

Abstract
Glioblastomas (GBMs) preferentially generate acetyl-CoA from acetate as a fuel source to promote tumor growth. O-GlcNAcylation has been shown to be elevated by increasing O-GlcNAc transferase (OGT) in many cancers and reduced O-GlcNAcylation can block cancer growth. Here, we identify a novel mechanism whereby OGT regulates acetate-dependent acetyl-CoA and lipid production by regulating phosphorylation of acetyl-CoA synthetase 2 (ACSS2) by cyclin-dependent kinase 5 (CDK5). OGT is required and sufficient for GBM cell growth and regulates acetate conversion to acetyl-CoA and lipids. Elevating O-GlcNAcylation in GBM cells increases phosphorylation of ACSS2 on Ser-267 in a CDK5-dependent manner. Importantly, we show that ACSS2 Ser-267 phosphorylation regulates its stability by reducing polyubiquitination and degradation. ACSS2 Ser-267 is critical for OGT-mediated GBM growth as overexpression of ACSS2 Ser-267 phospho-mimetic rescues growth in vitro and in vivo. Importantly, we show that pharmacologically targeting OGT and CDK5 reduces GBM growth ex vivo. Thus, the OGT/CDK5/ACSS2 pathway may be a way to target altered metabolic dependencies in brain tumors.
INTRODUCTION
Glioblastoma (GBM) is the most common type of primary malignant brain tumor [1]. GBMs, along with brain metastasis, oxidize acetate via the metabolic enzyme acetyl-CoA synthetase short chain family member 2 (ACSS2), which catalyzes the ATP-dependent conversion of acetate and coenzyme A (CoA) into acetyl-CoA [2]. ACSS2 is required for the majority of acetate conversion to acetyl-CoA [3] which can be utilized for de novo biosynthesis of fatty acids which is essential for tumor growth and survival [4, 5]. Reducing ACSS2 levels can block cancer cell growth both in vitro and in vivo [3, 4, 6]. While ACSS2 is emerging as a novel therapeutic target for brain cancer, little is known regarding its regulation.
The hexosamine biosynthetic pathway utilizes major metabolites to generate UDP-N-acetylglucosamine (UDP-GlcNAc) which serves as a substrate for both N- and O-Glycosylation that regulate cellular behaviors in response to nutrient availability [7]. UDP-GlcNAc is also a substrate for O-linked β-N-acetylglucosamine (O-GlcNAc) transferase (OGT) which catalyzes the addition of O-GlcNAc moieties onto serine and threonine residues of nuclear and cytoplasmic proteins. Removal of O-GlcNAcylation is catalyzed by the glycoside hydrolase O-GlcNAcase (OGA) [8]. This modification alters protein functions, can regulate protein-protein interactions and phosphorylation states [9]. OGT and O-GlcNAcylation are elevated in most cancers [10], and targeting this modification inhibits tumor cell growth in vitro and in vivo [11, [12]. Recently, metabolomic profiling of breast cancer cells depleted of OGT revealed significant changes in lipid metabolites [13]. Thus, we hypothesized that OGT may regulate lipid-dependent cancers including GBMs.
Cyclin-dependent kinase 5 (CDK5) is an atypical member of the cyclin-dependent kinase family predominantly expressed in the brain and activated by non-cyclin activators such as p35, or its truncated product p25 [14]. While CDK5 is not known to be mutated in cancers [14], deregulation of CDK5 stimulates hyper-activation of downstream proteins such as RB, STAT3, FAK, vimentin [14–19] and regulates tumorigenesis through cell proliferation and metastatic invasion [14, 20]. While CDK5 levels have been found to be elevated in GBM tissue [21–23], its functional roles in this cancer are not known.
Here, we present evidence that OGT and O-GlcNAcylation levels are elevated in GBM tissues and cells and that OGT is required for GBM cell growth in vitro and in vivo. OGT regulates acetate metabolism via regulation of ACSS2 protein levels. Mechanistically, we show that increased OGT activity results in CDK5-mediated ACSS2 phosphorylation on Ser-267, stabilizing ACSS2 protein levels, and reducing polyubiquitination and degradation. OGT regulation of ACSS2 requires CDK5 and its effect on GBM growth requires Ser-267 phosphorylation of ACSS2. Importantly, ACSS2 Ser-267 phosphorylation is elevated in human GBM and pharmacologically targeting OGT or CDK5 reduces GBM tumors ex vivo suggesting that this pathway is required for GBM growth and may serve as potential therapeutic targets for treating brain cancer.
RESULTS
OGT and O-GlcNAc levels are elevated in glioblastoma and are required for tumor growth in vitro and in vivo
OGT and O-GlcNAcylation have been shown to be elevated in multiple cancers [10]. However, the expression and possible role of OGT in GBMs has not been investigated. We first examined levels of OGT from normal glial tissue through progressing stages of glioma including GBM (Grade IV). Using immunohistochemistry (IHC) analysis, we observed an increase in OGT-positive staining that correlates with disease progression (Fig. 1a). Notably, normal glial tissue exhibits minimal OGT-positive staining compared to Grade IV glioma (Fig. 1a). Moreover, analysis of 69 Grade IV GBM patient tissue samples for global O-GlcNAcylation via IHC showed 82% of these samples expressed medium to high O-GlcNAc (Fig. 1b). We also found that primary GBM cells contain elevated OGT and total level of proteins containing O-GlcNAcylation, using a pan-O-GlcNAc antibody, compared to normal human astrocytes (Fig. 1c). Similar elevation of OGT and O-GlcNAcylation was detected in established GBM U87-MG and T98G cell lines (Fig. 1d). Together, these results show that human GBMs contain elevated levels of OGT and O-GlcNAcylation.
Fig. 1. OGT and O-GlcNAcylation levels are elevated in human glioblastoma.
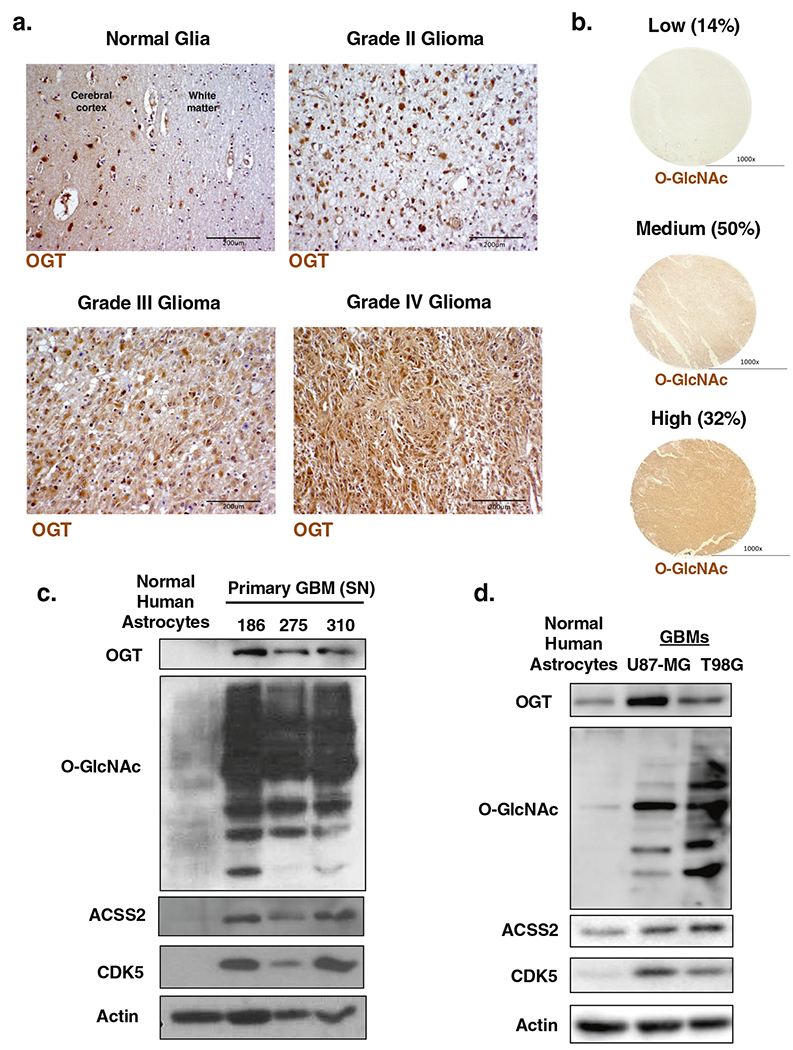
a Formaldehyde-fixed/paraffin-embedded clinical tissue samples representing WHO grades of gliomas were tested for OGT expression by immunohistochemistry (×20, scale bar 200 μm). b Immunohistochemical staining for O-GlcNAc on a tissue microarray (n = 69) of Grade IV glioblastoma patient biopsies (×4, scale bar 1000 μm). c Cell lysate from normal human astrocytes and primary cells isolated from three different GBM patients were collected for immunoblot analysis with the indicated antibodies. d Cell lysates of normal human astrocytes, U87-MG and T98G glioblastoma cell lines were collected for immunoblot analysis with the indicated antibodies.
To examine whether OGT is required for GBM growth, we targeted OGT with shRNA in GBM cell lines. We observed that suppression of OGT was sufficient to impede cell growth in both U87-MG (Fig. 2a, b) and T98G (Fig. 2c, d) cells, as indicated by crystal violet staining. We also tested whether altering OGT expression could impact anchorage-independent growth. Reducing OGT expression with stable expression of RNAi (Fig. 2a, c), we observed a significant reduction in the anchorage-independent growth in both U87-MG and T98G (Fig. 2e, Supplementary Fig. S1a) compared to controls. Additionally, we found that reducing OGT levels in two different primary GBM cell lines SN310 and SN186 (Fig. 2f) significantly blocked neurosphere formation (Fig. 2g, h). To ensure OGT contribution to GBM growth required its catalytic function, we also examined effects of treating GBM cells with a pharmacological inhibitor of O-GlcNAcylation Ac-5S-GlcNAc [24–26]. Treating U87-MG cells with Ac-5S-GlcNAc reduced total O-GlcNAcylation and also reduced cell growth (Supplementary Fig. S1b). Similar results were seen in T98G cells (Supplementary Fig. S1c) and primary GBM SN-186 cell lines Supplementary Fig. S1d). To test whether reducing OGT expression could block GBM cell growth in vivo, we utilized an intracranial orthotopic xenograft model. Reduction of OGT expression in U87-MG cells expressing luciferase (Supplementary Fig. S1e) resulted in a significant decrease in tumor growth in vivo as measured by bioluminescence (Fig. 2i). Representative images of H&E sections show a striking reduction in tumor growth at Day 21 in mice injected with OGT depleted cells (Fig. 2i). Importantly, mice that were grafted with GBM cells where OGT expression was reduced also had significantly extended survival (Fig. 2j), displaying a median survival of 42 days, compared to 20 days for the control group. Altogether, these data indicate that OGT and O-GlcNAc levels are elevated in human GBM and OGT contributes to GBM cell growth in vitro and in vivo.
