

Abstract
Background
Posterior reversible encephalopathy syndrome (PRES) is a disease characterised by reversible subcortical vasogenic oedema, neurological symptoms and abnormal findings on head imaging. It is recognised as one of the most prominent organ disorders in hypertensive emergencies but is rarely associated with thrombotic microangiopathy (TMA).
Case presentation
A woman in her 40s with untreated hypertension had occasional headaches in the past 4 months. The headaches worsened during the 3 weeks prior to admission. On the day of admission, the patient presented with severe headache accompanied by frequent vomiting. MRI of the head revealed oedematous changes in the brainstem, including the subcortical, cerebellum and pons. Fundus examination revealed hypertensive retinopathy with papilloedema. Blood tests indicated thrombocytopenia, renal dysfunction and haemolytic anaemia, and a blood smear confirmed fragmented erythrocytes. Coombs’ test, and tests for ADAMTS13 activity and infectious and autoimmune diseases were negative. The patient was diagnosed with PRES, secondary to malignant hypertension (MH) and associated with TMA. Antihypertensive therapy promptly improved the clinical symptoms, blood pressure, and the abnormal MRI and blood test findings. The patient was discharged from the hospital 20 days after admission.
Conclusions
We report a rare case of PRES that was associated with TMA and triggered by MH. Antihypertensive therapy was effective in alleviating the associated adverse clinical symptoms. Differentiation of underlying diseases is essential for early intervention, since treatment depends on factors causing TMA.
Keywords: cerebral blood flow, hypertensive encepha, haematology, thrombophilia, headache
Background
Posterior reversible encephalopathy syndrome (PRES), so-called reversible posterior leucoencephalopathy syndrome, is a disease predominantly but not exclusively affecting the posterior part of the brain (occipital lobe, parietal lobe and cerebellum), and it also affects some areas including frontal and temporal lobes. PRES is characterised by subcortical-dominant vasogenic oedema and reversible neurological symptoms such as headache, altered consciousness, visual disturbances, including cortical blindness, and seizures.1 PRES may be caused by rapid increases in blood pressure; renal insufficiency; use of immunosuppressive, cytotoxic or anticancer drugs; sepsis; autoimmune diseases; eclampsia and pre-eclampsia. In juvenile cases, PRES may also result from haematological and renal diseases such as Henoch-Schönlein purpura, glomerulonephritis and haemolytic uraemic syndrome.2 3
The precise pathophysiology of PRES remains unknown; however, cerebral endothelial damage and disruption of the blood–brain barrier (BBB) are known to cause vasogenic oedema.4 Cerebral endothelial cells can be damaged by cytotoxic substances including toxins, immunosuppressive or anticancer drugs such as antivascular endothelial growth factor drugs, and by underlying diseases such as autoimmune collagen disease and eclampsia.5 Since many patients with PRES have hypertension, it is possible that hypertension itself promotes damage to vascular endothelial cells. However, PRES also occurs in patients with normal or even low blood pressure, possibly via a mechanism mediated by an activated immune response.6
The area of cerebral oedema is sometimes larger in cases with normal to mildly elevated blood pressure than in cases with very high blood pressure, which might indicate that the extent of oedema may be related to the level of endothelial dysfunction and the severity of hypertension.7 In PRES, lesions tend to occur in the posterior region of the brain. The reason for this is unclear, although it is possible that sympathetic innervation of the posterior region is limited compared with the anterior region, resulting in insufficient blood pressure regulation and an increased susceptibility to hypertension-induced cellular damage.5 Additionally, although PRES was initially described as a disease in which imaging studies showed reversible oedematous changes mainly in the posterior white matter of the cerebrum,5 subsequent reports show that lesions can be found in areas besides the posterior brain region.8
We here report a case of PRES triggered by malignant hypertension (MH), in a patient presenting with thrombotic microangiopathy (TMA) and vasogenic oedema including the brainstem.
