

Abstract
Alzheimer’s disease (AD) is the most common cause of dementia in the general population and, to date, constitutes a major therapeutic challenge. In the pathogenesis of AD, aggregates of amyloid β (Aβ) and neurofibrillary tangles (NFTs) containing Tau-microtubule-associated protein (tau) are known to trigger a neuroinflammatory response with subsequent formation of an inflammasome. In particular, the NOD-like receptor pyrin domain-containing 3 (NLRP3) inflammasome is thought to play a crucial role in AD-related pathology. While the mechanisms for NLRP3 activation are not fully understood, it has been demonstrated that, after detection of protein aggregates, NLRP3 induces pro-inflammatory cytokines, such as interleukin 18 (IL-18) or interleukin 1β (IL-1β), that further potentiate AD progression. Specific inhibitors of NLRP3 that exhibit various mechanisms to attenuate the activity of NLRP3 have been tested in in vivo studies and have yielded promising results, as shown by the reduced level of tau and Aβ aggregates and diminished cognitive impairment. Herein, we would like to summarize the current state of knowledge on NLRP3 inflammasome priming, activation, and its actual role in AD pathogenesis, and to characterize the NLRP3 inhibitors that have been studied most and their impact on AD-related pathology.
Keywords: Alzheimer’s disease, amyloid β, neurofibrillary tangles, NOD-like receptor pyrin domain-containing 3, NOD-like receptor pyrin domain-containing 3 inflammasome, NOD-like receptor pyrin domain-containing 3 inhibitors, Alzheimer’s disease treatment
1. Introduction
Alzheimer’s disease (AD) is the most common cause of dementia and is estimated to be at least the sixth leading killer in America [1]. In general, it is regarded as a complicated and multifaceted neurodegenerative disorder of the central nervous system (CNS). AD starts with a long preclinical or pre-symptomatic stage, which advances to mild cognitive impairment (MCI), and ultimately leads to dementia and incapacitating memory impairment. Besides the severity of AD, to date there is no cure for this disease and current treatment options remain only symptomatic [2,3].
Mechanistically, AD is characterized by an accumulation of amyloid plaques and neurofibrillary tangles (NFTs) that contribute to neuronal cell death and disease progression. The amyloid plaques are fibrous aggregates composed of Aβ. The abnormally folded Aβ is a by-product of the Amyloid Precursor Protein (APP) metabolism of several sequential proteases: β-secretase and the intramembranous γ-secretase complex [4,5,6]. Physiologically, the APP is processed by the α-secretase and γ-secretase, which does not produce Aβ. There are several types of soluble and insoluble non-plaque Aβ aggregates. Protofibrils and oligomers are the intermediate species that may form amyloid fibrils. There are also different variants of Aβ peptides that result from alternative processing, which makes them prone to exhibiting specific aggregation properties [7]. The Aβ(1–40) peptide is the most abundant isoform, whereas the Aβ(1–42) peptide is present in certain forms of AD [8,9]. On the other hand, the NFTs are composed of hyperphosphorylated Tau-microtubule-associated protein that accumulates within the dystrophic neurites. Interestingly, synapse loss, as well as the clinical features and the progression of AD, are better associated with NFT pathology than with β-amyloid pathology [4]. The cortical density of NFTs correlates better with cognitive decline than senile plaques [10]. Furthermore, the two antibodies targeting Aβ in a clinical trial did not manage to improve cognitive function despite decreasing Aβ burden, as revealed by PET imaging [11].
A crucial aspect of inflammatory responses is the formation of the inflammasome—a multiprotein complex that mediates inflammation. Amongst others, the NOD-like receptor pyrin domain-containing 3 (NLRP3) inflammasome is thought to play the greatest role in neurodegeneration, particularly in AD-associated pathology [12,13]. Although the mechanisms of its activation are not fully understood [14], many studies have demonstrated that the microglial NLRP3 inflammasome, due to its ability to sense aggregated proteins such as Aβ, alters the generation of interleukin 18 and interleukin 1β (IL-18 and IL-1β, respectively). These inflammatory mediators are closely associated with the spread of Aβ within and between the cerebral areas specific to AD pathogenesis, and thus contribute to disease progression [15,16,17]. Moreover, several studies have proven that inhibition of the NLRP3 inflammasome reduced tau aggregation and decreased the amyloid burden, which in turn diminished cognitive impairment in AD mice [18,19].
Due to the failure of many clinical trials investigating potential therapies targeting Aβ, it has been hypothesized that AD progression is due to a more global neuronal dysfunction [20]. Currently, it is believed that neuroinflammation has a huge impact on the development and progression of AD, opening up the opportunities for research into new possible drugs [21]. Considering the importance of neuroinflammation over the course of AD progression, this review summarizes current knowledge regarding NLRP3 inflammasome inhibition and strategies of its modulation that may contribute to the amelioration of this disease.
2. NLRP3 Inflammasome Activation and Stimulation
The NLRP3 inflammasome, first described in 2002 [22,23], is involved in the activation of caspase-1, leading to the release of IL-18 and IL-1β and resulting in a strong inflammatory response that has been observed in multiple diseases, including AD [24].
NLRP3 is one of the inflammasomes that has been studied most, and its molecular structure has already been determined. It contains a cytosolic sensor molecule NLRP3, ASC (the adaptor molecule apoptosis-associated speck-like protein containing a caspase recruitment domain [CARD]), and protease pro-caspase-1, which is the effector molecule [23]. The NLRP3 protein consists of a conserved central nucleotide binding and oligomerization domain (NOD or NACHT), C-terminal leucine-rich repeat (LRR) domain, and an N-terminal pyrin domain (PYD) [25]. NOD exhibits ATPase activity and is necessary for the self-oligomerization of the molecule at the beginning of the inflammasome’s assembly [26]. The role of the LRR domain is to recognize pathogen-associated molecular patterns (PAMPs), damage-associated molecular patterns (DAMPs), and other ligands, and to maintain the NLRP inactive state [27,28,29], whereas PYD allows communication with ASC through PYD–PYD domain interaction [30]. ASC recruits pro-caspase-1 through CARD–CARD interaction, which enables the release of inflammatory cytokines, such as IL-18 and IL-β [31]. Upon activation, the NLRP3 also executes a form of cell death (pyroptosis), which occurs via the induction of gasdemin D (GSDMD) secretion. It has been shown that members of the gasdemin family can bind to membrane lipids and induce membrane-disrupting cytotoxicity through the formation of pores in the cell membrane, which facilitates the secretion of inflammatory cytokines [32,33,34].
It is assumed that two steps are necessary for the activation of the NLRP3 inflammasome. The first step involves a primer signal that is associated with the nuclear factor kappa B (NF-κB)-dependent transcription of NLRP3 and pro-IL-1β [35]. A large number of receptors may be responsible for triggering this process, including anaphylatoxin, cytokine receptors, and pattern recognition receptors (PRRs) such as Toll-like receptors (TLRs) [36]. After binding, the specific receptor triggers the transcription of NLRP3 by activating NF-κB through different pathways that may be associated with myeloid differentiation primary response 88 (MyD88), TIR-domain-containing adapter-inducing interferon-β (TRIF), Fas-associated protein with death domain (FADD), or caspase-8, as well as with reactive oxygen species (ROS) [35,36,37].
The second stage involves the activation and assembly of the NLRP3 inflammasome, which can be induced by many factors that are related to the disturbance of cell homeostasis [38]. Although the exact mechanism by which NLRP3 activation occurs is still unknown, there are several activation signals that are suggested to promote the second step of NLRP3 formation. One of these activation signals involves molecules such as silica, Aβ, and crystalline substances that can be phagocytized by cells such as microglia but are resistant to degradation [39,40,41,42,43,44,45]. This resistance later causes the destabilization of lysosomes, which leads to the release of various enzymes, such as cathepsins, into the cytosol [39,46,47]. This process drives the rise of cytosolic Ca2+ levels, which is involved in the activation of NLRP3. Moreover, it has been shown that a high extracellular K+ concentration decreases the rise in intracellular Ca2+ levels. Due to this fact, K+ efflux could promote Ca2+ influx and increase cytosolic Ca2+ concentrations, leading to NLRP3 activation [36,48,49] (Figure 1).
Figure 1.

The mechanisms of priming and activation of the NOD-like receptor pyrin domain-containing 3 (NLRP3) inflammasome. Firstly, protein aggregates (Aβ and tau) are recognized by microglial pattern recognition receptors, such as Toll-like receptors (e.g., TLR2, TLR4), or cluster of differentiation 36 (CD36). Afterwards, they induce activation of specific pathways like MYD88/NF-κB, which leads to transcription of pro-IL-β and NLRP3. One of the NLRP3 domains—the adaptor molecule apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC)—recruits pro-caspase-1, which enables the release of inflammatory cytokines, namely IL-18 and IL-β. Further, cell death in the form of pyroptosis occurs after the activation of NLRP3 via the induction of gasdemin D (GSDMD). GSDMD can induce membrane-disrupting cytotoxicity and facilitates the secretion of inflammatory cytokines via the creation of pores in the cell membrane.
Other circumstances that promote NLRP3 activation are ultraviolet radiation, the release of ATP by necrotic cells located nearby, or certain events that are related to mitochondrial damage, such as the loss of membrane potential, ROS production, the release of mitochondrial DNA (mtDNA), and the exposure of cardiolipin at the mitochondrial outer membrane. The latter is particularly and directly associated with NLRP3 activation [14,36,50,51,52]. The variety of mechanisms inducing NLRP3 illustrates that inflammasome activation is generally driven by the detection of homeostatic imbalance, not by a single pathway.