Fig. 2. OGT is required for glioblastoma growth in vitro and in vivo.
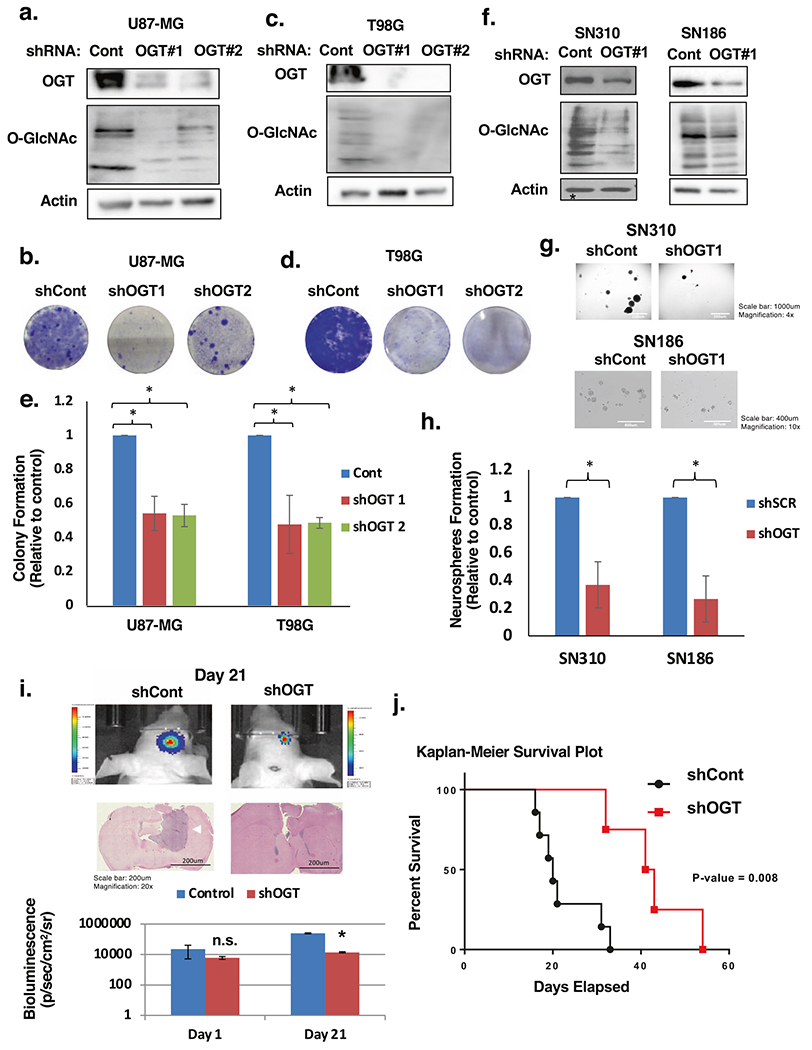
a Cell lysates from U87-MG cells expressing control or OGT shRNA were collected for immunoblot analysis with the indicated antibodies. b U87-MG cells infected with shControl or shOGT lentivirus effect on cell growth visualized with crystal violet staining. c Cell lysates from T98G cells expressing control or OGT shRNA were collected for immunoblot analysis with the indicated antibodies. d T98G cells infected with shControl or shOGT lentivirus effect on cell growth visualized with crystal violet staining. e Anchorage-independent growth assay comparing the colony formation of control or OGT shRNA expressing in indicated cells. Data are quantified and presented as average relative to control from three independent experiments. Student’s t-test reported as mean ± SD; *p < 0.05. f Cell lysates from SN310 (left) or SN186 (right) primary GBM cells expressing control or OGT shRNA were collected for immunoblot analysis with the indicated antibodies. g Cells from f were then plated for a neurosphere formation assay for 8 days and representative image taken. h Neurospheres were quantified for SN310 and SN186 cells and presented as average relative to control from three independent experiments. Student’s t-test reported as mean ± SD. *p-value < 0.05. i Representative images of bioluminescent (top) detection of tumors from mice injected with shControl and shOGT U87-MG cells 21 days post-injection. Representative images of H&E analysis (middle) on coronal sections from mice harboring shControl or shOGT tumors at Day 21. White arrow pointing to control tumor. Data are quantified and presented as average from mice injected with U87-MG cells expressing shControl (n = 5) or shOGT mice (n = 7). (bottom). Student’s t-test reported as mean ± SD; *p < 0.005. j Kaplan–Meier survival plot comparing overall survival of mice inoculated with shControl (n = 7) or shOGT (n = 4) U87-MG cells. Mantel–Cox log rank test P = 0.008.
OGT regulates lipid accumulation, acetyl-CoA, and acetate metabolism
Several groups have characterized the importance of lipid metabolism [5], and more specifically, acetate metabolism for supporting GBM growth [3, 4]. Thus, we investigated the effect of altering OGT on lipid accumulation, and acetate-dependent acetyl-CoA metabolism in GBM cells. Consistent with OGT being critical for GBM cell growth, overexpression of OGT in U87-MG cells increased O-GlcNAc modified proteins (Fig. 3a) and anchorage-independent growth (Fig. 3b). OGT overexpressing GBM cells also contained increased intracellular lipid droplet accumulation as measured by nile red staining (Fig. 3c), increased free fatty acids (Fig. 3d) and cellular acetyl-CoA levels (Fig. 3e). Conversely, stable knockdown of OGT in U87-MG (Supplementary Fig. S2a) cells contained reduced nile red staining (data not shown), free fatty acids (Supplementary Fig. S2b) and acetyl-CoA (Supplementary Fig. S2c) levels compared to control cells. Similar results were seen in T98G cells containing stable knockdown of OGT (Supplementary Fig. S2d–f) while treating T98G cells with OGA inhibitor increased acetyl-CoA levels (Supplementary Fig. S2g).
Fig. 3. OGT promotes acetate and lipid accumulation in glioblastoma cells.
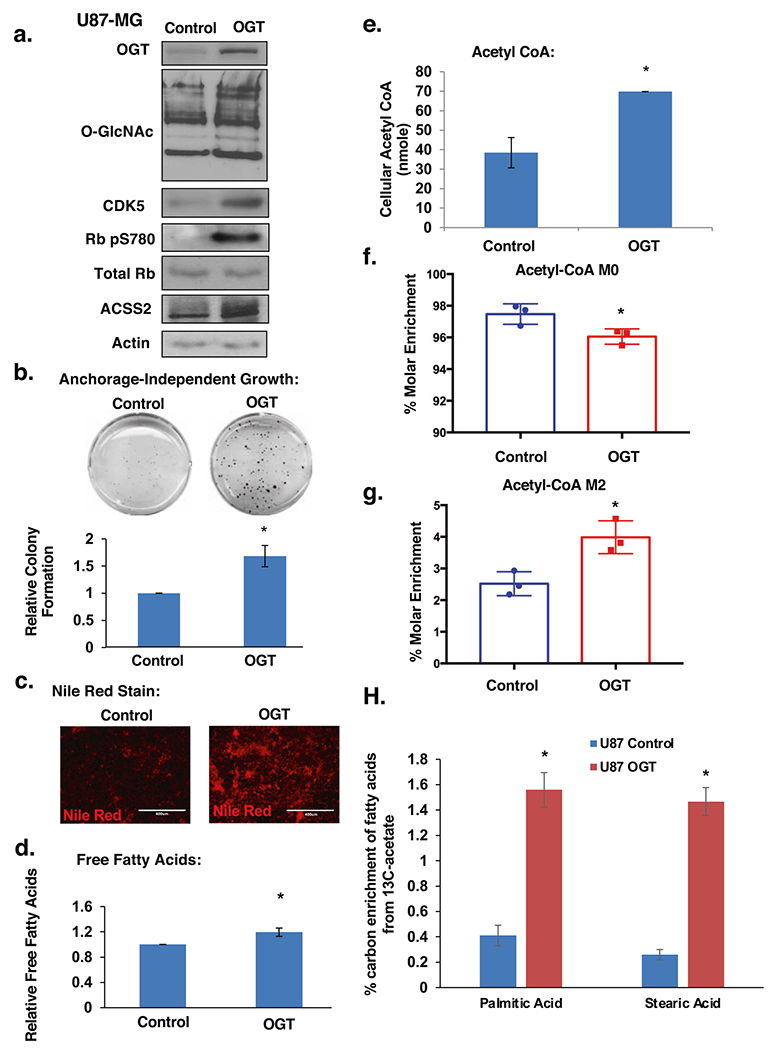
a Cell lysates from U87-MG cells stably overexpressing control or OGT were collected for immunoblot analysis with the indicated antibodies. b Control or stably overexpressing OGT U87-MG cells were placed in soft agar assay. Representative images from anchorage-independent growth assay (top) and colonies were counted and quantified (bottom) as average relative to control from three independent experiments. Student’s t-test reported as mean ± SD. *p-value < 0.005. c Representative images of nile red staining of U87-MG cells under same conditions as in a. d Measurement of relative free fatty acids in U87-MG cells stably overexpressing control or OGT and quantified as average relative to control. Student’s t-test reported as mean ± SD. *p-value < 0.05. e Measurement of acetyl-CoA extracted from U87-MG cells stably overexpressing control or OGT. Student’s t-test reported as mean ± SD. *p-value < 0.05. f Graphical representation of the percent molar enrichment of unlabeled acetyl-CoA in U87-MG cells stably overexpressing control or OGT for 4 h. Student’s t-test reported as mean ± SD. *p-value < 0.05. g U87-MG control and OGT overexpressing cells were labeled with 100 μM 13C2-acetate for 4 h. Measurement of the percent molar enrichment for labeled (M2) acetyl-CoA. Student’s t-test reported as mean ± SD. *p-value < 0.05. h U87-MG control and OGT overexpressing cells were labeled with 200 μM 13C2-acetate for 48 h. Graphical representation showing total carbon enrichment of palmitic acid and stearic acid. Student’s t-test reported as mean ± SD. *p-value < 0.05.
