

SUMMARY
MicroRNA (miRNA) abundance is tightly controlled by regulation of biogenesis and decay. Here, we show that the mir-35 miRNA family undergoes selective decay at the transition from embryonic to larval development in C. elegans. The seed sequence of the miRNA is necessary and largely sufficient for this regulation. Sequences outside the seed (3′ end) regulate mir-35 abundance in the embryo but are not necessary for sharp decay at the transition to larval development. Enzymatic modifications of the miRNA 3′ end are neither prevalent nor correlated with changes in decay, suggesting that miRNA 3′ end display is not a core feature of this mechanism and further supporting a seed-driven decay model. Our findings demonstrate that seed-sequence-specific decay can selectively and coherently regulate all redundant members of a miRNA seed family, a class of mechanism that has great biological and therapeutic potential for dynamic regulation of a miRNA family’s target repertoire.
In brief
Donnelly et al. show that sequence-specific miRNA decay contributes to the dynamic changes in miRNA repertoire during development. The seed sequence of the mir-35 family drives decay of these miRNAs at the end of embryogenesis, suggesting a selective decay mechanism that can co-regulate all redundant members of a miRNA family.
Graphical Abstract
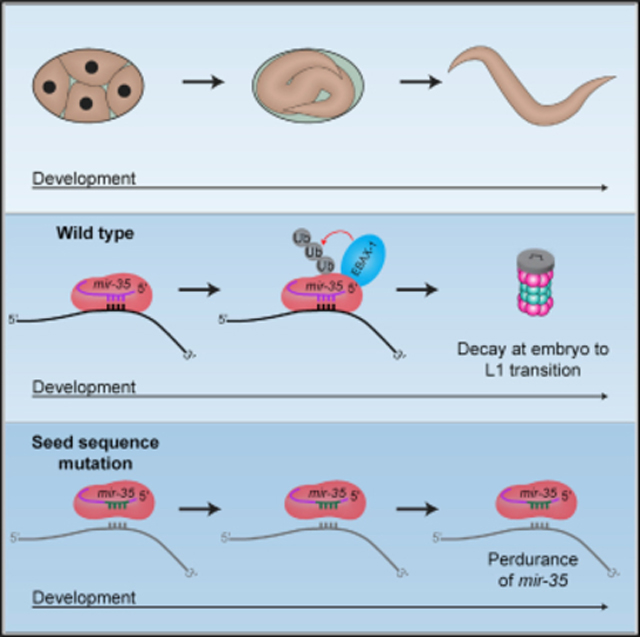
INTRODUCTION
MicroRNAs (miRNAs) are small non-coding RNAs that negatively regulate target mRNAs (Dallaire et al., 2018). The biogenesis of miRNAs begins with transcription of a primary miRNA: a long transcript containing a ~35 base pair stem-loop structure (Fang and Bartel, 2015; Han et al., 2006; Ma et al., 2013; Zeng et al., 2005). This structure is recognized and cleaved by the microprocessor complex into the miRNA precursor (Fang and Bartel, 2015; Han et al., 2006; Ma et al., 2013; Zeng et al., 2005) (Denli et al., 2004; Gregory et al., 2004; Han et al., 2004; Landthaler et al., 2004). The precursor is cleaved by Dicer into a ~22–23 nucleotide duplex that is loaded into Argonaute (Ago) (Bernstein et al., 2001; Grishok et al., 2001; Hutvágner et al., 2001; Ketting et al., 2001; Knight and Bass, 2001). The mature guide strand remains in Ago, while the star strand is ejected and degraded (Iwasaki et al., 2010, 2015). The bound miRNA guide strand targets complementary regions in the 3′ UTR of mRNAs to silence gene expression (Dexheimer and Cochella, 2020).
The interaction between the miRNA and mRNA target is primarily mediated through nucleotides 2–8 at the 5′ end of the miRNA (Brennecke et al., 2005; Lewis et al., 2003). This region, the seed sequence, defines a miRNA family: a group of miRNAs that act largely redundantly on an overlapping set of target genes due to their identical seed sequence (Alvarez-Saavedra and Horvitz, 2010; Parchem et al., 2015). Supplemental base pairing between the 3′ end of the miRNA and the target RNA occurs in some cases, conferring differences in target repertoire of miRNA family members (Brancati and Großhans, 2018; Broughton et al., 2016; Helwak et al., 2013; Isana Veksler-Lublinsky, 2022).
While much is known about the biogenesis and functions of miRNAs, little is known about the mechanisms of decay of mature miRNAs. While half-lives of miRNAs vary, what determines these differences in stability is, for the most part, unknown (Bail et al., 2010; Kingston and Bartel, 2019; Lehrbach et al., 2012; Marzi et al., 2016; Miki et al., 2014; Reichholf et al., 2019; Vieux et al., 2021). Thus far, multiple phenomena regulating miRNA stability have been observed, with different degrees of sequence specificity.
Some decay pathways are largely independent of miRNA sequence. In C. elegans, the 5′ to 3′ nuclease XRN-2, along with DCS-1, maintain wild-type miRNA levels by degrading many miRNAs (Bossé et al., 2013; Chatterjee et al., 2009). At the maternal-to-zygotic transition in Drosophila, terminal adenylation of maternal miRNAs by Wispy induces their wholesale clearance (Lee et al., 2014). In other species, 3′ nucleotide addition (tailing) has also been proposed to destabilize miRNAs in a sequence-independent manner (Boele et al., 2014; Katoh et al., 2015; Knouf et al., 2013; Lee et al., 2019; Shukla et al., 2019; Wyman et al., 2011; Yang et al., 2020a).
Other miRNA decay pathways are guided by moderate sequence specificity. One example is Tudor-SN-mediated miRNA decay (TumiD), in which the endonuclease Tudor-SN (TSN) cleaves a few dozen miRNAs at CA and UA dinucleotides (Elbarbary et al., 2017a; 2017b). A more specific phenomenon destabilizes several members of the extended miR-16 family; this decay is dependent on sequences in both the seed and the 3′ portion of the miRNA (Rissland et al., 2011).
The most sequence-specific mechanism of miRNA decay is target-directed miRNA degradation (TDMD). TDMD occurs when a high-abundance RNA (the TDMD “trigger”) binds to a miRNA with extensive complementarity to both the seed sequence and the 3′ half of the miRNA (Ameres et al., 2010; Baccarini et al., 2011; Bitetti et al., 2018; Cazalla et al., 2010; Ghini et al., 2018; Kleaveland et al., 2018; Libri et al., 2012; Marcinowski et al., 2012; la Mata et al., 2015; Piwecka et al., 2017). This extensive base pairing induces a conformational change that pulls the 3′ end of the miRNA out of the PAZ domain of Ago, making it accessible to modification by untemplated nucleotide additions (tailing) and exonucleolytic cleavage (trimming) (Sheu-Gruttadauria et al., 2019; Yang et al., 2020a). Recently, the Cullin-RING E3 ubiquitin ligase ZSWIM8 was identified as an effector of TDMD, leading to the model that the Ago and/or RNA conformation induced by extensive base pairing is recognized by ZSWIM8 for ubiquitylation and subsequent decay of the miRNA:Ago complex (Han et al., 2020; Shi et al., 2020).
The regulation of miRNA expression during development is crucial to ensure properly timed developmental transitions, but the extent to which miRNA decay contributes to ensuring proper temporal expression patterns of miRNAs and how development is coupled to timing of decay are not known.
In this work, we examine the mechanism of decay of the mir-35 family. The mir-35 family consists of 8 miRNAs, mir-35–42 (Alvarez-Saavedra and Horvitz, 2010). The mir-35 family members are maternally contributed as well as zygotically expressed in early embryogenesis, and they are sharply degraded at the transition from embryo to the first larval stage (L1; EtoL1) (Stoeckius et al., 2009; Wu et al., 2010). Understanding the mechanism of this decay will shed light on how selective miRNA decay occurs and how it is coupled to development.
The mir-35 family is one of two miRNA seed families that are necessary for C. elegans embryogenesis. Because of their identical seed sequences, the mir-35 family members are functionally redundant; deletion of any single miRNA has no detectable phenotypic consequences, whereas deletion of the whole family results in embryonic lethality (Alvarez-Saavedra and Horvitz, 2010). The mir-35 family miRNAs also play multiple roles in development, including promoting maximal fecundity, ensuring sex determination, and regulating cell death (Doll et al., 2019; Flamand et al., 2016, 2017; Kagias and Pocock, 2015; Liu et al., 2011; Massirer et al., 2012; McJunkin and Ambros, 2014, 2017; Tran et al., 2019; Yang et al., 2020b; Zhao et al., 2019).
How the mir-35 family is targeted for selective decay at the end of embryogenesis is not known. A recent study showed that the TDMD factor ZSWIM8 (known as EBAX-1 in C. elegans) drives instability of the mir-35 family, suggesting that the mir-35 family is subject to TDMD (Shi et al., 2020; Wang et al., 2013). However, positions in the 3′ portion of the miRNA that are usually involved in the base-pairing interactions that drive TDMD are highly degenerate across the mir-35 family members (Figure 1A), suggesting that the mechanism of mir-35 decay may differ from previously described examples of TDMD and may represent a novel type of selective miRNA decay mechanism.
Figure 1. mir-35 decay is seed-sequence dependent.
