

Abstract

Reaction of the dimers [(Cp*MCl)2(μ-Cl)2] (Cp* = η5-C5Me5) with Ph2PCH2CH2NC(NH(p-Tolyl))2 (H2L) in the presence of NaSbF6 affords the chlorido complexes [Cp*MCl(κ2N,P-H2L)][SbF6] (M = Rh, 1; Ir, 2). Upon treatment with aqueous NaOH, solutions of 1 and 2 yield the corresponding complexes [Cp*M(κ3N,N′,P-HL)][SbF6] (M = Rh, 3; Ir, 4) in which the ligand HL presents a fac κ3N,N′,P coordination mode. Treatment of THF solutions of complexes 3 and 4 with hydrogen gas, at room temperature, results in the formation of the metal hydrido-complexes [Cp*MH(κ2N,P-H2L)][SbF6] (M = Rh, 5; Ir, 6) in which the N(p-Tolyl) group has been protonated. Complexes 3 and 4 react with deuterated water in a reversible fashion resulting in the gradual deuteration of the Cp* group. Heating at 383 K THF/H2O solutions of the complexes 3 and 4 affords the orthometalated complexes [Cp*M(κ3C,N,P-H2L-H)][SbF6] [M = Rh, 7; Ir, 8, H2L-H = Ph2PCH2CH2NC(NH(p-Tolyl))(NH(4-C6H3Me))], respectively. At 333 K, complexes 3 and 4 react in THF with methanol, primary alcohols, or 2-propanol giving the metal-hydrido complexes 5 and 6, respectively. The reaction involves the acceptorless dehydrogenation of the alcohols at a relatively low temperature, without the assistance of an external base. The new complexes have been characterized by the usual analytical and spectroscopic methods including the X-ray diffraction determination of the crystal structures of complexes 1–5, 7, and 8. Notably, the chlorido complexes 1 and 2 crystallize both as enantiopure conglomerates and as racemates. Reaction mechanisms are proposed based on stoichiometric reactions, nuclear magnetic resonance studies, and X-ray crystallography as well as density functional theory calculations.
Short abstract
In solution, masked transition-metal frustrated Lewis pairs (TMFLPs) give rise to the corresponding TMFLP species which activate dihydrogen, water, and alcohols following FLP reaction pathways. When D2O or alcohols with deuterated OH groups were employed, H/D exchange at the Cp* ligand (involving C(sp3)−H activation) was observed. C(sp2)−H bond activation involving orthometalation of the p-Tolyl ring was also observed.
Introduction
In 2006, Stephan’s group reported that the phosphano-borane compound (C6H2Me3)2P(C6F4)B(C6F5)2 reacted reversibly with molecular hydrogen to give the phosphonium-borate species (C6H2Me3)2PH(C6F4)BH(C6F5)2. This reaction demonstrates that compounds of representative elements are capable of activating the dihydrogen molecule breaking the paradigm that hydrogen activation is an exclusive ability of transition-metal compounds.1 This novel reactivity is based on the cooperative behavior of an acidic (electron acceptor, boron) and a basic (electron donor, phosphorus) component that cannot form dative bonds due to geometry constrains. To highlight this feature, the term “frustrated Lewis pair” (FLP) was coined.2
Shortly afterward, the assortment of acidic and basic components was significantly expanded, and it was demonstrated that the resulting FLPs were capable of activating a variety of substrates including imines, olefins, alkynes, organic carbonyl compounds, carbon dioxide, azides, or nitric oxide. Subsequently, the FLP chemistry advanced by incorporating unusual stoichiometric reactions as well as catalytic processes such as hydrogenation (including enantioselective hydrogenation), hydrosilylation, hydroboration, or hydroamination.3
In addition, the potential of FLP systems increased considerably with the proposal of Wass’s group to incorporate components based on transition metals in their design, resulting in the so-called transition-metal frustrated Lewis pairs (TMFLPs).4 The incorporation of transition-metal fragments into FLP systems increases their structural and electronic diversity in such a way that it should allow them to efficiently promote the whole set of elementary reactions characteristic of catalytic processes. In this regard, Wass5 and Erker’s6 groups developed extensively the FLP chemistry of Zr/P systems and demonstrated their potential in the activation of small molecules as well as in catalysis. The area was quickly extended to new TMFLPs with various transition metals including bimetallic FLPs.7 In this context, it should be noted that the reactivity of TMFLP species can be framed in the broader field of metal–ligand cooperation.8
A further qualitative leap in the area of FLP systems occurred when it was discovered that some combinations of Lewis acids and Lewis bases exhibited FLP reactivity despite the fact that the formation of the corresponding classical Lewis adduct (CLA) was observed.9 In this regard, it was established that in order for the system to exhibit FLP behavior it is enough that an equilibrium exists between the CLA form and the dissociated form, that is, that the dissociated form is thermally accessible.10 To describe this type of system, the concept of “thermally induced frustration” was introduced11 and the terms “masked”12 and “dormant”13 have been used to refer to the involved FLPs.
The activation of the O–H bond of water is one of the steps in the search for efficient catalysts for water splitting on the route to renewable energy generation.14 Among the strategies employed to this end, metal–ligand cooperative chemistry14g,15 and FPLs, based on both representative elements16 and transition-metal components,7d,7 have been successfully applied. On the other hand, the Cp* ligand forms robust complexes with a large variety of elements of the periodic table and, usually, it is a nonreactive ligand. However, rare examples of cooperative metal–ligand reactivity involving this ligand have been reported. Indeed, hydrogen abstraction from Cp* methyls has been accomplished either by treatment with an external strong base17 or through an intramolecular pathway by means of a basic ligand.18 The C–H bond cleavage usually leads to tetramethylfulvene complexes in which the fulvene moiety may display distinct coordination modes.17b−17d When this activation was coupled with the activation of the O–D bond of deuterated water, in some instances, a very unusual H/D exchange of the Cp* methyl protons was observed.17e,18
The field of metal–ligand cooperation also includes some of the acceptorless alcohol dehydrogenation (AAD) processes catalyzed by metallic compounds. AAD is a dehydrogenative oxidation process with important applications in energy, green chemistry, and organic synthetic methods. Successful cases of AAD include the use of a variety of transition-metal complexes containing chelates, pincers, and related multidentate ligands as catalysts.19 Some of the ligands possess a basic site able to abstract a proton from the alcohol, and the resulting alkoxide transfers a hydride from the α-CH position to the metal directly or via β-elimination.19g,20
With these concerns in mind, in the present article, we report: (i) the preparation and characterization of the masked TMFLP compounds [Cp*M(κ3N,N′,P-HL)][SbF6] (Cp* = η5-C5Me5; H2L = N,N′-bis(p-Tolyl)-N″-(2-diphenylphosphanoethyl)guanidine; M = Rh, 3; Ir, 4; Chart 1); (ii) the reactivity of these complexes with H2 and H2O; (iii) the hydrogen abstraction from Cp* methyls in complexes 3 and 4 that results in an H/D gradual exchange when deuterated reagents were employed; (iv) the orthometalation reaction of one p-Tolyl ring of the phosphano-guanidine ligand, and (v) the acceptorless dehydrogenation of alcohols promoted by 3 and 4.
Chart 1. The Ligand H2L and the Complexes [Cp*M(κ3N,N′,P-HL)][SbF6].

Part of this work has been previously communicated.21 Herein, we extend our study to the iridium homologue complex 4. Moreover, the reaction of complexes 3 and 4 with alcohols as well as orthometalation reactions, involving new C(sp3)–H, O–H and C(sp2)–H activations, is also included.
Results and Discussion
Preparation of the Complexes [Cp*MCl(κ2N,P-H2L)][SbF6] (M = Rh, 1; Ir, 2)
Reaction of the dimers [(Cp*MCl)2(μ-Cl)2]22 with the phosphano-guanidine compound H2L(21) in the presence of NaSbF6 affords the chlorido complexes [Cp*MCl(κ2N,P-H2L)][SbF6] (M = Rh, 1; Ir, 2; eq 1).
 |
1 |
Compounds 1 and 2 were characterized by analytical and spectroscopic methods (see the Supporting Information) and by the X-ray determination of their crystal structures. The κ2N,P coordination of the H2L ligand renders the metal atom a stereogenic center. Consequently, the methylene protons of the phosphano-guanidine ligand are diastereotopic, and in the proton nuclear magnetic resonance (1H NMR) spectrum, they give four distinct resonances. Broad bands in the 3000–3400 cm–1 region of the IR spectra together with 1H NMR singlets at 9.24 and 7.34 ppm (1) and 8.93 and 7.16 ppm (2) are indicative of the presence of two nonequivalent NH groups in the molecule. The 31P{1H} NMR spectrum consists of a doublet centered at 51.37 ppm for the rhodium complex [J(RhP) = 142.4 Hz] and a singlet at 26.52 ppm for the iridium compound, proving the coordination of the phosphorus to the metal (δP free ligand: −21.14 ppm).
Slow evaporation of saturated solutions of 1 and 2 in CH2Cl2/Et2O/n-pentane mixtures gave rise to the simultaneous formation of single crystals of pure enantiomers (conglomerates23) and racemates, for both compounds. Enantiopure samples of 1 and 2 slowly racemize in solution. Thus, for example, starting from a dichloromethane solution of pure SRh-1, SRh-1/RRh-1 molar ratios of about 92/8 and 74/26 were measured, by circular dichroism (CD) spectroscopy, after 2 and 18 h at room temperature, respectively.
A view of the cation of both enantiomers of the rhodium complex 1 is depicted in Figure 1. Views of the rac-1 cations as well as of the cation of the iridium enantiomers RIr-2, SIr-2 and racemate rac-2 are included in the Supporting Information. Figure 2 shows the enantiomorphic relationship of the CD spectra of the two enantiomers of the rhodium complex 1.24
Figure 1.

View of the cations of the RRh (A) and SRh (B) enantiomers of the rhodium complex [Cp*RhCl(κ2N,P-H2L)][SbF6] (1). For clarity, hydrogen atoms (except those bonded to nitrogen atoms) have been omitted.
Figure 2.
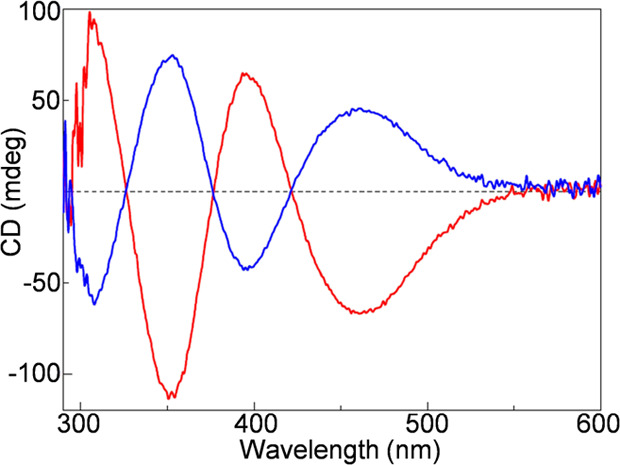
CD spectra of SRh-1 (blue) and RRh-1 (red) in CH2Cl2.
Table S1 (Supporting Information) collects the most relevant structural parameters for the cations of RRh-1, rac-1, RIr-2, SIr-2, and rac-2, comparable structural parameters being observed regardless of the configuration and the nature of the metal center. Hence, only the structural parameters found in the SRh-1 isomer will be discussed. Selected bond lengths and angles of the cation of SRh-1 are summarized in Table 1. The cation of this complex exhibits “three-legged piano-stool” geometry. An η5-C5Me5 group occupies three fac positions, and the κ2N,P chelating phosphano-guanidine ligand and a chlorine atom complete the coordination sphere of the metal. The absolute configuration of the metal center is S, according to the atom priority sequence η5-C5Me5 > Cl > P > N.25 The structural parameters of the CN3 guanidine moiety deserve some comments. The C–NH(p-Tolyl) bond distances, N(2)–C(25) 1.344(8), N(3)–C(25) 1.369(7) Å, indicate a slight partial double bond character for these bonds,26 while the N(1)–C(25) bond distance, involving the nitrogen coordinated to the metal atom, is found to be comparatively shorter, 1.310(7) Å, but also longer than typical N=C bond lengths (1.279(8) Å).26 The sum of the bond angles at the coordinated nitrogen is 359.6(7)° indicating that the C(12)N(1)Rh(1)C(25) fragment is essentially planar. Hydrogen bonds between the N(2)–H(2) proton and the chlorido ligand [N–H = 0.82(7) Å, H···Cl = 2.45(7) Å, N···Cl = 3.203(5) Å, N–H···Cl = 153(7)°] and between the N(3)–H(3) proton and one of the fluorine atoms of the SbF6 anion [N–H = 0.82(8) Å, H···F = 2.13(8) Å, N···F = 2.939(7) Å, N–H···F = 169(7)°] were observed (Figure 3).
