


Abstract
Tools for synthetically controlling gene expression are a cornerstone of genetic engineering. CRISPRi and CRISPRa technologies have been applied extensively for programmable modulation of gene transcription, but there are few such tools for targeted modulation of protein translation rates. Here, we employ CRISPR-Cas13 as a programmable activator of translation. We develop a novel variant of the catalytically-deactivated Cas13d enzyme dCasRx by fusing it to translation initiation factor IF3. We demonstrate dCasRx-IF3’s ability to enhance expression 21.3-fold above dCasRx when both are targeted to the start of the 5′ untranslated region of mRNA encoding red fluorescent protein in Escherichia coli. Activation of translation is location-dependent, and we show dCasRx-IF3 represses translation when targeted to the ribosomal binding site, rather than enhancing it. We provide evidence that dCasRx-IF3 targeting enhances mRNA stability relative to dCasRx, providing mechanistic insights into how this new tool functions to enhance gene expression. We also demonstrate targeted upregulation of native LacZ 2.6-fold, showing dCasRx-IF3’s ability to enhance expression of endogenous genes. dCasRx-IF3 requires no additional host modification to influence gene expression. This work outlines a novel approach, CRISPR-RNAa, for post-transcriptional control of translation to activate gene expression.
INTRODUCTION
Molecular tools derived from the prokaryotic adaptive immune system, CRISPR, have revolutionized the biological sciences. Exploitation of CRISPR-associated (Cas) nucleases alongside short, easily modifiable CRISPR RNA (crRNA) guides (1), enables researchers to target Cas proteins rapidly and reliably to specific genetic loci. By introducing mutations to catalytically inactivate these nucleases, and tethering additional functional domains, researchers have repurposed dCas proteins into a suite of tools for the precise manipulation of gene expression.
CRISPR interference (CRISPRi) allows for the targeted downregulation of gene expression through targeted DNA-binding and transcriptional inhibition, and is well established in both prokaryotic (2,3) and eukaryotic (4) organisms. Analogously, CRISPR activation (CRISPRa) tools enable the selective enhancement of transcription rates of target genes (5,6). In eukaryotes, fusing transcriptional regulators such as VP64 or VPR to dCas9 or dCpf1 allows for targeted gene expression enhancement via recruitment of RNA polymerase (7–9). In prokaryotes, researchers have similarly employed fusions of dCas9 to the omega subunit of RNA polymerase to enhance transcription (10). CRISPRa in prokaryotic systems has typically resulted in weaker effects on the level of ∼10-fold, and may require concurrent downregulation of native genes such as rpoZ to achieve substantial effects (11–18). While more recent work has improved the fold of gene-expression enhancement obtainable with CRISPRa in bacteria (11,12,19–24), efficacy has typically lagged behind the robust enhancement of gene expression in eukaryotic systems.
Recently, researchers uncovered a family of Type VI CRISPR nucleases (dubbed Cas13) that target RNA rather than DNA (25,26), facilitating the development of synthetic biology technologies for targeted manipulation of transcripts beyond targeted cleavage. By inactivating their nuclease activities and tethering them to functional domains, dCas13 (‘dead’ Cas13) proteins have been repurposed for a wide variety of applications, including editing RNA sequences (27–29), imaging RNA localization (30), exploring alternative splicing (31), and combating the SARS-CoV-2 pandemic (32,33). Researchers have also employed dCas13 for targeted repression of gene expression through inhibition of translation (34), in a fashion similar to CRISPRi transcriptional inhibition.
Because technologies for facile and programmable targeted activation of translation are lacking, we sought to convert dCas13 into a tool for programmable activation of translation to enhance protein expression. We hypothesized that tethering dCas13 to a functional domain involved in translation would create a tool for selectively increasing the translation rate of a target mRNA. Specifically, because the rate limiting step of translation is initiation in both prokaryotes (35,36) and eukaryotes (37–39), we reasoned that tethering translation initiation factors to dCas13 might facilitate enhanced translation initiation at a targeted ribosome binding site. In Escherichia coli, three initiation factors (IF1, IF2 and IF3, encoded by the genes infA, infB and infC respectively) are involved in commencing translation. IF1 and IF3 work together to perform the first step of translation initiation, in which they bind to the 30S subunit and prevent 70S ribosome assembly until the remaining machinery is in place (40,41). IF1 and IF3 are also substantially smaller (72 and 180 amino acids respectively) than IF2 (890 amino acids). We therefore hypothesized that either IF1 or IF3 fusions with dCas13 would be promising candidates for a tool to enhance translation.
We show here that both IF1 and IF3 fusions to dCas13 significantly activate expression of red fluorescent protein (RFP) in comparison to dCas13 alone, with the IF3 fusion providing elevated activation relative to IF1. We term this effect CRISPR-RNA activation (CRISPR-RNAa). Enhancement of gene expression is observed in the context of strains with unmodified translation machinery, exhibiting only a small increase in lag time with no other fitness impacts. We explore a library of 36 crRNAs to show that dCas13-IF3 activation is position-dependent, and is greater than the effects of crRNAs or dCas13 alone. We demonstrate a dose-dependent response of the tool, with greater IF-specific effects at higher target mRNA expression levels. Furthermore, we show that reducing temperature enhances the IF-specific effects further, allowing us to obtain levels of gene expression activation comparable to those obtained with analogous tools for transcriptional activation. We provide evidence that this fluorescence increase is correlated with an increase in mRNA stability, presumably by increasing translation initiation. Finally, we apply CRISPR-RNAa to activate expression of native β-galactosidase, highlighting its broader applicability. These results demonstrate how CRISPR-RNAa is a simple and modular tool for programmable activation of gene translation that, while still nascent, opens a new avenue for programmable enhancement of gene expression.
MATERIALS AND METHODS
Strains
Escherichia coli strains were used for all experiments and plasmid construction. For plasmid construction, transformations were performed using strain NEB® 5-alpha Competent E. coli (New England BioLabs). For β-galactosidase experiments, a derivative of strain DH1 with an intact native Lac operon, AG1 competent cells (Agilent), was used. For fluorescence experiments, a DH1 strain containing approximately sixty-six copies of rfp encoding Red Fluorescent Protein (RFP) under control of the IPTG-inducible lacUV5 integrated at the intA loci was used (42). This strain was recovered from the private instance of the Joint BioEnergy Institute Registry under the JBEI ID JBx_078907, and is also available in the public instance of the Joint BioEnergy Institute Registry under the ID JPUB_010181 (https://public-registry.jbei.org/folders/354). Plasmids encoding CRISPR machinery were transformed into these base strains to create the experimental strains used in this study. A list of all strains can be found in Supplementary Table S1.
Culture conditions
All strains were cultured in Luria-Bertani (LB) broth at 37°C supplemented with the appropriate antibiotics to maintain plasmid selection, unless otherwise stated. For cloning, all strains were selected on LB agar plates supplemented with the appropriate antibiotic at 37°C. Before all experiments, strains were streaked onto LB agar plates supplemented with the appropriate antibiotic and grown at 37°C to begin experiments from individual colonies. Bacteria were inoculated from individual colonies into 500 μl cultures in 96-well deep-well plates covered by a breathable seal, and grown at 37°C overnight. For Figures 1 and 2, Supplementary Figures S2, and S4, cultures were diluted 1:100 in fresh media excluding IPTG, and grown overnight again at 37°C. For kinetic experiments in Figure 2D, E, and Supplementary Figure S2B, growth was performed in 200 μl reactions in 96-well plates in a Synergy™ H1 Microplate Reader (BioTek) set to 37°C and continuous orbital shaking. For Figures 3–5, and Supplementary Figures S7–S13, cultures were diluted 1:100 in fresh media, grown for 10 h at 30°C or 37°C as indicated, diluted again 1:50 in fresh media, and grown overnight at 30°C or 37°C as indicated in Supplementary Figure S6. For Figure 4, volumes were increased in the 1:50 dilution from 500 μl to 1000 μl to ensure enough cells were collected for subsequent RNA analysis. Fluorescence and optical density measurements were then taken. For Figure 5B, the 1:100 dilution culture used to inoculate the 1:50 dilution was kept growing at 30°C and end measurements were also performed. For Supplementary Figure S11, the final 1:50 dilutions were also grown with kinetic measurements in 100 μl cultures as outlined above.
Figure 1.

Targeted activation of mRNA translation with dCasRx-IF fusions. (A) A two-plasmid system encoding aTc-inducible dCasRx or dCasRx-IF fusions on the first plasmid and a constitutively expressed crRNA on the second plasmid is expressed in E. coli harboring IPTG-inducible rfp. The crRNAs guide the dCasRx variants to the start of the 5′ UTR, which subsequently recruits ribosomal subunits to the target mRNA to presumably increase translation initiation. The target 20nt crRNA location is underlined. (B) RFP fluorescence of E. coli expressing rfp-targeting crRNA alongside dCasRx variants. Error bars represent the standard deviation of biological triplicates. Asterisks indicate P-values of two-tailed type II Student's t-tests. (C) Cultures of strains with rfp-targeting dCasRx-IF1 (left) and dCasRx-IF3 (middle) relative to dCasRx (right).
Figure 2.

