

Abstract
Head blight or scab caused by Fusarium graminearum (FG), once ranked as a minor disease in wheat, is now emerging as one of the economically important diseases in India. The present study represents the first in-depth population genetic analysis of the FG from the northern wheat belt of India. In this study, multiple conserved gene sequences comprised of β-tubulin (TUB), translation elongation factor 1-α (TEF), and histone-3 (HIS) regions were used for multi-locus phylogenetic analysis of 123 geographically distinct F. graminearum isolates collected from four different states (Haryana (HR), Punjab (PB), Rajasthan (RJ) and West Bengal (WB)) of India. The phylogenetic and haplotype analysis showed the presence of thirty haplotypes in all the analyzed populations. The haplotypic diversity in the RJ population (Hd = 0.981) was higher than in the HR (Hd = 0.972), PB (Hd = 0.965) and WB population (Hd = 0.962). Recombination events (Rm = 12) and mutation events (485) were also detected. Analysis of molecular variance (AMOVA) indicated that genetic diversity was exclusively due to the differences within populations. The haplotype network was widely dispersed and not associated with specific populations, as a single common haplotype was not detected. The PB population contained both unique (H9, H10 and H11) and shared haplotypes (27 haplotypes) in a higher number in comparison to other geographical locations. Except for haplotype H22 (contains highly aggressive isolates), there was no specific linkage noticed between the isolate aggressiveness and haplotype. The concatenated sequences of all the three genes demonstrated a low level of genetic differentiation (Fst = −0.014 to 0.02) in the analyzed population. Positive values for the neutrality tests in PB, HR and RJ reveal a balancing selection mechanism behind the FG population structure. The WB population showed both positive and negative values of neutrality indices, indicating the role of both population expansion as well as balancing selection in structuring the FG population.
Keywords: aggressiveness, head scab multi-locus sequence typing, mutation, phylogeny, population structure, recombination
1. Introduction
Fusarium head blight (FHB) incited by Fusarium graminearum (FG) fungus is ranked as the one of the prime annihilating fungal diseases of wheat (Triticum aestivum L.) globally [1]. The published literature indicated that this disease drastically reduces the crop yield, leading to huge economic losses [2,3,4]. It has been noticed that yield losses are primarily linked with poor quality seed production. The contamination of infected seed grains with mycotoxins has been observed [5]. In India, the typical symptoms produced by the fungus appear majorly on the glumes and rachis of wheat plants in the form of water-soaked lesions. Later on, the fungus spreads within the wheat ear heads, resulting in the partial bleaching to complete blighting of attacked ear heads (Figure 1). Historically, the disease was first noticed in India in the Siang District of Arunachal Pradesh in the year 1974 [6]. Later on, the disease has been reported by other workers from Wellington [7] and Gurdaspur (Punjab, India) [8]. Bagga et al. [9] documented that the disease heavily attacked the wheat cultivar PDW 274 in Dera Baba Nanak region of Gurdaspur in Punjab district of India and resulted in noteworthy yield reduction. In addition to India, chronic appearance of the disease has been observed in different corners of the world and major regions including China, Brazil, USA, Canada, the former USSR, Eastern and Western Europe, Romania, etc. which account for more than 50% of global production [10,11]. In recent reports, it has been cautioned that Fusarium head blight is liable to enhance under reduced tillage-based wheat cultivation and further aggravated with climate shift especially in the northern part of India, which is recognized as the main wheat basket of India [4].
Figure 1.

Typical symptoms of wheat head scab disease (Fusarium graminareum) on wheat spike.
FG showed a broad host range, and it has the ability to infect different plants such as maize, sorghum, millets, rye, triticale, oats, etc. [12]. Various research evidence indicated that FHB disease is highly prone to humid to semi-humid areas of the world, especially where heavy and frequent rainfall with a high level of moisture exists in the atmosphere throughout the wheat cultivation season [13,14]. Unfortunately, such type of weather is prevalent in the northern part of India, especially during and after the anthesis stage of wheat, which directly affects the crop yield [4]. It has been observed that the fungus in the off-season survive in infected wheat straw in different grasses of the wheat family origin and in crop residues that remain in the soil after wheat harvest [15,16]. The incidence and severity of the FHB disease is determined by numerous factors such as quantity of airborne inocula (both inside and outside of the field), and the prevalence of humidity during and after the anthesis period [14,17]. Currently, fungicide (e.g., Tebuconazole, Triazoles, Prochloraz, etc.) sprays are important methods to preclude and conquer FHB disease in a short time and lessen Fusarium toxins production, despite agro-ecological and resistance development problems [2,18,19,20]. Hence, the deployment of FHB-resistant varieties is a sustainable, cost-effective, and environmentally friendly approach to FHB management. Unfortunately, the majority of the popular varieties cultivated in India are prone to FHB disease [4]. Moreover, the effectiveness of resistant varieties and implementation of different methods singly or in an integrated manner for the control of Fusarium spp. still exist as a daunting task because of the complex nature of FG and fungicide-resistant development in natural FG populations [21]. Therefore, an appraisal of genetic diversity and population structure of FG becomes mandatory to understand its evolutionary relationships with respect to environmental change, selection pressures, and other forces (e.g., mutation, genetic drift, gene flow, etc.) linked with evolutionary change [22,23]. Different research groups also advocated the essentiality of genetic structure analysis to decode the modes of recombination and distribution of isolates within and among populations [24,25]. Most importantly, FHB-resistant genotypes could be judiciously deployed if the pathogen population is known. At present, limited information is available regarding the distribution, genetic diversity and population structure of the FG population in India.
Molecular marker-driven technologies play a significant role in species identification because of their potential usage in exploring the population structure and genetic diversity within the fungal species and their isolates [26,27,28,29,30,31,32,33,34]. It is worth noting that high-throughput sequencing of amplicon markers from conserved genomic regions has provided new opportunities to decipher Fusarium diversity in agricultural crops in recent times. The molecular identification of fungi is primarily based on the internal transcribed spacer region (ITS) [35,36]. However, it has been observed that a number of species of Fusarium genus comprise orthologous regions and as a consequence, ITS region-based identification resulted in erratic and unreliable results. In this context, genomic regions representing translocation elongation factor 1-α (TEF), β-tubulin (TUB), histones (HIS) and calmodulin (CAL) have been widely used by different group of researchers to distinguish Fusarium spp. [33,37,38,39,40,41,42,43,44,45,46]. Yli-Mattila et al. [47] demonstrated the application if ITS, IGS, mtSSU, and TUB genomic region comparisons predicting the variation of Fusarium spp. Similarly, TEF-1α, β-tubulin and histone 3 regions have been explored by Webb et al. [48] and Taha et al. [49] for dissecting the genetic variability among F. oxysporum isolates. Recently, Fulton et al. [50] employed two different housekeeping genes (BT and TEF) for analyzing the phylogenetic kinship of F. oxysporum f. sp. niveum isolates from the major watermelon-producing regions in north, central, and south Florida. A flooding of reports are available that highlight the significance of TUB, TEF and HIS in resolving boundaries of fungal species and further revealed species identification and discrimination based on the combined sequences of gene loci as a more valuable genomic resource than single gene loci [51,52]. Unfortunately, not a single report regarding the dissection of genetic variation and population structure of FG in the major wheat-growing belt of North India using combined housekeeping gene sequences is available.
The history of FG in wheat in India is not too old, and therefore, the northern plains, a major wheat-growing belt of India, offers a paragon site to pinpoint the prime evolutionary mechanism acting on a newly developing FG isolate as it dispersed topically and regionally within the Indo-gangetic plains of India. Therefore, the current research was planned with an intention to obtain the answers to the question of whether FG contains distinct evolutionary lineages in the northern plains of India. The other questions include: (i) Is the phylogenetic and evolutionary structure of FG indicative of recombination or mutation?; and (ii) Is there a biogeographic relationship with the evolutionary structure of FG? The answers to these questions are essential to improve the understanding of the ecology of the plant pathogen and devise a better management strategy for the management of FHB disease in wheat.
2. Materials and Methods
2.1. Sampling and Pathogen Isolation
One hundred and twenty three isolates of FG were used in the present study (Table 1). These isolates were collected during field surveys conducted from 2017 to 2022 in different wheat-growing fields in the four different states of India. These include: Punjab (PB; N = 47), Haryana (HR; N = 27), Rajasthan (RJ; N = 28) and West Bengal (WB; N = 21) (Figure 2). Disease wheat ear heads samples were showing typical premature bleaching with orange spore masses of the fungus on the infected spikelets and glumes (Figure 1). The symptomatic samples in the form of wheat ear heads were gathered in plastic bags and were taken into the laboratory. The detailed information of samples has been provided in Table 1. The isolation of the fungi was made by adopting the following procedure. Briefly, infected wheat samples were sliced into minute pieces of 2–3 mm and later surface-sterilized with ethanol (70%) for 30 s followed by NaOCl (1%) treatment for 1 min. After this, treated samples were washed twice with sterilized double-distilled water. After air drying, the treated wheat samples were placed on the Petri plates containing potato dextrose agar (PDA; Hi-Media India) and ampicillin (0.1 g 1−1). The Petri plates were incubated at 25 ± 2 °C. After five days of cultivation, the hyphal tip of fungus coming out from wheat tissue was placed onto other PDA-amended Petri plates and incubated at 25 ± 2 °C for conidia production. A single spore isolation methodology was adopted to raise the pure cultures of each FG isolate, which were stored at 4 °C as per the protocol of Kumar et al. [53].
Table 1.
