

Abstract
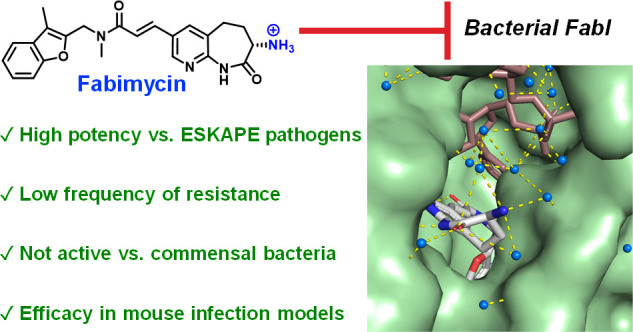
Genomic studies and experiments with permeability-deficient strains have revealed a variety of biological targets that can be engaged to kill Gram-negative bacteria. However, the formidable outer membrane and promiscuous efflux pumps of these pathogens prevent many candidate antibiotics from reaching these targets. One such promising target is the enzyme FabI, which catalyzes the rate-determining step in bacterial fatty acid biosynthesis. Notably, FabI inhibitors have advanced to clinical trials for Staphylococcus aureus infections but not for infections caused by Gram-negative bacteria. Here, we synthesize a suite of FabI inhibitors whose structures fit permeation rules for Gram-negative bacteria and leverage activity against a challenging panel of Gram-negative clinical isolates as a filter for advancement. The compound to emerge, called fabimycin, has impressive activity against >200 clinical isolates of Escherichia coli, Klebsiella pneumoniae, and Acinetobacter baumannii, and does not kill commensal bacteria. X-ray structures of fabimycin in complex with FabI provide molecular insights into the inhibition. Fabimycin demonstrates activity in multiple mouse models of infection caused by Gram-negative bacteria, including a challenging urinary tract infection model. Fabimycin has translational promise, and its discovery provides additional evidence that antibiotics can be systematically modified to accumulate in Gram-negative bacteria and kill these problematic pathogens.
Short abstract
Fabimycin is a bacterial FabI inhibitor that possesses potent antibiotic activity against multiple ESKAPE pathogens and efficacy in several mouse infection models, while largely sparing human commensal bacteria.
Introduction
Novel antibiotic classes for infections caused by Gram-positive pathogens have been a success story over the last 20 years, with drugs in the oxazolidinone, mutilin, and lipopeptide classes all having notable clinical or veterinary impact.1−3 Further, there are additional antibiotics moving through clinical trials for Gram-positive infections, including new compound classes and antibiotics that engage unexploited biological targets.4 In contrast, there has not been a novel class of antibiotics FDA approved for treatment of Gram-negative pathogens contained within the group of high-priority antibiotic-resistant, nosocomoial pathogens (ESKAPE pathogens5,6) in over 50 years; this situation has led to increased mortality, with these Gram-negative bacteria representing four of the top six pathogens causing antibiotic-associated deaths, and some studies showing that 75% of deaths from drug-resistant pathogens are now caused by Gram-negative bacteria.7,8 This discovery void is largely due to the low likelihood that a given compound will accumulate inside Gram-negative bacteria, as their dense lipopolysaccharide outer membrane and promiscuous efflux pumps work in concert to prevent candidate antibiotics from reaching their target. Recent studies reveal that compounds capable of accumulating inside Gram-negative bacteria often possess certain physicochemical properties,9−11 explaining why high-throughput screens of millions of compounds have failed to identify Gram-negative active antibiotics.12,13
Encouragingly, the same biological processes that are exploited through antibiotic intervention against Gram-positive bacteria can typically be leveraged to kill Gram-negative bacteria; inhibitors of protein translation, DNA replication, and cell wall biosynthesis have broad-spectrum activity (Gram-positive and Gram-negative) provided they can enter the cell and reach their target. However, many other promising biological targets have not yet been leveraged to kill Gram-negative organisms, as the antibiotics that engage these targets do not accumulate in Gram-negative bacteria. One such outstanding target is the enoyl-acyl carrier protein reductase enzyme FabI, which catalyzes the rate-determining step in bacterial fatty acid biosynthesis.14 A lead compound15,16 identified from a biochemical high-throughput screen for FabI inhibition was optimized into Debio-1452 (Figure 1A), the phosphonoxy methyl prodrug version of which (called afabicin) is in Phase 2 clinical trials for infections caused by Staphylococcus aureus.17 While FabI is a promising exploitable target for problematic Gram-negative ESKAPE pathogens, including Escherichia coli, Klebsiella pneumoniae, and Acinetobacter baumannii, Debio-1452 does not accumulate inside these cells and is consequently inactive against these bacteria.18
Figure 1.