IF3 fusion to dCasRx drives maximal translation. (A) Effect of varying the length of the 20nt crRNA target ‘RFP-1’ used in Figure 1, from 15 to 25 nts. mRNA structure and crRNA target location are shown above, and normalized fluorescence measurements below. crRNA length is shown in subscripts next to each crRNA name. (B) Effect of varying the target location of the crRNA across the mRNA. Distance between the start of the ‘UTR. (C) Effect of shifting the crRNA target location 1 nt at a time away from the start of the 5′ UTR. Distance between the RBS and the 5′ rfp mRNA target location are shown in subscripts next to each crRNA name. In A–C, two variants of GFP-targeting crRNAs are included as controls. Target distances from the start of the rfp mRNA 5′ start are shown in subscripts next to each crRNA name. Kinetic (D) growth and (E) fluorescence measurements of strains expressing the noted CRISPR machinery. All error bars represent the standard deviation of biological triplicates.
Figure 3.
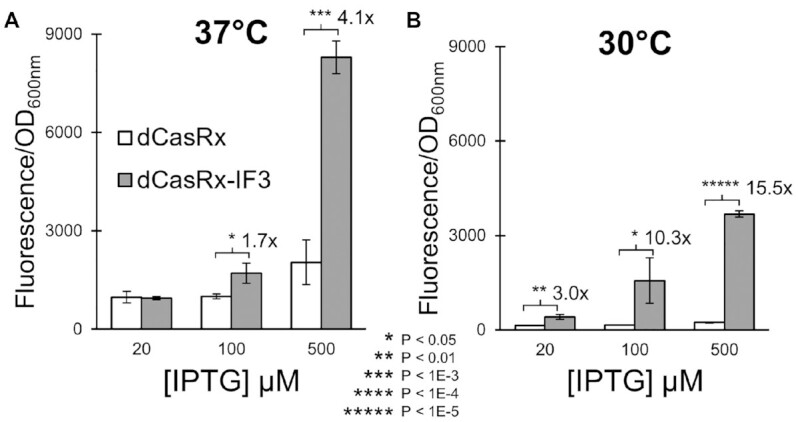
Increasing [IPTG] and reducing temperature enhances IF3-specific effect. A schematic of the revised experimental setup is presented in Supplementary Figure S6. (A) Normalized fluorescence of strains harboring either dCasRx (white bars) or dCasRx-IF3 (grey bars) alongside the RFP-1 20nt crRNA, when the experiment was performed at 37°C. (B) The same experiment, but performed at 30°C. All error bars represent standard deviation of biological triplicates. Asterisks indicate P-values of two-tailed type II Student's t-tests.
Figure 4.

Influence of CRISPR-RNAa on RNA stability over time. (A) Fluorescence of dCasRx-IF3 strains relative to dCasRx strains when targeting rfp mRNA with crRNA RFP-1, using the experimental setup outlined in Supplementary Figure S7 (with volume increased to 1 ml to ensure adequate cell count for measuring RNA). Fluorescence was measured at 960 min in the final cultures, with pre-culturing induction using either 500 μM or 0 μM IPTG induction of rfp mRNA transcription. (B) RT-qPCR measurement of rfp mRNA in dCasRx-IF3 strains relative to dCasRx strains, quantified at the noted time post-dilution (Supplementary Figure S7 step 4). Samples were either induced with 500 μM IPTG (left) or IPTG was excluded (right) during pre-culture. All error bars represent standard deviations of four biological replicates. P-values were calculated using two-tailed type II Student's t-tests.
Figure 5.

Expanding CRISPR-RNAa to enhance translation of the native E. coli gene lacZ, encoding β-galactosidase. (A) Target location of three crRNAs to enhance lacZ translation. Established transcription start sites (TSSs) are indicated, ranked in order of most common (TSS1) to least common (TSS4). (B) LacZ activity as measured using a β-galactosidase (‘Miller’) assay, performed at 30°C with 500 μM IPTG induction (as in Supplementary Figure S7, excluding step 4). (C) The same experiment, including step 4 (dilution and removal of IPTG) to enhance differences between dCasRx and dCasRx-IF3. All error bars represent standard deviation of biological triplicates. Asterisks indicate P-values of two-tailed type II Student's t-tests.
Carbenicillin (100 μg/ml) was used to maintain selection of crRNA plasmids, while kanamycin (50 μg/ml) was used to maintain selection of dCasRx plasmids. For induction of dCasRx variants, anhydrotetracycline (aTc) was utilized, at a concentration of 20 ng/ml for Figures 3 and 5, Supplementary Figures S3 and S7–S13 or at a concentration of 500 ng/ml for Figures 1 and 2, Supplementary Figures S4, and S5. For all other figures, aTc concentrations are listed within the figure. For induction of RFP and β-galactosidase, isopropyl β-d-1-thiogalactopyranoside (IPTG) was utilized, at a concentration of 100 μM for Figures 1 and 2, Supplementary Figures S4 and S5, or at a concentration of 500 μM for Figures 4 and 5, Supplementary Figures S3 and S7–S12. For all other figures, IPTG concentrations are listed within the figure. Unless otherwise noted, inducer was included at the time of culture inoculation or dilution.
For experiments in Figure 2 and Supplementary Figure S5A, only carbenicillin antibiotic selection was included for all cultures, while in Supplementary Figure S3 no antibiotics were included, to facilitate comparisons between strains with and without plasmid variants.
For Figure 4, five identical technical replicates 1 mL cultures were inoculated in the final 1:50 dilution as outlined in Supplementary Figure S6 step 5. One of each was used to collect RNA at the indicated timepoints, while the pre-dilution culture was used to collect RNA for time 0. For collecting RNA, cultures were immediately transferred into 1.5 ml Eppendorf tubes chilled on ice, and moved to a 4°C cold room. Samples were centrifuged at maximum speed for 2 min, the supernatant decanted, the samples spun again briefly, and the remaining supernatant removed with a pipette. Samples were then flash frozen in a dry ice-ethanol bath, and kept at –80°C for <48 h before all samples were processed simultaneously for RNA recovery.
Plasmid construction
A list of plasmids used in this study are included in Supplementary Table S2, and are available through Addgene (https://www.addgene.org) or the authors upon reasonable request. A list of primers used for plasmid construction and validation are included in Supplementary Table S3. Plasmids encoding dCasRx (pEJC 1.5 CasRx) and a base crRNA cloning vector (pEJC 2.5.1 empty) were built in a previous publication (34), and used to construct plasmids used in this study. Codon-optimized sequences of native infA and infC (excluding the methionine start codon) were ordered as gBlocks (IDT) and used for insertion onto the C-terminus of dCasRx, linked by a flexible 3x(GGGGS) linker domain. Four to five DNA fragments were generated via PCR using primers PBO.ACT001 to PBO.ACT012, and Gibson Assembly was used to stitch these fragments together using the NEBuilder® HiFi DNA Assembly Cloning Kit (New England BioLabs). Because the E. coli strains for expressing rfp contained chloramphenicol resistance, the chloramphenicol resistance marker on plasmid pEJC 1.5 was replaced with kanamycin resistance using plasmid pGI132 from our previous publication (43). This created plasmids pPBO.ACT001, pPBO.ACT002 and pPBO.ACT003 for expressing dCasRx, dCasRx-IF1 and dCasRx-IF3 respectively. We also used primers PBO.ACT023-24 to remove dCasRx from plasmid pPBO.ACT003, to create plasmid pPBO.ACT042 expressing aTc-inducible IF3.
A base plasmid for cloning constitutively expressed crRNA guides, PBO.ACT004 was created from pEJC 2.5.1 using primers PBO.ACT013 to PBO.ACT016. We noted the predicted crRNAs contained a substantial extended 3′ fragment (see Supplementary Figure S4) (44). We thus created plasmid PBO.ACT005, expressing a crRNA with a significantly shorter 3′ fragment predicted for the crRNAs, using primers PBO.ACT015 to PBO.ACT018. To facilitate cloning of new guide targets, we also created PBO.ACT006 containing eGFP (enhanced Green Fluorescent Protein) at the target crRNA cloning site using primers PBO.ACT014, PBO.ACT015 and PBO.ACT019 to PBO.ACT022. For creating new crRNA target plasmids, oligos were ordered and duplexed as previously described (45) and assembled with PBO.ACT004-PBO.ACT006 using BsmBI Golden Gate Assembly. This created plasmids PBO.ACT007-041 for constitutively expressing crRNAs guides. We also used primers PBO.ACT025-30 to convert pPBO.ACT012 into plasmid pPBO.ACT043 with crystal-violet inducible crRNAs with target RFP-1 (46). Sequencing primers used to verify all constructs, PBO.SEQ001-009, are listed in Supplementary Table S3.
Growth and fluorescence measurements
Optical density (OD) and fluorescence measurements were performed using a Synergy™ H1 Microplate Reader (BioTek). Endpoint fluorescence was measured using 590nm excitation and 635nm emission, and normalized to optical density 600 nm measurements where noted. For kinetic experiments, measurements were performed at 20-min intervals.
Flow cytometry
Flow cytometry experiments were performed using an autosampler-equipped Accuri™ C6 Plus Flow Cytometer system (BD Biosciences). The four biological replicates collected in Supplementary Figure S8A were used for flow cytometry analysis. 90 μl of culture was transferred to 10 μl of 37% formaldehyde solution (Sigma-Aldrich) and incubated at room temperature for 15 min, after which 20 μl of samples were diluted into 180 μl of phosphate-buffered saline. Samples were then analyzed on the Accuri system, with a minimum of ∼500 000 ungated events were collected. Data was analyzed using FlowJo® version 10.8.1.