Recently, it has been shown that human monocytes can form the NLRP3 inflammasome, bypassing the priming step that has long been viewed as essential. Numerous studies have also emphasized the role of transcription-independent priming in relation to post-translational modifications (PTMs) of NLRP3, such as ubiquitination and phosphorylation [53,54]. These changes are known to exert a significant impact on inflammasome functionality [55,56,57]. The role of other PTMs, such as methylation or acetylation, has not yet been thoroughly investigated, however the possibility of their involvement in NLRP3 activation cannot be excluded. It has also been suggested that, although priming may not always be obligatory for assembling a functional NLRP3 inflammasome, it may be required to boost inflammasome activation and the overall inflammation process [58].
3. NLRP3 Inflammasome in AD
NLRP3 inflammasome activation has been identified in AD patients by increased IL-1β and active caspase-1 production [19,59]. Transgenic animal models of AD, such as APP/PS1 mice [19] or Tg2576 mice [60], have also demonstrated NLRP3 activation through the presence of increased IL-1β and caspase-1 levels. Furthermore, the monocytes of patients with AD that were LPS-primed and Aβ42-stimulated have shown an increased expression of NLRP3 inflammasome components such as NLRP3, PYCARD, caspase-1, and its effectors: IL-18 and IL-1β [61]. Heneka et al. have found that AD mice with a double knockout of Nlrp3 or Casp1 (APP/PS1/NLRP3−/− and APP/PS1/Casp-1−/−) demonstrate improved memory function, as well as decreased levels of caspase-1, Il-1β, and Aβ deposits compared to APP/PS1 mice. However, contrary to the previous reports, a new study by Tang et al. that investigated specific NLRP3 inflammasome markers in post-mortem brain tissues indicated that NLRP3 activation may in fact not take place in AD patients’ brains [62]. Thus, the actual effect of NLRP3 activation on the AD course is yet to be determined.
Of note, it has been shown that the AD-specific aggregates, Aβ and NFTs, are able to induce NLRP3 inflammasome priming through a number of mechanisms [63,64,65,66]. Aβ and tau are mostly recognized by microglial pattern recognition receptors, such as Toll-like receptors (e.g., TL2, TLR4), cluster of differentiation 14 (CD14), cluster of differentiation 47 (CD47), cluster of differentiation 36 (CD36), and α6β1 integrin. Then, they induce inflammation through specific pathways such as MYD88 or NF-κB [67,68], leading to the transcription of pro-IL-β and NLRP3 [38,69].
The second step of NLRP3 inflammasome activation in AD may occur through lysosomal damage, the release of enzymes, such as cathepsin B, and the overproduction of mitochondrial ROS processes described above [50,70]. These events lead to the production of IL-1β, caspase 1 activation, pyroptosis, and neuroinflammation. Increased IL-1β and caspase-1 levels have been observed in patients with early or mild stages of AD, which suggests that NLRP3 inflammasome activation may contribute to early pathogenic events that stimulate the progression of AD [19,67,71,72,73]. APP/PS1 mice that are deficient in caspase-1 or NLRP3 have been shown to exhibit reduced spatial memory loss, decreased impairment of hippocampal synaptic plasticity, and amelioration of other AD-related symptoms [21,40] (Figure 2).
Figure 2.

The factors inducing NOD-like receptor pyrin domain-containing 3 (NLRP3) inflammasome activation in the pathogenesis of AD. The recognition of pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) by specific cellular receptors may bring NLRP3 into the active state. Activation may also be triggered by factors related to cell damage, which involve mitochondrial disruption with the overproduction of reactive oxygen species (ROS) and mitochondrial DNA (mtDNA) release, necrosis with an increased level of ATP released from damaged cells, ion dysregulation (K+ efflux and Ca2+ influx), and lysosomal destabilization resulting from impaired microglial phagocytosis. Furthermore, protein deposits of Tau and amyloid β (Aβ) may drive lysosomal dysfunction due to their resistance to degradation, and may also directly induce NLRP3 activation. Additionally, UV radiation constitutes another independent factor for activation of the NLRP3 inflammasome.
4. Neuroinflammation in the Pathogenesis and Progression of AD
Numerous diseases linked to immune system dysfunction and the overactivation of NLRP3 have been recognized as risk factors for developing AD. These disorders include autoimmune rheumatic diseases [74], traumatic brain injury [75], type 2 diabetes mellitus [76], obesity [77], cerebrovascular diseases [78], dyslipidemia [79], and hypertension [80,81]. The latter is strongly related to atherosclerosis, which is both an inflammatory disease involving NLRP3 activation and a vascular disease that may promote AD development by chronic hypoperfusion and the stiffening of arterial walls in the brain [80]. Hypertension is associated with increased Aβ and NFT deposits, and it has been established that midlife stage 1 and stage 2 systolic hypertension increases the risk of developing AD by 18 and 25%, respectively [81]. Aging, as the greatest risk factor for developing AD, is responsible for more than 95% cases of late-onset Alzheimer’s disease (LOAD). Senescence is strongly linked to immune system dysfunction: it may promote autoimmunity or constitutive low-grade inflammation, which is characteristic for AD. It was also found that different cell types, including astrocytes and microglia, present the accumulation of senescence-associated proteins, such as p16, p53, and p21 in AD patients’ brains, which impairs their proper functioning [82,83].
The NLRP3 inflammasome is present in microglia and astrocytes in the CNS [84,85,86]. Several studies have indicated that it can also be expressed in neurons and oligodendrocytes [87,88].
Microglia, which are macrophages found in the CNS, are crucial in maintaining brain homeostasis throughout life. They are involved in the innate immune response as a first-line defense against invading pathogens. Not so long ago, microglia were shown to exhibit the capacity to develop innate immune memory (IIM), which has changed our perspective on inborn immunity. The IIM indicates that microglial activity can be molecularly reprogrammed, leading to the suppression (immune tolerance characteristic of the M2 microglial phenotype) or enhancement of the immune response (trained immunity concerning the M1 phenotype) [89,90,91]. Similarly to microglia, astrocytes can also be divided into two phenotypes: the harmful A1 cells and the protective A2 cells, which express neurotrophic factors such as brain derived neurotrophic factor (BDNF) [92]. This dichotomic classification, although simplified, is still useful for conveying the idea of the specific nature of glial cells, depending on the circumstances [69].
There is growing evidence to suggest that microglial cells, upon activation, may lose their ability to retransform to a naïve status. Additionally, they are thought to remain “post-activated”, which may lead to some neuropathological changes [93]. IIM may exert an impact on the progression of neurodegeneration through an enhanced immune response and the polarization of microglia to the more active M1 phenotype. Moreover, it was shown that polarization to the immune-suppressive M2 phenotype can reduce neuroinflammation and presumably ameliorate neurodegeneration [94,95,96,97].
Genome studies have allowed for the detection of more than 40 gene variants that are associated with the development of late-onset AD [98]. Among these, there are some genes that are related to the mediation of inflammation, particularly microglial immunoreceptors such as TREM2, PILRA, and CD33. TREM2 is responsible for the enhancement of microglial phagocytosis and the secretion of inflammatory cytokines in response to specific stimuli, involving NFTs and Aβ [99]. It is important to note that, during the primal stages of AD, a lack of TREM2 ameliorates amyloid pathology and synaptic deficit, whereas it has the opposite affect at later stages (a detrimental effect due to the absence of Aβ phagocytosis) [100,101]. This conclusion is consistent with the two-peak theory of microglial activation in the progression of AD. In this theory, the first peak involves the phagocytosis of Aβ by the protective M2 phenotype and the second peak is driven by NFTs that activate the pro-inflammatory M1 phenotype [102,103].
The dual nature of neuroinflammation in AD suggests that an approach based simply on suppressing glial function may in fact be daunting. A more rational treatment option should probably focus on analyzing whether the regions of a patient’s brain are in the early or late stages of AD, which could help to distinguish between the neurotoxic or neuroprotective phenotypes of microglia [102]. In this case, selective inhibition of the more reactive and neurotoxic microglial phenotype should also be considered.
5. NLRP3 Inhibitors
Many preclinical models imply that small-molecule NLRP3 inhibitors may be practically useful against numerous diseases that involve neuroinflammation. In contrast to antibodies or vaccines, they possess the unquestionable advantage of being able to cross the brain blood barrier (BBB), which has been exploited in a number of studies of neurodegenerative disorders and other pathologies related to the CNS [104,105,106] (Table 1).
Table 1.
