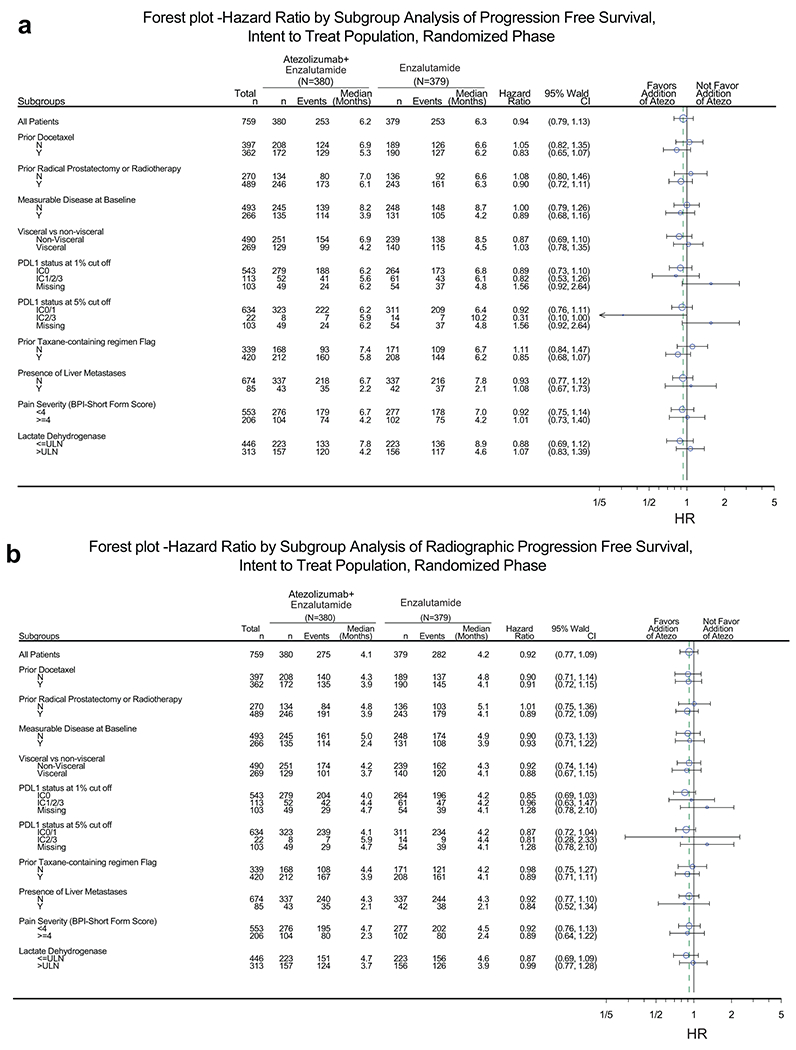
(a) PFS and (b) rPFS in the intention-to-treat (ITT) population. P value and HR are from the unstratified Cox regression model. Prior local therapy included prior radical prostatectomy or radiotherapy. PD-L1–positive immune cells (IC) defined as: IC0, <1%; IC1/2/3, ≥1%; IC2/3, ≥5%.
Extended Data Fig. 2. Forest plot of known biomarkers in urothelial carcinoma, renal cell carcinoma and prostate cancer.