RNA extraction and reverse transcription
From the same four biological replicates that were used to measure fluorescence in Supplementary Figure S8A, 300 μl were collected at the same time point and immediately processed. For the same four biological replicates that were used to measure fluorescence in Figure 4A, the five separate 1 ml technical replicate cultures were collected at each noted timepoint (as well as starting culture at time 0) and immediately frozen, for subsequent processing. These samples were thawed on ice for subsequent RNA collection within 48 h. RNA was collected using the GeneJET RNA Purification Kit (Thermo Scientific™) supplemented with lysozyme (Thermo Scientific™), following the manufacturer's instructions for bacterial total RNA purification. DNase I (Thermo Scientific™) treatment was subsequently used to remove genomic DNA following the manufacturer's instructions, with a final RNA concentration of approximately 5, 23 or 500 ng/μl. DNase I was diluted as appropriate to ensure no more than 1 unit per μg of input RNA, as recommended by the manufacturer. RNA was then converted into cDNA using the RevertAid First Strand cDNA Synthesis Kit (Thermo Scientific™) following the manufacturer's instructions for 10 μl reactions containing 0.5 μl random hexamer mix, 2 μl 5× reaction buffer, 0.5 μl RiboLock, 1 μl 10 mM dNTP and 0.5 μl Reverse Transcriptase. Samples were heated at 25°C for 5 min, followed by 42°C incubation for 60 min and heat inactivation at 70°C for 5 min. cDNA was diluted to approximately 2.5 or 5.0 (Figure 4) or 6.25 ng/μl (Supplementary Figure S8), stored at –80°C for less than 2 months and used for downstream qPCR analysis. RNA and DNA quality and quantity were assessed using a NanoDrop™ 2000 (Thermo Scientific™).
Genomic DNA extraction
From the same four biological replicates that were used to measure fluorescence in Supplementary Figure S8C, 200 μl of culture was collected at the same time point for extracting genomic DNA for quantification in Supplementary Figure S8D. The Wizard® Genomic DNA Purification Kit (Promega) was used to extract genomic DNA, following the manufacturer's instructions for Gram-negative bacteria. Background RNA was removed using the manufacturer's recommended 20 min RNase A incubation. gDNA concentrations were adjusted to 8 ng/μl, and used immediately for downstream qPCR analysis.
Quantitative PCR
Quantitative Real-Time PCR (qPCR) was performed in accordance with the Minimum Information for Publication of Quantitative Real-Time PCR Experiments (MIQE). Relative nucleic acid concentrations were quantified using PowerUp™ SYBR™ Green 2x Universal Master Mix (Applied Biosystems™), using the manufacturer's recommendations with no modifications or supplements. qPCR was performed manually in-house on a CFX96 or CFX384 Real-Time PCR Detection System (BioRad). For all samples, 10 μl technical duplicate reactions were performed containing 2.5 or 5.0 cDNA (for Figure 4, with 5.0 ng for time points 30, 60, and 120 min and 2.5 ng for the remaining time points), or 20 μl technical duplicate reactions containing 13 ng cDNA or 16 ng gDNA (for Supplementary Figure S8). Copy numbers of rfp were normalized to the housekeeping gene, hcaT, previously shown to be a good reference gene for E. coli (47). 500 nM unmodified primers produced with standard purification (IDT) were used, and primers are listed in Supplementary Table S3. Primers were designed based on the E. coli DH1 genome (Accession Number AP012030) with BLAST analysis to determine negligible off-targets. Amplicon lengths are 114 and 90 bp for rfp and hcaT, respectively. Reactions were performed with an initial 50°C 2-min incubation for uracil–DNA glycosylase activation, followed by a 95°C 2-min incubation for polymerase activation. Forty amplification cycles were subsequently performed using a 95°C 15 s denaturation step and 60°C 1-min annealing/extension step. CFX manager software (version 3.1) was used to calculate instrument-reported CT values of technical duplicates were averaged and used to calculate 2–ΔCT and 2–ΔΔCT values of rfp copy number relative to hcaT. DNA contamination was assessed using a no reverse transcriptase control, as well as a no cDNA control for each primer pair. A melt curve was performed after each amplification, and a single melt curve peak (around 82.5°C and 83.5°C for rfp and hcaT, respectively) was used to verify primer specificity. Primer efficiencies were calculated as 98.3% (standard curve slope = –3.4, intercept = 16.5, R2 = 0.97) and 107.8% (standard curve slope = –3.1, intercept = 24.5, R2 = 0.96) for rfp and hcaT primers respectively. No CT was observed for the NTC with hcaT primers, while the CT for the NTC with rfp primers was 35. Neither the linear dynamic range nor limit of detection were explored, nor was the intra-assay variation. No outliers were removed. Statistical significance was calculated using Student's two-tailed type II t-test comparing ΔCT values between strains harboring dCasRx and dCasRx-IF3 with the noted crRNA.
β-Galactosidase assay
Measurement of LacZ activity via the β-Galactosidase (also known as Miller) assay was performed as outlined previously (48). Measurements were performed using a Synergy™ H1 Microplate Reader (BioTek).
RESULTS
Fusion of translation initiation factors to dCasRx enables targeted translational activation
For exploring targeted activation of translation, we utilized a catalytically deactivated variant of Ruminococcus flavefaciens XPD30002 Cas13d, dubbed ‘dCasRx’ (31). dCasRx is a Cas13 ortholog that does not possess target sequence constraints due to its lack of a protospacer flanking sequence (PFS) requirement. We cloned onto the C-terminus a flexible 3x(GGGGS) protein linker domain, followed by genes infA or infC encoding E. coli translation initiation factors IF1 and IF3 respectively. These fusions replaced an original 2× FLAG-tag at the protein's C-terminus in its original design, which itself was used as a control for comparison of translational activation. As a target for gene activation, we utilized a strain of E. coli with ∼66 copies of rfp integrated into its genome at the intA loci under control of the IPTG-inducible LacUV5 promoter (42). A guide crRNA was constructed to target the first 20 nts of rfp mRNA, exactly 46–26 nts upstream of the ribosomal binding site (RBS) (Figure 1A).
We moved on to test the impact these constructs had on fluorescence by co-transforming each alongside rfp-targeting crRNAs. Our initial attempts yielded small (1.3-fold) yet statistically significant (P = 0.02) increases in fluorescence of dCasRx-IF3 strains relative to the dCasRx control strains (Supplementary Figure S1). We saw no substantial impact on growth (Supplementary Figure S2). We found that tuning the induction conditions of the experiment substantially influenced these differences. Our most optimal experimental setup for differentiating dCasRx from the dCasRx-IF fusions involved first culturing samples overnight in the presence of both aTc and IPTG, followed by a 1:100 dilution of cultures into media containing aTc but no IPTG, thus abrogating production of target rfp mRNA (Supplementary Figure S1E).
With these tuned experimental conditions, we found strains expressing rfp-targeting crRNAs and harboring either dCasRx-IF1 or dCasRx-IF3 to fluoresce significantly brighter than strains harboring dCasRx (Figure 1B). dCasRx-IF1 enhanced fluorescence 2.2 ± 0.2 fold (P = 3E–4) and dCasRx-IF3 enhanced fluorescence 3.3 ± 0.5 fold (P = 1E–3) respectively, relative to dCasRx strains. These fluorescence differences are visually distinct, and observable to the human eye (Figure 1C). We confirmed that overexpression of IF3 alone in the absence of dCasRx did not result in this substantial increase in fluorescence (Supplementary Figure S3). As the fusion to IF3 consistently resulted in the highest levels of fluorescence, we chose to focus on comparisons between dCasRx-IF3 and dCasRx for subsequent experiments.
Fusion of IF3 to dCasRx provides maximal expression enhancement, and the degree of enhancement is dependent on target-location on mRNA
As small RNAs have been shown to enhance protein expression encoded by a complementary strand of RNA under the right conditions (49,50), we next sought to demonstrate that the guide crRNAs alone were not responsible for these results. Additionally, as researchers have shown that CRISPR tools for activating transcription are heavily influenced by the target location of crRNA guides (10), we wanted to systematically explore a library of different target locations to determine good design principles for using CRISPR-RNAa. For this, we compared the fluorescence of strains harboring only the crRNA guides with those harboring both crRNAs and either dCasRx or dCasRx-IF3. We first explored varying the length of the crRNA from our original 20 nt construct (RFP-1) to 15–25 nts (RFP-2 to RFP-11) (Figure 2A). Two variants of a crRNA targeting gfp were used as controls, both demonstrative of basal fluorescence levels. Negligible fluorescence differences were observed between strains targeting these gfp controls and harboring either crRNA alone, in the presence of dCasRx, or dCasRx-IF3. The latter gfp-targeting crRNA (GFP-2) was used for comparison against other strains.
All crRNA variants of RFP-1 to RFP-11 were able to increase expression of rfp alone, without coupling with dCasRx or dCasRx-IF3. The fold-level of activation of these crRNAs was between 1.5 ± 0.1 (RFP-6, P = 1E–3) and 3.6 ± 0.7 (RFP-7, P = 2E–3). Taken together, these crRNA-only results demonstrate that trans-activating RNA–RNA interactions played a significant role in modulating RFP expression.
Notably, introduction of dCasRx into crRNA-expressing strains substantially enhanced fluorescence beyond any enhancement observed by crRNAs alone (Figure 2A). There were no substantial differences with the two shortest crRNAs (RFP-2 and RFP-3) with and without dCasRx proteins. However, with a length of 17nts (RFP-4) the effect of addition of dCasRx (2.1 ± 0.6, P = 0.03) and dCasRx-IF3 (2.0 ± 0.2, P = 8E–5) relative to the crRNA alone strain emerged. Increasing the crRNA target length further enhanced the activation effect of both proteins, in particular for dCasRx-IF3. With crRNA lengths of 19 nts and above, the fold increase of fluorescence of strains harboring dCasRx-IF3 was statistically greater than strains harboring dCasRx (excluding RFP-8 crRNA), ranging between 1.3 ± 0.1, (RFP-7 P = 4E–3) to 2.0 ± 0.2 (RFP-10, P = 0.02). For both proteins, the 23nt RFP-9 crRNA appeared to provide maximum gene expression activation, relative to GFP-2 crRNA (7.6 ± 0.6, P = 2E–5 and 12.5 ± 0.5, P = 4E–7 for dCasRx and dCasRx-IF3, respectively).
