

Abstract
Neuroinflammation is a central mechanism involved in neurodegeneration as observed in Alzheimer’s disease (AD), the most prevalent form of neurodegenerative disease. Apolipoprotein E4 (APOE4), the strongest genetic risk factor for AD, directly influences disease onset and progression by interacting with the major pathological hallmarks of AD including amyloid-β plaques, neurofibrillary tau tangles, as well as neuroinflammation. Microglia and astrocytes, the two major immune cells in the brain, exist in an immune-vigilant state providing immunological defense as well as housekeeping functions that promote neuronal well-being. It is becoming increasingly evident that under disease conditions, these immune cells become progressively dysfunctional in regulating metabolic and immunoregulatory pathways, thereby promoting chronic inflammation-induced neurodegeneration. Here, we review and discuss how APOE and specifically APOE4 directly influences amyloid-β and tau pathology, and disrupts microglial as well as astroglial immunomodulating functions leading to chronic inflammation that contributes to neurodegeneration in AD.
Keywords: Alzheimer’s disease, Apolipoprotein E, Inflammation, Microglia, Astrocytes, Neurodegeneration
1. Introduction
Inflammation is an important factor that can drive neurodegeneration, both in triggering neurodegeneration and in providing promising therapeutic avenues to limit neurodegeneration. In fact, genome-wide association studies (GWAS) have identified several innate immune related genes linked to increased risk of developing neurodegenerative disorders, suggesting that immune cells play a key role in the pathogenesis of neurodegeneration. One of the genes that has disease-associated variants is apolipoprotein E (APOE). The apolipoprotein E4 (APOE4) allele, is a major shared risk factor for several neurodegenerative diseases including Alzheimer’s disease (AD) and the APOE2 allele decreases risk for AD[1], [2]. APOE4 is also the strongest genetic risk factor for late onset AD, likely in part due to its role in lipid metabolism and related inflammation [3]. AD is the most common cause of dementia and is pathologically characterized by the presence of proteopathic aggregates in the brain including extracellular amyloid-β (Aβ)-containing plaques as well as intracellular neurofibrillary tangles containing hyperphosphorylated, aggregated tau. APOE4 carriers in AD show earlier Aβ deposition and clinical disease onset as well as faster disease progression, heavier Aβ plaque burden and increased brain atrophy compared to non-APOE4 carriers, highlighting a prominent role of APOE4 in AD pathogenesis [4], [5]. APOE2 carriers have later Aβ deposition, clinical onset and increased longevity relative to non-APOE2 carriers [6]. To date, there are clues to the cause(s) of AD, but there is still a lot that is not yet well understood. Early onset familial AD, also called autosomal dominant AD, which accounts for less than 1% of AD cases, is primarily caused by overproduction of longer forms of Aβ relative to other forms resulting from mutations in the amyloid precursor protein (APP) gene or genes encoding presenilin 1 (PSEN1) or 2 (PSEN) [7]. Less common forms are caused by mutations in APP in the coding sequence of Aβ that do not influence Aβ production but result in a more amyloidogenic form of Aβ that leads to earlier seeding. The majority of AD cases occur later in life over the age of 65 years and is therefore commonly referred to as late onset AD. Accumulating genetic and functional evidence strongly indicates an active role of the brain’s innate immunity in AD pathogenesis and progression [8]. Until recently, inflammatory processes driven by microglia and astrocytes were considered merely pathological bystanders in AD. However, an abundance of evidence now suggests that these immune cells adapt a dynamic disease-associated inflammatory profile in response to pathological aggregates [9], [10]. This immune response may serve as a defense mechanism that initially protects the brain by promoting tissue repair and removing both cellular debris and Aβ aggregates. Under other conditions, it may contribute to disease progression by sustained chronic inflammation that provokes synapse loss and neuronal cell death, as observed in AD (Figure 1). In this review, we highlight and discuss the significance of APOE in modulating immune responses that contribute to neurodegeneration in AD.
Figure 1. ApoE4 as a risk factor for AD.
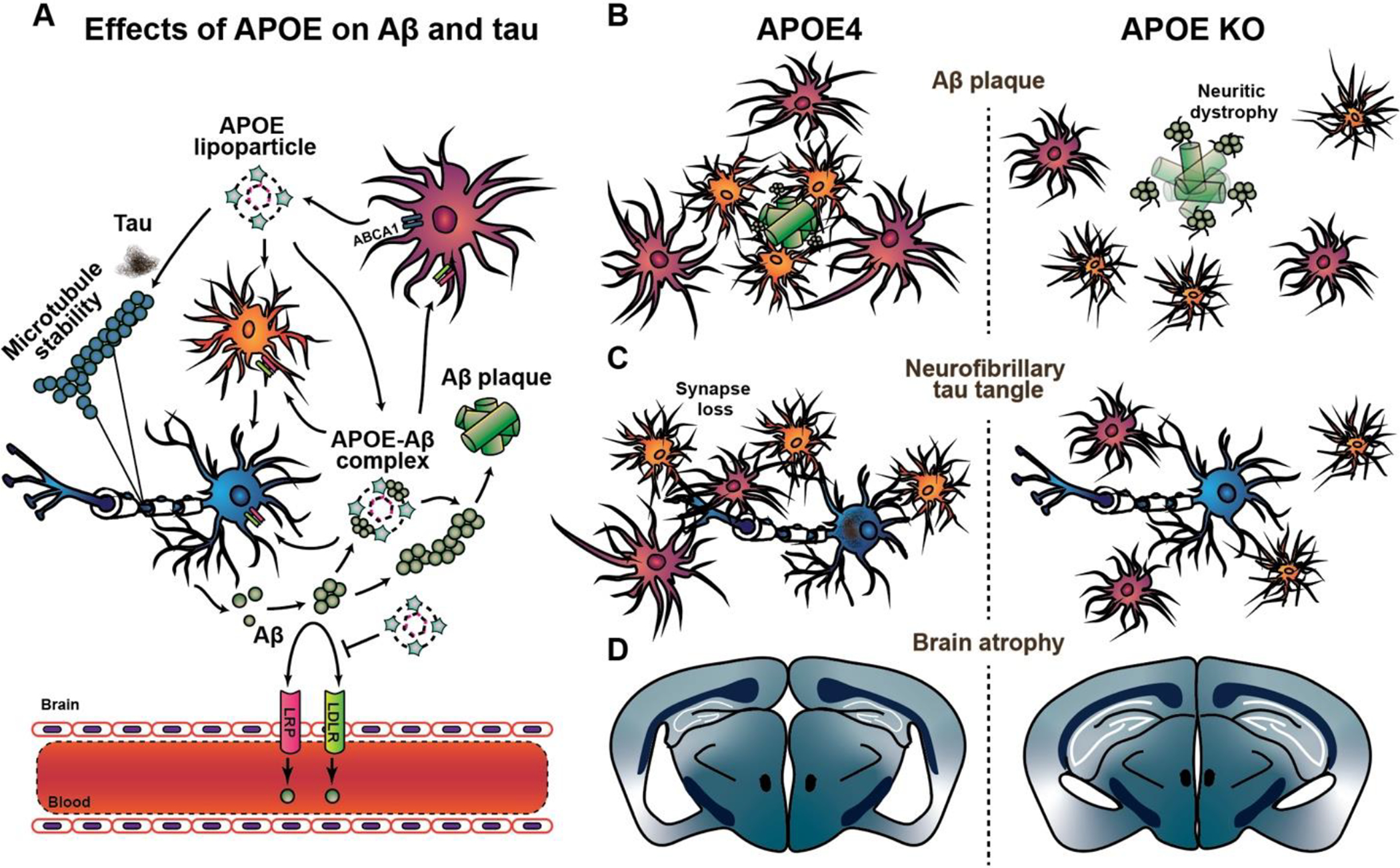
A. Under physiological conditions, APOE is mainly produced and lipidated by ABCA1, and thereafter secreted into the interstitial fluid by glia. LDLR and LRP clear Aβ into neurons and glia and facilitate transport across the BBB, which is impaired by APOE4. Amyloidogenic APP processing leads to Aβ production and release from neurons into the interstitial fluid (ISF). APOE likely binds to smaller oligomeric soluble Aβ species to facilitate its fibrilization into Aβ plaques in an isoform-dependent manner. Similarly, APOE, especially E4, enhances tau pathogenesis, but whether this interaction influences tau aggregation, microtubule instability, or both, is unclear. B. APOE4 promotes a disease-associated microglial and astroglial reactivity to dense core Aβ plaques, where these glial cells are commonly observed clustering around plaques in attempt to phagocytose them. The absence of APOE leads to diffused plaques with reduced peri-plaque microglial clustering as well as increased dystrophic neurites with enhanced plaque-associated tau pathology. C. APOE4 increases tau pathogenesis and leads to increased astroglial and microglial-mediated inflammation, with enhanced synaptic engulfment, whereas the absence of APOE in APOE KO mice blocks this glial-mediated inflammation D. A progressively persistent inflammatory response over time leads to significant neurodegeneration in the presence of APOE4, as marked by brain atrophy and ventricular enlargement. The absence of APOE blocks both the inflammation and the brain atrophy.
2. APOE in the central nervous system
APOE is considered the primary apolipoprotein lipid and cholesterol transporter in the central nervous system (CNS) [11], [12]. Native APOE is primarily lipidated by adenosine triphosphate-binding cassette transporter A1 (ABCA1) in the brain[13], [14]. Additionally, APOE facilitates transport of lipids among different cell types by serving as a ligand for the low-density lipoprotein receptor (LDLR) and lipoprotein receptor-related protein (LRP), the two major metabolic receptors for ApoE [15]. Compared to mice that express only a single type of ApoE protein, there are three major isoforms for human APOE encoded by E2, E3 and E4 alleles. Human APOE includes two separate N- and C-terminal domains joined by a flexible hinge. The N-terminus includes the receptor binding regions, which differs in the APOE isoform residues (APOE4, arginine 112; APOE3 and APOE2, cysteine 112). The C-terminal domain contains the lipid-binding region (residues 244–272) [16]. The cysteine residue at 112 found in both APOE2 and APOE3 may form disulfide-linked dimers. Additionally, while APOE3 and APOE4 that have arginine at residue 158 display normal LDLR binding activity, APOE2 with cysteine at residue 158 shows impaired LDLR binding abilities and is associated with the recessive form of type III hyperlipoproteinemia [17]–[20]. This suggests that the differences in one or two amino acids in the N- and C-terminal domains of APOE significantly alter their interaction and preference to bind lipid and receptor proteins involved in cholesterol uptake [21] as well as Aβ peptides [22], [23]. In contrast to APOE4, APOE2 confers protection against AD. Rare variants in APOE3 with mutations in the lipid binding region [24] or lipoprotein receptor binding region [25] decrease the risk of AD to a similar extent as APOE2. To date, it is not clear how the function of APOE as a lipid redistributor versus other effects it has is mechanistically related to AD. Based on different literature, some of the ways that the E4 allele disease risk association with AD and other diseases may be occurring are likely via Aβ aggregation and clearance as well as by influencing neuroinflammation, impairments in blood brain barrier integrity, synaptic plasticity, and tau hyperphosphorylation[21], [26], [27].