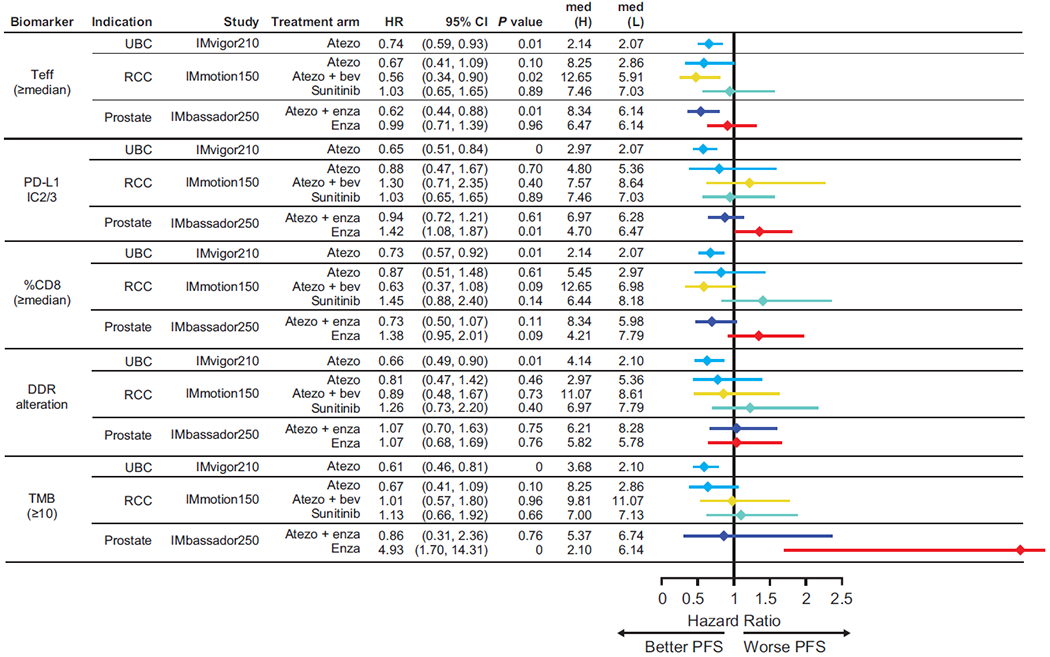
Biomarkers shown among urothelial carcinoma (IMvigor210), renal cell carcinoma (IMmotion150), and prostate cancer (IMbassador250) for PFS. DDR, DNA damage response; PD-L1, programmed death- ligand 1; PFS, progression-free survival; Teff, effector T cell, TMB, tumour mutational burden. Med refers to median PFS in months. HRs and CIs were calculated using Cox proportional hazards regression model, and P values were calculated using unstratified log-rank test without adjustment for multiplicity.
Extended Data Fig. 3. Exploratory analysis of DNA alterations in progression free survival.
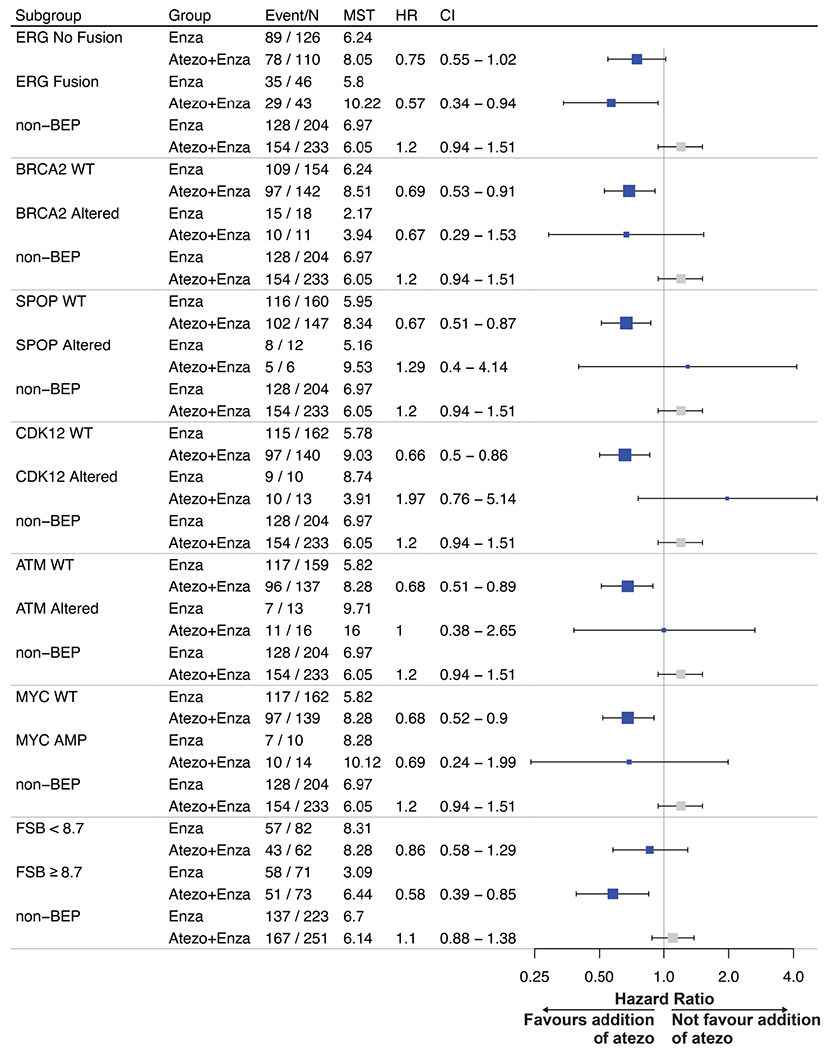
PFS in the atezolizumab + enzalutamide vs enzalutamide treatment arms. 325 samples were included for analysis. DNA alteration biomarkers included Androgen Receptor (AR) amplification status, v-ets erythroblastosis virus E26 oncogene homolog (ERG) fusions, alterations of TP53, BRCA2, SPOP, CDK12 (at least a frameshift, nonsense or splice-site alteration) and ATM. In addition, frameshift mutation burden (FSB) was included. FSB was calculated by the number of frameshift mutations divided by length of genome examined. It was reported as the number of frameshift mutations per megabase (mut/Mb). The cutoff of FSB (8.7 mut/Mb) was previously established in prostate cancer. HRs and CIs were calculated using Cox proportional hazards regression model, and P values were calculated using log-rank test. MST refers to median survival time (PFS) in months.