Case presentation
A woman in her 40s, with a history of untreated hypertension, presented with severe headache, nausea and fatigue. Her headaches, which started 4 months prior to admission, were localised to the bitemporal and occipital regions. They were pulsating with a gradual onset and resolved spontaneously within 30–40 min. On the day of admission, she had a severe headache, and no abdominal symptoms such as diarrhoea or abdominal pain. Her vital signs were as follows: blood pressure, 230/150 mm Hg; heart rate, 83 beats per minute; oxygen saturation, 99% on room air; and Glasgow Coma Scale, 15/15. She was slightly drowsy but able to follow commands. Physical examination revealed slight purpura on the extremities but was otherwise unremarkable. Neurological examination revealed exaggerated deep tendon reflexes. Although there were no obvious visual changes, bilateral papilloedema and attenuation of the retinal arteries were found on funduscopy. The pre-admission laboratory test results, which were mostly normal with only a few measurements that were slightly outside the normal range, were as follows (table 1): red blood cells (RBCs), 3.34×1012/L (3.7–5.0); haemoglobin, 91 g/L (115–150); platelets, 67×109/µL (125–375); lactate dehydrogenase (LDH), 673 U/L (119–229); total bilirubin, 1.0 mg/dL (0.3–1.2); blood urea nitrogen, 33 mg/dL (8–22); serum creatinine, 2.02 mg/dL (0.4–0.7); serum potassium, 3.2 mmol/L (3.6–4.9); serum sodium, 132 mmol/L (138–146); prothrombin time, 11.7s (10.5–13); activated partial thromboplastin time, 23.0s (24.6–33.5); serum D-dimer, 1.1 mg/dL (0–1); fibrinogen, 481 mg/dL (200–400); C3, 103 mg/dL (86–160); C4, 16 mg/dL (17.0–45.0); and total complement activity, 61 U/mL (30–45). Both the direct and indirect Coombs’ test results were negative. In the absence of IgG directed against ADAMTS13, serum ADAMTS13 activity was 92% (60%–130%). All hepatitis serological test results were negative. Liver function markers, folic acid, vitamin B12 and screening immunology workup were all within normal limits. Peripheral blood smear test revealed a small number of schistocytes: 1/high power field. Urinalysis revealed albuminuria (2+), amorphous urates (2+), granular casts (1+), neutrophils: 20–25/high power field, RBC: 10–12/high power field. Since there was no history of sexual activity within the past year, urine pregnancy test was not performed.
Table 1.
Laboratory results
| Patient results | Normal range | Interpretation of the tests: elevated (↑), low (↓) | |
| RBC | 334 | 370–500 ×104 /µL | ↓ |
| Hb | 9.1 | 11.5–15 g/dL | ↓ |
| Ht | 27.6 | 34–45 % | ↓ |
| Mean cell volume | 82.7 | 88–99 fL | ↓ |
| Mean cell haemoglobin | 27.2 | 26–34 pg | – |
| Plt | 6.7×104 | 12.5–37.5 /µL | ↓ |
| D-dimer | 1.1 | 0–1 µg/mL | ↑ |
| Fibrinogen | 481 | 200–400 mg/dL | ↑ |
| Antithrombin Ⅲ (AT-Ⅲ) | 133 | 83–118 % | ↑ |
| Fibrin Degradation Products (FDP) | 4.4 | 0–5 µg/mL | – |
| Mg | 2.4 | 1.8–2.4 mg/dL | – |
| Na | 132 | 138–146 mmol/L | ↓ |
| K | 3.2 | 3.6–4.9 mmol/L | ↓ |
| T-Bil | 1.0 | 0.3–1.2 mg/dL | – |
| LDH | 673 | 119–229 U/L | ↑ |
| BUN | 33 | 8–22 mg/dL | ↑ |
| Cr | 2.02 | 0.4–0.7 mg/dL | ↑ |
| C3 | 103 | 86–160 mg/dL | – |
| C4 | 16 | 17–45 mg/dL | ↓ |
| CH50 | 61 | 30–45 U/mL | ↑ |
| ADAMTS13 level | 90 | 60–130 % | ↓ |
| Anti-GBM Ab | 2.0> | 0–2.9 U/mL | – |
| Cardiolipin IgG | 2.0> | 0–9 U/mL | – |
| Anti-nuclear Ab | 40> | 0–39 U | – |
| Anti-cardiolipin・β2GP1 Ab | 1.2> | 0–3.4 U/mL | – |
| Direct Coomb’s test | Negative | ||
| Indirect Coomb’s test | Negative |
Ab, antibodies; BUN, blood urea nitrogen; CH50, total complement activity; Cr, creatinine; Hb, haemoglobin; Ht, haematocrit; K, potassium; LDH, lactate dehydrogenase; Mg, magnesium; Na, sodium; Plt, platelets; RBC, red blood cell; T-Bil, total bilirubin.