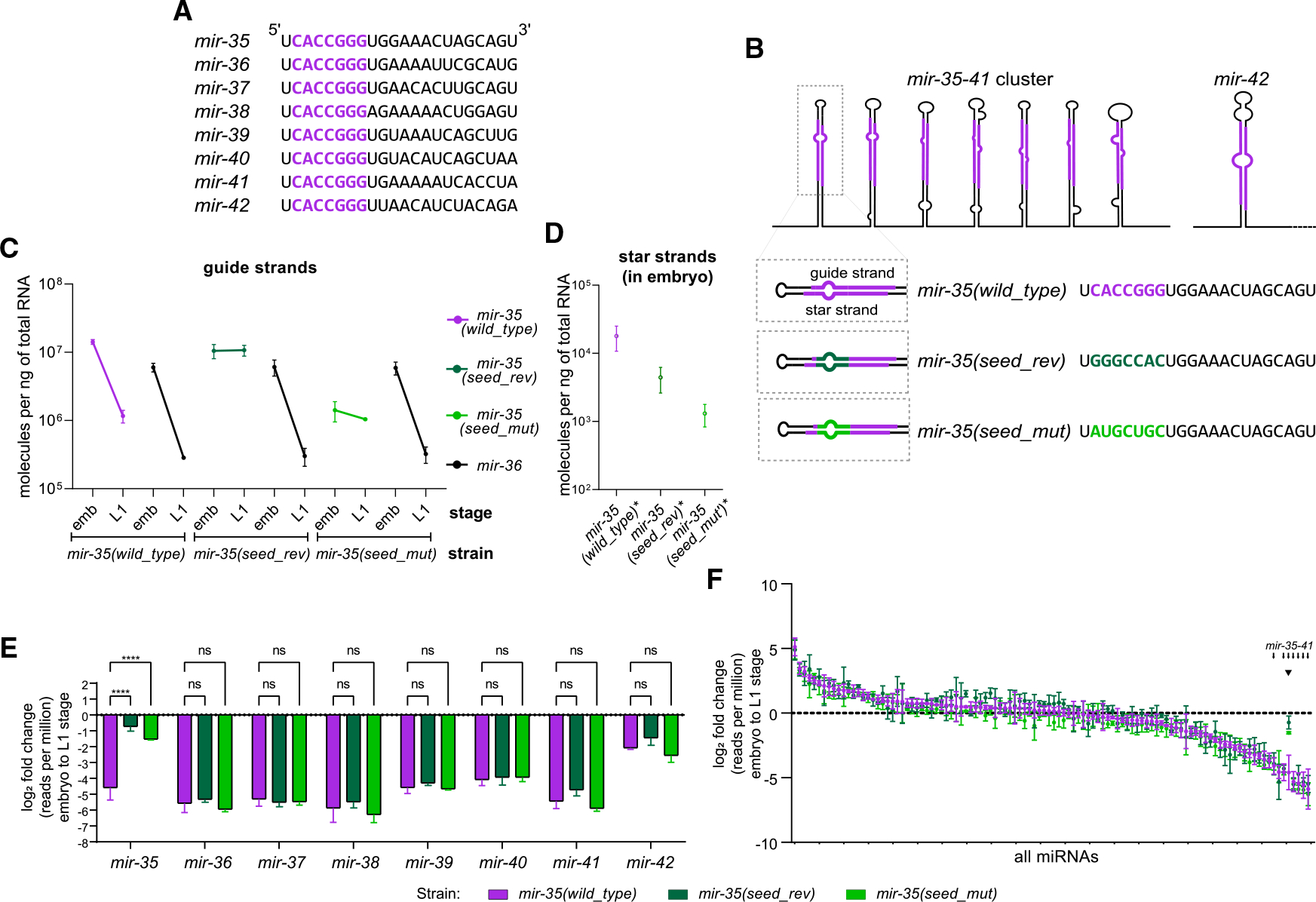
(A) Sequences of mir-35–42. Seed sequence in purple.
(B) Schematic of the mir-35–41 cluster and mir-42 with sequences of mir-35 and variants.
(C and D) Absolute quantification of mir-35 and mir-36 guide strands (C) or star strands (D).
(E and F) Log2(fold change) from embryo to L1, calculated from deep sequencing for either the mir-35–42 family (E) or all miRNAs >50 RPM in wild type (F). Note that color of bar indicates strain, not necessarily a mutant miRNA; only mir-35 is mutated in the indicated mutant strains.
(E) Two-way ANOVA, followed by Dunnett’s multiple comparisons test. ****p value < 0.0001.
(F) Small arrows indicate positions of mir-35–41 on ranked x axis, and arrowhead indicates mir-35 and mutant variants.
(C–F) Mean and SEM of three biological replicates are shown.
Here, we show that the mir-35 family is regulated at the level of decay at the EtoL1 transition in C. elegans. We demonstrate that the seed sequence of mir-35 is necessary and largely sufficient for this developmentally timed decay. This decay is not correlated with high levels of miRNA 3′ tailing and trimming. Together, these data suggest that this miRNA family is regulated by a mechanism distinct from - but possibly related to - TDMD. Seed-specific decay mechanisms such as this are likely to be more widespread in biological systems since they have potential to co-regulate all members of a redundant miRNA family, potentially allowing dynamic derepression of the miRNA family’s target genes.
RESULTS
mir-35 decay is seed-sequence dependent
The mir-35 family is decayed at EtoL1 (Stoeckius et al., 2009; Wu et al., 2010). We wondered (1) if this decay is a selectively regulated process or, alternatively, just a result of transcriptional shutoff in late embryogenesis and (2) whether the seed sequence plays a role in this decay. To this end, we used CRISPR to mutate the locus that encodes mir-35–41 on a single transcript (Figure 1B). We made mutations to the seed sequence of the first hairpin in the mir-35–41 cluster (mir-35) using CRISPR. This approach leaves the remainder of the mir-35–41 cluster intact, which serves two purposes: (1) mir-35 loss-of-function phenotypes are not induced since the other family members remain wild type, and 2) mir-36–41 serve as internal controls derived from the same transcript as mir-35. Both strands of the mir-35 hairpin were mutated to preserve secondary structure for efficient processing (Figure 1B). One mutation reversed the seed sequence, (mir-35(seed_rev)), whereas the other mutation replaced the mir-35 seed sequence with random nucleotides (mir-35(seed_mut)) (Figure 1B; Table S1).
To determine if these mutations affect biogenesis of mir-35, we quantified mir-35 and the mutant variants using miRNA-Taqman qPCR, along with synthetic RNA oligonucleotides to generate standard curves for absolute quantification (Figure S1A). The embryo concentration of mir-35(seed_rev) is similar to wild-type mir-35 (0.7-fold change), while mir-35(seed_mut) is 10-fold lower (Figure 1C). To determine if the changes in the amount of the mir-35 variants were at the level of biogenesis or post-biogenesis, we examined the abundance of their star strands in the embryo. Changes in abundance of the mir-35 variant star strands are similar to those in the respective guide strands (Figure 1D); these coupled changes suggest that the decreased abundance of mir-35(seed_mut) is due to loss of efficiency in biogenesis.
Next, we examined whether the decay of mir-35 at EtoL1 is altered by seed mutations. (Because we use arrested L1 samples, post-embryogenesis growth has not begun, so any decreases in miRNA abundance must be attributed to decay rather than dilution caused by growth.) As expected, a strong reduction in wild-type mir-35 occurred at EtoL1, with 12-fold lower abundance in L1 (Figure 1C). However, the decay of mutant mir-35(seed_rev) and mir-35(seed_mut) at EtoL1 was greatly attenuated to essentially no change and 1.3-fold lower in L1 than embryo, respectively (Figure 1C). mir-35(seed_rev) derived from a second CRISPR allele with altered precursor structure also showed attenuated decay (Figures S1B–S1C). Therefore, the decay of mir-35 depends on its seed sequence. Importantly, the decay of mir-36 was not altered by the mutations in mir-35 (Figure 1C). This decoupling of the behavior of mutant mir-35 and wild-type mir-36—which share a primary transcript—further shows that mir-35 family decay is regulated post-transcriptionally.
To confirm and extend these findings, we performed deep sequencing to profile all miRNAs. mir-35 was the only miRNA altered by these mutations in embryo or L1 samples (Figure S2A; Tables S1 and S2). Consistent with the qPCR, wild-type mir-35 displayed sharp decay at EtoL1, and the mir-35 seed mutants were resistant to this decay (Figure 1E; Table S2). The decay of the other members of the mir-35 family was not affected by mir-35 seed mutations, despite most members sharing the mir-35–41 primary transcript (Figure 1E). Global analysis further demonstrated the selectivity of the decay of the mir-35 family at EtoL1: mir-35–41 represent seven of the eight most sharply downregulated miRNAs at this time point in wild type (Figure 1F; Table S2). This analysis reiterates the specificity of the effect of the mir-35 seed mutations (Figure 1F).
Together, these results show that the decay of the mir-35 family at this developmental transition is a selectively regulated decay process (rather than simply the result of synchronous decay after transcriptional shutoff) since the behavior of miRNAs derived from the same transcript can be de-coupled. Furthermore, these results show that the mir-35 seed sequence is required for this regulated decay.
mir-35 3′ end mutants undergo efficient decay at the embryo-to-L1 transition
The necessity of the seed sequence for mir-35 decay (Figure 1) and the recent implication of the TDMD factor EBAX-1 in regulating stability of the mir-35 family (Shi et al., 2020) together suggest a TDMD-like decay mechanism. However, the degeneracy of sequences in the 3′ region of the miRNA across the mir-35 family members (Figure 1A) suggests that the mechanism may differ from previous examples of TDMD since multiple trigger RNAs would be necessary to bind with extensive complementarity to all family members. (Note that results from mir-35(seed_rev) and mir-35(seed_mut) rule out an antisense RNA from the mir-35–41 cluster acting as a TDMD trigger RNA since mutations at the genomic locus would not disrupt base-pairing with an antisense transcript.)