Extended Data Fig. 4. Distribution of biomarkers and PD-L1 IC status.
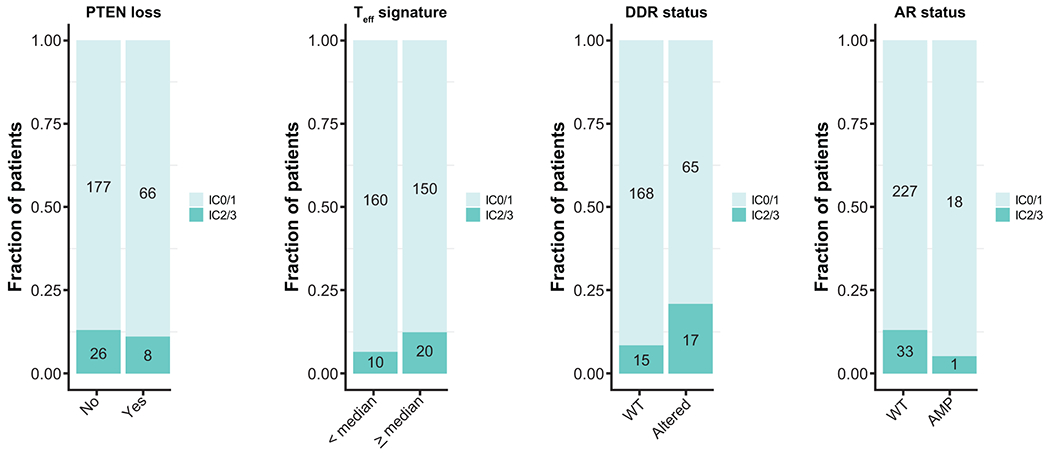
Analysis of PTEN loss, Teff signature, DNA Damage Response (DDR) alterations and Androgen Receptor (AR) amplification status and PD-L1 IC status. IC0/1 were considered low IC whereas IC2/3 were considered high IC. Numbers on the bars indicate the number of patients being analysed.
Supplementary Material
Acknowledgements
The study was supported by F. Hoffmann-La Roche Ltd./Genentech, Inc., a member of the Roche Group. Disclosure forms, online Methods section, and a data sharing statement provided by the authors are available within the full text of this article at nature.com/nm/. Gary L. Buchschacher Jr. MD, PhD, is partially funded by the Kaiser Permanente NCI National Community Oncology Research Program (NCORP) grant (SUG1CA189821-08). We thank the patients who participated in the trial and the clinical site investigators. Medical writing assistance for this manuscript was provided by Priscilla Hong, PharmD, of Health Interactions, Inc., and funded by F. Hoffmann-La Roche Ltd.