Since GBM cells are highly dependent on acetate metabolism to generate acetyl-CoA and regulate growth [4], we hypothesized that OGT may regulate acetate utilization. To test this, we exposed U87-MG control cells and cells overexpressing OGT with stable isotope labeled sodium acetate (1,2-13C2-acetate) to track the incorporation of acetate into acetyl-CoA. Cells overexpressing OGT displayed significantly increased acetate uptake as labeled acetate was depleted from the media (Supplementary Fig. S2h). Moreover, within the context of the increased pool of acetyl-CoA, we detect a 2% reduction in molar enrichment of unlabeled acetyl-CoA M+0 (Fig. 3f) and a corresponding increase in molar enrichment of acetyl-CoA M2 (Fig. 3g) in OGT overexpressing cells compared to control cells indicating an increased utilization of acetate in generating acetyl-CoA. Importantly, we show a significant increase in acetate-dependent labeling of fatty acids (Fig. 3h; Supplementary Fig. S2i, S2j) in OGT overexpressing cells compared to controls. Consistent with the idea that OGT regulation of acetate conversion to lipids may be critical for cell growth, exogenous addition of fatty acid oleic acid rescued OGT knockdown mediated anti-growth effects (Supplementary Fig. S1f). These data implicate a role for OGT in regulating the metabolism of acetate into acetyl-CoA and lipids and contributing to GBM lipid synthesis and growth.
O-GlcNAcylation regulates ACSS2 S267 phosphorylation in a CDK5-dependent manner
ACSS2 is a critical regulator of acetate conversion to acetyl-CoA and for growth of GBM cells [3, 27]. Therefore, we examined ACSS2 expression in the context of altered O-GlcNAcylation. Consistent with previous findings, we found ACSS2 levels to be elevated in both primary GBM (Fig. 1c) and established GBM cell lines (Fig. 1d) compared to normal astrocytes. U87-MG cells stably expressing OGT shRNA contained lower levels of ACSS2 protein (Supplementary Fig. S2a). RNA levels of ACSS2 were not reduced in OGT depleted U87-MG cells as measured by quantitative RT-PCR (qRT-PCR) (Supplementary Fig. S3a). Similar decreases in ACSS2 protein levels were seen in OGT depleted primary GBM cells SN310 and SN186 (Fig. 4a) and T98G (Supplementary Fig. S2d) cells. Conversely, we observe stabilization of ACSS2 protein when U87-MG cells (Fig. 3a; Supplementary Fig. S3b) or primary GBM SN310 cells (Supplementary Fig. S3c) were stably overexpressing OGT or U87-MG cells were treated with an OGA inhibitor NButGT (Supplementary Fig. S3d). ACSS2 protein does not appear to be directly modified by O-GlcNAc as exogenous ACSS2 was not detected using click chemistry (Supplementary Fig. S3e) while endogenous OGT, which is O-GlcNAcylated [28], was detected to be O-GlcNAcylated in GBM cells. Thus, O-GlcNAcylation likely regulates ACSS2 protein stability via an indirect mechanism.
Fig. 4. Ser267-ACSS2 phosphorylation is mediated by O-GlcNAcylation in a CDK5-dependent manner.
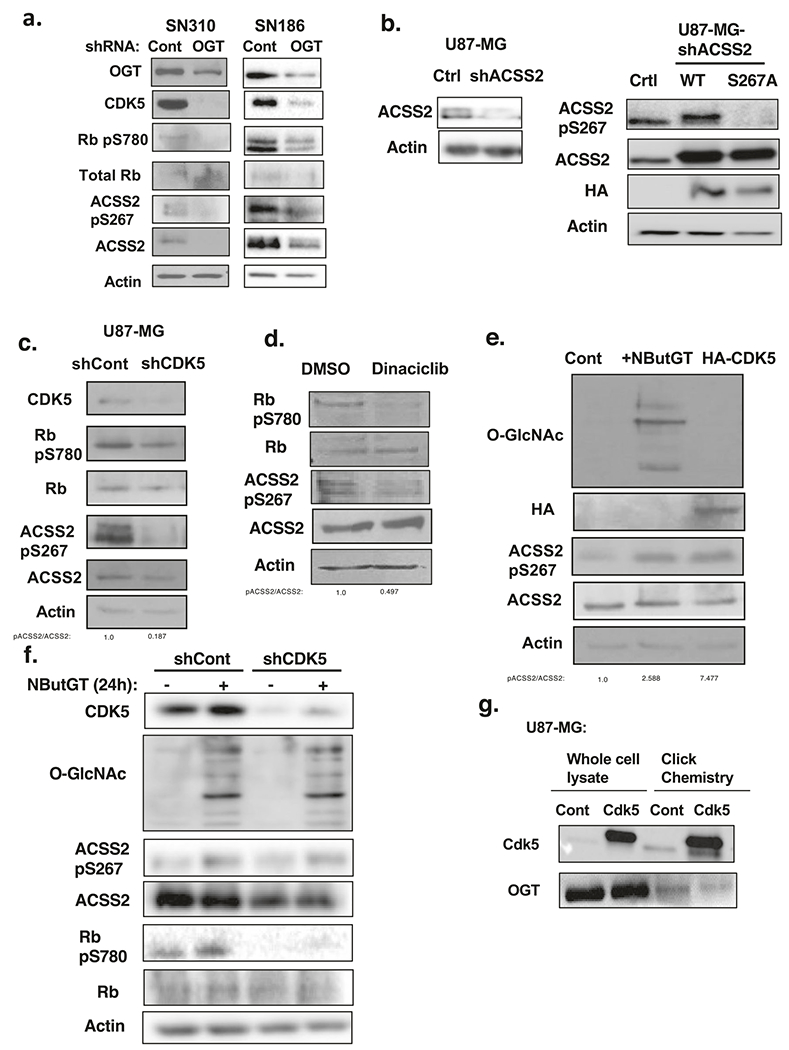
a Cell lysates from SN310 (left) or SN186 (right) primary GBM cells expressing control or OGT shRNA were collected for immunoblot analysis with the indicated antibodies. b Cell lysates from U87-MG cells stably expressing shRNA against endogenous ACSS2 (left) and overexpressing control, wild-type ACSS2 or ACSS2 S267A mutant (right) were collected for immunoblot analysis with the indicated antibodies. c Cell lysates from of U87-MG cells stably expressing shRNA against control or CDK5 were collected for immunoblot analysis with the indicated antibodies. d Cell lysates from U87-MG cells treated for 48 h with pan-CDK inhibitor dinaciclib (5 nM) were collected for immunoblot analysis with the indicated antibodies. e Cell lysate from U87-MG cells that were transfected with control plasmid with and without treatment of 100 μM OGA inhibitor (NButGT) for 24 h or HA-CDK5 plasmid were collected for immunoblot analysis with the indicated antibodies. f Cell lysate from U87-MG cells stably expressing control or CDK5 shRNA and treated with DMSO or 100 μM NButGT for 24 h were collected for immunoblot analysis with the indicated antibodies. g Whole lysate and avidin pulldown from CuAAC “click” labeling assay of U87-MG cells overexpressing control and CDK5 blotted with anti-CDK5 antibody and positive control OGT to determine O-GlcNAcylation.
O-GlcNAcylation can alter the phosphorylation state of proteins [9]. To analyze potential changes in ACSS2 phosphorylation regulated by O-GlcNAcylation, we analyzed changes in exogenous immunoprecipitated ACSS2 phosphorylation via mass-spectrometry in GBM cells treated with control or OGA inhibitor (Supplementary Fig. S3f). Proteomic analysis identified only Ser-267 of ACSS2 as being phosphorylated in NButGT-treated conditions (data not shown). Predictive analyses for phosphorylation sites (ScanSite; [29]) for ACSS2 revealed Ser-267 as a putative CDK5 phosphorylation site (Supplementary Fig. S3g). Alignment analysis revealed the consensus CDK5 motif containing Ser-267 (SQSPPIKR) as being evolutionarily conserved in mammals (data not shown). Consistent with our data, PhosphoSitePlus [30] lists 13 studies that have identified human ACSS2 Ser-267 as being phosphorylated in different cancers including breast, lung, cervical, ovarian, and skin cancers. However, the functional role of this phosphorylation is not known. Therefore, these data suggest that elevated O-GlcNAcylation increases ACSS2 phosphorylation on Ser-267, a putative CDK5 site.