Characterization of the most common NLRP3 inhibitors used in AD research.
| Compound Code of Inhibitor | Chemical Name ofInhibitor | Chemical Structure of Inhibitor | Mechanism of Action | Research Models |
|---|---|---|---|---|
|
MCC950 (also known as CRID-3 or CP-456773) |
1-(1,2,3,5,6,7-Hexahydro-s-indacen-4-yl)-3-[4-(1-hydroxy-1-methyl-ethyl)-furan-2-sulfonyl]urea |
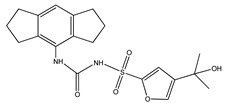
|
inhibition of NACHT domain of NLRP3 | |
| JC124 | (5-Chloro-2-methoxy-N-(4-(N-methylsulfamoyl) phenethyl)benzamide |
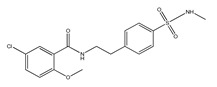
|
inhibition of caspase-1 activation, IL-1β secretion, and ASC aggregation | |
|
VX-765 (also known as Belnacasan) |
N-(4-Amino-3-chlorobenzoyl)-3-methyl-L-valyl-N-[(2R,3S)-2-ethoxytetrahydro-5-oxo-3-furanyl]-L-prolinamide |
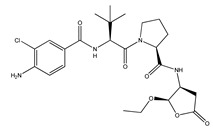
|
inhibition of caspase-1 and caspase-4 | |
|
OLT1177 (also known as Dapansutrile) |
3-(Methane sulfonyl)propanenitrile |
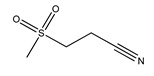
|
inhibition of NLRP-ASC and NLRP3-caspase-1 interaction |
|
5.1. MCC950
MCC950 (1-(1,2,3,5,6,7-Hexahydro-s-indacen-4-yl)-3-[4-(1-hydroxy-1-methyl-ethyl)-furan-2-sulfonyl]urea, also known as CRID3 and CP-456773) is one of the small-molecule inhibitors of the NLRP3 inflammasome. MCC950 interacts with the Walker B motif of the NLRP3 NACHT domain, that is, the Mg2+-binding ATPase specific P-loop. This interaction results in the blocking of ATP hydrolysis, the inhibition of NLRP3 activation, and oligomerization [104,107,108,109]. Studies on APP/PS1 mouse models of AD have shown that MCC950 is able to ameliorate cognitive function, presumably due to the drug’s ability reduce Aβ accumulation by stimulating its phagocytosis in vitro [110].
MCC950 has also been shown to inhibit microglial training induced by NLRP3 stimulation in sporadic AD mouse models. Therefore, it protects against pathologies such as Aβ accumulation or neuronal loss [111]. Another study demonstrated that MCC950 inhibits neuronal pyroptosis and significantly reduces Aβ neurotoxicity in human primary neurons in vitro. The treatment of AD mice with MCC950 positively impacted the histological morphology of senescence-accelerated mouse prone 8 (SAMP8) mice brains and improved the spatial memory of treated animals. Additionally, MCC950 reduced the overexpression of GSDMD, caspase-1, and NLRP3-response factors, which are strongly involved in neuronal pyroptosis in SAMP8 mice [112]. Although MCC950 exhibits high target selectivity, its therapeutic development is limited by its toxicokinetic properties [82]. It has been suggested that MCC950 could have unwanted effects by inhibiting carbonic anhydrase 2 and other carbonic anhydrases, which are widely expressed in human cells and involved in maintaining fluid balance and pH levels. For that reason, the focus has been on the development of less toxic MCC950 analogues, such as OLT1177, which will be described later in this review [113]. Despite this, MCC950 and Inzomelid (a related NLRP3 inhibitor), have now passed through phase 1b clinical trials and will soon move to phase 2 for a range of CNS degenerative disorders, such as AD or PD [114].
5.2. JC124
JC124 (5-chloro-2-methoxy-N-(4-(N-methylsulfamoyl)phenethyl)benzamide) is another small-molecule inhibitor of the NLRP3 inflammasome, which functions by blocking caspase-1 activation, IL-1β secretion, and ASC aggregation [115]. JC124 has been shown to protect mice brains from excessive inflammation after a traumatic brain injury (TBI). Post-injury treatment with the drug leads to reduced neuronal degeneration and inflammatory cell responses, as well as a decreased expression of molecules such as NLRP3, ASC, caspase-1, and IL-1β [116]. Moreover, JC124 has been shown to reduce Aβ load and neuroinflammation in APP/PS1 mice, leading to ameliorated synaptic plasticity and an improvement in cognitive function [117]. Another study has examined the effects of JC124 on AD-related pathologies in CRND8 APP transgenic mice (TgCRND8). JC124 treatment reduced the levels of both soluble and insoluble Aβ, decreased β-cleavage of APP, and increased astrocytosis, all whilst decreasing microglial activity at the same time. JC124 also reduces oxidative stress, indicating its neuroprotective effects. Moreover, the same study showed that even at a later point of amyloidosis progression, inhibition of the NLRP3 inflammasome can efficiently reduce the effects of amyloid accumulation and oxidative stress [118]. The above-mentioned findings make JC124 and its analogues promising drug candidates for the potential treatment of AD.
5.3. VX-765
VX-765 (N-(4-Amino-3-chlorobenzoyl)-3-methyl-L-valyl-N-[(2R,3S)-2-ethoxytetrahydro-5-oxo-3-furanyl]-L-prolinamide, also known as Belnacasan) is a small caspase-1 and caspase-4 inhibitor. Caspase-1-mediated inflammation is strongly involved in the activation and functioning of the NLRP3 inflammasome and is believed to play a crucial role in the onset of AD. VX-765 is BBB permeable, nontoxic, and has already been approved by the Food and Drug Administration (FDA) for clinical trials involving humans. This molecule has displayed the ability to reverse episodic and spatial memory impairment in a J20 mouse model by halting the progression of Aβ deposition. It also managed to reverse neuroinflammation and stabilize synaptophysin protein levels in the mouse hippocampus [119]. Another study has demonstrated that, in Swedish/Indiana mutant amyloid precursor protein (APPSw/Ind) J20 and wild-type mice, VX-765 postponed either AD- or age-related spatial memory deficits after pre-symptomatic treatment [120]. VX-765 was also studied in the context of neuroinflammation occurring after spinal cord injury (SCI) in mice, where it was shown to inhibit the secretion of caspase-1, IL-1β, and IL-18. Moreover, VX-765 decreased total macrophage infiltration, Th1 and Th17 differentiation, and M1 microglia activation, while promoting the differentiation of type 2 helper T cells (Th2) and regulatory T cells (Treg) and the activation of M2 microglia. Further, administration of the drug led to the alleviation of neural injury and white matter demyelination, thereby improving functional recovery from the trauma [121]. In rat models of temporal lobe epilepsy or stroke, VX-765 was also shown to decrease pyroptosis in cerebral ischemia and was shown to have an overall neuroprotective effect [122,123,124].
5.4. OLT1177
OLT1177 (3-(Methane sulfonyl)propanenitrile, also known as Dapansutrile) is an orally active β-sulfonyl nitrile molecule and a specific inhibitor of the NLRP3 inflammasome that has been proven to be safe for humans. Mechanistically, it inhibits the properties of NLRP3 that are associated with ATPase activity that is vital for ASC recruitment, thus preventing NLRP3-ASC formation and inflammasome activation. Besides inhibiting NLRPR3-ASC, OLT1177 also affects NLRP3-caspase-1 interaction. In vitro, OLT117 reduced the release of IL-1β and IL-18 resulting from NLRP3 inflammasome activation. In vitro, the inhibitor reduced IL-1β levels at concentrations more than 100-fold lower than the effective plasma concentrations of IL-1β that have been safely reached in humans. Moreover, it was shown that OLT1177 inhibits the accumulation of succinate in muscles, the level of which is correlated with oxidative stress driving mitochondrial uncoupling [125]. OLT117 was also studied in mouse models of multiple sclerosis (MS), wherein it improved the disease symptoms by reducing the infiltration of macrophages and CD4 T cells in the spinal cord, as well as decreasing levels of IL-1β and IL-18 [126]. In APP/PS1 mice, OLT1177 ameliorated synaptic plasticity, inhibited the activation of microglia, and decreased the number of Aβ plaques in the cortex. Furthermore, the study has shown that the levels of metabolic markers of AD (e.g., carboxylic acids, 5-oxoproline, kynurenine deaminated purines, glutaminolysis) in plasma were restored, depending on the administered dose of OLT117 [127].
5.5. NSAIDs
Recently, it has been suggested that nonsteroidal anti-inflammatory drugs (NSAIDs), due to their anti-inflammatory characteristics, may also inhibit neuroinflammation occurring at the onset of AD, and hence they may play a role in decreasing the risk of developing AD [128,129,130,131]. NSAIDs are one of the most commonly used medications and they appear on the World Health Organization (WHO) Model List of Essential Medicines. NSAIDs inhibit the cyclooxygenase-1/2 (COX-1 and COX-2) enzymes and are also able to selectively inhibit the NLRP3 inflammasome by interacting with volume-regulated anion channels independently of COX enzymes [132,133,134,135]. However, placebo-controlled clinical trials with NSAIDs in AD patients have shown negative outcomes. Neither rofecoxib in patients with MCI, nor naproxen and celecoxib administered to elderly patients have exhibited positive results [136]. Even though some studies have demonstrated a reduced risk of AD mortality with aspirin or other NSAIDs [137], which supports the hypothesis of the neuroprotective role of NSAIDs in AD development, thorough studies and meta-analyses indicate that there is no evidence to encourage the use of NSAIDs of any class for the prevention of dementia [136,138,139]. Furthermore, studies also highlight the adverse health effects that may result from sustained treatment with NSAIDs [140], for example, gastric irritation or the higher risk of ulcer formation [141], that are triggered by selective COX-1 inhibition.
5.6. Colchicine
Interestingly, colchicine, an alkaloid derived from autumn crocus (Colchicum autumnale), has already been shown to inhibit NLRP3 inflammasome oligomerization in patients with atherosclerosis [142,143]. However, the drug was able to induce AD-like symptoms, such as dementia or cognitive impairment, in rodents by single intracerebroventricular injection, as evaluated through the use of Morris water maze and elevated plus-maze tests [142,144,145]. This could be explained by colchicine’s ability to promote neuroinflammation via the activation of a number of other pathways and effectors, such as COX-2 [146,147], TNF-α, nitrite, or ROS in the hippocampus [148]. Moreover, it was found that colchicine directly destroys hippocampal granule cells and mossy fibers via the blockage of axoplasmic transport, which eventually leads to cholinergic neurodegeneration and AD symptoms [149].