3. APOE and non-glial AD pathology
3.1. APOE and Aβ pathology
In addition to being a constituent of amyloid plaques, ApoE is involved in several aspect of Aβ pathology including Aβ seeding/fibrillization, and clearance [23], [28]–[30]. Extensive imaging and cerebrospinal fluid (CSF) biomarker studies have consistently linked APOE4 with greater Aβ deposition, not only in AD cases, but also in individuals with mild cognitive impairment and cognitively healthy elderly [31]–[33]. APOE4 dose-dependently leads to earlier onset of Aβ deposition the brain of humans and animal models and APOE2 leads to later onset of Aβ deposition than APOE3 [30]. A large body of evidence suggests that ApoE influences Aβ aggregation in an isoform-dependent manner, with ApoE4 most actively promoting Aβ fibril formation compared to ApoE3 and ApoE2, respectively [29], [34]–[37]. Interestingly, the Aβ:ApoE ratio was found to reflect that found in Aβ plaques in the AD brain [36]. However, evidence suggests that the affinity for ApoE to bind to Aβ highly depends on ApoE lipidation state, Aβ species, as well as pH levels of the in vitro model system utilized [22], [38]–[40]. Additionally, ApoE4 appears to greatly influence oligomeric Aβ stabilization [41], [42], which may be essential in accelerating early seeding of Aβ pathology [43]. This is supported by in vivo findings where APOE isoforms were linked with an increased rate of longitudinal Aβ accumulation (E4>E3>E2) in amyloid-negative cognitively healthy individuals compared to amyloid-positive cases [44]. Interestingly, compared to APOE3 carriers, cases with the rare APOE3 V236E Jacksonville variant (APOE3-Jac) showed reduced fibrillar Aβ plaques [24] as well as a reduced risk of developing AD. Consequently, APOE3-Jac was far less prone to aggregate suggesting that changes in APOE aggregation propensity may directly influence Aβ pathology. Moreover, fibrillar amyloid deposits are observed nearly 20 years earlier in cognitively normal APOE4 carriers, compared to APOE4 non-carriers [45]. These studies further support a pivotal role of APOE in increasing AD risk by altering the early phase of amyloid deposition and metabolism (Figure 1).
The effects of ApoE on Aβ deposition and aggregation in vivo have been widely reported using amyloid-depositing transgenic mice lacking ApoE expression or additionally expressing human APOE isoforms. Ablating murine ApoE expression in PDAPP or Tg2576 mouse models of cerebral amyloidosis dramatically reduces Aβ deposits compared with mice endogenously expressing ApoE [46]–[48]. This observed reduction in Aβ plaques was shown to be ApoE gene dose-dependent, further strengthening the relationship between ApoE expression and Aβ deposition. Lack of ApoE expression in amyloid mouse models also appears to specifically and robustly abolish the presence of fibrillar Aβ plaques in both brain parenchyma and in the form of cerebral amyloid angiopathy (CAA), suggesting that the ApoE co-deposited with Aβ plaques in mouse models as well as AD brain, may actively promote Aβ aggregation [49]. APOE4 expression in APP transgenic mice also leads to a robust increase in fibrillar plaque burden (Figure 1) compared to mice expressing APOE3 or APOE2, respectively [28], [50]–[52]. This increase has been confirmed in APOE4 carriers that show an increase in both vascular and parenchymal Aβ plaques [53]–[56]. The exact mechanism through which APOE4 promotes Aβ deposition is not fully understood. One possible interpretation of the data presented above is an increased affinity for APOE4 to bind some form of aggregated Aβ such as oligomers may result in a shift that facilitates soluble Aβ pools to fibrillar Aβ plaques. This may in turn cause earlier disease onset and increase the rate of Aβ plaque formation by stabilizing progressively accumulating oligomeric Aβ species to enhance their neurotoxic effects. However, APOE also binds the diffuse plaques observed in Down’s syndrome brains [57], [58], which suggests that APOE may first bind to nascent diffuse plaques and help convert bound Aβ to β-sheet structures observed in dense cored plaques.
An alternative mechanism through which APOE influences Aβ pathology is through clearance, particularly of soluble Aβ monomers and oligomers (Figure 1) that may otherwise further promote early stages of Aβ seeding [28], [29]. APP transgenic mice lacking endogenous ApoE but expressing APOE3 or APOE4 isoform deposit less Aβ plaques compared to those expressing murine ApoE, suggesting that human APOE isoforms influence clearance [28]. It has also been shown that lipidated APOE has minimal to no binding to cell produced monomeric Aβ and that lipidated APOE competes with Aβ for the cellular update of Aβ via receptors that could slow monomeric Aβ clearance [40]. It is not yet known exactly how APOE influences Aβ clearance, though several mechanisms have been proposed including, lysosomal and enzymatic degradation [59], cellular uptake [60]–[62], interaction with receptors and transporters on cell surface [30], [40], transport across the blood brain barrier [63], and interstitial fluid (ISF) flow [29], [64].
While ApoE is understood to be largely lipidated in the brain, altering the amount of lipidated extracellular ApoE in the brain has a profound impact on Aβ fibrillization and related inflammation. For example, LDLR-deficient mice that show increased ApoE levels in the brain and cerebrospinal fluid (CSF) [51], display more thioflavine S-positive fibrillar plaque burden in APP transgenic mice compared to non-transgenic controls [65], [66]. In line with this, overexpression of LDLR in the brain of mice that develop amyloidosis not only dramatically reduced the amount of soluble ApoE by 90 percent due to enhanced LDLR-mediated degradation [67], but also inhibited plaque deposition and increased Aβ clearance [15]. Furthermore, an increase in the amount of lipidation per ApoE protein through ABCA1 overexpression reduced Aβ deposition in PDAPP mice [68], with an opposite effect on plaque burden in ABCA1-deficient APP mice [14], [69]. The protective effects of APOE3-Jac on reducing AD risk are also attributed to the variant’s effects on enhanced APOE lipidation and lipid-loading properties [24]. Viral-mediated brain expression of APOE3-Jac decreased fibrillar Aβ deposition, plaque-associated ApoE as well as neuritic dystrophy compared to APOE3-expressing APP mice. Interestingly, introducing the V236E substitution in APOE4-expressing cells was sufficient to reduce APOE4 aggregation and increase cholesterol efflux [24], highlighting the importance of APOE structure in the effects of APOE on critical disease-related processes. Altogether, these studies suggest that APOE exhibits effects on both Aβ aggregation as well as on clearance to modulate AD pathogenesis (Figure 1).
In contrast to APOE4-mediated effects on enhancing Aβ burden, reducing ApoE levels in the brain or increasing its lipidation state dramatically decreases plaque burden, further supporting a direct interaction between ApoE and Aβ plaque pathology in AD. Besides genetic ablation of ApoE in APP transgenic mice, immunotherapy studies using anti-ApoE antibody have shown to reduce parenchymal plaques [70]–[72] as well as CAA [73] burden in an antibody dose-dependent manner. Interestingly, the most effective antibody only binds to non-lipidated ApoE present in amyloid plaques and CAA and the effects require microglial-mediated phagocytosis of Aβ [71], [73]. From a therapeutic perspective, it is of note that the anti-ApoE antibody did not cause an amyloid-associated microhemorrhages as occurred with an anti-Aβ antibody [73]. Similarly, knocking down ApoE levels by ~50% in the brain with anti-sense oligonucleotides (ASO) from just after birth decreased Aβ plaque pathology [74]. However, the effect was lost if the treatment started after onset of plaque formation, affirming that ApoE plays an important role in modifying early stages of Aβ aggregation and accumulation. Of note, knocking down ApoE after plaque onset resulted in decreased neuritic dystrophy around plaques, suggesting that even though knocking down ApoE did not affect Aβ plaque accumulation, modulating ApoE after plaque onset may still influence the plaque-associated damage response in the brain. In line with this, homozygous APOE3 R136S Christchurch (APOE3ch) variant appeared to confer strong protection against developing cognitive decline in a patient with an autosomal dominant AD mutation (PSEN1-E280 carrier) in spite of high amyloid burden in the brain [25]. This particular individual only developed mild cognitive impairment three decades after the expected age of clinical onset, with limited changes to tau pathology, glucose metabolism and hippocampal atrophy compared to PSEN1-E280 carriers of a similar age. Compared to APOE3, APOE3ch impaired APOE binding to heparan sulfate proteoglycans, an interaction that is known to promote neuronal uptake of extracellular tau. Thus, while the Aβ-overproducing effects of PSEN1-E280 was not dampened or prevented by APOE3ch, the variant may potentially protect against Aβ-associated cognitive decline, as well as tau spreading and tau-mediated neurodegeneration. Whether this variant may additionally alter Aβ or tau-associated inflammatory response remains unknown.
3.2. APOE and tau pathology
A plethora of studies have thus far reported the interaction between ApoE and Aβ in AD, although until recently there has been a lack of evidence in understanding how ApoE isoforms may interact with tau or tau-mediated neurodegeneration. APOE4 is associated with an increase in tau pathology particularly when amyloid pathology is also present [75]. However, APOE4-positive AD cases were recently found to have a greater baseline Aβ and tau burden, with a higher tau accumulation rate compared to E4-negative cases [76], suggesting an Aβ-independent effect of APOE4 on vulnerability to progressive tau accumulation in AD. A growing body of research provide novel evidence that APOE4 is associated with increased tau pathology in the medial temporal lobe independently of age, clinical status, sex and Aβ [53], [77]–[79]. Interestingly, APOE mRNA expression is highest in the medial temporal lobes [80]. Given the topographical concordance between tau pathology and neurodegeneration [81]–[83], these findings point to tau pathology as a possible culprit responsible for the medial temporal neurodegeneration observed in APOE4 carriers. APOE4 has also been closely linked to increased CSF tau levels in AD, particularly in women compared to men [84], suggesting a sex-dependent effect in APOE4-mediated toxicity.
Transgenic mouse models of tauopathy further support an interaction between APOE and tau pathology [85]–[89]. Using the P301S mouse model of tauopathy expressing either no ApoE or each of the human isoforms, Shi et al. reported that APOE exacerbates tau burden and the subsequent pathogenic cascade underlying neurodegeneration in an isoform-dependent fashion, independently of Aβ [85]. The presence of APOE4 resulted in massive atrophy in the hippocampus, entorhinal cortex, piriform cortex and amygdala, with concomitant enlargement of the ventricular system (Figure 1). In contrast, little to no injury was observed in the absence of APOE, despite the fact that the P301S mice lacking APOE still accumulated some p-tau and insoluble tau in the brain. Additionally, APOE4 expression exacerbated tau accumulation, redistribution to neuronal cell bodies, as well as neuroinflammation, suggesting that APOE plays a pivotal role in neurodegeneration from soluble tau accumulation to brain atrophy. In support of these findings, LDLR overexpression in P301S tauopathy mice, which dramatically reduces ApoE levels in the brain, halved the normally observed phosphorylated tau levels, synaptic loss, and hippocampal atrophy in P301S mice [87]. Furthermore, knocking down ApoE levels similarly protected these mice from damage induced by tau in P301S mice, irrespective of Aβ. A 50% reduction of APOE4 protein using an ASO directed against the human APOE4 gene in P301S mice, similarly protected against tau pathology associated neurodegeneration, synaptic loss as well as neuroinflammation [88], supporting the conclusion that APOE4 directly influences neurodegeneration via gain of toxic functions in tauopathy. A similar effect of APOE4 on increased tau pathology, synaptic loss as well as neurodegeneration was recently reported in cerebral organoid models using induced pluripotent stem cells (iPSC) from AD patients [90]. Intriguingly, the APOE4-related phenotypes observed in AD patient-derived organoids were largely reversed through genome editing to APOE3. The mechanism through which APOE interacts with tau directly or indirectly and whether the ApoE lipidation status may influence said interaction remains to be clarified.
