


Abstract
Defining the impact of missense mutations on the recognition of DNA motifs is highly dependent on bioinformatic tools that define DNA binding elements. However, classical motif analysis tools remain limited in their capacity to identify subtle changes in complex binding motifs between distinct conditions. To overcome this limitation, we developed a new tool, MoMotif, that facilitates a sensitive identification, at the single base-pair resolution, of complex, or subtle, alterations to core binding motifs, discerned from ChIP-seq data. We employed MoMotif to define the previously uncharacterized recognition motif of CTCF zinc-finger 1 (ZF1), and to further define the impact of CTCF ZF1 mutation on its association with chromatin. Mutations of CTCF ZF1 are exclusive to breast cancer and are associated with metastasis and therapeutic resistance, but the underlying mechanisms are unclear. Using MoMotif, we identified an extension of the CTCF core binding motif, necessitating a functional ZF1 to bind appropriately. Using a combination of ChIP-Seq and RNA-Seq, we discover that the inability to bind this extended motif drives an altered transcriptional program associated with the oncogenic phenotypes observed clinically. Our study demonstrates that MoMotif is a powerful new tool for comparative ChIP-seq analysis and characterising DNA-protein contacts.
INTRODUCTION
Aberrant transcription factor (TF) activities or non-coding mutations located at promoters, enhancers or chromatin domain boundaries drive diverse pathologies, including a range of cancers (1–4). Biological investigation into the pathology of such events necessitates high-throughput sequencing based epigenomic approaches such as ChIP-Seq (5) and Hi-C (6). These epigenomic endeavors are expensive and require substantial quantities of biological samples (7). However, the development and fine-tuning of complementary bioinformatic analyses allow us to infer biological impact and subsequently predict sensitivity to personalized therapies. In particular, identifying context-dependent modifications of DNA-binding motifs specific to TFs is important for our understanding of cancer biology as motifs are frequently mutated, and mutated TFs may recognize altered motifs.
For the task of identifying DNA motifs, motif discovery tools, such as GADEM (8) or MEME (9), coupled with DNA motif databases for TFs, such as JASPAR (10), CisBP (11) and UniPROBE (12), are widely used. By comparing the immediate DNA sequence surrounding an oncogenic, non-coding, mutation to an online TF motif database, one can predict which TF or family of TFs is likely to experience hindered DNA binding at this locus and from such predictions, the mechanisms of oncogenic progression may be surmised. For example, multiple oncogenic non-coding variants were identified to colocalize with the core recognition motif of CCCTC-binding factor (CTCF) (13–17), a multifunctional 11-Zinc Finger DNA binding protein involved in transcriptional regulation through the organization of 3D chromatin structure (18,19). When coupled with available Hi-C datasets, motif driven hypotheses provide mechanistic insights into the role of these non-coding variants through altered chromatin looping at key, actionable, oncogenes (20). Although current tools are well-suited to detect the presence of a known binding motif in the examples above, they are limited in their capacity to detect changes in motif recognition upon introduction of variables into a system. This is especially true when subtle modifications or extensions of known binding motifs are involved. Further, defining subtle motif variances between sites located proximal different structural features, such as comparing TF binding motifs proximal to topologically associated domain (TAD) boundaries versus those proximal to transcription start sites, also represents a challenge.
Motif discovery is studied in diverse biochemical environments, each approach with their pros and cons. In silico DNA motif discovery tools can identify binding motifs by computing a position weight matrix (PWM), derived from normalized relative frequencies of each nucleic acid base, within the aligned TF binding sites identified by experiments such as ChIP-seq (21). Compared to in vitro techniques, such as protein binding microarrays (PBM), which test for motifs directly involved in TF-DNA interaction (22), motifs discovered by in silico analysis of ChIP-Seq datasets are influenced by cellular conditions. For instance, both methods will identify a similar motif for a TF whose binding is primarily driven by direct DNA-protein interactions. Alternatively, if a TF motif is acutely influenced by chromatin state (23), cofactor interaction (24) or recruitment by another TF (25), then the identified motif will be markedly different if discovered from a ChIP-Seq or a PBM experiment. As such, these two complementary approaches of motif discovery are competent to predict the primary recognition motif of a given TF, be it a direct or indirect DNA interaction. However, both fail to identify underrepresented motif variability. Subtle changes, or extensions to a core motif are statistically overlooked by the strict thresholding required for motif discovery from ChIP-Seq. Further, condition-dependent motif alterations cannot be detected in vitro, as current tools are programmed to identify motifs within a given group of sequences, compared to background or a complementary set of sequences, but not to compare the motifs, and surrounding nucleotides, themselves. Thus, discerning subtle motif alterations or extensions influencing TF binding after introducing variables, such as mutations, post-transcriptional modifications, small molecules or ligands, represents a major challenge.
For instance, the biological impact of the mutation of the first zinc finger (ZF1) of the epigenetic regulatory protein CTCF, such as the H284N mutation, exclusive to breast cancer and prevalent in hormone resistant breast tumors (26), has remained elusive. Interestingly, CTCF mutations are among the most enriched in metastatic breast tumors compared to primary tumors, behind only ESR1 mutations (27). In contrast to oncogenic mutations located within CTCF ZF3-7 (28), involved in CTCF’s ability to bind its core motif (29,30) present in ∼90% of CTCF binding sites (CBS) (31–33), CTCF ZF1 remains uncharacterised because its crystal structure has not be obtained (34). Although the truncation of CTCF ZF1 was shown to alter RNA dependent binding of CTCF to specific sites, the H284N mutation did not display such function (35). Also, CTCF ZF1 displays the weakest affinity for DNA of all CTCF zinc fingers and is not required for the binding of CTCF to its core binding motif (29,34). It is known that bases outside the core binding motif modulate CTCF binding (29,36), but it remains unknown whether CTCF ZF1 mutations (ZF1M) regulate binding to an extended motif, or alternatively influence CTCF binding affinity through impeding its interaction with non-coding RNAs (35). However, computational tools designed to directly compare motifs between discriminative conditions are lacking. Therefore, we would expect current bioinformatic approaches to fall short in identifying possible motifs variations associated to differential binding of ZF1 mutated CTCF, because subtle changes would be ‘drowned’ by the highly conserved elements of CTCF core binding motif. As such, new tools are required to predict the pathogenic mechanism of mutated DNA binding proteins, such as CTCF ZF1 mutations in breast cancer.
To meet this challenge, we developed a new R pipeline, in which we designed a new tool, MoMotif (modification of motif analysis at single base-pair resolution). Our R pipeline incorporates, and builds upon, the three central analysis steps to mine ChIP-Seq data for DNA-binding motifs that discriminate between biological conditions. First, csaw (37) is used for the identification of differentially bound sites. Second, rGADEM (8) allows for the discovery of enriched motifs from the given list of binding sequences. Third, our novel tool MoMotif is capable of detecting small or subtle variations around identified motifs, essential to analyze complex motif modifications and allowing for high-resolution identification of any discriminative motifs. Typically, each step requires multiple independent software applications (38–40). However, this R-based analysis pipeline allows us to streamline the complete analysis sequence and improve result visualization and interpretation of discriminative motifs between biological conditions.
In this study, we profile the potential of MoMotif by identifying the protein–DNA affinity changes conferred by the CTCF H284 mutation and characterizing altered motif recognition ability of CTCF ZF1M. As a model we engineered untransformed mammary epithelial cells to carry solely H284N mutated CTCF. Next, we employed MoMotif to best discriminate between lost or gained CTCF sites, compared to invariant sites, upon mutation of ZF1. MoMotif revealed an extension of the CTCF motif which requires an intact ZF1 for CTCF to bind. Further, we discovered that the loss of binding, driven by mutant CTCF ZF1 drives changes in gene expression characteristic of the clinical phenotypes of CTCF mutated breast tumors.
MATERIALS AND METHODS
Cell culture
The mammary epithelial cell line MCF10A and mutant derivatives were maintained in DMEM/F12 50/50 (Wisent, #319-085-CL) supplemented with EGF (100 μg/ml, Wisent, #511-110-UM), Insulin (10 mg/ml, Wisent, #H511-016-U6), hydrocortisone (1 mg/ml, Sigma, #H0888-1G), horse serum (2%, Wisent, #065150) and Choleratoxin (1 mg/ml, Sigma, #C8052-2MG) in an incubator at 37°C and 5% CO2.
CRISPR/Cas9 editing
CTCF H284N knock-in was performed similarly to those we previously described in Hilmi et al. (41). sgRNA guides targeting the genomic region around the nucleotide triplet coding for CTCF H284 were inserted into the vector backbone pSpCas9(BB)-2A-GFP (PX458) (Addgene, #48138) (Supplementary Table S1). A 250 base pair DNA donor, homologous to the region, but replacing the CAC, coding for H284, by AAC, coding for H284N, were also designed and ordered with IDT (Supplementary Table S1). Introduction of plasmids and donor to 1 × 106 MCF10A cells was carried out in a 6 cm dish using Lipofectamine 3000 (Invitrogen, # L3000001), 6 μg of pCas9+ guide and 12 μl of 10mM DNA Donor. Two days later, GFP-positive cells were selected by fluorescence-activated cell sorting of individual cells into 96-well plates. To screen for CTCF H284N mutant cell clones, we isolated genomic DNA of each clone and amplified proximal sequences surrounding the Cas9 targets by polymerase chain reaction. Positive clones were first identified using the SURVEYOR Assay Kit (IDT, #706020). Then, individual alleles of positive clones were validated by Sanger Sequencing (GenomeQuebec) following PCR amplification and Zero Blunt TOPO PCR insertion and Cloning (Invitrogen, #45-0245). Genomic DNA sequences were also compared to CTCF coding sequence using BlastX (blast.ncbi.nlm.nih.gov), to validate the presence of a mutation at the H284 position (Supplementary Figure S1A).