Therefore, we next investigated whether the 3′ portion of the miRNA plays a role in mir-35 family decay and if the mir-35 seed sequence is sufficient for decay. To test this, we used CRISPR to generate two mir-35 mutant strains in which the non-seed (hereafter referred to as the 3′ end) residues of the miRNA are mutated. The first mutant is comprised of the mir-35 seed sequence with a 3′ end containing nucleotides that are not present or are rare among all mir-35 family members at a given position while preserving overall GC content (mir-35(mut_3′)) (Figure 2A). The second mutant is a mir-35/mir-82 hybrid composed of the mir-35 seed sequence and the mir-82 3′ end (mir-35(mir-82_3′)) (Figure 2A). The mir-82 sequence was chosen for the non-seed region of mir-35 because mir-82 expression is steady rather than downregulated at EtoL1 (Kato et al., 2009).
Figure 2. mir-35 3′ end mutants do not alter decay.
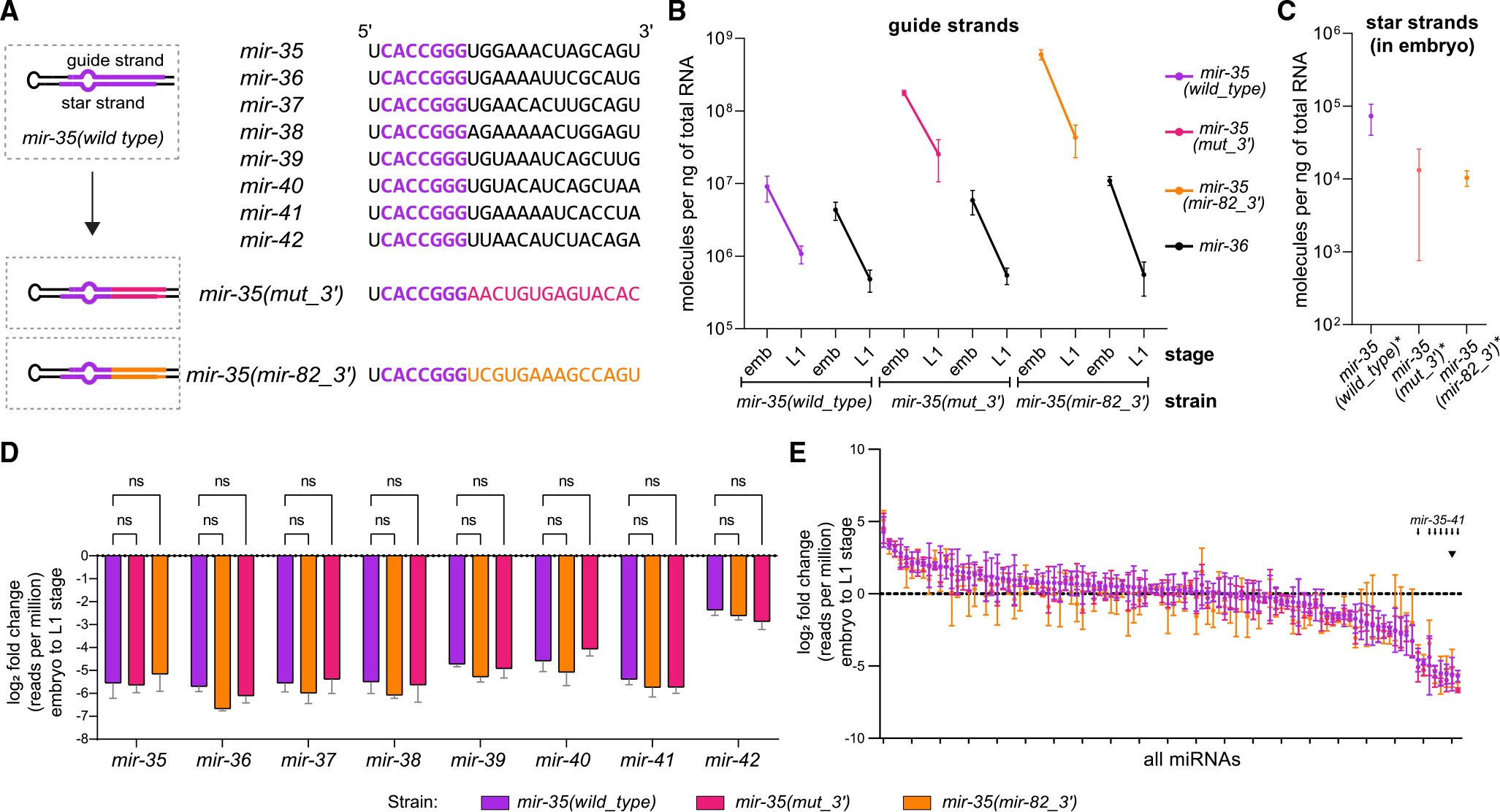
(A) Sequences of mir-35–42 with the identical seed sequences shown in purple (top). Schematic of the mir-35 3′ end mutants (bottom).
(B) Absolute quantification of mir-35 and mir-36 in embryos and L1. Mean and SEM of two to three biological replicates.
(C) Absolute quantification of star strands of mir-35 and mutant variants in embryos.
(D and E) Log2(fold change) from embryo to L1, calculated from normalized deep-sequencing reads for either the mir-35–42 family (D) or all miRNAs with >50 RPM in wild type (E). Note that color of bar indicates strain, not necessarily a mutant miRNA; only mir-35 is mutated in the indicated mutant strains.
(D) Two-way ANOVA was performed, followed by Dunnett’s multiple comparisons test.
(E) Small arrows indicate positions of mir-35–41 on ranked x axis, and arrowhead indicates mir-35 and mutant variants.
(C–E) Mean and SEM of three biological replicates.
Again, we performed miRNA-Taqman qPCR of mir-35 and its mutant variants using absolute quantification (Figure S1A). In embryos, the quantity of mir-35(mut_3′) and mir-35(mir-82_3′) were increased 20- and 139-fold relative to wild-type mir-35, respectively, while star-strand abundances did not reflect these changes (Figure 2B and 2C). While potential changes in strand selection or stability of the star strands may confound interpretation of the guide:star ratio of the mutant duplexes, the large overall increase in the number of molecules deriving from either strand of the 3′ end mutant precursors supports the model that biogenesis- or decay-level effects are contributing to the high abundance of mir-35(mut_3′) and mir-35(mir-82_3′) in embryos. Despite the caveats to interpreting the guide:star ratio, we currently favor the model that decay is disrupted because the apparent strand specificity of the effect is consistent across both mutant variants. Overall, we postulate that a second regulatory mechanism acts via 3′ end sequence to limit abundance of mir-35 in the embryo (Figure S3A).
We next measured the decay of the mir-35 3′ end variants at EtoL1. Unlike the seed mutants, the change in the mir-35 3′ end mutants at EtoL1 was similar to that of wild-type mir-35 (7-fold for the mir-35(mut_3′), 14-fold for mir-35(mir-82_3′), and 8-fold for wild type) (Figure 2B). Likewise, mir-36 was not affected by the mutations (Figure 2B). Deep sequencing confirmed that the 3′ end variants showed a similar depletion at EtoL1 as wild-type mir-35 and that no other miRNAs in the mir-35 family or otherwise were affected (Figures 2D–2E and S2B; Tables S1 and S2). Thus, the sequence of the 3′ end of the miRNA outside the seed did not affect the decay at this developmental transition.
Overall, we observed that seed mutations do not generally impact embryonic mir-35 abundance but strongly inhibit its decay at EtoL1, whereas 3′ end mutations strongly impact embryonic abundance of mir-35 but do not affect its decay at EtoL1. Taken together, we propose that two mechanisms regulate mir-35 abundance: a 3′ end-dependent mechanism limits abundance in embryos, while a seed-dependent mechanism drives decay at EtoL1 (Figure S3A). Given that all positions 3′ of the seed sequence are mutated in the 3′ end mutants, the seed sequence of mir-35 is not only necessary but also largely sufficient to drive its selective decay at EtoL1. Notably, this working model assumes that the 3′ end mutant variants are decayed by the same mechanism as wild-type mir-35 at EtoL1; alternatively, if the 3′ end mutant variants are decayed by a novel mechanism, then the 3′ end sequence could still play a role in EtoL1 decay of wild-type mir-35.
EBAX-1 regulates mir-35 family abundance in embryos and at the embryo-to-L1 transition
Given the model that the mir-35 family is regulated in two phases (Figure S3A), we asked whether EBAX-1 regulates mir-35 abundance in either of these developmental windows. To compare wild-type and ebax-1(null) animals, we performed both qPCR in bulk embryos and L1s and deep sequencing in hand-picked staged embryos and L1s.
Both assays showed a modest upregulation of mir-35 family members in embryos: a 1.4- and 1.3-fold increase was observed in mir-35 and mir-36, respectively, by bulk sample qPCR (Figure S3B). These changes were reflected in the star strands as well, which were each increased 1.4-fold (Figure S3B). The deep sequencing of staged embryos also showed a modest increase in ebax-1(null), especially in the comma stage, where mir-35–41 were upregulated 2.3-fold on average (Figure S3C; Table S3). (Data for star strands were sparse and noisy in this deep sequencing and therefore likely unreliable to interpret.) Together, these data suggest that EBAX-1 has a modest effect in regulating mir-35 family abundance in the embryo, possibly impacting transcription or biogenesis.