FabI inhibitors. (A) Debio-1452 is highly potent against S. aureus; Debio-1452-NH3 retains this potency and gains modest activity against many Gram-negative pathogens. (B) The structure–activity relationship (SAR) of Debio-1452 showing regions amenable to substitution (highlighted in green) and those critical for antibacterial activity (highlighted in red).
We recently applied an emerging understanding of the correlation between the physicochemical traits of small molecules and their accumulation in Gram-negative bacteria to design a version of Debio-1452 that is effective against these pathogens.19 This compound, Debio-1452-NH3 (Figure 1A), has antibacterial activity against Gram-negative clinical isolates and efficacy in mouse infection models, and as such is the first member of this class to have notable Gram-negative antibacterial activity.19 In efforts to develop a more promising clinical candidate for Gram-negative infections, we now report the use of iterative compound synthesis, clinical isolate testing, and X-ray crystallography to identify fabimycin, a FabI inhibitor with enhanced antibacterial potency, improved in vivo tolerability, and high specificity for pathogenic versus commensal bacteria. Fabimycin shows efficacy in multiple mouse infection models, including a challenging mouse model of urinary tract infection (UTI), acute pneumonia models, and several neutropenic soft tissue models of infection with Gram-negative bacteria.
Results
Identification of Fabimycin and Its Activity against Drug-Resistant Gram-Negative Pathogens
Aided by recently described guidelines for design of Gram-negative penetrant compounds (the eNTRy rules9,11,20), Debio-1452-NH3 was discovered through a focused synthetic campaign and subsequent evaluation of just a handful of compounds.19 However, to advance as a lead candidate, its therapeutic window would need to be widened, as efficacy of Debio-1452-NH3 in murine infection models with Gram-negative pathogens was observed at the maximal tolerated dose (MTD).19 An objective was set to identify next-generation FabI inhibitors that exhibit greater potency against Gram-negative clinical isolates and better in vivo tolerability, expecting that such compounds could then be efficacious even in very challenging models and those of high translational relevance, such as a UTI model.
Our critical path for advancement included the synthesis of compounds and subsequent evaluation of antibacterial activity against Gram-negative clinical isolates to prioritize leads, followed by evaluation of toxicity, pharmacokinetics, and ultimately efficacy in mouse infection models. Compound design was guided by the co-crystal structure of Debio-1452 with FabI (from S. aureus)21 and the established structure–activity relationship (SAR) for this compound class, both of which point to the immutability of the N-methyl acrylamide (E configuration) and the H-bond donor/acceptor pair on the naphthyridinone ring system that interacts with key amino acid residues within the FabI active site (Figure 1B).22−24 In contrast, the position adjacent to the carbonyl of the naphthyridinone ring was judged to be highly solvent-exposed; additionally, other ring systems were considered as replacements for the benzofuran (Figure 1B). This analysis was instrumental for informing the proper placement of a positively charged amine to facilitate accumulation in Gram-negative bacteria but not disrupt target engagement. Given the X-ray and SAR data, and lessons learned from previous identification of Debio-1452-NH3, priority compounds were envisioned with a variety of amines and ring systems proximal to the carbonyl of the lactam.