6. Conclusions
It is clear that the NLRP3/caspase-1/IL-1β axis is involved in crucial pathological events that are associated with AD. According to the latest research, it is the neuroinflammatory response to amyloid plaques that triggers the development and progression of AD, not the presence of the aggregates themselves. This review summarizes our current understanding of the NLRP3 inflammasome, the processes of its activation and stimulation, and its involvement in AD pathogenesis and neuroinflammation. We also reviewed the most popular NLRP3 inhibitors, which exhibit different mechanisms of attenuating NLRP3 activity and decreasing inflammation. MCC950 and OLT1177 block the ATPase activity that is essential for NLRP3 activation, and JC124 inhibits caspase-1 activation, IL-1β secretion, and ASC aggregation, whereas VX-765 prevents the activation of caspases 1 and 4. However, many important questions prevail, for example, which disease stage inhibitors of NLRP3 may be useful, or what mechanism of their action is most beneficial for ameliorating the progression of AD? AD is a complex disorder and so much remains to be explored in order to deepen our understanding of the complexity of neuroinflammation in CNS pathologies. The phenotypic changes taking place in the glial cells during the pathogenesis of these diseases remain particularly intriguing, as well as the possibility to target the innate immune stem cells and the IIM. Considering the positive findings regarding the amelioration of cognitive function in animal models that were treated with NLRP3 inflammasome inhibitors, as well as the first attempts to undertake clinical trials with the afore-mentioned drugs, this area of research sheds a light on the development of new potential approaches to AD treatment. However, there is still much to be investigated before efficient drugs that inhibit NLRP3 are officially approved for the treatment of AD. Nevertheless, the modulation of neuroinflammation remains a promising target of potential molecular therapy for this incurable disease.
Acknowledgments
Figures were created with BioRender.com (accessed on 9 December 2021).
Author Contributions
Conceptualization, I.M. and J.B.; formal analysis, I.M. and W.R-K.; writing—original draft preparation, J.B., N.S., W.L. and W.R.-K.; writing—review and editing, I.M., E.K. and W.R.-K.; visualization, N.S.; supervision, I.M.; project administration, I.M.; funding acquisition, I.M. All authors have read and agreed to the published version of the manuscript.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
Funding Statement
This research was funded by grant of Medical University of Lodz, Poland No. 503/5-108-05/503-51-001-19-00.
Footnotes
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Alzheimer’s Association 2021 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2021;17:327–406. doi: 10.1002/alz.12328. [DOI] [PubMed] [Google Scholar]
- 2.Abe T., Barber G.N. Cytosolic-DNA-mediated, STING-dependent proinflammatory gene induction necessitates canonical NF-kappaB activation through TBK1. J. Virol. 2014;88:5328–5341. doi: 10.1128/JVI.00037-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rozpedek W., Pytel D., Poplawski T., Walczak A., Gradzik K., Wawrzynkiewicz A., Wojtczak R., Mucha B., Diehl J.A., Majsterek I. Inhibition of the PERK-dependent unfolded protein response signaling pathway involved in the pathogenesis of Alzheimer’s disease. Curr. Alzheimer Res. 2019;16:209–218. doi: 10.2174/1567205016666190228121157. [DOI] [PubMed] [Google Scholar]
- 4.Lane C.A., Hardy J., Schott J.M. Alzheimer’s disease. Eur. J. Neurol. 2018;25:59–70. doi: 10.1111/ene.13439. [DOI] [PubMed] [Google Scholar]
- 5.O’Brien R.J., Wong P.C. Amyloid precursor protein processing and Alzheimer’s disease. Annu. Rev. Neurosci. 2011;34:185–204. doi: 10.1146/annurev-neuro-061010-113613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rozpedek W., Markiewicz L., Diehl J.A., Pytel D., Majsterek I. Unfolded Protein Response and PERK Kinase as a New Therapeutic Target in the Pathogenesis of Alzheimer’s Disease. Curr. Med. Chem. 2015;22:3169–3184. doi: 10.2174/0929867322666150818104254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thal D.R., Walter J., Saido T.C., Fandrich M. Neuropathology and biochemistry of Abeta and its aggregates in Alzheimer’s disease. Acta Neuropathol. 2015;129:167–182. doi: 10.1007/s00401-014-1375-y. [DOI] [PubMed] [Google Scholar]
- 8.Schmidt M., Sachse C., Richter W., Xu C., Fandrich M., Grigorieff N. Comparison of Alzheimer Abeta(1–40) and Abeta(1–42) amyloid fibrils reveals similar protofilament structures. Proc. Natl. Acad. Sci. USA. 2009;106:19813–19818. doi: 10.1073/pnas.0905007106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rozpedek-Kaminska W., Siwecka N., Wawrzynkiewicz A., Wojtczak R., Pytel D., Diehl J.A., Majsterek I. The PERK-Dependent Molecular Mechanisms as a Novel Therapeutic Target for Neurodegenerative Diseases. Int. J. Mol. Sci. 2020;21:2108. doi: 10.3390/ijms21062108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee H.G., Won S.M., Gwag B.J., Lee Y.B. Microglial P2X(7) receptor expression is accompanied by neuronal damage in the cerebral cortex of the APPswe/PS1dE9 mouse model of Alzheimer’s disease. Exp. Mol. Med. 2011;43:7–14. doi: 10.3858/emm.2011.43.1.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rinne J.O., Nagren K. Positron emission tomography in at risk patients and in the progression of mild cognitive impairment to Alzheimer’s disease. J. Alzheimer’s Dis. 2010;19:291–300. doi: 10.3233/JAD-2010-1224. [DOI] [PubMed] [Google Scholar]
- 12.Sita G., Graziosi A., Hrelia P., Morroni F. NLRP3 and Infections: β-Amyloid in Inflammasome beyond Neurodegeneration. Int. J. Mol. Sci. 2021;22:6984. doi: 10.3390/ijms22136984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Milner M.T., Maddugoda M., Gotz J., Burgener S.S., Schroder K. The NLRP3 inflammasome triggers sterile neuroinflammation and Alzheimer’s disease. Curr. Opin. Immunol. 2021;68:116–124. doi: 10.1016/j.coi.2020.10.011. [DOI] [PubMed] [Google Scholar]
- 14.Kelley N., Jeltema D., Duan Y., He Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019;20:3328. doi: 10.3390/ijms20133328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang Y., Zhao Y., Zhang J., Yang G. Mechanisms of NLRP3 Inflammasome Activation: Its Role in the Treatment of Alzheimer’s Disease. Neurochem. Res. 2020;45:2560–2572. doi: 10.1007/s11064-020-03121-z. [DOI] [PubMed] [Google Scholar]
- 16.Freeman L.C., Ting J.P. The pathogenic role of the inflammasome in neurodegenerative diseases. J. Neurochem. 2016;136:29–38. doi: 10.1111/jnc.13217. [DOI] [PubMed] [Google Scholar]
- 17.Feng Y.S., Tan Z.X., Wu L.Y., Dong F., Zhang F. The involvement of NLRP3 inflammasome in the treatment of Alzheimer’s disease. Ageing Res. Rev. 2020;64:101192. doi: 10.1016/j.arr.2020.101192. [DOI] [PubMed] [Google Scholar]
- 18.Fulp J., He L., Toldo S., Jiang Y., Boice A., Guo C., Li X., Rolfe A., Sun D., Abbate A., et al. Structural Insights of Benzenesulfonamide Analogues as NLRP3 Inflammasome Inhibitors: Design, Synthesis and Biological Characterization. J. Med. Chem. 2018;61:5412–5423. doi: 10.1021/acs.jmedchem.8b00733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Heneka M.T., Kummer M.P., Stutz A., Delekate A., Schwartz S., Vieira-Saecker A., Griep A., Axt D., Remus A., Tzeng T.C., et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature. 2013;493:674–678. doi: 10.1038/nature11729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Canter R.G., Penney J., Tsai L.H. The road to restoring neural circuits for the treatment of Alzheimer’s disease. Nature. 2016;539:187–196. doi: 10.1038/nature20412. [DOI] [PubMed] [Google Scholar]
- 21.Heneka M.T., Carson M.J., El Khoury J., Landreth G.E., Brosseron F., Feinstein D.L., Jacobs A.H., Wyss-Coray T., Vitorica J., Ransohoff R.M., et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015;14:388–405. doi: 10.1016/S1474-4422(15)70016-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Martinon F., Burns K., Tschopp J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell. 2002;10:417–426. doi: 10.1016/S1097-2765(02)00599-3. [DOI] [PubMed] [Google Scholar]
- 23.Agostini L., Martinon F., Burns K., McDermott M.F., Hawkins P.N., Tschopp J. NALP3 forms an IL-1β-processing inflammasome with increased activity in Muckle-Wells autoinflammatory disorder. Immunity. 2004;20:319–325. doi: 10.1016/S1074-7613(04)00046-9. [DOI] [PubMed] [Google Scholar]