4. Systemic inflammation and glia-dependent effects of APOE on AD pathology
In addition to clearance or response to misfolded proteins, such as Aβ and tau, an overwhelming body of evidence demonstrate a critical role for APOE at the interface of inflammation and neurodegeneration via glial-mediated mechanisms. While ApoE is mainly produced by astrocytes and disease-associated microglia (DAM) in the CNS, in the periphery ApoE is predominantly expressed by leukocytes and hepatocytes, particularly a type of resident hepatic macrophage called Kuppfer cells [91]. Though the role of peripheral ApoE in AD is not as thoroughly investigated as in the CNS, studies suggest that the two sources and metabolism of each pool of ApoE function independently from one another [92], [93]. Genetic restoration of peripheral ApoE expression in mice lacking ApoE expression in the brain rescues learning and memory deficits observed in global ApoE KO mice [94], suggesting that peripheral ApoE may have an effect on CNS functions through the vasculature [95], [96]. Similarly to the CNS, APOE4 is also associated with an increased inflammatory response in the periphery as evidenced by APOE4 carriers that have increased proinflammatory IL-8 and TNFα cytokine levels after cardiopulmonary bypass surgery [97]. Furthermore, peripheral chronic low-grade inflammation in APOE4 carriers was associated with increased risk of AD with earlier disease onset [98]. Immune-challenged APOE4 mice peripherally injected with lipopolysaccharide (LPS) produce higher levels of proinflammatory cytokines such as TNFα, IL-6, as well as damage-inducing nitric oxide [99], [100]. APOE4 polymorphism has additionally been associated with inflammation-related metabolic disorders such as diet-induced adipose tissue inflammation [101], and more recently disease-associated changes in the gut microbiome [102]. These studies highlight the diverse effects of APOE4 in systemic inflammation in general as well as in AD, and suggest the APOE4 allele may contribute to AD pathology through an altered inflammatory state. An important remaining question is how APOE4 synergizes with immune cell function leading to AD-related neurodegeneration.
4.1. APOE and astrocytes
Astrocytes are required for neuronal survival and the loss of physiological astrocyte function can be a primary contributor to neurodegeneration [89]. This is not only observed in AD but also, Alexander disease and hepatic encephalopathy, where changes in expression of key astrocytic proteins such as the glutamate transporter 1 (GLT-1), aquaporin 4 (Aqp4), the glucose transporter GLUT1, and glial fibrillary acidic protein (GFAP) render astrocytes dysfunctional and unable to maintain CNS homeostasis, leading to chronic neuroinflammation and neurodegeneration [103]. Astrocytes constitute approximately 20% of the glial cells in the brain, and as a highly heterogenous population that regulate a wide range of functions including maintenance of the BBB, synaptic function, as well as lipid and glucose metabolism. Given that astrocytes express the majority of ApoE in the brain, APOE4 expression may influence these abilities required to support energy demanding neurons with aging.
In AD, reactive GFAP-positive astroglia are commonly found surrounding Aβ plaques as well as in brain regions with increased accumulation of aggregated, hyper-phosphorylated tau (Figure 1). Astrocytes show dynamic plastic phenotypes such as migratory activity in response to injury as well as phagocytic and proteolytic degradation of Aβ. Adult mouse astrocytes expressing endogenous ApoE internalize and degrade human Aβ peptides effectively in vitro [104]. In contrast, cultured astrocytes from ApoE KO mice were unable to digest Aβ deposits, suggesting that ApoE plays an important role in Aβ clearance under these conditions [105]. In fact, APOE modulates Aβ burden in an isoform-dependent manner that is linked to impaired clearance of soluble Aβ in the brain ISF, with APOE4 leading to greatest impairment compared to other APOE isoforms [29]. Interesting, the authors also showed that expressing human APOE isoforms did not affect the rates of Aβ synthesis in PDAPP mice, but rather modulated the onset of Aβ accumulation that was linked to differential regulation of Aβ clearance. These findings suggest that APOE4 likely increases the risk for AD by both, increasing Aβ aggregation as well as impairing its clearance [106], [107]. The mechanism through which APOE4 impairs clearance is not known, yet it is conceivable that multiple astrocytic functions may be involved. For example, astrocytic clearance of ApoE and ApoE-containing lipoproteins is regulated by LDLR and LRP1 [108], both of which facilitate Aβ uptake and clearance across the BBB (Figure 1) [15]. Astrocytes are involved in the clearance of Aβ through the BBB via the AQP4 channels expressed in their end feet [96], [104], [109]. Furthermore, dysfunctional astroglia may affect levels of neprilysin and insulin-degrading enzyme levels, both of which are known to help degrade and clear Aβ [110].
The idea of astrocyte reactivity is currently under great discussion as recent evidence suggests that the all-or-none phenotype of activation may greatly vary depending on disease and disease stage-specific manner. To this end, several groups have independently identified the existence of molecularly diverse subtypes of reactive astrocytes in response to injury, Aβ or tau pathology [85], [89], [111]–[113]. Using two different models of injury, focal cerebral ischaemia and systemic LPS injection, Zamanian et al found that astrogliosis not only consisted of a rapid change in astrocytic function-specific genes, but also this phenotype varied according to the type of insult [111]. Interestingly, while ischaemia induced a reactive astrocyte phenotype that was considered more protective, LPS stimulation induced astroglial genes that were neurotoxic. Such differences in astroglial reactivity are now referred to different states of reactive astrogliosis [114]. In certain states of reactive astrocytes, the cells can have strongly upregulated genes related to classical complement activation pathway, such as C1q and C3, IL-1α, and TNFα, all of which detrimental to synapses. In contrast, in other states of reactive astrocytes, there is upregulation of anti-inflammatory genes that support neuronal survival and growth, and are therefore considered protective. The major damaging state of reactive astrocytes have been shown to generate neurotoxic factors that were found to mediate death of axotomized neurons [115]. A follow up of this work has shown that upon optic nerve crush in mice, long chain saturated lipids carried by ApoE and ApoJ containing lipoproteins mediate neurotoxicity through lipoapoptotic pathways that facilitate cell death [116]. This type of reactive astrocyte change appears to be found in post-mortem tissue of several neurodegenerative diseases, including AD, supporting a common mechanism through which these type of reactive astrocyte changes may promote neurodegeneration. It is not known whether an AD-specific reactive astrogliosis signature is possible given the astroglial diversity and dynamic pathologies involved in AD. Nonetheless, it opens important novel avenues into understanding astrocyte reactivity in the context of persistent inflammation that may lead to neurodegeneration. Further research is required to understand whether a more neurotoxic reactive astroglial state forms in an attempt to remove irreversibly damaged neurons, or whether a more damaging reactive astrocyte state malevolently removes healthy functioning neurons by inappropriately targeting them, similarly to that observed in chronic inflammation.
APOE4 expression in P301S tauopathy mice induces a strong activation of a more damaging related reactive astroglial signature compared to P301S mice expressing APOE3 or no ApoE [85]. Of note, the increase in genes such as Serping1, Gfap, and Ggta1 highly correlated with brain atrophy, suggesting these reactive astrocytes play a detrimental role in facilitating neurodegeneration. The in vivo findings were further supported by P301S expressing neurons co-cultured with E4 glia that displayed similar neuronal damage, compared to the protective effects seen in the absence of ApoE, highlighting that APOE4 from astrocytes may not only increase neurodegeneration through its effect on phosphorylated tau, but also through enhancing neuroinflammation that together with tau further exacerbates neurodegeneration. As such, P301S mice lacking ApoE show minimal gliosis with little to no brain atrophy [85], [89]. Similarly, reducing APOE4 levels in the brain via ASOs markedly reduces gliosis as well as tau-mediated neurodegeneration in P301S/APOE4 mice [88].
Astrocytes express very high levels of ApoE in the normal brain with little expression by microglia. However, reactive microglia express similar levels of ApoE as do astrocytes [89]. Under the in vitro conditions studied, it has been found that astrocyte-derived ApoE particles are significantly larger compared with ApoE-containing lipoproteins secreted by microglia, likely due to differences in lipidation [117]. This points to differential roles of ApoE based on glial origin. To distinguish the effects of astrocytic ApoE on tau-mediated neurodegeneration, Wang et al. used P301S mice and combined genetic as well as tamoxifen-based strategy to selectively modulate astrocytic APOE and found that a reduction in astrocyte-derived APOE4 over a period of 4 months, rescued the brain atrophy typically observed in P301S/APOE4 at 9.5 months of age [89]. A corresponding reduction in aggregated tau pathology was observed, specifically in females, suggesting not only a sex-dependent effect as seen in AD [84], but also an immune modulating effect of astroglial APOE4 on neurodegeneration. Using single cell nuclei sequencing, the authors found that in the setting of tauopathy, APOE4 expression has drastic consequences for astrocytes, as well as affects gene expression in non-astrocytic cells, such as an APOE4-dependent increase in Rorb-positive neurons, a distinct neuronal subpopulation that show selective vulnerability in AD [118]. Intriguingly, astrocytic ApoE removal decreased the disease-associated gene signature in not only astrocytes, but also neurons, oligodendrocytes, and microglia, strongly supporting crosstalk between glia and neurons to modulate key neurodegenerative pathways in the brain. In addition to APOE4-dependent protective effects on neurons, removal of astrocytic APOE4 reduced microglial engulfment PSD95-positive synapses, another key feature that contributes to AD neurodegeneration. These findings highlight that astrocyte-derived APOE4 is a major regulator of tauopathy-mediated neurodegeneration and represents a toxic gain of function.
Given the important role of APOE in glial lipid metabolism, it is conceivable that the lipidation status of APOE4 or APOE4’s effect on glial lipid metabolism is a key mediator of both, inter- and intracellular effects that contribute to neurodegeneration. In fact, lipid accumulation in glia is a pathologically defining feature of AD [119], which has been closely linked to reactive oxygen species (ROS)-induced inflammation and neurotoxicity. Recent studies using iPSC-derived astrocytes generated from APOE4 or APOE3 carriers have demonstrated that compared to astrocytes from APOE3 carriers, APOE4 promotes both, an increase in lipid droplets as well as an accumulation of unsaturated fatty acids [120]. An APOE4-dependent accumulation of lipid droplet further renders astrocytes incapable of supporting metabolic and synaptic functions for neurons [121]. Moreover, APOE4 iPSC-derived glia accumulate unesterified cholesterol after excessive uptake of extracellular lipids, which triggers secretion of inflammatory chemokines and cytokines, suggesting that APOE4 results in dysregulated cholesterol metabolism that further promotes a proinflammatory transcriptome similar to that observed in AD [122], [123]. Interestingly, lack of APOE expression also leads to excess lipid droplet formation in a cerebral organoid model [90], and increased cholesterol ester accumulation in microglia [124]. These findings suggest that APOE-dependent cholesterol dyshomeostasis result in widespread proinflammatory responses that drive both astrocytes and microglia toward a neurodegenerative state. Future studies exploring the role of altered APOE lipidation and how APOE influences lipid metabolism in astrocytes, microglia, and oligodendrocytes may provide invaluable insights into neurodegenerative pathways.
4.2. APOE and microglia
Microglia constitute approximately 10% of the total glial population in the healthy adult brain. Together with meningeal, choroid plexus, and perivascular macrophages, microglia compromise the brain myeloid cell population that plays a pivotal role in the CNS immune response and homeostasis. As the main innate immune cells of the CNS, microglia represent the first line of immunological defense by constantly surveying their microenvironment for signs of damage or debris allowing them to respond rapidly to focal injury or invading pathogens. Comprehensive single cell RNA sequencing analysis of AD and aging mouse models show that microglia are present in a continuum of distinct phenotypes that are highly dependent on their spatiotemporal context [9], [89], [112], [113]. Microgliosis is commonly observed across several neurodegenerative diseases, including AD and in mouse models with AD-like pathology, where microglia are often found surrounding Aβ plaques as well as in brain regions with high tau accumulation and neurodegeneration (Figure 1) [87], [89], [125]–[127]. These subsets of microglia upregulate specific genes during disease conditions such as Apoe, Trem2, Clec7a, Cd68 and Cst3, and are called disease-associated microglia (DAM) [9] or microglia of neurodegenerative phenotype (MGnD) [10]. Compared to microglia in non-disease conditions, DAM downregulate homeostatic genes such as P2ry12, Tmem119, and Cx3cr1. Such transitioning changes in microglial gene expression orchestrate their diverse functions involving axon growth and synaptogenesis, synaptic pruning, recognizing, and responding to abnormal events including focal injury or neuropathological insults such as proteopathic aggregates, as well as phagocytosis and apoptosis [128]. These functions are central to disease progression and modulation because chronic glial activation is a prominent feature of aging and often goes hand-in-hand with pathological protein accumulation as well as cell death, both of which constitute central characteristics of neurodegeneration.