Western blotting
Western blots were carried out as previously described (42). Cells are lysed in whole-cell lysis buffer [20 mM Tris (pH 7.5), 420 mM NaCl, 2 mM MgCl2, 1 mM EDTA, 10% glycerol, 0.5% NP-40, 0.5% Triton X-100, supplemented with fresh 1 mM dithiothreitol, phenylmethylsulfonyl fluoride, protease inhibitor cocktail (Roche) and phosphatase inhibitors, bis-glycerol phosphate, and NaF] for 15 min, then spined at 13 000 rpm at 4°C for 15 min to pellet cellular debris. Then, the protein concentration of the supernatant is assessed using a Bradford assay (Fisher, #1856209). 40 μg of proteins are loaded on 8% acrylamide gel and electrophoresed 120 V for ∼1 h. Gel-separated proteins are transferred to nitrocellulose membranes (Pall, #66485) at 4°C, 34 V, overnight. The membrane is then blocked with 5% milk in TBST [20 mM Tris base, 137 mM NaCl and 0.1% Tween 20] for 3 h at 4°C. The membrane is subsequently incubated with primary antibodies (mouse anti-CTCF, BD, #612149; mouse anti-β-Actin, Sigma, #A5316) overnight at 4°C. Membranes are rinsed and washed for 10 min twice with TBST prior to secondary antibody incubation with goat anti-rabbit (SeraCare, #5220-0458) or anti-mouse (SeraCare, #5450-0011) diluted 1/10 000 or 1/20 000 in 5% milk in TBST. Membranes are washed again for 10 min in TBST 3 times, then revealed using ECL (Biorad, #170-5061).
ChIP-seq
70–80% confluent cells are first fixed 10 minutes in 4% formaldehyde and stored at −80°C. The pellets are resuspended in 1ml of ChIP-buffer [0.25% NP-40, 0.25% Triton X-100, 0.25% sodium deoxycholate, 0.005% SDS, 50 nM Tris (pH 8), 100 mM NaCl, 5 mM EDTA, 1× PMSF, 2 mM NaF, 1× P8340 Cocktail Inhibitor (Roche)] and sonicated with a probe sonicator (Fisher Scientific Sonic Dismembrator Model 500) using the following cycles: 5 cycles at 20% power, 5 cycles at 25% power, and 5 cycles at 30% power. Each cycle is fixed at 10 s, and the samples are kept on ice between each cycle to avoid overheating. The samples are then spined and protein concentration is measured using Bradford assay, as described above. Samples are diluted to 2 mg/ml protein in ChIP-buffer and 50 ul/ml of Protein G Plus-Agarose Suspension Beads (Calbiochem, IP04-1.5ML) are added for 3 h to preclear. 2% of the sample is collected as input and kept at −20°C until DNA purification. Immunoprecipitation is done at 4°C overnight with 1ml of samples, 60 ul of beads and 2 μl of anti-CTCF antibody (Millipore, #07-729). The beads are then washed once with Wash1, Wash2, and Wash3, varying in their NaCl content [0.10% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris (pH 8), 150/200/500 mM NaCl for Wash 1, 2, 3, respectively]. Subsequently, beads are washed with Wash LiCl [0.25M LiCl, 1% NP-40, 1% sodium deoxycholate, 1 mM EDTA, 10 mM Tris (pH 8)] and finally, twice with TE buffer [10 mM Tris (pH 8), 1 mM EDTA]. Next, beads are resuspended in elution buffer [1% SDS, 0.1 M NaHCO3]. The samples are then decrosslinked overnight at 65C. 1 μg of Proteinase K (Sigma, # 39450-01-6) is added for 1 h at 42°C and the samples DNA is purified using BioBasic DNA collection column (BioBasic, #SD5005). DNA concentration is assessed via Picogreen assay (Invitrogen, #P7589). Minimally 15 ng of each ChIP samples was sent to GenomeQuebec for library preparation and next-generation sequencing.
ChIP-seq quality control and genome alignment
Quality control of reads and sequencing was assessed before and after trimming by FastQC (Babraham Bioinformatics). Reads were trimmed with Trimmomatics (43) using the following parameters: ILLUMINACLIP:$Adapters:2:30:10, LEADING:30, TRAILING:30, SLIDINGWINDOW:4:30, MINLEN:30. Aligned on hg19 human genome was performed using BWA (44). Sam file generated by BWA is converted to bam format using Samtools (44). Genomic distribution and Reactome Pathway enrichment were performed using clusterProfiler pipeline (45).
ChIP-Seq heatmaps, profile plot, tracks
Heatmaps, profile plot and tracks were generate using deepTools (46). Heatmaps and Profile plot were generated using 3kb regions centered around the differential peakset. Both the computeMatrix and plotHeatmaps were used with default parameter; yMax, zMax and colors were adjusted in each condition to better represent the results. Tracks were generated as profile plot of the single genomic regions of interest with a gene annotation track from IGV (47) under each figure to represent the relative location of the gene of interest.
ChIP-Seq MACS2 and DiffBind
Comparative peak calling was performed with MACS2 (48) under default conditions, with normalization on the respective Input datasets of each cell lines. Differentially binding regions were quantified using DiffBind 3.0 (49). Bam and narrowPeak files for each samples and bam files of the corresponding input were used. CTCF normalization and analysis was performed with the following parameters: normalize = DBA_NORM_DEFAULT, library = DBA_LIBSIZE_PEAKREADS, background = F, bREtrieve = F. Threshold of significance were set at FDR ≤ 0.05.
MoMotif analysis pipeline
The analysis sequence for the discovery of modification of motif is comprised of three principal steps: Step1: identification of sites of differential DNA binding; Step 2: discovery of motifs enriched within DNA binding sites that are either gained, lost or stable binding under experimental conditions; and Step 3: learning the discriminative motifs. These steps are conducted using three R packages: csaw, rGADEM and MoMotif, as illustrated in Figure 2. The first two packages have been widely utilized by the scientific community, but MoMotif, written in R, was developed specifically for this project.
Figure 2.

Flowchart representation of an R pipeline utilizing newly developed software MoMotif to identify complex DNA binding motifs based on ChIP-seq profiling.
Step 1: Differentially binding analysis: csaw: The first step involves quantifying binding intensity/counts from the aligned ChIP-Seq reads and de novo detection of differentially bound regions while controlling the genome-wide false discovery rates (FDR). For these processes, we rely on an existing R package, csaw (37). csaw uses a sliding window-based approach to summarize read counts across the genome. It examines the differential binding at the window level using quasi-likelihood F-tests with empirical Bayes-based dispersion estimations, which naturally handle low, over dispersed counts with a limited number of replicates (50). csaw then aggregates adjacent windows into regions for output. The P-values for the aggregated regions are calculated using Simes’ method (51), which correctly controls FDR at the region level. Our detailed steps for this differential binding analysis are summarized in Supplementary Figure S2A. We used a window of size of 10 bp with spacing of 50 bp to count the aligned reads. The differentially bound regions were detected using an FDR cut-off of 0.05. The outputs from this csaw pipeline are three sets of genomic regions (of varying lengths); experimentally-induced (i) gain of binding, (ii) lost binding and (iii) binding regions with no statistically significant differences between control and experimental conditions. Hereafter we refer to these three sets of genomic sequences as gained, lost and constant clusters.
Step 2: de novo motif discovery, rGADEM: Once lists of binding regions are returned by csaw, the next step of our new pipeline involves discovering enriched motif models. For this step, we rely on another existing R package rGADEM (52) (Droit A, et al. R package version 2.42.0), built upon the GADEM algorithm (8). GADEM is an efficient de novo motif discovery method that combines the two commonly used techniques for pattern matching; word enumeration and probabilistic local search. Enumerative methods identify motifs by counting all m-letter patterns, such as the method Drim (53). Probabilistic approaches model starting positions of motif patterns as latent variables and infer the final motif models using the Expectation-Maximization (EM) algorithm; such methods include MEME (54,55) and fdrMotif (56). Specifically, GADEM constructs spaced dyads by enumerating candidate words (4 to 6 nucleotides), and then uses them as starting positions to guide an EM algorithm for unbiased motif discovery.
We applied rGADEM to the three clusters of sequences obtained from the differential binding analysis step. To ease the computational burden and to focus on the most robust differentially bound motifs, we performed the motif discovery analysis exclusively on the top 1000 regions in the gained and lost clusters, and the bottom 1000 regions (with the largest adjusted P-values) in the stable cluster, separately. The main outputs include the enriched motif models for each cluster, represented by either position weight matrices or consensus logos. Along with a specific motif, rGADEM also reports other helpful information, including all sequences in the input data incorporating this motif and the location of the identified motif patterns in the original sequence data. This information is subsequently employed as the input for the following discriminative motif analysis step.
Step 3: Discriminative motif analysis and result visualization, MoMotif: To detect small or subtle variations built upon a primary known motif, we have developed a new discriminative motif analysis tool, MoMotif, that represents the concluding step in our pipeline. This approach starts with the short core motif reported by rGADEM, which incorporates the core pattern of our primary known motif. We then retrieve and align all sequences carrying this core motif, referred to as core sequences, for each cluster. For a comprehensive characterization of subtle variability occurring within and around the core motif, we extend both ends of the core sequences by several base pairs (a user-chosen parameter permitting versatility). This strategy results in a set of adequately aligned long sequences of the same lengths, which allows us to compare the nucleotide distribution at each single base-pair to see which base pairs seem to distinguish clusters.
Next, we are able to compare the extended sequences in the lost or gained cluster to the stable cluster by assessing the statistical significance of differences in nucleotide frequency at each position. We used the Pearson's chi-square test to assess the statistical significance of the difference in nucleotide distribution at one position between two sets of aligned sequences (lost vs. stable or gained vs. stable). For a given position, let 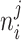 be the number of sequences in Group
be the number of sequences in Group 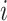 that have nucleotide
that have nucleotide 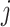 at this position, where
at this position, where  and
and 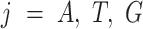 and
and 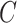 . Let
. Let 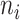 be the total number of sequences in Group
be the total number of sequences in Group 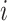 ,
, 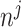 be the number of sequences with nucleotide
be the number of sequences with nucleotide 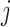 at this position in both groups, and
at this position in both groups, and 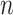 be the total number of considered sequences, i.e.
be the total number of considered sequences, i.e. 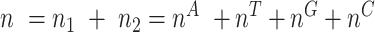 . These notations are summarized in the following contingency table:
. These notations are summarized in the following contingency table:
| A | T | G | C | Total | |
|---|---|---|---|---|---|
| Group 1 |
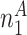
|
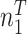
|
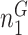
|
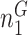
|
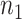
|
| Group 2 |
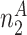
|
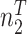
|
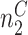
|
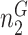
|
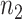
|
| Total |
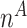
|
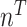
|
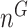
|
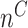
|
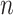
|
Specifically, the chi-square test compares the observed frequencies in each subcategory with the frequencies one would expect if the two groups had the same nucleotide distribution. The expected frequencies, denoted as 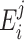 , are of the form:
, are of the form:
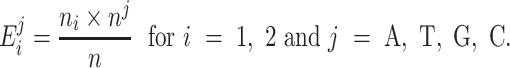 |
Then the observed chi-squared test statistics can be calculated as
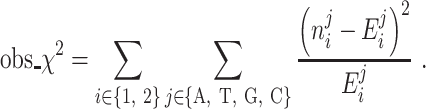 |
The P-value for the chi-squared test is thus defined as the right-tailed probability in a 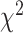 distribution with degrees of freedom 3, i.e.
distribution with degrees of freedom 3, i.e.