At EtoL1, qPCR of bulk samples showed stark stabilization of mir-35 and mir-36 in ebax-1(null); decay was 10- and 12-fold for mir-35 and mir-36 in wild type, whereas no measurable decay was observed in ebax-1(null) (Figure S3B). Deep-sequencing results corroborated the impact of ebax-1(null) on decay at EtoL1; however, the amplitude of this effect was slightly inconsistent. For instance, mir-35 and mir-36 were decayed 17- and 8-fold from the comma stage to L1 in wild type, and this was reduced to 6- and 5-fold, respectively, in ebax-1(null) (Figure S3C). These discrepancies may arise from differences in the two methodologies used. Nonetheless, both experiments support a role for EBAX-1 in mir-35 family decay at EtoL1.
mir-35 variants are tailed and trimmed similarly to wild-type mir-35
While the seed dependence and EBAX-1 dependence of mir-35 regulation suggest a TDMD-like mechanism, the dispensability of the 3′ end for EtoL1 decay suggests an alternative mechanism. TDMD is often accompanied by high tailing and trimming during the decay process due to conformational changes induced by extensive base-pairing that expose the 3′ end of the miRNA. Thus, we examined trimming and tailing of mir-35.
We first examined the level of background in tailing measurements in our experimental and computational pipeline. To this end, synthetic miRNAs were spiked into total RNA after purification, and the amount of tailing called on these miRNAs is considered background since these miRNAs were never present in the context of cellular lysate, so any apparent “tailing” must derive from errors introduced in cloning or sequencing. Tailing was below 1.5% in 98% of spike-in measurements, so tailing below 1.5% is considered background in these datasets (dashed line on all tailing plots).
In embryos and L1s, we observed that miRNAs are generally not highly tailed (Figure 3A and S4A). Tailing was mostly mono-U, with some miRNAs displaying A- or C-tailing, as previously observed (Figures 3A and S4A) (Vieux et al., 2021). Overall tailing and miRNA abundance were not correlated, and the mir-35 family members were generally high in abundance, with a wide range of tailing frequencies observed across different members (Figures 3B; Table S2).
Figure 3. Changes in tailing and trimming of mir-35 variants do not correlate with changes in decay.

(A) Tailing of each miRNA >50 RPM in embryo.
(B) Total tailing (sum of all single nucleotide tails) versus abundance (RPM) for all miRNAs >50 RPM in embryo.
(C and D) Tailing in the embryo and L1.
(E and F) Length distribution (excluding tail) of mir-35 in wild-type embryo and L1 (E) or mir-35 variants in embryo (F).
(A–F) Mean and SEM shown. Wild-type and mutant samples have six and three biological replicates, respectively.
(C–F) For each nucleotide, one-way ANOVA was performed, followed by Sidak’s multiple comparison test. *p < 0.05, **p < 0.01, ****p < 0.0001. For (D), p values are described in the text and are not on the graph for simplicity.
We and others previously observed slight increases in tailed and trimmed miRNAs as miRNAs approach decay (Baccarini et al., 2011; Kingston and Bartel, 2019; Vieux et al., 2021). In TDMD, miRNAs often experience very high levels of tailing and/or trimming (generally ≥20%–40% tailed or trimmed isoforms) (Ameres et al., 2010; Baccarini et al., 2011; Bitetti et al., 2018; Cazalla et al., 2010; Ghini et al., 2018; Kleaveland et al., 2018; Li et al., 2021; Marcinowski et al., 2012). We hypothesized that the prevalence of tailed isoforms might increase at EtoL1 as the mir-35 family members undergo decay. Small increases in miRNA tailing and trimming were observed, but in most cases, these were not statistically significant, and the prevalence of modified isoforms remained modest (Figures 3C and 3E; Table S2).
We next examined tailing in the context of mutant versions of mir-35. Significant changes in tailing were observed, but these did not correlate with changes in rates of decay (Figure 3D; Table S2). For instance, mir-35(mut_3′) was more C- and U-tailed than wild-type mir-35 in embryo and L1, and mir-35(mir-82_3′) was more A-, C-, and U-tailed than wild-type mir-35 in both stages (Figure 3D). However, these two mir-35 variants displayed decay similar to that of wild-type mir-35 at EtoL1 (Figure 2B). In contrast, mir-35(seed_rev) and mir-35(seed_mut) show similar tailing to the wild-type mir-35 (Figure 3D) despite these variants’ dramatically altered decay at EtoL1 (Figure 1C). Oligonucleotide tails were much less frequent than single nucleotide tails, with di-nucleotide tails occurring about 10-fold less than single-nucleotide tails; again, mir-35(seed_rev) showed very similar oligonucleotide tails to wild-type mir-35 (Table S4). Thus, changes in tailing did not correlate with changes in decay.
We next examined trimming of mir-35 variants. Like tailing, trimming varied widely among mir-35 variants but not in a manner that correlated with the rate of decay. For instance, trimming increased most for mir-35(seed_mut) despite the enhanced stability of this variant (Figures 3F and S4B). In contrast, mir-35(seed_rev)—which shows similarly enhanced stability—had no change in trimming (Figures 3F and S4B). This isoform analysis also showed that mir-35(mut_3′) yields two major isoforms from biogenesis, the canonical 22-nt isoform and a 23-nt isoform that is extended by 1 nt at the 3′ end (Figures 3F and S4B). Deep-sequencing data showed that both isoforms are decayed similarly at EtoL1 (Figure S4C). Overall, changes in trimming did not correlate with changes in decay.
All together, these data show that the tailing and trimming of the mir-35 family are much lower than in most known instances of TDMD and that the incidence of trimmed and tailed isoforms across mir-35 variants did not correlate with rate of decay at EtoL1. Together with the dispensability of the 3′ end sequences of mir-35 for decay, this suggests that the mechanism of decay of mir-35 differs from previously described examples of TDMD.
Reintroducing miRNA-target interactions does not restore decay of seed mutant variants of mir-35
To further investigate the mechanism of mir-35 family decay at EtoL1, we examined the involvement of complementary RNA molecules as in TDMD. Decay of mir-35 at EtoL1 is dependent on its seed sequence but not its 3′ end, and canonical targets were previously shown to regulate miRNA stability in C. elegans (Chatterjee et al., 2011). We therefore asked whether canonical miRNA:target interactions might play a role in mediating this decay. The mir-35(seed_rev) variant is predicted to have fewer target molecules in embryos compared with those of wild-type mir-35: the mir-35(seed_rev) target pool is ~59% that of wild type, based on target prediction and relative expression according to RNA sequencing (RNA-seq) (Agarwal et al., 2015; Grün et al., 2014). The lower dose of canonical target interactions may influence decay, or, alternatively, wild-type mir-35 targets may have unknown properties required for decay. We therefore restored canonical target interactions for mir-35(seed_rev) to determine whether this restored developmentally timed decay.
We sought to alter a similar stoichiometric proportion of the pool of mir-35 family miRNAs and the pool of mir-35 family targets. mir-35 makes up 20% of the mir-35–42 miRNA molecules in embryos (Dexheimer et al., 2020), so we selected three target genes that together make up 20% of the target molecules (as estimated from embryo RNA-seq) (Grün et al., 2014). These genes—egl-1, nhl-2, and sup-26—were are all validated targets that influence physiology downstream of mir-35–42 (Kagias and Pocock, 2015; McJunkin and Ambros, 2017; Sherrard et al., 2017; Tran et al., 2019; Wu et al., 2010; Yang et al., 2020b). Using CRISPR, we made mutations to the mir-35 family binding site in the 3′ UTR of these genes. These mutations enable binding by mir-35(seed_rev) rather than wild-type mir-35, and we have previously shown that mir-35(seed_rev) represses such targets (Figure 4A) (Yang et al., 2020b).
Figure 4. Reintroducing miRNA-target interactions does not restore decay of a seed mutant variant of mir-35.
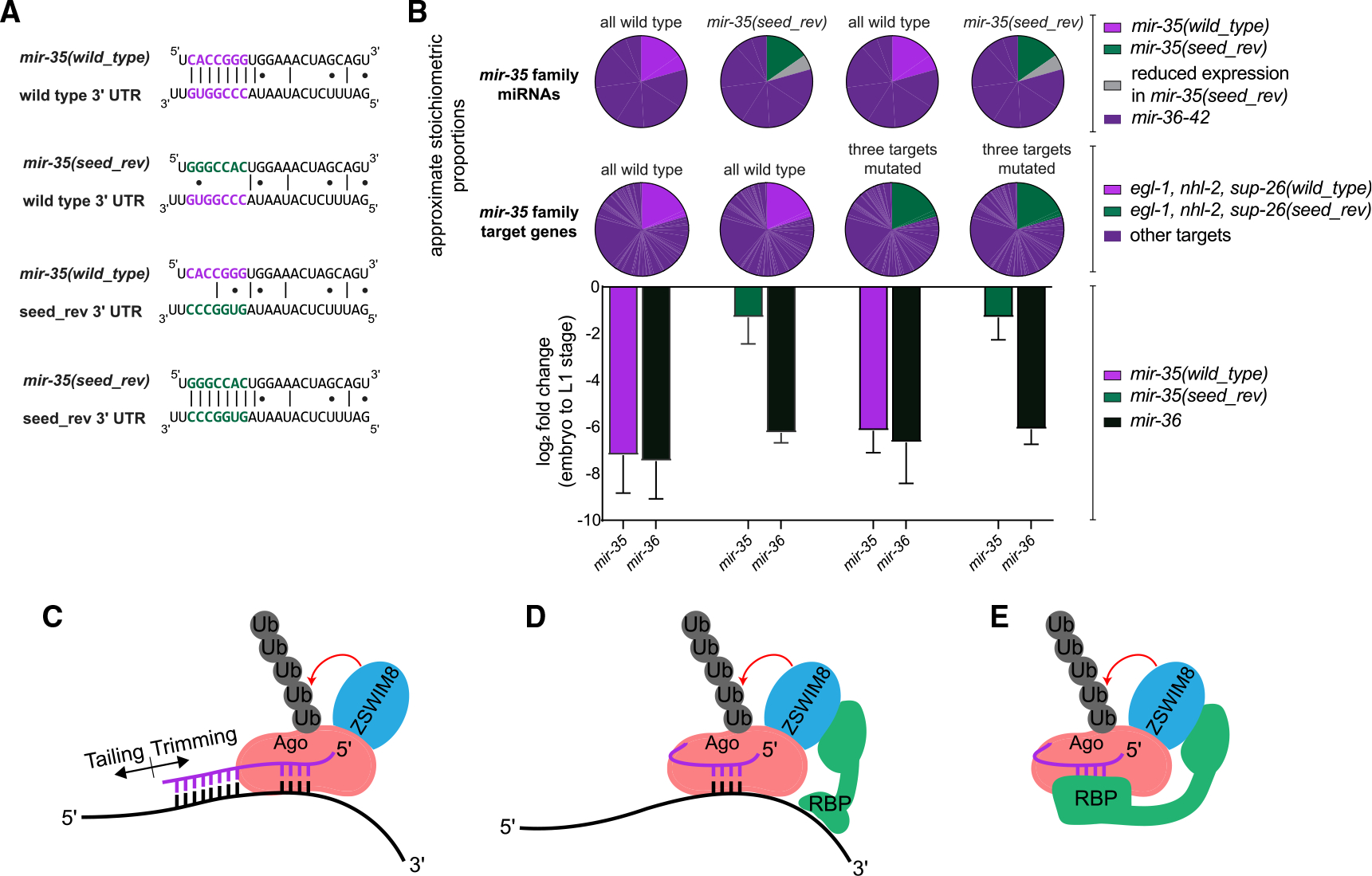
(A) Representative miRNA-target interactions at the egl-1 3′ UTR.