Given that ApoE is upregulated in DAM, APOE isoform-dependent effects on plaque-associated microglia have yielded conflicting results [129]–[131], which may partially be owed to differences in biochemical composition of fibrillar plaques across different APP transgenic mouse models as an additional confounding variable in microglial response to plaques. In human iPSC-derived microglia, isogenic conversion of APOE3 to APOE4 transformed the microglial transcriptome phenotype to DAM, with significant overlap of microglial gene expression profile observed in human AD brain [123]. This suggests that APOE4 may promote a DAM phenotype in different situations such as in AD. Ulrich et al showed that lack of ApoE expression in APP/PS1 transgenic mice reduced expression of Clec7a, Cst7 and Itgax among other microglial genes that are important for migration and chemotaxis, with a concomitant reduction in number of fibrillar plaque-associated microglia [62]. Although the removal of ApoE in this model resulted in a marked reduction of fibrillar plaques, the plaques that did form in ApoE KO mice showed increased neuritic dystrophy surrounding them. This suggests that while ApoE facilitates microglial functions such as migration to plaques to potentially phagocytose them, this microglia subset may additionally have a protective role in decreasing plaque-induced neuronal process damage. Furthermore, microglial depletion in APP transgenic mice reduces plaque-associated ApoE, with or without changes in plaque burden [126], [132], [133]. This discrepancy may be explained by differences in methods used to deplete microglia, the duration of microglial depletion and repopulation, as well as microglial removal before or after plaque onset. Of note, microglia depletion before plaque onset resulted in dramatically decreased parenchymal plaque burden; however, this resulted in increased CAA in the 5xFAD Tg6799 amyloid mouse model where CAA is not typically observed [132]. This suggests that ApoE-expressing microglia may be critical in early plaque deposition and may also exhibit mechanistic heterogeneity in amyloid fibril formation. Interestingly, a reduction in plaque-associated ApoE, either by genetic ablation or microglial depletion, consistently increases dystrophic neurites around plaques, illustrating an innate immune-based role of ApoE in modulating fibrillar plaques to become less neurotoxic, despite its role in promoting plaque fibrillogenesis. This may largely be due to DAM clustering around plaques essentially forming a barrier that protects against further damage [134]. However, genetic removal of murine ApoE from microglial using a CSF1R-Cre promoter neither altered plaque pathology, plaque-associated microgliosis nor the typical DAM signature associated with heightened inflammation [135]. Additionally, selective removal of microglial ApoE via this approach reduced neuritic dystrophy around plaques, contrary to that observed in global ApoE KO or microglia depletion. It is conceivable that murine ApoE, though to some extent comparable to APOE4, may have differential effects compared to human APOE isoforms. Furthermore, given the heterogeneity of microglial subset-specific functions, microglial ApoE may display CSF1R-independent interactions that may otherwise be recapitulated in a Cre-inducible model similar to that shown in astrocytes [89]. Altogether, these findings suggest that DAM may interact with other cellular sources of ApoE, likely astroglial-derived, through either uptake of secreted ApoE, or phagocytosis of plaques, which may result in changes that contribute to neuronal damage in AD.
Although removing microglia-derived ApoE did not alter changes in GFAP-positive astroglial reactivity [135], selective removal of astrocytic APOE4 in P301S tau transgenic mice considerably changed the DAM profile, in support of the notion that APOE may influence crosstalk among immune cells and neurons contributing to neurodegeneration [89]. While DAM genes were significantly upregulated in the presence of astroglial APOE4 expression, genetic deletion of astrocytic APOE4 decreased DAM and simultaneously attenuated microglial phagocytosis of PSD-95-positive synapses, demonstrating an important role of astrocyte-derived ApoE4 that contributes to microglia-dependent synaptic loss in tauopathy. Whether a crosstalk between cell-dependent sources of ApoE influence Aβ pathology remains to be explored.
Microglia drive APOE-dependent neurodegeneration in the presence of tau pathology [85], [86], [88], [89], [136]. While pathological tau is directly linked to neurodegeneration, degenerating neurons as well as tau also further exacerbate microgliosis leading to a vicious cycle promoting chronic neuroinflammation. For example, APOE4 expression in P301S mouse model of tauopathy not only increased pathological tau burden and tau-mediated neurodegeneration, but also upregulated a set of microglial genes reminiscent of DAM [85], [86]. Consequently, APOE4 expression downregulated microglial genes involved in homeostatic cell function once tau pathology was present. Of note, lack of ApoE expression in age-matched P301S mice rescued tau-mediated neurodegeneration and corresponding induction of DAM (Figure 1). Furthermore, depleting microglia through selectively inhibiting colony stimulating factor 1 receptor (CSF1R), a marker crucial for microglial survival, completely blocked neurodegeneration in P301S/APOE4 mice [86] as well as blocking tau-mediated neurodegeneration in another P301S model [136], strongly supporting a role of microglia, specifically DAM, in regulating tau-mediated neurodegeneration [86], [136]. Sustained inhibition of CSF1R signaling depletes a majority of microglia in the brain, though not all [132], [137], [138]. The remaining CSF1R-resistant microglia show a DAM-like phenotype [139], which have recently been reported to contribute to plaque-associated neuritic dystrophy [133]. Interestingly, P301S/ApoE KO mice showed increased microglial resistance to CSF1R inhibition, wherein a large proportion of the surviving microglia were CD68-negative, indicative of their homeostatic status [86]. The enhanced survival of non-activated microglia in the absence of ApoE suggests that ApoE-expressing DAM may be more susceptible to CSF1R signaling-mediated apoptosis. Consequently, it is tempting to speculate that a higher baseline of microglial activation contributing to DAM maybe more burned-out owing to accelerated disease progression in P301S/APOE4 mice, and as a result more vulnerable to apoptosis due to CSF1R inhibition. This suggests a bidirectional effect of tau pathology and microgliosis on neurodegeneration. A reduction in ApoE by LDLR overexpression [87] or ApoE4-targeting ASOs [88] show a similar decrease in microgliosis, which concurrently prevents tau-mediated neurodegeneration, supporting a role for ApoE-expressing activated microglia in facilitating neuronal damage. Additionally, reducing ApoE by LDLR overexpression does not affect tau pathology prior to DAM onset, suggesting that its protective effect on tau pathology likely occurs at a later stage by modulating or decreasing microglial reactivity under neurodegeneration. Altogether these findings indicate that microglia-derived ApoE may shift microglia to a neurodegenerative transcriptional state. However, it may be difficult to distinguish whether the observed changes on neurodegeneration is selectively due to microglial ApoE, rather than a combination of CSF1R-dependent DAM functions that contribute to neuronal dysfunction, or a reduction in total ApoE levels coming from other cells such as astrocytes that underlie these findings.
5. ApoE and other immune risk genes
Given the heterogenous nature of immune cells, it is highly likely that ApoE interacts with several other immune genes to regulate their diverse set of physiological functions, both in the presence and absence of AD pathogenesis. Recent GWAS have identified more than 30 genetic loci involved in increasing AD risk, half of which are related to immune response and microglia, affirming that the innate immune cells in the brain, play an important role in the overall response during AD [8]. One of these genes, the triggering receptor expressed on myeloid cells 2 (TREM2), has shown to interact with APOE and alter inflammatory functions involving response to injury and microglial phagocytic ability [9], [10]. Both TREM2 and APOE directly influence DAM, Aβ and tau pathology, as well as tau-induced neurodegeneration [140]. This supports the hypothesis that the difference in AD and healthy aging may be the failure of microglia to appropriately respond to pathogenic insults contributing to neuronal injury. Other risk genes include SPI1/PU.1, which was recently reported as a master regulator of several AD risk genes that affect microglial development and homeostasis, including ABCA7, CD33, MS4A4A, MS4A6A, CLU, as well as TREM2 and APOE [141]. Most of these genes have shown to interact with APOE to functionally modify DAM, or in regulating the homeostasis of phospholipids and cholesterol [9], [142]. For example, several studies support a role for ABCA7 and CLU in regulation of cholesterol and Aβ metabolism in AD pathogenesis [143]–[147]. Furthermore, the presence of APOE4 in cases with either CLU or ABCA7 variants enhances the risk for AD [148]. Clarifying the interactions between APOE and other immune risk genes will likely unravel pathomechanisms underlying AD as well as provide novel insights into refined therapeutic strategies.
6. Future directions and considerations
The role of APOE4 in mediating AD risk is multifactorial and complex. The pleiotropic effects of the APOE involving a diverse array of cell types as well as cell type-specific functions must be considered for an attempt to therapeutically modulate APOE or related microglial and astroglial functions. In that case, is APOE a good therapeutic target and can targeting APOE mitigate AD pathology? A wide variety of therapeutic strategies reducing APOE4 in animal models with AD-like pathology appear to be beneficial in moderating parenchymal and cerebrovascular amyloid plaque and tau pathology, associated inflammation as well as tau-mediated neurodegeneration, highlighting a protective role of reducing APOE to lessen AD pathology. In support of this, the AD-associated APOE3 Jac and Christchurch variants demonstrate that functional APOE is required for full pathological and clinical development of AD. However, while reducing APOE may protect from sustained inflammation that contributes to tau-induced neurodegeneration, APOE also regulates multiple microglial and astroglial functions involving response to Aβ and tau, migration, clearance and phagocytosis, all of which play a crucial role in modulating AD pathogenesis (Figure 1). Additionally, how different interventions will alter peripheral lipid homeostasis and vascular function would need to be determined.
The pre-clinical stage of AD begins decades before clinical symptoms, also referred to as the cellular phase of AD pathology [149]. During this phase, extensive changes occur in glial cells and vasculature, which may orchestrate subsequent neuronal deficits. Recent advances in transcriptomics have deepened our understanding of the plasticity of microglia and astrocytes, which allow them to rapidly react based on what is required of them from their immediate vicinity, and release soluble factors such as chemokines and cytokines, as a means of communication inter- and intracellularly to amplify or dampen their responses. Keren-Shaul et al demonstrated that microglia alter their gene expression in a linear manner based on age and presence or absence of amyloid pathology, wherein APOE is required as an initial trigger [9]. A similar phenomenon is also observed with astrocytes across disease models [86], [87], [89], [115]. This strongly suggests that microglia and astrocytes are not binary, altering their gene expression in an “on” or “off” switch, but rather co-exist in dynamic, assorted populations that simultaneously regulate varying degrees of both DAM and homeostatic genes. This likely allows the immune cells to ramp up and modulate their functions corresponding to the level of threat in the brain. Such plasticity also argues for a threshold that allows microglia and astrocytes to carry out specific functions based on the predominant set of genes expressed. Moreover, an early stimulation of the immune system, both innate and adaptive, may induce long-lasting changes in glial response, which may dictate how microglia and astrocytes respond to stressors later with aging. Wendeln et al demonstrated that systemic immune challenges essentially create an innate immunological memory that have lasting effects on microglial identity and function in response to amyloid pathology [150]. This study highlights that microglia in the brain are not only able to respond to emerging threats, but are also able to learn and modify their behavior and be primed in preparation for future immunological challenges, similar to adaptive immune responses.