A modular and convergent synthesis was designed leveraging a Heck coupling of acrylamide 1 with bromo functionalized acylaminopyridine rings which upon deprotection provided the final compounds (Figure 2). Brominated coupling partners were designed to give final compounds that maintained the necessary N/NH arrangement for engagement with FabI and contained the basic amine required for Gram-negative accumulation. To quickly interrogate the solvent-exposed region of the scaffold adjacent to the lactam carbonyl, ring size and shape were altered. Specifically, a series comprising four-membered ring spirocycles (2–4), six-membered ring spirocycles (5, 6), and azepanones (7–9) was constructed.
Figure 2.
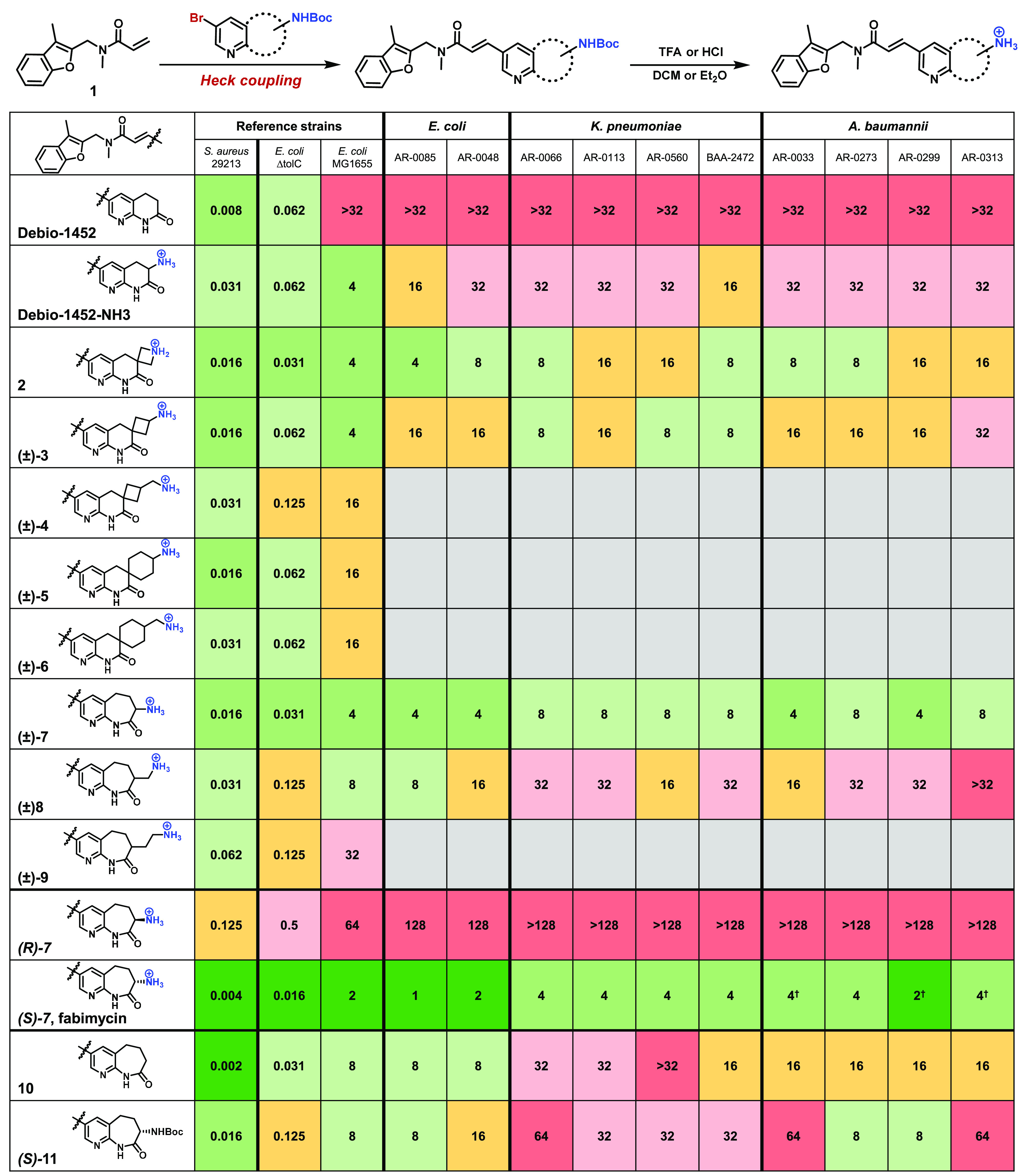
Debio-1452 analogue synthesis and antibacterial activity. The general synthetic route utilized to synthesize amine-containing compounds, and their antimicrobial activities against Gram-positive and Gram-negative bacteria. (†) indicates dose-independent trailing growth observed; see Supporting Information, Extended Data Figure S1. MIC values were determined using the microdilution broth method, as outlined by CLSI. All experiments were performed in biological triplicate. E. coli ΔtolC = JW5503.