(B) Top: pie charts represent the proportion of the mir-35 miRNA and target molecules that are mutated in each strain. Bottom: log2(fold change) from embryo to L1 in the indicated strains, as measured by Taqman qPCR. Mean and SEM of three biological replicates.
(C) Model of conventional TDMD.
(D and E) Alternative models for regulation of mir-35 family decay, in which the seed sequence is recognized by a complementary RNA (D) or an RNA-binding protein (RBP) (E).
Again, we observed decay of wild-type mir-35 at EtoL1 and attenuated decay of mir-35(seed_rev) by qPCR (Figure 4B). Wild-type mir-35 decay was not affected by the mutations of the target sites (Figure 4B). When mir-35(seed_rev) was combined with the mutant targets containing complementary binding sites, decay was similar to mir-35(seed_rev) without engineered target interactions (Figure 4B). Thus, restoring interactions with canonical target genes is not sufficient to restore decay of the mir-35 seed mutant.
DISCUSSION
Here, we investigate the regulation of the embryonically expressed mir-35 family during development. We show that the decay of these miRNAs at EtoL1 is regulated post-transcriptionally, since mutations in the seed sequence of mir-35 decouple its regulation from that of its clustermates on the same transcript, strongly supporting a selective decay mechanism.
The seed sequence of mir-35 is not only necessary for this selective decay but is also largely sufficient since mutations in the 3′ end of the miRNA do not disrupt decay at EtoL1. This model assumes that the 3′ end mutant variants are decayed by the same mechanism as wild-type mir-35 in this developmental window. The 3′ end regulates mir-35 abundance in the embryo, in what may be a decay-level effect. We postulate that whereas a seed-dependent decay mechanism enacts developmentally timed decay, a 3′ end-dependent mechanism limits mir-35 abundance in the embryo (Figure S3A).
Our data suggest that the mechanisms of mir-35 regulation differ from previously-described instances of TDMD in key aspects (Figures 4C–4E). First, the decay at EtoL1 does not require the 3′ end sequences that would be involved in base-pairing to a typical TDMD trigger RNA. Second, the decay is not accompanied by high levels of tailing or trimming, and seed mutations that reduce decay do not reduce tailing or trimming. Together, these data suggest that the mir-35 family is post-transcriptionally regulated by a novel seed-dependent mechanism, possibly a variant of TDMD.
We propose a model for mir-35 family decay wherein EBAX-1 is recruited in a seed-dependent manner that does not require extensive 3′ end base-pairing. How is the seed recognized, and how is EBAX-1 recruited? Like TDMD, a trigger RNA may base pair with the mir-35 family seed sequence and recruit an RNA-binding protein, which can in turn recruit EBAX-1 (Figure 4D). Alternatively, the trigger RNA could induce conformational changes in Ago that directly recruit EBAX-1. A third possibility is that no trigger RNA is involved in seed recognition for decay; in this case, an RNA-binding protein (RBP) could bind the mir-35 seed to recruit EBAX-1 or induce Ago conformational changes (Figure 4E). Because of the large number of possible trigger RNAs or RBPs, further elucidating this mechanism will require large-scale screens. Better understanding the mechanism of mir-35 family recognition will further test the model of the sufficiency of the seed sequence for this selective regulation.
Understanding the seed-sequence-specific decay mechanism regulating mir-35 will have broad impact. Such seed-specific mechanisms are likely to be present in other biological systems because they allow for simultaneous regulation of redundant miRNA paralogs, enabling dynamic regulation of a miRNA seed family’s targets. Outside functioning in normal physiology, seed-specific decay mechanisms could be an attractive avenue for modulating abundance of specific miRNA families and their target genes in disease.
Limitations of the study
Only mir-35 was modified; whether the strength of the impact of these mutations is similar in the context of mir-36–42 will be a matter of future investigation. Having not examined processing intermediates, we have as yet an incomplete understanding of how the mutations introduced in mir-35 affect its biogenesis. We have not directly measured the decay of mir-35 3′ end variants in the embryo stage, leaving ambiguity as to the molecular basis of upregulation of mir-35 3′ end mutant variants in the embryo. Furthermore, the decay of the 3′ end mutant variants of mir-35 at EtoL1 occurs at a similar rate as wild type; our interpretation is that these variants are subject to the same mechanism that targets wild-type mir-35 at EtoL1. However, if the 3′ end variants are targeted by a distinct mechanism of decay in the same developmental time window, this could obscure the 3′ end’s role in the regulation of wild-type mir-35.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Katherine McJunkin (mcjunkin@nih.gov).
Materials availability
Key C. elegans strains generated in this study have been deposited at the Caenorhabditis Genetics Center (CGC). Others are deposited in the McJunkin Lab strain collection and are available from the lead contact upon request.
Data and code availability
All raw sequence data have been deposited in NCBI Sequence Read Archive (SRA) under accession number SRA: PRJNA782102. Analyzed data appear in Tables S2–S4.
This paper does not report original code.
Any additional information required to analyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
The C. elegans strains generated and used in this study are found in Table S1 and the key resources table. Allele information is found in Table S2.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Bacterial and virus strains | ||
|
| ||
| E. coli OP50 | CGC | https://cgc.umn.edu/strain/OP50 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Sodium Hypochlorite Solution (5.65–6%/Laboratory) | Fisher Scientific | Cat#SS290-1 |
| Sodium Hydroxide | Fisher Scientific | Cat#AC206060010 |
| Cholesterol | Fisher Scientific | Cat#AAA1147018 |
| Alt-R® S.p. Cas9 Nuclease V3, 100 μg | IDT | Cat#1081058 |
| Nuclease Free Duplex Buffer | IDT | Cat#11-01-03-01 |
| Alt-R® CRISPR-Cas9 tracrRNA, 5 nmol | IDT | Cat#1072532 |
| Edit-R CRISPR-Cas9 Synthetic tracrRNA | Horizon Discovery | Cat#U-002005-20 |
| RNAse H | NEB | Cat#M0297L |
| TRIzol™ LS Reagent | ThermoFisher | CAT#10296010 |
| TRIzol Reagent | ThermoFisher | Cat#15596018 |
| TaqMan™ Universal Master Mix II, with UNG | ThermoFisher | Cat#4440038 |
|
| ||
| Critical commercial assays | ||
|
| ||
| TaqMan™ MicroRNA Reverse Transcription Kit | ThermoFisher | Cat#4366596 |
| NEBNext®Multiplex Small RNA Library Prep Kit for Illumina® | NEB | Cat#E7560S |
| TaqMan™ MicroRNA Assay | ThermoFisher | Cat#4440886 |
|
| ||
| Deposited data | ||
|
| ||
| Raw sequence data | This study | Deposited in NCBI Sequence Read Archive (SRA) under accession number SRA:PRJNA782102. |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| See Table S2 for oligonucleotides. | N/A | N/A |
| C. elegans:mir-35(seed_rev): mir-35(cdb2 cdb4) II | Yang et al. (2020b) Genes and Development | MCJ11 |
| C. elegans:mir-35(seed_mut): mir-35(cdb2 cdb6) II | This paper | MCJ13 |
| C. elegans:mir-35(mut_3′): mir-35(cdb2 cdb72) II | This paper | MCJ180 |
| C. elegans:mir-35(mir-82_3′): mir-35(cdb2 cdb78) II | This paper | MCJ191 |
| C. elegans: mir-35(seed_rev_no_bulge): mir-35(cdb2 cdb95) II | This paper | MCJ211 |
| C. elegans: mir-35(seed_rev); egl-1, nhl-2, sup-26(seed_rev): mir-35(cdb2 cdb4) II; sup-26(cdb99) nhl-2(cdb100) III; egl-1(cdb97) V | This paper | MCJ218 |
| C. elegans: mir-35(wild_type); egl-1, nhl-2, sup-26(seed_rev): sup-26(cdb99) nhl-2(cdb100) III; egl-1(cdb97) V | This paper | MCJ219 |
| C. elegans: ebax-1(null): ebax-1(tm2321) IV | Wang et al. (2013) Neuron | CZ9907 |
| See Table S2 for allele information. | N/A | N/A |
|
| ||
| Oligonucleotides | ||
|
| ||
| See Table S2 for oligonucleotides. | N/A | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| SnapGene 5 | SnapGene | www.snapgene.com |
| Adobe Illustrator 26 | Adobe Inc. | https://www.adobe.com/products/illustrator.html |
| Prism 9 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| Cutadapt 3.4 | Martin (2011) | https://cutadapt.readthedocs.io/en/stable/ |
| bowtie2 2.4.4 | Langmead et al. (2009) | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| samtools 1.13 | Danecek et al. (2021) | https://sourceforge.net/projects/samtools/files/samtools/ |
| htseq 0.13.5 | Anders et al. (2015) | https://pypi.python.org/pypi/HTSeq |
| Tailor Package | Chou et al. (2015) | https://github.com/jhhung/Tailor |
| TargetScan 7.0 | Agarwal et al. (2015) | https://www.targetscan.org/vert_70/ |
| DESeq2 | Love et al. (2014) | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
METHOD DETAILS
General C. elegans culture and maintenance
C. elegans were maintained at 20°C on NGM seeded with OP50. For large scale harvest of embryos, 8,000 starved L1s were plated onto a 10cm plate with a large lawn of OP50. The worms were re-fed with concentrated OP50 48 h later. At 96 h after initial plating, the gravid adults were harvested by bleaching to collect large quantities of embryos.