It is conceivable that healthy glial cells are capable of dynamically altering their gene expression that allow these cells to transition between phenotypes. This may potentially prolong their life span and turnover rate as shown in the absence of disease [151], [152]. However, the long-lived populations of microglia may also be primed and therefore inherently better equipped to face future challenges to mitigate AD pathology [150]. With accumulating proteopathic insults and heightened inflammation, the cellular energy demand may render these immune cells dysfunctional and unable to metabolically cope with additional stressors that eventually lead to neurodegeneration. Interestingly, microglial turnover occurs more rapidly in the hippocampus and cerebellum in mice, compared to cortex [153]. This brain region-dependent selectivity in microglia turnover correlates to higher immune surveillance and bioenergetics reported for glia residing in these compartments [154]. Given the role of APOE in facilitating microglial response to AD pathology, supporting cholesterol and lipid metabolism, as well as in aiding astrocytes in regulating energy homeostasis, it is tempting to speculate that APOE isoforms may be key to maintaining said threshold-dependent states, which may or may not be reversible based on the extent of damage. Therefore, APOE-dependent inflammation, irrespective of cellular source, is more complex than simply protective or detrimental, but rather may depend on a number of factors including, age, sex, underlying pathology as well as disease stage; a combination of which may drive chronic inflammation-induced neurodegeneration.
Acknowledgements
S.P. wrote the draft of the manuscript. D.M.H. reviewed and edited the manuscript.
Funding
This work was supported by the National Institutes of Health grants U19AG069701 (DMH); 2RF1AG047644 (DMH); RF1NS090934; Tau Consortium (DMH); Cure Alzheimer’s Fund (DMH), the JPB Foundation (DMH); and Alzheimer’s Association [AARF-21-850865] (SP).
Footnotes
Declaration of interests
D.M.H. is as an inventor on a patent licensed by Washington University to C2N Diagnostics on the therapeutic use of anti-tau antibodies. D.M.H. co-founded and is on the scientific advisory board of C2N Diagnostics. C2N Diagnostics has licensed certain anti-tau antibodies to AbbVie for therapeutic development. D.M.H. is on the scientific advisory board of Denali and consults for Genentech, Merck, and Cajal Neuroscience. S.P. declares no competing interests.
Citations
- [1].Strittmatter WJ et al. , “Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease.,” Proc. Natl. Acad. Sci. U. S. A, vol. 90, no. 5, p. 1977, Mar. 1993, doi: 10.1073/PNAS.90.5.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Corder EH et al. , “Protective effect of apolipoprotein E type 2 allele for late onset Alzheimer disease,” Nat. Genet, vol. 7, no. 2, pp. 180–184, Jun. 1994, doi: 10.1038/NG0694-180. [DOI] [PubMed] [Google Scholar]
- [3].Chen Y, Strickland MR, Soranno A, and Holtzman DM, “Apolipoprotein E: Structural Insights and Links to Alzheimer Disease Pathogenesis,” Neuron, vol. 109, no. 2, pp. 205–221, Jan. 2021, doi: 10.1016/J.NEURON.2020.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Corder EH et al. , “Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families,” Science, vol. 261, no. 5123, pp. 921–923, 1993, doi: 10.1126/SCIENCE.8346443. [DOI] [PubMed] [Google Scholar]
- [5].Schmechel DE et al. , “Increased amyloid beta-peptide deposition in cerebral cortex as a consequence of apolipoprotein E genotype in late-onset Alzheimer disease,” Proc. Natl. Acad. Sci. U. S. A, vol. 90, no. 20, pp. 9649–9653, Oct. 1993, doi: 10.1073/PNAS.90.20.9649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Shinohara M et al. , “APOE2 is associated with longevity independent of Alzheimer’s disease.,” Elife, vol. 9, 2020, doi: 10.7554/eLife.62199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Hardy J and Selkoe DJ, “The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics,” Science, vol. 297, no. 5580. 2002, doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- [8].Pimenova AA, Raj T, and Goate AM, “Untangling Genetic Risk for Alzheimer’s Disease,” Biol. Psychiatry, vol. 83, no. 4, pp. 300–310, Feb. 2018, doi: 10.1016/J.BIOPSYCH.2017.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Keren-Shaul H et al. , “A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease,” Cell, vol. 169, no. 7, 2017, doi: 10.1016/j.cell.2017.05.018. [DOI] [PubMed] [Google Scholar]
- [10].Krasemann S et al. , “The TREM2-APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases,” Immunity, vol. 47, no. 3, p. 566, Sep. 2017, doi: 10.1016/J.IMMUNI.2017.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Boyles JK, Pitas RE, Wilson E, Mahley RW, and Taylor JM, “Apolipoprotein E associated with astrocytic glia of the central nervous system and with nonmyelinating glia of the peripheral nervous system,” J. Clin. Invest, vol. 76, no. 4, pp. 1501–1513, 1985, doi: 10.1172/JCI112130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Mahley R, “Apolipoprotein E: cholesterol transport protein with expanding role in cell biology.,” Science (80-.)., vol. 240, 1988. [DOI] [PubMed] [Google Scholar]
- [13].Wahrle SE et al. , “Deletion of Abca1 increases Aβ deposition in the PDAPP transgenic mouse model of Alzheimer disease,” J. Biol. Chem, vol. 280, no. 52, pp. 43236–43242, Dec. 2005, doi: 10.1074/jbc.M508780200. [DOI] [PubMed] [Google Scholar]
- [14].Hirsch-Reinshagen V et al. , “The absence of ABCA1 decreases soluble ApoE levels but does not diminish amyloid deposition in two murine models of Alzheimer disease.,” J. Biol. Chem, vol. 280, no. 52, pp. 43243–56, Dec. 2005, doi: 10.1074/jbc.M508781200. [DOI] [PubMed] [Google Scholar]
- [15].Kim J et al. , “Overexpression of low-density lipoprotein receptor in the brain markedly inhibits amyloid deposition and increases extracellular A beta clearance,” Neuron, vol. 64, no. 5, pp. 632–644, Dec. 2009, doi: 10.1016/J.NEURON.2009.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Bu G, “Apolipoprotein E and its receptors in Alzheimer’s disease: pathways, pathogenesis and therapy,” Nat. Rev. Neurosci, vol. 10, no. 5, pp. 333–344, May 2009, doi: 10.1038/NRN2620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Ahn K, Song JH, Kim DK, Park MH, Jo SA, and Koh YH, “Ubc9 gene polymorphisms and late-onset Alzheimer’s disease in the Korean population: a genetic association study,” Neurosci. Lett, vol. 465, no. 3, pp. 272–275, Nov. 2009, doi: 10.1016/J.NEULET.2009.09.017. [DOI] [PubMed] [Google Scholar]
- [18].Weisgraber KH and Shinto LH, “Identification of the disulfide-linked homodimer of apolipoprotein E3 in plasma. Impact on receptor binding activity.,” J. Biol. Chem, vol. 266, no. 18, pp. 12029–34, Jun. 1991, [Online]. Available: http://www.ncbi.nlm.nih.gov/pubmed/2050696. [PubMed] [Google Scholar]
- [19].Weisgraber KH, Roses AD, and Strittmatter WJ, “The role of apolipoprotein E in the nervous system.,” Curr. Opin. Lipidol, vol. 5, no. 2, pp. 110–6, Apr. 1994, doi: 10.1097/00041433-199404000-00007. [DOI] [PubMed] [Google Scholar]
- [20].Weisgraber KH, “Apolipoprotein E distribution among human plasma lipoproteins: role of the cysteine-arginine interchange at residue 112.,” J. Lipid Res, vol. 31, no. 8, pp. 1503–11, Aug. 1990, [Online]. Available: http://www.ncbi.nlm.nih.gov/pubmed/2280190. [PubMed] [Google Scholar]
- [21].Mahley RW and Huang Y, “Apolipoprotein e sets the stage: response to injury triggers neuropathology,” Neuron, vol. 76, no. 5, pp. 871–885, Dec. 2012, doi: 10.1016/J.NEURON.2012.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Sanan DA et al. , “Apolipoprotein E associates with beta amyloid peptide of Alzheimer’s disease to form novel monofibrils. Isoform apoE4 associates more efficiently than apoE3,” J. Clin. Invest, vol. 94, no. 2, pp. 860–869, 1994, doi: 10.1172/JCI117407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Strittmatter WJ et al. , “Binding of human apolipoprotein E to synthetic amyloid beta peptide: isoform-specific effects and implications for late-onset Alzheimer disease.,” Proc. Natl. Acad. Sci. U. S. A, vol. 90, no. 17, pp. 8098–102, Sep. 1993, doi: 10.1073/pnas.90.17.8098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Liu CC et al. , “APOE3-Jacksonville (V236E) variant reduces self-aggregation and risk of dementia,” Sci. Transl. Med, vol. 13, no. 613, p. 9375, Sep. 2021, doi: 10.1126/SCITRANSLMED.ABC9375/SUPPL_FILE/SCITRANSLMED.ABC9375_DATA_FILE_S1.ZIP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Arboleda-Velasquez JF et al. , “Resistance to autosomal dominant Alzheimer’s disease in an APOE3 Christchurch homozygote: a case report,” Nat. Med. 2019 2511, vol. 25, no. 11, pp. 1680–1683, Nov. 2019, doi: 10.1038/S41591-019-0611-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Holtzman DM and Fagan AM, “Potential role of apoE in structural plasticity in the nervous system; implications for disorders of the central nervous system,” Trends Cardiovasc. Med, vol. 8, no. 6, pp. 250–255, 1998, doi: 10.1016/S1050-1738(98)00017-6. [DOI] [PubMed] [Google Scholar]
- [27].Kim J, Basak JM, and Holtzman DM, “The role of apolipoprotein E in Alzheimer’s disease,” Neuron, vol. 63, no. 3, pp. 287–303, Aug. 2009, doi: 10.1016/J.NEURON.2009.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Holtzman DM et al. , “Apolipoprotein E isoform-dependent amyloid deposition and neuritic degeneration in a mouse model of Alzheimer’s disease,” Proc. Natl. Acad. Sci. U. S. A, vol. 97, no. 6, 2000, doi: 10.1073/pnas.050004797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Castellano JM et al. , “Human apoE isoforms differentially regulate brain amyloid-β peptide clearance,” Sci. Transl. Med, vol. 3, no. 89, 2011, doi: 10.1126/scitranslmed.3002156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Verghese PB, Castellano JM, and Holtzman DM, “Apolipoprotein E in Alzheimer’s disease and other neurological disorders,” Lancet. Neurol, vol. 10, no. 3, pp. 241–252, Mar. 2011, doi: 10.1016/S1474-4422(10)70325-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Gonneaud J et al. , “Relative effect of APOE ε4 on neuroimaging biomarker changes across the lifespan,” Neurology, vol. 87, no. 16, pp. 1696–1703, Oct. 2016, doi: 10.1212/WNL.0000000000003234. [DOI] [PubMed] [Google Scholar]