CT of the head and neck showed no intracranial haemorrhage or mass lesions. Fluid-attenuated inversion recovery (FLAIR) images from brain MRI revealed extensive confluent pontine and middle cerebellar peduncle oedema with effacement of the fourth ventricle. Extensive but less confluent vasogenic oedema was seen in the supratentorial brain, predominantly frontally (figure 1). Susceptibility weighted imaging (SWI) demonstrated numerous thalamic, callosal and subcortical white matter micro-haemorrhages (figure 2). Magnetic resonance angiography confirmed the absence of vasoconstriction and occlusion, and the results of thoraco-abdominal CT, abdominal MRI and renal vascular ultrasound sonography were unremarkable. Anaemia was recognised as a secondary haemolytic phenomenon. The RBC and platelet depletion, elevated LDH and total indirect bilirubin levels, decreased haptoglobin levels and the presence of schistocytes in a smear of peripheral blood prompted suspicion of TMA. Based on the physical symptoms manifested and the results of the laboratory tests, disseminated intravascular coagulation, thrombotic thrombocytopenic purpura (TTP) and Shiga toxin-mediated TMA were deemed unlikely. MH was identified as the likely direct cause of TMA. Additional tests were performed to determine potential secondary causes of hypertension. Hyperaldosteronism was considered since the patient was hypokalaemic at admission. However, plasma renin activity, serum aldosterone, morning cortisol, serum metanephrine and serum normetanephrine levels were within the normal ranges, and analysis of 24-hour urine samples also revealed normal levels of catecholamine and its metabolites. As a result, endocrine disorders were considered unlikely.
Figure 1.

FLAIR images at onset. Extensive confluent pontine and middle cerebellar peduncle oedema with effacement of the fourth ventricle. Extensive but less confluent vasogenic oedema was seen in the supratentorial brain, predominantly frontally. FLAIR, fluid-attenuated inversion recovery.
Figure 2.
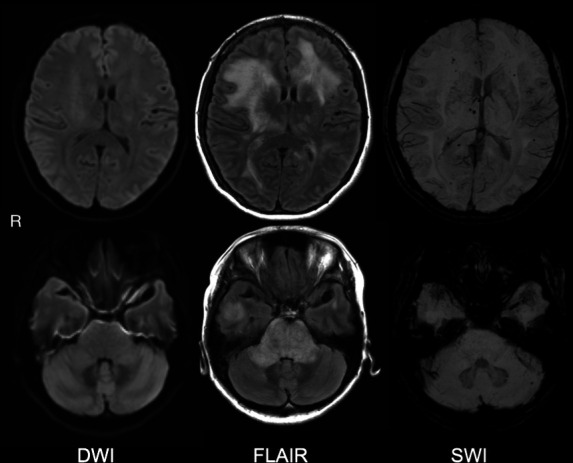
DWI, FLAIR and SWI on brain MRI at the onset of PRES. FLAIR: oedema in pontine, middle cerebellar peduncle (lower panel) and supratentorial, predominantly anterior (upper panel). SWI: numerous thalamic, callosal and subcortical white matter micro-haemorrhages. DWI, diffusion-weighted MRI; FLAIR, fluid-attenuated inversion recovery; PRES, posterior reversible encephalopathy syndrome; SWI, susceptibility weighted imaging.