Table 1. Selected Bonds Lengths (Å) and Angles (°) for Complex SRh-1.
| Rh–Cl | 2.4199(14) | Cl-Rh–Cta | 121.8(1) |
| Rh–P | 2.2916(15) | P–Rh–N(1) | 83.31(13) |
| Rh–N(1) | 2.123(5) | P–Rh–Cta | 130.4(2) |
| Rh–Cta | 1.8218(1) | N(1)–Rh–Cta | 131.2(2) |
| N(1)–C(25) | 1.310(7) | Rh–N(1)–C(12) | 118.3(3) |
| N(2)–C(25) | 1.344(8) | Rh–N(1)–C(25) | 122.8(4) |
| N(3)–C(25) | 1.369(7) | C(12)–N(1)–C(25) | 118.5(5) |
| Cl–Rh–P | 90.54(5) | Σ°N(1)b | 359.6(7) |
| Cl–Rh–N(1) | 85.15(14) |
Ct represents the centroid of the η5-C5Me5 ligand.
Σ°N(1) is the sum of bond angles around N(1) atom.
Figure 3.
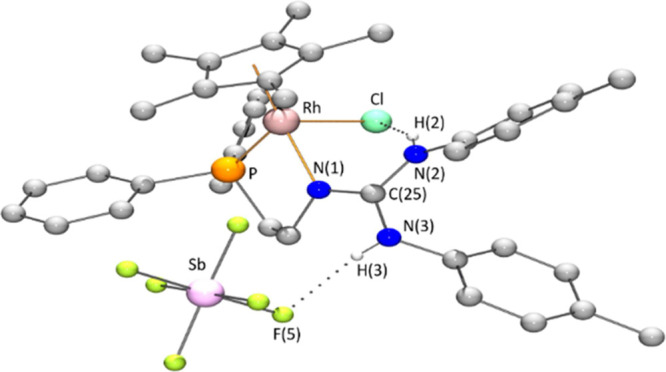
H-bond interactions in complex SRh-1. For clarity, only hydrogen atoms of N–H fragments have been depicted.
Preparation of the Complexes [Cp*M(κ3N,N′,P-HL)][SbF6] (M = Rh, 3; Ir, 4)
Solutions of 1 and 2 in 1:1 (v/v) THF/toluene were treated with aqueous NaOH for 1.5 h affording the corresponding complexes [Cp*M(κ3N,N′,P-HL)][SbF6] (M = Rh, 3; Ir, 4) through base-induced elimination of HCl and subsequent coordination of the deprotonated nitrogen (eq 2).
 |
2 |
The compounds were characterized by analytical and spectroscopic methods (see the Supporting Information) and by the X-ray diffraction determination of their crystal structures. A weak IR band at 3377 and 3362 cm–1 for 3 and 4, respectively, and a broad singlet in the proton NMR spectrum at 7.89 (3) and 8.00 ppm (4) are attributed to the NH functionality. As a consequence of the stereogenicity at the metal, the PCH2CH2N methylene protons are asynchronous and give rise to four resonances at the expected chemical shifts and with the awaited multiplicities (see the Supporting Information). A doublet centered at 48.27 ppm [J(RhP) = 159.0 Hz] and a singlet at 27.75 ppm in the 31P{1H} NMR spectrum are assigned to the phosphorus nucleus of the PPh2 group of the phosphano-guanidine ligand.
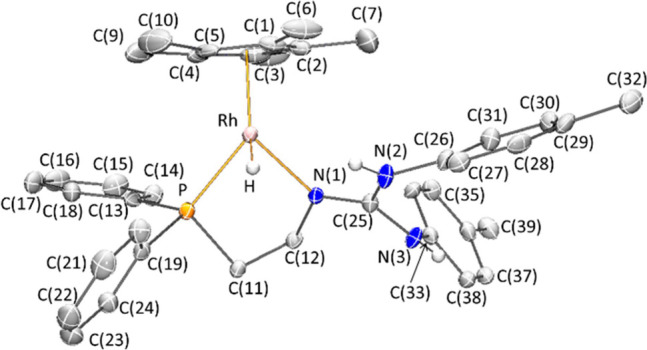
The molecular structure of 3 and 4 has been determined by X-ray diffraction means. There is no significant chemical difference to be remarked when comparing the structural parameters of the cations of the two complexes. For more detailed data about the molecular structure of the rhodium complex 3, see ref (21). Figure 4 shows a view of the two crystallographically independent, but chemically equivalent, cations (A and B) found in the asymmetric unit of iridium complex 4. Table 2 collects selected bond lengths and angles of both cations. The molecular structure reveals that the ligand HL presents a fac κ3N,N′,P coordination mode. This type of coordination renders the metal and the central nitrogen atom of the ligand stereogenic. The configuration at metal induces the configuration at nitrogen in such a way that only the RM,SN diastereomer and its SM,RN enantiomer form, both of them being present in the centrosymmetric unit cell of 4·C4H8O. In Figure 4, a view of the two independent cations of the RM,SN diastereomer is depicted.
Figure 4.

View of the two independent molecules of the cation of complex RM,SN-4. For clarity, only the ipso carbon of the phenyl rings of the PPh2 group is shown, and hydrogen atoms (except the NH proton) have been omitted.
Table 2. Selected Bond Lengths (Å) and Angles (°) for the Two Independent Cations of Complex RM,SN-4.
| cation A | cation B | ||
|---|---|---|---|
| Ir(1)–P(1) | 2.2984(12) | Ir(51)–P(51) | 2.2843(12) |
| Ir(1)–N(1) | 2.110(4) | Ir(51)–N(51) | 2.124(4) |
| Ir(1)–N(2) | 2.121(4) | Ir(51)–N(52) | 2.130(4) |
| Ir(1)–Cta | 1.8276(1) | Ir(51)–Cta | 1.8338(1) |
| N(1)–C(25) | 1.357(6) | N(51)–C(75) | 1.362(6) |
| N(2)–C(25) | 1.329(5) | N(52)–C(75) | 1.324(5) |
| N(3)–C(25) | 1.364(6) | N(53)–C(75) | 1.361(6) |
| P(1)–Ir(1)–N(1) | 80.02(11) | P(51)–Ir(51)–N(51) | 80.31(11) |
| P(1)–Ir(1)–N(2) | 90.53(11) | P(51)–Ir(51)–N(52) | 91.49(11) |
| P(1)–Ir(1)–Cta | 134.64(1) | P(51)–Ir(51)–Cta | 133.98(1) |
| N(1)–Ir(1)–N(2) | 62.33(14) | N(51)–Ir(51)–N(52) | 62.03(14) |
| N(1)–Ir(1)–Cta | 131.10(1) | N(51)–Ir(51)–Cta | 133.21(1) |
| N(2)–Ir(1)–Cta | 130.92(1) | N(52)–Ir(51)–Cta | 129.55(1) |
| Ir(1)–N(1)–C(24) | 118.3(3) | Ir(51)–N(51)–C(74) | 118.5(3) |
| Ir(1)–N(1)–C(25) | 93.9(3) | Ir(51)–N(51)–C(75) | 93.8(3) |
| C(25)–N(1)–C(24) | 116.6(4) | C(75)–N(51)–C(74) | 114.9(4) |
| Σ°N(1)b | 328.8(6) | Σ°N(51)b | 327.2(6) |
| Ir(1)–N(2)–C(25) | 94.3(3) | Ir(51)–N(52)–C(75) | 94.7(3) |
| N(1)–C(25)–N(2) | 109.2(4) | N(51)–C(75)–N(52) | 109.4(4) |
Ct represents the centroid of the η5-C5Me5 ligand.
Σ°N(1) and Σ°N(51) stand for the sum of bond angles around N(1) and N(51) atoms, respectively.
Focusing the discussion on cation A, the fac κ3N,N′,P coordination mode of the HL ligand forces the central N(1) atom to adopt a pyramidal geometry [Σ°N(1) = 328.8(6)°]. This geometry together with the N(1)–C(25) bond length [1.357(6) Å] contrasts with the structural features of the corresponding nitrogen atom in the precursor complex 2 where the H2L ligand coordinates in a chelate κ2N,P manner (for the corresponding parameters of compound 2, see Table S1, Supporting Information). Remarkably, the bond angles N(1)–Ir(1)–N(2) and N(1)–C(25)–N(2), 62.33(14) and 109.2(4)°, respectively, are far from the hybridization ideal values. All these features will most likely lead to a strong strain within the Ir–N–C–N four-membered metalacycle.
Reaction of Complexes 3 and 4 with Molecular Hydrogen
The structural parameters found in compounds 3 and 4, and in particular the envisaged strain within the four-membered metalacycle M–N–C–N led us to hypothesize that these compounds could behave like masked FLPs: the heterolytic cleavage of one of its M–N bonds could generate a TMFLP in which the metal and the nitrogen would play the role of the acid and basic center, respectively. As reported for compound 3,21 these assumptions prompted us to try the reaction of complex 4 with molecular hydrogen.
Indeed, treatment of THF solutions of complexes 3 and 4 with hydrogen gas (4 bar), at room temperature, resulted in the formation of the metal hydrido-complexes [Cp*MH(κ2N,P-H2L)][SbF6] (M = Rh, 5; Ir, 6) in which the N(p-Tolyl) group has been protonated (eq 3). Formally, the heterolytic breakage of the molecule of hydrogen gives rise to hydridic M–H and protic N–H bonds. Complete conversion to complex 5 was obtained after 4 h of reaction under the above mentioned conditions. Conversion to the iridium complex 6 was complete after 24 h at 373 K. The reaction is reversible but to achieve appreciable dehydrogenation rates it is necessary to heat THF solutions above 373 K. Indeed, heating at 393 K a solution of the hydride 5 for 30 min, a conversion of 30% to the rhodium compound 3 was observed. After heating at the same temperature a solution of the iridium complex 6 for 2.5 h, a conversion of 50% to the dehydrogenated compound 4 was measured.
 |
3 |
A doublet of doublets centered at −10.79 ppm [J(PH) = 38.8 Hz, J(RhH) 22.6 Hz] for complex 5 and a doublet centered at −10.16 ppm [J(PH) = 32.3 Hz] for complex 6 are attributed to the M–H functionality in the cations. The presence of two peaks attributed to NH protons (see the Supporting Information) is indicative of the protonation of the N(p-Tolyl) group. The 31P{1H} NMR spectrum consists of a doublet centered at 61.77 ppm [J(RhP) = 143.9 Hz] for the rhodium complex and a singlet at 27.88 ppm for the iridium one.
The molecular structure of complex 5 corroborates all these features.21 The compound crystallizes as a racemate in the P21/n space group of the monoclinic system with one solvent molecule in the asymmetric unit (5·CD4O). The RRh enantiomer is depicted in Figure 5. Selected bond lengths and angles are shown in Table 3. The phosphano-guanidine ligand displays a κ2N,P coordination mode. A Cp* ligand, formally occupying three coordination sites, and a hydrido ligand [Rh–H = 1.56(5) Å] complete the coordination sphere of the metal. The observed RhH···HN (2) separation, 2.20(7) Å, is shorter than twice the hydrogen Van der Waals radius, 2.4 Å, indicating a significant H···H interaction between the protic NH and hydridic RhH functionalities. The structural parameters of the CN3 guanidino fragment are comparable to those found for the chlorido compound 1, that is, a greater double bond character for the CN bond involving the nitrogen atom coordinated to the metal [N(1)–C(25) 1.309(6) Å] when compared with the remaining CN bonds [N(2)–C(25) 1.359(6) Å, N(3)–C(25) 1.369(6) Å] and a planar geometry at the N(1) atom [Σ°N(1) = 358.9(6)°].
Figure 5.
Molecular structure of the cation of complex 5·CD4O. For clarity, hydrogen atoms (except the Rh–H and N–H protons) have been omitted.
Table 3. Selected Bond Lengths (Å) and Angles (°) for the Cation of Complex 5·CD4O.
| Rh–P | 2.2419(12) | P–Rh–H | 82(2) |
| Rh–N(1) | 2.110(4) | N(1)–Rh–Cta | 129.39(1) |
| Rh–Cta | 1.8697(1) | N(1)–Rh–H | 88(2) |
| Rh–H | 1.56(5) | Cta–Rh–H | 124 |
| N(1)–C(25) | 1.309(6) | Rh–N(1)–C(12) | 115.9(3) |
| N(2)–C(25) | 1.359(6) | Rh–N(1)–C(25) | 124.7(3) |
| N(3)–C(25) | 1.369(6) | C(12)–N(1)–C(25) | 118.3(4) |
| P–Rh–N(1) | 82.85(11) | Σ°N(1)b | 358.9(6) |
| P–Rh–Cta | 134.74(1) |
Ct represents the centroid of the η5-C5Me5 ligand.
Σ°N(1) stands for the sum of bond angles around N(1) atom.
Probably, the structural relaxation within the four-membered Ir–N–C–N metalacycle facilitates the reaction from 3 to 5 as well as from 4 to 6, which, in turn, results in the change in the coordination mode of the phosphano-guanidine ligand from κ3N,N′,P to κ2N,P with the concomitant change of the geometry at the N(1) atom from pyramidal to planar.
Water Activation by Complexes 3 and 4
As previously reported,21 the rhodium complex 3 reacts with deuterated water in a reversible fashion resulting in the gradual deuteration of the Cp* group. At 293 K, 1H NMR measurements and mass spectrometry analysis show that deuteration of this group is complete after 15 h in [D8]THF/D2O (78%/22%, v/v) solution. Also, deuteration was evidenced by the determination of the crystal structure of 3-d15 by low-temperature single crystal neutron-diffraction experiments.21 During the deuteration process, only isotopologues of compound 3 at different degrees of deuteration are detected by NMR spectroscopy.