- [32].Kantarci K et al. , “Multimodality imaging characteristics of dementia with Lewy bodies,” Neurobiol. Aging, vol. 33, no. 9, pp. 2091–2105, Sep. 2012, doi: 10.1016/J.NEUROBIOLAGING.2011.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Morris JC et al. , “APOE predicts amyloid-beta but not tau Alzheimer pathology in cognitively normal aging,” Ann. Neurol, vol. 67, no. 1, 2010, doi: 10.1002/ana.21843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Ma J, Yee A, Brewer HB, Das S, and Potter H, “Amyloid-associated proteins α1-antichymotrypsin and apolipoprotein E promote assembly of Alzheimer β-protein into filaments,” Nat. 1994 3726501, vol. 372, no. 6501, pp. 92–94, 1994, doi: 10.1038/372092a0. [DOI] [PubMed] [Google Scholar]
- [35].Wisniewski T and Frangione B, “Apolipoprotein E: A pathological chaperone protein in patients with cerebral and systemic amyloid,” Neurosci. Lett, vol. 135, no. 2, pp. 235–238, Feb. 1992, doi: 10.1016/0304-3940(92)90444-C. [DOI] [PubMed] [Google Scholar]
- [36].Castano EM et al. , “Fibrillogenesis in Alzheimer’s disease of amyloid beta peptides and apolipoprotein E.,” Biochem. J, vol. 306 (Pt 2, pp. 599–604, Mar. 1995, doi: 10.1042/bj3060599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Fitz NF et al. , “Abca1 deficiency affects Alzheimer’s disease-like phenotype in human ApoE4 but not in ApoE3-targeted replacement mice,” J. Neurosci, vol. 32, no. 38, pp. 13125–13136, Sep. 2012, doi: 10.1523/JNEUROSCI.1937-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].LaDu MJ, Pederson TM, Frail DE, Reardon CA, Getz GS, and Falduto MT, “Purification of apolipoprotein E attenuates isoform-specific binding to β-amyloid,” J. Biol. Chem, vol. 270, no. 16, pp. 9039–9042, 1995, doi: 10.1074/jbc.270.16.9039. [DOI] [PubMed] [Google Scholar]
- [39].Tokuda T et al. , “Lipidation of apolipoprotein E influences its isoform-specific interaction with Alzheimer’s amyloid beta peptides.,” Biochem. J, vol. 348 Pt 2, pp. 359–65, Jun. 2000, [Online]. Available: http://www.ncbi.nlm.nih.gov/pubmed/10816430. [PMC free article] [PubMed] [Google Scholar]
- [40].Verghese PB et al. , “ApoE influences amyloid-β (Aβ) clearance despite minimal apoE/Aβ association in physiological conditions.,” Proc. Natl. Acad. Sci. U. S. A, vol. 110, no. 19, pp. E1807–16, May 2013, doi: 10.1073/pnas.1220484110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Hashimoto T et al. , “Apolipoprotein E, especially apolipoprotein E4, increases the oligomerization of amyloid β peptide.,” J. Neurosci, vol. 32, no. 43, pp. 15181–92, Oct. 2012, doi: 10.1523/JNEUROSCI.1542-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Garai K, Verghese PB, Baban B, Holtzman DM, and Frieden C, “The binding of apolipoprotein E to oligomers and fibrils of amyloid-β alters the kinetics of amyloid aggregation.,” Biochemistry, vol. 53, no. 40, pp. 6323–31, Oct. 2014, doi: 10.1021/bi5008172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Liu CC et al. , “ApoE4 Accelerates Early Seeding of Amyloid Pathology,” Neuron, vol. 96, no. 5, pp. 1024–1032. e3, Dec. 2017, doi: 10.1016/J.NEURON.2017.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Lim YY and Mormino EC, “APOE genotype and early β-amyloid accumulation in older adults without dementia,” Neurology, vol. 89, no. 10, pp. 1028–1034, Sep. 2017, doi: 10.1212/WNL.0000000000004336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Fleisher AS et al. , “Apolipoprotein E ε4 and age effects on florbetapir positron emission tomography in healthy aging and Alzheimer disease,” Neurobiol. Aging, vol. 34, no. 1, pp. 1–12, Jan. 2013, doi: 10.1016/J.NEUROBIOLAGING.2012.04.017. [DOI] [PubMed] [Google Scholar]
- [46].Bales KR et al. , “Lack of apolipoprotein E dramatically reduces amyloid β-peptide deposition,” Nat. Genet, vol. 17, no. 3, pp. 263–264, 1997, doi: 10.1038/NG1197-263. [DOI] [PubMed] [Google Scholar]
- [47].Irizarry MC, Cheung BS, Rebeck GW, Paul SM, Bales KR, and Hyman BT, “Apolipoprotein E affects the amount, form, and anatomical distribution of amyloid beta-peptide deposition in homozygous APP(V717F) transgenic mice,” Acta Neuropathol, vol. 100, no. 5, pp. 451–458, 2000, doi: 10.1007/S004010000263. [DOI] [PubMed] [Google Scholar]
- [48].Holtzman DM et al. , “Expression of human apolipoprotein E reduces amyloid-beta deposition in a mouse model of Alzheimer’s disease.,” J. Clin. Invest, vol. 103, no. 6, pp. R15–R21, Mar. 1999, doi: 10.1172/JCI6179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Holtzman DM et al. , “Apolipoprotein E facilitates neuritic and cerebrovascular plaque formation in an Alzheimer’s disease model.,” Ann. Neurol, vol. 47, no. 6, pp. 739–47, Jun. 2000, [Online]. Available: http://www.ncbi.nlm.nih.gov/pubmed/10852539. [PubMed] [Google Scholar]
- [50].Fagan AM, Watson M, Parsadanian M, Bales KR, Paul SM, and Holtzman DM, “Human and murine ApoE markedly alters A beta metabolism before and after plaque formation in a mouse model of Alzheimer’s disease.,” Neurobiol. Dis, vol. 9, no. 3, pp. 305–18, Apr. 2002, doi: 10.1006/nbdi.2002.0483. [DOI] [PubMed] [Google Scholar]
- [51].Fryer JD et al. , “Human Apolipoprotein E4 Alters the Amyloid-β 40:42 Ratio and Promotes the Formation of Cerebral Amyloid Angiopathy in an Amyloid Precursor Protein Transgenic Model,” J. Neurosci, vol. 25, no. 11, p. 2803, Mar. 2005, doi: 10.1523/JNEUROSCI.5170-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Bales KR et al. , “Human APOE Isoform-Dependent Effects on Brain β-Amyloid Levels in PDAPP Transgenic Mice,” J. Neurosci, vol. 29, no. 21, pp. 6771–6779, May 2009, doi: 10.1523/JNEUROSCI.0887-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Filippini N et al. , “Distinct patterns of brain activity in young carriers of the APOE-ε4 allele,” Proc. Natl. Acad. Sci, vol. 106, no. 17, pp. 7209–7214, Apr. 2009, doi: 10.1073/PNAS.0811879106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Nelson PT et al. , “APOE-ε2 and APOE-ε4 correlate with increased amyloid accumulation in cerebral vasculature,” J. Neuropathol. Exp. Neurol, vol. 72, no. 7, pp. 708–715, Jul. 2013, doi: 10.1097/NEN.0B013E31829A25B9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Ghebremedhin E et al. , “Gender and age modify the association between APOE and AD-related neuropathology,” Neurology, vol. 56, no. 12, pp. 1696–1701, Jun. 2001, doi: 10.1212/WNL.56.12.1696. [DOI] [PubMed] [Google Scholar]
- [56].Nagy ZS et al. , “Influence of the apolipoprotein E genotype on amyloid deposition and neurofibrillary tangle formation in Alzheimer’s disease,” Neuroscience, vol. 69, no. 3, pp. 757–761, 1995, doi: 10.1016/0306-4522(95)00331-C. [DOI] [PubMed] [Google Scholar]
- [57].Lemere CA, Blusztajn JK, Yamaguchi H, Wisniewski T, Saido TC, and Selkoe DJ, “Sequence of deposition of heterogeneous amyloid beta-peptides and APO E in Down syndrome: implications for initial events in amyloid plaque formation,” Neurobiol. Dis, vol. 3, no. 1, pp. 16–32, 1996, doi: 10.1006/NBDI.1996.0003. [DOI] [PubMed] [Google Scholar]
- [58].Arai Y, Mizuguchi M, Ikeda K, and Takashima S, “Developmental changes of apolipoprotein E immunoreactivity in Down syndrome brains,” Brain Res. Dev. Brain Res, vol. 87, no. 2, pp. 228–232, Jul. 1995, doi: 10.1016/0165-3806(95)00066-M. [DOI] [PubMed] [Google Scholar]
- [59].Jiang Q et al. , “ApoE promotes the proteolytic degradation of Abeta,” Neuron, vol. 58, no. 5, pp. 681–693, Jun. 2008, doi: 10.1016/J.NEURON.2008.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Thal DR, Schultz C, Dehghani F, Yamaguchi H, Braak H, and Braak E, “Amyloid beta-protein (Abeta)-containing astrocytes are located preferentially near N-terminal-truncated Abeta deposits in the human entorhinal cortex,” Acta Neuropathol, vol. 100, no. 6, pp. 608–617, 2000, doi: 10.1007/S004010000242. [DOI] [PubMed] [Google Scholar]
- [61].Yeh FL, Wang Y, Tom I, Gonzalez LC, and Sheng M, “TREM2 Binds to Apolipoproteins, Including APOE and CLU/APOJ, and Thereby Facilitates Uptake of Amyloid-Beta by Microglia,” Neuron, vol. 91, no. 2, pp. 328–340, Jul. 2016, doi: 10.1016/J.NEURON.2016.06.015. [DOI] [PubMed] [Google Scholar]
- [62].Ulrich JD et al. , “ApoE facilitates the microglial response to amyloid plaque pathology,” J. Exp. Med, vol. 215, no. 4, pp. 1047–1058, Apr. 2018, doi: 10.1084/JEM.20171265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Cirrito JR et al. , “Synaptic activity regulates interstitial fluid amyloid-beta levels in vivo,” Neuron, vol. 48, no. 6, pp. 913–922, Dec. 2005, doi: 10.1016/J.NEURON.2005.10.028. [DOI] [PubMed] [Google Scholar]
- [64].Cirrito JR et al. , “In vivo assessment of brain interstitial fluid with microdialysis reveals plaque-associated changes in amyloid-beta metabolism and half-life,” J. Neurosci, vol. 23, no. 26, pp. 8844–8853, Oct. 2003, doi: 10.1523/JNEUROSCI.23-26-08844.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Katsouri L and Georgopoulos S, “Lack of ldl receptor enhances amyloid deposition and decreases glial response in an alzheimer’s disease mouse model,” PLoS One, vol. 6, no. 7, 2011, doi: 10.1371/journal.pone.0021880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Cao D, ichiro Fukuchi K, Wan H, Kim H, and Li L, “Lack of LDL receptor aggravates learning deficits and amyloid deposits in Alzheimer transgenic mice,” Neurobiol. Aging, vol. 27, no. 11, pp. 1632–1643, 2006, doi: 10.1016/J.NEUROBIOLAGING.2005.09.011. [DOI] [PubMed] [Google Scholar]
- [67].Heeren J, Beisiegel U, and Grewal T, “Apolipoprotein E recycling: implications for dyslipidemia and atherosclerosis,” Arterioscler. Thromb. Vasc. Biol, vol. 26, no. 3, pp. 442–448, Mar. 2006, doi: 10.1161/01.ATV.0000201282.64751.47. [DOI] [PubMed] [Google Scholar]
- [68].Wahrle SE et al. , “Overexpression of ABCA1 reduces amyloid deposition in the PDAPP mouse model of Alzheimer disease,” J. Clin. Invest, vol. 118, no. 2, pp. 671–682, Feb. 2008, doi: 10.1172/JCI33622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Koldamova R, Staufenbiel M, and Lefterov I, “Lack of ABCA1 considerably decreases brain ApoE level and increases amyloid deposition in APP23 mice,” J. Biol. Chem, vol. 280, no. 52, pp. 43224–43235, Dec. 2005, doi: 10.1074/JBC.M504513200. [DOI] [PubMed] [Google Scholar]