FG isolates used in the current study.
| Isolate | Region/State | Wheat Cultivar | Year | NCBI Gene Accession | Aggressiveness | ||
|---|---|---|---|---|---|---|---|
| TUB | TEF | HIS | |||||
| NFG1 | Punjab | PBW343 | 2018 | OM169181 | ON215826 | ON215856 | HA |
| NFG2 | Punjab | HD2967 | 2018 | OM169182 | ON215827 | ON215857 | MA |
| NFG3 | Punjab | HD2967 | 2018 | OM169183 | ON215828 | ON215858 | HA |
| NFG4 | Punjab | PBW 550 | 2018 | OM169184 | ON215829 | ON215859 | HA |
| NFG5 | Punjab | PBW343 | 2018 | OM169185 | ON215830 | ON215860 | LA |
| NFG6 | Punjab | PBW752 | 2018 | OM169186 | ON215831 | ON215861 | LA |
| NFG7 | Punjab | PBW502 | 2018 | OM169187 | ON215832 | ON215862 | HA |
| NFG8 | Punjab | PBW502 | 2019 | OM169188 | ON215833 | ON215863 | MA |
| NFG9 | Punjab | PBW343 | 2019 | OM169189 | ON215834 | ON215864 | MA |
| NFG10 | Punjab | DBW187 | 2019 | OM169190 | ON215835 | ON215865 | MA |
| NFG11 | Punjab | HD2967 | 2019 | OM169191 | ON215836 | ON215866 | LA |
| NFG12 | Punjab | PBW343 | 2019 | OM169192 | ON215837 | ON215867 | HA |
| NFG13 | Punjab | PBW 757 | 2019 | OM169193 | ON215838 | ON215868 | LA |
| NFG14 | Punjab | PBW502 | 2019 | OM169194 | ON215839 | ON215869 | HA |
| NFG15 | Punjab | HD2967 | 2019 | OM169195 | ON215840 | ON215870 | HA |
| NFG16 | Punjab | DBW187 | 2019 | OM169196 | ON215841 | ON215871 | MA |
| NFG17 | Punjab | DBW187 | 2019 | OM169197 | ON215842 | ON215872 | LA |
| NFG18 | Punjab | PBW 757 | 2019 | OM169198 | ON215843 | ON215873 | LA |
| NFG19 | Punjab | HD2967 | 2019 | OM169199 | ON215844 | ON215874 | MA |
| NFG20 | Punjab | DBW187 | 2019 | OM169200 | ON215845 | ON215875 | MA |
| NFG21 | Haryana | UP 2338 | 2019 | OM169201 | ON215846 | ON215876 | LA |
| NFG22 | Haryana | HD2967 | 2019 | OM169202 | ON215847 | ON215877 | HA |
| NFG23 | Haryana | DBW187 | 2019 | OM169203 | ON215848 | ON215878 | LA |
| NFG24 | Haryana | HD2967 | 2019 | OM169204 | ON215849 | ON215879 | LA |
| NFG25 | Haryana | HD2967 | 2019 | OM169205 | ON215850 | ON215880 | HA |
| NFG26 | Haryana | DBW187 | 2019 | OM169206 | ON215851 | ON215881 | LA |
| NFG27 | Haryana | DBW187 | 2019 | OM169207 | ON215852 | ON215882 | HA |
| NFG28 | Haryana | HD2967 | 2019 | OM169208 | ON215853 | ON215883 | HA |
| NFG29 | Haryana | HD2967 | 2019 | OM169209 | ON215854 | ON215884 | MA |
| NFG30 | Rajasthan | HD 2824 | 2019 | OM169210 | ON215855 | ON215885 | HA |
| NFG31 | Rajasthan | HD 2824 | 2019 | ON215733 | ON215979 | ON215886 | LA |
| NFG32 | Rajasthan | DBW187 | 2019 | ON215734 | ON215980 | ON215887 | HA |
| NFG33 | Rajasthan | HD 3118 | 2019 | ON215735 | ON215981 | ON215888 | MA |
| NFG34 | Rajasthan | PBW343 | 2019 | ON215736 | ON215982 | ON215889 | HA |
| NFG35 | Rajasthan | HD2967 | 2019 | ON215737 | ON215983 | ON215890 | HA |
| NFG36 | Rajasthan | HD2967 | 2019 | ON215738 | ON215984 | ON215891 | HA |
| NFG37 | Rajasthan | RAJ 4079 | 2020 | ON215739 | ON215985 | ON215892 | LA |
| NFG38 | Rajasthan | HD 2824 | 2020 | ON215740 | ON215986 | ON215893 | HA |
| NFG39 | Punjab | PBW502 | 2020 | ON215741 | ON215987 | ON215894 | LA |
| NFG40 | Punjab | WB 2 | 2020 | ON215742 | ON215988 | ON215895 | LA |
| NFG41 | Punjab | DBW187 | 2020 | ON215743 | ON215989 | ON215896 | HA |
| NFG42 | Punjab | WB 2 | 2020 | ON215744 | ON215990 | ON215897 | MA |
| NFG43 | Punjab | DBW187 | 2020 | ON215745 | ON215991 | ON215898 | HA |
| NFG44 | Punjab | HD2967 | 2020 | ON215746 | ON215992 | ON215899 | HA |
| NFG45 | Haryana | UP 2338 | 2021 | ON215747 | ON215993 | ON215900 | HA |
| NFG46 | Haryana | UP 2338 | 2021 | ON215748 | ON215994 | ON215901 | LA |
| NFG47 | Haryana | DBW303 | 2021 | ON215749 | ON215995 | ON215902 | HA |
| NFG48 | Haryana | DBW303 | 2021 | ON215750 | ON215996 | ON215903 | HA |
| NFG49 | Haryana | DBW187 | 2021 | ON215751 | ON215997 | ON215904 | HA |
| NFG50 | Haryana | DBW187 | 2021 | ON215752 | ON215998 | ON215905 | MA |
| NFG51 | Haryana | HD2967 | 2021 | ON215753 | ON215999 | ON215906 | MA |
| NFG52 | Haryana | DBW303 | 2021 | ON215754 | ON216000 | ON215907 | HA |
| NFG53 | Rajasthan | HD3086 | 2021 | ON215755 | ON216001 | ON215908 | LA |
| NFG54 | Rajasthan | UP 2338 | 2021 | ON215756 | ON216002 | ON215909 | LA |
| NFG55 | Rajasthan | PBW343 | 2021 | ON215757 | ON216003 | ON215910 | HA |
| NFG56 | Rajasthan | HD3086 | 2021 | ON215758 | ON216004 | ON215911 | LA |
| NFG57 | West Bengal | DBW187 | 2021 | ON215759 | ON216005 | ON215912 | HA |
| NFG58 | West Bengal | Shatabadi | 2021 | ON215760 | ON216006 | ON215913 | LA |
| NFG59 | West Bengal | HD2967 | 2021 | ON215761 | ON216007 | ON215914 | HA |
| NFG60 | West Bengal | Shatabadi | 2021 | ON215762 | ON216008 | ON215915 | LA |
| NFG61 | West Bengal | DBW187 | 2021 | ON215763 | ON216009 | ON215916 | HA |
| NFG62 | West Bengal | Prodip | 2021 | ON215764 | ON216010 | ON215917 | HA |
| NFG63 | West Bengal | HD3086 | 2021 | ON215765 | ON216011 | ON215918 | HA |
| NFG64 | West Bengal | Prodip | 2021 | ON215766 | ON216012 | ON215919 | LA |
| NFG65 | Punjab | DBW303 | 2021 | ON215767 | ON216013 | ON215920 | LA |
| NFG66 | Punjab | DBW303 | 2021 | ON215768 | ON216014 | ON215921 | LA |
| NFG67 | Punjab | PBW550 | 2021 | ON215769 | ON216015 | ON215922 | HA |
| NFG68 | Punjab | PBW502 | 2021 | ON215770 | ON216016 | ON215923 | HA |
| NFG69 | Punjab | DBW187 | 2021 | ON215771 | ON216017 | ON215924 | MA |
| NFG70 | Punjab | PBW 757 | 2021 | ON215772 | ON216018 | ON215925 | HA |
| NFG71 | Punjab | HD2967 | 2021 | ON215773 | ON216019 | ON215926 | MA |
| NFG72 | Punjab | HD2967 | 2021 | ON215774 | ON216020 | ON215927 | LA |
| NFG73 | Punjab | PBW550 | 2021 | ON215775 | ON216021 | ON215928 | HA |
| NFG74 | Punjab | DBW222 | 2021 | ON215776 | ON216022 | ON215929 | HA |
| NFG75 | Rajasthan | DBW187 | 2021 | ON215777 | ON216023 | ON215930 | MA |
| NFG76 | Rajasthan | HD3086 | 2021 | ON215778 | ON216024 | ON215931 | HA |
| NFG77 | Rajasthan | DBW187 | 2021 | ON215779 | ON216025 | ON215932 | LA |
| NFG78 | Rajasthan | PBW343 | 2021 | ON215780 | ON216026 | ON215933 | HA |
| NFG79 | Rajasthan | HD3086 | 2021 | ON215781 | ON216027 | ON215934 | MA |
| NFG80 | Rajasthan | HD2967 | 2021 | ON215782 | ON216028 | ON215935 | LA |
| NFG81 | Rajasthan | RAJ 4252 | 2021 | ON215783 | ON216029 | ON215936 | MA |
| NFG82 | Rajasthan | HD 2864 | 2021 | ON215784 | ON216030 | ON215937 | HA |
| NFG83 | Rajasthan | DBW222 | 2021 | ON215785 | ON216031 | ON215938 | MA |
| NFG84 | West Bengal | HD2733 | 2022 | ON215786 | ON216032 | ON215939 | HA |
| NFG85 | West Bengal | DBW187 | 2022 | ON215787 | ON216033 | ON215940 | LA |
| NFG86 | West Bengal | DBW 107 | 2022 | ON215788 | ON216034 | ON215941 | HA |
| NFG87 | West Bengal | DBW303 | 2022 | ON215789 | ON216035 | ON215942 | MA |
| NFG88 | West Bengal | DBW187 | 2022 | ON215790 | ON216036 | ON215943 | LA |
| NFG89 | West Bengal | DBW 107 | 2022 | ON215791 | ON216037 | ON215944 | HA |
| NFG90 | West Bengal | HD3086 | 2022 | ON215792 | ON216038 | ON215945 | LA |
| NFG91 | West Bengal | Shatabadi | 2022 | ON215793 | ON216039 | ON215946 | MA |
| NFG92 | West Bengal | HD2733 | 2022 | ON215794 | ON216040 | ON215947 | HA |
| NFG93 | West Bengal | HD2967 | 2022 | ON215795 | ON216041 | ON215948 | MA |
| NFG94 | West Bengal | DBW303 | 2022 | ON215796 | ON216042 | ON215949 | MA |
| NFG95 | West Bengal | HD2967 | 2022 | ON215797 | ON216043 | ON215950 | HA |
| NFG96 | West Bengal | DBW 107 | 2022 | ON215798 | ON216044 | ON215951 | MA |
| NFG97 | Punjab | DBW187 | 2022 | ON215799 | ON216045 | ON215952 | HA |
| NFG98 | Punjab | DBW222 | 2022 | ON215800 | ON216046 | ON215953 | MA |
| NFG99 | Punjab | HD 3226 | 2022 | ON215801 | ON216047 | ON215954 | HA |
| NFG100 | Punjab | HD3086 | 2022 | ON215802 | ON216048 | ON215955 | MA |
| NFG101 | Punjab | DBW222 | 2022 | ON215803 | ON216049 | ON215956 | LA |
| NFG102 | Haryana | HD3086 | 2022 | ON215804 | ON216050 | ON215957 | MA |
| NFG103 | Haryana | DBW303 | 2022 | ON215805 | ON216051 | ON215958 | HA |
| NFG104 | Haryana | DBW303 | 2022 | ON215806 | ON216052 | ON215959 | MA |
| NFG105 | Haryana | DBW303 | 2022 | ON215807 | ON216053 | ON215960 | LA |
| NFG106 | Rajasthan | DBW222 | 2022 | ON215808 | ON216054 | ON215961 | HA |
| NFG107 | Rajasthan | DBW222 | 2022 | ON215809 | ON216055 | ON215962 | La |
| NFG108 | Rajasthan | HD 2864 | 2022 | ON215810 | ON216056 | ON215963 | LA |
| NFG109 | Rajasthan | PBW343 | 2022 | ON215811 | ON216057 | ON215964 | HA |
| NFG110 | Rajasthan | HD 2824 | 2022 | ON215812 | ON216058 | ON215965 | LA |
| NFG111 | Rajasthan | HD 2864 | 2022 | ON215813 | ON216059 | ON215966 | LA |
| NFG112 | Haryana | DBW303 | 2022 | ON215814 | ON216060 | ON215967 | HA |
| NFG113 | Haryana | DBW303 | 2022 | ON215815 | ON216061 | ON215968 | LA |
| NFG114 | Haryana | KRL210 | 2022 | ON215816 | ON216062 | ON215969 | LA |
| NFG115 | Haryana | DBW187 | 2022 | ON215817 | ON216063 | ON215970 | LA |
| NFG116 | Haryana | DBW222 | 2022 | ON215818 | ON216064 | ON215971 | HA |
| NFG117 | Haryana | HD 3226 | 2022 | ON215819 | ON216065 | ON215972 | MA |
| NFG118 | Punjab | HD2967 | 2022 | ON215820 | ON216066 | ON215973 | LA |
| NFG119 | Punjab | HD3086 | 2022 | ON215821 | ON216067 | ON215974 | LA |
| NFG120 | Punjab | DBW222 | 2022 | ON215822 | ON216068 | ON215975 | HA |
| NFG121 | Punjab | DBW222 | 2022 | ON215823 | ON216069 | ON215976 | MA |
| NFG122 | Punjab | HD 3226 | 2022 | ON215824 | ON216070 | ON215977 | LA |
| NFG123 | Punjab | DBW303 | 2022 | ON215825 | ON216071 | ON215978 | LA |
HA: Highly aggressive; MA: Moderately aggressive; LA: Least aggressive.