To establish ACSS2 as a putative substrate for CDK5 phosphorylation, we performed an in vitro kinase assay. We observed a protein concentration-dependent increase in ACSS2 phosphorylation by CDK5 (Supplementary Fig. S3h), confirming ACSS2 as a bona-fide substrate of CDK5. Importantly, ACSS2 Ser-267 to alanine (S267A) mutant contained reduced phosphorylation by CDK5 in vitro (Supplementary Fig. S3i). To test whether Ser-267 was also phosphorylated in GBM cells, we knocked down endogenous ACSS2 with stable expression of shRNA against ACSS2 targeting the 3′ UTR and overexpressed wild-type ACSS2 or ACSS2 S267A mutant. Mutation of ACSS2 S267 into alanine abrogated ACSS2 phosphorylation, which was detected using an antibody specifically recognizing ACSS2 pS267 (Fig. 4b). To further confirm the specificity of the phospho-ACSS2-S267 antibody, we immunoprecipitated wild-type ACSS2 and ACSS2-S267A mutant from U87-MG cells and found that the ACSS2 p267 antibody only detected wild-type ACSS2 protein (Supplementary Fig. S3j). Phosphorylation of ACSS2 on Ser-267 in GBM cells is dependent on CDK5 activity as stable expression of CDK5 shRNA reduces phosphorylation of ACSS2 on Ser-267 (Fig. 4c) as well as phosphorylation of RB on Ser-780 (Fig. 4c), a CDK5-specific phosphorylation site [31, 32]. Similar results were seen in cells treated with the pan-CDK inhibitor dinaciclib that targets CDK5 (Fig. 4d) [33]. Conversely, overexpression of HA-CDK5 in U87-MG cells increased ACSS2 Ser-267 phosphorylation (Fig. 4e). In addition, we found that CDK5 and ACSS2 interact in vitro (Supplementary Fig. S3k). However, in GBM cells this interaction was only detected in conditions of elevated O-GlcNAcylation as immunoprecipitation of CDK5 showed an interaction with ACSS2 only in cells treated with OGA inhibitor (Supplementary Fig. S3l). CDK5 was also found to interact with phosphorylated ACSS2-S267, determined by immunoprecipitating with antibody specifically recognizing ACSS2 pS267, and this interaction was increased under conditions of elevated O-GlcNAcylation (Supplementary Fig. S4a). Consistent with the mass-spectrometry results, we detected an increase of ACSS2 Ser-267 phosphorylation in cells treated with an OGA inhibitor (Fig. 4e, f). Conversely, inhibition of O-GlcNAcylation by treating cells with OGT inhibitor (Ac-5S-GlcNAc) reduced ACSS2 Ser-267 phosphorylation in U87-MG (Supplementary Fig. S1b), T98G (Supplementary Fig. S1c) and SN-186 cells (Supplementary Fig. S1d). We also tested whether OGT-mediated phosphorylation of ACSS2 occurred in normal brain cells or tissue. Increased O-GlcNAcylation in normal human astrocytes treated with an OGA inhibitor did not alter ACSS2 Ser-267 phosphorylation compared to GBM cells (Supplementary Fig. S4b). Using an ex vivo brain slice model [34], treating tumor-free brain tissue with an OGA inhibitor elevated O-GlcNAcylation but had no effect on ACSS2 Ser-267 phosphorylation (Supplementary Fig. S4c) suggesting that OGT-dependent regulation of ACSS2 Ser-267 phosphorylation may be cancer cell-specific. Increased O-GlcNAcylation also increased CDK5 activity as measured by phosphorylation of RB on Ser-780 (Figs. 3a and 4f). Importantly, O-GlcNAc regulation of ACSS2 Ser-267 phosphorylation was reduced in cells stably expressing CDK5 shRNA (Fig. 4f, Supplementary Fig. S4d). Consistent with these results, cells overexpressing OGT contain increased CDK5 levels and activity (Fig. 3a) and GBM cells stably expressing OGT RNAi contain reduced levels of CDK5 and activity and ACSS2 ser-267 phosphorylation (Fig. 4a; Supplementary Fig. S2a and d). A previous study has shown CDK5 to be O-GlcNAcylated in neurons [35], thus we tested whether CDK5 can be O-GlcNAcylated in GBM cells. We show using click chemistry that endogenous and exogenous CDK5 is highly O-GlcNAcylated in GBM cells (Fig. 4g). These results indicate that O-GlcNAc regulates ACSS2 phosphorylation on Ser-267 in a CDK5-dependent manner, possibly via direct CDK5 O-GlcNAcylation.
ACSS2 S267 phosphorylation blocks degradation of ACSS2 and is required for GBM growth
Since we observed stabilization of ACSS2 protein when GBM cells were overexpressing OGT (Fig. 3a; Supplementary Fig. S3b and c) or following OGA inhibition (Supplementary Fig. S3d), we hypothesized that OGT and CDK5 regulate ACSS2 stability via phosphorylation of Ser-267. To test whether OGT regulates ACSS2 degradation, U87-MG cells stably expressing control shRNA or OGT shRNA were treated with the proteasome inhibitor lactacystin. OGT shRNA-mediated inhibition of ACSS2 protein levels could be reversed by treatment with lactacystin (Fig. 5a) suggesting ACSS2 is being regulated at the proteasomal level. To test whether ACSS2 Ser-267 phosphorylation alters polyubiquitination of ACSS2, U87-MG cells stably expressing wild-type ACSS2 and S267 phosphosite mutants were transfected with ubiquitin (WT-Ub) or ubiquitin-K48R (K48R-Ub) mutant. Following immunoprecipitation of ACSS2, we show that wild-type ACSS2 is polyubiquitinated and that ACSS2-S267A mutant contains significantly increased polyubiquitination compared to wild-type ACSS2 and ACSS2-S267D phospho-mimetic mutant in cells transfected with WT-Ub (Fig. 5b; Supplementary Fig. S4e). Since mutation of lysine 48 to arginine renders ubiquitin (Ub) unable to form poly-Ub chains via lysine 48 linkages with other Ub molecules [36], we tested effect of K48R-Ub on polyubiquitination of ACSS2-S267A. K48 polyubiquitination of ACSS2-S267A was significantly reduced in cells transfected with ubiquitin K48R mutant (K48R-Ub) compared to WT-Ub (Fig. 5b; Supplementary Fig. S4e). This data suggests that Ser-267 phosphorylation alters polyubiquitination of ACSS2. Consistent with the idea that ACSS2 phosphorylation of Ser-267 stabilizes and blocks degradation of ACSS2, we show that the protein half-life of ACSS2 S267A mutant is significantly reduced compared to wild-type ACSS2 or ACSS2 S267D phospho-mimetic mutant (Fig. 5c) in cells treated with cyclohexamide. To test whether phosphorylation of ACSS2 Ser-267 plays a role in tumorigenesis in GBM cells, we stably reduced endogenous ACSS2 expression utilizing a shRNA targeting the 3′UTR (Fig. 5d) and then stably re-expressed either wild-type ACSS2, ACSS2 S267A or ACSS2 S267D phospho-mimetic mutant (Fig. 5d). U87-MG cells overexpressing wild-type ACSS2 or phospho-mimetic S267D mutant, but not ACSS2 S267A mutant, were able to rescue anchorage-independent growth (Fig. 5e). Consistent with results seen in vitro, U87-MG cells expressing wild-type ACSS2, but not cells containing ACSS2 S267A mutant, were able to form tumors in vivo (Fig. 5f; Supplementary Fig. S4f). A previous study has shown that AMPK phosphorylation of ACSS2 on S659 increases nuclear localization of ACSS2 [37]. Phosphorylation of ACSS2 on S267 had no effect on changes in localization as wild type, S267A and S267D mutants all had similar cellular localization (data not shown). These results indicate that phosphorylation of ACSS2 on S267 is critical for its protein stability, polyubiquitination and for GBM cell growth in vitro and in vivo.
Fig. 5. Phosphorylation of Ser267 enhances stability of ACSS2 and is required for GBM growth.
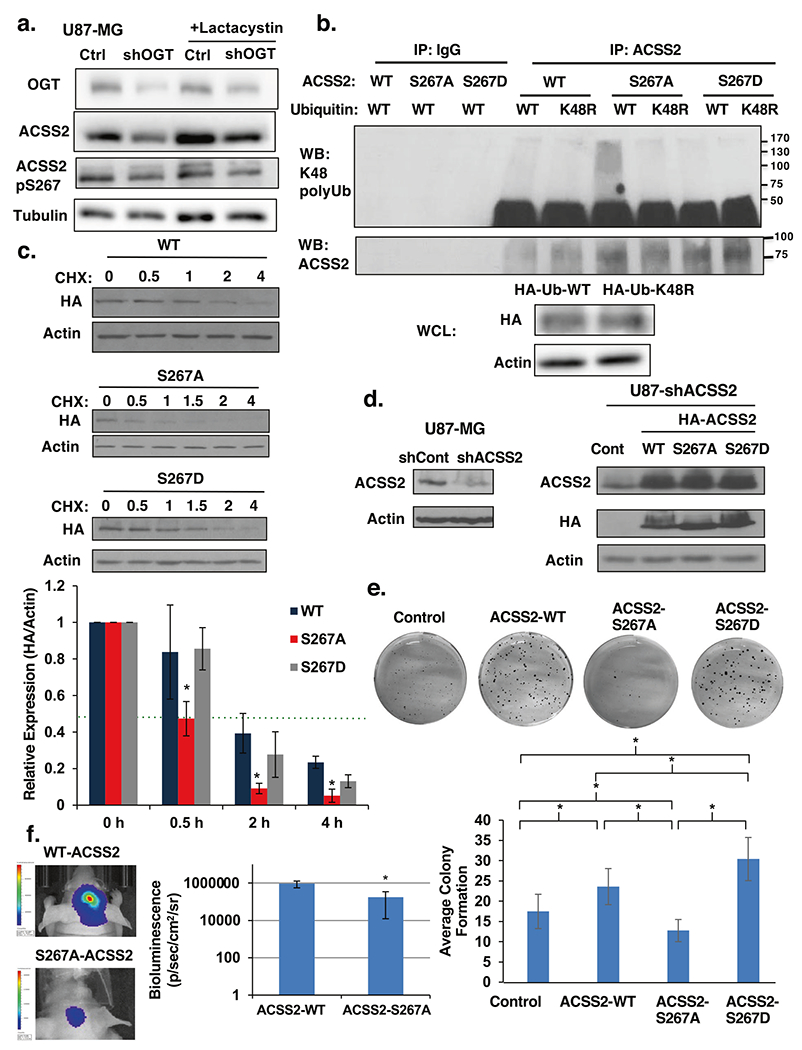
a Cell lysate from U87-MG cells stably expressing control or OGT shRNA and treated with the proteasomal inhibitor 10 μM lactacystin for 6 h were collected for immunoblot analysis with the indicated antibodies. b Immunoprecipitation was performed with the indicated antibodies from U87-MG cell lysates stably expressing wild-type (WT)-, S267A-, or S267D- HA-ACSS2 and transfected with Ub-WT or Ub-K48. c Cell lysate from U87-MG cells stably expressing wild-type (WT)-, S267A-, or S267D-HA-ACSS2 in U87-MG glioblastoma cells treated with 10 μg/μl cycloheximide for indicated time (hours) were collected for immunoblot analysis with the indicated antibodies (top). Densitometry quantification of three independent time-course experiments presented as relative to expression (HA/actin) to 0 h (bottom). Green dotted line represents 0.5 relative expression. Student’s t-test reported as mean ± SD. *p-value < 0.05. d Cell lysates from U87-MG cells stably expressing ACSS2 shRNA against endogenous 3′ UTR of ACSS2 (left) and stably overexpressing wild-type ACSS2, ACSS2-S267A and ACSS2-S267D mutant (right) were collected for immunoblot analysis with the indicated antibodies. e Representative image of cells in d seeded into an anchorage-independent growth assay and imaged at day 14 (top). Data are quantified and presented as average from at least three independent experiments (bottom). Student’s t-test reported as mean ± SD. *p-value < 0.05. f Representative images of tumor growth detected via bioluminescence at Day 16 following injection of U87-MG-luciferase cells WT-ACSS2 or S267A-ACSS2 (left). Quantification of tumor size (WT n = 4, S267A n = 4) (right). Student’s t-test reported as mean ± SD. *p-value < 0.05.