Extensive testing effectively ruled out other causes of TMA. Therefore, the patient was diagnosed with PRES due to MH, and treatment with continuous intravenous injection of nicardipine and osmotic diuretics (glycerol at 600 mL/day) was started immediately. The next day after initiating the treatment, the patient’s blood pressure decreased to 143/97 mm Hg, and her clinical symptoms were improved, followed by complete remission on the second day of hospitalisation. Intravenous medication for blood pressure control was replaced with oral medication (80 mg telmisartan and 2 mg doxazosin mesilate daily) gradually. FLAIR brain MRI, which was repeated once weekly, showed gradual improvements in the signal hyperintensity (figure 3). The micro-haemorrhages on SWI remained. There was no improvement in the renal function, which suggested a gradual progression of impaired renal function prior to admission. The patient did not present any new neurological symptoms and was discharged on day 20, after the blood pressure was controlled with oral antihypertensive drugs.
Figure 3.
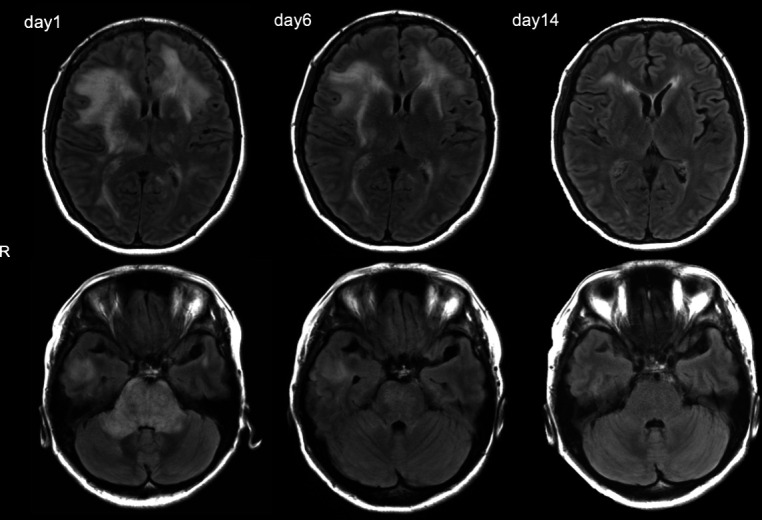
Follow-up supratentorial and pontocerebellar axial FLAIR images on the 1st, 6th and 14th day of admission. Supratentorial and infratentorial oedema improved over time. FLAIR, fluid-attenuated inversion recovery.
Discussion and conclusions
Here, we report a rare case of PRES that was triggered by MH and associated with TMA. To our knowledge, only one other case has been reported with a similar combination of these conditions.9
TMA is a pathological diagnosis that is characterised by vascular endothelial cell damage and microvascular thrombosis. Clinically, TMA is associated with thrombocytopenia, organ damage and microangiopathic haemolytic anaemia, a condition in which the RBCs are fragmented. Although TMA usually progresses to end-stage renal failure and is associated with a high mortality rate, early diagnosis and appropriate therapeutic intervention improve prognosis. TMA can be classified into several categories depending on the contributing factors.10 Identification of the specific category of TMA is essential for early intervention since treatment may vary depending on the contributing factors. The clinical symptoms and laboratory findings can help with prompt identification of the specific category of TMA. For example, the presence of gastrointestinal symptoms such as diarrhoea suggests the presence of Escherichia coli-producing Shiga toxin as a contributing factor.11 However, if ADAMTS13 activity is less than 10%, TTP may be suspected, but if the activity is more than 10%, another complement factor or underlying disease may be involved.12 Although careful and prompt consideration is required for differentiating TTP, ADAMTS13 activity could take time to achieve results depending on institution settings. In our case, we also took into account the fact that there was no significant increase of schistocytes in blood smear, and that the reduction in platelets in TTP is reported to be mostly in the range between 1×104/µL and 3×104/µL domestically,13 which was not as significant in this case.