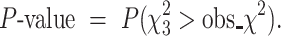 |
We repeated the test for all positions in the extended sequences and reported the p-values for each position. To control the family-wise error rate at a 5%, we suggest a stringent P-value threshold of 1 × 10–10 for declaring significance of a single position, which was derived from the approximate total number of 50M nucleotides in a small human chromosome. We also provided visualization to compare the significance level at each position relative to the overall significance level in the extended region. Therefore, discriminative motif models are then identified as the smallest sub-region containing all sites reaching our stringent threshold of significance.
In addition, the MoMotif package contains functions for various output visualizations, including bar-plots showing the frequency for each nucleotide in a given set of sequences, sequence logo for the identified discriminative motif models and their position to the core motif of our interest. In our data analysis, we treated the 10th nucleotide in the canonical CTCF motif, shown in Figure 3A, as the center and extended by 30 bp on both directions.
Figure 3.

MoMotif identifies a unique motif enriched for CBS compromised upon mutation of ZF1 (A) Classical CTCF motif outputted by rGADEM. (B) Frequency of overlap with CTCF-Like motif in each 1000 sites subset. (C) MoMotif analysis of base frequency difference and p-value of bases distribution difference around CTCF-Like motif in common lost and gain CBS subsets compared to common constant subset. The purple line represents the middle of the CTCF Motif. The dotted line represented the selected region shown in (D) (D) MoMotif results depiction as the height of each nucleotide representing the Shannon Entropy of its occurrence frequency at each position in each subset. Highlighting the extended motif (40A, 43G, 46C) in the lost subset. (E) Bar chart representing the relative presence of each individual and combined element of the extended motif in each subset. Showing an enrichment of the partial or complete extended motif in the lost subset, while the complete or partial extended motif is absent from the gain sites. Highlighting a role for CTCF ZF1 in the recognition of this sequence.
MEME suite—SEA (simple enrichment analysis)
The same subset of the top 1000 constant and lost sites from CTCF ZF1M/ZF1M used for MoMotif analysis were used for SEA analysis. SEA was run on the MEME suite web tool (https://meme-suite.org/meme/tools/sea), using the option ‘Shuffled Input Sequences’ for the motif enrichment in mutant cell line alone and ‘User-provided Sequences’ for the comparative enrichment analysis of lost sites against constant sites.
Individual zinc finger motif prediction
Human CTCF amino acid sequence from Ensembl (https://useast.ensembl.org) was inputted in Perkov et al. (57,58) webtool (http://zf.princeton.edu/b1h/index.html). 3bp predicted sequence from the F2 model were used for our analysis.
RNA-seq
RNA-Seq sample preparation and sequencing
Total RNA was extracted according to Sigma RNA Extraction Kit (Sigma, #RTN350-1KT) protocol. RNA quantity and quality was measured using Nanodrop. RNA was sent to Genome Quebec for polyA RNA library preparation using NEBNext Ultra II Directional RNA Library Prep Kit for Illumina and sequencing of 50M 100 bp Paired-End reads per replicate on Illumina NovaSeq 6000 platform.
RNA-Seq data processing and analysis
The overall quality of reads and sequencing was assessed before and after trimming using the FastQC package (Babraham Bioinformatics). Prior to mapping, reads were trimmed with Trimmomatics (43) using the following condition: ILLUMINACLIP:$Adapters:2:30:10:8:true, HEADCROP:4, SLIDINGWINDOW:4:30, LEADING:3, TRAILING:3, MINLEN:30. Alignment on hg19 human genome was performed with STAR (59) default parameters, and converted into bam format using Samtools (44). Differential expression analysis was generated using FeatureCounts count matrix (60) followed by DESEQ2 analysis (61), using default parameters and prefiltering, for comparison across samples.
RNA-Seq dot plot
Dot plot representation of the RNA-Seq results was generated using the DESEQ2 calculated log2FC and −log(adjusted P-value) of the respective mutant MCF10A compared to CTL MCF10A for every gene with a basemean >100. Genes with P-value <0.05 were represented in grey. Genes with log2FC > 1 were represented in orange. Genes with log2FC <−1 were represented in purple.
RNA-Seq GSEA pathway analysis
Pathway analysis was performed using GSEA tools (62) default setting on the read count matrix of all genes (basemean > 10). All gene sets shown were significant for both P-value (<0.001) and FDR (<0.25). Pathway names were shortened for esthetic purposes in the Figure 5B, with the full name of each pathway being written in Figure 5C.
Figure 5.

CTCF ZF1M drives oncogenic transcription profiles. (A) Dot plot representation of the RNA-Seq Log2FC of the individual mutant to control MCF10A on each axis. Showing a strong correlation and reproducibility between the samples (with Pearson correlation and test P-value displayed). (B) GSEA enrichment representation of significantly upregulated and downregulated pathways. Heatmap of the Log2FC with control MCF10A of significantly altered genes in these pathways. Showing an upregulation of genes related to drug metabolism and downregulation of genes related to ECM. (C) Top 10 up and downregulated pathways (sorted by GSEA FDR) in Gene Ontology and Reactome Databases. Filled orange bars are linked to drug metabolism and filled purple bars are linked to ECM organization. Showing an over-representation of these pathways among the top altered pathways in diverse databases. (D) CTCF ChIP-Seq track around altered genes from the RNA-Seq in MCF10A CTCF ZF1M versus CTL and in TCGA Breast Tumor CTCF ZF1M versus CTCF WT related to Xenobiotic metabolism and extracellular matrix organization. Showing a significant loss of CTCF binding in proximity to ADAMTS1 promoter (P = 8.91 × 10–5 and 0.003054 for ZF1M/ZF1M and ZF1M/- respectively) and within SLC20A1 (P = 7.28 × 10–5 and 0.001778 for ZF1M/ZF1M and ZF1M/- respectively). (E) Pie chart showing a majority of genes significantly altered in the MCF10A models are also significantly altered in the same direction in breast tumors data from TCGA database when comparing changes in gene expression associated to CTCF ZF1M. Significance of the correlation between the alteration of gene expression of the two datasets is also shown.
RNA-Seq heatmaps
Heatmaps were generated using the log2FC with CTL MCF10A of genes with the highest absolute log2FC from the following significantly altered pathways: ‘GOBP_RESPONSE_TO_XENOBIOTIC_STIMULUS’; ‘REACTOME_EXTRACELLULAR_MATRIX_ORGANIZATION’
TCGA RNA-Seq analysis
Average gene expression of each gene in breast cancer patient with CTCF ZF1M was compared to average gene expression in CTCF WT breast tumors. Log2FC of significantly altered genes in patients were then compared to log2FC of significantly altered genes in MCF10A CTCF ZF1M/ZF1M and CTL.
Hi-C
Hi-C Sample preparation and sequencing
Hi-C data was generated from two biological replicates of 1M CTL MCF10A cells, using the Arima-HiC kit, according to the manufacturer's protocols (Arima Genomics). Library preparation was performed using KAPA Hyper Prep Kit (#07962312001) following Arima protocol for library preparation. Libraries were sent at TCAG at SickKids Hospital for next-generation sequencing using Illumina Novaseq S1 flowcell (paired-end 50 bp sequencing, ∼300M reads per replicate)
Hi-C data processing and analysis
Quality control of reads and sequencing was assessed by FastQC (Babraham Bioinformatics). Raw sequencing read were mapped, filtered and binned using the runHiC pipeline (63). Contact matrix were binned at 5 and 10 kb resolution and stored in ‘.cool’ format.
Hierarchical TAD calling
Hierarchical TAD calling was performed using the hiTAD function of the TADLib package (63) and SpectralTAD package (64), using the 10 kb resolution contact matrix and default settings.
Colocalization analysis
TSS of altered genes (FDR < 0.05) and altered CTCF sites were mapped to TAD/subTAD boundaries (±1 resolution bin/10 kb) or within each TAD/subTAD. The distribution of strongly gained and lost gene (abs(logFC) > 1) compared to all mapped genes was measured and compared using a ChiSQ test in each distribution of: TAD/subTAD, TSS location and CTCF status.
Quantification and statistical analysis
Unless stated otherwise, all graphical representations display the mean and SEM of the sample's distribution and P-values are determined with Student's one-tailed T-test. Unless stated otherwise, graphics were generated using GraphPad Prism 9.1, GraphPad Software, San Diego, CA, USA, www.graphpad.com. CTCF visual model and figures were drawn and arranged using Inkscape (https://inkscape.org/).
RESULTS
CTCF ZF1M is associated with CTCF LOH in breast cancer
To gain insight into the biological importance of CTCF ZF1 mutation, we first sought to interrogate the clinical correlation between ZF1 mutation and CTCF Loss of Heterozygosity (LOH). CTCF LOH is observed in a majority of breast tumors and we investigated a potential association or exclusivity of CTCF ZF1M and CTCF LOH to identify the most common clinical genotypes of CTCF ZF1M in breast tumors. Using copy number variation data from cancer patients within the TCGA 2018 dataset, we detect a significant downregulation of copy number in patients with CTCF ZF1M, of which CTCF H284N was the most common, compared with patients with other CTCF mutations or with WT CTCF (Figure 1A). Among CTCF mutations across tumor types, the association between ZF1 mutation and CTCF LOH is the most pronounced, especially in breast tumors (Figure 1A). Indeed, ∼83% of breast tumors with CTCF ZF1M co-occur with CTCF LOH (Figure 1B). Comparatively, CTCF LOH is detected in ∼52% of breast tumors and ∼16% of other types of tumors when WT CTCF is expressed from the second allele. Therefore, we conclude that a significant co-occurrence of CTCF ZF1M and CTCF LOH is found within breast tumors.