Kinetic measurements indicate that the deuteration process obeys a pseudo-first-order rate law with kobs values from 3.31 × 10–6 to 4.99 × 10–4 s–1, in the 298–333 K temperature range. The formation of 3-d15 from 3 is reversible, and at 313 K, a [D8]THF/H2O (78%/22%, v/v) solution of 3-d15 evolves to 3 with an observed pseudo-first-order rate constant of 3.89 × 10–5 s–1. The measured ratio kH/kD (2.44) indicates that the rate-determining step for the exchange process is the C–H(D) bond cleavage.21
Based on density functional theory (DFT) calculations, we previously reported21 that the H/D exchange relies on the activation of the water O–H bond at IRh rendering the key hydroxo intermediate IIRh (Scheme 1). IIRh ultimately promotes the reversible hydrogen abstraction from Cp* (TS_IIRh-IIIRh) and affords the rhodium(I)-fulvene complex IIIRh, which should undergo a reversible H2O/D2O exchange, yielding the progressive hydrogen exchange/deuteration of the Cp* ligand (Scheme 1).
Scheme 1. Reaction Sequence for the Hydrogen Exchange at 3.

The Cp* ligand of the iridium complex 4 undergoes an H/D exchange process similar to that described for the rhodium analogue 3 but at a much slower rate. Indeed, at 293 K the 1H NMR spectrum of [D8]THF/D2O (78%/22%, v/v) solutions of the iridium complex 4 does not change over time. It is necessary to heat the reaction mixture at 343 K to observe the H/D exchange at an appreciable rate. After 4 days at this temperature, the Cp* ligand is deuterated at about 50%, on average. Apart from the isotopologues of 4 derived from the H/D exchange process, the formation of a new iridium complex, labeled as 8 (vide infra), was detected by NMR spectroscopy. The overlapping of the 1H NMR signals prevents the detailed study of the evolution of both the H/D exchange process and the reaction of formation of complex 8. The complete characterization of 8 will be discussed in the next subsection.
For the sake of comparison, the Gibbs free energy profiles of the hydrogen exchange for both 3 and 4 were calculated at the level wB97XD/def2tzvp//wB97XD/def2svp, using the SMD model for the solvent, at 298 K. Figure 6 shows the calculated intermediates and transition states along with the relative Gibbs free energies.
Figure 6.

Gibbs free energy profile (kcal·mol–1) for the hydrogen exchange at 3 (black) and 4 (gray) in the presence of water [wB97XD/def2tzvp//wB97XD/def2svp, in THF (SMD model), 298 K, 1 atm].
DFT calculations indicate that for both 3 and 4 the hydroxo intermediates IIRh and IIIr, respectively, are obtained stepwise by reaction of water with 3 or 4. Actually, the dissociation of the terminal M–N bond of 3 or 4 renders the true FLP complex, namely IRh and IIr, which interacts with one water molecule affording IRh·H2O or IIr·H2O. Neither IRh·H2O nor IIr·H2O contains a metal-oxygen bond, rather the incoming water molecule forms an N···H–O bond (Figure 7).27 Subsequent coordination of oxygen to the metal center and the concomitant H–OH bond rupture yield IIRh or IIIr in which a hydrogen bond still exists between the newly formed MOH and NH moieties (Figure 7). Once IIRh and IIIr form, hydrogen abstraction from its Cp* ligand gives the η4-tetramethylfulvene ligand in IIIRh and IIIIr and one weakly bonded water molecule (Figure 7). The hydrogen abstraction (IIRh → IIIRh; IIIr → IIIIr) entails the formal reduction of the metal center from the oxidation state +3 to +1. Accordingly, the metal centers of IIIRh and IIIIr feature a distorted square planar geometry in which two coordination sites are occupied by fulvene, whereas the phosphorus and nitrogen atoms from the phosphano-guanidine ligand complete the coordination sphere of the metal center (Figure 7). Notably, the metal-oxygen distance (IIIRh, 3.562; IIIIr, 3.616 Å) rules out the existence of a metal-oxygen bond. In addition, the water molecule is weakly bonded to the metal complex by means of the N2–H2···O hydrogen bond and an additional C2···H1–O short contact (Figure 7). Finally, the exchange of this weakly bonded water molecule with water (or D2O) solvent molecules should cause the progressive hydrogen exchange (deuteration) of the Cp* ligand. In view of the Gibbs free energy profiles given in Figure 6, it should be noted that intermediates 3·H2O (or 4·H2O) and IIRh (or IIIr), even if thermally accessible, are less stable than the starting complex 3 (or 4), which nicely agrees with the fact that 3 (or 4) are the only detected species in the course of the H/D exchange, and none of the intermediates has been observed by NMR spectroscopy.
Figure 7.
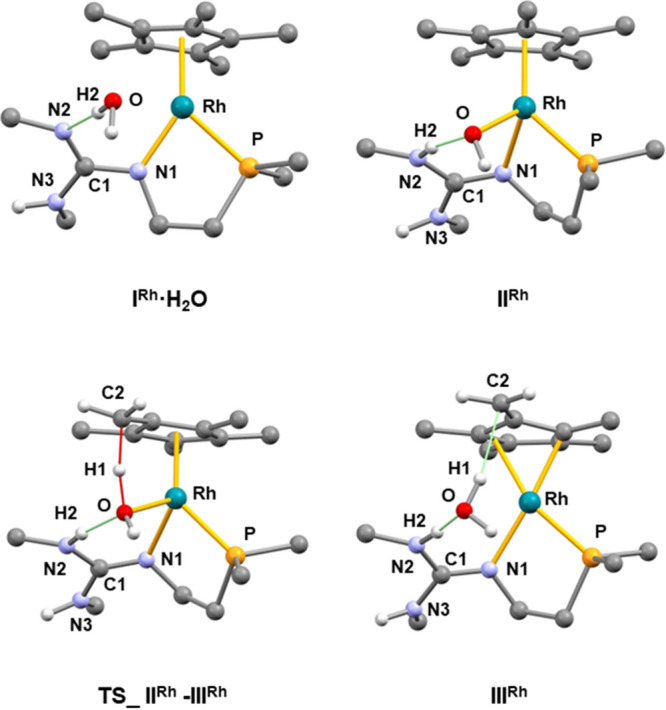
Calculated structures of IRh·H2O, IIRh, IIIRh, and TS_IIRh-IIIRh with the numbering scheme adopted. The calculated structures of IIr·H2O, IIIr, IIIIr, and TS_IIIr-IIIIr are similar and are not reported for the sake of brevity, the same numbering scheme being adopted. For clarity, most hydrogens are omitted, and only ipso carbon atoms of Tolyl and Phenyl groups are shown. Selected bond lengths/interatomic distances (Å) and angles (°) are: IRh·H2O, N2–H2 1.836, O–H2 0.988, N2–O 2.820, N2–H2–O 173.4, Rh–O 3.651; IIr·H2O, N2–H2 1.842, O–H2 0.986, N2–O 2.826, N2–H2–O 176.0, Ir–O 3.685; IIRh, Rh–O 2.078, O–H2 1.642, N2–H2 1.055, N2–O 2.655, N2–H2–O 159.4; IIIr, Ir–O 2.095, O–H2 1.647, N2–H2 1.051, N2–O 2.649, N2–H2–O 157.5; TS_IIRh-IIIRh, Rh–O 2.219, O–H2 1.787, N2–H2 1.031, N2–O 2.787, N2–H2-O 162.2; C2–H1 1.429, O–H1 1.207, O–H1–C2 158.2; TS_IIIr-IIIIr, Ir–O 2.255, O–H2 1.807, N2–H2 1.209, N2–O 2.798, N2–H2–O 160.4; C2–H1 1.485, O–H1 1.169, O–H1–C2 159.1; IIIRh, Rh–O 3.562, O–H2 1.847, N2–H2 1.029, N2–O 2.866, N2–H2–O 170.1; C2–H1 2.267, O–H1 0.972, O–H1–C2 167.8; IIIIr, Ir–O 3.616, O–H2 1.849, N2–H2 1.031, N2–O 2.865, N2–H2–O 167.7; C2–H1 2.196, O–H1 0.974, O–H1–C2 170.2.
As for the Gibbs free energy variation along the reaction sequence 3/4 + H2O ⇆ IIIRh/IIIIr, hydrogen exchange at 4 exhibits a significantly higher activation barrier (ΔGact = +29.9 kcal mol–1) when compared with 3 (ΔGact = +22.8 kcal mol–1), which perfectly fits in with the experimental conditions required for the H/D exchange of 3 and 4, and with the observed degree of deuteration (vide supra).
Finally, as far as the CN3 core is concerned, despite the fact that some degree of delocalization is expected to occur within the three carbon-nitrogen bonds, in the course of the hydrogen exchange a considerable electronic rearrangement takes place and it is reasonably beneficial to the accomplishment of the hydrogen exchange itself. Indeed, the analysis of the calculated carbon-nitrogen bond lengths for complexes 3, 4, IM·H2O, IIM, TS_IIM-IIIM, and IIIM (M = Rh, Ir, Scheme 2) points out that in the course of the hydrogen exchange the C1–N3 bond essentially holds its single bond character, whereas the C1–N1 and C1–N2 bonds switch from single to double and vice versa in the course of the sequence IM·H2O ⇆ IIM ⇆ IIIM, the formal protonation of N2 triggering the switch from one electronic distribution to the other.
Scheme 2. Calculated Carbon-Nitrogen Bond Lengths (Å) in 3, 4, IM·H2O, IIM, and IIIM (M = Rh, Ir).

Orthometalation Reactions
Heating THF/H2O (4/1, v/v) solutions of 3 or 4 at 383 K affords the orthometalated complexes 7 and 8, respectively (eq 4).
 |
4 |
Single crystals of 7 and 8 were grown from THF/Et2O (7), CH3OH/Acetone/Et2O/n-pentane (8a, from here on) and CH2Cl2 (8b, from here on) solutions, 8a and 8b featuring different crystal structures. Figure 8 shows the ORTEP plot of the cations [Cp*Ir(κ3C,N,P-H2L-H)]+ in 8a and 8b [H2L-H = Ph2PCH2CH2NC(NH(p-Tolyl))(NH(4-C6H3Me))], and Table 4 contains selected bond lengths and angles. The rhodium compound 7 exhibits a crystal structure virtually superimposable to 8a, so only 8a will be discussed in detail and selected data of 7 are included in the Supporting Information.
Figure 8.

ORTEP plot of the cations of complexes 8a and 8b. Thermal ellipsoids are at 50% probability, and most hydrogen atoms have been omitted for clarity.
Table 4. Selected Bond Lengths (Å) and Angles (°) of 8a and 8b.
| 8a | 8b | 8a | 8b | ||
|---|---|---|---|---|---|
| Ir–P | 2.2621(10) | 2.2598(10) | P–Ir–Cta | 133.81(3) | 135.48(3) |
| Ir–N(1) | 2.081(3) | 2.095(3) | N(1)–Ir–C(27) | 84.25(14) | 82.48(13) |
| Ir–C(27) | 2.065(4) | 2.069(4) | N(1)–Ir–Cta | 126.22(9) | 128.99(9) |
| Ir–Cta | 1.8559(2) | 1.8809(2) | C(27)–Ir–Cta | 125.66(10) | 119.32(10) |
| N(1)–C(25) | 1.312(5) | 1.304(5) | Ir–N(1)–C(12) | 118.0(2) | 118.6(2) |
| N(2)–C(25) | 1.355(5) | 1.356(5) | Ir–N(1)–C(25) | 122.2(3) | 119.9(3) |
| N(3)–C(25) | 1.363(5) | 1.374(5) | C(12)–N(1)–C(25) | 119.5(3) | 120.4(3) |
| P–Ir–N(1) | 77.05(9) | 81.44(9) | Σ°N(1)b | 359.7(5) | 358.9(5) |
| P–Ir–C(27) | 92.47(11) | 93.21(11) | N(1)–C(25)–N(3)–C(33) | 153.8(4) | –43.0(6) |
Ct represents the centroid of the η5-C5Me5 ligand.
Σ°N(1) stands for the sum of bond angles around the N(1) atom.
Both 8a and 8b exhibit a three-legged-piano stool geometry with an η5 coordinated Cp* ligand. The metalated phosphano-guanidine ligand occupies three mutually cis positions at the metal center [8a, P–Ir–N(1) 77.05(9), P–Ir–C(27) 92.47(11), N(1)–Ir–C(27) 84.25(14)°; 8b, P–Ir–N(1) 81.44(9), P–Ir–C(27) 93.21(11), N(1)–Ir–C(27) 82.48(13)°] rendering two fused metalacycles, namely, the five-membered ring Ir–P–C(11)–C(12)–N(1) and the six-membered ring Ir–C(27)–C(26)–N(2)–C(25)–N(1).