Figure 2.
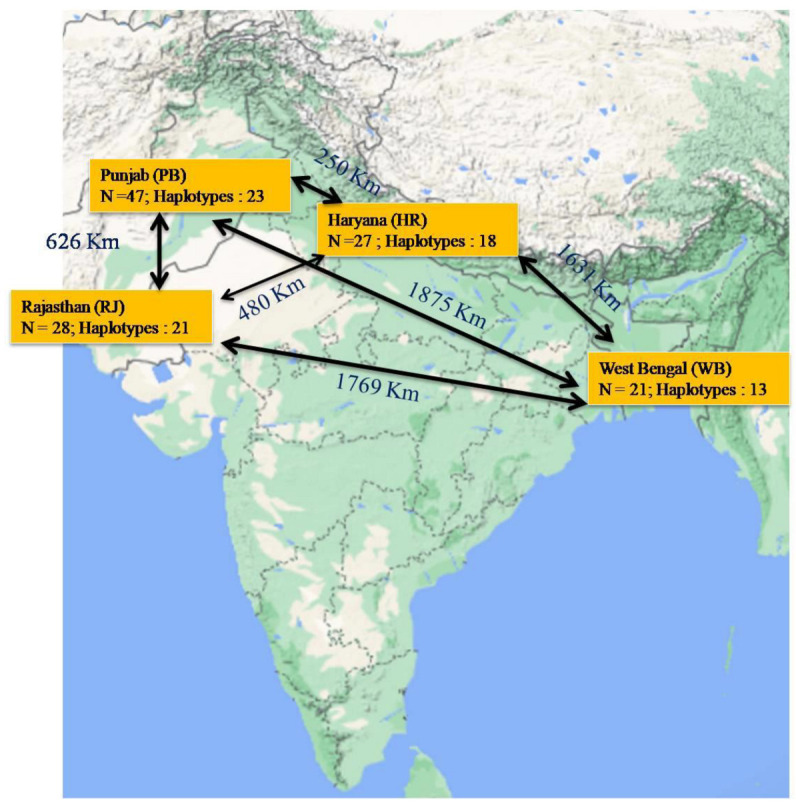
Map showing the sample collection sites in Northern wheat belt of India. N= Number of samples; Distance between two sample collecting states is mentioned over the black line.
2.2. Total Genomic DNA Extraction and Sequencing
Fungal isolates were grown for five days in shake cultures (200 rpm) at 25 ± 2 °C in 100 mL of potato dextrose broth (PDB; Himedia, India). The resulting mycelium was filtered through Whatman filter paper and thoroughly washed with distilled water. About 25–30 mg of mycelium was ground with the help of liquid nitrogen and used to extract the total genomic DNA as per the methodology of Kumar et al. [54]. The determination of total genomic DNA concentration was performed by using an Analytik Jena ScanDrop² instrument. Biometra Trios (Analytic Jena, Jena, Germany) machine, which along with the different primers mentioned in Table 2 were employed to amplify the fungal genomic DNA. The PCR master mix (25 μL) used in the thermal cycler comprised the following components: Go Taq Green master mix (12.5 μL; Promega Biotech India Pvt. Ltd. Jasola, New Delhi, India), fungal DNA (1 μL of 50 ng μL−1 concentration) and each primer pair (1 μL of 10 μM concentraion). In addition, nuclear-free water was used to make the total PCR master mix volume at 25 µL. The detailed information of the temperature profile used in the PCR for amplification of target genes (TUB, TEF and HIS) has been depicted in Table 2. Agarose gel electrophoresis was performed by using an E-BOX gel documentation system to see the PCR amplified amplicons of ≈500 bp ≈ 700 bp, and ≈350 bp fragments for TUB, TEF and HIS gene loci, respectively. The generated amplicons by each set of primers were freeze dried and sent to DNA sequencing analysis to Eurofins Genomics sequencing services, India. The obtained sequences of FG isolates were matched with F. graminareum isolates in National Center for Biotechnology Information (NCBI; https://www.ncbi.nlm.nih.gov/ (accessed on 11 June, 2021)), and we obtained the gene accession numbers.
Table 2.
Gene regions and primer pairs used in the current study.
| Gene Region | Sequence (5′-3′) | Product Size (bp) | Optimized PCR Conditions | Reference |
|---|---|---|---|---|
| Translation elongation factor 1 alpha (TEF) | TEF1: ATGGGTAAGGAGGACAAGAC | ≈700 | 95 °C: 5 min, (95 °C: 30 s, 56 °C: 30 s, 72 °C: 1 min) × 35 cycles 72 °C: 10 min | [55] |
| TEF2: GGAAGTACCAGTGATCAT GTT | ||||
| Histone (HIS) | CYLH3F: AGGTCC ACTGGTGGCAAG | ≈500 | 95 °C: 5 min, (95 °C: 30 s, 55 °C: 50 s, 72 °C: 1 min) × 35 cycles 72 °C: 5 min | [56] |
| H3-1b: GCGGGCGAGCTGGATGTCCTT | ||||
| Beta-tubulin (TUB) | BT2a:GGTAACCAAATCGGTGCTGCTTTC | 94 °C: 5 min, (94 °C: 30 s, 54 °C: 50 s, 72 °C: 1 min) × 35 cycles 72 °C: 10 min | [56] | |
| Bt2b: ACCCTCAGTGTAGTGACCCTTGGC | ≈350 |
2.3. Phylogenetic and Network Analysis
Nucleotide sequences of each gene loci (TUB, TEF and HIS) were matched with the gene sequences of respective loci available in the NCBI databank by basic local alignment search tool (BLAST; http://blast.ncbi.nlm.nih.gov/Blast.cgi (accessed on 11 June 2021)). Editing of gene sequences was conducted by using BioEdit 7.4.0.1 software [57]. ClustalW, a multiple sequence alignment program, was used for gene sequence alignment [58]. The DnaSP version 5 bioinformatic tool [59] was used to calculate the numbers of variable sites, parsimony informative sites, haplotypes and haplotype diversity. During the analysis, all positions containing gaps were removed. The DnaSP version 5 tool was also employed to determine the partitioning between populations from different studied sites and pair-wise comparisons of the nearest neighbor statistic, Snn [60] were calculated with 1000 permutations. Combined data sets of all three genes were employed to build a phylogenetic tree with the help of MEGA7 software [61], and the bootstrap value was adjusted at 1000 replications. A median joining haplotype network [62] was constructed for each of the four different populations based on different states and three population-based virulence features of isolates independently in PopART 1.7 [63]. The default epsilon value was fixed at zero. Analysis of molecular variance (AMOVA) was performed for each isolate independently in PopART 1.7 [63] to decipher the geographical grouping of genetic diversity. All the analyses were executed on a concatenated alignment of TUB, TEF and HIS data using PopART and were based on geographical locations and virulence features of the FG isolates. During the analysis, an experimental run was performed using complete deletion parameters.
2.4. Isolate Aggressiveness Analysis
Aggressiveness analysis of F. graminearum isolates was studied by inoculating susceptible wheat cultivar (cv. PBW343) with each isolate during 2021–2022 under a greenhouse. The mass production of FG isolates was completed by cultivating them on PDA media. After 15 days of incubation at 25 ± 2 °C, the inoculum was collected by rinsing the Petri plates with sterile distilled water containing Tween 20. Afterwards, the scraped mycelial mat with spores was passed through a double-layer sterile cheesecloth. Final spore inocolum concentration was adjusted at 5 × 106 spore ml−1 with the help of a hemocytometer. The cotton wool ball technique [64] was used to inoculate FG isolates at the wheat anthesis stage. Five spikes per isolate were used. A perforated plastic bag was put over each of the inoculated wheat spike to avoid cross-contamination. Misting was performed to maintain the desired level of humidity (RH > 90%) in the greenhouse. Phenotypic disease data were collected by visual inspection of each inoculated wheat spike. The data pertaining to the healthy and infected spikes as well as infected spikelets per spike in each plant was taken at 15 days post inoculation. Disease severity (%) or percentage of infected spikelets were determined by following the below mentioned formula:
| Disease severity (%) = [Total infected spikelets/Total spikelets per spike)] × 100. |
The aggressiveness of all the FG isolates was categorized into four classes as highly aggressive (HA; FHB infection of more than 50%), moderately aggressive (MA; FHB infection ranged between 25–50%) and Least or weakly aggressive (LA; FHB infection below 25%).