Figure 1.

H284N mutation of CTCF ZF1 alters a subset of DNA binding sites. (A) Enrichment of copy number loss of CTCF in ZF1M in tumors of all origin (N = 13, P = 0.0018) and ZF1M in breast tumors (N = 6, P < 0.0001) compared to Non-WT Non-ZF1M CTCF tumors (N = 258). (B) Bar chart representation of the increased frequency of CTCF LOH in CTCF ZF1M in BRCA (N = 5) compared to CTCF WT BRCA (N = 1045) and CTCF WT tumors of all cancer (N = 10 607). (C) CTCF ChIP-Seq heatmaps of commonly constant, gained and lost CBS (csaw, FDR < 0.05). (D–F) Pie Charts of the number of CBS commonly altered or uniquely altered CBS in each clone, coupled with profile plot representation of read density at these specific sites. Beside the 1013 uniquely lost in ZF1M/-, all groups of altered CBS display nearly identical changes in read density in both mutant cell lines.
In light of these observations, we decided to explore the biological impact of CTCF ZF1M in breast epithelium using two relevant models. First, the ZF1M/- model, in which the CTCF H284N mutation is inserted into one allele while the second allele of CTCF is knocked-out, similar to the most commonly observed genotype in the clinic. Second, the ZF1M/ZF1M model, in which a biallelic insertion of the CTCF H284N mutation results in the sole expression of the mutated form of CTCF at the same expression level as the control cell line, to account for any biological effects of the lower CTCF protein levels in the ZF1M/- cell line. Using CRISPR-Cas9, we generated clonal lines for each of these genotypes, by knocking-in the CTCF H284N mutation or knocking-out CTCF in MCF10A cells (Supplementary Figure S1A, B). MCF10A were chosen as they are immortalized, but not transformed, mammary epithelial cells, suitable to study the impact of the CTCF ZF1M in early events of breast cancer formation, without confounding effects of complex oncogenic mutations carried in breast cancer cell models.
CTCF H284N mutation leads to altered DNA binding
Next, to clarify the debated importance of ZF1 for co-ordinating CTCF-DNA interaction, we tested the hypothesis that the H284N mutation might alter CTCF binding to the DNA. Towards this goal, we carried out ChIP-Seq for CTCF using MCF10A CTL, ZF1M/ZF1M and ZF1M/-. 48 340 CTCF binding sites (CBS) were identified in CTCF CTL cells, consistent with other studies (65). Following csaw differential binding analysis, we identified 27997 constant CBS between all three conditions, 3812 gained CBS in both ZF1M/- and ZF1M/ZF1M and 6556 commonly lost CBS (FDR ≤ 0.05) (Figure 1C/D). Interestingly, the genomic distribution of the altered CBS was not prominently different from the constant CBS, beside a slight enrichment of altered CBS on distal intergenic elements (Supplementary Figure S1C). Overall, the changes in CTCF were consistent between the two mutant cell lines. CBS gained in the ZF1M/- cell line were also gained in ZF1M/ZF1M cells, however, without reaching a threshold for significance (Figure 1E). On the other hand, sites gained in the ZF1M/ZF1M cell line appeared as low signal CBS in CTL displaying a slightly increased read density in both mutants, but only reaching the significance threshold in ZF1M/ZF1M, likely due to the higher availability of CTCF in this cell line, compared to the ZF1M/- cells (Figure 1E). Similarly to gained CBS, lost CBS within the ZF1M/ZF1M cell line were likewise frequently lost in the ZF1M/- cells (Figure 1F), indicating a high degree of similarity between ZF1M/ZF1M and ZF1M/- cells. The only subset of altered CBS that did not display a strong similarity between the 2 mutant cell lines were the 1013 CBS uniquely lost in ZF1M/-, which are likely caused by the lower levels of CTCF (Figure 1F).
Independent ChIP analysis from previously published reports help validate our findings. We compared the CTCF binding profile of our mutant cell lines with a CTCF WT ChIP-Seq dataset from an independent study (66). Here, the altered CBS called by csaw were markedly consistent (Supplementary Figure S1D), with our own MCF10A dataset, suggesting that changes in CTCF binding are intrinsic to the mutant clones. Further, the changes in CTCF binding in our mutant MCF10As were also consistent when the datasets were analyzed with a different pipeline, using MACS2 (48) for peak calling and DiffBind (49) for differential binding analysis (Supplementary Figure S1E). Therefore, these results indicate that the CTCF H284N, ZF1 mutation, likely induces a shift in the ability of CTCF to recognize or bind DNA. Also, due to the strong similarity between our models, the influence of the CTCF mutation on DNA binding seems to be largely independent of varying CTCF expression levels, hinting at a molecular mechanism underpinning the altered binding that does not include a stochastic loss in the general ability of CTCF to bind DNA.
Classical motif enrichment analysis
Following the identification of differentially bound sites, our next goal (and subsequent step of our motif discovery pipeline, represented in Figure 2) was the identification of enriched motifs. To do so, we first constructed representative subsets of each cluster by selecting the 1000 most significantly altered sites in the Gained and Lost clusters, based on the FDR-adjusted q-values. In contrast, to characterize the ‘Constant subset’, of unchanged CBS, we selected the 1000 least significantly changed binding regions. Analysing these subsets, as opposed to the entire cluster, focuses the analysis on the most relevant sites, thereby filtering out less significant differentially bound sites that might arise stochastically. Also, the selection of subsets reduced the computational burden, optimizing the analysis time and making the pipeline we developed more amenable for individuals working on less powerful hardware.
Once subsets were defined, we performed motif discovery analysis on these three clusters using rGADEM. We additionally compared the identified motif patterns to the JASPAR database and reported the significant matching motifs. Not surprisingly, rGADEM identified the CTCF motif as the most represented motif in all three clusters (Figure 3A). Indeed, the core CBS is found in 78% to 93% of all CTCF binding sites, depending on the cell line being probed (Figure 3B). However, as expected, the tools used for standard motif discovery analysis were unable to identify changes in motifs associated with altered binding affinity. This is expected since subtle changes would be drowned by the high representation of the CTCF core binding motif. Therefore, we continued our motif analysis using MoMotif.
Novel MoMotif analysis
To detect single nucleotide changes in the binding sequences of Lost or Gained CTCF sites, we aligned and extended the CTCF-like motifs to a 61-bp sequence centred at the mid-point of the canonical CTCF motif (represented by the purple dotted line in Figure 3C). The extension of the sequence allows us to focus on single nucleotide changes, within and outside of the classical ∼15 bp CTCF motif, that potentially influence CTCF binding affinity. Then, using MoMotif, we calculated frequency differences and P-values at each nucleotide within the extension, comparing the Lost and Gained subsets to the Constant subset, within the common altered sites (Figure 3C) and in each mutant cell line individually (Supplementary Figure S3A, B). We defined a section of the extended sequences containing every position reaching the required statistical threshold (P < 1 × 10–10) and a frequency differences greater than 0.1, in the lost or gained sites. Specifically, from position 25 to 48, as indicated by the black dashed line in Figure 3C, which encompass a downstream extended CTCF core binding motif. We therefore defined this subsection of the original 61 bp sequence as our newly-identified nucleotide region capable of influencing CTCF binding affinity in the context of the H284N mutation. Akin to the alteration of CTCF binding between our two mutants cell lines, the changes in nucleotides frequency were also markedly consistent (Supplementary Figure S3A, B).
By depicting these new motifs with the height of each nucleotide representing the Shannon Entropy of its occurrence frequency at each position (Figure 3D), we visually highlight the unique extended motif enriched at each position. This reveals an extended motif specific to the lost sites defined by an A at position 40, a G at position 43 and a C at position 46. Interestingly, the G at position 43 also displays the lowest p-value and highest frequency difference when comparing lost sites to constant sites in all conditions (Figure 3C, Supplementary Figure S3A, B). Furthermore, the extended motif identified with MoMotif is homologous to the previously defined module 4 of CBS, carrying a very weak consensus, identified by ChIP-exo (36) (30). Although a mechanism explaining how CTCF recognizes this motif was not revealed in prior publications, module 4 of the CTCF binding motif has been associated with a stronger DNA-binding affinity of WT CTCF. This conclusion is supported by our observations (Supplementary Figure S4A) and these results, from independent studies, validate the predictive value of MoMotif.
The extended motif influencing the association of CTCF to DNA through ZF1 appears to be mediated by three nucleotides at position 40, 43, 46. The enrichment of the extended motif in the sites lost in cells carrying the H284N mutant becomes even more prominent when investigating the proportion of the 1000 sites that display a combination of two or three of these specific nucleotides. Indeed, 24% of common sites lost across both our mutant cell lines, co-localizing with a CTCF-like motif, displayed the three defining nucleotides of the extended sequence. In contrast, only 6% and 2% of Stable and Gained sites, respectively, carried this motif. Furthermore, the combination of at least two of these nucleotides was found in 66% of Lost sites, compared to 33% and 8% of Stable and Gained Sites, respectively. The exclusion of this extended sequence in the Gained Sites is also represented in the proportion of CTCF-like sites that do not include any of the three nucleotides, being 54% in the Gained Sites, compared to only 6% in the Lost Sites (Figure 3E).
As a comparison, we analysed the lost sites using the classical motif enrichment tool SEA, from the MEME Suite. SEA identified the CTCF core binding motif as the most enriched motif in the lost sites (Supplementary Figure S4B), similarly as earlier steps in our pipeline. When using the MEME suite software to carry out motif enrichment analysis comparing the lost CTCF sites with constant sites, SEA identified differentially enriched motifs in a small subset of lost sites, with low frequency of True Positives (TP) below 10% for each motif. These marginally differentially enriched motifs are also located in regions surrounding the center of the sequences, where a consensus CTCF motif is located (Supplementary Figure S4C), inconsistent with a ZF1-specific effect. However, software from the MEME suite, such as SEA, does not identify unique motifs, differentially enriched between conditions, or motifs only partially present in both, a necessity to output a single nucleotide analysis of the modification of a specific motif between the conditions. Therefore, classical motif enrichment analysis is competent to identify TFs showing differential binding between conditions, but can not precisely identify changes to a specific motif under variable conditions, as summarized in Supplementary Figure S4D.