Interestingly, the metal center in both 8a and 8b is stereogenic. Nonetheless, as a consequence of the centrosymmetric space group P21/c of 8a and 8b, both enantiomers, namely, SIr-8a/b (shown in Figure 8) and RIr-8a/b, are present in the crystal.28 When dealing with the differences between 8a and 8b, the arrangement of the exocyclic N(3)H(p-Tolyl) moiety with respect to the IrCp*(P)(N1)(C27) core is worth a mention. As a matter of fact, the dihedral angle N(1)–C(25)–N(3)–C(33) is 153.8(4)° in 8a and–43.0(6)° in 8b indicating that the N(3)–C(25) bond adopts a conformation close to s-trans in 8a and close to s-cis in 8b (Figure 8). As a final remark, when comparing 8a/8b with 4, reasonably as a consequence of the formation of the less strained six membered ring Ir–C(27)–C(26)–N(2)–C(25)–N(1) instead of the four membered ring Ir–N(1)–C(25)–N(2), the nitrogen atom N(1) exhibits a planar geometry both in 8a [Σ°N(1) = 359.7(5)°] and 8b [Σ°N(1) = 358.9(5)°]. On this ground, N(1) should adopt a sp2 hybridization in 8a and 8b. Accordingly, the N(1)–C(25) bond length is shorter [8a, 1.312(5); 8b, 1.304(5) Å] than the N(2)–C(25) [8a, 1.355(5); 8b, 1.356(5) Å] and N(3)–C(25) bond lengths [8a, 1.363(5); 8b, 1.374(5) Å], suggesting that, despite some degree of delocalization over the CN3 core, the N(1)–C(25) bond should exhibit a higher double bond character when compared with N(2)–C(25) and N(3)–C(25).
No significant differences between the solution NMR spectra of both iridium rotamers 8a and 8b have been found in the 293–233 K temperature range indicating that, under these conditions, rotation around the N(3)–C(25) bond is free. Most probably, crystal packing accounts for the two dispositions encountered in the solid state.
The presence of two 1H peaks attributed to NH protons, at 8.41 and 8.36 ppm for 7, and at 8.43 and 8.34 ppm for 8, is indicative of the protonation of the N(p-Tolyl) group. The orthometalated carbon nucleus gives a doublet of doublets at 141.15 ppm [J(RhC) = 31.7 Hz, J(PC) = 13.8 Hz] for complex 7 and a doublet at 123.61 ppm (J(PC) = 9.1 Hz) for complex 8. The 31P{1H} NMR spectrum consists of a doublet centered at 46.45 ppm [J(RhP) = 153.3 Hz] and one singlet at 19.01 ppm for 8.
The energy profile E vs dihedral angle NCNC (α) for the rotation around the exocyclic C–N bond (Figure 9) was calculated for the rhodium complexes 7a/7b showing that the barrier for the rotation is about 10 kcal mol–1. In addition, the Gibbs free energy differences between isomers 7a and 7b as well as between isomers 8a and 8b are small (G7a-G7b = +1.4 kcal mol–1, G8a-G8b = +0.1 kcal mol–1). On these grounds, both isomers for each metal should be present in solution at room temperature and the interconversions 7a ⇆ 7b and 8a ⇆ 8b should be fast at that temperature, which fits in with the observation of averaged NMR spectra for 7a and 7b as well as for 8a and 8b, and with the isolation of crystals of each isomer using different crystallization mixtures of solvents.
Figure 9.
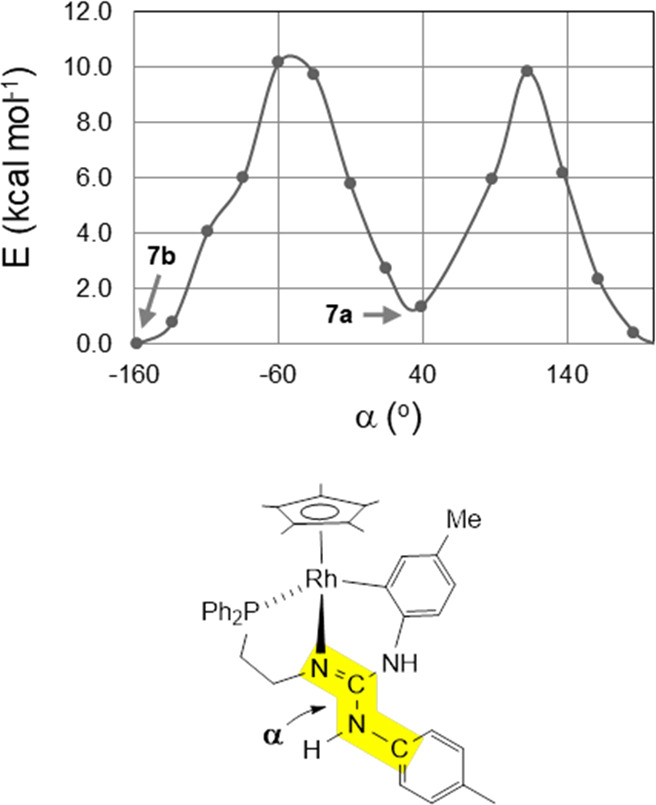
Energy profile (E vs α, wB97XD/def2svp, 298 K) for the rotation around the exocyclic C–N bond.
Reaction of 3 or 4 with Alcohols
At 333 K, complexes 3 and 4 react in THF with methanol, primary alcohols, and 2-propanol cleanly giving the metal-hydrido complexes 5 and 6, respectively (eq 5). The reaction involves the dehydrogenation of the alcohols at a relatively low19,20 temperature and without the assistance of an external base. 1H NMR signals assigned to methyl formate19h [δH 8.07, brq; 3.76, brd], acetaldehyde [δH 9.67, q; 2.07, d (J = 2.8 Hz)], propionaldehyde [δH 9.56 t (J = 1.3 Hz)], benzaldehyde (δH 10.02, s), and acetone (δH 2.04, s) were detected after the reaction with methanol, ethanol, n-propanol, benzyl alcohol, and 2-propanol, respectively.
 |
5 |
As the reactions with the iridium complex 4 were much slower than those with the rhodium compound 3, kinetic studies were carried out only with 3. Table 5 collects the values of the kinetic constants measured at 333 K (see the Supporting Information). The dehydrogenation rate is greater for methanol and primary alcohols (entries 1–4) than for the secondary alcohol 2-propanol (entry 5).19h To obtain information about the mechanism, in independent experiments the reaction was carried out with CH3OD (entry 6) or CD3OH (entry 7). In the reaction with CD3OH, the metal-hydrido region of the 1H NMR spectrum of the resulting product was silent, but when CH3OD was used as a reagent, a Rh–H 1H resonance was observed in the product. Notably, values of 5.21 and 8.29 were obtained for the kobs(CH3OH)/kobs(CH3OD) and kobs(CH3OH)/kobs(CD3OH) ratios, respectively. A detailed kinetic study was not carried out with CD3OD because the reaction rate in this solvent is very low. Indeed, a conversion of only about 4% was measured after 60 h of reaction at 333 K.
Table 5. Kinetic Constant for the Reaction of Complex 3 with Alcohols at 333 Ka.
| entry | alcohol | 106kobs/s–1 |
|---|---|---|
| 1 | MeOH | 8.12 ± 0.07 |
| 2 | EtOH | 11.6 ± 0.1 |
| 3 | nPrOH | 4.0 ± 0.2 |
| 4 | BnOH | 24.9 ± 0.5 |
| 5 | iPrOH | 1.12 ± 0.04 |
| 6 | CH3OD | 1.56 ± 0.03 |
| 7 | CD3OH | 1.0 ± 0.1 |
See the SI for experimental details.
As it was observed in the reaction of 3 or 4 with deuterated water, mass and 1H NMR spectra of solutions of compound 3 in alcohols with deuterated hydroxo groups indicate that the progressive deuteration of the methyl groups of the Cp* ring occurs. Kinetic measurements establish that the deuteration process obeys a pseudo-first order rate law. Table 6 collects the values of the kinetic constants measured at 313 K, and for comparative purposes, kobs obtained for D2O21 was also included. In general, the kobs for the H/D exchange process is greater than that measured for the alcohol dehydrogenation. For example, for CH3OD, the kobs for the Cp* deuteration is 6.39 ± 0.08 × 10–4 s–1 at 323 K (see the Supporting Information), and that for the dehydrogenation process is 1.56 ± 0.03 × 10–6 s–1, at 333 K (entry 6, Table 5), that is, the latter is about 400 times lower than the former despite being measured at a temperature 10 K higher. Based on the Eyring plot [ln(kobs/T) vs 1/T] ΔG≠293 of around 24 kcal·mol–1 was calculated in all cases (see the Supporting Information).
Table 6. Kinetic Constants for the H/D Exchange at 313 Ka.
| entry | R–OD | 105kobs/s–1 |
|---|---|---|
| 1 | D–OD | 9.5 ± 0.2 |
| 2 | CH3–OD | 28.2 ± 0.6 |
| 3 | CD3–OD | 16.9 ± 0.2 |
| 4 | Et–OD | 7.73 ± 0.08 |
| 5 | iPr–OD | 1.92 ± 0.04 |
See the SI for experimental details.
The mechanism of the reactions of 3 and 4 with methanol was explored by DFT calculations in order to shed light on the deuteration of 3 and 4 in the presence of CH3OD as well as on the formation of 5 and 6, respectively, as a result of the dehydrogenation of methanol. For both reactions, the energy profiles for 3 and 4 were elucidated by means of DFT computational methods at the level wB97XD/def2tzvp//wB97XD/def2svp using the SMD model for the solvent (THF).
As for the H/D exchange, the calculated reaction sequence is reminiscent of that previously discussed for the reaction of 3 or 4 with water (Figure 10). As a matter of fact, methanol reacts with 3 or 4 yielding IRh·MeOH or IIr·MeOH, respectively, in which a N···HO hydrogen bond brings together the dissociated form of 3 or 4, namely, IRh and IIr, with a methanol molecule, no metal-oxygen bond being observed (vide infra). In the following, the rupture of the O–H bond affords the methoxo derivatives IVRh and IVIr featuring an intramolecular NH···O hydrogen bond between the newly formed NH group and the methoxo ligand. Similar to IIRh and IIIr (Figure 6), the Cp* ligand in IVRh and IVIr undergoes a hydrogen abstraction yielding the tetramethylfulvene metal(I) complexes VRh and VIr, respectively, in which the resulting methanol molecule is still involved in an NH···O hydrogen bond with the NH group. Also, a short OH···CH2fulvene contact is observed between the fulvene ligand and the methanol molecule. Like for IIIRh and IIIIr, no metal-oxygen bond exists in VRh and VIr, and the exchange of the weakly bonded methanol molecule with methanol/methanol-d1 solvent molecules triggers the hydrogen exchange/deuteration of the Cp* ligand. Similar to the reaction of 3 or 4 with water, the activation barrier for 3 + MeOH ⇆ VRh (+21.5 kcal·mol–1) is significantly lower than that for 4 + MeOH ⇆ VIr (+29.0 kcal mol–1), which nicely fits in with the experimental conditions and the outcome of the deuteration reaction with 3 or 4.
Figure 10.

Gibbs free energy profile (kcal·mol–1) for the hydrogen exchange at 3 (black) and 4 (gray) in the presence of methanol [wB97XD/def2tzvp//wB97XD/def2svp, in THF (SMD model), 298 K, 1 atm].
As for the dehydrogenation of methanol rendering 5 or 6, DFT calculations suggest that IRh·MeOH and IIr·MeOH are again key intermediates (Figure 11). As a matter of fact, they convert into 5·CH2O or 6·CH2O, respectively, via the concerted transition state TS_IRh·MeOH-5·CH2O or TS_IIr·MeOH-6·CH2O. Notably, the elimination of CH2O results from the simultaneous migration of one CH hydrogen atom to the metal center and of the OH hydrogen atom to a nitrogen atom of the guanidine moiety (cf. TS_IRh·MeOH-5·CH2O, Figure 11). Accordingly, the carbon–oxygen bond shortens from 1.396 Å (av.) to 1.206 Å (av.) on going from IRh·MeOH and IIr·MeOH to 5·CH2O and 6·CH2O, respectively (Figure 11).
Figure 11.

Gibbs free energy profile for the reaction 3 (or 4) + MeOH → 5 (or 6) + CH2O [wB97XD/def2tzvp//wB97XD/def2svp, in THF (SMD model), 298 K]. View of the IRh·MeOH, 5·CH2O, and TS_IRh·MeOH-5·CH2O with the numbering scheme adopted. The calculated structures of IIr·MeOH, 6·CH2O, and TS_IIr·MeOH-6·CH2O are similar and are not reported for the sake of brevity, the same numbering scheme being adopted. Selected bond lengths/interatomic distances (Å) and angles (°) are IRh·MeOH, N2–H2 1.817, O–H2 0.988, N2–O 2.800, N2–H2–O 173.0, Rh–O 3.660, C2–O 1.397; IIr·MeOH, N2–H2 1.821, O–H2 0.986, N2–O 2.802, N2–H2–O 172.8, Rh–O 3.673, C2–O 1.396; TS_IRh·MeOH-5·CH2O, Rh–H1 1.832, H1–C1 1.198, C2–O 1.322, O–H2 1.279, N2–H2 1.206; TS_IIr·MeOH-6·CH2O, Ir–H1 1.863, H1–C1 1.211, C2–O 1.319, O–H2 1.268, N2–H2 1.216; 5·CH2O, H3–O 2.447, H1–C2 2.433, C2–O 1.206; 6·CH2O, H3–O 2.436, H1–C2 2.413, C2–O 1.205.