3. Results
3.1. Molecular Identity Confirmation of F. graminareum Isolates
All the isolates based on the comparison of genomics regions for all the three loci (700 bp TEF, 500 bp TUB, and 350 bp HIS) confirmed their identity as F. graminareum. The sequences of all three genes of all 123 FG isolates reflected 99–100% similarity (Table 1). The sequences identified in this study have been deposited in GenBank, and the obtained accession numbers are mentioned in Table 1. After sequence alignment, the final combined dataset of TUB, TEF and HIS had 2045 characters (Table 3). The percentage of sequence similarity for FG isolates (N = 123) was also performed by making comparative analysis of the sequences of the FUSARIUM-ID database. The sequence similarity for FG isolates between 98.3 and 100% was found to be within the threshold value documented by O’Donnell et al. [40]. In addition, the maximum likelihood phylogenetic analysis also displayed strong support for different lineage. It has been noticed that all the 123 FG isolates were grouped into two major clusters: Cluster I (115 isolates) and Cluster II (8 isolates) (Figure 3).
Table 3.
DNA polymorphism data for F. graminearum isolates based on tubulin (TUB), translation elongation factor (TEF) and histone (HIS) gene sequence comparisons.
| Parameters | TUB | TEF | HIS | Combined |
|---|---|---|---|---|
| Number of sites | 823 | 993 | 460 | 2045 |
| Theta (per site) from Eta | 0.012 | 0.120 | 0.003 | 0.083 |
| Theta (per sequence) from Eta | 1.857 | 38.995 | 1.300 | 90.059 |
| Total number of mutations (Eta) | 10.000 | 210.000 | 7.000 | 485.000 |
| Fu and Li’s F * | 1.333 | 1.184 | 1.703 | 2.718 |
| Fu and Li’s D * | 1.361 | 2.697 | 1.178 | 2.817 |
| Average number of pairwise nucleotide differences, k | 2.367 | 25.969 | 2.408 | 133.948 |
| Total number of mutations, Eta | 10 | 210 | 7 | 485 |
| Minimum number of Recombination events, Rm | 4 | 4 | 4 | 12 |
| Tajima’s D | 0.6814 | −1.1005 | 1.93605 | 1.62305 |
* indicates neutrality statistics without an outgroup.
Figure 3.

Phylogenetic relationship determined by using combined sequence of three gene loci (TUB, TEF and HIS sequences) and neighbor-joining (NJ) method. The percentage of replicate tree in the bootstrap test is 1000 replicates. The evolutionary distances were computed using the Kimura 2-parameter method. All positions containing gaps and missing data were eliminated. The tree was rooted with Diaporthe alleghaniensis strain CBS 495.72 [KC343733.1].
3.2. Haplotypic Diversity
The analysis of the combined gene loci reflected that both haplotype and nucleotide diversity were high (Hd = 0.974 and π = 0.083) (Table 4). The haplotype diversity ranged from 0.962 (WB) to 0.981 (RJ) (Table 4). A consensus maximum parsimony (MP) tree was generated for all the haplotype sequences observed in the study and shown in Figure 4. It is important to mention that MP analysis was preferred over ML owing to the fact that the TCS haplotype network was also based on parsimony-based statistics and hence will be a better option to make comparisons of groupings with strong bootstrap support. Figure 4 showed >90% bootstrap score and supported haplotype positioning with reference to distinct groupings of FG sequences. It has been observed that the network is reticulate type at the interior and star-like at the tip. Moreover, there was an absence of clearly evident centrally located single haplotype, from where various haplotypes came out (Figure 5). Isolates collected from the PB region were partitioned in 22 haplotypes, which is approximately 73.33% of the total haplotypes observed in the total analyzed population of the north plains of India. There was some evidence of geographic structure in the distribution of haplotypes. Punjab had a majority of the unique (H9, H10 and H11) and shared haplotypes (27 haplotypes) among all the studied populations. The predominant haplotypes were identified as H1, H2 and H3 and were observed in PB, RJ and WB. Sequences of isolates from PB belonged exclusively to three haplotypes (H9, H10 and H11 and absent in other populations. Interestingly, PB, RJ and WB shared seven haplotypes (H1–H6 and H30), while PB, HR and RJ shared three haplotypes (H15, H16, H17, H18, H19, and H20). Similarly, HR, RJ and WB shared three haplotypes (H24, H26 and H26). The population haplotype network for the 123 FG isolates was performed based on combined gene loci (Figure 5).
Table 4.
DNA polymorphism data for F. graminearum isolates based on beta-tubulin (TUB), translation elongation factor 1 alpha (TEF) and histone (HIS) gene sequence comparisons.
| Gene Loci | Region of Collection | Number of Isolates (N) | Number of Segregating Sites (S) | Number of Haplotypes (H) | Haplotype Diversity (Hd) | Nucleotide Diversity (π) |
|---|---|---|---|---|---|---|
| TUB | PB | 47 | 5 | 7 | 0.816 | 0.013 |
| HR | 27 | 6 | 10 | 0.900 | 0.019 | |
| RJ | 28 | 6 | 11 | 0.865 | 0.015 | |
| WB | 21 | 6 | 8 | 0.838 | 0.012 | |
| Total | 123 | 6 | 11 | 0.846 | 0.015 | |
| TEF | PB | 47 | 197 | 12 | 0.891 | 0.056 |
| HR | 27 | 195 | 11 | 0.883 | 0.128 | |
| RJ | 28 | 197 | 13 | 0.939 | 0.089 | |
| WB | 21 | 196 | 7 | 0.671 | 0.061 | |
| Total | 123 | 197 | 15 | 0.888 | 0.080 | |
| HIS | PB | 47 | 7 | 14 | 0.920 | 0.006 |
| HR | 27 | 7 | 10 | 0.855 | 0.005 | |
| RJ | 28 | 7 | 11 | 0.857 | 0.004 | |
| WB | 21 | 7 | 10 | 0.919 | 0.006 | |
| Total | 123 | 7 | 17 | 0.893 | 0.893 | |
| Combined loci | PB | 47 | 384 | 23 | 0.965 | 0.072 |
| HR | 27 | 381 | 18 | 0.972 | 0.097 | |
| RJ | 28 | 385 | 21 | 0.981 | 0.098 | |
| WB | 21 | 381 | 13 | 0.962 | 0.070 | |
| Total | 123 | 385 | 30 | 0.974 | 0.083 |
Haplotype diversity and nucleotide diversity are important indicators of population genetic variation. Haplotype and nucleotide diversities are generally considered to be low where haplotype diversity (Hd) and nucleotide diversity (π) are less than 0.5.
Figure 4.

Unrooted maximum parsimony (MP) tree of haplotypes.
Figure 5.

Median joining network of different haplotypes of FG population. Size of the circle is related with frequency of haplotypes. Colors indicate the proportion of individuals sampled in different populations within the study area.
The haplotypes calculated on the basis of individual locus, i.e., TUB, TEF, and HIS, were 11, 15, and 17, respectively (Table 4). There were a total of 30 haplotypes observed in all the 123 FG isolates. Haplotype frequency was varies from four to five, where the majority of the haplotypes were composed of at least four isolates (Table 5). Haplotypes with higher frequencies were observed in PB from where the maximum number of isolates (23 haplotypes out of 47 isolates) was collected (Table 4). H10 and H11 had the maximum frequency (4) in the PB population (N = 47). Similarly, the RJ population shows a maximum frequency of six haplotypes (H16–H20 and H22). The frequency of H22 was found to be maximum in the HR population and included one isolate (NFG82) which belonged to the RJ population. WB populations have a maximum frequency of seven haplotypes (H1–H4, H27–H29 and H30) (Table 5). A single common haplotype was absent in all the analyzed populations. However, PB populations show some unique haplotypes (H9, H10 and H11) that were absent in other analyzed populations. The structure of the haplotype network matched with the MP topological tree.
Table 5.
Haplotype frequency (Freq) and distribution for F. graminareum isolates based on combined gene loci.
| Haplotype | Frequency | Region (Isolates) |
|---|---|---|
| Hap_1 | 5 | Punjab (NFG1, NFG121); Rajasthan (NFG31); West Bengal (NFG61, NFG91) |
| Hap_2 | 5 | Punjab (NFG2 and NFG122); Rajasthan (NFG32); West Bengal (NFG62, NFG92) |
| Hap_3 | 5 | Punjab (NFG3, NFG123); Rajasthan (NFG33); West Bengal (NFG63, NFG93) |
| Hap_4 | 4 | Punjab (NFG4); Rajasthan (NFG34); West Bengal (NFG64, NFG94) |
| Hap_5 | 4 | Punjab (NFG5, NFG65); Rajasthan (NFG35); West Bengal (NFG95) |
| Hap_6 | 4 | Punjab (NFG6, NFG96); Rajasthan (NFG36); West Bengal NFG66, |
| Hap_7 | 4 | Punjab (NFG7, NFG97 NFG67); Rajasthan (NFG37) |
| Hap_8 | 4 | Punjab (NFG8, NFG98, NFG68); Rajasthan (NFG38) |
| Hap_9 | 4 | Punjab (NFG9, NFG39, NFG69, NFG99) |
| Hap_10 | 4 | Punjab (NFG10, NFG40, NFG70, NFG100) |
| Hap_11 | 4 | Punjab (NFG11, NFG41, NFG71, NFG101) |
| Hap_12 | 4 | Punjab (NFG12, NFG42, NFG72); Haryana (NFG102) |
| Hap_13 | 4 | Punjab (NFG13, NFG43, NFG73); Haryana (NFG103) |
| Hap_14 | 4 | Punjab (NFG14, NFG44, NFG74); Haryana (NFG104) |
| Hap_15 | 4 | Punjab (NFG15); Haryana (NFG45, NFG105); Rajasthan (NFG75) |
| Hap_16 | 4 | Punjab (NFG16); Haryana (NFG46); Rajasthan (NFG76, NFG106) |
| Hap_17 | 4 | Punjab (NFG17); Haryana (NFG47); Rajasthan (NFG77, NFG107) |
| Hap_18 | 4 | Punjab (NFG18); Haryana (NFG48); Rajasthan (NFG78, NFG108) |
| Hap_19 | 4 | Punjab (NFG19); Haryana (NFG49); Rajasthan (NFG79, NFG109) |
| Hap_20 | 4 | Punjab (NFG20); Haryana (NFG50); Rajasthan (NFG80, NFG110) |
| Hap_21 | 4 | Haryana (NFG21, NFG51); Rajasthan (NFG81, NFG111) |
| Hap_22 | 4 | Haryana (NFG22, NFG52, NFG112); Rajasthan (NFG82) |
| Hap_23 | 4 | Haryana (NFG23, NFG113); Rajasthan (NFG53, NFG83) |
| Hap_24 | 4 | Haryana (NFG24, NFG114), Rajasthan (NFG54);West Bengal (NFG84) |
| Hap_25 | 4 | Haryana (NFG25 (NFG115); Rajasthan (NFG55); West Bengal (NFG85) |
| Hap_26 | 4 | Haryana (NFG26 NFG116); Rajasthan (NFG56); West Bengal (NFG86) |
| Hap_27 | 4 | Haryana (NFG27, NFG117); West Bengal (NFG57, NFG87) |
| Hap_28 | 4 | Haryana (NFG28); West Bengal (NFG58, NFG88); Punjab (NFG118) |
| Hap_29 | 4 | Punjab (NFG119); Haryana (NFG29); West Bengal (NFG59, NFG89) |
| Hap_30 | 4 | Punjab (NFG120); Rajasthan (NFG30), West Bengal (NFG60, NFG90) |
3.3. Nucleotide Diversity
The nucleotide diversity among the four FG populations was low and lies between 0.070 (WB population) and 0.098 (RJ) (Table 4). The pair-wise genetic distances between different FG populations are shown in Table 6. The pairwise Fst for different populations is shown in Table 6. All the analyzed populations showed low Fst values (−0.012 to 0.025; p > 0.05). Negative Fst values were observed for PB and HR and WB and HR with p > 0.05 (Table 6). The results also indicated low nucleotide diversity for all the populations, i.e., WB (π = 0.070), PB (π = 0.072), RJ (π = 0.098), and HR (π =0.097) (Table 6).