- 24.Henao-Mejia J., Elinav E., Thaiss C.A., Flavell R.A. Inflammasomes and metabolic disease. Annu. Rev. Physiol. 2014;76:57–78. doi: 10.1146/annurev-physiol-021113-170324. [DOI] [PubMed] [Google Scholar]
- 25.Jha S., Ting J.P. Inflammasome-associated nucleotide-binding domain, leucine-rich repeat proteins and inflammatory diseases. J. Immunol. 2009;183:7623–7629. doi: 10.4049/jimmunol.0902425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Duncan J.A., Ting J.P. Rebuilding host-pathogen interaction from the ground up: In vitro reconstitution of the inflammasome. Cell Host Microbe. 2007;1:7–9. doi: 10.1016/j.chom.2007.02.007. [DOI] [PubMed] [Google Scholar]
- 27.Meng G., Grabiec A., Vallon M., Ebe B., Hampel S., Bessler W., Wagner H., Kirschning C.J. Cellular recognition of tri-/di-palmitoylated peptides is independent from a domain encompassing the N-terminal seven leucine-rich repeat (LRR)/LRR-like motifs of TLR2. J. Biol. Chem. 2003;278:39822–39829. doi: 10.1074/jbc.M304766200. [DOI] [PubMed] [Google Scholar]
- 28.O’Connor W., Jr., Harton J.A., Zhu X., Linhoff M.W., Ting J.P. Cutting edge: CIAS1/cryopyrin/PYPAF1/NALP3/CATERPILLER 1.1 is an inducible inflammatory mediator with NF-kappa B suppressive properties. J. Immunol. 2003;171:6329–6333. doi: 10.4049/jimmunol.171.12.6329. [DOI] [PubMed] [Google Scholar]
- 29.Hoffman H.M., Scott P., Mueller J.L., Misaghi A., Stevens S., Yancopoulos G.D., Murphy A., Valenzuela D.M., Liu-Bryan R. Role of the leucine-rich repeat domain of cryopyrin/NALP3 in monosodium urate crystal-induced inflammation in mice. Arthritis Rheum. 2010;62:2170–2179. doi: 10.1002/art.27456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dowds T.A., Masumoto J., Zhu L., Inohara N., Nunez G. Cryopyrin-induced interleukin 1beta secretion in monocytic cells: Enhanced activity of disease-associated mutants and requirement for ASC. J. Biol. Chem. 2004;279:21924–21928. doi: 10.1074/jbc.M401178200. [DOI] [PubMed] [Google Scholar]
- 31.Schroder K., Zhou R., Tschopp J. The NLRP3 inflammasome: A sensor for metabolic danger? Science. 2010;327:296–300. doi: 10.1126/science.1184003. [DOI] [PubMed] [Google Scholar]
- 32.Kesavardhana S., Kanneganti T.D. Mechanisms governing inflammasome activation, assembly and pyroptosis induction. Int. Immunol. 2017;29:201–210. doi: 10.1093/intimm/dxx018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Aglietti R.A., Estevez A., Gupta A., Ramirez M.G., Liu P.S., Kayagaki N., Ciferri C., Dixit V.M., Dueber E.C. GsdmD p30 elicited by caspase-11 during pyroptosis forms pores in membranes. Proc. Natl. Acad. Sci. USA. 2016;113:7858–7863. doi: 10.1073/pnas.1607769113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Song L., Pei L., Yao S., Wu Y., Shang Y. NLRP3 Inflammasome in Neurological Diseases, from Functions to Therapies. Front. Cell. Neurosci. 2017;11:63. doi: 10.3389/fncel.2017.00063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bauernfeind F.G., Horvath G., Stutz A., Alnemri E.S., MacDonald K., Speert D., Fernandes-Alnemri T., Wu J., Monks B.G., Fitzgerald K.A., et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J. Immunol. 2009;183:787–791. doi: 10.4049/jimmunol.0901363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Groslambert M., Py B.F. Spotlight on the NLRP3 inflammasome pathway. J. Inflamm. Res. 2018;11:359–374. doi: 10.2147/JIR.S141220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gurung P., Anand P.K., Malireddi R.K., Vande Walle L., Van Opdenbosch N., Dillon C.P., Weinlich R., Green D.R., Lamkanfi M., Kanneganti T.D. FADD and caspase-8 mediate priming and activation of the canonical and noncanonical Nlrp3 inflammasomes. J. Immunol. 2014;192:1835–1846. doi: 10.4049/jimmunol.1302839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee J.H., Kim H.J., Kim J.U., Yook T.H., Kim K.H., Lee J.Y., Yang G. A Novel Treatment Strategy by Natural Products in NLRP3 Inflammasome-Mediated Neuroinflammation in Alzheimer’s and Parkinson’s Disease. Int. J. Mol. Sci. 2021;22:1324. doi: 10.3390/ijms22031324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hornung V., Bauernfeind F., Halle A., Samstad E.O., Kono H., Rock K.L., Fitzgerald K.A., Latz E. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat. Immunol. 2008;9:847–856. doi: 10.1038/ni.1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Halle A., Hornung V., Petzold G.C., Stewart C.R., Monks B.G., Reinheckel T., Fitzgerald K.A., Latz E., Moore K.J., Golenbock D.T. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat. Immunol. 2008;9:857–865. doi: 10.1038/ni.1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mariathasan S., Weiss D.S., Newton K., McBride J., O’Rourke K., Roose-Girma M., Lee W.P., Weinrauch Y., Monack D.M., Dixit V.M. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature. 2006;440:228–232. doi: 10.1038/nature04515. [DOI] [PubMed] [Google Scholar]
- 42.Martinon F., Petrilli V., Mayor A., Tardivel A., Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440:237–241. doi: 10.1038/nature04516. [DOI] [PubMed] [Google Scholar]
- 43.Demento S.L., Eisenbarth S.C., Foellmer H.G., Platt C., Caplan M.J., Mark Saltzman W., Mellman I., Ledizet M., Fikrig E., Flavell R.A., et al. Inflammasome-activating nanoparticles as modular systems for optimizing vaccine efficacy. Vaccine. 2009;27:3013–3021. doi: 10.1016/j.vaccine.2009.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Duncan J.A., Gao X., Huang M.T., O’Connor B.P., Thomas C.E., Willingham S.B., Bergstralh D.T., Jarvis G.A., Sparling P.F., Ting J.P. Neisseria gonorrhoeae activates the proteinase cathepsin B to mediate the signaling activities of the NLRP3 and ASC-containing inflammasome. J. Immunol. 2009;182:6460–6469. doi: 10.4049/jimmunol.0802696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shi F., Yang L., Kouadir M., Yang Y., Wang J., Zhou X., Yin X., Zhao D. The NALP3 inflammasome is involved in neurotoxic prion peptide-induced microglial activation. J. Neuroinflamm. 2012;9:73. doi: 10.1186/1742-2094-9-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sharp F.A., Ruane D., Claass B., Creagh E., Harris J., Malyala P., Singh M., O’Hagan D.T., Petrilli V., Tschopp J., et al. Uptake of particulate vaccine adjuvants by dendritic cells activates the NALP3 inflammasome. Proc. Natl. Acad. Sci. USA. 2009;106:870–875. doi: 10.1073/pnas.0804897106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bruchard M., Mignot G., Derangere V., Chalmin F., Chevriaux A., Vegran F., Boireau W., Simon B., Ryffel B., Connat J.L., et al. Chemotherapy-triggered cathepsin B release in myeloid-derived suppressor cells activates the Nlrp3 inflammasome and promotes tumor growth. Nat. Med. 2013;19:57–64. doi: 10.1038/nm.2999. [DOI] [PubMed] [Google Scholar]
- 48.Lee G.S., Subramanian N., Kim A.I., Aksentijevich I., Goldbach-Mansky R., Sacks D.B., Germain R.N., Kastner D.L., Chae J.J. The calcium-sensing receptor regulates the NLRP3 inflammasome through Ca2+ and cAMP. Nature. 2012;492:123–127. doi: 10.1038/nature11588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Munoz-Planillo R., Kuffa P., Martinez-Colon G., Smith B.L., Rajendiran T.M., Nunez G. K+ efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity. 2013;38:1142–1153. doi: 10.1016/j.immuni.2013.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhou R., Yazdi A.S., Menu P., Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469:221–225. doi: 10.1038/nature09663. [DOI] [PubMed] [Google Scholar]
- 51.Zhou R., Tardivel A., Thorens B., Choi I., Tschopp J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 2010;11:136–140. doi: 10.1038/ni.1831. [DOI] [PubMed] [Google Scholar]
- 52.Korhonen E., Piippo N., Hytti M., Hyttinen J.M.T., Kaarniranta K., Kauppinen A. Only IL-1β release is inflammasome-dependent upon ultraviolet B irradiation although IL-18 is also secreted. FASEB J. 2020;34:6437–6448. doi: 10.1096/fj.201902355RR. [DOI] [PubMed] [Google Scholar]