In this connection, previously reported studies already indicated that mono-29 and dinuclear30 iridium complexes as well as ruthenium derivatives31 are able to perform the acceptorless dehydrogenation of methanol via a concerted transition state taking advantage of the bifunctional character of the metal–ligand platform.
The calculated barriers for the rhodium complex 3 (+22.7 kcal·mol–1 at 298 K, +24.3 kcal·mol–1 at 333 K) and for the iridium complex 4 (+24.5 kcal·mol–1 at 298 K, +26.1 kcal·mol–1 at 333 K) underpin the experimental conditions. It is worth mentioning that according to the Gibbs free energy profile given in Figure 11 the above mentioned overall reactions are slightly endergonic (+2.9 kcal mol–1, M = Rh; +0.6 kcal mol–1, M = Ir). Nonetheless, in this regard, as mentioned before, methyl formate was detected as a product in the reaction of 3 or 4 with methanol, and its formation from formaldehyde was calculated to be exergonic (CH2O → 1/2 HCOOCH3, ΔGr = −9.7 kcal mol–1) which compensate the above mentioned positive ΔGr. On the other hand, when ethanol, 2-propanol, and benzyl alcohol were used, the overall dehydrogenation reaction CHR2OH + 3 (or 4) → CR2O + 5 (or 6) was calculated to be exergonic (ΔGr = from −10.8 to −4.5 kcal·mol–1) in agreement with the observation of acetaldehyde, acetone, and benzaldehyde, respectively, in the reaction mixture.
Conclusions
Compounds 3 and 4 behave like masked TMFLPs. The fac κ3N,N′,P coordination of the phosphano-guanidine ligand forces the central nitrogen atom to adopt an sp3 hybridization thus generating a strong strain within the M–N–C–N four-membered metalacycle. This structural stress makes the “unmasked” TMFLP thermally accessible (eq 6). The metal (acidic center) and the iminic nitrogen (basic center) synergistically cooperate in the reversible activation of molecular hydrogen as well as in the activation of the O–H bond of water and alcohols. The resulting nucleophilic M–OH and M–OR fragments are able to reversibly dehydrogenate the Cp* methyl groups giving rise to sequential and complete H/D exchange of the Cp* protons when deuterated D–OD or R–OD solvents were employed. On the other hand, the “unmasked” TMFLP is also able to dehydrogenate alcohols affording metal hydrido derivatives via a concerted transition state involving simultaneously the acidic and the basic sites.
 |
6 |
In this respect, the greater reactivity of the aldehyde or ketone products of the alcohol dehydrogenation versus the starting alcohol together with the reversibility of the hydrogenation reaction of complexes 3 and 4 paves the way to the potential application of these complexes to catalyzed reactions of alcohols using borrowing hydrogen methodology. On the other hand, judicious design of tridentate ligands capable of a fac κ3N,N′,P coordination as well as incorporation of d6 ions of precious and nonprecious metals would dramatically expand the applicability of the derived TMFLP species both in small-molecule activation chemistry and in the development of new catalytic processes.
Further work in this area is in progress and will be reported in due course.
Acknowledgments
We thank the Ministerio de Ciencia, Innovación y Universidades (MCIU) of Spain, Agencia Estatal de Investigación (AEI) of Spain, Fondo Europeo de Desarrollo Regional (FEDER) (CTQ2018-095561-BI00), and Gobierno de Aragón (Grupo de Referencia: Catálisis Homogénea Enantioselectiva E05-20R) for financial support. V.P. acknowledges the resources from the supercomputer “Memento,” and the technical expertise and assistance provided by the Institute for Biocomputation and Physics of Complex Systems (BIFI) Universidad de Zaragoza. F.J.L. and P.G.-O. acknowledge the Thematic network “CrysFact,” ref. RED2018-102574-T (MCIU/AEI) for its support.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.inorgchem.2c01902.
The synthesis and characterization of the complexes 1–8; 1H, 31P{1H}, and 13C{1H} spectra for the complexes 2, 4, 6, 7, and 8; CD of complexes 1 and 2; dehydrogenation reaction of complexes 5 and 6; kinetic studies for the H/D exchange at Cp* of complex 3; kinetic studies for acceptorless alcohol dehydrogenation; crystal structure determination of 1, 2, 4, 7, 8a, and 8b; DFT calculations, and energies and coordinates of calculated structures (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Welch G. C.; Juan R. R. S.; Masuda J. D.; Stephan D. W. Reversible, Metal-Free Hydrogen Activation. Science 2006, 314, 1124–1126. 10.1126/science.1134230. [DOI] [PubMed] [Google Scholar]
- McCahill J. S. J.; Welch G. C.; Stephan D. W. Reactivity of “Frustrated Lewis Pairs:” Three-Component Reactions of Phosphines, a Borane, and Olefins. Angew. Chem., Int. Ed. 2007, 46, 4968–4971. 10.1002/anie.200701215. [DOI] [PubMed] [Google Scholar]
- a Lam J.; Szkop K. M.; Mosaferi E.; Stephan D. W. FLP Catalysis: Main Group Hydrogenations of Organic Unsaturated Substrates. Chem. Soc. Rev. 2019, 48, 3592–3612. 10.1039/C8CS00277K. [DOI] [PubMed] [Google Scholar]; b Jupp A. R.; Stephan D. W. New Directions for Frustrated Lewis Pair Chemistry. Trends Chem. 2019, 1, 35–48. 10.1016/j.trechm.2019.01.006. [DOI] [Google Scholar]; c Paradies J. From Structure to Novel Reactivity in Frustrated Lewis Pairs. Coord. Chem. Rev. 2019, 380, 170–183. 10.1016/j.ccr.2018.09.014. [DOI] [Google Scholar]; d Scott D. J.; Fuchter M. J.; Ashley A. E. Designing Effective ‘Frustrated Lewis Pair’ Hydrogenation Catalysts. Chem. Soc. Rev. 2017, 46, 5689–5700. 10.1039/C7CS00154A. [DOI] [PubMed] [Google Scholar]; e Stephan D. W. The Broadening Reach of Frustrated Lewis Pair Chemistry. Science 2016, 354, aaf7229. 10.1126/science.aaf7229. [DOI] [PubMed] [Google Scholar]; f Stephan D. W. Frustrated Lewis Pairs. J. Am. Chem. Soc. 2015, 137, 10018–10032. 10.1021/jacs.5b06794. [DOI] [PubMed] [Google Scholar]; g Stephan D. W.; Erker G. Frustrated Lewis Pair Chemistry: Development and Perspectives. Angew. Chem., Int. Ed. 2015, 54, 6400–6441. 10.1002/anie.201409800. [DOI] [PubMed] [Google Scholar]; h Stephan D. W. Frustrated Lewis Pairs: From Concept to Catalysis. Acc. Chem. Res. 2015, 48, 306–316. 10.1021/ar500375j. [DOI] [PubMed] [Google Scholar]; (i) Frustrated Lewis Pairs II: Expanding the Scope; Erker G.; Stephan D. W., Eds.; Topics in Current Chemistry; Springer: Berlin Heidelberg, 2013; [Google Scholar]; (j) Frustrated Lewis Pairs I: Uncovering and Understanding; Erker G.; Stephan D. W., Eds.; Topics in Current Chemistry; Springer Berlin Heidelberg: Berlin, Heidelberg, 2013; [Google Scholar]; k Stephan D. W.; Erker G. Frustrated Lewis Pairs: Metal-Free Hydrogen Activation and More. Angew. Chem., Int. Ed. 2010, 49, 46–76. 10.1002/anie.200903708. [DOI] [PubMed] [Google Scholar]
- a Flynn S. R.; Wass D. F. Transition Metal Frustrated Lewis Pairs. ACS Catal. 2013, 3, 2574–2581. 10.1021/cs400754w. [DOI] [Google Scholar]; b Chapman A. M.; Haddow M. F.; Wass D. F. Frustrated Lewis Pairs beyond the Main Group: Synthesis, Reactivity, and Small Molecule Activation with Cationic Zirconocene–Phosphinoaryloxide Complexes. J. Am. Chem. Soc. 2011, 133, 18463–18478. 10.1021/ja207936p. [DOI] [PubMed] [Google Scholar]; c Chapman A. M.; Haddow M. F.; Wass D. F. Frustrated Lewis Pairs beyond the Main Group: Cationic Zirconocene–Phosphinoaryloxide Complexes and Their Application in Catalytic Dehydrogenation of Amine Boranes. J. Am. Chem. Soc. 2011, 133, 8826–8829. 10.1021/ja201989c. [DOI] [PubMed] [Google Scholar]
- a Hamilton H. B.; King A. M.; Sparkes H. A.; Pridmore N. E.; Wass D. F. Zirconium–Nitrogen Intermolecular Frustrated Lewis Pairs. Inorg. Chem. 2019, 58, 6399–6409. 10.1021/acs.inorgchem.9b00569. [DOI] [PubMed] [Google Scholar]; b Flynn S. R.; Metters O. J.; Manners I.; Wass D. F. Zirconium-Catalyzed Imine Hydrogenation via a Frustrated Lewis Pair Mechanism. Organometallics 2016, 35, 847–850. 10.1021/acs.organomet.6b00027. [DOI] [Google Scholar]; c Metters O. J.; Flynn S. R.; Dowds C. K.; Sparkes H. A.; Manners I.; Wass D. F. Catalytic Dehydrocoupling of Amine–Boranes Using Cationic Zirconium(IV)–Phosphine Frustrated Lewis Pairs. ACS Catal. 2016, 6, 6601–6611. 10.1021/acscatal.6b02211. [DOI] [Google Scholar]; d Metters O. J.; Forrest S. J. K.; Sparkes H. A.; Manners I.; Wass D. F. Small Molecule Activation by Intermolecular Zr(IV)-Phosphine Frustrated Lewis Pairs. J. Am. Chem. Soc. 2016, 138, 1994–2003. 10.1021/jacs.5b12536. [DOI] [PubMed] [Google Scholar]; e Chapman A. M.; Flynn S. R.; Wass D. F. Unexpected Formation of Early Late Heterobimetallic Complexes from Transition Metal Frustrated Lewis Pairs. Inorg. Chem. 2016, 55, 1017–1021. 10.1021/acs.inorgchem.5b01424. [DOI] [PubMed] [Google Scholar]; f Forrest S. J. K.; Clifton J.; Fey N.; Pringle P. G.; Sparkes H. A.; Wass D. F. Cooperative Lewis Pairs Based on Late Transition Metals: Activation of Small Molecules by Platinum(0) and B(C6F5)3. Angew. Chem., Int. Ed. 2015, 54, 2223–2227. 10.1002/anie.201409872. [DOI] [PubMed] [Google Scholar]; g Chapman A. M.; Haddow M. F.; Wass D. F. Cationic Group 4 Metallocene–(O-Phosphanylaryl)Oxido Complexes: Synthetic Routes to Transition-Metal Frustrated Lewis Pairs. Eur. J. Inorg. Chem. 2012, 2012, 1546–1554. 10.1002/ejic.201100968. [DOI] [Google Scholar]; h Chapman A. M.; Wass D. F. Cationic Ti(IV) and Neutral Ti(III) Titanocene–Phosphinoaryloxide Frustrated Lewis Pairs: Hydrogen Activation and Catalytic Amine-Borane Dehydrogenation. Dalton Trans. 2012, 41, 9067–9072. 10.1039/c2dt30168g. [DOI] [PubMed] [Google Scholar]