- [70].Liao F et al. , “Anti-ApoE Antibody Given after Plaque Onset Decreases Aβ Accumulation and Improves Brain Function in a Mouse Model of Aβ Amyloidosis,” J. Neurosci, vol. 34, no. 21, pp. 7281–7292, May 2014, doi: 10.1523/JNEUROSCI.0646-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Liao F et al. , “Targeting of nonlipidated, aggregated apoE with antibodies inhibits amyloid accumulation,” J. Clin. Invest, vol. 128, no. 5, pp. 2144–2155, May 2018, doi: 10.1172/JCI96429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Kim J et al. , “Anti-apoE immunotherapy inhibits amyloid accumulation in a transgenic mouse model of Aβ amyloidosis,” J. Exp. Med, vol. 209, no. 12, pp. 2149–2156, Nov. 2012, doi: 10.1084/JEM.20121274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Xiong M et al. , “APOE immunotherapy reduces cerebral amyloid angiopathy and amyloid plaques while improving cerebrovascular function,” Sci. Transl. Med, vol. 13, no. 581, Feb. 2021, doi: 10.1126/scitranslmed.abd7522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Huynh TPV, Davis AA, Ulrich JD, and Holtzman DM, “Apolipoprotein E and Alzheimer’s disease: The influence of apolipoprotein E on amyloid-β and other amyloidogenic proteins,” J. Lipid Res, vol. 58, no. 5, pp. 824–836, May 2017, doi: 10.1194/JLR.R075481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Farfel JM, Yu L, De Jager PL, Schneider JA, and Bennett DA, “Association of APOE with tau-tangle pathology with and without β-amyloid,” Neurobiol. Aging, vol. 37, pp. 19–25, Jan. 2016, doi: 10.1016/j.neurobiolaging.2015.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Baek MS, Cho H, Lee HS, Lee JH, Ryu YH, and Lyoo CH, “Effect of APOE ε4 genotype on amyloid-β and tau accumulation in Alzheimer’s disease,” Alzheimer’s Res. Ther, vol. 12, no. 1, pp. 1–12, Dec. 2020, doi: 10.1186/S13195-020-007106/FIGURES/5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Geroldi C et al. , “APOE-epsilon4 is associated with less frontal and more medial temporal lobe atrophy in AD,” Neurology, vol. 53, no. 8, pp. 1825–1832, Nov. 1999, doi: 10.1212/WNL.53.8.1825. [DOI] [PubMed] [Google Scholar]
- [78].Therriault J et al. , “Association of Apolipoprotein E ε4 With Medial Temporal Tau Independent of Amyloid-β,” JAMA Neurol, vol. 77, no. 4, pp. 470–479, Apr. 2020, doi: 10.1001/JAMANEUROL.2019.4421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Donix M et al. , “Longitudinal changes in medial temporal cortical thickness in normal subjects with the APOE-4 polymorphism,” Neuroimage, vol. 53, no. 1, p. 37, Oct. 2010, doi: 10.1016/J.NEUROIMAGE.2010.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Gryglewski G et al. , “Spatial analysis and high resolution mapping of the human whole-brain transcriptome for integrative analysis in neuroimaging,” Neuroimage, vol. 176, pp. 259–267, Aug. 2018, doi: 10.1016/J.NEUROIMAGE.2018.04.068. [DOI] [PubMed] [Google Scholar]
- [81].C X, SJ M, and C C, “Association of in vivo [18 F]AV-1451 tau PET imaging results with cortical atrophy and symptoms in typical and atypical Alzheimer disease.,” JAMA Neurol, vol. 74, no. 4, pp. 427–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].H B and E B, “Neuropathological stageing of Alzheimer-related changes.,” Acta Neuropathol, vol. 82, no. 4, pp. 239–259. [DOI] [PubMed] [Google Scholar]
- [83].Ossenkoppele R et al. , “Tau PET patterns mirror clinical and neuroanatomical variability in Alzheimer’s disease,” Brain, vol. 139, no. Pt 5, pp. 1551–1567, May 2016, doi: 10.1093/BRAIN/AWW027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Hohman TJ et al. , “Sex-Specific Association of Apolipoprotein E With Cerebrospinal Fluid Levels of Tau,” JAMA Neurol, vol. 75, no. 8, p. 989, Aug. 2018, doi: 10.1001/jamaneurol.2018.0821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Shi Y et al. , “ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy,” Nature, 2017, doi: 10.1038/nature24016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Shi Y et al. , “Microglia drive APOE-dependent neurodegeneration in a tauopathy mouse model,” J. Exp. Med, vol. 216, no. 11, 2019, doi: 10.1084/jem.20190980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Shi Y et al. , “Overexpressing low-density lipoprotein receptor reduces tau-associated neurodegeneration in relation to apoE-linked mechanisms,” Neuron, vol. 109, no. 15, pp. 2413–2426. e7, Aug. 2021, doi: 10.1016/J.NEURON.2021.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Litvinchuk A et al. , “Apolipoprotein E4 Reduction with Antisense Oligonucleotides Decreases Neurodegeneration in a Tauopathy Model,” Ann. Neurol, vol. 89, no. 5, pp. 952–966, May 2021, doi: 10.1002/ANA.26043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Wang C et al. , “Selective removal of astrocytic APOE4 strongly protects against tau-mediated neurodegeneration and decreases synaptic phagocytosis by microglia,” Neuron, vol. 109, no. 10, pp. 1657–1674. e7, May 2021, doi: 10.1016/J.NEURON.2021.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Zhao J et al. , “APOE4 exacerbates synapse loss and neurodegeneration in Alzheimer’s disease patient iPSC-derived cerebral organoids,” Nat. Commun. 2020 111, vol. 11, no. 1, pp. 1–14, Nov. 2020, doi: 10.1038/s41467-020-19264-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Consortium Tabula Muris et al. , “Single-cell transcriptomics of 20 mouse organs creates a Tabula Muris.,” Nature, vol. 562, no. 7727, pp. 367–372, 2018, doi: 10.1038/s41586-018-0590-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Baker-Nigh AT et al. , “Human Central Nervous System (CNS) ApoE Isoforms Are Increased by Age, Differentially Altered by Amyloidosis, and Relative Amounts Reversed in the CNS Compared with Plasma.,” J. Biol. Chem, vol. 291, no. 53, pp. 27204–27218, 2016, doi: 10.1074/jbc.M116.721779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Linton MF et al. , “Phenotypes of apolipoprotein B and apolipoprotein E after liver transplantation,” J. Clin. Invest, vol. 88, no. 1, pp. 270–281, 1991, doi: 10.1172/JCI115288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Lane-Donovan C et al. , “Genetic Restoration of Plasma ApoE Improves Cognition and Partially Restores Synaptic Defects in ApoE-Deficient Mice.,” J. Neurosci, vol. 36, no. 39, pp. 10141–50, 2016, doi: 10.1523/JNEUROSCI.1054-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Zerbi V et al. , “Resting-state functional connectivity changes in aging apoE4 and apoE-KO mice.,” J. Neurosci, vol. 34, no. 42, pp. 13963–75, Oct. 2014, doi: 10.1523/JNEUROSCI.0684-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Achariyar TM et al. , “Glymphatic distribution of CSF-derived apoE into brain is isoform specific and suppressed during sleep deprivation.,” Mol. Neurodegener, vol. 11, no. 1, p. 74, 2016, doi: 10.1186/s13024-016-0138-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Grocott HP, Newman MF, El-Moalem H, Bainbridge D, Butler A, and Laskowitz DT, “Apolipoprotein E genotype differentially influences the proinflammatory and anti-inflammatory response to cardiopulmonary bypass,” J. Thorac. Cardiovasc. Surg, vol. 122, no. 3, pp. 622–623, 2001, doi: 10.1067/MTC.2001.115152. [DOI] [PubMed] [Google Scholar]
- [98].Tao Q et al. , “Association of Chronic Low-grade Inflammation With Risk of Alzheimer Disease in ApoE4 Carriers,” JAMA Netw. Open, vol. 1, no. 6, p. e183597, Oct. 2018, doi: 10.1001/jamanetworkopen.2018.3597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Vitek MP, Brown CM, and Colton CA, “APOE genotype-specific differences in the innate immune response,” Neurobiol. Aging, vol. 30, no. 9, pp. 1350–1360, Sep. 2009, doi: 10.1016/j.neurobiolaging.2007.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Lynch JR et al. , “APOE Genotype and an ApoE-mimetic Peptide Modify the Systemic and Central Nervous System Inflammatory Response,” J. Biol. Chem, vol. 278, no. 49, pp. 48529–48533, Dec. 2003, doi: 10.1074/jbc.M306923200. [DOI] [PubMed] [Google Scholar]
- [101].Cash JG et al. , “Apolipoprotein E4 Impairs Macrophage Efferocytosis and Potentiates Apoptosis by Accelerating Endoplasmic Reticulum Stress,” J. Biol. Chem, vol. 287, no. 33, pp. 27876–27884, Aug. 2012, doi: 10.1074/jbc.M112.377549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Tran TTT et al. , “APOE genotype influences the gut microbiome structure and function in humans and mice: relevance for Alzheimer’s disease pathophysiology,” FASEB J, vol. 33, no. 7, pp. 8221–8231, Jul. 2019, doi: 10.1096/fj.201900071R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Sofroniew MV, “Astrocyte Reactivity: Subtypes, States, and Functions in CNS Innate Immunity,” Trends Immunol, vol. 41, no. 9, pp. 758–770, Sep. 2020, doi: 10.1016/J.IT.2020.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Wyss-Coray T et al. , “Adult mouse astrocytes degrade amyloid-beta in vitro and in situ,” Nat. Med, vol. 9, no. 4, pp. 453–457, Apr. 2003, doi: 10.1038/NM838. [DOI] [PubMed] [Google Scholar]
- [105].Koistinaho M et al. , “Apolipoprotein E promotes astrocyte colocalization and degradation of deposited amyloid-beta peptides,” Nat. Med, vol. 10, no. 7, pp. 719–726, Jul. 2004, doi: 10.1038/NM1058. [DOI] [PubMed] [Google Scholar]
- [106].Deane R et al. , “apoE isoform-specific disruption of amyloid beta peptide clearance from mouse brain,” J. Clin. Invest, vol. 118, no. 12, pp. 4002–4013, Dec. 2008, doi: 10.1172/JCI36663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Simonovitch S et al. , “Impaired Autophagy in APOE4 Astrocytes,” J. Alzheimers. Dis, vol. 51, no. 3, pp. 915–927, Mar. 2016, doi: 10.3233/JAD-151101. [DOI] [PubMed] [Google Scholar]
- [108].Liu CC et al. , “Astrocytic LRP1 Mediates Brain Aβ Clearance and Impacts Amyloid Deposition,” J. Neurosci, vol. 37, no. 15, pp. 4023–4031, Apr. 2017, doi: 10.1523/JNEUROSCI.3442-16.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Igarashi H, Suzuki Y, Kwee IL, and Nakada T, “Neurological Research,” Neurol. Res, vol. 36, pp. 1094–1098, doi: 10.1179/1743132814Y.0000000434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Miners JS, Baig S, Tayler H, Kehoe PG, and Love S, “Neprilysin and InsulinDegrading Enzyme Levels Are Increased in Alzheimer Disease in Relation to Disease Severity,” J. Neuropathol. Exp. Neurol, vol. 68, no. 8, pp. 902–914, Aug. 2009, doi: 10.1097/NEN.0B013E3181AFE475. [DOI] [PubMed] [Google Scholar]