These results indicate that dCasRx interaction with the mRNA provides some mechanism for enhancing fluorescent protein levels independent of RNA–RNA effects or IF3 effects alone (potentially by providing protection from RNase E, see Discussion for details). The addition of IF3 to dCasRx appears to provide some additional benefit to enhancing gene expression, possibly by increasing translation initiation as we intended or mRNA stabilization (see Discussion).
Next, we explored the effect of varying the target crRNA location across the mRNA (Figure 2B). Strikingly, shifting the crRNA target from the start of the 5′ UTR to just 10 nts downstream (RFP-12) removed virtually all activation activity. Progressing any further towards the RBS resulted in repression of RFP relative to GFP-targeting crRNAs, regardless of if the crRNA was alone (0.78 ± 0.06, P = 7E–3), coupled with dCasRx (0.71 ± 0.03, P = 4E–4), or coupled with dCasRx-IF3 (0.51 ± 0.06, P = 1E–4). Maximum repression was obtained when targeting directly to the RBS (RFP-15), again with the crRNA alone (0.69 ± 0.10, P = 8E–3), coupled with dCasRx (0.38 ± 0.11, P = 8E–4), or coupled with dCasRx-IF3 (0.27 ± 0.03, P = 4E–6). A slight inhibitory effect was also seen when targeting with the start of the ORF (RFP-16). No effect was observed when targeting in the middle of the ORF (330–350nts from the translation start site, RFP-17) or when targeting the 3′ UTR (RFP-18). These results suggest that, despite being designed for translational activation, dCasRx-IF3 efficiently represses translation when occluding the RBS and could therefore be employed in multiplexing strategies for simultaneous knockdown and overexpression.
At this point, we recognized that our crRNAs used a sub-optimal transcription termination sequence, leading to a 300nt 3′ overhang between the end of our crRNAs and the actual transcription termination (Supplementary Figure S4A). To ensure this did not influence our results, we redesigned our crRNA base-cloning construct to remove this overhang. The names of all crRNAs using this shortened terminator are annotated as ending in ‘T’ to indicate this change. We explored 3 of our previous crRNAs (RFP-1, RFP-8 and RFP-10) with and without the terminator change. We found the effect was either negligible, or slightly improved activation efficiency (particularly of the crRNA alone) (Supplementary Figure S4B). Our highest fold-activation of dCasRx-IF3 relative to the GFP-2 targeting crRNA control (13.7 ± 0.6, P = 4E–7) was obtained using the RFP-8T crRNA, indicating that use of these shorter terminators is preferable.
We therefore proceeded to explore our next question with crRNAs using this shorter terminator. We wanted to systematically explore the space between RFP-1 and RFP-12 step-by-step, using crRNAs RFP-19T to RFP-27T respectively (Figure 2C). We found that stepping 2 nts downstream from the start of the 5′ UTR slightly increased dCasRx-IF3 activation relative to GFP-2 dCasRx-IF3 strains to 11.9 ± 0.4 fold (P = 1E–8). A large drop in activation was then seen when transitioning further downstream, with the exception of RFP-23T targeting 5 nts downstream of the 5′ UTR start where both dCasRx and dCasRx-IF3 still maintained substantial activation potential (7.5 ± 2.5, P = 0.01 and 7.6 ± 0.4, P = 7E–6 for dCasRx and dCasRx-IF3 respectively).
We repeated this experiment with a subset of crRNAs alone, and in combination with dCasRx and dCasRx-IF3 (Supplementary Figure S5). These results largely confirmed the reproducibility of the results demonstrated in Figure 2A-C. In one of these experiments, we were able to obtain a slightly higher fold-activation of dCasRx-IF3 relative to the GFP-2 targeting crRNA control (16.0 ± 1.5 fold, P = 9.6E–6) when using RFP-11 (Supplementary Figure S5A). This is comparable to the 23-fold activation first reported with transcription-based CRISPRa in bacteria, which required deletion of native rpoZ to achieve (10).
Finally, we also performed kinetic experiments in parallel to provide an insight into the dynamics of fluorescence changes over time, using our longest crRNA RFP-11. All strains exhibited nearly identical growth profiles (Figure 2D). While there were no significant differences (P = 0.13) in growth rates between dCasRx (0.42 ± 0.04 h–1) and dCasRx-IF3 (0.36 ± 0.05 h–1) strains, there was a slight yet noticeable increase in the lag time of dCasRx-IF3 strains relative to dCasRx strains. This suggests that dCasRx-IF3 does introduce a small influence in the shift into exponential growth. Fluorescence of these strains remained relatively similar in early growth, yet diverged markedly towards later growth (Figure 2E).
Enhancing the IF3-specific activation of CRISPR-RNAa
While the results above show that dCasRx-IF3 enhances expression more than dCasRx alone, the greatest difference observed was only 3.3-fold. We therefore sought to determine parameters which increase the IF3-specific effect of CRISPR-RNAa. We reduced the abundance of dCasRx and dCasRx-IF3 molecules by reducing aTc induction concentration from 500 to 20 ng/μl, to ensure the Cas proteins were not saturating the target mRNAs. We also delayed induction of the target rfp mRNA production from the start of the pre-culture, to 5 h after inoculation, a common strategy for enhancing expression of difficult proteins (51).
Using these slightly altered experimental parameters (as outlined in Supplementary Figure S6), we explored varying the concentration of IPTG induction. We first performed this experiment at 37°C (Figure 3A). Reducing IPTG concentration to 20 μM eliminated differences in fluorescence between these strains (P = 0.21), while increasing IPTG concentration increased the fold change in fluorescence activation from 1.7 ± 0.3-fold (P = 0.02) to 4.1 ± 1.4-fold (P = 2E–4). This indicates a dose-dependent response tied to the copy number of rfp mRNAs.
Next, we repeated the experiment at 30°C (Figure 3B). Low temperatures are frequently used to slow protein production rates, providing time for proper folding and to discourage protein aggregation (51). We found that this lower temperature further accentuated the differences between dCasRx and dCasRx-IF3. The fold difference between dCasRx-IF3 and dCasRx was 3.0 ± 0.6 (P = 5E–3) at the lowest IPTG concentration, 10.3 ± 4.8 (P = 0.03) at the intermediate IPTG concentration, and 15.5 ± 1.0 (P = 5E–7) at the highest IPTG concentration.
These trends were largely recapitulated when the experiment was performed with IPTG induction included at the start of the experiment, rather than after 5 h of growth (Supplementary Figure S6). In this case, lower IPTG concentrations resulted in less fluorescence differences overall between dCasRx and dCasRx-IF3. We also explored the 22nt crRNA variant RFP-8T using this new experimental setup. (Supplementary Figure S7). The trends presented in Figure 3 were largely recapitulated with this slightly altered guide RNA; at 30°C, the fold difference between dCasRx-IF3 and dCasRx was 2.1 ± 1.1 (P = 0.03) at the lowest IPTG concentration, 6.9 ± 1.6 (P = 0.003) at the intermediate IPTG concentration, and 9.3 ± 7.2 (P = 2E–3) at the highest IPTG concentration.
Quantifying copy number differences
The reporter E. coli strain used in this work encodes multiple copies of rfp integrated into its genome. This strain was built specifically to maintain stable copy number, and it has been reported to maintain 71% of its original copies of rfp after a billion-fold outgrowth in the absence of antibiotics (42). However, to ensure that copy number variations emerging during strain construction did not lead to the results presented here, we explored the copy number of rfp at both the RNA and DNA level.
We first quantified target RNA abundance in the strains at the time of fluorescence measurements, using the protocol outlined in Supplementary Figure S6 (at 30°C) for strains harboring dCasRx variants and RFP-1 and RFP-8T crRNAs. Fold-fluorescence enhancement of dCasRx-IF3 relative to dCasRx strains was 7.2 ± 0.5 (P = 7E–8) and 3.9 ± 0.3 (P = 4E–7) when harboring either the 20 nt RFP-1 or 22 nt RFP-8T crRNA respectively (Supplementary Figure S8A). Using flow cytometry, we noted a uniform population distribution in both cell shape and fluorescence distribution, indicating no obvious emergence of sub-populations due to copy number loss during the experiment (Supplementary Figure S9 and Supplementary Table S4). Using RT-qPCR, we found that both dCasRx-IF3 strains harbored more copies of rfp mRNA than their corresponding dCasRx strains. Specifically, rfp mRNA abundance was 2.0 ± 0.4 (P = 0.001) and 4.5 ± 1.9 (P = 4E–4) fold higher in the dCasRx-IF3 strains relative to the dCasRx strains harboring either the 20 nt RFP-1 or 22 nt RFP-8T crRNA, respectively (Supplementary Figure S8B).