- a Jian Z.; Daniliuc C. G.; Kehr G.; Erker G. Frustrated Lewis Pair vs Metal–Carbon σ-Bond Insertion Chemistry at an o-Phenylene-Bridged Cp2Zr+/PPh2 System. Organometallics 2017, 36, 424–434. 10.1021/acs.organomet.6b00828. [DOI] [Google Scholar]; b Xu X.; Kehr G.; Daniliuc C. G.; Erker G. Frustrated Lewis Pair Behavior of [Cp2ZrOCR2CH2PPh2]+ Cations. Organometallics 2015, 34, 2655–2661. 10.1021/om501312a. [DOI] [Google Scholar]; c Normand A. T.; Richard P.; Balan C.; Daniliuc C. G.; Kehr G.; Erker G.; Le Gendre P. Synthetic Endeavors toward Titanium Based Frustrated Lewis Pairs with Controlled Electronic and Steric Properties. Organometallics 2015, 34, 2000–2011. 10.1021/acs.organomet.5b00250. [DOI] [Google Scholar]; d Normand A. T.; Daniliuc C. G.; Wibbeling B.; Kehr G.; Le Gendre P.; Erker G. Phosphido- and Amidozirconocene Cation-Based Frustrated Lewis Pair Chemistry. J. Am. Chem. Soc. 2015, 137, 10796–10808. 10.1021/jacs.5b06551. [DOI] [PubMed] [Google Scholar]; e Xu X.; Kehr G.; Daniliuc C. G.; Erker G. Stoichiometric Reactions and Catalytic Hydrogenation with a Reactive Intramolecular Zr+/Amine Frustrated Lewis Pair. J. Am. Chem. Soc. 2015, 137, 4550–4557. 10.1021/jacs.5b01623. [DOI] [PubMed] [Google Scholar]; f Xu X.; Kehr G.; Daniliuc C. G.; Erker G. Reactions of a Cationic Geminal Zr+/P Pair with Small Molecules. J. Am. Chem. Soc. 2013, 135, 6465–6476. 10.1021/ja3110076. [DOI] [PubMed] [Google Scholar]; g Xu X.; Kehr G.; Daniliuc C. G.; Erker G. 1,1-Carbozirconation: Unusual Reaction of an Alkyne with a Methyl Zirconocene Cation and Subsequent Frustrated Lewis Pair Like Reactivity. Angew. Chem., Int. Ed. 2013, 52, 13629–13632. 10.1002/anie.201307493. [DOI] [PubMed] [Google Scholar]
- See, for example:; a Osipova E. S.; Gulyaeva E. S.; Gutsul E. I.; Kirkina V. A.; Pavlov A. A.; Nelyubina Y. V.; Rossin A.; Peruzzini M.; Epstein L. M.; Belkova N. V.; Filippov O. A.; Shubina E. S. Bifunctional Activation of Amine-Boranes by the W/Pd Bimetallic Analogs of “Frustrated Lewis Pairs”. Chem. Sci. 2021, 12, 3682–3692. 10.1039/D0SC06114J. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Hidalgo N.; Romero-Pérez C.; Maya C.; Fernández I.; Campos J. Reactivity of [Pt(PtBu3)2] with Zinc(I/II) Compounds: Bimetallic Adducts, Zn–Zn Bond Cleavage, and Cooperative Reactivity. Organometallics 2021, 40, 1113–1119. 10.1021/acs.organomet.1c00088. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Bajo S.; Alférez M. G.; Alcaide M. M.; López-Serrano J.; Campos J. Metal-only Lewis Pairs of Rhodium with s, p and d -Block Metals. Chem. – Eur. J. 2020, 26, 16833–16845. 10.1002/chem.202003167. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Tinnermann H.; Fraser C.; Young R. D. Zero Valent Iron Complexes as Base Partners in Frustrated Lewis Pair Chemistry. Dalton Trans. 2020, 49, 15184–15189. 10.1039/D0DT03551C. [DOI] [PubMed] [Google Scholar]; e Mistry K.; Pringle P. G.; Sparkes H. A.; Wass D. F. Transition Metal Cooperative Lewis Pairs Using Platinum(0) Diphosphine Monocarbonyl Complexes as Lewis Bases. Organometallics 2020, 39, 468–477. 10.1021/acs.organomet.9b00568. [DOI] [Google Scholar]; f Hidalgo N.; Moreno J. J.; Pérez-Jiménez M.; Maya C.; López-Serrano J.; Campos J. Evidence for Genuine Bimetallic Frustrated Lewis Pair Activation of Dihydrogen with Gold(I)/Platinum(0) Systems. Chem. – Eur. J. 2020, 26, 5982–5993. 10.1002/chem.201905793. [DOI] [PubMed] [Google Scholar]; g Hidalgo N.; Moreno J. J.; Pérez-Jiménez M.; Maya C.; López-Serrano J.; Campos J. Tuning Activity and Selectivity during Alkyne Activation by Gold(I)/Platinum(0) Frustrated Lewis Pairs. Organometallics 2020, 39, 2534–2544. 10.1021/acs.organomet.0c00330. [DOI] [PMC free article] [PubMed] [Google Scholar]; h Hidalgo N.; Maya C.; Campos J. Cooperative Activation of X–H (X = H, C, O, N) Bonds by a Pt(0)/Ag(I) Metal-Only Lewis Pair. Chem. Commun. 2019, 55, 8812–8815. 10.1039/C9CC03008E. [DOI] [PubMed] [Google Scholar]; i Hidalgo N.; Bajo S.; Moreno J. J.; Navarro-Gilabert C.; Mercado B. Q.; Campos J. Reactivity of a Gold(I)/Platinum(0) Frustrated Lewis Pair with Germanium and Tin Dihalides. Dalton Trans. 2019, 48, 9127–9138. 10.1039/C9DT00702D. [DOI] [PubMed] [Google Scholar]; j Zwettler N.; Mösch-Zanetti N. C. Interaction of Metal Oxido Compounds with B(C6F5)3. Chem. Eur. J. 2019, 25, 6064–6076. 10.1002/chem.201805148. [DOI] [PubMed] [Google Scholar]; k Sinopalnikova I. S.; Peganova T. A.; Belkova N. V.; Deydier E.; Daran J.; Shubina E. S.; Kalsin A. M.; Poli R. Ruthenium p-Cymene Iminophosphonamide Complexes: Activation under Basic Conditions and Transfer Hydrogenation Catalysis. Eur. J. Inorg. Chem. 2018, 2018, 2285–2299. 10.1002/ejic.201701344. [DOI] [Google Scholar]; l Zwettler N.; Walg S. P.; Belaj F.; Mösch-Zanetti N. C. Heterolytic Si–H Bond Cleavage at a Molybdenum-Oxido-Based Lewis Pair. Chem. – Eur. J. 2018, 24, 7149–7160. 10.1002/chem.201800226. [DOI] [PMC free article] [PubMed] [Google Scholar]; m Chang K.; Wang X.; Fan Z.; Xu X. Reactions of Neutral Scandium/Phosphorus Lewis Pairs with Small Molecules. Inorg. Chem. 2018, 57, 8568–8580. 10.1021/acs.inorgchem.8b01292. [DOI] [PubMed] [Google Scholar]; n Campos J. Dihydrogen and Acetylene Activation by a Gold(I)/Platinum(0) Transition Metal Only Frustrated Lewis Pair. J. Am. Chem. Soc. 2017, 139, 2944–2947. 10.1021/jacs.7b00491. [DOI] [PubMed] [Google Scholar]; o Zhang S.; Appel A. M.; Bullock R. M. Reversible Heterolytic Cleavage of the H–H Bond by Molybdenum Complexes: Controlling the Dynamics of Exchange Between Proton and Hydride. J. Am. Chem. Soc. 2017, 139, 7376–7387. 10.1021/jacs.7b03053. [DOI] [PubMed] [Google Scholar]; p Bullock R. M.; Chambers G. M. Frustration across the Periodic Table: Heterolytic Cleavage of Dihydrogen by Metal Complexes. Philos. Trans. R. Soc. A. 2017, 375, 20170002. 10.1098/rsta.2017.0002. [DOI] [PMC free article] [PubMed] [Google Scholar]; q Arndt S.; Rudolph M.; Hashmi A. S. K. Gold-Based Frustrated Lewis Acid/Base Pairs (FLPs). Gold Bull. 2017, 50, 267–282. 10.1007/s13404-017-0219-7. [DOI] [Google Scholar]; r Barnett B. R.; Neville M. L.; Moore C. E.; Rheingold A. L.; Figueroa J. S. Oxidative-Insertion Reactivity Across a Geometrically Constrained Metal Borane Interaction. Angew. Chem., Int. Ed. 2017, 56, 7195–7199. 10.1002/anie.201702151. [DOI] [PubMed] [Google Scholar]; s Simonneau A.; Turrel R.; Vendier L.; Etienne M. Group 6 Transition-Metal/Boron Frustrated Lewis Pair Templates Activate N2 and Allow Its Facile Borylation and Silylation. Angew. Chem., Int. Ed. 2017, 56, 12268–12272. 10.1002/anie.201706226. [DOI] [PubMed] [Google Scholar]; t Wang Z.; Ying A.; Fan Z.; Hervieu C.; Zhang L. Tertiary Amino Group in Cationic Gold Catalyst: Tethered Frustrated Lewis Pairs That Enable Ligand-Controlled Regiodivergent and Stereoselective Isomerizations of Propargylic Esters. ACS Catal. 2017, 7, 3676–3680. 10.1021/acscatal.7b00626. [DOI] [Google Scholar]; u Chang K.; Xu X. Frustrated Lewis Pair Behavior of a Neutral Scandium Complex. Dalton Trans. 2017, 46, 4514–4517. 10.1039/C7DT00676D. [DOI] [PubMed] [Google Scholar]; v Rahman M. M.; Smith M. D.; Amaya J. A.; Makris T. M.; Peryshkov D. V. Activation of C–H Bonds of Alkyl- and Arylnitriles by the TaCl5–PPh3 Lewis Pair. Inorg. Chem. 2017, 56, 11798–11803. 10.1021/acs.inorgchem.7b01800. [DOI] [PubMed] [Google Scholar]; w Jamali S.; Abedanzadeh S.; Khaledi N. K.; Samouei H.; Hendi Z.; Zacchini S.; Kia R.; Shahsavari H. R. A Cooperative Pathway for Water Activation Using a Bimetallic Pt0–CuI System. Dalton Trans. 2016, 45, 17644–17651. 10.1039/C6DT03305A. [DOI] [PubMed] [Google Scholar]; x Lambic N. S.; Sommer R. D.; Ison E. A. Transition-Metal Oxos as the Lewis Basic Component of Frustrated Lewis Pairs. J. Am. Chem. Soc. 2016, 138, 4832–4842. 10.1021/jacs.6b00705. [DOI] [PubMed] [Google Scholar]; y Cui P.; Comanescu C. C.; Iluc V. M. Frustrated Lewis Pair-like Reactions of Nucleophilic Palladium Carbenes with B(C6F5)3. Chem. Commun. 2015, 51, 6206–6209. 10.1039/C5CC00868A. [DOI] [PubMed] [Google Scholar]; z Jiang Y.; Blacque O.; Fox T.; Berke H. Catalytic CO2 Activation Assisted by Rhenium Hydride/B(C6F5)3 Frustrated Lewis Pairs—Metal Hydrides Functioning as FLP Bases. J. Am. Chem. Soc. 2013, 135, 7751–7760. 10.1021/ja402381d. [DOI] [PubMed] [Google Scholar]
- a Elsby M. R.; Baker R. T. Strategies and Mechanisms of Metal–Ligand Cooperativity in First-Row Transition Metal Complex Catalysts. Chem. Soc. Rev. 2020, 49, 8933–8987. 10.1039/D0CS00509F. [DOI] [PubMed] [Google Scholar]; b Higashi T.; Kusumoto S.; Nozaki K. Cleavage of Si–H, B–H, and C–H Bonds by Metal–Ligand Cooperation: Focus Review. Chem. Rev. 2019, 119, 10393–10402. 10.1021/acs.chemrev.9b00262. [DOI] [PubMed] [Google Scholar]; c Alig L.; Fritz M.; Schneider S. First-Row Transition Metal (De)Hydrogenation Catalysis Based On Functional Pincer Ligands. Chem. Rev. 2019, 119, 2681–2751. 10.1021/acs.chemrev.8b00555. [DOI] [PubMed] [Google Scholar]; d Dub P. A.; Gordon J. C. The Role of the Metal-Bound N–H Functionality in Noyori-Type Molecular Catalysts. Nat. Rev. Chem. 2018, 2, 396–408. 10.1038/s41570-018-0049-z. [DOI] [Google Scholar]; e Khusnutdinova J. R.; Milstein D. Metal-Ligand Cooperation. Angew. Chem., Int. Ed. 2015, 54, 12236–12273. 10.1002/anie.201503873. [DOI] [PubMed] [Google Scholar]; f Annibale V. T.; Song D. Multidentate Actor Ligands as Versatile Platforms for Small Molecule Activation and Catalysis. RSC Adv. 2013, 3, 11432. 10.1039/c3ra40618k. [DOI] [Google Scholar]; g Askevold B.; Roesky H. W.; Schneider S. Learning from the Neighbors: Improving Homogeneous Catalysts with Functional Ligands Motivated by Heterogeneous and Biocatalysis. ChemCatChem 2012, 4, 307–320. 10.1002/cctc.201100347. [DOI] [Google Scholar]
- Spies P.; Erker G.; Kehr G.; Bergander K.; Fröhlich R.; Grimme S.; Stephan D. W. Rapid Intramolecular Heterolytic Dihydrogen Activation by a Four-Membered Heterocyclic Phosphane–Borane Adduct. Chem. Commun. 2007, 47, 5072–5074. 10.1039/B710475H. [DOI] [PubMed] [Google Scholar]