Table 6.
Neutrality tests statistics observed in current study.
| Test Method | PB | HR | RJ | WB | ||||
|---|---|---|---|---|---|---|---|---|
| S * | p | S | p | S | p | S | p | |
| Tajima’s D | 0.809 | p > 0.10 | 0.037 | p > 0.10 | 0.614 | p > 0.10 | −0.978 | p > 0.10 |
| Fu and Li’s D | 2.176 | p < 0.02 | 1.857 | p < 0.02 | 1.638 | p < 0.02 | 0.356 | p > 0.10 |
| Fu and Li’s F | 1.988 | p < 0.02 | 1.486 | p < 0.10 | 1.535 | p < 0.10 | −0.064 | p > 0.10 |
| Fu’s Fs | 24.788 | 11.400 | 6.389 | 13.859 | ||||
S * represents statistics of Tajima’s D, Fu and Li’s D, Fu and Li’s F and Fu’s Fs; p < 0.05 indicates significant differences, rejecting the null hypothesis; p > 0.10 indicates no significant differences, following the neutrality model; PB: Punjab; HR: Haryana; RJ: Rajasthan; and WB: West Bengal.
3.4. Gene Flow and Genetic Divergence
The results (Table 7) indicated that Nm values for the RJ and PB (Nm = 88.51), RJ and HR (Nm = 61.87) and RJ and WB (Nm = 11.89) populations were more than 4. The genetic differentiation fixation coefficient was found to be non-significant among all the analyzed populations (Table 7). Snn values (Table 8) were found to be non- significant. When all the three loci were concatenated into a single sequence, a total of 12 recombination events were detected (Table 8).
Table 7.
Pair-wise Fst (above diagonal) and Nm (below diagonal) of F. graminareum populations from four states of northern plains of India.
| Population | PB | HR | RJ | WB |
|---|---|---|---|---|
| PB | NA | −0.012 * | 0.009 * | −0.014 * |
| HR | −21.32 | NA | 0.002 * | −0.019 * |
| RJ | 88.51 | 61.87 | NA | 0.025 * |
| WB | −16.92 | −11.45 | 11.89 | NA |
Fst: Genetic differentiation coefficient. When Fst ranges from 0 to 0.05, the genetic differentiation is low (Rousset 1997); Nm > 4 indicates that gene flow is frequent between populations, while negative value indicates an absence of gene flow; * Not statistically significant with p > 0.05. N/A: Not applicable.
Table 8.
Snn statistics of four different population of F. graminareum isolates of northern plains of India.
| Population | PB | HR | RJ | WB |
|---|---|---|---|---|
| PB | - | |||
| HR | 0.699 | |||
| RJ | 0.535 | 0.412 | ||
| WB | 0.636 | 0.657 | 0.549 | - |
Rm indicates the minimum number of recombination events; Snn value represents how often the nearest neighbor sequences are found in the same locality.
3.5. Neutrality Test and Demographic History
The results of neutrality tests (Table 7) reflected non-significant negative values for both Tajima’s D and Fu’s FS statistic, indicating toward the spread of the FG population with supernumerary alleles. Furthermore, it has been noticed that the null hypothesis of neutrality for the combined gene loci based inference of FG populations was not jilted due to the existence of non-significant values of Tajima’s D and Fu’s FS. In addition, the analysis of the demographic population history performed on the basis of neutrality statistics for all the four sets of populations revealed a non-significant negative Tajima’s D value, indicating the presence of low-frequency polymorphism in the analyzed populations.
3.6. Analysis of Molecular Variance (AMOVA)
The results of AMOVA analysis demonstrated a variance of 100.84 within the FG populations. However, a negative percentage of variations (−0.84%) was recorded among FG populations (Table 9). The fixation indices (FI) among the populations were negative (FI = −0.0084), indicating a lack of genetic differentiation among the populations.
Table 9.
Hierarchical analysis of molecular variance (AMOVA) in FG populations.
| Source of Variation | Degree of Freedom | Sum of Squares | Variance Component | Variation (%) | p-Value | Fixation Index (FI) |
|---|---|---|---|---|---|---|
| Among populations | 3 | 29,143.70 | −122.70 | −0.84 | 0.61 | −0.0084 |
| Within populations | 119 | 1,754,661.87 | 14,745.06 | 100.84 |
Statistical significance calculated at p > 0.05; Negative values for variations among populations are regarded as zero. Genetic structure for F. graminareum population was not detected through AMOVA. Negative FI values can be inferred as no genetic differences between the populations compared.
3.7. Aggressiveness of Isolates
The FG isolates were evaluated for their aggressiveness on wheat cultivar (cv. PBW343), and results in terms of HA, MA and LA are mentioned in Table 1. The results indicated that the aggressiveness of FG isolates varies from 42.28% isolates as highly aggressive to least aggressive (34.15%) and moderately aggressive (23.58%) (Table 1; Figure 6). The network also revealed no significant correlation between the genetic variability of FG isolates and aggressiveness levels. Furthermore, haplotype network analysis does not show any significant correlation with the FG isolates except for H22 haplotypes, which contained HA isolates.
Figure 6.

Median joining network according to different categories of aggressiveness of FG haplotypes. Size of the circle is related with aggressivity of the haplotypes. Colors indicate the proportion of individuals depicting the same level of the study area. HA: Highly aggressive; MA: Moderately aggressive and LA: Least aggressive.
4. Discussion
FG is one of the emerging and economically important diseases affecting wheat in India [4,65]. Diversity analysis is essential to infer the population genetics of such an important disease for framing cost-effective management tactics. In this context, a comprehensive understanding of genetic variation of the Indian FG population becomes necessity. Conserved region-based DNA markers are reported as one of the potential tools for determining the genetic variation, speciation of fungal pathogens and inferring their ancestral background [35,66]. FG diversity analysis based on TEF, HIS and TUB genes has discovered the existence of different putative subspecies of Fusarium in the Asian and sub-Saharan Africa terrains [67]. As Indian FG populations have not been analyzed so far to determine their diversity and population structure, therefore, the current investigation presented for the first time effort to quantify genetic variability in FG in northern plains of India using three highly versatile molecular markers, i.e., TUB, TEF and HIS. In this study, attempts have been made to (i) determine the identity of FG infecting wheat in the northern plains of India; (ii) confirm the level of DNA polymorphism of TUB, TEF and HIS sequences; (iii) decode the haplotype diversity and their distribution; and (iv) work out the phylogenetic kinship between FG isolates of Indian origin.
Previous published literature indicated that TEF is highly recommended for species delineation and rejected ITS sequences in offering superior taxa resolution for several of the fungal genera (e.g., Trichoderma and Fusarium) of the ascomycetes group [31,40]. From earlier published reports, it is cleared that when the distance between different Fusarium spp. to the nearest neighbor fungal species was deduced on the basis of ITS regions, very low genetic distance values were obtained, which ultimately resulted in the inferior resolution and poor taxon placement in phylogenetic lineages [68,69]. It is important to mention that slow evolving genes, for instance, TEF and HIS, serve excellent options in inferring phylogeny-based relationships. Contrarily, more recent evolutionary and speciation events can be captured from the gene sequences (e.g., TUB) displaying high evolutionary rates. Therefore, all the three genes were satisfying all the basic requirements needed for the phylogenetic analysis, as the concatenated sequences were composed of both variable introns and conserved exons [70] and hence selected for the present research investigation. According to the results of the current study and others [40], it is crucial to ascertain the DNA polymorphism in the sequences to be employed for the identification of intraspecies kinships and inferring phylogenetic lineages because of their direct influence on the correct and precise identity with strong phylogenetic arrangement and signal strength. Additionally, its influence on genetic diversity indices such as haplotype diversity and their dispersion have been reported [40]. The low level of genetic diversity noticed for wheat-associated FG populations in the northern plains of India based on concatenated gene sequences is not a rare event, as similar observations regarding the existence of a low level of phylogenetic diversity for FG populations in wheat has been documented by Castellá and Cabañes [71].
The significance of evolutionary forces on the FG population has been studied by conducting neutrality tests, which provides evidence regarding the divergence of combined gene loci sequence data from neutrality statistics. Moreover, the significant positive Tajima’s D values highlighted that all the three gene loci are experiencing population bottlenecks in PB, HR and RJ, where the FG population appears to be uniform and only a few sequences are in a deciding role for the development of the nascent population. This indicates that FG biology and the colonization pattern could act as one important factor behind population bottlenecks. Contrarily, significant negative values of Tajima’s D and F statistics in case of the WB population showed a strong purifying selection. This observation has been matched with the earlier report of Zhao et al. [72], where strong purifying selection in accordance with their important biological roles in cellular processes in fungi has been noticed.