We next measured rfp copy number at the DNA level to determine if potential copy number changes might contribute to increased rfp transcript abundance. We repeated the experiment, this time including the GFP-2 control. Fold-enhancement of fluorescence in the case of dCasRx-IF3 relative to dCasRx strains was 7.7 ± 1.1 (P = 1E–9) and 8.7 ± 4.6 (P = 7E–7) when harboring either the 20 nt RFP-1 or 22 nt RFP-8T crRNA, respectively (Supplementary Figure S8C). There was a slight drop in fluorescence for strains harboring dCasRx-IF3 relative to dCasRx when harboring the GFP-targeting crRNA (fold change = 0.89 ± 0.05, P = 4E–3), possibly due to the lag time effect noted earlier. We found no difference in the copy number of rfp in the 20 nt RFP-1 strains, where the relative rfp copy number of dCasRx-IF3 to dCasRx was 1.0 ± 0.2 (P = 0.29) (Supplementary Figure S8D). However, for both of the other strains, there was a slight increase in the copy number of the dCasRx-IF3 relative to the dCasRx strains. The relative rfp copy number of dCasRx-IF3 to dCasRx was 1.6 ± 0.3 (P = 5E–3) when harboring the 22 nt RFP-8T crRNA, and 1.9 ± 0.5 (P = 5E–3) when harboring the GFP-targeting crRNA. Notably, while strains of dCasRx-IF3 with these two crRNAs exhibited slightly higher copy numbers at the DNA level, only in targeting rfp did this result in enhanced fluorescence. These results suggest that while copy number differences were indeed possible at both the DNA and RNA level, they do not appear to have substantially influenced the enhanced mRNA copy numbers.
We next revisited the differences in mRNA copy numbers, this time focusing on quantifying relative RNA copy numbers over time between dCasRx and dCasRx-IF3 strains both harboring RFP-1 crRNA targets (Figure 4). We repeated the experiment as outlined in Figure 3A, except this time we specifically fixed the endpoint measurement to 16 h, while also collecting timepoints at 0, 30, 60, 120 and 240 min post-dilution (IE after Supplementary Figure S6, step 4). We also included controls with no IPTG induction at any point, to ensure leaky expression was not the cause of these differences. We measured endpoint fluorescence and optical densities of these cultures at 960 min (Supplementary Figure S10). We also setup a kinetic experiment in parallel to capture growth and fluorescence profiles over time of these cultures (Supplementary Figure S11). We found that the relative normalized fluorescence of dCasRx-IF3 to dCasRx strains without IPTG induction was 1.7 ± 0.1 (P = 2E–6), indicating a slight effect of leaky expression through the IPTG-inducible promoter (Figure 4A). However, we found a much greater difference when IPTG was included 21.3 ± 0.9 (P = 3E–14), indicating this was not the sole reason for differences in these strains.
We next explored the copy numbers of rfp mRNA over time in these conditions. At the time of culture dilution, we found that there were roughly half as many copies of rfp mRNA in the IPTG-induced dCasRx-IF3 strains relative to dCasRx strains (0.46-fold, P = 3E–3). There were no differences in rfp mRNA copy number in the uninduced cultures. Within the first 60 min of culture dilution, we could find no difference in copy number. However, by 120 min, we found 4.9-fold more rfp mRNA in the dCasRx-IF3 strain (P = 3E–5). By 240 min, this fold-difference was increased to 9.8 (P = 1E–7). By the end of the experiment, the relative rfp copy numbers had normalized between these strains, both with and without IPTG induction. Raw CT values are presented in Supplementary Figure S12.
Collectively, these results suggest that rfp mRNA was more stable in dCasRx-IF3 strains for at least the first 4 h post-culture dilution. This could be due to an increase in translation initiation caused by IF3 leading to greater ribosome saturation of the mRNA, thereby extending mRNA half-life. Indeed, increasing translation initiation by varying the strength of the RBS has been shown to not only increase gene expression, but copy number of the target mRNA up to 11.8-fold (52), lending credence to this hypothesis.
Activation of native β-galactosidase
To further validate the application of dCasRx-IF3 for enhancing gene expression, we explored activation of an endogenous gene. We turned to the well-characterized lacZ gene of E. coli, encoding β-galactosidase. While the transcription start site (TSS) of lacZ is canonically at the beginning of the LacO binding region, transcription can also begin at three other locations (numbered in order of decreasing strength from TSS1 to TSS4) (53,54). We designed three 25 nt crRNAs (with the aforementioned shortened terminator sequence) to target the 5′UTR of lacZ, corresponding to these three less-frequent TSSs (Figure 5A). TSS1 was not targeted, as this would encroach onto the RBS (25 bp upstream), as evidenced by our previous results with crRNA RFP-13 (26 bp upstream). We transformed these constructs into AG1, a base strain of DH1 E. coli with the native operon of lacZ kept intact. Experiments were conducted using the protocol developed in Supplementary Figure S6, with the temperature maintained at 30°C and 500 μM IPTG used to induce the lac operon. A β-Galactosidase assay was performed to quantify β-galactosidase expression both before and after dilution and removal of IPTG (48).
Before dilution, strains harboring the LacZ1 crRNA alongside both dCasRx and dCasRx-IF3 enhanced β-galactosidase activity (Figure 5B). The relative Miller units between strains harboring dCasRx and either the LacZ1 or control GFP-2 crRNAs was 2.2 ± 0.4 (P = 5E–4). Likewise, the relative Miller units between strains harboring dCasRx and either the LacZ1 or control GFP crRNAs was 2.6 ± 0.5 (P = 4E–4). Strains harboring dCasRx-IF3 and the LacZ1 crRNA showed a 1.2 ± 0.1 (P = 0.02) fold increase in β-galactosidase activity relative to strains harboring dCasRx and the LacZ1 crRNA. There were no significant differences in any of the remaining strains.
Samples were diluted 1:50 and IPTG was removed, and β-galactosidase activity was again measured at the end of growth. While the strains harboring LacZ1 crRNAs again produced the most Miller units, there was no significant difference between the strain harboring dCasRx and LacZ1 crRNA and the strain harboring dCasRx and a GFP crRNA (1.2 ± 0.2, P = 0.24). However, there was a significant enhancement between the strain harboring dCasRx-IF3 and LacZ1 and the strain harboring dCasRx-IF3 and a GFP crRNA (2.1 ± 0.4, P = 0.02). The difference between dCasRx and dCasRx-IF3 was also enhanced. The LacZ1 crRNA strain harboring dCasRx-IF3 produced 1.5 ± 0.3 (P = 0.03) fold more Miller units than that harboring dCasRx. Again, no significant differences were observed between any of the remaining strains. Collectively, these results further validate the application of CRISPRa for activating protein expression at the translational level.
DISCUSSION
Even before researchers first co-opted CRISPR-Cas machinery for genetic manipulation, they pondered tethering of translation initiation factors to RNA-targeting proteins for translation efficiency enhancement (55). While scientists have proposed reconfiguring CRISPR machinery to enhance translation (56), it has not been demonstrated in practice (with the exception of CRISPR YTHDF1-mediated 1.4-fold activation in human HEK293T cells (57)), and efforts have largely been focused on controlling transcription.
Here we fill this conspicuous gap by developing CRISPR-RNAa, an approach in which RNA-targeting dCas13-IF fusions are targeted to 5′ UTRs to increase protein production by enhancing translation of the target mRNA-encoded gene. We first show that dCasRx-IF1 and dCasRx-IF3 induce stronger fluorescence than dCasRx when targeted to the start of the 5′UTR of rfp mRNA. We next decouple the individual effects of the guide crRNA, the binding of dCasRx, and IF3 itself. We show that crRNAs alone can slightly enhance gene expression (1.5–3.7-fold), likely through previously established RNA-RNA activating interactions (49,50). Addition of dCasRx increases this effect 2.4-fold. As RNase E, the primary enzyme responsible for mRNA degradation in E. coli, has a preference for 5′ monophosphorylated RNA (58–60), it is possible that dCasRx binding sterically hinders RNase E recognition of the 5′ region and consequently preserves the target mRNA’s half-life (61). This is supported by our observations in Figure 2 that shifting the target location of dCasRx 10 nt downstream of the 5′ UTR failed to activate (or inhibit) gene expression, and also the observation that activation decreased as we shifted the target location 1nt at a time downstream from the start of the 5′UTR.
We proceed to show that tethering translation initiation factors IF1 and IF3 to dCasRx further enhances gene expression, which we theorize is due to the IF’s recruitment of 30S ribosome subunits to the RBS of target mRNA-encoded genes. For evidence of this, we note the increased RFP expression of dCasRx-IF3 relative to dCasRx strains in Figure 2. This is further supported by our results in Figure 4, where rfp mRNA appears more stable in dCasRx-IF3 strains relative to dCasRx strains within the first 4 h of removal of IPTG induction. Enhanced translation initiation rates have been directly tied to higher mRNA levels due to protection of the mRNA by ribosomes from RNases, particularly through preventing RNase E endonucleolytic cleavage (52,62,63). Furthermore, researchers have argued that a competition between translation initiation rates and mRNA decay is the major determinant of mRNA half-life in yeast (64).
In a similar vein as RNA half-life, the influence of growth phase on CRISPR-RNAa is of interest. Free 70S ribosomes are prolific during exponential growth (upwards of 70 000 units) and are thought not to be rate-limiting to translation (65). However, during stationary phase, they can be reduced to as little as 2000 units as they transition into functionally inactive 100S dimers, promoting hibernation (66). This stationary-phase effect is accompanied by a similar reduction in IF3 levels (67) and an overall downregulation of translation initiation (68). Providing excess IF3 has been shown to help dissociate 100S ribosomes into functional 70S ribosomes (69). The influence of 100S ribosome formation and related hibernation factors such as RMF, HPF, and YfiA on CRISPR-RNAa therefore warrants further investigation (70).