COMPETING INTERESTS
T.P. received honoraria from advisory/consultancy roles with AstraZeneca, BMS, Exelixis, Incyte, Ipsen, Merck, MSD, Novartis, Pfizer, Seattle Genetics, Merck Serono (EMD Serono), Astellas, Johnson & Johnson, Eisai, and Roche; institutional research funding support from AstraZeneca, Roche, BMS, Exelixis, Ipsen, Merck, MSD, Novartis, Pfizer, Seattle Genetics, Merck Serono (EMD Serono), Astellas, and Johnson & Johnson; and travel, accommodation, and expenses support from Roche, Pfizer, MSD, AstraZeneca, and Ipsen. S.G. received honoraria from Janssen; advisory/consultancy fees to the institution from Active Biotech, Astellas Pharma, Bayer, Bristol Myers Squibb, Clovis Oncology, CureVac, Ferring, Innocrin, Janssen, Menarini Silicon Biosystems, and Novartis; advisory/consultancy fees from Advanced Accelerator Applications, Amgen, MaxiVax; Orion Pharma, Roche, and Sanofi; and travel, accommodations, and expenses from Nektar and ProteoMediX. SG also holds a patent involving a method for biomarker (WO 3752009138392 A1). K.C.Y. is an employee of Genentech and has stock ownership of Roche. E.E.K. is an employee of Genentech and has stock ownership of Roche, Clinuvel, Epizyme, Mannkind, and Merck. D.R. received advisory/consultancy fees from AstraZeneca, Bayer, Genentech, and Janssen; and institutional research funding support from AstraZeneca, Celgene, Ferring, Genentech/Roche, Janssen, Medivation, Millennium, Novartis, Taiho Pharmaceutical, Takeda, and TRACON. N.M. received advisory/consultancy fees from Janssen, MSD, Chugai, and Sanofi; and institutional research funding support from Janssen, MSD, Chugai, Astellas, Eli Lilly, Taiho, and Pfizer. C.D. received advisory/consultancy fees from AstraZeneca/MedImmune, Bristol Myers Squibb, Compugen, Janssen Oncology, Merck, Pfizer, Pierre Fabre, Potenza Therapeutics, Roche/Genentech, and Tizona Therapeutics; received institutional research funding support from Bristol Myers Squibb; owns stock and other ownership interests in Compugen, Harpoon Therapeutics, Kleo Pharmaceuticals, and Tizona Therapeutics; and received travel, accommodations, and expenses support from AACR, ASCO, Merck Sharp & Dohme, Pfizer, and Roche/Genentech. CD also licenses patents through the institution to Bristol Myers Squibb and Potenza Therapeutics. K.F. received honoraria from Astellas Pharma, Janssen, and Sanofi; received advisory/consultancy fees from Amgen, Astellas Pharma, AstraZeneca, Bayer, CureVac, ESSA Pharma, Janssen Oncology, Orion Pharma, Roche/Genentech, and Sanofi; and travel, accommodations, and expenses support from Amgen and Janssen. J.M.P. received advisory/consultancy fees from Astellas Pharma, Bristol Myers Squibb, Clovis Oncology, Janssen Oncology, Merck Sharp & Dohme, Roche/Genentech, and VCN Biosciences; research funding support from AstraZeneca/MedImmune, Bristol Myers Squibb, Incyte, Janssen Oncology, Merck Sharp & Dohme, and Pfizer/EMD Serono; and travel, accommodations, and expenses support from Bristol Myers Squibb, Janssen Oncology, and Roche. P.J.W. received honoraria from advisory/consultancy roles with Astra Zeneca, Astellas, Bayer, Bristol Myers Squibb, Immunicom, Janssen, MSD, Merck, Novartis, Pfizer, Pierre Fabre, Roche, and Sanofi. G.L.B. declares no conflict of interest. B.A. received grants from P. Herzen Oncology Research Institute during the conduct of the study; grants from AstraZeneca, Bayer, Bristol Myers Squibb, Janssen, Astellas, MSD, Eisai, and Roche; personal fees from AstraZeneca, Bayer, Bristol Myers Squibb, Janssen, Astellas, MSD, Sanofi, Ferring, Ipsen, Eisai, and Roche; and nonfinancial support from AstraZeneca, Bayer, Bristol Myers Squibb, Janssen, Astellas, MSD, Sanofi, Ferring, and Roche. B.M. received advisory/consultancy fees to the institution from Amgen, Pfizer, and Roche; advisory/consultancy fees from Astellas Pharma, AstraZeneca, Bayer, Janssen, Pfizer, and Roche; research funding support from Bayer, Janssen, and Roche; and travel, accommodations, and expenses support from Janssen-Cilag and Roche. B.K. declares no conflict of interest. J.D. is an employee of Genentech and has stock ownership of Roche. G.R. is an employee of Roche and has stock ownership of Roche. A.D. is an employee of Roche and has stock ownership of Roche. S.M. is an employee of Genentech and has stock ownership of Roche. P.W. is an employee of Genentech and has stock ownership of Roche. C.J.S. received advisory/consultancy fees from Astellas, Pfizer, Janssen, Dendreon, Bayer, Genentech, and Glaxo; and institutional research funding support from Astellas, Pfizer, Janssen, Dendreon, Bayer, and Sanofi.