In sum, MoMotif can be used to facilitate the discovery of subtle motif changes after the introduction of experimental variables. As will be detailed below, MoMotif may also be used to compare DNA motifs within subsets of single datasets, including ChIP-seq and Hi-C. Regarding CTCF, we used MoMotif to define a unique DNA motif that requires CTCF ZF1 for recognition. This motif is strongly associated with the sites lost upon ZF1 mutation and was ignored by classical motif analysis tools. These data suggest a model where the CTCF ZF1 mutation induces a loss of function rendering the mutant CTCF unable to bind, or recognize, the extended sequence, leading to its stochastic redistribution on CBS without this sequence, specifically those that do not require ZF1 to bind appropriately.
Structural analysis of CTCF zinc finger-DNA contacts suggests conformation changes imparted by zinc finger 1 mutation
CTCF is known to use variable combinations of zinc fingers to flexibly bind diverse sites on the DNA (67). Therefore, we investigated whether the modified CTCF motif identified by MoMotif was recognized by a specific combination of CTCF zinc fingers requiring ZF1. We used per-domain predictions of CTCF ZFs DNA-binding specificity using the software and databases from Persikov et al. (57,58), to identify 3bp sequences that are recognized by individual CTCF zinc finger (Figure 4A). CTCF ZF3 to ZF7 are known to mediate strong binding to the CTCF core binding motif (29,30). When aligning the ZF3-7 consensus motif with the motifs identified in the constant and gained clusters, a majority of the bases identified at each position match between motifs (92.8% and 85.7% against the constant and gain motifs respectively) (Figure 4B). These associations indicate that CTCF recognizes the motif identified within the constant and gained sites independently of CTCF ZF1 and is therefore not directly hindered by the mutation of ZF1. In contrast, the extended motif enriched in the lost cluster aligns with a different combination of ZFs. Indeed, although ZF7 to ZF4 match similarly to the first half of the extended motif (Figure 4C), the primary DNA base matches with ZF3 at the constant sites is replaced by secondary matches at lost sites. Further, a strong de novo primary match motif is observed at both ZF2 and ZF1 within the sites lost in ZF1M cells (Figure 4C). These results hint at an enrichment, at lost sites, of sequences that require the combination of ZF4-7 and ZF1-2, with a possible variation in ZF3 binding, to be appropriately recognized and bound by CTCF. As CTCF H284 is necessary for the coupling of the zinc ion, crucial to the ZF structure, it is expected that ZF1M structure would be aberrant and therefore, unable to carry out its function. In turn, blocking the ability of the ZF1-2 tandem to properly recognize the A and G of the extended motif, resulting in a dissociation specifically at these sites. However, the zinc finger structure alone can not explain the presence of an extended motif from position 46 to 48, primarily defined by a C at position 46, hinting that a secondary binding mechanism of ZF1, beyond its binding of 3 core bp is at play, or alternatively, a protein, or RNA co-factor may influence the DNA recognition by CTCF ZF1. Overall, this analysis strongly supports our model that ZF1-mutation of CTCF is unable to bind an extended motif at a subset of CBS, and this pool of CTCF is then redistributed to gained sites stochastically, where ZF1 binding is not required.
Figure 4.

Extended Motif of CTCF is associated to an altered binding conformation. (A) Predicted 3bp sequences recognized by each ZF of CTCF by Persikov et al. (57,58). (B) Alignment of the predicted motif to the motif identified by MoMotif for Constant and Gain CTCF binding sites. (C) Alignment of the predicted motif to the extended motif identified by MoMotif for Lost CTCF binding sites. For (B) and (C), colored vertical bars represent a match between the primary called base at each position and grey vertical bars represent a match between a secondary called base and a primary base.
MoMotif reveals increased stability of the core CTCF binding motif at domain boundaries
MoMotif is a versatile computational tool that may not only be used to compare DNA-binding motifs across ChIP-seq samples, but can also be used to compare complex DNA motifs present within subsets of a single ChIP-seq dataset.
CTCF plays an essential role in the organization of chromatin conformation, in part by defining the boundaries of Topologically Associated Domains (TADs). Therefore, we asked whether ZF1 mutation impacted differentially the binding of CTCF at sites maintaining 3D chromatin organization. Towards this goal, we used the genomic coordinates of TADs and subTADs (defined as self-associating domains within TADs), binned at 10kb, using our Hi-C datasets from CTL MCF10A to provide topological context to our CTCF ChIP-Seq. TAD and subTADs were called using the hierarchical TAD caller hiTAD (63), ranked best TAD caller in term of average concordance over normalizations and resolutions in Zufferey et al. 2018 (68). Further, as different TAD callers may output variable boundaries from a same sample, we also used SpectralTAD (64) to call and compare boundaries at 10kb resolution. Overall, ∼97% of boundaries called by hiTAD in our CTL MCF10A were called in the same region (±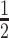 bin/5kb) by SpectralTAD (Supplementary Figure S5A), confirming the reproducibility of the topological context we provided.
bin/5kb) by SpectralTAD (Supplementary Figure S5A), confirming the reproducibility of the topological context we provided.
Next, we categorized all CBS of the CTL MCF10A cells based on their co-localization with a TAD boundary, a subTAD boundary, or not on a domain boundary, independently of whether they are constant or lost in the ZF1M lines. Overall, 10 276 and 4915 CBSs colocalized with a TAD or a subTAD boundary, respectively, compared to 36 029 CBS that did not colocalized with any boundaries. These ratios are consistent with multiple previous investigation of CTCF and TAD colocalization (69,70).
Next, we used MoMotif to identity any discriminative modifications of the CTCF motif comparing sites at subTAD boundaries, TAD boundaries, or not at boundaries (Supplementary Figure S5B, C). We found that the CTCF core binding motif is exquisitely consistent on subTAD and TAD boundaries (Supplementary Figure S5B, C). However, when comparing the CBS motif found at TAD boundaries to CBS outside TAD and subTAD boundaries, MoMotif detected an increased variability around ZF3 and ZF2 and to, a lesser extend, between ZF6 and ZF7. However, no specific enrichment for a particular base was observed at these positions. Instead, the bases recognized by these ZFs displayed a reduced Shannon Entropy, hinting at an increased motif disparity for CBS found within domains compared to CBS found at their boundaries, perhaps highlighting their diverse roles. Interestingly, the extended motif associated to lost CBS in CTCF ZF1M mutated cell lines is equally present on CBS colocalizing or not with a boundary. Supporting this conclusion, when comparing the genomic localization of constant and lost CBS between CTL and ZF1M MCF10A lines, the sites are distributed equally among domain boundaries or within domains (Supplementary Figure S5D). These results demonstrate, in a unique context, the sensitivity of MoMotif to identify precise regions of variability around a given motif, while showing that CTCF extended motif and its associated lost binding sites of CTCF ZF1M are not enriched in specific topological contexts.
Gene expression changes induced by CTCF ZF1M concur with observed clinical phenotypes
Next, we investigated whether the changes in CTCF binding might be associated with transcriptional changes that might underpin the clinical phenotypes observed in CTCF mutated breast tumors (26,27). First, we used RNA-Seq to define the differences in steady state RNA levels between MCF10A CTL, CTCF ZF1M/ZF1M and CTCF ZF1M/-. Overall, the changes in gene expression observed were highly conserved in both mutant cell lines, highlighting the impact of the H284N mutation on regulating gene expression. Indeed, when correlating the respective log2FC of both mutant lines with MCF10A CTL, the lines carrying the H284N mutation displayed a strong correlation (r = 0.7811 and P-value < 0.0001) (Figure 5A). Approximately 95% of significantly altered genes (FDR ≤ 0.05) in ZF1M/ZF1M cells were altered in the same direction in ZF1M/-, while 69% and 76% of strongly up and downregulated genes (abs(log2FC) ≥ 1) in ZF1M/- were strongly altered in both cell lines. Similar to the ChIP-Seq distributions and MoMotif nucleotide frequency, the effect of the mutation appears to be dominant over any effects of the LOH.
Next, we used GSEA to run pathway analysis of altered genes in both mutant cell lines. Interestingly, pathways associated with drug metabolism were consistently among the top upregulated pathways (Figure 5B, C). Our RNA-seq analysis also revealed that pathways involved in extracellular matrix (ECM) organization were among the top downregulated pathways (Figure 5B, C). Multiple genes involved in these pathways, such as ADAMST1 and SLC20A1, are proximal to lost sites of CTCF in ZF1M/ZF1M or ZF1M/- cell lines (Figure 5D). These genes are also within the majority of genes that were significantly altered in the same direction in our model and in patient's CTCF ZF1M breast tumors compared to CTCF WT breast tumors from TCGA datasets (Figure 5E). Consistent with our data, CTCF H284N mutations are frequently enriched in hormone resistant breast tumors (26). We propose that the upregulation of metabolic pathways that target xenobiotics may explain this phenomenon. We also propose that changes to the ECM may underlie the increased metastatic abilities of CTCF mutated breast tumors (27), also consistent with previous reports (71–74).
Loss of CTCF binding within TADs is associated with the changes in gene expression
We next sought to determine the mechanisms underlying the transcriptional changes apparent in H284N-carrying cells. Because CTCF modulation of transcription may be highly dependent on the topological organization of the chromatin (19), an altered CBS could influence the expression of a gene thousands of kilobases away. Thus, we used the genomic coordinates of TAD and subTAD from our Hi-C datasets from CTL MCF10A to provide topological context to our RNA-Seq and ChIP-Seq results.
TAD boundaries are known to be highly conserved between cell types, often colocalizing with ubiquitously expressed genes, while CTCF-mediated interactions within TADs are prone to changes and less conserved between cell types (75). Therefore, we expect that genes most strongly deregulated by ZF1 mutated CTCF would likely be located within TADs and not at their boundaries. To study this hypothesis, we divided the TADs into two groups; TADs in which the TSS of all altered genes (FDR < = 0.05) are localized exclusively within their boundaries (±1 resolution bin/10 kb) (termed TAD-B) and TADs in which the TSS of all altered genes are found exclusively within the domains, and not at boundaries (TAD-I). Then, we computed and compared the distribution of strongly altered genes (abs(log2FC) ≥ 1) in each condition. As predicted, the TAD-I group is significantly associated with strong alteration of gene expression, while the TAD-B group was not enriched for significant changes in gene expression (Figure 6A, B).