Many components of our current implementation of CRISPR-RNAa could be optimized in the future. Here we explore only one form of protein linker, and only through tethering to the C-terminus. Varying the linker location, as well as its structure, could improve functionality. Further investigating the IF domains is also of interest. For instance, IF3 is a two-part protein consisting of an essential C-terminal domain (that performs the bulk of its functions) connected by a flexible hydrophilic linker to a non-essential N-terminal domain (71,72). Future iterations of CRISPR-RNAa could explore these domains individually to reduce size or improve efficiency.
The crRNA might also be made to be inducible, to provide further tunable control. We explore this briefly in Supplementary Figure S13 by placing the crRNA under control of a crystal-violet inducible promoter (46). However, this modification substantially extends the 5′ end of the crRNA and has strong secondary structure. The secondary structure of the 5′ stem has been shown to be critical for CasRx targeting efficacy (73). As many inducible promoters necessitate an element downstream of the +1 transcription start site, the fact that slight modifications to the crRNA 5′ stem can have large effects on CasRx activity suggests that detailed optimization may be required for obtaining the ideal crRNA structure with each different inducible promoter. Future iterations of CRISPR-RNAa using inducible guide RNAs could include elements such as the hammerhead ribozyme to tightly control the 5′ sequence of dCasRx crRNAs (74).
Of note, the target gene and its mRNA copy number might constrain the efficiency of translational activation. We saw significantly greater success of CRISPR-RNAa in activating a heterologous gene expressed at a high DNA copy number (rfp) than a native gene expressed from a single copy (lacZ). CRISPR-RNAa is an overexpression method, and factors peculiar to the target gene product that normally effect any overexpression experiment, such as folding rates or co-translational regulation, should still be expected to have an influence. Relatedly, we determined that lowering the temperature, a common tactic for slowing production rates to improve folding of heterologous proteins (51), enhanced the difference between dCasRx and dCasRx-IF3.
Expansion of CRISPR-RNAa into other organisms is also of interest. To this end, it is important to note the highly conserved nature of the components and processes of translation across all kingdoms of life (75). Within prokaryotes, IF3 is widely conserved, with a fascinatingly unique non-AUG start codon (40). Indeed, it is one of only two genes in E. coli (alongside the nonessential pcnB) to start translation from an AUU codon (76), which has been implicated to be a result of IF3’s capacity for self-regulation (77–79). IF3 discrimination against its own AUU start codon reduces its own translation initiation at high IF3 concentrations, which has been correlated with a reduction in infC coding mRNA in B. subtills (80).
While eukaryotes require additional IFs, forms of prokaryotic IF1, IF2 and IF3 are preserved in some form (81,82). The C-terminal domain of IF3 in particular bears similarity in both form and function to eukaryotic initiation factor eIF1 (83) and archaeal initiation factor aIF1 (84,85). Strikingly, IF3 and eIF1 both bind to both the 30S and 40S subunits in the same region (86). We suspect this conservation in binding will be of great interest in expanding dCasRx-IF3-mediated translational activation into organisms in diverse kingdoms of life.
Finally, we highlight the limitations of CRIPSR-RNAa in its current form. The strong differences between dCasRx and dCasRx-IF3 in activating gene expression required a carefully constructed setup, notably the removal of the inducer driving target mRNA transcription. In contexts more applicable to real-world use of such a tool, such as Supplementary Figure S3 or 5A where inducer was kept constant, fold-differences were 3.3 or 2.6 above basal levels and only 1.3 or 1.2 above enhancement caused by dCasRx-crRNA. Additionally, our data show that mRNAs with short 5′UTR may not be amenable to CRISPR-RNAa. The length of the 5′UTR in which RFP and LacZ were successfully activated were 59 and 53 nts respectively. The median 5′ UTR length of the 3746 mRNAs in E. coli is 153 nts, of which 29% are shorter than 53 nts and may not be amenable (87). Furthermore, given the apparent importance of targeting the start of the 5′UTR as shown in Figure 2, the ability to activate downstream genes with substantially longer 5′UTRs (such as those encoded downstream within an operon) remains to be seen. There is wide latitude for further optimizing CRISPR-RNAa, and future work will be required to address and overcome these limitations.
DATA AVAILABILITY
The authors commit to providing the underlying data generated in this study upon reasonable request to the corresponding author.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Garima Goyal and Nathan J. Hilson for providing access to E. coli strains with rfp integrated in the genome, and for Gina Geiselman for providing supply support. We thank the Joint BioEnergy Institute (JBEI) for the use of its plate readers and flow cytometer. The authors are thankful to Emeric Charles and David Savage for construction of plasmids used as templates for this study.
Author contributions: P.B.O. conceived of the project, built constructs, designed and conducted experiments, performed analysis and composed the manuscript. B.F.C. assisted with strain construction. B.F.C, J.A.D. and J.S.S. provided critical input for the manuscript and provided resources. J.S.S. guided the scope of the project. All authors read and approved the final manuscript.
Notes
Present address: Peter Otoupal, Spotlight Therapeutics, Hayward, CA 94545, USA.
Contributor Information
Peter B Otoupal, Biomanufacturing and Biomaterials Department, Sandia National Laboratories, Livermore, CA, USA; Deconstruction Division, Joint BioEnergy Institute, Lawrence Berkeley National Laboratory, Emeryville, CA, USA; Agile BioFoundry, Department of Energy, Emeryville, CA, USA.
Brady F Cress, Innovative Genomics Institute, University of California, Berkeley, CA, USA; Department of Molecular and Cell Biology, University of California, Berkeley, CA, USA.
Jennifer A Doudna, Innovative Genomics Institute, University of California, Berkeley, CA, USA; Department of Molecular and Cell Biology, University of California, Berkeley, CA, USA; California Institute for Quantitative Biosciences, University of California, Berkeley, CA, USA; Department of Chemistry, University of California, Berkeley, CA, USA; Howard Hughes Medical Institute, University of California, Berkeley, CA, USA; Molecular Biophysics & Integrated Bioimaging Division, Lawrence Berkeley National Laboratory, Berkeley, CA, USA; Gladstone Institutes, University of California, San Francisco, CA, USA.
Joseph S Schoeniger, Systems Biology Department, Sandia National Laboratories, Livermore, CA, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
U. S. Department of Energy, Office of Science, through the Genomic Science Program, Office of Biological and Environmental Research, under the Secure Biosystems Design Initiative project Intrinsic Control for Genome and Transcriptome Editing in Communities (InCoGenTEC); Sandia National Laboratories is managed by National Technology and Engineering Solutions of Sandia, LLC, a wholly owned subsidiary of Honeywell International, Inc., for the U.S. Department of Energy [DE-NA-0003525].
Conflict of interest statement. The authors P.B.O. and J.S.S. have filed a provisional application (serial no. 63/296,943) for a patent regarding this technology. The remaining authors declare no competing financial interests.