- 53.Juliana C., Fernandes-Alnemri T., Kang S., Farias A., Qin F., Alnemri E.S. Non-transcriptional priming and deubiquitination regulate NLRP3 inflammasome activation. J. Biol. Chem. 2012;287:36617–36622. doi: 10.1074/jbc.M112.407130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Seok J.K., Kang H.C., Cho Y.Y., Lee H.S., Lee J.Y. Regulation of the NLRP3 Inflammasome by Post-Translational Modifications and Small Molecules. Front. Immunol. 2020;11:618231. doi: 10.3389/fimmu.2020.618231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yang J., Liu Z., Xiao T.S. Post-translational regulation of inflammasomes. Cell. Mol. Immunol. 2017;14:65–79. doi: 10.1038/cmi.2016.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Stutz A., Kolbe C.C., Stahl R., Horvath G.L., Franklin B.S., van Ray O., Brinkschulte R., Geyer M., Meissner F., Latz E. NLRP3 inflammasome assembly is regulated by phosphorylation of the pyrin domain. J. Exp. Med. 2017;214:1725–1736. doi: 10.1084/jem.20160933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Duong B.H., Onizawa M., Oses-Prieto J.A., Advincula R., Burlingame A., Malynn B.A., Ma A. A20 restricts ubiquitination of pro-interleukin-1beta protein complexes and suppresses NLRP3 inflammasome activity. Immunity. 2015;42:55–67. doi: 10.1016/j.immuni.2014.12.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gritsenko A., Yu S., Martin-Sanchez F., Diaz-Del-Olmo I., Nichols E.M., Davis D.M., Brough D., Lopez-Castejon G. Priming Is Dispensable for NLRP3 Inflammasome Activation in Human Monocytes In Vitro. Front. Immunol. 2020;11:565924. doi: 10.3389/fimmu.2020.565924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bai H., Zhang Q. Activation of NLRP3 Inflammasome and Onset of Alzheimer’s Disease. Front. Immunol. 2021;12:701282. doi: 10.3389/fimmu.2021.701282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Apelt J., Schliebs R. Beta-amyloid-induced glial expression of both pro- and anti-inflammatory cytokines in cerebral cortex of aged transgenic Tg2576 mice with Alzheimer plaque pathology. Brain Res. 2001;894:21–30. doi: 10.1016/S0006-8993(00)03176-0. [DOI] [PubMed] [Google Scholar]
- 61.Saresella M., La Rosa F., Piancone F., Zoppis M., Marventano I., Calabrese E., Rainone V., Nemni R., Mancuso R., Clerici M. The NLRP3 and NLRP1 inflammasomes are activated in Alzheimer’s disease. Mol. Neurodegener. 2016;11:23. doi: 10.1186/s13024-016-0088-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tang H., Harte M. Investigating Markers of the NLRP3 Inflammasome Pathway in Alzheimer’s Disease: A Human Post-Mortem Study. Genes. 2021;12:1753. doi: 10.3390/genes12111753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hanslik K.L., Ulland T.K. The Role of Microglia and the Nlrp3 Inflammasome in Alzheimer’s Disease. Front. Neurol. 2020;11:570711. doi: 10.3389/fneur.2020.570711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Venegas C., Heneka M.T. Inflammasome-mediated innate immunity in Alzheimer’s disease. FASEB J. 2019;33:13075–13084. doi: 10.1096/fj.201900439. [DOI] [PubMed] [Google Scholar]
- 65.Ising C., Venegas C., Zhang S., Scheiblich H., Schmidt S.V., Vieira-Saecker A., Schwartz S., Albasset S., McManus R.M., Tejera D., et al. NLRP3 inflammasome activation drives tau pathology. Nature. 2019;575:669–673. doi: 10.1038/s41586-019-1769-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Stancu I.C., Cremers N., Vanrusselt H., Couturier J., Vanoosthuyse A., Kessels S., Lodder C., Brone B., Huaux F., Octave J.N., et al. Aggregated Tau activates NLRP3-ASC inflammasome exacerbating exogenously seeded and non-exogenously seeded Tau pathology in vivo. Acta Neuropathol. 2019;137:599–617. doi: 10.1007/s00401-018-01957-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yang J., Wise L., Fukuchi K.I. TLR4 Cross-Talk With NLRP3 Inflammasome and Complement Signaling Pathways in Alzheimer’s Disease. Front. Immunol. 2020;11:724. doi: 10.3389/fimmu.2020.00724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Huang N.Q., Jin H., Zhou S.Y., Shi J.S., Jin F. TLR4 is a link between diabetes and Alzheimer’s disease. Behav. Brain Res. 2017;316:234–244. doi: 10.1016/j.bbr.2016.08.047. [DOI] [PubMed] [Google Scholar]
- 69.Leng F., Edison P. Neuroinflammation and microglial activation in Alzheimer disease: Where do we go from here? Nat. Rev. Neurol. 2021;17:157–172. doi: 10.1038/s41582-020-00435-y. [DOI] [PubMed] [Google Scholar]
- 70.Campden R.I., Zhang Y. The role of lysosomal cysteine cathepsins in NLRP3 inflammasome activation. Arch. Biochem. Biophys. 2019;670:32–42. doi: 10.1016/j.abb.2019.02.015. [DOI] [PubMed] [Google Scholar]
- 71.Swardfager W., Lanctot K., Rothenburg L., Wong A., Cappell J., Herrmann N. A meta-analysis of cytokines in Alzheimer’s disease. Biol. Psychiatry. 2010;68:930–941. doi: 10.1016/j.biopsych.2010.06.012. [DOI] [PubMed] [Google Scholar]
- 72.Brosseron F., Krauthausen M., Kummer M., Heneka M.T. Body fluid cytokine levels in mild cognitive impairment and Alzheimer’s disease: A comparative overview. Mol. Neurobiol. 2014;50:534–544. doi: 10.1007/s12035-014-8657-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sudduth T.L., Schmitt F.A., Nelson P.T., Wilcock D.M. Neuroinflammatory phenotype in early Alzheimer’s disease. Neurobiol. Aging. 2013;34:1051–1059. doi: 10.1016/j.neurobiolaging.2012.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lin T.M., Chen W.S., Sheu J.J., Chen Y.H., Chen J.H., Chang C.C. Autoimmune rheumatic diseases increase dementia risk in middle-aged patients: A nationwide cohort study. PLoS ONE. 2018;13:e0186475. doi: 10.1371/journal.pone.0186475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ramos-Cejudo J., Wisniewski T., Marmar C., Zetterberg H., Blennow K., de Leon M.J., Fossati S. Traumatic Brain Injury and Alzheimer’s Disease: The Cerebrovascular Link. EBioMedicine. 2018;28:21–30. doi: 10.1016/j.ebiom.2018.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.De Nazareth A.M. Type 2 diabetes mellitus in the pathophysiology of Alzheimer’s disease. Dement. Neuropsychol. 2017;11:105–113. doi: 10.1590/1980-57642016dn11-020002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Whitmer R.A., Gunderson E.P., Barrett-Connor E., Quesenberry C.P., Jr., Yaffe K. Obesity in middle age and future risk of dementia: A 27 year longitudinal population based study. BMJ. 2005;330:1360. doi: 10.1136/bmj.38446.466238.E0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Love S., Miners J.S. Cerebrovascular disease in ageing and Alzheimer’s disease. Acta Neuropathol. 2016;131:645–658. doi: 10.1007/s00401-015-1522-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Reitz C. Dyslipidemia and the risk of Alzheimer’s disease. Curr. Atheroscler. Rep. 2013;15:307. doi: 10.1007/s11883-012-0307-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Eglit G.M.L., Weigand A.J., Nation D.A., Bondi M.W., Bangen K.J. Hypertension and Alzheimer’s disease: Indirect effects through circle of Willis atherosclerosis. Brain Commun. 2020;2:fcaa114. doi: 10.1093/braincomms/fcaa114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lennon M.J., Makkar S.R., Crawford J.D., Sachdev P.S. Midlife Hypertension and Alzheimer’s Disease: A Systematic Review and Meta-Analysis. J. Alzheimer’s Dis. 2019;71:307–316. doi: 10.3233/JAD-190474. [DOI] [PubMed] [Google Scholar]
- 82.Sadighi Akha A.A. Aging and the immune system: An overview. J. Immunol. Methods. 2018;463:21–26. doi: 10.1016/j.jim.2018.08.005. [DOI] [PubMed] [Google Scholar]
- 83.Liu R.M. Aging, Cellular Senescence, and Alzheimer’s Disease. Int. J. Mol. Sci. 2022;23:1989. doi: 10.3390/ijms23041989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Cho M.H., Cho K., Kang H.J., Jeon E.Y., Kim H.S., Kwon H.J., Kim H.M., Kim D.H., Yoon S.Y. Autophagy in microglia degrades extracellular beta-amyloid fibrils and regulates the NLRP3 inflammasome. Autophagy. 2014;10:1761–1775. doi: 10.4161/auto.29647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lu M., Sun X.L., Qiao C., Liu Y., Ding J.H., Hu G. Uncoupling protein 2 deficiency aggravates astrocytic endoplasmic reticulum stress and nod-like receptor protein 3 inflammasome activation. Neurobiol. Aging. 2014;35:421–430. doi: 10.1016/j.neurobiolaging.2013.08.015. [DOI] [PubMed] [Google Scholar]
- 86.Zendedel A., Johann S., Mehrabi S., Joghataei M.T., Hassanzadeh G., Kipp M., Beyer C. Activation and Regulation of NLRP3 Inflammasome by Intrathecal Application of SDF-1a in a Spinal Cord Injury Model. Mol. Neurobiol. 2016;53:3063–3075. doi: 10.1007/s12035-015-9203-5. [DOI] [PubMed] [Google Scholar]
- 87.Maturana C.J., Aguirre A., Saez J.C. High glucocorticoid levels during gestation activate the inflammasome in hippocampal oligodendrocytes of the offspring. Dev. Neurobiol. 2017;77:625–642. doi: 10.1002/dneu.22409. [DOI] [PubMed] [Google Scholar]