CDK5 regulates acetate metabolism and GBM growth via ACSS2-S267 phosphorylation
CDK5 has been implicated in the development and progression of multiple cancers [14]. However, its role in GBM has not been demonstrated. CDK5 levels have been shown to be elevated in GBM tissue [23]. A high level of CDK5 expression predicted poor prognosis in GBM patients (Supplementary Fig. S5a). Consistent with this data, we detect an increase in CDK5 levels in primary (Fig. 1c) and established GBM cell lines (Fig. 1d) compared to normal human astrocytes. Since CDK5 phosphorylates ACSS2 S267 in vitro and in cells, we tested the role of CDK5 on acetate metabolism and GBM cell growth. To test whether CDK5 regulates acetate metabolism and GBM cell growth we reduced CDK5 levels via shRNA which reduced ACSS2 phosphorylation on Ser-267 (Fig. 6a). We observed that suppression of CDK5 was sufficient to impede the growth of U87-MG cells as indicated by crystal violet staining (Fig. 6b) and anchorage-independent growth (Supplementary Fig. S5b). Reducing CDK5 levels in T98G cells also blocked growth (Supplementary Fig. S5c) and anchorage-independent growth (Supplementary Fig. S5d) and knockdown in primary GBM cells reduced ACSS2 phosphorylation and (Fig. 6c; Supplementary Fig. S5e) and significantly blocked neurosphere formation (Fig. 6c; Supplementary Fig. S5f). Consistent with the idea that CDK5 regulates ACSS2, we also found that depleting CDK5 levels in GBM cells reduced acetyl-CoA levels (Fig. 6d) and nile red staining (data not shown). Importantly, we show that reduction in CDK5 expression in U87-MG cells resulted in a significant decrease of 1,2-13C2-acetate incorporation into acetyl-CoA (Fig. 6e). Consistent with the idea that CDK5 regulation of acetate conversion to acetyl-CoA may be critical for cell growth, exogenous addition of fatty acid oleic acid partly rescued CDK5 knockdown mediated anti-growth effects (Supplementary Fig. S5g). Moreover, we show that reduction of CDK5 expression in U87-MG cells resulted in a significant decrease in tumor growth in vivo as measured by bioluminescence and histology (Fig. 6f). To test whether OGT and O-GlcNAc-mediated growth in GBM cells was dependent on CDK5 expression we tested the effect of reducing CDK5 in OGT overexpressing cells. The increase in anchorage-independent growth observed in U87-MG cells stably overexpressing OGT was reduced as a result of CDK5 knockdown cells (Supplementary Fig. S6a and b). Consistent with OGT regulating ACSS2, the observed OGT-mediated increase in ACSS2-S267 phosphorylation (Supplementary Fig. S6c) and acetyl-CoA (Supplementary Fig. S6d) was reduced in CDK5 knockdown cells. We detected similar decrease of ACSS2-S267 phosphorylation (Supplementary Fig. S6e) and acetyl-CoA (Supplementary Fig. S6f) in U87-MG cells treated with OGA inhibitor containing CDK5 knockdown. To test whether ACSS2 phosphorylation was required for CDK5 depletion-mediated effects, we overexpressed wild-type HA-ACSS2, HA-ACSS2 S267A and HA-ACSS2 S267D mutant in the context of CDK5 knockdown (Fig. 6g). Cells stably overexpressing exogenous wild-type ACSS2 and ACSS2 S267D mutant, but not ACSS2 S267A mutant, partly restored growth (Fig. 6h) and nile red staining (Supplementary Fig. S6g) in CDK5 knockdown cells. Thus, these data suggest that CDK5 can regulate acetate metabolism and GBM growth in vitro and in vivo and that OGT-mediated cell growth and acetyl-CoA regulation is partly dependent on CDK5. In addition, we show that CDK5-mediated growth in GBM cells is partly dependent on phosphorylation of ACSS2 Ser267.
Fig. 6. CDK5 is a critical regulator of GBM growth and requires ACSS2 phosphorylation.
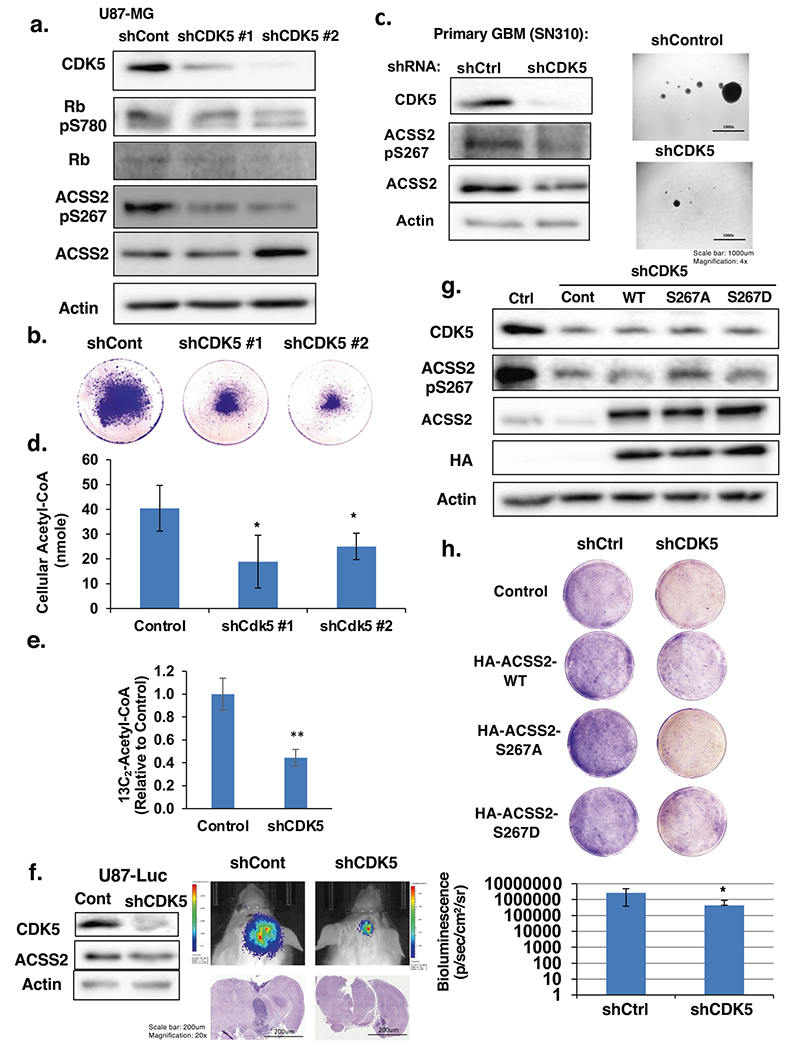
a Cell lysates from of U87-MG cells stably expressing shRNA against control or CDK5 were collected for immunoblot analysis with the indicated antibodies. b Representative images of U87-MG cells stained with crystal violet stably expressing of shRNA targeting control or CDK5. c Cell lysates from SN310 primary GBM cells expressing control or CDK5 shRNA were collected for immunoblot analysis with the indicated antibodies (left). Representative images from neurosphere growth assay (right) at day 6 comparing the neurosphere formation of control or CDK5 shRNA expressing SN310 cells. d Measurement of acetyl-CoA extracted from U87-MG cells stably overexpressing control or CDK5 shRNA. Student’s t-test reported as mean ± SD. *p-value < 0.05. e U87-MG cells stably expressing shRNA against control or CDK5 were labeled with 13C2-acetate for 4 h and 13C2-acetyl-CoA concentrations were measured and shown. Student’s t-test reported as mean ± SD. **p-value < 0.0001 f Cell lysate from U87-MG-Luciferase cells stably expressing control or CDK5 shRNA were collected for immunoblot analysis with the indicated antibodies (left). Representative images of tumor growth detected via bioluminescence and H&E staining at Day 16 following injection of U87-MG-luciferase cells expressing control or CDK5 shRNA (middle). Quantification of tumor size at Day 16 (shControl n = 5, shCDK5 n = 5) (right). Student’s t-test reported as mean ± SD. *p-value < 0.05. g Cell lysates from U87-MG cells stably expressing shRNA against control or CDK5 and overexpressing HA-ACSS2-WT (wild type), HA-ACSS2-S267A, or HA-ACSS2-S267D mutants were collected for immunoblot analysis with the indicated antibodies. h Representative images of U87-MG cells stained with crystal violet containing shRNA targeting control or CDK5 and overexpressing ACSS2-WT (wild type), ACSS2-S267A, or ACSS2-S267D mutants.