Footnotes
The investigators in the IMbassador250 study are listed in the Supplementary Appendix, available at nature.com/nm/.
Data Availability
Qualified researchers may request access to individual patient-level data through the clinical study data request platform (https://vivli.org/). Further details on Roche’s criteria for eligible studies are available here (https://vivli.org/members/ourmembers/). For further details on Roche’s Global Policy on the Sharing of Clinical Information and how to request access to related clinical study documents, see here (https://www.roche.com/research_and_development/who_we_are_how_we_work/clinical_trials/our_commitment_to_data_sharing.htm). IMbassador250 raw data analysed in this study has been submitted to the European Genome-Phenome Archive (EGA) with accession number EGAS00001004852. Raw RNAseq data from IMmotion150 and IMvigor210 has been submitted to EGA with accession number EGAS00001004386.
REFERENCES
- 1.Kirby M, Hirst C & Crawford ED Characterising the castration-resistant prostate cancer population: a systematic review. Int. J. Clin. Pract 65, 1180–1192 (2011). [DOI] [PubMed] [Google Scholar]
- 2.Logothetis CJ, et al. Effect of abiraterone acetate and prednisone compared with placebo and prednisone on pain control and skeletal-related events in patients with metastatic castration-resistant prostate cancer: exploratory analysis of data from the COU-AA-301 randomised trial. Lancet Oncol. 13, 1210–1217 (2012). [DOI] [PubMed] [Google Scholar]
- 3.Basch E, et al. Development of the National Cancer Institute’s patient-reported outcomes version of the Common Terminology Criteria for Adverse Events (PRO-CTCAE). J. Natl. Cancer Inst 106 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fizazi K, et al. Effect of enzalutamide on time to first skeletal-related event, pain, and quality of life in men with castration-resistant prostate cancer: results from the randomised, phase 3 AFFIRM trial. Lancet Oncol. 15, 1147–1156 (2014). [DOI] [PubMed] [Google Scholar]
- 5.Ryan CJ, et al. Abiraterone in metastatic prostate cancer without previous chemotherapy. N. Engl. J. Med 368, 138–148 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Beer TM, et al. Enzalutamide in metastatic prostate cancer before chemotherapy. N. Engl. J. Med. 371, 424–433 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Scher HI, et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N. Engl. J. Med 367, 1187–1197 (2012). [DOI] [PubMed] [Google Scholar]
- 8.Antonarakis ES, Armstrong AJ, Dehm SM & Luo J Androgen receptor variant-driven prostate cancer: clinical implications and therapeutic targeting. Prostate Cancer Prostatic Dis. 19, 231–241 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Donahue RN, et al. Abstract 4901: Short-course enzalutamide reveals immune activating properties in patients with biochemically recurrent prostate cancer. Cancer Res. 76, 4901 (2016). [Google Scholar]
- 10.Bishop JL, et al. PD-L1 is highly expressed in enzalutamide resistant prostate cancer. Oncotarget 6, 234–242 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Graff JN, et al. Early evidence of anti-PD-1 activity in enzalutamide-resistant prostate cancer. Oncotarget 7, 52810–52817 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kantoff PW, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N. Engl. J. Med 363, 411–422 (2010). [DOI] [PubMed] [Google Scholar]
- 13.Kwon ED, et al. Ipilimumab versus placebo after radiotherapy in patients with metastatic castration-resistant prostate cancer that had progressed after docetaxel chemotherapy (CA184–043): a multicentre, randomised, double-blind, phase 3 trial. Lancet Oncol. 15, 700–712 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Beer TM, et al. Randomized, double-blind, phase iii trial of ipilimumab versus placebo in asymptomatic or minimally symptomatic patients with metastatic chemotherapy-naive castration-resistant prostate cancer. J. Clin. Oncol 35, 40–47 (2017). [DOI] [PubMed] [Google Scholar]
- 15.TECENTRIQ (atezolizumab). Prescribing information. Genentech, Inc.; 2020. [Google Scholar]
- 16.TECENTRIQ (atezolizumab). Summary of product characteristics. Roche Registration Limited; 2020. [Google Scholar]
- 17.Petrylak DP, et al. Safety and clinical activity of atezolizumab in patients with metastatic castration-resistant prostate cancer: a phase I study. Clin. Cancer Res Preprint at: doi: 10.1158/1078-0432.CCR-20-1981 (2021). [DOI] [PubMed] [Google Scholar]
- 18.Hansen AR, et al. Pembrolizumab for advanced prostate adenocarcinoma: findings of the KEYNOTE-028 study. Ann. Oncol 29, 1807–1813 (2018). [DOI] [PubMed] [Google Scholar]