Liquid culture
For experiments in which bulk sample deep sequencing was performed (Figures 1 and 2), worms were grown in liquid culture as previously described and harvested with some modifications (Zanin et al., 2011). Briefly, the gravid worms were harvested by centrifugation at 3000×g for 2 min in 50mL conical tubes. They were washed once with room temperature water and then pelleted. The volume of the sample was brought up to 28mL with water, and then 4mL of 5M NaOH and 8mL of 4% sodium hypochlorite were added. The tubes were immediately shaken vigorously for 2 min and allowed to rest on the bench for 1 min, and this shaking and resting was repeated three times. The worms were immediately centrifuged at 3000×g for 2 min. The supernatant was decanted, and the worms were washed four times with 45mL of water. The synchronized embryos were either collected for the embryo samples, or M9 with cholesterol was added, and the worms were placed on a rocker at 20°C overnight to obtain a population of synchronized, starved L1 worms.
CRISPR/Cas9-mediated genome editing
For all CRISPR experiments, pre-assembled Cas9 RNPs were injected into germlines along with short homology-directed repair templates with ~35-nt homology arms (Paix et al., 2014). For all CRISPR injections, one of the guide RNAs used targeted dpy-10 as a visible marker to select plates with efficient genome editing (Arribere et al., 2014). crRNAs and tracrRNA were ordered from IDT (Alt-R) or Dharmacon (Edit-R), and annealed at 10μM in IDT duplex buffer by heating to 95°C for 5 min and then cooling to room temperature. Injection mixes contained 2–4μM Cas9, 4μM total of pre-annealed gRNAs (comprised of gRNAs targeting dpy-10 and the site of interest), 0.8μM of the dpy-10 donor oligonucleotide, and the homology directed donor at 40–100ng/μL (Table S1).
Mutations to mir-35 were made by two rounds of CRISPR. First, as previously described (Yang et al., 2020b), two gRNAs recognizing the protospacers TTTCCATTAGAACTATCACC and ATTGCTGGTTTCTTCCACAG were used to create a 50bp deletion at the mir-35 locus. This allele is mir-35(cdb2):
GCTGGTTTCTTCCACAGT-50bp_del-CTTTTCCACTTGCTCCAC. The strain carrying mir-35(cdb2) was then injected with homology-directed repair donors, along with a gRNA (GGAGCAAGTGGAAAAGACTG) recognizing a sequence which is created by the mir35(cdb2) mutation. See Table S1 for allele and strain details and Table S1 for all donor oligonucleotides.
Deep sequencing library preparation and data analysis
Library preparation was performed using the NEBNext Small RNA Library Prep Set for Illumina with modifications as previously described (Vieux et al., 2021). Briefly, size selection was performed only after reverse transcription, using 8% urea gels to purify ~65–75nt RT products. Prior to loading on the gel, each RT reaction was treated with 5000units of RNAse H (New England Biolabs) for 30 min at 37°C. For bulk embryo and L1 samples, 15 PCR cycles were performed. For samples of 20 staged embryos or 20 corresponding L1s, 15–20 PCR cycles were performed. Sequence analysis was performed on the NIH High Performance Computing Cluster. The 3′ adapter sequence was trimmed using Cutadapt 3.4 (Martin, 2011). The reads were mapped to a custom genome file which was comprised of C. elegans genome WS280 with an additional chromosome containing the sequences of the spike-in miRNAs and the mutant mir-35 precursors with flanking genomic sequence. Mapping was performed using bowtie2 2.4.4 (Langmead et al., 2009) with the following settings: –no-unal –end-to-end –sensitive. BAM files were sorted and indexed using samtools 1.13 (Danecek et al., 2021). Reads were assigned to miRNAs using htseq 0.13.5 (Anders et al., 2015) with the following settings: –mode union –nonunique fraction -a 0. The htseq analysis was performed using a gff file modified from mirGeneDB (Fromm et al., 2015) by replacing mirGeneDB IDs with miRbase IDs (Kozomara and Griffiths-Jones, 2014) and adding the intervals corresponding to the spike-in miRNAs and the mir-35 mutant miRNAs in the custom genome file. miRNA differential gene expression was analyzed using DESeq2 analysis with default settings (Love et al., 2014). For analysis of tailing and trimming, the Tailor package (Chou et al., 2015) was used with the genome file described above and FASTA files derived from mirGeneDB, but with IDs replaced by miRbase IDs and sequences for spike-in miRNAs and the mir-35 mutant miRNAs appended.
To assess the stoichiometry of the potential targets of mir-35(seed_rev), TargetScan 7.0 was used to predict binding sites for mir-35(seed_rev) (Agarwal et al., 2015). Expression data from (Grün et al., 2014) was used to infer relative expression of predicted target genes.
RNA isolation
Total RNA was isolated from bulk samples by resuspending the sample in the recommended volume of Trizol reagent (Life Technologies), followed by vortexing at room temperature for 15 min, followed by preparation according to the Trizol manufacturer’s instructions. After preparation, ten spike-in oligos (see Table S1) were added at a final concentration of 1pg/μL each in 100ng/μL total RNA prior to deep sequencing library preparation.
For staged embryo samples, 20 staged embryos or 20 L1s (synchronized by starvation for 24h) were collected by hand. Samples were snap frozen in Trizol LS reagent. Prior to purification, 0.9pg of each spike-in oligo (Table S1) was added to each sample. Trizol LS-resuspended samples were subjected to three freeze-thaw cycles to promote lysis and then vortexed for 15 min at room temperature prior to purification according to the Trizol LS manufacturer’s instructions.
Taqman miRNA qPCR
For all miRNA qPCR, 5μL reverse transcription reactions were performed using the TaqMan MicroRNA Reverse Transcription kit (ThermoFisher). For all samples, 1.66μL of total RNA at 6ng/μL was used in the reverse transcription. RT reactions were diluted 1:4 and 1.33μL was used in a 5μL qPCR reaction prepared using Taqman miRNA probes with the Taqman Universal Mastermix II with UNG (ThermoFisher). Reactions were run in triplicate on the Applied Biosystems QuantStudio Pro 6.
QUANTIFICATION AND STATISTICAL ANALYSIS
Quantification and statistical analysis can be found in figure legends.
Supplementary Material
Highlights.
The mir-35 family undergoes selective decay at the end of C. elegans embryogenesis
The seed sequence of mir-35 is necessary and largely sufficient for mir-35 decay
The sequence 3′ of the seed limits mir-35 abundance in the embryo
The TDMD factor EBAX-1 contributes to multiple phases of mir-35 regulation
ACKNOWLEDGMENTS
This work was funded by the NIDDK Intramural Research Program (ZIA DK075147). We thank WormBase, the NIDDK Genomics Core, the NCI Genomics Core, NIH High Performance Computing, and the CGC. The CGC is funded by the NIH Office of Research Infrastructure Programs (P40 OD010440). Thank you to Yishi Jin for the ebax-1(null) strain. Thank you to members of the McJunkin lab, Eric Miska, Kenneth Murfitt, Michael Lichten, Joana Vidigal, John Kim, and Leemor Joshua-Tor for helpful discussions.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
INCLUSION AND DIVERSITY
One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in science. One or more of the authors of this paper self-identifies as a member of the LGBTQ+ community.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.111154.