- 88.Von Herrmann K.M., Salas L.A., Martinez E.M., Young A.L., Howard J.M., Feldman M.S., Christensen B.C., Wilkins O.M., Lee S.L., Hickey W.F., et al. NLRP3 expression in mesencephalic neurons and characterization of a rare NLRP3 polymorphism associated with decreased risk of Parkinson’s disease. NPJ Parkinson’s Dis. 2018;4:24. doi: 10.1038/s41531-018-0061-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Schaafsma W., Zhang X., van Zomeren K.C., Jacobs S., Georgieva P.B., Wolf S.A., Kettenmann H., Janova H., Saiepour N., Hanisch U.K., et al. Long-lasting pro-inflammatory suppression of microglia by LPS-preconditioning is mediated by RelB-dependent epigenetic silencing. Brain Behav. Immun. 2015;48:205–221. doi: 10.1016/j.bbi.2015.03.013. [DOI] [PubMed] [Google Scholar]
- 90.Wendeln A.C., Degenhardt K., Kaurani L., Gertig M., Ulas T., Jain G., Wagner J., Hasler L.M., Wild K., Skodras A., et al. Innate immune memory in the brain shapes neurological disease hallmarks. Nature. 2018;556:332–338. doi: 10.1038/s41586-018-0023-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Neher J.J., Cunningham C. Priming Microglia for Innate Immune Memory in the Brain. Trends Immunol. 2019;40:358–374. doi: 10.1016/j.it.2019.02.001. [DOI] [PubMed] [Google Scholar]
- 92.Jeon S.J., Rhee S.Y., Ryu J.H., Cheong J.H., Kwon K., Yang S.I., Park S.H., Lee J., Kim H.Y., Han S.H., et al. Activation of adenosine A2A receptor up-regulates BDNF expression in rat primary cortical neurons. Neurochem. Res. 2011;36:2259–2269. doi: 10.1007/s11064-011-0550-y. [DOI] [PubMed] [Google Scholar]
- 93.Hanisch U.K., Kettenmann H. Microglia: Active sensor and versatile effector cells in the normal and pathologic brain. Nat. Neurosci. 2007;10:1387–1394. doi: 10.1038/nn1997. [DOI] [PubMed] [Google Scholar]
- 94.Jin J., Guo J., Cai H., Zhao C., Wang H., Liu Z., Ge Z.M. M2-Like Microglia Polarization Attenuates Neuropathic Pain Associated with Alzheimer’s Disease. J. Alzheimer’s Dis. 2020;76:1255–1265. doi: 10.3233/JAD-200099. [DOI] [PubMed] [Google Scholar]
- 95.Lajqi T., Stojiljkovic M., Williams D.L., Hudalla H., Bauer M., Witte O.W., Wetzker R., Bauer R., Schmeer C. Memory-Like Responses of Brain Microglia Are Controlled by Developmental State and Pathogen Dose. Front. Immunol. 2020;11:546415. doi: 10.3389/fimmu.2020.546415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Colonna M., Butovsky O. Microglia Function in the Central Nervous System During Health and Neurodegeneration. Annu. Rev. Immunol. 2017;35:441–468. doi: 10.1146/annurev-immunol-051116-052358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Tang Y., Le W. Differential Roles of M1 and M2 Microglia in Neurodegenerative Diseases. Mol. Neurobiol. 2016;53:1181–1194. doi: 10.1007/s12035-014-9070-5. [DOI] [PubMed] [Google Scholar]
- 98.Kamboh M.I. A Brief Synopsis on the Genetics of Alzheimer’s Disease. Curr. Genet. Med. Rep. 2018;6:133–135. doi: 10.1007/s40142-018-0155-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Xue F., Du H. TREM2 Mediates Microglial Anti-Inflammatory Activations in Alzheimer’s Disease: Lessons Learned from Transcriptomics. Cells. 2021;10:321. doi: 10.3390/cells10020321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sheng L., Chen M., Cai K., Song Y., Yu D., Zhang H., Xu G. Microglial Trem2 induces synaptic impairment at early stage and prevents amyloidosis at late stage in APP/PS1 mice. FASEB J. 2019;33:10425–10442. doi: 10.1096/fj.201900527R. [DOI] [PubMed] [Google Scholar]
- 101.Zheng H., Cheng B., Li Y., Li X., Chen X., Zhang Y.W. TREM2 in Alzheimer’s Disease: Microglial Survival and Energy Metabolism. Front. Aging Neurosci. 2018;10:395. doi: 10.3389/fnagi.2018.00395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hampel H., Caraci F., Cuello A.C., Caruso G., Nistico R., Corbo M., Baldacci F., Toschi N., Garaci F., Chiesa P.A., et al. A Path Toward Precision Medicine for Neuroinflammatory Mechanisms in Alzheimer’s Disease. Front. Immunol. 2020;11:456. doi: 10.3389/fimmu.2020.00456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Fan Z., Brooks D.J., Okello A., Edison P. An early and late peak in microglial activation in Alzheimer’s disease trajectory. Brain. 2017;140:792–803. doi: 10.1093/brain/aww349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Corcoran S.E., Halai R., Cooper M.A. Pharmacological Inhibition of the Nod-Like Receptor Family Pyrin Domain Containing 3 Inflammasome with MCC950. Pharmacol. Rev. 2021;73:968–1000. doi: 10.1124/pharmrev.120.000171. [DOI] [PubMed] [Google Scholar]
- 105.Ismael S., Zhao L., Nasoohi S., Ishrat T. Inhibition of the NLRP3-inflammasome as a potential approach for neuroprotection after stroke. Sci. Rep. 2018;8:5971. doi: 10.1038/s41598-018-24350-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Jiao J., Zhao G., Wang Y., Ren P., Wu M. MCC950, a Selective Inhibitor of NLRP3 Inflammasome, Reduces the Inflammatory Response and Improves Neurological Outcomes in Mice Model of Spinal Cord Injury. Front. Mol. Biosci. 2020;7:37. doi: 10.3389/fmolb.2020.00037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Coll R.C., Hill J.R., Day C.J., Zamoshnikova A., Boucher D., Massey N.L., Chitty J.L., Fraser J.A., Jennings M.P., Robertson A.A.B., et al. MCC950 directly targets the NLRP3 ATP-hydrolysis motif for inflammasome inhibition. Nat. Chem. Biol. 2019;15:556–559. doi: 10.1038/s41589-019-0277-7. [DOI] [PubMed] [Google Scholar]
- 108.Tapia-Abellan A., Angosto-Bazarra D., Martinez-Banaclocha H., de Torre-Minguela C., Ceron-Carrasco J.P., Perez-Sanchez H., Arostegui J.I., Pelegrin P. MCC950 closes the active conformation of NLRP3 to an inactive state. Nat. Chem. Biol. 2019;15:560–564. doi: 10.1038/s41589-019-0278-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Vande Walle L., Stowe I.B., Sacha P., Lee B.L., Demon D., Fossoul A., Van Hauwermeiren F., Saavedra P.H.V., Simon P., Subrt V., et al. MCC950/CRID3 potently targets the NACHT domain of wild-type NLRP3 but not disease-associated mutants for inflammasome inhibition. PLoS Biol. 2019;17:e3000354. doi: 10.1371/journal.pbio.3000354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Dempsey C., Rubio Araiz A., Bryson K.J., Finucane O., Larkin C., Mills E.L., Robertson A.A.B., Cooper M.A., O’Neill L.A.J., Lynch M.A. Inhibiting the NLRP3 inflammasome with MCC950 promotes non-phlogistic clearance of amyloid-beta and cognitive function in APP/PS1 mice. Brain Behav. Immun. 2017;61:306–316. doi: 10.1016/j.bbi.2016.12.014. [DOI] [PubMed] [Google Scholar]
- 111.He X.F., Xu J.H., Li G., Li M.Y., Li L.L., Pei Z., Zhang L.Y., Hu X.Q. NLRP3-dependent microglial training impaired the clearance of amyloid-beta and aggravated the cognitive decline in Alzheimer’s disease. Cell Death Dis. 2020;11:849. doi: 10.1038/s41419-020-03072-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Li J., Zhuang L., Luo X., Liang J., Sun E., He Y. Protection of MCC950 against Alzheimer’s disease via inhibiting neuronal pyroptosis in SAMP8 mice. Exp. Brain Res. 2020;238:2603–2614. doi: 10.1007/s00221-020-05916-6. [DOI] [PubMed] [Google Scholar]
- 113.El-Sharkawy L.Y., Brough D., Freeman S. Inhibiting the NLRP3 Inflammasome. Molecules. 2020;25:5533. doi: 10.3390/molecules25235533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Onyango I.G., Jauregui G.V., Carna M., Bennett J.P., Jr., Stokin G.B. Neuroinflammation in Alzheimer’s Disease. Biomedicines. 2021;9:524. doi: 10.3390/biomedicines9050524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Wang Z., Zhang S., Xiao Y., Zhang W., Wu S., Qin T., Yue Y., Qian W., Li L. NLRP3 Inflammasome and Inflammatory Diseases. Oxid. Med. Cell. Longev. 2020;2020:4063562. doi: 10.1155/2020/4063562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Kuwar R., Rolfe A., Di L., Xu H., He L., Jiang Y., Zhang S., Sun D. A novel small molecular NLRP3 inflammasome inhibitor alleviates neuroinflammatory response following traumatic brain injury. J. Neuroinflammation. 2019;16:81. doi: 10.1186/s12974-019-1471-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kuwar R., Rolfe A., Di L., Blevins H., Xu Y., Sun X., Bloom G.S., Zhang S., Sun D. A Novel Inhibitor Targeting NLRP3 Inflammasome Reduces Neuropathology and Improves Cognitive Function in Alzheimer’s Disease Transgenic Mice. J. Alzheimer’s Dis. 2021;82:1769–1783. doi: 10.3233/JAD-210400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Yin J., Zhao F., Chojnacki J.E., Fulp J., Klein W.L., Zhang S., Zhu X. NLRP3 Inflammasome Inhibitor Ameliorates Amyloid Pathology in a Mouse Model of Alzheimer’s Disease. Mol. Neurobiol. 2018;55:1977–1987. doi: 10.1007/s12035-017-0467-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Flores J., Noel A., Foveau B., Lynham J., Lecrux C., LeBlanc A.C. Caspase-1 inhibition alleviates cognitive impairment and neuropathology in an Alzheimer’s disease mouse model. Nat. Commun. 