- a Arsenault N. E.; Xu Z.; Wolf M. O. Solvent- and Temperature-Responsive Platinum(II)-Functionalized Flexible Lewis Pairs. Inorg. Chem. 2019, 58, 65–68. 10.1021/acs.inorgchem.8b03053. [DOI] [PubMed] [Google Scholar]; b Hou Q.; Liu L.; Mellerup S. K.; Wang N.; Peng T.; Chen P.; Wang S. Stimuli-Responsive B/N Lewis Pairs Based on the Modulation of B–N Bond Strength. Org. Lett. 2018, 20, 6467–6470. 10.1021/acs.orglett.8b02774. [DOI] [PubMed] [Google Scholar]; c Johnstone T. C.; Wee G. N. J. H.; Stephan D. W. Accessing Frustrated Lewis Pair Chemistry from a Spectroscopically Stable and Classical Lewis Acid-Base Adduct. Angew. Chem., Int. Ed. 2018, 57, 5881–5884. 10.1002/anie.201802385. [DOI] [PubMed] [Google Scholar]; d Han Y.; Zhang S.; He J.; Zhang Y. Switchable C–H Silylation of Indoles Catalyzed by a Thermally Induced Frustrated Lewis Pair. ACS Catal. 2018, 8, 8765–8773. 10.1021/acscatal.8b01847. [DOI] [Google Scholar]; e Wu L.; Chitnis S. S.; Jiao H.; Annibale V. T.; Manners I. Non-Metal-Catalyzed Heterodehydrocoupling of Phosphines and Hydrosilanes: Mechanistic Studies of B(C6F5)3-Mediated Formation of P–Si Bonds. J. Am. Chem. Soc. 2017, 139, 16780–16790. 10.1021/jacs.7b09175. [DOI] [PubMed] [Google Scholar]; f Zheng J.; Lin Y.-J.; Wang H. Synthesis of 2-(Lutidinyl)Organoboranes and Their Reactivities against Dihydrogen and Pinacol Borane. Dalton Trans. 2016, 45, 6088–6093. 10.1039/C5DT03815D. [DOI] [PubMed] [Google Scholar]; g Hoshimoto Y.; Kinoshita T.; Ohashi M.; Ogoshi S. A Strategy to Control the Reactivation of Frustrated Lewis Pairs from Shelf-Stable Carbene Borane Complexes. Angew. Chem., Int. Ed. 2015, 54, 11666–11671. 10.1002/anie.201505974. [DOI] [PMC free article] [PubMed] [Google Scholar]; h Mahdi T.; Stephan D. W. Enabling Catalytic Ketone Hydrogenation by Frustrated Lewis Pairs. J. Am. Chem. Soc. 2014, 136, 15809–15812. 10.1021/ja508829x. [DOI] [PubMed] [Google Scholar]; i Wang X.; Kehr G.; Daniliuc C. G.; Erker G. Internal Adduct Formation of Active Intramolecular C4-Bridged Frustrated Phosphane/Borane Lewis Pairs. J. Am. Chem. Soc. 2014, 136, 3293–3303. 10.1021/ja413060u. [DOI] [PubMed] [Google Scholar]; j Houghton A. Y.; Hurmalainen J.; Mansikkamäki A.; Piers W. E.; Tuononen H. M. Direct Observation of a Borane–Silane Complex Involved in Frustrated Lewis-Pair-Mediated Hydrosilylations. Nat. Chem 2014, 6, 983–988. 10.1038/nchem.2063. [DOI] [PubMed] [Google Scholar]; k Hounjet L. J.; Bannwarth C.; Garon C. N.; Caputo C. B.; Grimme S.; Stephan D. W. Combinations of Ethers and B(C6F5)3 Function as Hydrogenation Catalysts. Angew. Chem., Int. Ed. 2013, 52, 7492–7495. 10.1002/anie.201303166. [DOI] [PubMed] [Google Scholar]; l Stute A.; Kehr G.; Daniliuc C. G.; Fröhlich R.; Erker G. Electronic Control in Frustrated Lewis Pair Chemistry: Adduct Formation of Intramolecular FLP Systems with −P(C6F5)2 Lewis Base Components. Dalton Trans. 2013, 42, 4487–4499. 10.1039/c2dt32806b. [DOI] [PubMed] [Google Scholar]; m Kolychev E. L.; Bannenberg T.; Freytag M.; Daniliuc C. G.; Jones P. G.; Tamm M. Reactivity of a Frustrated Lewis Pair and Small-Molecule Activation by an Isolable Arduengo Carbene–B{3,5-(CF3)2C6H3}3 Complex. Chem. – Eur. J. 2012, 18, 16938–16946. 10.1002/chem.201202840. [DOI] [PubMed] [Google Scholar]; n Holtrichter-Rößmann T.; Rösener C.; Hellmann J.; Uhl W.; Würthwein E.-U.; Fröhlich R.; Wibbeling B. Generation of Weakly Bound Al–N Lewis Pairs by Hydroalumination of Ynamines and the Activation of Small Molecules: Phenylethyne and Dicyclohexylcarbodiimide. Organometallics 2012, 31, 3272–3283. 10.1021/om3001179. [DOI] [Google Scholar]; o Ullrich M.; Lough A. J.; Stephan D. W. Dihydrogen Activation by B(p-C6F4H)3 and Phosphines. Organometallics 2010, 29, 3647–3654. 10.1021/om100563m. [DOI] [Google Scholar]; p Spies P.; Kehr G.; Bergander K.; Wibbeling B.; Fröhlich R.; Erker G. Metal-Free Dihydrogen Activation Chemistry: Structural and Dynamic Features of Intramolecular P/B Pairs. Dalton Trans. 2009, 9, 1534–1541. [DOI] [PubMed] [Google Scholar]; q Geier S. J.; Stephan D. W. Lutidine/B(C6F5)3: At the Boundary of Classical and Frustrated Lewis Pair Reactivity. J. Am. Chem. Soc. 2009, 131, 3476–3477. 10.1021/ja900572x. [DOI] [PubMed] [Google Scholar]
- Rokob T. A.; Hamza A.; Stirling A.; Pápai I. On the Mechanism of B(C6F5)3-Catalyzed Direct Hydrogenation of Imines: Inherent and Thermally Induced Frustration. J. Am. Chem. Soc. 2009, 131, 2029–2036. 10.1021/ja809125r. [DOI] [PubMed] [Google Scholar]
- Roters S.; Appelt C.; Westenberg H.; Hepp A.; Slootweg J. C.; Lammertsma K.; Uhl W. Dimeric Aluminum–Phosphorus Compounds as Masked Frustrated Lewis Pairs for Small Molecule Activation. Dalton Trans. 2012, 41, 9033–9045. 10.1039/c2dt30080j. [DOI] [PubMed] [Google Scholar]
- Boudjelel M.; Sosa Carrizo E. D.; Mallet-Ladeira S.; Massou S.; Miqueu K.; Bouhadir G.; Bourissou D. Catalytic Dehydrogenation of (Di)Amine-Boranes with a Geometrically Constrained Phosphine-Borane Lewis Pair. ACS Catal. 2018, 8, 4459–4464. 10.1021/acscatal.8b00152. [DOI] [Google Scholar]
- See, for example:; a Funes-Ardoiz I.; Garrido-Barros P.; Llobet A.; Maseras F. Single Electron Transfer Steps in Water Oxidation Catalysis. Redefining the Mechanistic Scenario. ACS Catal. 2017, 7, 1712–1719. 10.1021/acscatal.6b03253. [DOI] [Google Scholar]; b Michaelos T. K.; Shopov D. Y.; Sinha S. B.; Sharninghausen L. S.; Fisher K. J.; Lant H. M. C.; Crabtree R. H.; Brudvig G. W. A Pyridine Alkoxide Chelate Ligand That Promotes Both Unusually High Oxidation States and Water-Oxidation Catalysis. Acc. Chem. Res. 2017, 50, 952–959. 10.1021/acs.accounts.6b00652. [DOI] [PubMed] [Google Scholar]; c Blakemore J. D.; Crabtree R. H.; Brudvig G. W. Molecular Catalysts for Water Oxidation. Chem. Rev. 2015, 115, 12974–13005. 10.1021/acs.chemrev.5b00122. [DOI] [PubMed] [Google Scholar]; d Kärkäs M. D.; Verho O.; Johnston E. V.; Åkermark B. Artificial Photosynthesis: Molecular Systems for Catalytic Water Oxidation. Chem. Rev. 2014, 114, 11863–12001. 10.1021/cr400572f. [DOI] [PubMed] [Google Scholar]; e Parent A. R.; Crabtree R. H.; Brudvig G. W. Comparison of Primary Oxidants for Water-Oxidation Catalysis. Chem. Soc. Rev. 2013, 42, 2247–2252. 10.1039/C2CS35225G. [DOI] [PubMed] [Google Scholar]; f Piers W. E. Future Trends in Organometallic Chemistry: Organometallic Approaches to Water Splitting. Organometallics 2011, 30, 13–16. 10.1021/om100910d. [DOI] [Google Scholar]; g Kohl S. W.; Weiner L.; Schwartsburd L.; Konstantinovski L.; Shimon L. J. W.; Ben-David Y.; Iron M. A.; Milstein D. Consecutive Thermal H2 and Light-Induced O2 Evolution from Water Promoted by a Metal Complex. Science 2009, 324, 74–77. 10.1126/science.1168600. [DOI] [PubMed] [Google Scholar]
- a Zhang J.; Foley B. J.; Bhuvanesh N.; Zhou J.; Janzen D. E.; Whited M. T.; Ozerov O. V. Synthesis and Reactivity of Pincer-Type Cobalt Silyl and Silylene Complexes. Organometallics 2018, 37, 3956–3962. 10.1021/acs.organomet.8b00594. [DOI] [Google Scholar]; b Grünwald A.; Orth N.; Scheurer A.; Heinemann F. W.; Pöthig A.; Munz D. An Isolable Terminal Imido Complex of Palladium and Catalytic Implications. Angew. Chem., Int. Ed. 2018, 57, 16228–16232. 10.1002/anie.201809152. [DOI] [PubMed] [Google Scholar]; c Dub P. A.; Wang H.; Matsunami A.; Gridnev I. D.; Kuwata S.; Ikariya T. C–F Bond Breaking through Aromatic Nucleophilic Substitution with a Hydroxo Ligand Mediated via Water Bifunctional Activation. BCSJ 2013, 86, 557–568. 10.1246/bcsj.20120359. [DOI] [Google Scholar]; d Gutsulyak D. V.; Piers W. E.; Borau-García J.; Parvez M. Activation of Water, Ammonia, and Other Small Molecules by PCcarbeneP Nickel Pincer Complexes. J. Am. Chem. Soc. 2013, 135, 11776–11779. 10.1021/ja406742n. [DOI] [PubMed] [Google Scholar]
- a Vasko P.; Fuentes M. Á.; Hicks J.; Aldridge S. Reversible O–H Bond Activation by an Intramolecular Frustrated Lewis Pair. Dalton Trans. 2019, 48, 2896–2899. 10.1039/C9DT00228F. [DOI] [PubMed] [Google Scholar]; b Mo Z.; Szilvási T.; Zhou Y.; Yao S.; Driess M. An Intramolecular Silylene Borane Capable of Facile Activation of Small Molecules, Including Metal-Free Dehydrogenation of Water. Angew. Chem., Int. Ed. 2017, 56, 3699–3702. 10.1002/anie.201700625. [DOI] [PubMed] [Google Scholar]; c Ghattas G.; Bizzarri C.; Hölscher M.; Langanke J.; Gürtler C.; Leitner W.; Subhani M. A. Interaction of Formaldehyde with a Water-Tolerant Frustrated Lewis Pair. Chem. Commun. 2017, 53, 3205–3208. 10.1039/C6CC08044H. [DOI] [PubMed] [Google Scholar]; d Wang T.; Kehr G.; Liu L.; Grimme S.; Daniliuc C. G.; Erker G. Selective Oxidation of an Active Intramolecular Amine/Borane Frustrated Lewis Pair with Dioxygen. J. Am. Chem. Soc. 2016, 138, 4302–4305. 10.1021/jacs.6b00325. [DOI] [PubMed] [Google Scholar]; e Rochette É.; Courtemanche M.-A.; Pulis A.; Bi W.; Fontaine F.-G. Ambiphilic Frustrated Lewis Pair Exhibiting High Robustness and Reversible Water Activation: Towards the Metal-Free Hydrogenation of Carbon Dioxide. Molecules 2015, 20, 11902–11914. 10.3390/molecules200711902. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Klahn M.; Spannenberg A.; Rosenthal U. Tri- Tert- Butylphosphonium Hydroxytris(Pentafluorophenyl)Borate. Acta Crystallogr. E Struct. Rep. Online 2012, 68, o1549. 10.1107/S1600536812018144. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Di Saverio A.; Focante F.; Camurati I.; Resconi L.; Beringhelli T.; D’Alfonso G.; Donghi D.; Maggioni D.; Mercandelli P.; Sironi A. Oxygen-Bridged Borate Anions from Tris(Pentafluorophenyl)Borane: Synthesis, NMR Characterization, and Reactivity. Inorg. Chem. 2005, 44, 5030–5041. 10.1021/ic0502168. [DOI] [PubMed] [Google Scholar]; h Roesler R.; Piers W. E.; Parvez M. Synthesis, Structural Characterization and Reactivity of the Amino Borane 1-(NPh2)-2-[B(C6F5)2]C6H4. J. Organomet. Chem. 2003, 680, 218–222. 10.1016/S0022-328X(03)00384-X. [DOI] [Google Scholar]; i Bergquist C.; Bridgewater B. M.; Harlan C. J.; Norton J. R.; Friesner R. A.; Parkin G. Aqua, Alcohol, and Acetonitrile Adducts of Tris(Perfluorophenyl)Borane: Evaluation of Brønsted Acidity and Ligand Lability with Experimental and Computational Methods. J. Am. Chem. Soc. 2000, 122, 10581–10590. 10.1021/ja001915g. [DOI] [Google Scholar]