As for this case, the aetiology of PRES was determined to be MH. The diagnosis of MH is dependent on the diastolic blood pressure, fundus findings and progressive organ damage.14 An excessive increase in blood pressure causes vascular endothelial cell injury and stimulation of the renin–angiotensin–aldosterone system, resulting in a deleterious cycle.15 The mechanism by which MH leads to TMA is believed to involve MH-induced endothelial cell damage, which causes RBC rupture, intravascular haemolysis, thrombocytopenia and organ damage.16
MH often leads to organ damage at the level of small arteries, with outcomes including decreased endothelial function, hyperproliferation of vascular smooth muscle cells, increased vascular permeability and extravasation of fibrin substances. The precise mechanism by which chronic hypertension causes sudden, uncontrollable hypertension is unclear. However, two potential mechanisms have been proposed: (1) increased systemic vascular resistance and (2) uncontrolled cerebral blood flow (CBF) autoregulation.17 In patients with chronic hypertension, the cerebral perfusion pressure control range tends to be elevated compared with the CBF.18 This, in turn, may cause exhaustion of continuous arteriolar vasoconstriction, thus leading to endothelial damage. When blood pressure exceeds the physiological autoregulatory capacity of blood vessels in the brain, cerebral vascular endothelial cells and the BBB are mechanically damaged. The subsequent increased vascular permeability and cerebral oedema lead to the development of PRES.5
There have been 18 previously reported cases of PRES associated with TMA, including six cases with lesions in the brainstem (table 2). In selecting these, reports that were unclear about the course of the disease, the diagnosis process, or the treatment and its effects were excluded. In all cases, the patients were females. Although these cases all originated from a variety of underlying diseases, only one case was reported with MH as the trigger.5 The findings from these previous reports suggest that gender-specific hormone regulation of blood pressure may play a role in this disorder, since all patients, except those with HELLP (haemolysis, elevated liver enzymes and low platelets) syndrome, were approaching menopause.
Table 2.
Six cases of PRES including brainstem lesions associated with TMA, in addition to our case
| Cases | Principal conditions | Age, sex |
BP at admission (mm Hg) |
Lesions in MRI | Details |
| Sakai, 2016 |
Haemodialysis patient with ANCA(Antineutrophil Cytoplasmic Antibodies)-associated vasculitis | 50 years, F | 185/106 | Right midbrain, periventricular area bilateral deep cerebral white matter left lens nucleus | Three months after introduction of dialysis, developed vision loss and nausea |
| Omoto, 2017 | Multiple sclerosis Long-term interferon-β1b treatment |
42 years, F | 226/138 | Bilateral basal ganglia, brainstem, cerebellum | Onset after long-term interferon-β1b treatment, improvement with drug discontinuation |
| Yamamoto, 2021 | Anti-melanoma differentiation-associated gene 5 antibody-positive dermatomyositis | 56 years, F | SBP 140 | Bilateral basal ganglia, thalamus and brainstem | After onset of unconsciousness, died of heart failure on the following day |
| Deguchi, 2017 |
Malignant hypertension | 42 years, F | 240/120 | Brainstem and cerebellum | Rapid improvement with antihypertensive treatment |
| Hara, 2007 |
HELLP | 35 years, F | 220/119 | Pons, bilateral caudate nuclei, right lateral capsule, left thalamus | Treated with haemodialysis and plasma exchange |
| Takahata, 2017 | HELLP-sustained acute kidney dysfunction | 33 years, F | 200/137 | Bilateral occipital parietal lobes, basal ganglia, cerebellum, brainstem | Treated with haemodialysis and plasma exchange |
| This case | Malignant hypertension | 40s, F | 230/150 | Bilateral subcortical matter, midbrain, pons, superior medulla | Rapid improvement with antihypertensive treatment |
BP, blood pressure; HELLP, haemolysis, elevated liver enzymes and low platelets; PRES, posterior reversible encephalopathy syndrome; SBP, systolic BP; TMA, thrombotic microangiopathy.
Despite the many uncertainties and hypothetical mechanisms, we speculate that brainstem lesions are more common in PRES caused by chronic hypertension. In chronic hypertension, endothelium-dependent vasodilation is impaired, and the resistance of cerebral arteries is increased due to a decrease in agonist-induced nitric oxide (NO) release.19 NO, which is predominantly clustered in the basilar artery, is an essential neurotransmitter with vasodilator effects.20 Limited to speculation, we estimate that during chronic hypertension, as in the case reported here, the decreased NO release and impaired vasorelaxation in the basilar arteries could lead to a breakdown in arterial autoregulation, which possibly result in vasogenic oedema and endothelial damage in posterior circulatory loci, particularly in the brainstem. It is worth noting that NO production is stimulated by oestrogen,21 which suggests a possible association between decreased NO production and menopause. Although accounting for the fact that oestrogen replacement therapy was not effective in reducing hypertension, other age-related factors or factors affected by the reduced NO production during menopause may contribute to chronic hypertension at this age.22 We note again that our speculative is hypothetical, available information is sufficient and requires further research and evaluation.