2018;9:3916. doi: 10.1038/s41467-018-06449-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Flores J., Noel A., Foveau B., Beauchet O., LeBlanc A.C. Pre-symptomatic Caspase-1 inhibitor delays cognitive decline in a mouse model of Alzheimer disease and aging. Nat. Commun. 2020;11:4571. doi: 10.1038/s41467-020-18405-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Chen J., Chen Y.Q., Shi Y.J., Ding S.Q., Shen L., Wang R., Wang Q.Y., Zha C., Ding H., Hu J.G., et al. VX-765 reduces neuroinflammation after spinal cord injury in mice. Neural Regen. Res. 2021;16:1836–1847. doi: 10.4103/1673-5374.306096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Li J., Hao J.H., Yao D., Li R., Li X.F., Yu Z.Y., Luo X., Liu X.H., Wang M.H., Wang W. Caspase-1 inhibition prevents neuronal death by targeting the canonical inflammasome pathway of pyroptosis in a murine model of cerebral ischemia. CNS Neurosci. Ther. 2020;26:925–939. doi: 10.1111/cns.13384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Dong Y., Li X., Cheng J., Hou L. Drug Development for Alzheimer’s Disease: Microglia Induced Neuroinflammation as a Target? Int. J. Mol. Sci. 2019;20:558. doi: 10.3390/ijms20030558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Noe F.M., Polascheck N., Frigerio F., Bankstahl M., Ravizza T., Marchini S., Beltrame L., Bandero C.R., Loscher W., Vezzani A. Pharmacological blockade of IL-1β/IL-1 receptor type 1 axis during epileptogenesis provides neuroprotection in two rat models of temporal lobe epilepsy. Neurobiol. Dis. 2013;59:183–193. doi: 10.1016/j.nbd.2013.07.015. [DOI] [PubMed] [Google Scholar]
- 125.Marchetti C., Swartzwelter B., Gamboni F., Neff C.P., Richter K., Azam T., Carta S., Tengesdal I., Nemkov T., D’Alessandro A., et al. OLT1177, a β-sulfonyl nitrile compound, safe in humans, inhibits the NLRP3 inflammasome and reverses the metabolic cost of inflammation. Proc. Natl. Acad. Sci. USA. 2018;115:E1530–E1539. doi: 10.1073/pnas.1716095115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Sanchez-Fernandez A., Skouras D.B., Dinarello C.A., Lopez-Vales R. OLT1177 (Dapansutrile), a Selective NLRP3 Inflammasome Inhibitor, Ameliorates Experimental Autoimmune Encephalomyelitis Pathogenesis. Front. Immunol. 2019;10:2578. doi: 10.3389/fimmu.2019.02578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Lonnemann N., Hosseini S., Marchetti C., Skouras D.B., Stefanoni D., D’Alessandro A., Dinarello C.A., Korte M. The NLRP3 inflammasome inhibitor OLT1177 rescues cognitive impairment in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA. 2020;117:32145–32154. doi: 10.1073/pnas.2009680117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Moore A.H., Bigbee M.J., Boynton G.E., Wakeham C.M., Rosenheim H.M., Staral C.J., Morrissey J.L., Hund A.K. Non-Steroidal Anti-Inflammatory Drugs in Alzheimer’s Disease and Parkinson’s Disease: Reconsidering the Role of Neuroinflammation. Pharmaceuticals. 2010;3:1812–1841. doi: 10.3390/ph3061812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Dill J., Patel A.R., Yang X.L., Bachoo R., Powell C.M., Li S. A molecular mechanism for ibuprofen-mediated RhoA inhibition in neurons. J. Neurosci. 2010;30:963–972. doi: 10.1523/JNEUROSCI.5045-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Zhou Y., Su Y., Li B., Liu F., Ryder J.W., Wu X., Gonzalez-DeWhitt P.A., Gelfanova V., Hale J.E., May P.C., et al. Nonsteroidal anti-inflammatory drugs can lower amyloidogenic Abeta42 by inhibiting Rho. Science. 2003;302:1215–1217. doi: 10.1126/science.1090154. [DOI] [PubMed] [Google Scholar]
- 131.Fu Q., Hue J., Li S. Nonsteroidal anti-inflammatory drugs promote axon regeneration via RhoA inhibition. J. Neurosci. 2007;27:4154–4164. doi: 10.1523/JNEUROSCI.4353-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Bindu S., Mazumder S., Bandyopadhyay U. Non-steroidal anti-inflammatory drugs (NSAIDs) and organ damage: A current perspective. Biochem. Pharmacol. 2020;180:114147. doi: 10.1016/j.bcp.2020.114147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Daniels M.J., Rivers-Auty J., Schilling T., Spencer N.G., Watremez W., Fasolino V., Booth S.J., White C.S., Baldwin A.G., Freeman S., et al. Fenamate NSAIDs inhibit the NLRP3 inflammasome and protect against Alzheimer’s disease in rodent models. Nat. Commun. 2016;7:12504. doi: 10.1038/ncomms12504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Cole G.M., Frautschy S.A. Mechanisms of action of non-steroidal anti-inflammatory drugs for the prevention of Alzheimer’s disease. CNS Neurol. Disord. Drug Targets. 2010;9:140–148. doi: 10.2174/187152710791011991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Shoaib M., Kamal M.A., Rizvi S.M.D. Repurposed Drugs as Potential Therapeutic Candidates for the Management of Alzheimer’s Disease. Curr. Drug Metab. 2017;18:842–852. doi: 10.2174/1389200218666170607101622. [DOI] [PubMed] [Google Scholar]
- 136.Imbimbo B.P. An update on the efficacy of non-steroidal anti-inflammatory drugs in Alzheimer’s disease. Expert Opin. Investig. Drugs. 2009;18:1147–1168. doi: 10.1517/13543780903066780. [DOI] [PubMed] [Google Scholar]
- 137.Benito-Leon J., Contador I., Vega S., Villarejo-Galende A., Bermejo-Pareja F. Non-steroidal anti-inflammatory drugs use in older adults decreases risk of Alzheimer’s disease mortality. PLoS ONE. 2019;14:e0222505. doi: 10.1371/journal.pone.0222505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Karceski S., Karceski S. Can naproxen slow the progression of Alzheimer disease? Neurology. 2019;92:e2181–e2184. doi: 10.1212/WNL.0000000000007418. [DOI] [PubMed] [Google Scholar]
- 139.Li X., Kaida-Yip F., Zabel M. NSAID Use and the Prevention of Alzheimer’s Disease: A Meta-Analysis (P6.184) Neurology. 2018;90:P6.184. [Google Scholar]
- 140.Meyer P.F., Tremblay-Mercier J., Leoutsakos J., Madjar C., Lafaille-Maignan M.E., Savard M., Rosa-Neto P., Poirier J., Etienne P., Breitner J., et al. INTREPAD: A randomized trial of naproxen to slow progress of presymptomatic Alzheimer disease. Neurology. 2019;92:e2070–e2080. doi: 10.1212/WNL.0000000000007232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Laine L., Wogen J., Yu H. Gastrointestinal health care resource utilization with chronic use of COX-2-specific inhibitors versus traditional NSAIDs. Gastroenterology. 2003;125:389–395. doi: 10.1016/S0016-5085(03)00900-4. [DOI] [PubMed] [Google Scholar]
- 142.Silvis M.J.M., Fiolet A.T.L., Opstal T.S.J., Dekker M., Suquilanda D., Zivkovic M., Duyvendak M., The S.H.K., Timmers L., Bax W.A., et al. Colchicine reduces extracellular vesicle NLRP3 inflammasome protein levels in chronic coronary disease: A LoDoCo2 biomarker substudy. Atherosclerosis. 2021;334:93–100. doi: 10.1016/j.atherosclerosis.2021.08.005. [DOI] [PubMed] [Google Scholar]
- 143.Martinez G.J., Celermajer D.S., Patel S. The NLRP3 inflammasome and the emerging role of colchicine to inhibit atherosclerosis-associated inflammation. Atherosclerosis. 2018;269:262–271. doi: 10.1016/j.atherosclerosis.2017.12.027. [DOI] [PubMed] [Google Scholar]
- 144.Nazem A., Sankowski R., Bacher M., Al-Abed Y. Rodent models of neuroinflammation for Alzheimer’s disease. J. Neuroinflamm. 2015;12:74. doi: 10.1186/s12974-015-0291-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Kumar A., Seghal N., Naidu P.S., Padi S.S., Goyal R. Colchicines-induced neurotoxicity as an animal model of sporadic dementia of Alzheimer’s type. Pharmacol. Rep. 2007;59:274–283. [PubMed] [Google Scholar]
- 146.Sil S., Ghosh T. Role of cox-2 mediated neuroinflammation on the neurodegeneration and cognitive impairments in colchicine induced rat model of Alzheimer’s Disease. J. Neuroimmunol. 2016;291:115–124. doi: 10.1016/j.jneuroim.2015.12.003. [DOI] [PubMed] [Google Scholar]
- 147.Sil S., Ghosh T. Cox-2 Plays a Vital Role in the Impaired Anxiety Like Behavior in Colchicine Induced Rat Model of Alzheimer Disease. Behav. Neurol. 2016;2016:1501527. doi: 10.1155/2016/1501527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Sil S., Goswami A.R., Dutta G., Ghosh T. Effects of naproxen on immune responses in a colchicine-induced rat model of Alzheimer’s disease. Neuroimmunomodulation. 2014;21:304–321. doi: 10.1159/000357735. [DOI] [PubMed] [Google Scholar]
- 149.Tilson H.A., Rogers B.C., Grimes L., Harry G.J., Peterson N.J., Hong J.S., Dyer R.S. Time-dependent neurobiological effects of colchicine administered directly into the hippocampus of rats. Brain Res. 1987;408:163–172. doi: 10.1016/0006-8993(87)90368-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.