OGT regulates acetate metabolism and GBM growth via ACSS2 S267 in vitro and in vivo
We have shown that GBM cell growth in vitro (Fig. 5e) and in vivo (Fig. 5f) requires ACSS2 Ser-267 phosphorylation and that CDK5-mediated regulation of cell growth also requires ACSS2 Ser-267 phosphorylation (Fig. 6h). To test whether OGT regulation of GBM lipid accumulation and growth also requires ACSS2 Ser-267 phosphorylation, we tested whether overexpression of wild-type ACSS2 or ACSS2 phospho-mimetic mutant would rescue the growth defects observed in OGT knockdown cells. Stable OGT knockdown reduced anchorage-independent growth in vitro could be rescued by overexpressing wild-type ACSS2 and ACSS2 S267D mutant, but not ACSS2 S267A mutant (Fig. 7a–c). In addition, we found that overexpressing wild-type ACSS2 and ACSS2 S267D mutant, but not ACSS2 S267A mutant, could partly rescue nile red staining (Supplementary Fig. S7a). Consistent with the idea that OGT regulates acetyl-CoA via ACSS2, the OGT RNAi-mediated decrease in acetyl-CoA was reversed by overexpressing the ACSS2-267D mutant (Supplementary Fig. S7b, c). Similar to results in vitro, U87-MG cells expressing the phospho-mimetic mutant ACSS2 S267D, but not cells containing an ASCC2 S267A mutant, were able to partly rescue the growth effects of OGT depletion in vivo (Fig. 7d, e; Supplementary Fig. S7d). Thus, OGT regulation of GBM cell growth in vivo is partly dependent on ACSS2 Ser-267 phosphorylation.
Fig. 7. Phosphorylation of S267-ACSS2 is required for OGT-mediated GBM growth.
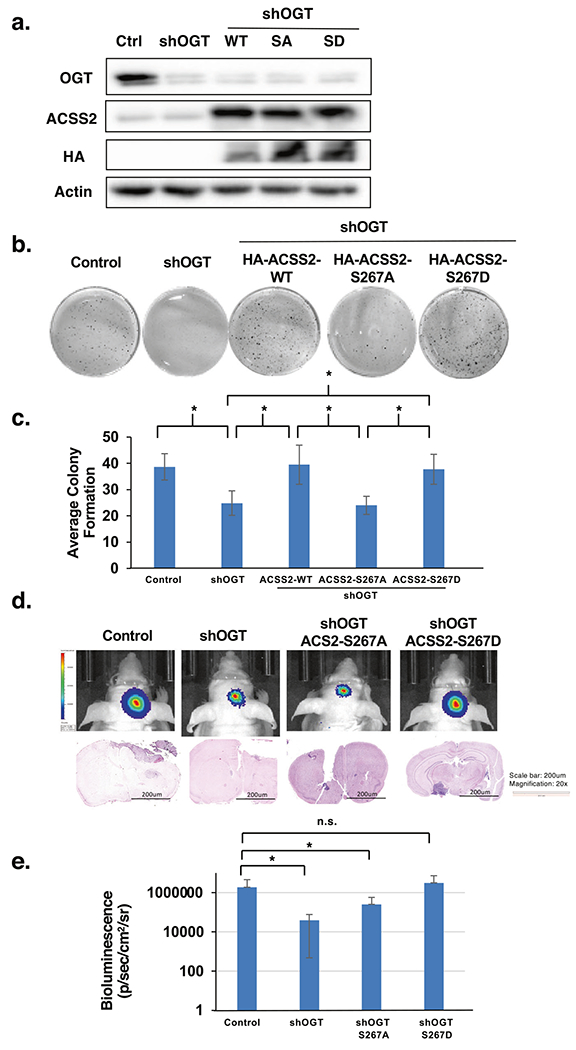
a Cell lysates from of U87-MG cells stably expressing shRNA against control or OGT and overexpressing HA-ACSS2-WT, HA-ACSS2-S267A, and HA-ACSS2-S267D were collected for immunoblot analysis with the indicated antibodies. b Representative images of cells generated in a and seeded into anchorage-independent growth assay and imaged at day 14. c Average colony formation quantified and presented as average from three independent experiments corresponding to b showing U87-MG cells stable expressing ACSS2-WT, ACSS2-S267A and ACSS2-S267D mutants and expressing shControl or shOGT as indicated. Student’s t-test reported as mean ± SD. *p-value < 0.05. d Representative images of tumor growth detected via bioluminescence (top) and H&E staining (bottom) U87-MG-luciferase cells subjected to the same treatment as in a and then used for orthotopic intracranial injections in mice. e Quantification of tumor size at Day 16 corresponding to d (shControl n = 5, shOGT n = 8, shOGT+ACSS2-S267A n = 4, shOGT+ACSS2-S267D n = 4). Student’s t-test reported as mean ± SD. *p-value < 0.05.
Pharmacologically targeting OGT and CDKs in GBM ex vivo
To test whether targeting OGT or CDK5 in a preformed GBM tumor can block cancer growth, we treated ex vivo brain slices containing GBM tumors with an OGT inhibitor or pan-CDK inhibitor. Twelve days following intracranial injections of U87-MG-luc cells, intact brains were excised and sectioned, then slices containing tumors were cultured and treated with vehicle or Ac-5S-GlcNAc. Tumors in brain slices treated with control continued growing ex vivo, while tumors exposed to OGT inhibitor treatment significantly decreased in size (Fig. 8a, b) and exhibited reduced detection of the proliferation marker Ki-67, ACSS2 Ser-267 staining, and increased cleaved-caspase-3 staining (Fig. 8a). Importantly, treating control brain slices with the OGT inhibitor reduced total O-GlcNAc levels (Supplementary Fig. S7e) but did not alter brain tissue viability compared to control (Supplementary Fig. S7f). Treatment of GBM primary cells SN310 with pan-CDK inhibitor dinaciclib, which targets CDK1, CDK2, CDK5 and CDK9 [38], blocked ACSS2 S267 phosphorylation (Supplementary Fig. S8a) and reduced neurosphere formation (Supplementary Fig. S8b). Treatment of U87-MG with dinaciclib also reduced ACSS2 S267 phosphorylation (Supplementary Fig. S8c) and also significantly decreased size of preformed tumors ex vivo (Fig. 8c, d) without causing loss of viability in brain slices (Supplementary Fig. S8d). These results demonstrate that targeting OGT and CDKs is efficacious in reducing preformed GBM tumor growth ex vivo and that this reduction is associated with reduced ACSS2 Ser-267 phosphorylation.
Fig. 8. Targeting OGT or CDKs blocks GBM growth ex vivo.
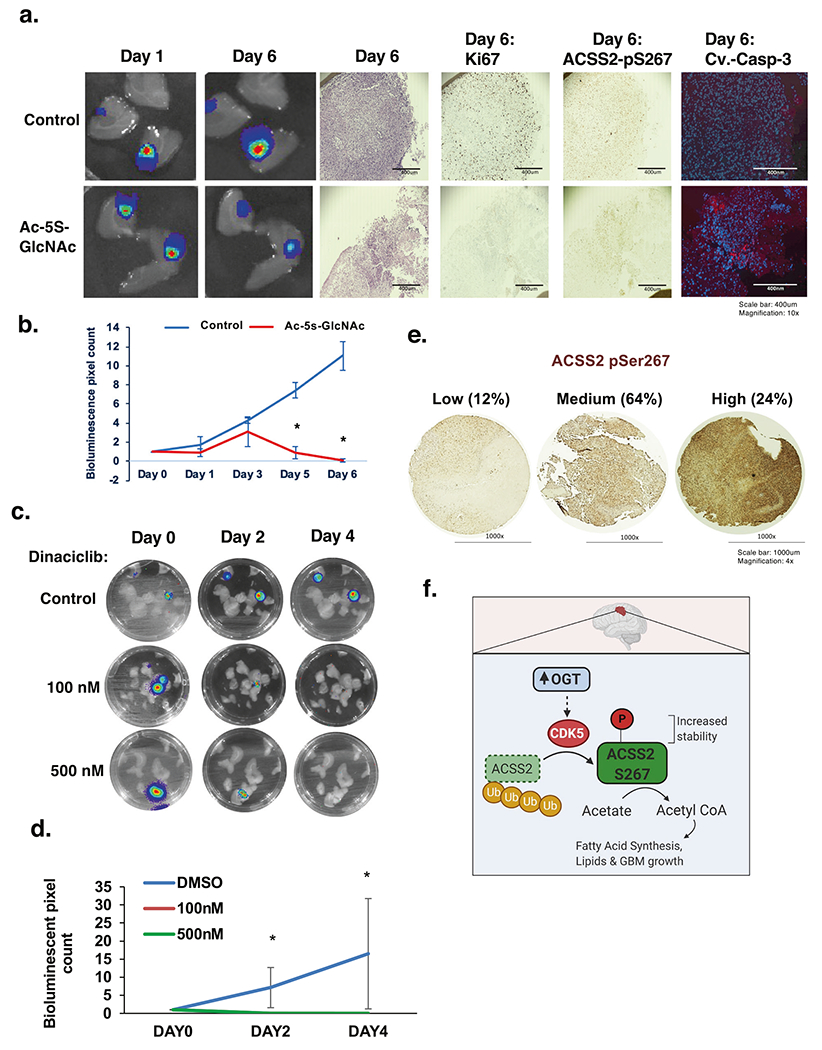
a Representative images depicting tumor growth in organotypic brain slices derived from mice intracranially injected with U87-MG-luc cells detected via bioluminescence. Brain slices containing tumors are treated with vehicle Control (DMSO) or Ac-5S-GlcNAc (200 μM) for indicated days (top) (image magnification ×10, scale bar: 400 μm). Slices were fixed and assayed for H&E, Ki-67, phospho-ACSS2-S267, cleaved-caspase-3 staining. b Quantification of tumor growth at indicated day (Control: DMSO n = 3, Ac-5S-GlcNAc n = 3) (bottom). Student’s t-test reported as mean ± SD. *p-value < 0.05. c Luciferase images of ex vivo brain explants containing preformed tumors U87-MG-luc treated with indicated dose of Dinaciclib for indicated days. d Quantification of tumor growth at indicated day treated with Control: DMSO (n = 3), dinaciclib 100 nM (n = 3) or 500 nM (n = 3). Student’s t-test reported as mean ± SD. *p-value < 0.05. e Immunohistochemical staining for ACSS2 pSer267 on a tissue microarray of Grade IV glioblastoma patient biopsies (n = 69). f Model. Increased levels and activity of OGT in GBM leads to a CDK5-depepndent phosphorylation of ACSS2 on Ser267 that increases its stability and blocks ubiquitination and increases acetate conversion to acetyl-CoA and contributes to growth and survival of GBM tumors.