Figure 6.

Loss of CTCF binding within TADs drives oncogenic transcription. (A, B) Impact on the distribution of altered genes TSS (DESEQ2, FDR < 0.05) and altered CBS (csaw, FDR < 0.05) in the context of TAD on the enrichment of strongly altered genes (ZF1M/ZF1M to CTL abs(log2FC) ≥ 1). Showing the most significant impact of the lost of CTCF at TADs encompassing genes within them (TAD-I), compared to gain of CTCF or at TAD encompassing genes at their boundaries only (TAD-B) (P-value were generated from chi-square test on distribution of altered genes, −log(P-values) depicting significantly less strongly altered genes were turned negative in (A) to ease comprehensiveness of the graph). (C) Top 3 pathway, sorted by P-value, of Reactome Pathway Enrichment Analysis of strongly upregulated and downregulated genes from the distribution highlighted in red in (B). Showing that lost of CTCF within TAD is driving the major changes in gene expression observed in global GSEA analysis of the RNA-Seq.
We then layered the CBS altered in ZF1-mutant cells onto our analysis to identify the cluster of CBS which was the most influential for altered gene expression. Within the TAD-I group, both the loss and the gain of CTCF within TADs was associated with RNA-Seq alterations. However, the association between lost CBS and changes to gene expression was markedly more significant than for the gained CBS (P-value = 0.0027 for CTCF Lost Sites, P-value = 0.028 for CTCF Gained Sites) (Figure 6A, B). Supporting the validity of these findings, TAD-I in which no CBS displayed significantly less changes in transcription. Although the TAD-B group was not associated with significant changes in gene expression, loss of CTCF binding at the boundaries of these TADs still led to increased transcriptional variability (Figure 6A, B). In contrast, gain of CTCF at TAD boundaries, likely brought about through a stochastic redistribution of the mutant CTCF to strongly conserved CBS, was significantly associated to a conservation, instead of an alteration, of gene expression (Figure 6A).
The distribution of CTCF and gene expression changes at subTADs also supported a model where lost CTCF sites are driving gene expression changes. When investigating subTADs with TSS of altered genes colocalizing exclusively at their boundaries (subTAD-B), the only changes in CTCF binding promoting upregulation or downregulation of gene expression were lost CBS located at the boundaries of these subTADs (Supplementary Figure S6A). Overall, these results suggest that changes to CTCF binding within TADS predicts the altered gene expression through reorganization of intra-TAD interactions.
Supporting this theory, pathway analysis of altered genes proximal to a lost site of CTCF within a TAD (TAD-I) reproduces the top pathways we identified in the global RNA-Seq, being dominated by drug metabolism and ECM related pathways (Figure 6C). Therefore, our contextual analysis of ChIP-Seq and RNA-Seq revealed that the loss of CTCF binding sites within TADs, including those sites at the boundaries of subTADs, are the main drivers of the changes in gene expression resultant from CTCF ZF1 mutation. This supports a model where the inability of CTCF to bind the extended recognition motif drives aberrant phenotypical changes.
MoMotif identifies promoter proximal variability of TF recognition motif
Next, we wanted to validate the capacity of MoMotif to be used as a computational tool to compare DNA binding motifs across ChIP-Seq datasets incorporating independent experimental variables. To this end, we used previously published ChIP-Seq datasets from diverse transcription factors and compared their recognition motif among promoters (±3 kb), non-coding intronic and distal intergenic regions.
First, we investigated ligand-dependant sites of Estrogen Receptor (ER) binding from Swinstead et al. (76). Of the 8173 ligand-dependant sites identified by csaw, 988 colocalized with promoter, while 6737 were found on non-coding regions. The ER recognition motif (shown from JASPAR database in Supplementary Figure S7A) was present in 48% of promoter proximal and 60% of non-coding binding sites (Supplementary Figure S7B). Interestingly, bases within the core recognition motif were slightly differently enriched following rGADEM motif discovery (Supplementary Figure S7C). These changes were validated and quantified by MoMotif, which also reveal that differential motif recognition at promoter and non-coding regions are limited within the core recognition motif of ER, as no extensions were detected, and the only noticeable change involves a background enrichment of C within the spacing region of the motif (Supplementary Figure S7D, E). These data support MoMotif as being amenable to motif discovery using diverse datasets, and also indicate that changes in DNA-binding motifs are not invariably identified, highlighting the robustness of both the tool and our CTCF ZF1 mutation data.
Secondly, we probed ZNF263 ChIP-seq data (77) using MoMotif, and again divided the called peaks by their proximity to promoters or non-coding regions. Of the 2202 ZNF263 binding regions common among the two published peaksets, 314 were promoter proximal, while 1729 were found in non-coding regions. ZNF263 recognizes a GA rich repetitive motif without a clear consensus (77,78). Using Perkisov et al. software and databases (57,58), we validated the specificity of ZNF263 zinc-fingers for G and A enriched motifs, as each of its zinc fingers recognizes primarily these two bases (Supplementary Figure S7F). This hints that the repetitive motif is likely directly recognized by ZNF263 and not artificial. Following rGADEM analysis, both groups display a G and A rich motif, with a slightly longer motif being found in promoter proximal ZNF263 binding sites (Supplementary Figure S7G). Due to the repetitive nature of the motif, direct comparison of both motifs at this step is arduous, as it is unknown how the two motifs align together and whether the bases present in the longer, promoter proximal, motif are also present outside of the identified non-coding regions motif. However, using MoMotif sequence alignment, extension, quantification, and analysis, revealed the exclusivity of the motif extension at both end of the promoter proximal motif and the strong enrichment of A at two positions within the non-coding sites motif, while offering detailed quantification and statistical analysis of the changes in bases frequencies (Supplementary Figure S7H/I). These observations promote the concept that ZNF263 binding on promoters is dependant on a longer combination of its zinc fingers, or cofactors beyond its zinc-fingers, while binding at non-coding region might be facilitated by a fewer, but more specific, combination of zinc-fingers.
Overall, these analysis provide examples of the power of MoMotif to expand classical motif analysis with discovery, validation and quantification of motif variability between experimental conditions or functional regions.
DISCUSSION
In this study, we demonstrate that single-base pair resolution analysis of changes to DNA binding motifs may provide insights into the oncogenic impact of recurrent cancer mutations. The role of CTCF ZF1 in directing DNA contacts was previously quite ambiguous, yet the clinical relevance of its mutation is emerging (26,27). Using MoMotif, we were able to associate the structural function of CTCF ZF1 to the recognition of a previously identified extended motif of CTCF. Simultaneously, we revealed a potential role of ZF1 in mediating the transcriptional control of drug metabolism and ECM related genes. Both pathways being involved in the clinical phenotypes of CTCF mutated breast tumors.
Despite the strong association between the mutation of CTCF ZF1 and its inability to bind or recognize the extended motif, the precise mechanism explaining this loss of function remains to be elucidated. Our analysis, based on the predicted recognition motif of each ZF, hints at a mechanism in which CTCF requires a specific conformation of ZF4-7 and ZF1-2 to bind the extended motif. These results explain, in part, the low affinity of CTCF ZF1 for DNA (29) because it specifically recognizes the extended motif, but is not directly involved in DNA binding at other sites. However, the presence of conserved bases immediately downstream of the 3 bp expected to be recognized by CTCF ZF1 could also indicate that indirect mechanisms are also play. Such mechanisms include recruitment by partner proteins or non-coding RNA. CTCF ZF1 is known to contribute to RNA binding (35). As such, its mutation could hinder the interaction with RNA-dependent co-factors (79) necessary for recognition of or recruitment at this extended motif. Future ‘enhanced CrossLink and ImmunoPrecipitation and Sequencing’ (eCLIP-Seq) (80,81) investigations of the altered RNA-binding properties of mutant CTCF could provide further insight into this relationship. We suggest that eCLIP-Seq analysis of mutated RNA-binding proteins would also benefit from MoMotif. Akin to identifying altered DNA recognition motifs, by identifying sequences of RNAs gaining or losing interaction with mutated proteins, MoMotif may be applied to deepen our biological understanding of the interactome of non-coding RNAs.
Similar to our analysis of mutated CTCF or motif variations at promoter proximal regions, MoMotif analysis of available genomics data on DNA binding proteins and their co-factors in varied conditions has the potential to identify modified motifs specific to a context-dependent combination of transcription factors (TF), mutations, co-factors and functions. For example, differences in TF recognition motifs when adjacent to TAD boundaries, transcription start sites or at enhancer elements may be explored. Further, we propose that mutated TFs may harbor unknown context specific binding motifs. Factors impacting both wild-type and mutated TF binding motifs may include proximity to transcription start sites, proximity of co-factor binding sites, chromatin states at binding sites or post-translational modifications. Mining available genomic databases using our pipeline will allow the identification and association of subtle motif disparities between various contexts greatly extending our compendium of knowledge regarding biological influencers of DNA binding. In turn, this knowledge may be helpful in identifying therapeutic vulnerabilities.
Overall, MoMotif is a powerful and polyvalent tool capable of providing additional depth to a diverse range of previously existing, or novel, genomic studies.
DATA AVAILABILITY
MoMotif is an open source software in the GitHub repository (https://github.com/kaiqiong/MoMotif).
RNA-Seq data from all samples and CTCF ChIP-Seq data from MCF10A CTCF ZF1M/- and ZF1M/ZF1M can be found on GEO under accession number GSE190118.
CTCF ChIP-Seq and Hi-C data from MCF10A CTL (CTCF WT) can be found on GEO under accession number GSE183381.
Supplementary Material
Contributor Information
Benjamin Lebeau, Department of Experimental Medicine, McGill University, Montréal, Québec H3A 0G4, Canada; Lady Davis Institute, Jewish General Hospital, Montréal, Québec H3T 1E2, Canada.