- See for example:; a Wei C. S.; Jiménez-Hoyos C. A.; Videa M. F.; Hartwig J. F.; Hall M. B. Origins of the Selectivity for Borylation of Primary over Secondary C–H Bonds Catalyzed by Cp*-Rhodium Complexes. J. Am. Chem. Soc. 2010, 132, 3078–3091. 10.1021/ja909453g. [DOI] [PubMed] [Google Scholar]; b Holland P. L.; Andersen R. A.; Bergman R. G.; Huang J.; Nolan S. P. Monomeric Cyclopentadienylnickel Methoxo and Amido Complexes: Synthesis, Characterization, Reactivity, and Use for Exploring the Relationship between H–X and M–X Bond Energies. J. Am. Chem. Soc. 1997, 119, 12800–12814. 10.1021/ja971829p. [DOI] [Google Scholar]; c Gusev O. V.; Sergeev S.; Saez I. M.; Maitlis P. M. Ring-Methyl Activation in Pentamethylcyclopentadienyl Complexes. 3. Synthesis and Reactions of (.Eta.4-Tetramethylfulvene)(.Eta.5-Cyclopentadienyl)Rhodium and -Iridium. Organometallics 1994, 13, 2059–2065. 10.1021/om00017a072. [DOI] [Google Scholar]; d Glueck D. S.; Bergman R. G. Deprotonation of a Cp* Methyl Group by an Iridium Anilide: Formation, Structure, and Solution Dynamics of an .Eta.4-Tetramethylfulvene Complex. Organometallics 1990, 9, 2862–2863. 10.1021/om00161a006. [DOI] [Google Scholar]; e Kang J. W.; Maitlis P. M. (Pentamethylcyclopentadienyl)Rhodium and -Iridium Complexes. V. Complexes with Oxy-Ligands and the Exchange of Methyl Protons by Deuterium under Basic Conditions. J. Organomet. Chem. 1971, 30, 127–133. 10.1016/S0022-328X(00)82604-2. [DOI] [Google Scholar]
- a Banerjee S.; Soldevila-Barreda J. J.; Wolny J. A.; Wootton C. A.; Habtemariam A.; Romero-Canelón I.; Chen F.; Clarkson G. J.; Prokes I.; Song L.; O’Connor P. B.; Schünemann V.; Sadler P. J. New Activation Mechanism for Half-Sandwich Organometallic Anticancer Complexes. Chem. Sci. 2018, 9, 3177–3185. 10.1039/C7SC05058E. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Ciancaleoni G.; Bolaño S.; Bravo J.; Peruzzini M.; Gonsalvi L.; Macchioni A. Counterion-Dependent Deuteration of Pentamethylcyclopentadiene in Water-Soluble Cationic Rh(III) Complexes Assisted by PTA. Dalton Trans. 2010, 39, 3366–3368. 10.1039/c003056m. [DOI] [PubMed] [Google Scholar]
- a Borthakur I.; Sau A.; Kundu S. Cobalt-Catalyzed Dehydrogenative Functionalization of Alcohols: Progress and Future Prospect. Coord. Chem. Rev. 2022, 451, 214257. 10.1016/j.ccr.2021.214257. [DOI] [Google Scholar]; b Trodden E. C.; Delve M. P.; Luz C.; Newland R. J.; Andresen J. M.; Mansell S. M. A Ruthenium Cis -Dihydride with 2-Phosphinophosphinine Ligands Catalyses the Acceptorless Dehydrogenation of Benzyl Alcohol. Dalton Trans. 2021, 50, 13407–13411. 10.1039/D1DT02508B. [DOI] [PubMed] [Google Scholar]; c Pradhan D. R.; Pattanaik S.; Kishore J.; Gunanathan C. Cobalt-Catalyzed Acceptorless Dehydrogenation of Alcohols to Carboxylate Salts and Hydrogen. Org. Lett. 2020, 22, 1852–1857. 10.1021/acs.orglett.0c00193. [DOI] [PubMed] [Google Scholar]; d Fuse H.; Mitsunuma H.; Kanai M. Catalytic Acceptorless Dehydrogenation of Aliphatic Alcohols. J. Am. Chem. Soc. 2020, 142, 4493–4499. 10.1021/jacs.0c00123. [DOI] [PubMed] [Google Scholar]; e Paul B.; Maji M.; Chakrabarti K.; Kundu S. Tandem Transformations and Multicomponent Reactions Utilizing Alcohols Following Dehydrogenation Strategy. Org. Biomol. Chem. 2020, 18, 2193–2214. 10.1039/C9OB02760B. [DOI] [PubMed] [Google Scholar]; f Zhang J.; Guo B.; Young D. J.; Li H.-X. Acceptorless Dehydrogenative Coupling with Ru-Based Catalysts for the Synthesis of N-Heteroaromatic Compounds. Dalton Trans. 2020, 49, 15527–15547. 10.1039/D0DT03282D. [DOI] [PubMed] [Google Scholar]; g Awasthi M. K.; Singh S. K. Ruthenium Catalyzed Dehydrogenation of Alcohols and Mechanistic Study. Inorg. Chem. 2019, 58, 14912–14923. 10.1021/acs.inorgchem.9b02691. [DOI] [PubMed] [Google Scholar]; h Crabtree R. H. Homogeneous Transition Metal Catalysis of Acceptorless Dehydrogenative Alcohol Oxidation: Applications in Hydrogen Storage and to Heterocycle Synthesis. Chem. Rev. 2017, 117, 9228–9246. 10.1021/acs.chemrev.6b00556. [DOI] [PubMed] [Google Scholar]; i Werkmeister S.; Neumann J.; Junge K.; Beller M. Pincer-Type Complexes for Catalytic (De)Hydrogenation and Transfer (De)Hydrogenation Reactions: Recent Progress. Chem. – Eur. J. 2015, 21, 12226–12250. 10.1002/chem.201500937. [DOI] [PubMed] [Google Scholar]; j Trincado M.; Banerjee D.; Grützmacher H. Molecular Catalysts for Hydrogen Production from Alcohols. Energy Environ. Sci. 2014, 7, 2464–2503. 10.1039/C4EE00389F. [DOI] [Google Scholar]; k Gunanathan C.; Milstein D. Applications of Acceptorless Dehydrogenation and Related Transformations in Chemical Synthesis. Science 2013, 341, 1229712. 10.1126/science.1229712. [DOI] [PubMed] [Google Scholar]; l Johnson T. C.; Morris D. J.; Wills M. Hydrogen Generation from Formic Acid and Alcohols Using Homogeneous Catalysts. Chem. Soc. Rev. 2010, 39, 81–88. 10.1039/B904495G. [DOI] [PubMed] [Google Scholar]
- a Yao W.; DeRegnaucourt A. R.; Shrewsbury E. D.; Loadholt K. H.; Silprakob W.; Qu F.; Brewster T. P.; Papish E. T. Reinvestigating Catalytic Alcohol Dehydrogenation with an Iridium Dihydroxybipyridine Catalyst. Organometallics 2020, 39, 3656–3662. 10.1021/acs.organomet.0c00398. [DOI] [Google Scholar]; b Gusev D. G. Revised Mechanisms of the Catalytic Alcohol Dehydrogenation and Ester Reduction with the Milstein PNN Complex of Ruthenium. Organometallics 2020, 39, 258–270. 10.1021/acs.organomet.9b00542. [DOI] [Google Scholar]; c Tseng K.-N. T.; Kampf J. W.; Szymczak N. K. Mechanism of N,N,N-Amide Ruthenium(II) Hydride Mediated Acceptorless Alcohol Dehydrogenation: Inner-Sphere β-H Elimination versus Outer-Sphere Bifunctional Metal–Ligand Cooperativity. ACS Catal. 2015, 5, 5468–5485. 10.1021/acscatal.5b00952. [DOI] [Google Scholar]; d Li H.; Wang Z. Computational Mechanistic Studies of Acceptorless Dehydrogenation Reactions Catalyzed by Transition Metal Complexes. Sci. China Chem. 2012, 55, 1991–2008. 10.1007/s11426-012-4713-8. [DOI] [Google Scholar]
- Carmona M.; Ferrer J.; Rodríguez R.; Passarelli V.; Lahoz F. J.; García-Orduña P.; Cañadillas-Delgado L.; Carmona D. Reversible Activation of Water by an Air- and Moisture-Stable Frustrated Rhodium Nitrogen Lewis Pair. Chem. −A Eur. J. 2019, 25, 13665–13670. 10.1002/chem.201902452. [DOI] [PubMed] [Google Scholar]
- White C.; Yates A.; Maitlis P. M.; Heinekey D. M.. (η5-Pentamethylcyclopentadienyl)Rhodium and -Iridium Compounds. In Inorganic Syntheses; Grimes R. N., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2007; pp. 228–234. [Google Scholar]
- Eliel E. L.; Wilen S. H.; Mander L. N.. Stereochemistry of Organic Compounds; Wiley: New York, 1994, p. 159. [Google Scholar]
- Racemic and enantiopure crystals of compounds 1 and 2 are isolated together and mixed. However, fortunately, its morphology and size allow their separation and individualized study. Indeed, single crystals of rac-1 and rac-2 are needle-shaped and both enantiomers of each of the two compounds crystallize in large parallelepipeds of up to about 10 mg in weight. Therefore, it is easy to manually separate racemic crystals from enantiopure crystals. For one of the enantiomers of each compound, the CD spectrum of exactly the crystal whose molecular structure was determined by X-ray diffraction was measured and taken as reference (see Supporting Information). A small portion (about 1 mg) was cut from a large enantiopure crystal (about 4–10 mg) and its CD spectrum was measured. By comparison with the above mentioned CD reference spectrum, the configuration was assigned to the remaining crystal (approximately 3–9 mg). By repeating the operation, samples of several tens of milligrams of each of the four enantiomers, RRh-1, SRh-1, RIr-2 and SIr-2, can be obtained. Their CD spectra are shown in the Supporting Information.
- a Cahn R. S.; Ingold C.; Prelog V. Specification of Molecular Chirality. Angew. Chem. Int. Ed. 1966, 5, 385–415. 10.1002/anie.196603851. [DOI] [Google Scholar]; b Prelog V.; Helmchen G. Basic Principles of the CIP-System and Proposals for a Revision. Angew. Chem. Int. Ed. 1982, 21, 567–583. 10.1002/anie.198205671. [DOI] [Google Scholar]; c Lecomte C.; Dusausoy Y.; Protas J.; Tirouflet J.; Dormond A. Structure cristalline et configuration relative d’un complexe du titanocene presentant une chiralite plane et une chiralite centreé sur l’atome de titane. J. Organomet. Chem. 1974, 73, 67–76. 10.1016/S0022-328X(00)80382-4. [DOI] [Google Scholar]
- Allen F. H.; Kennard O.; Watson D. G.; Brammer L.; Orpen A. G.; Taylor R. Tables of Bond Lengths Determined by X-Ray and Neutron Diffraction. Part 1. Bond Lengths in Organic Compounds. J. Chem. Soc., Perkin Trans. 2 1987, 12, S1–S19. 10.1039/p298700000s1. [DOI] [Google Scholar]
- The previously reported Gibbs free energy profile of the hydrogen exchange at 3[21] calculated at the level M06/def2tzvp//B3PW91-GD3BJ/def2svp is substantially identical to that given in Figure 6. Nonetheless, it is worth a mention that we were unable to identify the intermediate IRh·H2O, but a PES scan showed that water should approach the FLP IRh forming an OH···N hydrogen bond in the first place, followed by the coordination of oxygen to rhodium, finally affording the hydroxo derivative IIRh.
- N(2) adopts an almost planar geometry both in 8a [Σ°N(2) = 357(6)°] and 8b [Σ°N(2) = 353(6)°] reasonably as a consequence of some degree of delocalization of the lone pair at nitrogen N(2) either on the adjacent aromatic ring as well as on the CN3 core of the guanidine moiety.
- Li H.; Lu G.; Jiang J.; Huang F.; Wang Z.-X. Computational Mechanistic Study on Cp*Ir Complex-Mediated Acceptorless Alcohol Dehydrogenation: Bifunctional Hydrogen Transfer vs β-H Elimination. Organometallics 2011, 30, 2349–2363. 10.1021/om200089m. [DOI] [Google Scholar]
- Mena I.; Casado M. A.; Polo V.; García-Orduña P.; Lahoz F. J.; Oro L. A. The Dehydrogenation of Alcohols through a Concerted Bimetallic Mechanism Involving an Amido-Bridged Diiridium Complex. Angew. Chem., Int. Ed. 2012, 51, 8259–8263. 10.1002/anie.201202936. [DOI] [PubMed] [Google Scholar]
- de Zwart F. J.; Sinha V.; Trincado M.; Grützmacher H.; de Bruin B. Computational Mechanistic Studies of Ruthenium Catalysed Methanol Dehydrogenation. Dalton Trans. 2022, 51, 3019–3026. 10.1039/D1DT04168A. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.