- 19.Antonarakis ES, et al. Pembrolizumab for treatment-refractory metastatic castration-resistant prostate cancer: multicohort, open-label phase II KEYNOTE-199 study. J. Clin. Oncol 38, 395–405 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bou-Dargham MJ, Sha L, Sang QA & Zhang J Immune landscape of human prostate cancer: immune evasion mechanisms and biomarkers for personalized immunotherapy. BMC Cancer 20, 572 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Danaher P, et al. Pan-cancer adaptive immune resistance as defined by the tumor inflammation signature (TIS): results from The Cancer Genome Atlas (TCGA). J. Immunother. Cancer 6, 63 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Haffner MC, et al. Comprehensive evaluation of programmed death-ligand 1 expression in primary and metastatic prostate cancer. Am. J. Pathol 188, 1478–1485 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sharma P, et al. Nivolumab plus ipilimumab for metastatic castration-resistant prostate cancer: preliminary analysis of patients in the CheckMate 650 trial. Cancer Cell 38, 489–499.e483 (2020). [DOI] [PubMed] [Google Scholar]
- 24.Graff JN, et al. A phase II single-arm study of pembrolizumab with enzalutamide in men with metastatic castration-resistant prostate cancer progressing on enzalutamide alone. J. Immunother. Cancer 8, e000642 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mariathasan S, et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 554,544–548 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Feng Q & He B Androgen receptor signaling in the development of castration-resistant prostate cancer. Front Oncol. 9, 858 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cancer Genome Atlas Research Network. The molecular taxonomy of primary prostate cancer. Cell 163, 1011–1025 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Taylor BS, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell 18, 11–22 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Carver BS, et al. Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN-deficient prostate cancer. Cancer Cell 19, 575–586 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Peng W, et al. Loss of PTEN promotes resistance to t cell-mediated immunotherapy. Cancer discovery 6, 202–216 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jamaspishvili T, et al. Clinical implications of PTEN loss in prostate cancer. Nat. Rev. Urol 15, 222–234 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Herbst RS, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 515, 563–567 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen DS & Mellman I Elements of cancer immunity and the cancer-immune set point. Nature 541, 321–330 (2017). [DOI] [PubMed] [Google Scholar]
- 34.Mateo J, Seed G, Bertan C, et al. Genomics of lethal prostate cancer at diagnosis and castration resistance. J Clin Invest. 04 2020;130(4):1743–1751. doi: 10.1172/JCI132031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Powles T, et al. Atezolizumab versus chemotherapy in patients with platinum-treated locally advanced or metastatic urothelial carcinoma (IMvigor211): a multicentre, open-label, phase 3 randomised controlled trial. Lancet 391, 748–757 (2018). [DOI] [PubMed] [Google Scholar]
- 36.McDermott DF, et al. Clinical activity and molecular correlates of response to atezolizumab alone or in combination with bevacizumab versus sunitinib in renal cell carcinoma. Nat. Med 24, 749–757 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Powles T, et al. Atezolizumab (atezo) vs. chemotherapy (chemo) in platinum-treated locally advanced or metastatic urothelial carcinoma (mUC): immune biomarkers, tumor mutational burden (TMB), and clinical outcomes from the phase III IMvigor211 study. J. Clin. Oncol 36, 409 (2018). [Google Scholar]
- 38.Hegde PS, Karanikas V & Evers S The where, the when, and the how of immune monitoring for cancer immunotherapies in the era of checkpoint inhibition. Clin. Cancer Res 22, 1865–1874 (2016). [DOI] [PubMed] [Google Scholar]
- 39.Kowanetz M, et al. Differential regulation of PD-L1 expression by immune and tumor cells in NSCLC and the response to treatment with atezolizumab (anti-PD-L1). Proc. Natl. Acad. Sci. U S A 115, E10119–E10126 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Farhood B, Najafi M & Mortezaee K CD8. J. Cell. Physiol 234, 8509–8521 (2019). [DOI] [PubMed] [Google Scholar]
- 41.Fernandez-Poma SM, et al. Expansion of tumor-infiltrating CD8. Cancer Res. 77, 3672–3684 (2017). [DOI] [PubMed] [Google Scholar]