Additionally, it has been reported that CBF varies between patients with chronic hypertension and patients with normotension, depending on the brain region. CBF of patients with chronic hypertension is reduced in supratentorial areas including the cortex, striatum and thalamus, but not in the brainstem–cerebellum area.23 This implies that a mechanism of neurotransmitters likely involving NO, and endothelial damage rather than CBF itself, contributes to TMA-related PRES. Nevertheless, the specific differences in the neuromodulatory systems between the basilar artery and the anterior circulation, as well as the pathogenesis of PRES itself, remain unresolved and require further investigation.
Conclusions
In this report, we presented a woman in her 40s with a rare case of PRES that was associated with TMA and triggered by MH. We conclude that antihypertensive therapy may be effective in alleviating the associated adverse clinical symptoms. We also conclude that differentiation of the specific TMA category is essential for early intervention and to avoid unneeded and costly examinations, since treatment varies depending on the factors contributing to TMA.
Footnotes
Contributors: HO prepared the initial draft manuscript, which was critically edited by IA and YI. All authors have examined the patient. HO, IA and YI collected the case information, drafted the manuscript and conducted the literature research. HO and IA followed up with the patient. TS, JS, NN, YT and KT contributed to analysing the clinical data, diagnosis and treatment. All authors have read and approved the final version of the manuscript.
Funding: The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests: None declared.
Provenance and peer review: Not commissioned; externally peer reviewed.
Data availability statement
Data are available upon reasonable request. Additional images available through request to corresponding author.
Ethics statements
Patient consent for publication
Obtained.
Ethics approval
This study involves human participants and was approved by the Toyota Memorial Hospital Ethics Committee (reference number: S27). Participants gave informed consent to participate in the study before taking part.
References
- 1.Hinchey J, Chaves C, Appignani B, et al. A reversible posterior leukoencephalopathy syndrome. N Engl J Med 1996;334:494–500. 10.1056/NEJM199602223340803 [DOI] [PubMed] [Google Scholar]
- 2.Marra A, Vargas M, Striano P, et al. Posterior reversible encephalopathy syndrome: the endothelial hypotheses. Med Hypotheses 2014;82:619–22. 10.1016/j.mehy.2014.02.022 [DOI] [PubMed] [Google Scholar]
- 3.Bartynski WS, Boardman JF, Zeigler ZR, et al. Posterior reversible encephalopathy syndrome in infection, sepsis, and shock. AJNR Am J Neuroradiol 2006;27:2179–90. [PMC free article] [PubMed] [Google Scholar]
- 4.Stokum JA, Gerzanich V, Simard JM. Molecular pathophysiology of cerebral edema. J Cereb Blood Flow Metab 2016;36:513–38. 10.1177/0271678X15617172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fugate JE, Rabinstein AA. Posterior reversible encephalopathy syndrome: clinical and radiological manifestations, pathophysiology, and outstanding questions. Lancet Neurol 2015;14:914–25. 10.1016/S1474-4422(15)00111-8 [DOI] [PubMed] [Google Scholar]
- 6.Nelke C, Schulte-Mecklenbeck A, Pawlitzki M, et al. The innate immune response characterizes posterior reversible encephalopathy syndrome. J Clin Immunol 2021;41:1229–40. 10.1007/s10875-021-01033-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bartynski WS. Posterior reversible encephalopathy syndrome, part 2: controversies surrounding pathophysiology of vasogenic edema. AJNR Am J Neuroradiol 2008;29:1043–9. 10.3174/ajnr.A0929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Granata G, Greco A, Iannella G, et al. Posterior reversible encephalopathy syndrome--Insight into pathogenesis, clinical variants and treatment approaches. Autoimmun Rev 2015;14:830–6. 10.1016/j.autrev.2015.05.006 [DOI] [PubMed] [Google Scholar]