Geography and climate are two of the most important epidemic linked factors and strongly influence the prevalence and establishment of Fusarium spp. as pathogens in various types of plants [71,73]. Usually, in nature, a mixture of both old and new haplotypes in the form of living descendants in field populations exists. However, the relationship between the two types of haplotypes may be reticulate and non-bifurcated because of non-dichotomous historical events, etc. [74]. Similarly, in the present study, a minimum of recombination events based on the combined three gene loci has been observed in the FG populations. This clearly reflects the mechanism of intragenic recombination behind the genetic variability in the pathogen population in the regions. However, it is important to mention here that Ma et al. [75] also confirmed the significance of horizontal gene transfer in the evolution of Fusarium genomes. Thus, high-frequency haplotypes (H1, H2 and H3) have been present in the population for a long time and detected from PB, RJ and WB population dominantly. Contrarily, other seem to be less frequent haplotypes and indicate toward more recent mutation events. Moreover, the terminal haplotypes (e.g., H2, H3, H5, H13–H15, H21, H22, H24–H26, H29, and H30) located at the network tips in the present study indicate a holocene pedigree rather than old ancestry (H1, H18 and H28) as placed in the interiors of the haplotype network. Carbone and Kohn [76] reported that newer haplotypes generally show restricted geographical dispersion in comparison to ancestral haplotypes, which have a wide geographical distribution and show restricted gene flow. However, the current study indicated toward a reticulate network, where both ancestral and nascent haplotypes occupied an internal celestial point of the network. The pattern of haplotype network and haplotype distribution in the studied sites of the northern plains illustrate that the FG isolates were separated from other regional isolates by a series of mutational events rather than geographical location (e.g., PB, HR, RJ and WB). These points indicate toward the possibility of the operation of both direct and indirect factors in the distribution of FG fungus, which is seed-, soil- and air-borne in nature and further supported by the haplotype network. The possibility of human-driven transfer of fungi in contaminated plant material and the transfer of FG inocula belong to other regions such as PB (where the maximum number of haplotypes was observed) or vice versa, which is followed by host adaptation and panoramic spatiotemporal dispersion. It has been observed that a number of haplotypes displayed only a difference at one site in the present study when compared with their genetically closest haplotype. These findings clearly reflect the significance of mutation in developing haplotype diversity as observed by earlier researchers in case of other fungal crop plant pathogens [30,77,78].
Haplotype analysis provides important information on the existence of different types of haplotypes (h), their diversity (Hd) and frequency. Usually, Hd values varied between 0 and 1, which reflects zero to high level of haplotype diversity, respectively [79]. In the current investigation, Hd (0.962–0.981) values revealed high levels of genetic variability in all the states. PB reflected maximum diversity (i.e., 27 haplotypes from 47 FG isolates) followed by RJ (i.e., 21 haplotypes from 28 FG isolates). It is important to mention that several haplotypes have been shared among different populations that clearly hinted toward the significance of asexual reproduction and effective spore migration. Two other points may help to explain the observed level of high genetic variability among FG isolates. These include the possibility of the existence of multiple founder populations, which resulted in population admixture as well as dispersion due to the assemblage of different alleles in FG populations, as clearly extrapolated from mutation events (both cumulative and shared mutational events) recorded in the current research. This observation has been supported by the high level of population admixture noticed in network analysis in the current study. The results of the current study also pointed toward the mutation as an essential factor for the generation of diverse regional populations of FG that later resulted in the developments of a number of mutants from which new and virulent type isolates can arise. Moreover, sharing a number of haplotypes among FG populations supports multiple introductions of the identical haplotypes, which may be due to the asexual reproductive phase of FG. Overall, it seems that the analyzed FG populations are admixture ones and occupied haplotypes from different populations in the northern plains of India.
The population genetics analysis performed in the current study revealed that the FG population from RJ seems genetically similar with other populations of PB, HR and WB. This statement is clearly supported by the values of high gene flow (Nm = 11.89–88.51) and low genetic differentiation (Fst = 0.002−0.025). A similar observation of high gene flow among different subpopulations of F. graminearum in Canada and the USA has been documented [25,80]. Furthermore, the ANOVA analysis conducted in present study supports the previous statement, as a very high level of genetic variation (100%) among individuals within the population has been recorded. These observations further indicate toward the greater possibility of sexual reproduction in the regions and are well supported with earlier research findings related to the population genetic structure of FG populations from wheat in Canada and the USA [25]. There might be another plausible reason, i.e., infected wheat seed movement as a planting material among different regions as a prime cause of gene flow between isolates of distinct origins. However, the role of long-distance spore transfer of FG in determining the gene migration cannot be omitted because of the air-borne mode of dissemination of the fungus [81].
A high variation in the aggressiveness of FG isolates has been observed in FG isolates collections from different wheat-growing states. These results are in agreement with earlier workers’ findings in both within-field populations and crossing populations [82,83,84]. The study shows that highly aggressive isolates are widely distributed in the northern plains of India and reflects the robust genetic diversity among regional FG populations. However, an exhaustive understanding of localized FG populations and their potential to surmount disease management tactics is essential to secure the wheat farmers from this emerging disease threat in India.
Acknowledgments
The authors thank ICAR-IIWBR, Karnal (Haryana) for providing required support for conducting the research work and acknowledge the Amity Institute of Microbial technology, Amity University, Rajasthan, India. This work is the part of program of the first author.
Author Contributions
Conceptualization, P.L.K.; Data curation, N.K. and P.L.K.; Formal analysis, N.K. and P.L.K.; Funding acquisition, P.L.K. and G.P.S.; Investigation, N.K. and P.L.K.; Methodology, N.K., P.L.K. and S.K.; Project administration, P.L.K., D.S. and G.P.S.; Resources, P.L.K. and S.K.; Software, N.K. and P.L.K.; Supervision, P.L.K. and D.S.; Validation, N.K. and P.L.K.; Visualization, N.K. and P.L.K.; Writing—original draft, N.K. and P.L.K.; Writing—review and editing, S.K., D.S. and G.P.S. All authors have read and agreed to the published version of the manuscript.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data are contained within the article or gene sequences generated in this study were deposited in GenBank under the accession numbers listed in Table 1.
Conflicts of Interest
The authors declare no conflict of interest.
Funding Statement
This research was funded by Indian Council of Agricultural Research.
Footnotes
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Mengesha G.G., Abebe S.M., Fedilu K.B., Tadesse Y.B., Mekonnen A.A., Abate G., Lera Z.T., Shertore M.M., Cheleko D.C., W/Silassie A.B. Fusarium head blight progression and yield response of bread wheat as affected by fungicides and spray regimes under field condition in Southern Ethiopia. J. Crop Sci. Biotechnol. 2022 doi: 10.1007/s12892-022-00152-6. [DOI] [Google Scholar]
- 2.Mawcha K.T., Zhang N., Wang Y., Yang W. Advances in wheat breeding for resistance to Fusarium head blight. Czech J. Genet. Plant Breed. 2022 doi: 10.17221/1/2022-CJGPB. [DOI] [Google Scholar]
- 3.Kashyap P.L., Gupta V., Prakash Gupta O., Sendhil R., Gopalareddy K., Jasrotia P., Singh G.P. New Horizons in Wheat and Barley Research: Global Trends, Breeding and Quality Enhancement. Springer; Singapore: 2022. p. 842. [DOI] [Google Scholar]
- 4.Saharan M.S., Kumar H.M.A., Gurjar M.S., Aggarwal R. Fusarium head blight of wheat in india-variability in pathogens associated and sources of resistance: An overview. Indian Phytopathol. 2021;74:345–353. doi: 10.1007/s42360-021-00358-8. [DOI] [Google Scholar]
- 5.Qiu J.-B., Sun J.-T., Yu M.-Z., Xu J.-H., Shi J.-R. Temporal dynamics, population characterization and mycotoxins accumulation of Fusarium graminearum in Eastern China. Sci. Rep. 2016;6:36350. doi: 10.1038/srep36350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Roy A.K. Ear blight and scab of wheat in Arunachal Pradesh. Curr. Sci. 1973;43:162. [Google Scholar]
- 7.Brahma R.N., Singh S.D. Occurrence of scab of wheat in the Nilgiris hills. Curr. Sci. 1985;54:1184–1185. [Google Scholar]
- 8.Chahal S.S., Kaur J., Aulakh K.S. Innovative approaches in plant disease management; Proceedings of the Symposium held at the Punjab Agricultural University; Ludhiana, India. 1993. pp. 25–27. [Google Scholar]
- 9.Bagga P. Fusarium head blight (FHB) of wheat: Role of host resistance, wheat aphids, insecticide and strobilurin fungicide in disease control in Punjab, India. Cereal Res. Commun. 2008;6:667–670. doi: 10.1556/CRC.36.2008.Suppl.B.57. [DOI] [Google Scholar]