- 42.Wu YM, et al. Inactivation of CDK12 delineates a distinct immunogenic class of advanced prostate cancer. Cell 173, 1770–1782.e1714 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sokol ES, et al. Pan-cancer analysis of CDK12 loss-of-function alterations and their association with the focal tandem-duplicator phenotype. Oncologist 24, 1526–1533 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mateo J, et al. DNA-repair defects and olaparib in metastatic prostate cancer. N. Engl. J. Med 373, 1697–1708 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Le DT, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 357, 409–413 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hellmann MD, et al. Nivolumab plus ipilimumab in lung cancer with a high tumor mutational burden. N. Engl. J. Med 378, 2093–2104 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chan TA, et al. Development of tumor mutation burden as an immunotherapy biomarker: utility for the oncology clinic. Ann. Oncol 30, 44–56 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Konstantinopoulos PA, et al. Phase II study of avelumab in patients with mismatch repair deficient and mismatch repair proficient recurrent/persistent endometrial cancer. J. Clin. Oncol 37, 2786–2794 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Abida W, et al. Analysis of the prevalence of microsatellite instability in prostate cancer and response to immune checkpoint blockade. JAMA Oncol. 5, 471–478 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vidotto T, et al. Emerging role of PTEN loss in evasion of the immune response to tumours. Br. J. Cancer 122, 1732–1743 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ferraldeschi R, et al. PTEN protein loss and clinical outcome from castration-resistant prostate cancer treated with abiraterone acetate. Eur. Urol 67, 795–802 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Powles T, et al. An adaptive, biomarker-directed platform study of durvalumab in combination with targeted therapies in advanced urothelial cancer. Nat Med. 27, 793–801 (2021). [DOI] [PubMed] [Google Scholar]
- 53.Subramanian A, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U S A 102, 15545–15550 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fizazi K, et al. Final analysis of the ipilimumab versus placebo following radiotherapy phase III trial in postdocetaxel metastatic castration-resistant prostate cancer identifies an excess of long-term survivors. Eur. Urol. 78, 822–830 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sharma M, Yang Z & Miyamoto H Immunohistochemistry of immune checkpoint markers PD-1 and PD-L1 in prostate cancer. Medicine (Baltimore) 98, e17257 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Marabelle A, et al. Association of tumour mutational burden with outcomes in patients with advanced solid tumours treated with pembrolizumab: prospective biomarker analysis of the multicohort, open-label, phase 2 KEYNOTE-158 study. Lancet Oncol. 21, 1353–1365 (2020). [DOI] [PubMed] [Google Scholar]
- 57.Scher HI, et al. Trial Design and objectives for castration-resistant prostate cancer: updated recommendations from the Prostate Cancer Clinical Trials Working Group 3. J. Clin. Oncol 34, 1402–1418 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Qi Z, et al. Reliable gene expression profiling from small and hematoxylin and eosin-stained clinical formalin-fixed, paraffin-embedded specimens using the HTG EdgeSeq Platform. J. Mol. Diagn 21, 796–807 (2019). [DOI] [PubMed] [Google Scholar]
- 59.Chalmers ZR, et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 9, 34 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Trabucco SE et al. A Novel next-generation sequencing approach to detecting microsatellite instability and pan-tumor characterization of 1000 microsatellite instability-high cases in 67,000 patient samples. J. Mol. Diagn 21, 1053–1066 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mateo J, et al. Olaparib in patients with metastatic castration-resistant prostate cancer with DNA repair gene aberrations (TOPARP-B): a multicentre, open-label, randomised, phase 2 trial. Lancet Oncol. 21, 162–174 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Qualified researchers may request access to individual patient-level data through the clinical study data request platform (https://vivli.org/). Further details on Roche’s criteria for eligible studies are available here (https://vivli.org/members/ourmembers/). For further details on Roche’s Global Policy on the Sharing of Clinical Information and how to request access to related clinical study documents, see here (https://www.roche.com/research_and_development/who_we_are_how_we_work/clinical_trials/our_commitment_to_data_sharing.htm). IMbassador250 raw data analysed in this study has been submitted to the European Genome-Phenome Archive (EGA) with accession number EGAS00001004852. Raw RNAseq data from IMmotion150 and IMvigor210 has been submitted to EGA with accession number EGAS00001004386.