REFERENCES
- Agarwal V, Bell GW, Nam JW, and Bartel DP (2015). Predicting effective microRNA target sites in mammalian mRNAs. Elife 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez-Saavedra E, and Horvitz HR (2010). Many families of C. elegans microRNAs are not essential for development or viability. Curr. Biol. 20, 367–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ameres SL, Horwich MD, Hung J-H, Xu J, Ghildiyal M, Weng Z, and Zamore PD (2010). Target RNA-directed trimming and tailing of small silencing RNAs. Science 328, 1534–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Pyl PT, and Huber W (2015). HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arribere JA, Bell RT, Fu BXH, Artiles KL, Hartman PS, and Fire AZ (2014). Efficient marker-free recovery of custom genetic modifications with CRISPR/Cas9 in Caenorhabditis elegans. Genetics 198, 837–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baccarini A, Chauhan H, Gardner TJ, Jayaprakash AD, Sachidanandam R, and Brown BD (2011). Kinetic analysis reveals the fate of a microRNA following target regulation in mammalian cells. Curr. Biol. 21, 369–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bail S, Swerdel M, Liu H, Jiao X, Goff LA, Hart RP, and Kiledjian M (2010). Differential regulation of microRNA stability. RNA 16, 1032–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein E, Caudy AA, Hammond SM, and Hannon GJ (2001). Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature 409, 363–366. [DOI] [PubMed] [Google Scholar]
- Bitetti A, Mallory AC, Golini E, Carrieri C, Carreño Gutiérrez H, Perlas E, Pérez-Rico YA, Tocchini-Valentini GP, Enright AJ, Norton WHJ, et al. (2018). MicroRNA degradation by a conserved target RNA regulates animal behavior. Nat. Struct. Mol. Biol. 25, 244–251. [DOI] [PubMed] [Google Scholar]
- Boele J, Persson H, Shin JW, Ishizu Y, Newie IS, Søkilde R, Hawkins SM, Coarfa C, Ikeda K, Takayama K -i., et al. (2014). PAPD5-mediated 3’ adenylation and subsequent degradation of miR-21 is disrupted in proliferative disease. Proc. Natl. Acad. Sci. USA 111, 11467–11472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bossé GD, Rüegger S, Ow MC, Vasquez-Rifo A, Rondeau EL, Ambros VR, Grosshans H, and Simard MJ (2013). The decapping scavenger enzyme DCS-1 controls microRNA levels in Caenorhabditis elegans. Mol. Cell 50, 281–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brancati G, and Großhans H (2018). An interplay of miRNA abundance and target site architecture determines miRNA activity and specificity. Nucleic Acids Res. 46, 3259–3269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennecke J, Stark A, Russell RB, and Cohen SM (2005). Principles of microRNA-target recognition. PLoS Biol. 3, e85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broughton JP, Lovci MT, Huang JL, Yeo GW, and Pasquinelli AE (2016). Pairing beyond the seed supports MicroRNA targeting specificity. Mol. Cell 64, 320–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cazalla D, Yario T, Steitz JA, and Steitz J (2010). Down-regulation of a host microRNA by a Herpesvirus saimiri noncoding RNA. Science 328, 1563–1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee S, Fasler M, Büssing I, and Grosshans H (2011). Target-mediated protection of endogenous microRNAs in C. elegans. Dev. Cell 20, 388–396. [DOI] [PubMed] [Google Scholar]
- Chatterjee S, Grosshans H, and Großhans H (2009). Active turnover modulates mature microRNA activity in Caenorhabditis elegans. Nature 461, 546–549. [DOI] [PubMed] [Google Scholar]
- Chou M. Te, Han BW, Hsiao CP, Zamore PD, Weng Z, and Hung JH (2015). Tailor: a computational framework for detecting non-templated tailing of small silencing RNAs. Nucleic Acids Res. 43, e109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dallaire A, Frédérick PM, and Simard MJ (2018). Somatic and germline MicroRNAs form distinct silencing complexes to regulate their target mRNAs differently. Dev. Cell 47, 239–247.e4. [DOI] [PubMed] [Google Scholar]
- Danecek P, Bonfield JK, Liddle J, Marshall J, Ohan V, Pollard MO, Whitwham A, Keane T, McCarthy SA, Davies RM, and Li H (2021). Twelve years of SAMtools and BCFtools. GigaScience 10, giab008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denli AM, Tops BBJ, Plasterk RHA, Ketting RF, and Hannon GJ (2004). Processing of primary microRNAs by the Microprocessor complex. Nature 432, 231–235. [DOI] [PubMed] [Google Scholar]
- Dexheimer PJ, and Cochella L (2020). MicroRNAs: from mechanism to organism. Front. Cell Dev. Biol. 8, 409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dexheimer PJ, Wang J, and Cochella L (2020). Two microRNAs are sufficient for embryogenesis in C. elegans. Preprint at bioRxiv. 10.1101/2020.06.28.176024. [DOI] [Google Scholar]
- Doll MA, Soltanmohammadi N, and Schumacher B (2019). ALG-2/AGO-Dependent mir-35 family regulates DNA damage-induced apoptosis through MPK-1/ERK MAPK signaling downstream of the core apoptotic machinery in Caenorhabditis elegans. Genetics 213, 173–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbarbary RA, Miyoshi K, Hedaya O, Myers JR, and Maquat LE (2017b). UPF1 helicase promotes TSN-mediated miRNA decay. Genes Dev. 31, 1483–1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbarbary RA, Miyoshi K, Myers JR, Du P, Ashton JM, Tian B, and Maquat LE (2017a). Tudor-SN-mediated endonucleolytic decay of human cell microRNAs promotes G1/S phase transition. Science 356, 859–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang W, and Bartel DP (2015). The menu of features that define primary MicroRNAs and enable de novo design of MicroRNA genes. Mol. Cell 60, 131–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flamand MN, Gan HH, Mayya VK, Gunsalus KC, and Duchaine TF (2017). A non-canonical site reveals the cooperative mechanisms of microRNA-mediated silencing. Nucleic Acids Res. 45, 7212–7225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flamand MN, Wu E, Vashisht A, Jannot G, Keiper BD, Simard MJ, Wohlschlegel J, and Duchaine TF (2016). Poly(A)-binding proteins are required for microRNA-mediated silencing and to promote target deadenylation in C. elegans. Nucleic Acids Res. 44, 5924–5935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fromm B, Billipp T, Peck LE, Johansen M, Tarver JE, King BL, Newcomb JM, Sempere LF, Flatmark K, Hovig E, et al. (2015). A uniform system for the annotation of vertebrate microRNA genes and the evolution of the human microRNAome. Annu. Rev. Genet. 49, 213–242. 10.1146/Annurev-Genet-120213-092023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghini F, Rubolino C, Climent M, Simeone I, Marzi MJ, and Nicassio F (2018). Endogenous transcripts control miRNA levels and activity in mammalian cells by target-directed miRNA degradation. Nat. Commun. 9, 3119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory RI, Yan K-P, Amuthan G, Chendrimada T, Doratotaj B, Cooch N, and Shiekhattar R (2004). The Microprocessor complex mediates the genesis of microRNAs. Nature 432, 235–240. [DOI] [PubMed] [Google Scholar]
- Grishok A, Pasquinelli AE, Conte D, Li N, Parrish S, Ha I, Baillie DL, Fire A, Ruvkun G, and Mello CC (2001). Genes and mechanisms related to RNA interference regulate expression of the small temporal RNAs that control C. elegans developmental timing. Cell 106, 23–34. [DOI] [PubMed] [Google Scholar]
- Grün D, Kirchner M, Thierfelder N, Stoeckius M, Selbach M, and Rajewsky N (2014). Conservation of mRNA and protein expression during development of C. elegans. Cell Rep. 6, 565–577. [DOI] [PubMed] [Google Scholar]
- Han J, LaVigne CA, Jones BT, Zhang H, Gillett F, and Mendell JT (2020). A ubiquitin ligase mediates target-directed microRNA decay independently of tailing and trimming. Science 370, eabc9546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J, Lee Y, Yeom K-H, Kim Y-K, Jin H, and Kim VN (2004). The Drosha-DGCR8 complex in primary microRNA processing. Genes Dev. 18, 3016–3027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J, Lee Y, Yeom K-H, Nam J-W, Heo I, Rhee J-K, Sohn SY, Cho Y, Zhang B-T, and Kim VN (2006). Molecular basis for the recognition of primary microRNAs by the Drosha-DGCR8 complex. Cell 125, 887–901. [DOI] [PubMed] [Google Scholar]
- Helwak A, Kudla G, Dudnakova T, and Tollervey D (2013). Mapping the human miRNA interactome by CLASH reveals frequent noncanonical binding. Cell 153, 654–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutvágner G, McLachlan J, Pasquinelli AE, Bálint E, Tuschl T, and Zamore PD (2001). A cellular function for the RNA-interference enzyme Dicer in the maturation of the let-7 small temporal RNA. Science 293, 834–838. [DOI] [PubMed] [Google Scholar]
- Duan Y, Veksler-Lublinsky I, and Ambros V (2022). Critical contribution of 3’ non-seed base pairing to the in vivo function of the evolutionarily conserved let-7a microRNA. Cell Rep. 39, 110745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki S, Kobayashi M, Yoda M, Sakaguchi Y, Katsuma S, Suzuki T, and Tomari Y (2010). Hsc70/Hsp90 chaperone machinery mediates ATP-dependent RISC loading of small RNA duplexes. Mol. Cell 39, 292–299. [DOI] [PubMed] [Google Scholar]
- Iwasaki S, Sasaki HM, Sakaguchi Y, Suzuki T, Tadakuma H, and Tomari Y (2015). Defining fundamental steps in the assembly of the Drosophila RNAi enzyme complex. Nature 521, 533–536. [DOI] [PubMed] [Google Scholar]