- 10.Ghimire B., Mergoum M., Martinez-Espinoza A.D., Sapkota S., Pradhan S., Babar M.A., Bai G., Dong Y., Buck J.W. Genetics of Fusarium head blight resistance in soft red winter wheat using a genome-wide association study. Plant Genome. 2022:e20222. doi: 10.1002/tpg2.20222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Teli B., Purohit J., Rashid M.M., Jailani A.A.K., Chattopadhyay A. Omics insight on Fusarium head blight of wheat for translational research perspective. Curr. Genom. 2020;21:411–428. doi: 10.2174/1389202921999200620222631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wegulo S.N. Factors influencing deoxynivalenol accumulation in small grain cereals. Toxins. 2012;4:1157–1180. doi: 10.3390/toxins4111157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huang L., Li T., Ding C., Zhao J., Zhang D., Yang G. Diagnosis of the severity of Fusarium head blight of wheat ears on the basis of image and spectral feature fusion. Sensors. 2020;20:2887. doi: 10.3390/s20102887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Karasi M., Jorge D.S., Pierce A.l. Agriculture and Natural Resources. Ohio State University; Columbus, OH, USA: 2016. [(accessed on 25 May 2022)]. Fusarium head blight or head scab of wheat, barley and other small grain crops. Available online: https://ohioline.osu.edu/factsheet/plpath-cer-06. [Google Scholar]
- 15.Fernando W.G.D., Oghenekaro A.O., Tucker J.R., Badea A. Building on a foundation: Advances in epidemiology, resistance breeding, and forecasting research for reducing the impact of Fusarium head blight in wheat and barley. Can. J. Plant Pathol. 2021;43:495–526. doi: 10.1080/07060661.2020.1861102. [DOI] [Google Scholar]
- 16.Pereyra S.A., Dill-Macky R., Sims A.L. Survival and inoculum production of Gibberella Zeae in wheat residue. Plant Dis. 2004;88:724–730. doi: 10.1094/PDIS.2004.88.7.724. [DOI] [PubMed] [Google Scholar]
- 17.McMullen M., Bergstrom G., De Wolf E., Dill-Macky R., Hershman D., Shaner G., Van Sanford D. A unified effort to fight an enemy of wheat and barley: Fusarium head blight. Plant Dis. 2012;96:1712–1728. doi: 10.1094/PDIS-03-12-0291-FE. [DOI] [PubMed] [Google Scholar]
- 18.de Chaves M.A., Reginatto P., da Costa B.S., de Paschoal R.I., Teixeira M.L., Fuentefria A.M. Fungicide resistance in Fusarium graminearum species complex. Curr. Microbiol. 2022;79:62. doi: 10.1007/s00284-021-02759-4. [DOI] [PubMed] [Google Scholar]
- 19.Shude S.P.N., Yobo K.S., Mbili N.C. Progress in the management of Fusarium head blight of wheat: An overview. S. Afr. J. Sci. 2020;116:7854. doi: 10.17159/sajs.2020/7854. [DOI] [Google Scholar]
- 20.Wegulo S.N., Baenziger P.S., Hernandez Nopsa J., Bockus W.W., Hallen-Adams H. Management of Fusarium head blight of wheat and barley. Crop Prot. 2015;73:100–107. doi: 10.1016/j.cropro.2015.02.025. [DOI] [Google Scholar]
- 21.Rampersad S.N. Spatial pattern of genetic diversity in field populations of Fusarium incarnatum-equiseti species complex. Ecol. Evol. 2021;11:9010–9020. doi: 10.1002/ece3.7738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Crous P.W., Groenewald J.Z., Slippers B., Wingfield M.J. Global food and fibre security threatened by current inefficiencies in fungal identification. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2016;371:20160024. doi: 10.1098/rstb.2016.0024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McDonald B.A., Linde C. Pathogen population genetics, evolutionary potential, and durable resistance. Annu. Rev. Phytopathol. 2002;40:349–379. doi: 10.1146/annurev.phyto.40.120501.101443. [DOI] [PubMed] [Google Scholar]
- 24.Vanheule A., De Boevre M., Moretti A., Scauflaire J., Munaut F., De Saeger S., Bekaert B., Haesaert G., Waalwijk C., van der Lee T., et al. Genetic divergence and chemotype diversity in the Fusarium head blight pathogen Fusarium poae. Toxins. 2017;9:255. doi: 10.3390/toxins9090255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Oghenekaro A.O., Oviedo-Ludena M.A., Serajazari M., Wang X., Henriquez M.A., Wenner N.G., Kuldau G.A., Navabi A., Kutcher H.R., Fernando W.G.D. Population genetic structure and chemotype diversity of Fusarium graminearum populations from wheat in Canada and North Eastern United States. Toxins. 2021;13:180. doi: 10.3390/toxins13030180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hafez M., Gourlie R., Telfer M., Schatz N., Turkington T.K., Beres B., Aboukhaddour R. Diversity of Fusarium spp. associated with wheat node and grain in representative sites across the Western Canadian Prairies. Phytopathology. 2022;112:1003–1015. doi: 10.1094/PHYTO-06-21-0241-R. [DOI] [PubMed] [Google Scholar]
- 27.Kashyap P.L., Rai S., Kumar S., Srivastava A.K. Genetic diversity, mating types and phylogenetic analysis of Indian races of Fusarium Oxysporum f. sp. ciceris from chickpea. Arch. Phytopathol. Plant Prot. 2016;49:533–553. doi: 10.1080/03235408.2016.1243024. [DOI] [Google Scholar]
- 28.Kashyap P.L., Rai S., Kumar S., Srivastava A.K., Anandaraj M., Sharma A.K. Mating type genes and genetic markers to decipher intraspecific variability among Fusarium udum isolates from pigeonpea: Diversity analysis of Fusarium udum isolates. J. Basic Microbiol. 2015;55:846–856. doi: 10.1002/jobm.201400483. [DOI] [PubMed] [Google Scholar]
- 29.Kumar S., Rai S., Maurya D.K., Kashyap P.L., Srivastava A.K., Anandaraj M. Cross-species transferability of microsatellite markers from Fusarium oxysporum for the assessment of genetic diversity in Fusarium udum. Phytoparasitica. 2013;41:615–622. doi: 10.1007/s12600-013-0324-y. [DOI] [Google Scholar]
- 30.Kashyap P.L., Kumar S., Tripathi R., Kumar R.S., Jasrotia P., Singh D.P., Singh G.P. Phylogeography and population structure analysis reveal diversity by gene flow and mutation in Ustilago Segetum (Pers.) Roussel tritici causing loose smut of wheat. Front. Microbiol. 2019;10:1072. doi: 10.3389/fmicb.2019.01072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rai S., Kashyap P.L., Kumar S., Srivastava A.K., Ramteke P.W. Identification, characterization and phylogenetic analysis of antifungal trichoderma from tomato rhizosphere. Springerplus. 2016;5:1939. doi: 10.1186/s40064-016-3657-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang J.-H., Ndoye M., Zhang J.-B., Li H.-P., Liao Y.-C. Population structure and genetic diversity of the Fusarium graminearum species complex. Toxins. 2011;3:1020–1037. doi: 10.3390/toxins3081020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chandra N.S., Wulff E.G., Udayashankar A.C., Nandini B.P., Niranjana S.R., Mortensen C.N., Prakash H.S. Prospects of molecular markers in Fusarium species diversity. Appl. Microbiol. Biotechnol. 2011;90:1625–1639. doi: 10.1007/s00253-011-3209-3. [DOI] [PubMed] [Google Scholar]
- 34.Suga H., Karugia G.W., Ward T., Gale L.R., Tomimura K., Nakajima T., Miyasaka A., Koizumi S., Kageyama K., Hyakumachi M. Molecular characterization of the Fusarium graminearum species complex in Japan. Phytopathology. 2008;98:159–166. doi: 10.1094/PHYTO-98-2-0159. [DOI] [PubMed] [Google Scholar]
- 35.Kashyap P.L., Rai P., Kumar S., Chakdar H., Srivastava A.K. Fungal Biology. Springer International Publishing; Cham, Germany: 2017. DNA barcoding for diagnosis and monitoring of fungal plant pathogens; pp. 87–122. [DOI] [Google Scholar]
- 36.Schoch C.L., Seifert K.A., Huhndorf S., Robert V., Spouge J.L., Levesque C.A., Chen W., Fungal barcoding consortium. fungal barcoding consortium author list Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for fungi. Proc. Natl. Acad. Sci. USA. 2012;109:6241–6246. doi: 10.1073/pnas.1117018109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jabłońska E., Piątek K., Wit M., Mirzwa-Mróz E., Wakuliński W. Molecular diversity of the Fusarium fujikuroi species complex from maize. Eur. J. Plant Pathol. 2020;158:859–877. doi: 10.1007/s10658-020-02121-7. [DOI] [Google Scholar]
- 38.Araújo N.A.F., Pasqual M., Pio L.A.S., Alves E., de Matos Moura N., Costa S.D.S. Identification and aggressiveness of four isolates of Fusarium oxysporum f. sp. cubense from latundan banana in Brazil. J. Phytopathol. 2017;165:257–264. doi: 10.1111/jph.12557. [DOI] [Google Scholar]
- 39.Karlsson I., Edel-Hermann V., Gautheron N., Durling M.B., Kolseth A.-K., Steinberg C., Persson P., Friberg H. Genus-specific primers for study of Fusarium communities in field samples. Appl. Environ. Microbiol. 2015;82:491–501. doi: 10.1128/AEM.02748-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.O’Donnell K., Ward T.J., Robert V.A.R.G., Crous P.W., Geiser D.M., Kang S. DNA sequence-based identification of Fusarium: Current status and future directions. Phytoparasitica. 2015;43:583–595. doi: 10.1007/s12600-015-0484-z. [DOI] [Google Scholar]
- 41.Scauflaire J., Gourgue M., Munaut F. Fusarium temperatum sp. Nov. from maize, an emergent species closely related to Fusarium subglutinans. Mycologia. 2011;103:586–597. doi: 10.3852/10-135. [DOI] [PubMed] [Google Scholar]
- 42.Wulff E.G., Sørensen J.L., Lübeck M., Nielsen K.F., Thrane U., Torp J. Fusarium spp. associated with rice bakanae: Ecology, genetic diversity, pathogenicity and toxigenicity. Environ. Microbiol. 2010;12:649–657. doi: 10.1111/j.1462-2920.2009.02105.x. [DOI] [PubMed] [Google Scholar]
- 43.O’Donnell K., Sutton D.A., Fothergill A., McCarthy D., Rinaldi M.G., Brandt M.E., Zhang N., Geiser D.M. Molecular phylogenetic diversity, multilocus haplotype nomenclature, and in vitro antifungal resistance within the Fusarium solani species complex. J. Clin. Microbiol. 2008;46:2477–2490. doi: 10.1128/JCM.02371-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Geiser D.M., Jiménez-Gasco M., Kang S., Makalowska I., Veeraraghavan N., Ward T.J., O’Donnell K. FUSARIUMID v. 1.0: A DNA sequence database for identifying Fusarium. Eur. J. Plant Pathol. 2004;110:473–479. doi: 10.1023/B:EJPP.0000032386.75915.a0. [DOI] [Google Scholar]