REFERENCES
- 1. Jinek M., Chylinski K., Fonfara I., Hauer M., Doudna J.A., Charpentier E.. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012; 337:816–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Qi L.S., Larson M.H., Gilbert L.A., Doudna J.A., Weissman J.S., Arkin A.P., Lim W.A.. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell. 2013; 152:1173–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cress B.F., Toparlak Ö.D., Guleria S., Lebovich M., Stieglitz J.T., Englaender J.A., Jones J.A., Linhardt R.J., Koffas M.A.G.. CRISPathBrick: modular combinatorial assembly of type II-A CRISPR arrays for dCas9-mediated multiplex transcriptional repression in E. coli. ACS Synth. Biol. 2015; 4:987–1000. [DOI] [PubMed] [Google Scholar]
- 4. Gilbert L.A., Larson M.H., Morsut L., Liu Z., Brar G.A., Torres S.E., Stern-Ginossar N., Brandman O., Whitehead E.H., Doudna J.A.et al.. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell. 2013; 154:442–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gilbert L.A., Horlbeck M.A., Adamson B., Villalta J.E., Chen Y., Whitehead E.H., Guimaraes C., Panning B., Ploegh H.L., Bassik M.C.et al.. Genome-scale CRISPR-mediated control of gene repression and activation. Cell. 2014; 159:647–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Konermann S., Brigham M.D., Trevino A.E., Joung J., Abudayyeh O.O., Barcena C., Hsu P.D., Habib N., Gootenberg J.S., Nishimasu H.et al.. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature. 2015; 517:583–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Perez-Pinera P., Kocak D.D., Vockley C.M., Adler A.F., Kabadi A.M., Polstein L.R., Thakore P.I., Glass K.A., Ousterout D.G., Leong K.W.et al.. RNA-guided gene activation by CRISPR-Cas9-based transcription factors. Nat. Methods. 2013; 10:973–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Maeder M.L., Linder S.J., Cascio V.M., Fu Y., Ho Q.H., Joung J.K.. CRISPR RNA-guided activation of endogenous human genes. Nat. Methods. 2013; 10:977–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tak Y.E., Kleinstiver B.P., Nuñez J.K., Hsu J.Y., Horng J.E., Gong J., Weissman J.S., Joung J.K.. Inducible and multiplex gene regulation using CRISPR-Cpf1-based transcription factors. Nat. Methods. 2017; 14:1163–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bikard D., Jiang W., Samai P., Hochschild A., Zhang F., Marraffini L.A.. Programmable repression and activation of bacterial gene expression using an engineered CRISPR-Cas system. Nucleic Acids Res. 2013; 41:7429–7437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Fontana J., Sparkman-Yager D., Zalatan J.G., Carothers J.M.. Challenges and opportunities with CRISPR activation in bacteria for data-driven metabolic engineering. Curr. Opin. Biotechnol. 2020; 64:190–198. [DOI] [PubMed] [Google Scholar]
- 12. Dong C., Fontana J., Patel A., Carothers J.M., Zalatan J.G.. Synthetic CRISPR-Cas gene activators for transcriptional reprogramming in bacteria. Nat. Commun. 2018; 9:2489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Otoupal P.B., Erickson K.E., Escalas-Bordoy A., Chatterjee A.. CRISPR perturbation of gene expression alters bacterial fitness under stress and reveals underlying epistatic constraints. ACS Synth. Biol. 2017; 6:94–107. [DOI] [PubMed] [Google Scholar]
- 14. Otoupal P.B., Chatterjee A.. CRISPR gene perturbations provide insights for improving bacterial biofuel tolerance. Front. Bioeng. Biotechnol. 2018; 6:122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Otoupal P.B., Cordell W.T., Bachu V., Sitton M.J., Chatterjee A.. Multiplexed deactivated CRISPR-Cas9 gene expression perturbations deter bacterial adaptation by inducing negative epistasis. Commun. Biol. 2018; 1:129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Schilling C., Koffas M.A.G., Sieber V., Schmid J.. Novel prokaryotic CRISPR-Cas12a-based tool for programmable transcriptional activation and repression. ACS Synth. Biol. 2020; 9:3353–3363. [DOI] [PubMed] [Google Scholar]
- 17. Lu Z., Yang S., Yuan X., Shi Y., Ouyang L., Jiang S., Yi L., Zhang G.. CRISPR-assisted multi-dimensional regulation for fine-tuning gene expression in Bacillussubtilis. Nucleic Acids Res. 2019; 47:e40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Peng R., Wang Y., Feng W.-W., Yue X.-J., Chen J.-H., Hu X.-Z., Li Z.-F., Sheng D.-H., Zhang Y.-M., Li Y.-Z.. CRISPR/dCas9-mediated transcriptional improvement of the biosynthetic gene cluster for the epothilone production in Myxococcusxanthus. Microb. Cell Fact. 2018; 17:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ho H.-I., Fang J.R., Cheung J., Wang H.H.. Programmable CRISPR-Cas transcriptional activation in bacteria. Mol. Syst. Biol. 2020; 16:e9427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fontana J., Dong C., Kiattisewee C., Chavali V.P., Tickman B.I., Carothers J.M., Zalatan J.G.. Effective CRISPRa-mediated control of gene expression in bacteria must overcome strict target site requirements. Nat. Commun. 2020; 11:1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kiattisewee C., Dong C., Fontana J., Sugianto W., Peralta-Yahya P., Carothers J.M., Zalatan J.G.. Portable bacterial CRISPR transcriptional activation enables metabolic engineering in Pseudomonasputida. Metab. Eng. 2021; 66:283–295. [DOI] [PubMed] [Google Scholar]
- 22. Villegas Kcam M.C., Tsong A.J., Chappell J.. Uncovering the distinct properties of a bacterial type I-E CRISPR activation system. ACS Synth. Biol. 2022; 11:1000–1003. [DOI] [PubMed] [Google Scholar]
- 23. Villegas Kcam M.C., Tsong A.J., Chappell J.. Rational engineering of a modular bacterial CRISPR-Cas activation platform with expanded target range. Nucleic Acids Res. 2021; 49:4793–4802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Liu Y., Wan X., Wang B.. Engineered CRISPRa enables programmable eukaryote-like gene activation in bacteria. Nat. Commun. 2019; 10:3693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. East-Seletsky A., O’Connell M.R., Knight S.C., Burstein D., Cate J.H.D., Tjian R., Doudna J.A.. Two distinct RNase activities of CRISPR-C2c2 enable guide-RNA processing and RNA detection. Nature. 2016; 538:270–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Abudayyeh O.O., Gootenberg J.S., Konermann S., Joung J., Slaymaker I.M., Cox D.B.T., Shmakov S., Makarova K.S., Semenova E., Minakhin L.et al.. C2c2 is a single-component programmable RNA-guided RNA-targeting CRISPR effector. Science. 2016; 353:aaf5573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cox D.B.T., Gootenberg J.S., Abudayyeh O.O., Franklin B., Kellner M.J., Joung J., Zhang F.. RNA editing with CRISPR-Cas13. Science. 2017; 358:1019–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Qu L., Yi Z., Zhu S., Wang C., Cao Z., Zhou Z., Yuan P., Yu Y., Tian F., Liu Z.et al.. Programmable RNA editing by recruiting endogenous ADAR using engineered RNAs. Nat. Biotechnol. 2019; 37:1059–1069. [DOI] [PubMed] [Google Scholar]
- 29. Abudayyeh O.O., Gootenberg J.S., Franklin B., Koob J., Kellner M.J., Ladha A., Joung J., Kirchgatterer P., Cox D.B.T., Zhang F.. A cytosine deaminase for programmable single-base RNA editing. Science. 2019; 365:382–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yang L.-Z., Wang Y., Li S.-Q., Yao R.-W., Luan P.-F., Wu H., Carmichael G.G., Chen L.-L.. Dynamic imaging of RNA in living cells by CRISPR-Cas13 systems. Mol. Cell. 2019; 76:981–997. [DOI] [PubMed] [Google Scholar]
- 31. Konermann S., Lotfy P., Brideau N.J., Oki J., Shokhirev M.N., Hsu P.D.. Transcriptome engineering with RNA-targeting type VI-D CRISPR effectors. Cell. 2018; 173:665–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang L., Zhou J., Wang Q., Wang Y., Kang C.. Rapid design and development of CRISPR-Cas13a targeting SARS-CoV-2 spike protein. Theranostics. 2021; 11:649–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Blanchard E.L., Vanover D., Bawage S.S., Tiwari P.M., Rotolo L., Beyersdorf J., Peck H.E., Bruno N.C., Hincapie R., Michel F.et al.. Treatment of influenza and SARS-CoV-2 infections via mRNA-encoded Cas13a in rodents. Nat. Biotechnol. 2021; 39:717–726. [DOI] [PubMed] [Google Scholar]
- 34. Charles E.J., Kim S.E., Knott G.J., Smock D., Doudna J., Savage D.F.. Engineering improved Cas13 effectors for targeted post-transcriptional regulation of gene expression. 2021; bioRxiv doi:26 May 2021, preprint: not peer reviewed 10.1101/2021.05.26.445687. [DOI]
- 35. Myasnikov A.G., Simonetti A., Marzi S., Klaholz B.P.. Structure-function insights into prokaryotic and eukaryotic translation initiation. Curr. Opin. Struct. Biol. 2009; 19:300–309. [DOI] [PubMed] [Google Scholar]
- 36. Gualerzi C.O., Pon C.L.. Initiation of mRNA translation in prokaryotes. Biochemistry. 1990; 29:5881–5889. [DOI] [PubMed] [Google Scholar]
- 37. Shah P., Ding Y., Niemczyk M., Kudla G., Plotkin J.B.. Rate-limiting steps in yeast protein translation. Cell. 2013; 153:1589–1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sharma A.K., Sormanni P., Ahmed N., Ciryam P., Friedrich U.A., Kramer G., O’Brien E.P. A chemical kinetic basis for measuring translation initiation and elongation rates from ribosome profiling data. PloS Comput. Biol. 2019; 15:e1007070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Conte M.R., Pennell S.. Roberts G.C.K. Translation initiation: a eukaryotic perspective. Encyclopedia of Biophysics. 2013; Berlin, Heidelberg: Springer Berlin Heidelberg; 2644–2650. [Google Scholar]
- 40. Kozak M. Initiation of translation in prokaryotes and eukaryotes. Gene. 1999; 234:187–208. [DOI] [PubMed] [Google Scholar]
- 41. Laursen B.S., Sørensen H.P., Mortensen K.K., Sperling-Petersen H.U.. Initiation of protein synthesis in bacteria. Microbiol. Mol. Biol. Rev. 2005; 69:101–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Goyal G., Costello Z., Alonso-Gutierrez J., Kang A., Lee T.S., Garcia Martin H., Hillson N.J.. Parallel integration and chromosomal expansion of metabolic pathways. ACS Synth. Biol. 2018; 7:2566–2576. [DOI] [PubMed] [Google Scholar]