- [111].Zamanian JL et al. , “Genomic Analysis of Reactive Astrogliosis,” J. Neurosci, vol. 32, no. 18, pp. 6391–6410, May 2012, doi: 10.1523/JNEUROSCI.6221-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Habib N et al. , “Disease-associated astrocytes in Alzheimer’s disease and aging,” Nat. Neurosci. 2020 236, vol. 23, no. 6, pp. 701–706, Apr. 2020, doi: 10.1038/s41593-020-0624-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Mathys H et al. , “Single-cell transcriptomic analysis of Alzheimer’s disease,” Nat. 2019 5707761, vol. 570, no. 7761, pp. 332–337, May 2019, doi: 10.1038/S41586-019-1195-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Escartin C et al. , “Reactive astrocyte nomenclature, definitions, and future directions.,” Nat. Neurosci, vol. 24, no. 3, pp. 312–325, 2021, doi: 10.1038/s41593-020-00783-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Liddelow SA et al. , “Neurotoxic reactive astrocytes are induced by activated microglia,” Nature, vol. 541, no. 7638, p. 481, Jan. 2017, doi: 10.1038/NATURE21029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Guttenplan KA et al. , “Neurotoxic reactive astrocytes induce cell death via saturated lipids.,” Nature, vol. 599, no. 7883, pp. 102–107, Nov. 2021, doi: 10.1038/s41586-021-03960-y. [DOI] [PubMed] [Google Scholar]
- [117].Huynh TPV et al. , “Lack of hepatic apoE does not influence early Aβ deposition: observations from a new APOE knock-in model,” Mol. Neurodegener, vol. 14, no. 1, Oct. 2019, doi: 10.1186/S13024-019-0337-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Leng K et al. , “Molecular characterization of selectively vulnerable neurons in Alzheimer’s disease,” Nat. Neurosci, vol. 24, no. 2, pp. 276–287, Feb. 2021, doi: 10.1038/S41593-020-00764-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Alzheimer A, “Uber eine eigenartige Erkrankung der Hirnrinde.,” Allg Zeitschr f Psychiatr. u Psych-Gerichtl Med, vol. 64, no. 64, 1907. [Google Scholar]
- [120].Sienski G et al. , “APOE4 disrupts intracellular lipid homeostasis in human iPSC-derived glia.,” Sci. Transl. Med, vol. 13, no. 583, 2021, doi: 10.1126/scitranslmed.aaz4564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Qi G, Mi Y, Shi X, Gu H, Brinton RD, and Yin F, “ApoE4 Impairs Neuron-Astrocyte Coupling of Fatty Acid Metabolism,” Cell Rep, vol. 34, no. 1, p. 108572, Jan. 2021, doi: 10.1016/j.celrep.2020.108572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].TCW J et al. , “Cholesterol and matrisome pathways dysregulated in human APOE ε4 glia,” bioRxiv, p. 713362, Jan. 2019, doi: 10.1101/713362. [DOI] [Google Scholar]
- [123].Lin YT et al. , “APOE4 Causes Widespread Molecular and Cellular Alterations Associated with Alzheimer’s Disease Phenotypes in Human iPSC-Derived Brain Cell Types,” Neuron, vol. 98, no. 6, pp. 1141–1154. e7, Jun. 2018, doi: 10.1016/J.NEURON.2018.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Nugent AA et al. , “TREM2 Regulates Microglial Cholesterol Metabolism upon Chronic Phagocytic Challenge.,” Neuron, vol. 105, no. 5, pp. 837–854. e9, 2020, doi: 10.1016/j.neuron.2019.12.007. [DOI] [PubMed] [Google Scholar]
- [125].Meyer-Luehmann M et al. , “Rapid appearance and local toxicity of amyloid-beta plaques in a mouse model of Alzheimer’s disease,” Nature, vol. 451, no. 7179, pp. 720–724, Feb. 2008, doi: 10.1038/NATURE06616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Parhizkar S et al. , “Loss of TREM2 function increases amyloid seeding but reduces plaque-associated ApoE,” Nat. Neurosci. 2019 222, vol. 22, no. 2, pp. 191–204, Jan. 2019, doi: 10.1038/s41593-018-0296-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Pascoal TA et al. , “Microglial activation and tau propagate jointly across Braak stages,” Nat. Med. 2021 279, vol. 27, no. 9, pp. 1592–1599, Aug. 2021, doi: 10.1038/s41591-021-01456-w. [DOI] [PubMed] [Google Scholar]
- [128].Ransohoff RM, “How neuroinflammation contributes to neurodegeneration,” Science, vol. 353, no. 6301, pp. 777–783, Aug. 2016, doi: 10.1126/SCIENCE.AAG2590. [DOI] [PubMed] [Google Scholar]
- [129].Rodriguez GA, Tai LM, LaDu MJ, and Rebeck GW, “Human APOE4 increases microglia reactivity at Aβ plaques in a mouse model of Aβ deposition,” J. Neuroinflammation, vol. 11, p. 111, Jun. 2014, doi: 10.1186/1742-2094-11-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Stephen TL et al. , “APOE genotype and sex affect microglial interactions with plaques in Alzheimer’s disease mice,” Acta Neuropathol. Commun, vol. 7, no. 1, pp. 1–11, May 2019, doi: 10.1186/S40478-019-0729-Z/FIGURES/5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Serrano-Pozo A et al. , “Differential relationships of reactive astrocytes and microglia to fibrillar amyloid deposits in Alzheimer disease,” J. Neuropathol. Exp. Neurol, vol. 72, no. 6, pp. 462–471, Jun. 2013, doi: 10.1097/NEN.0B013E3182933788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Spangenberg E et al. , “Sustained microglial depletion with CSF1R inhibitor impairs parenchymal plaque development in an Alzheimer’s disease model,” Nat. Commun. 2019 101, vol. 10, no. 1, pp. 1–21, Aug. 2019, doi: 10.1038/s41467-019-11674-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Gratuze M et al. , “Activated microglia mitigate Aβ-associated tau seeding and spreading,” J. Exp. Med, vol. 218, no. 8, Aug. 2021, doi: 10.1084/JEM.20210542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Yuan P et al. , “TREM2 Haplodeficiency in Mice and Humans Impairs the Microglia Barrier Function Leading to Decreased Amyloid Compaction and Severe Axonal Dystrophy,” Neuron, 2016, doi: 10.1016/j.neuron.2016.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Henningfield CM, Arreola MA, Soni N, Spangenberg EE, and Green KN, “Microglia-specific ApoE knock-out does not alter Alzheimer’s disease plaque pathogenesis or gene expression,” Glia, 2021, doi: 10.1002/GLIA.24105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Mancuso R et al. , “CSF1R inhibitor JNJ-40346527 attenuates microglial proliferation and neurodegeneration in P301S mice,” Brain, vol. 142, no. 10, pp. 3243–3264, Oct. 2019, doi: 10.1093/BRAIN/AWZ241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Elmore MRP et al. , “Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain,” Neuron, vol. 82, no. 2, 2014, doi: 10.1016/j.neuron.2014.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Spangenberg EE et al. , “Eliminating microglia in Alzheimer’s mice prevents neuronal loss without modulating amyloid-beta pathology,” Brain, vol. 139, no. 4, pp. 1265–1281, Apr. 2016, doi: 10.1093/brain/aww016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Zhan L et al. , “A mac2-positive progenitor-like microglial population is resistant to csf1r inhibition in adult mouse brain,” Elife, vol. 9, pp. 1–22, Oct. 2020, doi: 10.7554/ELIFE.51796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Shi Y and Holtzman DM, “Interplay between innate immunity and Alzheimer disease: APOE and TREM2 in the spotlight,” Nat. Rev. Immunol, vol. 18, no. 12, pp. 759–772, Dec. 2018, doi: 10.1038/s41577-018-0051-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Pimenova AA et al. , “Alzheimer’s-associated PU.1 expression levels regulate microglial inflammatory response,” Neurobiol. Dis, vol. 148, p. 105217, Jan. 2021, doi: 10.1016/J.NBD.2020.105217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Hoogmartens J, Cacace R, and Van Broeckhoven C, “Insight into the genetic etiology of Alzheimer’s disease: A comprehensive review of the role of rare variants,” Alzheimer’s Dement. Diagnosis, Assess. Dis. Monit, vol. 13, no. 1, p. e12155, Jan. 2021, doi: 10.1002/DAD2.12155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Holtzman DM, “In vivo effects of ApoE and clusterin on amyloid-beta metabolism and neuropathology.,” J. Mol. Neurosci, vol. 23, no. 3, pp. 247–254, 2004, doi: 10.1385/JMN:23:3:247. [DOI] [PubMed] [Google Scholar]
- [144].DeMattos RB et al. , “ApoE and clusterin cooperatively suppress Abeta levels and deposition: evidence that ApoE regulates extracellular Abeta metabolism in vivo,” Neuron, vol. 41, no. 2, pp. 193–202, Jan. 2004, doi: 10.1016/S0896-6273(03)00850-X. [DOI] [PubMed] [Google Scholar]
- [145].Kim WS, Weickert CS, and Garner B, “Role of ATP-binding cassette transporters in brain lipid transport and neurological disease,” J. Neurochem, vol. 104, no. 5, pp. 1145–1166, Mar. 2008, doi: 10.1111/J.1471-4159.2007.05099.X. [DOI] [PubMed] [Google Scholar]
- [146].Wojtas AM et al. , “Astrocyte-derived clusterin suppresses amyloid formation in vivo,” Mol. Neurodegener, vol. 15, no. 1, p. 71, Dec. 2020, doi: 10.1186/s13024-020-00416-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Wojtas AM et al. , “Loss of clusterin shifts amyloid deposition to the cerebrovasculature via disruption of perivascular drainage pathways,” Proc. Natl. Acad. Sci, vol. 114, no. 33, pp. E6962–E6971, Aug. 2017, doi: 10.1073/pnas.1701137114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Dos Santos LR, Almeida JFF, Pimassoni LHS, Morelato RL, and de Paula F, “The combined risk effect among BIN1,CLU, and APOE genes in Alzheimer’sdisease,” Genet. Mol. Biol, vol. 43, no. 1, pp. 1–7, 2020, doi: 10.1590/1678-4685-GMB-2018-0320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].De Strooper B and Karran E, “The Cellular Phase of Alzheimer’s Disease,” Cell, vol. 164, no. 4, pp. 603–615, Feb. 2016, doi: 10.1016/J.CELL.2015.12.056. [DOI] [PubMed] [Google Scholar]
- [150].Wendeln A-C et al. , “Innate immune memory in the brain shapes neurological disease hallmarks.,” Nature, vol. 556, no. 7701, pp. 332–338, 2018, doi: 10.1038/s41586-018-0023-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Réu P et al. , “The Lifespan and Turnover of Microglia in the Human Brain.,” Cell Rep, vol. 20, no. 4, pp. 779–784, 2017, doi: 10.1016/j.celrep.2017.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Füger P et al. , “Microglia turnover with aging and in an Alzheimer’s model via long-term in vivo single-cell imaging,” Nat. Neurosci, vol. 20, no. 10, pp. 1371–1376, Oct. 2017, doi: 10.1038/nn.4631. [DOI] [PubMed] [Google Scholar]
- [153].Tay TL et al. , “A new fate mapping system reveals context-dependent random or clonal expansion of microglia,” Nat. Neurosci, vol. 20, no. 6, pp. 793–803, Jun. 2017, doi: 10.1038/nn.4547. [DOI] [PubMed] [Google Scholar]
- [154].Grabert K et al. , “Microglial brain region−dependent diversity and selective regional sensitivities to aging,” Nat. Neurosci, vol. 19, no. 3, pp. 504–516, Mar. 2016, doi: 10.1038/nn.4222. [DOI] [PMC free article] [PubMed] [Google Scholar]