- 9.Deguchi I, Uchino A, Suzuki H, et al. Malignant hypertension with reversible brainstem hypertensive encephalopathy and thrombotic microangiopathy. J Stroke Cerebrovasc Dis 2012;21:915.e17–915.e20. 10.1016/j.jstrokecerebrovasdis.2012.02.005 [DOI] [PubMed] [Google Scholar]
- 10.George JN, Nester CM. Syndromes of thrombotic microangiopathy. N Engl J Med 2014;371:654–66. 10.1056/NEJMra1312353 [DOI] [PubMed] [Google Scholar]
- 11.Tarr PI, Gordon CA, Chandler WL. Shiga-Toxin-Producing Escherichia coli and haemolytic uraemic syndrome. Lancet 2005;365:1073–86. 10.1016/S0140-6736(05)71144-2 [DOI] [PubMed] [Google Scholar]
- 12.George JN, Chen Q, Deford CC, et al. Ten patient stories illustrating the extraordinarily diverse clinical features of patients with thrombotic thrombocytopenic purpura and severe ADAMTS13 deficiency. J Clin Apher 2012;27:302–11. 10.1002/jca.21248 [DOI] [PubMed] [Google Scholar]
- 13.Matsumoto M, Bennett CL, Isonishi A, et al. Acquired idiopathic ADAMTS13 activity deficient thrombotic thrombocytopenic purpura in a population from Japan. PLoS One 2012;7:e33029. 10.1371/journal.pone.0033029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wong TY, Mitchell P. Hypertensive retinopathy. N Engl J Med 2004;351:2310–7. 10.1056/NEJMra032865 [DOI] [PubMed] [Google Scholar]
- 15.van den Born B-JH, Koopmans RP, van Montfrans GA. The renin-angiotensin system in malignant hypertension revisited: plasma renin activity, microangiopathic hemolysis, and renal failure in malignant hypertension. Am J Hypertens 2007;20:900–6. 10.1016/j.amjhyper.2007.02.018 [DOI] [PubMed] [Google Scholar]
- 16.Shantsila A, Dwivedi G, Shantsila E, et al. Persistent macrovascular and microvascular dysfunction in patients with malignant hypertension. Hypertension 2011;57:490–6. 10.1161/HYPERTENSIONAHA.110.166314 [DOI] [PubMed] [Google Scholar]
- 17.Tulman DB, Stawicki SPA, Papadimos TJ, et al. Advances in management of acute hypertension: a Concise review. Discov Med 2012;13:375–83. [PMC free article] [PubMed] [Google Scholar]
- 18.Owens WB. Blood pressure control in acute cerebrovascular disease. J Clin Hypertens 2011;13:205–11. 10.1111/j.1751-7176.2010.00394.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chillon J-M. Cerebral vessels during chronic hypertension: from arteries to therapeutics. J Hypertens 2002;20:817–8. 10.1097/00004872-200205000-00007 [DOI] [PubMed] [Google Scholar]
- 20.Tamayo A, Siepmann T. Regulation of blood flow in the cerebral posterior circulation by parasympathetic nerve fibers: physiological background and possible clinical implications in patients with vertebrobasilar stroke. Front Neurol 2021;12:660373. 10.3389/fneur.2021.660373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Weiner CP, Lizasoain I, Baylis SA, et al. Induction of calcium-dependent nitric oxide synthases by sex hormones. Proc Natl Acad Sci U S A 1994;91:5212–6. 10.1073/pnas.91.11.5212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reckelhoff JF. Gender differences in the regulation of blood pressure. Hypertension 2001;37:1199–208. 10.1161/01.HYP.37.5.1199 [DOI] [PubMed] [Google Scholar]
- 23.Fujishima M, Ibayashi S, Fujii K, et al. Cerebral blood flow and brain function in hypertension. Hypertens Res 1995;18:111–7. 10.1291/hypres.18.111 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data are available upon reasonable request. Additional images available through request to corresponding author.