- 43. Otoupal P.B., Ito M., Arkin A.P., Magnuson J.K., Gladden J.M., Skerker J.M.. Multiplexed CRISPR-Cas9-based genome editing of Rhodosporidiumtoruloides. Msphere. 2019; 4:e00099-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chappell J., Takahashi M.K., Lucks J.B.. Creating small transcription activating RNAs. Nat. Chem. Biol. 2015; 11:214–220. [DOI] [PubMed] [Google Scholar]
- 45. Ran F.A., Hsu P.D., Wright J., Agarwala V., Scott D.A., Zhang F.. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013; 8:2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ruegg T.L., Pereira J.H., Chen J.C., DeGiovanni A., Novichkov P., Mutalik V.K., Tomaleri G.P., Singer S.W., Hillson N.J., Simmons B.A.et al.. Jungle express is a versatile repressor system for tight transcriptional control. Nat. Commun. 2018; 9:3617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhou K., Zhou L., Lim Q.’En, Zou R., Stephanopoulos G., Too H.-P.. Novel reference genes for quantifying transcriptional responses of Escherichiacoli to protein overexpression by quantitative PCR. BMC Mol. Biol. 2011; 12:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Schaefer J., Jovanovic G., Kotta-Loizou I., Buck M.. Single-step method for β-galactosidase assays in Escherichiacoli using a 96-well microplate reader. Anal. Biochem. 2016; 503:56–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Isaacs F.J., Dwyer D.J., Ding C., Pervouchine D.D., Cantor C.R., Collins J.J.. Engineered riboregulators enable post-transcriptional control of gene expression. Nat. Biotechnol. 2004; 22:841–847. [DOI] [PubMed] [Google Scholar]
- 50. Li L.-C., Okino S.T., Zhao H., Pookot D., Place R.F., Urakami S., Enokida H., Dahiya R.. Small dsRNAs induce transcriptional activation in human cells. Proc. Natl. Acad. Sci. U.S.A. 2006; 103:17337–17342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Rosano G.L., Ceccarelli E.A.. Recombinant protein expression in Escherichiacoli: advances and challenges. Front. Microbiol. 2014; 5:172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Cetnar D.P., Salis H.M.. Systematic quantification of sequence and structural determinants controlling mRNA stability in bacterial operons. ACS Synth. Biol. 2021; 10:318–332. [DOI] [PubMed] [Google Scholar]
- 53. Xiong X.F., de la Cruz N., Reznikoff W.S.. Downstream deletion analysis of the lac promoter. J. Bacteriol. 1991; 173:4570–4577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Czarniecki D., Noel R.J., Reznikoff W.S.. The -45 region of the Escherichiacoli lac promoter: CAP-dependent and CAP-independent transcription. J. Bacteriol. 1997; 179:423–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Mackay J.P., Font J., Segal D.J.. The prospects for designer single-stranded RNA-binding proteins. Nat. Struct. Mol. Biol. 2011; 18:256–261. [DOI] [PubMed] [Google Scholar]
- 56. O’Connell M.R., Oakes B.L., Sternberg S.H., East-Seletsky A., Kaplan M., Doudna J.A.. Programmable RNA recognition and cleavage by CRISPR/Cas9. Nature. 2014; 516:263–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Rauch S., He E., Srienc M., Zhou H., Zhang Z., Dickinson B.. Programmable RNA-guided RNA effector proteins built from human parts. Cell. 2019; 178:122–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Mackie G.A. RNase E: at the interface of bacterial RNA processing and decay. Nat. Rev. Microbiol. 2013; 11:45–57. [DOI] [PubMed] [Google Scholar]
- 59. Schoenberg D.R. The end defines the means in bacterial mRNA decay. Nat. Chem. Biol. 2007; 3:535–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Koslover D.J., Callaghan A.J., Marcaida M.J., Garman E.F., Martick M., Scott W.G., Luisi B.F.. The crystal structure of the Escherichiacoli RNase E apoprotein and a mechanism for RNA degradation. Structure. 2008; 16:1238–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Celesnik H., Deana A., Belasco J.G.. Initiation of RNA decay in Escherichiacoli by 5′ pyrophosphate removal. Mol. Cell. 2007; 27:79–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Jeon H.J., Kang C., N M.P.A., Lee Y., Wang X., Chattoraj D.K., Lim H.M. Translation initiation control of RNase E-mediated decay of polycistronic gal mRNA. Front. Mol. Biosci. 2020; 7:586413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Baker K.E., Mackie G.A.. Ectopic RNase e sites promote bypass of 5′-end-dependent mRNA decay in Escherichiacoli. Mol. Microbiol. 2003; 47:75–88. [DOI] [PubMed] [Google Scholar]
- 64. Chan L.Y., Mugler C.F., Heinrich S., Vallotton P., Weis K.. Non-invasive measurement of mRNA decay reveals translation initiation as the major determinant of mRNA stability. Elife. 2018; 7:e32536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Kaczanowska M., Rydén-Aulin M.. Ribosome biogenesis and the translation process in Escherichiacoli. Microbiol. Mol. Biol. Rev. 2007; 71:477–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Yoshida H., Wada A.. The 100S ribosome: ribosomal hibernation induced by stress. Wiley Interdiscip. Rev. RNA. 2014; 5:723–732. [DOI] [PubMed] [Google Scholar]
- 67. Howe G., Hershey J.. Initiation factor and ribosome levels are coordinately controlled in Escherichiacoli growing at different rates. J. Biol. Chem. 1991; 258:1954–1959. [PubMed] [Google Scholar]
- 68. Dai X., Zhu M., Warren M., Balakrishnan R., Patsalo V., Okano H., Williamson J.R., Fredrick K., Wang Y.-P., Hwa T.. Reduction of translating ribosomes enables Escherichiacoli to maintain elongation rates during slow growth. Nat. Microbiol. 2016; 2:16231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Yoshida H., Ueta M., Maki Y., Sakai A., Wada A.. Activities of Escherichiacoli ribosomes in IF3 and RMF change to prepare 100S ribosome formation on entering the stationary growth phase. Genes Cells. 2009; 14:271–280. [DOI] [PubMed] [Google Scholar]
- 70. Polikanov Y.S., Blaha G.M., Steitz T.A.. How hibernation factors RMF, HPF, and YfiA turn off protein synthesis. Science. 2012; 336:915–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Ayyub S.A., Dobriyal D., Varshney U.. Contributions of the N- and C-terminal domains of initiation factor 3 to its functions in the fidelity of initiation and antiassociation of the ribosomal subunits. J. Bacteriol. 2017; 199:e00051-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Petrelli D., LaTeana A., Garofalo C., Spurio R., Pon C.L., Gualerzi C.O.. Translation initiation factor IF3: two domains, five functions, one mechanism?. EMBO J. 2001; 20:4560–4569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Wessels H.H., Méndez-Mancilla A., Guo X., Legut M., Daniloski Z., Sanjana N.E.. Massively parallel Cas13 screens reveal principles for guide RNA design. Nat. Biotechnol. 2020; 38:722–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Gao Y., Zhao Y.. Self-processing of ribozyme-flanked RNAs into guide RNAs in vitro and in vivo for CRISPR-mediated genome editing. J. Integr. Plant Biol. 2014; 56:343–349. [DOI] [PubMed] [Google Scholar]
- 75. Tuller T., Carmi A., Vestsigian K., Navon S., Dorfan Y., Zaborske J., Pan T., Dahan O., Furman I., Pilpel Y.. An evolutionarily conserved mechanism for controlling the efficiency of protein translation. Cell. 2010; 141:344–354. [DOI] [PubMed] [Google Scholar]
- 76. Binns N., Masters M.. Expression of the Escherichiacoli pcnB gene is translationally limited using an inefficient start codon: a second chromosomal example of translation initiated at AUU. Mol. Microbiol. 2002; 44:1287–1298. [DOI] [PubMed] [Google Scholar]
- 77. Butler J.S., Springer M., Dondon J., Graffe M., Grunberg-Manago M.. Escherichia coli protein synthesis initiation factor IF3 controls its own gene expression at the translational level in vivo. J. Mol. Biol. 1986; 192:767–780. [DOI] [PubMed] [Google Scholar]
- 78. Sacerdot C., Chiaruttini C., Engst K., Graffe M., Milet M., Mathy N., Dondon J., Springer M.. The role of the AUU initiation codon in the negative feedback regulation of the gene for translation initiation factor IF3 in Escherichiacoli. Mol. Microbiol. 1996; 21:331–346. [DOI] [PubMed] [Google Scholar]
- 79. McCarthy J.E., Brimacombe R.. Prokaryotic translation: the interactive pathway leading to initiation. Trends Genet. 1994; 10:402–407. [DOI] [PubMed] [Google Scholar]
- 80. Bruscella P., Shahbabian K., Laalami S., Putzer H.. RNase Y is responsible for uncoupling the expression of translation factor IF3 from that of the ribosomal proteins L35 and L20 in Bacillussubtilis. Mol. Microbiol. 2011; 81:1526–1541. [DOI] [PubMed] [Google Scholar]
- 81. Hussain T., Llácer J.L., Wimberly B.T., Kieft J.S., Ramakrishnan V.. Large-Scale movements of IF3 and tRNA during bacterial translation initiation. Cell. 2016; 167:133–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Gutu A., Nesbit A.D., Alverson A.J., Palmer J.D., Kehoe D.M.. Unique role for translation initiation factor 3 in the light color regulation of photosynthetic gene expression. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:16253–16258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Sonenberg N., Dever T.E.. Eukaryotic translation initiation factors and regulators. Curr. Opin. Struct. Biol. 2003; 13:56–63. [DOI] [PubMed] [Google Scholar]
- 84. Coureux P.-D., Lazennec-Schurdevin C., Monestier A., Larquet E., Cladière L., Klaholz B.P., Schmitt E., Mechulam Y.. Cryo-EM study of start codon selection during archaeal translation initiation. Nat. Commun. 2016; 7:13366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Schmitt E., Coureux P.-D., Kazan R., Bourgeois G., Lazennec-Schurdevin C., Mechulam Y.. Recent advances in archaeal translation initiation. Front. Microbiol. 2020; 11:584152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Lomakin I.B., Shirokikh N.E., Yusupov M.M., Hellen C.U.T., Pestova T.V.. The fidelity of translation initiation: reciprocal activities of eIF1, IF3 and YciH. EMBO J. 2006; 25:196–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Kim D., Hong J.S.-J., Qiu Y., Nagarajan H., Seo J.-H., Cho B.-K., Tsai S.-F., Palsson B.Ø.. Comparative analysis of regulatory elements between Escherichiacoli and Klebsiellapneumoniae by genome-wide transcription start site profiling. PloS Genet. 2012; 8:e1002867. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors commit to providing the underlying data generated in this study upon reasonable request to the corresponding author.