Lastly, we performed IHC analysis on GBM (Grade IV) patient samples and found that phosphorylation levels of ACSS2 S267 was highly elevated in 24% percent of GBM samples (Fig. 8e). These results support the idea that ACSS2 S267 phosphorylation is highly elevated in GBM patients.
DISCUSSION
Our studies reveal a novel role of O-GlcNAc in regulation of acetate conversion to acetyl-CoA and lipids in GBM cells via regulation of ACSS2. The brain has a unique ability to rewire its metabolism in response to changing metabolite availability [39]. Similarly, brain tumors must also be able to adapt in their ability to generate energy from non-glucose sources including acetate [40]. In the present study, we demonstrated that elevated OGT and O-GlcNAcylation helps GBMs rewire metabolism to utilize acetate that provides a survival and growth advantage in this unique environment. Specifically, we propose a previously unknown pathway by which OGT regulates phosphorylation of ACSS2 on serine 267 in a CDK5-dependent manner and that this phosphorylation stabilizes ACSS2 protein levels, reduces its polyubiquitination, and regulates acetate conversion into acetyl-CoA and lipids and contributes to GBM growth (Fig. 8f). To our knowledge, this is the first report to link CDK5 directly to regulation of cancer cell metabolism. Interestingly, CDK5 has previously been shown to be regulated by glucose [41]. In pancreatic beta-cells, increased concentrations of glucose result in increased CDK5 activity that regulates insulin gene expression [41]. However, the mechanism by which glucose regulates activation of CDK5 is not known. We speculate that increased glucose may lead to elevated O-GlcNAcylation of CDK5 and its activity. Our data suggest that CDK5 is O-GlcNAcylated in GBM cells which is consistent with studies in neurons that identified three residues on CDK5 to be dynamically O-GlcNAc modified during intracerebral hemorrhage [35]. It will be of interest to determine how O-GlcNAcylation of CDK5 regulates its activity in GBMs. Despite OGT inhibitors not being well developed to determine tumor efficacy or possible toxicities, pan-CDK inhibitors such as dinaciclib have shown promise as a primary therapy for multiple myeloma [42] and in preclinical models of pancreatic [43] and ovarian cancers [44]. Therefore, in light of our ex vivo results which suggest inhibition of GBM growth by dinaciclib, it would be of clinical interest to analyze the efficacy of these compounds, as well as more CDK5-specific inhibitors, for targeting GBM metabolism and growth.
Acetate conversion to acetyl-CoA in cancer can contribute to three major metabolic pathways: de novo fatty acid and isoprenoid synthesis, the TCA cycle and histone acetylation [2]. For example, nuclear ACSS2 has been described as having the ability to recapture acetate produced as the result of histone deacetylation and can utilize this acetate to maintain histone acetylation [45]. In addition, acetyl-CoA locally produced by ACSS2 using acetate generated from nuclear protein deacetylation can be used for acetylation of promoters critical for regulating gene expression of genes involved in cancer cell survival [37]. Although OGT-CDK5-mediated Ser267 phosphorylation did not alter the localization of ACSS2, it is possible that this phosphorylation be influence gene expression processes through ACSS2-dependent histone acetylation [37, 45]. Since OGT can also regulate glucose metabolism [46] it’s possible acetyl-CoA is generated from multiple sources in cancer with high levels of O-GlcNAcylation. Our data does suggest that the OGT-CDK5-ACSS2 pathway may contribute to lipid biosynthesis as OGT overexpression increased acetate conversion to lipids, OGT and CDK5 knockdown inhibition of intracellular lipid storage can be reversed by overexpressing ACSS2 phospho-mimetic mutant. Recent studies have shown that cancers in the brain must adapt to lack of lipid availability in the brain environment and thus are highly dependent on lipid metabolism and fatty acid synthesis for growth and survival [47] including tumors that metastasize to the brain [48]. Our study provides one potential pathway for a mechanism by which GBM cells can generate acetyl-CoA from acetate and contribute to lipid synthesis, via phosphorylation of ACSS2 by CDK5 and activation of nutrient sensor OGT. Future experiments investigating whether the OGT-CDK5-ACSS2 pathway contributes to other tumors that metastasize to the brain will be further explored.
MATERIALS AND METHODS
Cell culture
Details are provided in the Supplementary information. Primary GBM WHO grade IV cell lines SN186 (76-year-old male), SN275 (58-year-old male) and SN310 (78-year-old female) were provided as a kind gift by P. Hothi (Swedish Neuroscience Institute, Seattle, WA) and have been previously described [49].
Animal models of cancer
Details are provided in the Supplementary information.
Human samples
Details are provided in the Supplementary information For survival analysis Kaplan–Meier curves were generated using the online database UALCAN (http://ualcan.path.uab.edu/analysis.html) [50].
Reagents
Details are provided in the Supplementary information Ac-5S-GlcNAc and NButGT were a kind gift from David J. Vocadlo and previously described [24].
RNA interference
Stable cell lines for shRNA knockdowns were generated as previously described [11]. Details are provided in the Supplementary information.
mRNA expression
qRT-PCR was performed as previously described [26] Details are provided in the Supplementary information.
Immunoblotting
Immunoblotting protocols have been previously described [12]. Details are provided in the Supplementary information.
Immunohistochemical staining
Details are provided in the Supplementary information.
Immunoprecipitation
Details are provided in the Supplementary information.
LC-MS/MS analyses and data processing
Details are provided in the Supplementary information. Peptide sequences were identified using MaxQuant 1.5.2.8 [51].
Soft-agar colony formation assay and neurosphere assay
Soft agar assays have been previously described [12]. Details are provided in the Supplementary information.
In vitro kinase assay
Details are provided in the Supplementary information.
Nile red staining of cells
Details are provided in the Supplementary information.
Free fatty acid and acetyl-CoA extraction and quantification
Details are provided in the Supplementary information [52, 53].
13C2-acetate labeling
Details are provided in the Supplementary information.
Ex vivo brain slice model
Ex vivo tumor brain slice model was previously described [54]. Details are provided in the Supplementary information.
Click chemistry
Details are provided in the Supplementary information[55].
Statistical analysis and reproducibility
Details are provided in the Supplementary information.
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by PA CURE grant (to MJR, NWS and JGJ), UO1CA244303 (to MJR), Drexel University Dean’s Fellowship award (to ZAB and LC). KEW acknowledges R01CA228339. LTI is supported by 2-T32-CA-115299-14. The authors thank Chaitali Bhadiadra for technical assistance, Dr. Valerie Sodi and Dr. Edward Hartsough for helpful discussions. In memoriam Christos D. Katsetos.
Footnotes
COMPETING INTERESTS
The authors declare no competing interests.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41388-022-02237-6.
REFERENCES
- 1.Dunn GP, Rinne ML, Wykosky J, Genovese G, Quayle SN, Dunn IF, et al. Emerging insights into the molecular and cellular basis of glioblastoma. Genes Dev. 2012;26:756–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schug ZT, Vande Voorde J, Gottlieb E. The metabolic fate of acetate in cancer. Nat Rev Cancer. 2016;16:708–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Comerford SA, Huang Z, Du X, Wang Y, Cai L, Witkiewicz AK, et al. Acetate dependence of tumors. Cell. 2014;159:1591–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mashimo T, Pichumani K, Vemireddy V, Hatanpaa KJ, Singh DK, Sirasanagandla S, et al. Acetate is a bioenergetic substrate for human glioblastoma and brain metastases. Cell. 2014;159:1603–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lewis CA, Brault C, Peck B, Bensaad K, Griffiths B, Mitter R, et al. SREBP maintains lipid biosynthesis and viability of cancer cells under lipid- and oxygen-deprived conditions and defines a gene signature associated with poor survival in glioblastoma multiforme. Oncogene. 2015;34:5128–40. [DOI] [PubMed] [Google Scholar]
- 6.Yoshii Y, Furukawa T, Yoshii H, Mori T, Kiyono Y, Waki A, et al. Cytosolic acetyl-CoA synthetase affected tumor cell survival under hypoxia: the possible function in tumor acetyl-CoA/acetate metabolism. Cancer Sci. 2009;100:821–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zachara NE, Hart GW. O-GlcNAc modification: a nutritional sensor that modulates proteasome function. Trends Cell Biol. 2004;14:218–21. [DOI] [PubMed] [Google Scholar]
- 8.Gao Y, Wells L, Comer FI, Parker GJ, Hart GW. Dynamic O-glycosylation of nuclear and cytosolic proteins: cloning and characterization of a neutral, cytosolic beta-N-acetylglucosaminidase from human brain. J Biol Chem. 2001;276:9838–45. [DOI] [PubMed] [Google Scholar]
- 9.Bond MR, Hanover JA. A little sugar goes a long way: the cell biology of O-GlcNAc. J Cell Biol. 2015;208:869–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ferrer CM, Sodi VL, Reginato MJ. O-GlcNAcylation in Cancer Biology: Linking Metabolism and Signaling. J Mol Biol. 2016;428:3282–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Caldwell SA, Jackson SR, Shahriari KS, Lynch TP, Sethi G, Walker S, et al. Nutrient sensor O-GlcNAc transferase regulates breast cancer tumorigenesis through targeting of the oncogenic transcription factor FoxM1. Oncogene. 2010;29:2831–42. [DOI] [PubMed] [Google Scholar]