- 45.Roux J., Steenkamp E.T., Marasas W.F.O., Wingfield M.J., Wingfield B.D. Characterization of Fusarium graminearum from Acacia and Eucalyptus using β-tubulin and histone gene sequences. Mycologia. 2001;93:704–711. doi: 10.1080/00275514.2001.12063201. [DOI] [Google Scholar]
- 46.Steenkamp E.T., Wingfield B.D., Coutinho T.A., Wingfield M.J., Marasas W.F. Differentiation of Fusarium subglutinans f. sp. pini by histone gene sequence data. Appl. Environ. Microbiol. 1999;65:3401–3406. doi: 10.1128/AEM.65.8.3401-3406.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yli-Mattila T., Parikka P., Lahtinen T., Rämö S., Kokkonen M., Rizzo A., Jestoi M., Hietaniemi V. In: Current Advances in Molecular Mycology. Gherbawy Y., Mach R.L., Rai M., editors. Nova Science; Hauppauge, NY, USA: 2009. [Google Scholar]
- 48.Webb K.M., Covey P.A., Hanson L.E. Pathogenic and phylogenetic analysis of Fusarium oxysporum from sugarbeet in Michigan and Minnesota. J. SugarBeet Res. 2012;49:38–56. doi: 10.5274/jsbr.49.1.38. [DOI] [Google Scholar]
- 49.Taha E.M., Rabie W., Mousa A.S.M., Yasser M.M., Fahmy Z.M. Phylogenetic diversity among Egyptian isolates of Fusarium species from sugar beet. Int. J. Agric. Technol. 2016;12:365–381. [Google Scholar]
- 50.Fulton J.C., Amaradasa B.S., Ertek T.S., Iriarte F.B., Sanchez T., Ji P., Paret M.L., Hudson O., Ali M.E., Dufault N.S. Phylogenetic and phenotypic characterization of Fusarium oxysporum f. sp. Niveum isolates from Florida-grown watermelon. PLoS ONE. 2021;16:e0248364. doi: 10.1371/journal.pone.0248364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Żelechowski M., Molcan T., Bilska K., Myszczyński K., Olszewski J., Karpiesiuk K., Wyrębek J., Kulik T. Patterns of diversity of Fusarium fungi contaminating soybean grains. Toxins. 2021;13:884. doi: 10.3390/toxins13120884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chaisiri C., Liu X.-Y., Lin Y., Li J.-B., Xiong B., Luo C.-X. Phylogenetic analysis and development of molecular tool for detection of Diaporthe citri causing melanose disease of citrus. Plants. 2020;9:329. doi: 10.3390/plants9030329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kumar S., Kashyap P.L., Singh R., Srivastava A.K. Springer Protocols Handbooks. Springer; Berlin/Heidelberg, Germany: 2013. Preservation and maintenance of microbial cultures; pp. 135–152. [DOI] [Google Scholar]
- 54.Kumar S., Singh R., Kashyap P.L., Srivastava A.K. Rapid detection and quantification of Alternaria solani in tomato. Sci. Hortic. 2013;151:184–189. doi: 10.1016/j.scienta.2012.12.026. [DOI] [Google Scholar]
- 55.O’Donnell K., Cigelnik E., Casper H.H. Molecular phylogenetic, morphological, and mycotoxin data support reidentification of the quorn mycoprotein fungus as Fusarium venenatum. Fungal Genet. Biol. 1998;23:57–67. doi: 10.1006/fgbi.1997.1018. [DOI] [PubMed] [Google Scholar]
- 56.Glass N.L., Donaldson G.C. Development of primer sets designed for use with the PCR to amplify conserved genes from filamentous ascomycetes. Appl. Environ. Microbiol. 1995;61:1323–1330. doi: 10.1128/aem.61.4.1323-1330.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hall T.A. BioEdit: A User-friendly biological sequence alignment editor and analysis program for windows 95/98/NT. Nucleic Acids Symp. Ser. 1999;41:95–98. [Google Scholar]
- 58.Thompson J.D., Higgins D.G., Gibson T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Librado P., Rozas J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25:1451–1452. doi: 10.1093/bioinformatics/btp187. [DOI] [PubMed] [Google Scholar]
- 60.Hudson R.R. A new statistic for detecting genetic differentiation. Genetics. 2000;155:2011–2014. doi: 10.1093/genetics/155.4.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kumar S., Stecher G., Tamura K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016;33:1870–1874. doi: 10.1093/molbev/msw054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bandelt H.J., Forster P., Rohl A. Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 1999;16:37–48. doi: 10.1093/oxfordjournals.molbev.a026036. [DOI] [PubMed] [Google Scholar]
- 63.Leigh J.W., Bryant D. Popart: Full-feature software for haplotype network construction. Methods Ecol. Evol. 2015;6:1110–1116. doi: 10.1111/2041-210X.12410. [DOI] [Google Scholar]
- 64.Singh R.P., Ma H., Rajaram S. Genetic analysis of resistance to scab in spring wheat cultivar Frontana. Plant Dis. 1995;79:238–240. doi: 10.1094/PD-79-0238. [DOI] [Google Scholar]
- 65.Kumar S., Saharan M.S., Panwar V., Chatrath R., Singh G.P. Genetics of Fusarium head blight resistance in three wheat genotypes. Indian J. Genet. Plant Breed. 2019;79:614–617. doi: 10.31742/IJGPB.79.3.11. [DOI] [Google Scholar]
- 66.Ramdial H., Latchoo R.K., Hosein F.N., Rampersad S.N. Phylogeny and haplotype analysis of fungi within the Fusarium incarnatum-equiseti species complex. Phytopathology. 2017;107:109–120. doi: 10.1094/PHYTO-05-16-0209-R. [DOI] [PubMed] [Google Scholar]
- 67.Periasamy M., Schafleitner R., Muthukalingan K., Ramasamy S. Phylogeographical structure in mitochondrial DNA of Legume Pod Borer (Maruca vitrata) population in tropical Asia and Sub-Saharan Africa. PLoS ONE. 2015;10:e0124057. doi: 10.1371/journal.pone.0124057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Vitale S., Santori A., Wajnberg E., Castagnone-Sereno P., Luongo L., Belisario A. Morphological and molecular analysis of Fusarium lateritium, the cause of gray necrosis of hazelnut fruit in Italy. Phytopathology. 2011;101:679–686. doi: 10.1094/PHYTO-04-10-0120. [DOI] [PubMed] [Google Scholar]
- 69.O’Donnell K., Rooney A.P., Proctor R.H., Brown D.W., McCormick S.P., Ward T.J., Frandsen R.J.N., Lysøe E., Rehner S.A., Aoki T., et al. Phylogenetic analyses of RPB1 and RPB2 support a middle cretaceous origin for a clade comprising all agriculturally and medically important Fusaria. Fungal Genet. Biol. 2013;52:20–31. doi: 10.1016/j.fgb.2012.12.004. [DOI] [PubMed] [Google Scholar]
- 70.Stielow J.B., Lévesque C.A., Seifert K.A., Meyer W., Iriny L., Smits D., Renfurm R., Verkley G.J.M., Groenewald M., Chaduli D., et al. One fungus, which genes? Development and assessment of universal primers for potential secondary fungal DNA Barcodes. Persoonia. 2015;35:242–263. doi: 10.3767/003158515X689135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Castellá G., Cabañes F.J. Phylogenetic diversity of Fusarium incarnatum-equiseti species complex isolated from Spanish Wheat. Antonie Van Leeuwenhoek. 2014;106:309–317. doi: 10.1007/s10482-014-0200-x. [DOI] [PubMed] [Google Scholar]
- 72.Zhao Z., Liu H., Luo Y., Zhou S., An L., Wang C., Jin Q., Zhou M., Xu J.-R. Molecular evolution and functional divergence of tubulin superfamily in the fungal tree of life. Sci. Rep. 2015;4:6746. doi: 10.1038/srep06746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Panwar V., Aggarwal A., Paul S., Kumar J., Saharan M.S. Distribution dynamics of Fusarium spp. causing fusarium head blight (FHB) in wheat at different geographical locations in India. S. Asian J. Exp. Biol. 2017;6:167–177. doi: 10.38150/sajeb.6(5).p167-177. [DOI] [Google Scholar]
- 74.Posada D., Crandall K.A. Evaluation of methods for detecting recombination from DNA wequences: Computer simulations. Proc. Natl. Acad. Sci. USA. 2001;98:13757–13762. doi: 10.1073/pnas.241370698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ma L.-J., Geiser D.M., Proctor R.H., Rooney A.P., O’Donnell K., Trail F., Gardiner D.M., Manners J.M., Kazan K. Fusarium pathogenomics. Annu. Rev. Microbiol. 2013;67:399–416. doi: 10.1146/annurev-micro-092412-155650. [DOI] [PubMed] [Google Scholar]
- 76.Carbone I., Kohn L. Applied Mycology and Biotechnology. Volume 4 Elsevier Science B.V.; Amsterdam, The Netherlands: 2004. Inferring process from pattern in fungal population genetics. [Google Scholar]
- 77.Yang B.J., Zhong S.B. Fourteen polymorphic microsatellite markers for the fungal banana pathogen Mycosphaerella Fijiensis: Permanent genetic resources. Mol. Ecol. Resour. 2008;8:910–912. doi: 10.1111/j.1755-0998.2008.02113.x. [DOI] [PubMed] [Google Scholar]
- 78.Brunner P.C., Stefanato F.L., Mcdonald B.A. Evolution of the CYP51 gene in Mycosphaerella Graminicola: Evidence for intragenic recombination and selective replacement. Mol. Plant Pathol. 2008;9:305–316. doi: 10.1111/j.1364-3703.2007.00464.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Nei M., Tajima F. Genetic drift and estimation of effective population size. Genetics. 1981;98:625–640. doi: 10.1093/genetics/98.3.625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Guo X.W., Fernando W.G.D., Seow-Brock H.Y. Population structure, chemotype diversity, and potential chemotype shifting of Fusarium graminearum in wheat fields of Manitoba. Plant Dis. 2008;92:756–762. doi: 10.1094/PDIS-92-5-0756. [DOI] [PubMed] [Google Scholar]
- 81.Keller M.D., Bergstrom G.C., Shields E.J. The Aerobiology of Fusarium graminearum. Aerobiologia. 2014;30:123–136. doi: 10.1007/s10453-013-9321-3. [DOI] [Google Scholar]
- 82.Dusabenyagasani M., Dostaler D., Hamelin R.C. Genetic diversity among Fusarium graminearum strains from Ontario and Quebec. Can. J. Plant Pathol. 1999;21:308–314. doi: 10.1080/07060669909501196. [DOI] [Google Scholar]
- 83.Akinsanmi O.A., Mitter V., Simpfendorfer S., Backhouse D., Chakraborty S. Identity and pathogenicity of Fusarium spp. isolated from wheat fields in Queensland and Northern New South Wales. Aust. J. Agric. Res. 2004;55:97. doi: 10.1071/AR03090. [DOI] [Google Scholar]
- 84.Miedaner T., Cumagun C.J.R., Chakraborty S. Population genetics of three important head blight pathogens Fusarium graminearum, F. pseudograminearum and F. culmorum. J. Phytopathol. 2008;156:129–139. doi: 10.1111/j.1439-0434.2007.01394.x. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data are contained within the article or gene sequences generated in this study were deposited in GenBank under the accession numbers listed in Table 1.