Kaiqiong Zhao, Lady Davis Institute, Jewish General Hospital, Montréal, Québec H3T 1E2, Canada; Department of Epidemiology, Biostatistics and Occupational Health, McGill University, Montreal, Québec H3A 1A2, Canada.
Maika Jangal, Lady Davis Institute, Jewish General Hospital, Montréal, Québec H3T 1E2, Canada.
Tiejun Zhao, Lady Davis Institute, Jewish General Hospital, Montréal, Québec H3T 1E2, Canada.
Maria Guerra, Lady Davis Institute, Jewish General Hospital, Montréal, Québec H3T 1E2, Canada.
Celia M T Greenwood, Lady Davis Institute, Jewish General Hospital, Montréal, Québec H3T 1E2, Canada; Department of Epidemiology, Biostatistics and Occupational Health, McGill University, Montreal, Québec H3A 1A2, Canada; Department of Human Genetics, McGill University, Montreal, Québec H3A 0C7, Canada; Gerald Bronfman Department of Oncology, McGill University, Montreal, Québec H4A 3T2, Canada.
Michael Witcher, Department of Experimental Medicine, McGill University, Montréal, Québec H3A 0G4, Canada; Lady Davis Institute, Jewish General Hospital, Montréal, Québec H3T 1E2, Canada.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Canadian Institutes of Health Research (CIHR) [159759, 159663 to M.W.]; B.L. is supported by a Lady Davis Institute TD Bank Studentship Award; M.W. is supported by a Fonds de Recherche du Québec Santé (FRQ-S), Senior Chercheur Boursier award. Funding for open access charge: Canadian Institute of Health Research [159759].
Conflict of interest statement. None declared.
REFERENCES
- 1. Bushweller J.H. Targeting transcription factors in cancer - from undruggable to reality. Nat. Rev. Cancer. 2019; 19:611–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Akdemir K.C., Le V.T., Chandran S., Li Y., Verhaak R.G., Beroukhim R., Campbell P.J., Chin L., Dixon J.R., Futreal P.A.et al.. Disruption of chromatin folding domains by somatic genomic rearrangements in human cancer. Nat. Genet. 2020; 52:294–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rheinbay E., Nielsen M.M., Abascal F., Wala J.A., Shapira O., Tiao G., Hornshoj H., Hess J.M., Juul R.I., Lin Z.et al.. Analyses of non-coding somatic drivers in 2,658 cancer whole genomes. Nature. 2020; 578:102–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lee T.I., Young R.A.. Transcriptional regulation and its misregulation in disease. Cell. 2013; 152:1237–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Robertson G., Hirst M., Bainbridge M., Bilenky M., Zhao Y., Zeng T., Euskirchen G., Bernier B., Varhol R., Delaney A.et al.. Genome-wide profiles of STAT1 DNA association using chromatin immunoprecipitation and massively parallel sequencing. Nat. Methods. 2007; 4:651–657. [DOI] [PubMed] [Google Scholar]
- 6. Lieberman-Aiden E., van Berkum N.L., Williams L., Imakaev M., Ragoczy T., Telling A., Amit I., Lajoie B.R., Sabo P.J., Dorschner M.O.et al.. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009; 326:289–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yan H., Tian S., Slager S.L., Sun Z.. ChIP-seq in studying epigenetic mechanisms of disease and promoting precision medicine: progresses and future directions. Epigenomics. 2016; 8:1239–1258. [DOI] [PubMed] [Google Scholar]
- 8. Li L. GADEM: a genetic algorithm guided formation of spaced dyads coupled with an EM algorithm for motif discovery. J. Comput. Biol. 2009; 16:317–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bailey T.L., Johnson J., Grant C.E., Noble W.S.. The MEME suite. Nucleic Acids Res. 2015; 43:W39–W49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fornes O., Castro-Mondragon J.A., Khan A., van der Lee R., Zhang X., Richmond P.A., Modi B.P., Correard S., Gheorghe M., Baranasic D.et al.. JASPAR 2020: update of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 2020; 48:D87–D92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Weirauch M.T., Yang A., Albu M., Cote A.G., Montenegro-Montero A., Drewe P., Najafabadi H.S., Lambert S.A., Mann I., Cook K.et al.. Determination and inference of eukaryotic transcription factor sequence specificity. Cell. 2014; 158:1431–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hume M.A., Barrera L.A., Gisselbrecht S.S., Bulyk M.L.. UniPROBE, update 2015: new tools and content for the online database of protein-binding microarray data on protein-DNA interactions. Nucleic Acids Res. 2015; 43:D117–D122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dai J., Zhu M., Wang C., Shen W., Zhou W., Sun J., Liu J., Jin G., Ma H., Hu Z.et al.. Systematical analyses of variants in CTCF-binding sites identified a novel lung cancer susceptibility locus among Chinese population. Sci. Rep. 2015; 5:7833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Liu Y., Walavalkar N.M., Dozmorov M.G., Rich S.S., Civelek M., Guertin M.J.. Identification of breast cancer associated variants that modulate transcription factor binding. PLoS Genet. 2017; 13:e1006761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Liu E.M., Martinez-Fundichely A., Diaz B.J., Aronson B., Cuykendall T., MacKay M., Dhingra P., Wong E.W.P., Chi P., Apostolou E.et al.. Identification of cancer drivers at CTCF insulators in 1,962 whole genomes. Cell Syst. 2019; 8:446–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fang R., Wang C., Skogerbo G., Zhang Z.. Functional diversity of CTCFs is encoded in their binding motifs. BMC Genomics. 2015; 16:649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Azazi D., Mudge J.M., Odom D.T., Flicek P.. Functional signatures of evolutionarily young CTCF binding sites. BMC Biol. 2020; 18:132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Filippova G.N., Lindblom A., Meincke L.J., Klenova E.M., Neiman P.E., Collins S.J., Doggett N.A., Lobanenkov V.V.. A widely expressed transcription factor with multiple DNA sequence specificity, CTCF, is localized at chromosome segment 16q22.1 within one of the smallest regions of overlap for common deletions in breast and prostate cancers. Genes Chromosomes Cancer. 1998; 22:26–36. [PubMed] [Google Scholar]
- 19. Braccioli L., de Wit E.. CTCF: a Swiss-army knife for genome organization and transcription regulation. Essays Biochem. 2019; 63:157–165. [DOI] [PubMed] [Google Scholar]
- 20. Liu Y., Li C., Shen S., Chen X., Szlachta K., Edmonson M.N., Shao Y., Ma X., Hyle J., Wright S.et al.. Discovery of regulatory noncoding variants in individual cancer genomes by using cis-X. Nat. Genet. 2020; 52:811–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Stormo G.D. DNA motif databases and their uses. Curr. Protoc. Bioinformatics. 2015; 51:2.15.1–2.15.6. [DOI] [PubMed] [Google Scholar]
- 22. Berger M.F., Philippakis A.A., Qureshi A.M., He F.S., Estep P.W. 3rd, Bulyk M.L.. Compact, universal DNA microarrays to comprehensively determine transcription-factor binding site specificities. Nat. Biotechnol. 2006; 24:1429–1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Meers M.P., Janssens D.H., Henikoff S.. Pioneer factor-nucleosome binding events during differentiation are motif encoded. Mol Cell. 2019; 75:562–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hansen A.S., Hsieh T.S., Cattoglio C., Pustova I., Saldana-Meyer R., Reinberg D., Darzacq X., Tjian R.. Distinct classes of chromatin loops revealed by deletion of an RNA-Binding region in CTCF. Mol. Cell. 2019; 76:395–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Partridge E.C., Chhetri S.B., Prokop J.W., Ramaker R.C., Jansen C.S., Goh S.T., Mackiewicz M., Newberry K.M., Brandsmeier L.A., Meadows S.K.et al.. Occupancy maps of 208 chromatin-associated proteins in one human cell type. Nature. 2020; 583:720–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Razavi P., Chang M.T., Xu G., Bandlamudi C., Ross D.S., Vasan N., Cai Y., Bielski C.M., Donoghue M.T.A., Jonsson P.et al.. The genomic landscape of endocrine-resistant advanced breast cancers. Cancer Cell. 2018; 34:427–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rinaldi J., Sokol E.S., Hartmaier R.J., Trabucco S.E., Frampton G.M., Goldberg M.E., Albacker L.A., Daemen A., Manning G.. The genomic landscape of metastatic breast cancer: insights from 11,000 tumors. PLoS One. 2020; 15:e0231999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Marshall A.D., Bailey C.G., Champ K., Vellozzi M., O’Young P., Metierre C., Feng Y., Thoeng A., Richards A.M., Schmitz U.et al.. CTCF genetic alterations in endometrial carcinoma are pro-tumorigenic. Oncogene. 2017; 36:4100–4110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nakahashi H., Kieffer Kwon K.R., Resch W., Vian L., Dose M., Stavreva D., Hakim O., Pruett N., Nelson S., Yamane A.et al.. A genome-wide map of CTCF multivalency redefines the CTCF code. Cell Rep. 2013; 3:1678–1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yin M., Wang J., Wang M., Li X., Zhang M., Wu Q., Wang Y.. Molecular mechanism of directional CTCF recognition of a diverse range of genomic sites. Cell Res. 2017; 27:1365–1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kim T.H., Abdullaev Z.K., Smith A.D., Ching K.A., Loukinov D.I., Green R.D., Zhang M.Q., Lobanenkov V.V., Ren B.. Analysis of the vertebrate insulator protein CTCF-binding sites in the human genome. Cell. 2007; 128:1231–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cuddapah S., Jothi R., Schones D.E., Roh T.Y., Cui K., Zhao K.. Global analysis of the insulator binding protein CTCF in chromatin barrier regions reveals demarcation of active and repressive domains. Genome Res. 2009; 19:24–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Schmidt D., Schwalie P.C., Wilson M.D., Ballester B., Goncalves A., Kutter C., Brown G.D., Marshall A., Flicek P., Odom D.T.. Waves of retrotransposon expansion remodel genome organization and CTCF binding in multiple mammalian lineages. Cell. 2012; 148:335–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hashimoto H., Wang D., Horton J.R., Zhang X., Corces V.G., Cheng X.. Structural basis for the versatile and methylation-dependent binding of CTCF to DNA. Mol. Cell. 2017; 66:711–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Saldana-Meyer R., Rodriguez-Hernaez J., Escobar T., Nishana M., Jacome-Lopez K., Nora E.P., Bruneau B.G., Tsirigos A., Furlan-Magaril M., Skok J.et al.. RNA interactions are essential for CTCF-Mediated genome organization. Mol Cell. 2019; 76:412–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rhee H.S., Pugh B.F.. Comprehensive genome-wide protein-DNA interactions detected at single-nucleotide resolution. Cell. 2011; 147:1408–1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lun A.T., Smyth G.K.. csaw: a bioconductor package for differential binding analysis of chip-seq data using sliding windows. Nucleic Acids Res. 2016; 44:e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wu D.Y., Bittencourt D., Stallcup M.R., Siegmund K.D.. Identifying differential transcription factor binding in chip-seq. Front. Genet. 