- 12.Lynch TP, Ferrer CM, Jackson SR, Shahriari KS, Vosseller K, Reginato MJ. Critical role of O-Linked β-N-acetylglucosamine transferase in prostate cancer invasion, angiogenesis, and metastasis. J Biol Chem. 2012;287:11070–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sodi VL, Bacigalupa ZA, Ferrer CM, Lee JV, Gocal WA, Mukhopadhyay D, et al. Nutrient sensor O-GlcNAc transferase controls cancer lipid metabolism via SREBP-1 regulation. Oncogene. 2018;37:924–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pozo K, Bibb JA. The emerging role of Cdk5 in cancer. Trends Cancer. 2016;2:606–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Antoniou X, Gassmann M, Ogunshola OO. Cdk5 interacts with Hif-1alpha in neurons: a new hypoxic signalling mechanism? Brain Res. 2011;1381:1–10. [DOI] [PubMed] [Google Scholar]
- 16.Fu AK, Fu WY, Ng AK, Chien WW, Ng YP, Wang JH, et al. Cyclin-dependent kinase 5 phosphorylates signal transducer and activator of transcription 3 and regulates its transcriptional activity. Proc Natl Acad Sci USA. 2004;101:6728–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Futatsugi A, Utreras E, Rudrabhatla P, Jaffe H, Pant HC, Kulkarni AB. Cyclin-dependent kinase 5 regulates E2F transcription factor through phosphorylation of Rb protein in neurons. Cell Cycle. 2012;11:1603–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lindqvist J, Imanishi SY, Torvaldson E, Malinen M, Remes M, Orn F, et al. Cyclin-dependent kinase 5 acts as a critical determinant of AKT-dependent proliferation and regulates differential gene expression by the androgen receptor in prostate cancer cells. Mol Biol Cell. 2015;26:1971–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sharma S, Zhang T, Michowski W, Rebecca VW, Xiao M, Ferretti R, et al. Targeting the cyclin-dependent kinase 5 in metastatic melanoma. Proc Natl Acad Sci USA. 2020;117:8001–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lenjisa JL, Tadesse S, Khair NZ, Kumarasiri M, Yu M, Albrecht H, et al. CDK5 in oncology: recent advances and future prospects. Future Med Chem. 2017;9:1939–62. [DOI] [PubMed] [Google Scholar]
- 21.Yushan R, Wenjie C, Suning H, Yiwu D, Tengfei Z, Madushi WM, et al. Insights into the clinical value of cyclin-dependent kinase 5 in glioma: a retrospective study. World J Surgical Oncol. 2015;13:223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Catania A, Urban S, Yan E, Hao C, Barron G, Allalunis-Turner J. Expression and localization of cyclin-dependent kinase 5 in apoptotic human glioma cells. Neuro Oncol. 2001;3:89–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu R, Tian B, Gearing M, Hunter S, Ye K, Mao Z. Cdk5-mediated regulation of the PIKE-A-Akt pathway and glioblastoma cell invasion. Proc Natl Acad Sci USA. 2008;105:7570–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gloster TM, Zandberg WF, Heinonen JE, Shen DL, Deng L, Vocadlo DJ. Hijacking a biosynthetic pathway yields a glycosyltransferase inhibitor within cells. Nat Chem Biol. 2011;7:174–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ferrer CM, Lynch TP, Sodi VL, Falcone JN, Schwab LP, Peacock DL, et al. O-GlcNAcylation regulates cancer metabolism and survival stress signaling via regulation of the HIF-1 pathway. Mol Cell. 2014;54:820–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sodi VL, Khaku S, Krutilina R, Schwab LP, Vocadlo DJ, Seagroves TN, et al. mTOR/MYC Axis Regulates O-GlcNAc Transferase Expression and O-GlcNAcylation in Breast Cancer. Mol Cancer Res. 2015;13:923–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schug ZT, Peck B, Jones DT, Zhang Q, Grosskurth S, Alam IS, et al. Acetyl-CoA synthetase 2 promotes acetate utilization and maintains cancer cell growth under metabolic stress. Cancer Cell. 2015;27:57–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kreppel LK, Blomberg MA, Hart GW. Dynamic glycosylation of nuclear and cytosolic proteins. Cloning and characterization of a unique O-GlcNAc transferase with multiple tetratricopeptide repeats. J Biol Chem. 1997;272:9308–15. [DOI] [PubMed] [Google Scholar]
- 29.Obenauer JC, Cantley LC, Yaffe MB. Scansite 2.0: Proteome-wide prediction of cell signaling interactions using short sequence motifs. Nucleic Acids Res. 2003;31:3635–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hornbeck PV, Zhang B, Murray B, Kornhauser JM, Latham V, Skrzypek E. PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res 2015;43:D512–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lin H, Chen MC, Chiu CY, Song YM, Lin SY. Cdk5 regulates STAT3 activation and cell proliferation in medullary thyroid carcinoma cells. J Biol Chem. 2007;282:2776–84. [DOI] [PubMed] [Google Scholar]
- 32.Piedrahita D, Hernandez I, Lopez-Tobon A, Fedorov D, Obara B, Manjunath BS, et al. Silencing of CDK5 reduces neurofibrillary tangles in transgenic alzheimer’s mice. J Neurosci. 2010;30:13966–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Parry D, Guzi T, Shanahan F, Davis N, Prabhavalkar D, Wiswell D, et al. Dinaciclib (SCH 727965), a novel and potent cyclin-dependent kinase inhibitor. Mol Cancer Ther. 2010;9:2344–53. [DOI] [PubMed] [Google Scholar]
- 34.Jackson JG, O’Donnell JC, Takano H, Coulter DA, Robinson MB. Neuronal activity and glutamate uptake decrease mitochondrial mobility in astrocytes and position mitochondria near glutamate transporters. J Neurosci. 2014;34:1613–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ning X, Tao T, Shen J, Ji Y, Xie L, Wang H, et al. The O-GlcNAc Modification of CDK5 Involved in Neuronal Apoptosis Following In Vitro Intracerebral Hemorrhage. Cell Mol Neurobiol. 2017;37:527–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chau V, Tobias JW, Bachmair A, Marriott D, Ecker DJ, Gonda DK, et al. A multiubiquitin chain is confined to specific lysine in a targeted short-lived protein. Science. 1989;243:1576–83. [DOI] [PubMed] [Google Scholar]
- 37.Li X, Yu W, Qian X, Xia Y, Zheng Y, Lee JH, et al. Nucleus-Translocated ACSS2 Promotes Gene Transcription for Lysosomal Biogenesis and Autophagy. Mol Cell. 2017;66:684–97 e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Parry D, Guzi T, Shanahan F, Davis N, Prabhavalkar D, Wiswell D, et al. Dinaciclib (SCH 727965), a novel and potent cyclin-dependent kinase inhibitor. Mol Cancer Ther. 2010;9:2344–53. [DOI] [PubMed] [Google Scholar]
- 39.Magistretti PJ, Allaman I. A cellular perspective on brain energy metabolism and functional imaging. Neuron. 2015;86:883–901. [DOI] [PubMed] [Google Scholar]
- 40.Schild T, Low V, Blenis J, Gomes AP. Unique metabolic adaptations dictate distal organ-specific metastatic colonization. Cancer Cell. 2018;33:347–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ubeda M, Kemp DM, Habener JF. Glucose-induced expression of the cyclin-dependent protein kinase 5 activator p35 involved in Alzheimer’s disease regulates insulin gene transcription in pancreatic beta-cells. Endocrinology. 2004;145:3023–31. [DOI] [PubMed] [Google Scholar]
- 42.Kumar SK, LaPlant B, Chng WJ, Zonder J, Callander N, Fonseca R, et al. Dinaciclib, a novel CDK inhibitor, demonstrates encouraging single-agent activity in patients with relapsed multiple myeloma. Blood. 2015;125:443–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Feldmann G, Mishra A, Bisht S, Karikari C, Garrido-Laguna I, Rasheed Z, et al. Cyclin-dependent kinase inhibitor Dinaciclib (SCH727965) inhibits pancreatic cancer growth and progression in murine xenograft models. Cancer Biol Ther. 2011;12:598–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen XX, Xie FF, Zhu XJ, Lin F, Pan SS, Gong LH, et al. Cyclin-dependent kinase inhibitor dinaciclib potently synergizes with cisplatin in preclinical models of ovarian cancer. Oncotarget. 2015;6:14926–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bulusu V, Tumanov S, Michalopoulou E, van den Broek NJ, MacKay G, Nixon C, et al. Acetate recapturing by nuclear acetyl-CoA synthetase 2 prevents loss of histone acetylation during oxygen and serum limitation. Cell Rep. 2017;18:647–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ferrer CM, Lynch TP, Sodi VL, Falcone JN, Schwab LP, Peacock DL, et al. O-GlcNAcylation regulates cancer metabolism and survival stress signaling via regulation of the HIF-1 pathway. Mol Cell. 2014;54:820–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jin X, Demere Z, Nair K, Ali A, Ferraro GB, Natoli T, et al. A metastasis map of human cancer cell lines. Nature 2020;588:331–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ferraro GB, Ali A, Luengo A, Kodack DP, Deik A, Abbott KL, et al. Fatty Acid Synthesis Is Required for Breast Cancer Brain Metastasis. Nat Cancer. 2021;2:414–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hothi P, Martins TJ, Chen L, Deleyrolle L, Yoon JG, Reynolds B, et al. High-throughput chemical screens identify disulfiram as an inhibitor of human glioblastoma stem cells. Oncotarget. 2012;3:1124–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chandrashekar DS, Bashel B, Balasubramanya SAH, Creighton CJ, Ponce-Rodriguez I, Chakravarthi B, et al. UALCAN: a portal for facilitating tumor subgroup gene expression and survival analyses. Neoplasia. 2017;19:649–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol. 2008;26:1367–72. [DOI] [PubMed] [Google Scholar]
- 52.Zhao S, Torres A, Henry RA, Trefely S, Wallace M, Lee JV, et al. ATP-Citrate Lyase Controls a Glucose-to-Acetate Metabolic Switch. Cell Rep. 2016;17:1037–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Trefely S, Ashwell P, Snyder NW. FluxFix: automatic isotopologue normalization for metabolic tracer analysis. BMC Bioinforma. 2016;17:485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ciraku L, Moeller RA, Esquea EM, Gocal WA, Hartsough EJ, Simone NL, et al. An Ex Vivo Brain Slice Model to Study and Target Breast Cancer Brain Metastatic Tumor Growth. J Vis Exp. 2021;175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Thompson JW, Griffin ME, Hsieh-Wilson LC. Methods for the Detection, Study, and Dynamic Profiling of O-GlcNAc Glycosylation. Methods Enzymol. 2018;598:101–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.