- Kagias K, and Pocock R (2015). microRNA regulation of the embryonic hypoxic response in Caenorhabditis elegans. Sci. Rep. 5, 11284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato M, de Lencastre A, Pincus Z, and Slack FJ (2009). Dynamic expression of small non-coding RNAs, including novel microRNAs and piRNAs/21U-RNAs, during Caenorhabditis elegans development. Genome Biol. 10, R54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh T, Hojo H, and Suzuki T (2015). Destabilization of microRNAs in human cells by 3′ deadenylation mediated by PARN and CUGBP1. Nucleic Acids Res. 43, 7521–7534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ketting RF, Fischer SE, Bernstein E, Sijen T, Hannon GJ, and Plasterk RH (2001). Dicer functions in RNA interference and in synthesis of small RNA involved in developmental timing in C. elegans. Genes Dev. 15, 2654–2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingston ER, and Bartel DP (2019). Global analyses of the dynamics of mammalian microRNA metabolism. Genome Res. 29, 1777–1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleaveland B, Shi CY, Stefano J, and Bartel DP (2018). A network of noncoding regulatory RNAs acts in the mammalian brain. Cell 174, 350–362.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight SW, and Bass BL (2001). A role for the RNase III enzyme DCR-1 in RNA interference and germ line development in Caenorhabditis elegans. Science 293, 2269–2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knouf EC, Wyman SK, and Tewari M (2013). The human TUT1 nucleotidyl transferase as a global regulator of microRNA abundance. PLoS One 8, e69630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozomara A, and Griffiths-Jones S (2014). MiRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 42, D68–D73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- la Mata M, Gaidatzis D, Vitanescu M, Stadler MB, Wentzel C, Scheiffele P, Filipowicz W, and Großhans H (2015). Potent degradation of neuronal mi RNA s induced by highly complementary targets. EMBO Rep. 16, 500–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landthaler M, Yalcin A, and Tuschl T (2004). The human DiGeorge syndrome critical region gene 8 and its D. melanogaster homolog are required for miRNA biogenesis. Curr. Biol. 14, 2162–2167. [DOI] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, and Salzberg SL (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee D, Park D, Park JH, Kim JH, and Shin C (2019). Poly(A)-specific ribonuclease sculpts the 3′ ends of microRNAs. RNA 25, 388–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M, Choi Y, Kim K, Jin H, Lim J, Nguyen TA, Yang J, Jeong M, Giraldez AJ, Yang H, et al. (2014). Adenylation of maternally inherited microRNAs by Wispy. Mol. Cell 56, 696–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehrbach NJ, Castro C, Murfitt KJ, Abreu-Goodger C, Griffin JL, and Miska EA (2012). Post-developmental microRNA expression is required for normal physiology, and regulates aging in parallel to insulin/IGF-1 signaling in C. elegans. RNA 18, 2220–2235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis BP, Shih I.h., Jones-Rhoades MW, Bartel DP, and Burge CB (2003). Prediction of mammalian microRNA targets. Cell 115, 787–798. [DOI] [PubMed] [Google Scholar]
- Li L, Sheng P, Li T, Fields CJ, Hiers NM, Wang Y, Li J, Guardia CM, Licht JD, and Xie M (2021). Widespread microRNA degradation elements in target mRNAs can assist the encoded proteins. Genes Dev. 35, 1595–1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libri V, Helwak A, Miesen P, Santhakumar D, Borger JG, Kudla G, Grey F, Tollervey D, and Buck AH (2012). Murine cytomegalovirus encodes a miR-27 inhibitor disguised as a target. Proc. Natl. Acad. Sci. USA 109, 279–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Liu P, Zhang L, Cai Q, Gao G, Zhang W, Zhu Z, Liu D, and Fan Q (2011). mir-35 is involved in intestine cell G1/S transition and germ cell proliferation in C. elegans. Cell Res. 21, 1605–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma H, Wu Y, Choi J-G, and Wu H (2013). Lower and upper stem-single-stranded RNA junctions together determine the Drosha cleavage site. Proc. Natl. Acad. Sci. USA 110, 20687–20692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcinowski L, Tanguy M, Krmpotic A, Rädle B, Lisnić VJ, Tuddenham L, Chane-Woon-Ming B, Ruzsics Z, Erhard F, Benkartek C, et al. (2012). Degradation of cellular mir-27 by a novel, highly abundant viral transcript is important for efficient virus replication in vivo. PLoS Pathog. 8, e1002510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 17, 10–12. [Google Scholar]
- Marzi MJ, Ghini F, Cerruti B, De Pretis S, Bonetti P, Giacomelli C, Gorski MM, Kress T, Pelizzola M, Muller H, et al. (2016). Degradation dynamics of micrornas revealed by a novel pulse-chase approach. Genome Res. 26, 554–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massirer KB, Perez SG, Mondol V, and Pasquinelli AE (2012). The miR-35–41 family of microRNAs regulates RNAi sensitivity in Caenorhabditis elegans. PLoS Genet. 8, e1002536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McJunkin K, and Ambros V (2014). The embryonic mir-35 family of microRNAs promotes multiple aspects of fecundity in Caenorhabditis elegans. G3 (Bethesda) 4, 1747–1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McJunkin K, and Ambros V (2017). A microRNA family exerts maternal control on sex determination in C. elegans. Genes Dev. 31, 422–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miki TS, Rüegger S, Gaidatzis D, Stadler MB, and Großhans H (2014). Engineering of a conditional allele reveals multiple roles of XRN2 in Caenorhabditis elegans development and substrate specificity in microRNA turnover. Nucleic Acids Res. 42, 4056–4067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paix A, Wang Y, Smith HE, Lee CYS, Calidas D, Lu T, Smith J, Schmidt H, Krause MW, and Seydoux G (2014). Scalable and versatile genome editing using linear DNAs with microhomology to Cas9 sites in Caenorhabditis elegans. Genetics 198, 1347–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parchem RJ, Moore N, Fish JL, Parchem JG, Braga TT, Shenoy A, Oldham MC, Rubenstein JLR, Schneider RA, and Blelloch R (2015). miR-302 is required for timing of neural differentiation, neural tube closure, and embryonic viability. Cell Rep. 12, 760–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piwecka M, Glažar P, Hernandez-Miranda LR, Memczak S, Wolf SA, Rybak-Wolf A, Filipchyk A, Klironomos F, Cerda Jara CA, Fenske P, et al. (2017). Loss of a mammalian circular RNA locus causes miRNA deregulation and affects brain function. Science 357, eaam8526. [DOI] [PubMed] [Google Scholar]
- Reichholf B, Herzog VA, Fasching N, Manzenreither RA, Sowemimo I, and Ameres SL (2019). Time-resolved small RNA sequencing unravels the molecular principles of MicroRNA homeostasis. Mol. Cell 75, 756–768.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rissland OS, Hong S-J, and Bartel DP (2011). MicroRNA destabilization enables dynamic regulation of the miR-16 family in response to cell-cycle changes. Mol. Cell 43, 993–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherrard R, Luehr S, Holzkamp H, McJunkin K, Memar N, and Conradt B (2017). Mirnas cooperate in apoptosis regulation during c. Genes Dev. 31, 209–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheu-Gruttadauria J, Pawlica P, Klum SM, Wang S, Yario TA, Schirle Oakdale NT, Steitz JA, and MacRae IJ (2019). Structural basis for target-directed MicroRNA degradation. Mol. Cell 75, 1243–1255.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi CY, Kingston ER, Kleaveland B, Lin DH, Stubna MW, and Bartel DP (2020). The ZSWIM8 ubiquitin ligase mediates target-directed microRNA degradation. Science 370, eabc9359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla S, Bjerke GA, Muhlrad D, Yi R, and Parker R (2019). The RNase PARN controls the levels of specific miRNAs that contribute to p53 regulation. Mol. Cell 73, 1204–1216.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoeckius M, Maaskola J, Colombo T, Rahn H-P, Friedländer MR, Li N, Chen W, Piano F, and Rajewsky N (2009). Large-scale sorting of C. elegans embryos reveals the dynamics of small RNA expression. Nat. Methods 6, 745–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran AT, Chapman EM, Flamand MN, Yu B, Krempel SJ, Duchaine TF, Eroglu M, and Derry WB (2019). MiR-35 buffers apoptosis thresholds in the C. elegans germline by antagonizing both MAPK and core apoptosis pathways. Cell Death Differ. 26, 2637–2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieux K-F, Prothro KP, Kelley LH, Palmer C, Maine EM, Veksler-Lublinsky I, and McJunkin K (2021). Screening by deep sequencing reveals mediators of microRNA tailing in C. elegans. Nucleic Acids Res. 49, 11167–11180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Hou Y, Guo X, vanderVoet M, Boxem M, Dixon JE, Chisholm AD, and Jin Y (2013). The EBAX-type cullin-RING E3 ligase and Hsp90 guard the protein quality of the SAX-3/robo receptor in developing neurons. Neuron 79, 903–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu E, Thivierge C, Flamand M, Mathonnet G, Vashisht AA, Wohlschlegel J, Fabian MR, Sonenberg N, and Duchaine TF (2010). Pervasive and cooperative deadenylation of 3’UTRs by embryonic microRNA families. Mol. Cell 40, 558–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyman SK, Knouf EC, Parkin RK, Fritz BR, Lin DW, Dennis LM, Krouse MA, Webster PJ, and Tewari M (2011). Post-transcriptional generation of miRNA variants by multiple nucleotidyl transferases contributes to miRNA transcriptome complexity. Genome Res. 21, 1450–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang A, Shao T-J, Bofill-De Ros X, Lian C, Villanueva P, Dai L, and Gu S (2020a). AGO-bound mature miRNAs are oligouridylated by TUTs and subsequently degraded by DIS3L2. Nat. Commun. 11, 2765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang B, Schwartz M, and McJunkin K (2020b). In vivo CRISPR screening for phenotypic targets of the mir-35–42 family in C. elegans. Genes Dev. 34, 1227–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanin E, Dumont J, Gassmann R, Cheeseman I, Maddox P, Bahmanyar S, Carvalho A, Niessen S, Yates JR, Oegema K, and Desai A (2011). Affinity purification of protein complexes in C. elegans. Methods Cell Biol. 106, 289–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng Y, Yi R, and Cullen BR (2005). Recognition and cleavage of primary microRNA precursors by the nuclear processing enzyme Drosha. EMBO J. 24, 138–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Jin L, Wang Y, Kong Y, and Wang D (2019). Prolonged exposure to multi-walled carbon nanotubes dysregulates intestinal mir-35 and its direct target MAB-3 in nematode Caenorhabditis elegans. Sci. Rep. 9, 12144. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All raw sequence data have been deposited in NCBI Sequence Read Archive (SRA) under accession number SRA: PRJNA782102. Analyzed data appear in Tables S2–S4.
This paper does not report original code.
Any additional information required to analyze the data reported in this paper is available from the lead contact upon request.