2015; 6:169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ruan S., Stormo G.D.. Comparison of discriminative motif optimization using matrix and DNA shape-based models. BMC Bioinf. 2018; 19:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Song T., Gu H.. Discriminative motif discovery via simulated evolution and random under-sampling. PLoS One. 2014; 9:e87670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hilmi K., Jangal M., Marques M., Zhao T., Saad A., Zhang C., Luo V.M., Syme A., Rejon C., Yu Z.et al.. CTCF facilitates DNA double-strand break repair by enhancing homologous recombination repair. Sci. Adv. 2017; 3:e1601898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Marques M., Jangal M., Wang L.C., Kazanets A., da Silva S.D., Zhao T., Lovato A., Yu H., Jie S., Del Rincon S.et al.. Oncogenic activity of poly (ADP-ribose) glycohydrolase. Oncogene. 2019; 38:2177–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bolger A.M., Lohse M., Usadel B.. Trimmomatic: a flexible trimmer for illumina sequence data. Bioinformatics. 2014; 30:2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R.Genome Project Data Processing, S. . The sequence alignment/map format and SAMtools. Bioinformatics. 2009; 25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yu G., Wang L.G., Han Y., He Q.Y.. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012; 16:284–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ramirez F., Dundar F., Diehl S., Gruning B.A., Manke T.. deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res. 2014; 42:W187–W191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Robinson J.T., Thorvaldsdottir H., Winckler W., Guttman M., Lander E.S., Getz G., Mesirov J.P.. Integrative genomics viewer. Nat. Biotechnol. 2011; 29:24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nusbaum C., Myers R.M., Brown M., Li W.et al.. Model-based analysis of chip-Seq (MACS). Genome Biol. 2008; 9:R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ross-Innes C.S., Stark R., Teschendorff A.E., Holmes K.A., Ali H.R., Dunning M.J., Brown G.D., Gojis O., Ellis I.O., Green A.R.et al.. Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature. 2012; 481:389–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Robinson M.D., McCarthy D.J., Smyth G.K.. edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010; 26:139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. SIMES R.J. An improved bonferroni procedure for multiple tests of significance. Biometrika. 1986; 73:751–754. [Google Scholar]
- 52. Jayaram N., Usvyat D., AC R.M.. Evaluating tools for transcription factor binding site prediction. BMC Bioinf. 2016; 17:547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Eden E., Lipson D., Yogev S., Yakhini Z.. Discovering motifs in ranked lists of DNA sequences. PLoS Comput Biol. 2007; 3:e39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bailey T.L., Elkan C.. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1994; 2:28–36. [PubMed] [Google Scholar]
- 55. Bailey T.L., Boden M., Buske F.A., Frith M., Grant C.E., Clementi L., Ren J., Li W.W., Noble W.S.. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 2009; 37:W202–W208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Li L., Bass R.L., Liang Y.. fdrMotif: identifying cis-elements by an EM algorithm coupled with false discovery rate control. Bioinformatics. 2008; 24:629–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Persikov A.V., Rowland E.F., Oakes B.L., Singh M., Noyes M.B.. Deep sequencing of large library selections allows computational discovery of diverse sets of zinc fingers that bind common targets. Nucleic Acids Res. 2014; 42:1497–1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Persikov A.V., Wetzel J.L., Rowland E.F., Oakes B.L., Xu D.J., Singh M., Noyes M.B.. A systematic survey of the cys2his2 zinc finger DNA-binding landscape. Nucleic Acids Res. 2015; 43:1965–1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R.. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013; 29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Liao Y., Smyth G.K., Shi W.. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014; 30:923–930. [DOI] [PubMed] [Google Scholar]
- 61. Love M.I., Huber W., Anders S.. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014; 15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S.et al.. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Wang X.T., Cui W., Peng C.. HiTAD: detecting the structural and functional hierarchies of topologically associating domains from chromatin interactions. Nucleic Acids Res. 2017; 45:e163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Cresswell K.G., Stansfield J.C., Dozmorov M.G.. SpectralTAD: an R package for defining a hierarchy of topologically associated domains using spectral clustering. BMC Bioinf. 2020; 21:319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Wang H., Maurano M.T., Qu H., Varley K.E., Gertz J., Pauli F., Lee K., Canfield T., Weaver M., Sandstrom R.et al.. Widespread plasticity in CTCF occupancy linked to DNA methylation. Genome Res. 2012; 22:1680–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Fritz A.J., Ghule P.N., Boyd J.R., Tye C.E., Page N.A., Hong D., Shirley D.J., Weinheimer A.S., Barutcu A.R., Gerrard D.L.et al.. Intranuclear and higher-order chromatin organization of the major histone gene cluster in breast cancer. J. Cell Physiol. 2018; 233:1278–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Filippova G.N., Fagerlie S., Klenova E.M., Myers C., Dehner Y., Goodwin G., Neiman P.E., Collins S.J., Lobanenkov V.V.. An exceptionally conserved transcriptional repressor, CTCF, employs different combinations of zinc fingers to bind diverged promoter sequences of avian and mammalian c-myc oncogenes. Mol Cell Biol. 1996; 16:2802–2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Zufferey M., Tavernari D., Oricchio E., Ciriello G.. Comparison of computational methods for the identification of topologically associating domains. Genome Biol. 2018; 19:217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Kentepozidou E., Aitken S.J., Feig C., Stefflova K., Ibarra-Soria X., Odom D.T., Roller M., Flicek P.. Clustered CTCF binding is an evolutionary mechanism to maintain topologically associating domains. Genome Biol. 2020; 21:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Dixon J.R., Selvaraj S., Yue F., Kim A., Li Y., Shen Y., Hu M., Liu J.S., Ren B.. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012; 485:376–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Shah R., Smith P., Purdie C., Quinlan P., Baker L., Aman P., Thompson A.M., Crook T.. The prolyl 3-hydroxylases P3H2 and P3H3 are novel targets for epigenetic silencing in breast cancer. Br. J. Cancer. 2009; 100:1687–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Loftus P.G., Watson L., Deedigan L.M., Camarillo-Retamosa E., Dwyer R.M., O’Flynn L., Alagesan S., Griffin M., O’Brien T., Kerin M.J.et al.. Targeting stromal cell syndecan-2 reduces breast tumour growth, metastasis and limits immune evasion. Int. J. Cancer. 2021; 148:1245–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Odagiri H., Kadomatsu T., Endo M., Masuda T., Morioka M.S., Fukuhara S., Miyamoto T., Kobayashi E., Miyata K., Aoi J.et al.. The secreted protein ANGPTL2 promotes metastasis of osteosarcoma cells through integrin alpha5beta1, p38 MAPK, and matrix metalloproteinases. Sci. Signal. 2014; 7:ra7. [DOI] [PubMed] [Google Scholar]
- 74. Lehner A., Magdolen V., Schuster T., Kotzsch M., Kiechle M., Meindl A., Sweep F.C., Span P.N., Gross E.. Downregulation of serine protease HTRA1 is associated with poor survival in breast cancer. PLoS One. 2013; 8:e60359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Hanssen L.L.P., Kassouf M.T., Oudelaar A.M., Biggs D., Preece C., Downes D.J., Gosden M., Sharpe J.A., Sloane-Stanley J.A., Hughes J.R.et al.. Tissue-specific CTCF-cohesin-mediated chromatin architecture delimits enhancer interactions and function in vivo. Nat. Cell Biol. 2017; 19:952–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Swinstead E.E., Miranda T.B., Paakinaho V., Baek S., Goldstein I., Hawkins M., Karpova T.S., Ball D., Mazza D., Lavis L.D.et al.. Steroid receptors reprogram foxa1 occupancy through dynamic chromatin transitions. Cell. 2016; 165:593–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Frietze S., Lan X., Jin V.X., Farnham P.J.. Genomic targets of the KRAB and SCAN domain-containing zinc finger protein 263. J. Biol. Chem. 2010; 285:1393–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Kennedy B.A., Lan X., Huang T.H., Farnham P.J., Jin V.X.. Using chipmotifs for de novo motif discovery of OCT4 and ZNF263 based on chip-based high-throughput experiments. Methods Mol. Biol. 2012; 802:323–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Kung J.T., Kesner B., An J.Y., Ahn J.Y., Cifuentes-Rojas C., Colognori D., Jeon Y., Szanto A., del Rosario B.C., Pinter S.F.et al.. Locus-specific targeting to the X chromosome revealed by the RNA interactome of CTCF. Mol. Cell. 2015; 57:361–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Van Nostrand E.L., Pratt G.A., Shishkin A.A., Gelboin-Burkhart C., Fang M.Y., Sundararaman B., Blue S.M., Nguyen T.B., Surka C., Elkins K.et al.. Robust transcriptome-wide discovery of RNA-binding protein binding sites with enhanced CLIP (eCLIP). Nat. Methods. 2016; 13:508–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Hollin T., Abel S., Le Roch K.G. Genome-wide analysis of RNA-Protein interactions in plasmodium falciparum using eCLIP-Seq. Methods Mol. Biol. 2021; 2369:139–164. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
MoMotif is an open source software in the GitHub repository (https://github.com/kaiqiong/MoMotif).
RNA-Seq data from all samples and CTCF ChIP-Seq data from MCF10A CTCF ZF1M/- and ZF1M/ZF1M can be found on GEO under accession number GSE190118.
CTCF ChIP-Seq and Hi-C data from MCF10A CTL (CTCF WT) can be found on GEO under accession number GSE183381.