The compounds synthesized, shown in Figure 2, were evaluated against Gram-positive and Gram-negative reference strains (S. aureus ATCC 29213 and E. coli MG1655, respectively), as well as an efflux-deficient E. coli strain (ΔtolC, JW5503). While compounds (±)-4, (±)-5, (±)-6, and (±)-9 all had reduced antibacterial activity against E. coli MG1655 relative to Debio-1452-NH3, compounds 2, (±)-3, (±)-7, and (±)-8 maintained good activity (Figure 2) and were thus chosen for further evaluation. A panel of 10 clinical isolates was selected where Debio-1452-NH3 had previously demonstrated only minimal potency (MIC values of 16 or 32 μg/mL), including two E. coli, four K. pneumoniae, and four A. baumannii strains.19 As expected, Debio-1452 has no activity against this Gram-negative “challenge panel”, and Debio-1452-NH3 has only minimal activity (Figure 2). Assessment of 2, (±)-3, (±)-7, and (±)-8 revealed more potent activity for all four compounds relative to Debio-1452-NH3, with the ε-caprolactam (±)-7 emerging as a promising candidate. This compound possessed superior activity against the clinical isolate challenge panel with all strains inhibited at ≤8 μg/mL. Two derivatives were constructed where the amine-decorated ε-caprolactam was coupled to alternative ring systems, substituting out the benzofuran, but neither of these compounds provided an improvement in activity (Extended Data, Figure S2).
The two enantiomers of (±)-7 were separated by chiral preparatory HPLC, and biological assessment revealed the (−) enantiomer to possess significantly greater antibacterial activity than the (+) enantiomer. X-ray crystallography studies, described later herein, were used to determine that the highly active (−) enantiomer possesses the S stereochemical configuration. As shown in Figure 2, (S)-7, coined fabimycin,25–265 has outstanding activity against S. aureus and E. coli ΔtolC, an MIC value of 2 μg/mL against E. coli MG1655, and very good activity against the clinical isolate challenge panel, whereas (R)-7 is significantly less active.
To probe the influence of the amine on antibacterial activity and compound accumulation, two additional derivatives were synthesized: compound 10, with the expanded ring system but lacking the amine, and compound (S)-11, a carbamylated version of fabimycin. Of note, 10 and (S)-11 both lack a primary amine. While both compounds display modest inhibition of purified bacterial FabI (Extended Data, Figure S3) and excellent antimicrobial activity against reference strains, they have diminished activity against Gram-negative clinical isolates (Figure 2). Furthermore, when assessed in a whole-cell accumulation assay27 in E. coli, 10 displayed reduced whole-cell accumulation consistent with its reduction in antibacterial activity (Extended Data, Figure S4). In contrast, other amine-containing compounds in Figure 2 show significant whole-cell accumulation in E. coli (Extended Data, Figure S4) (the limited aqueous solubility of (S)-11 prevented it from being assessed in this assay).
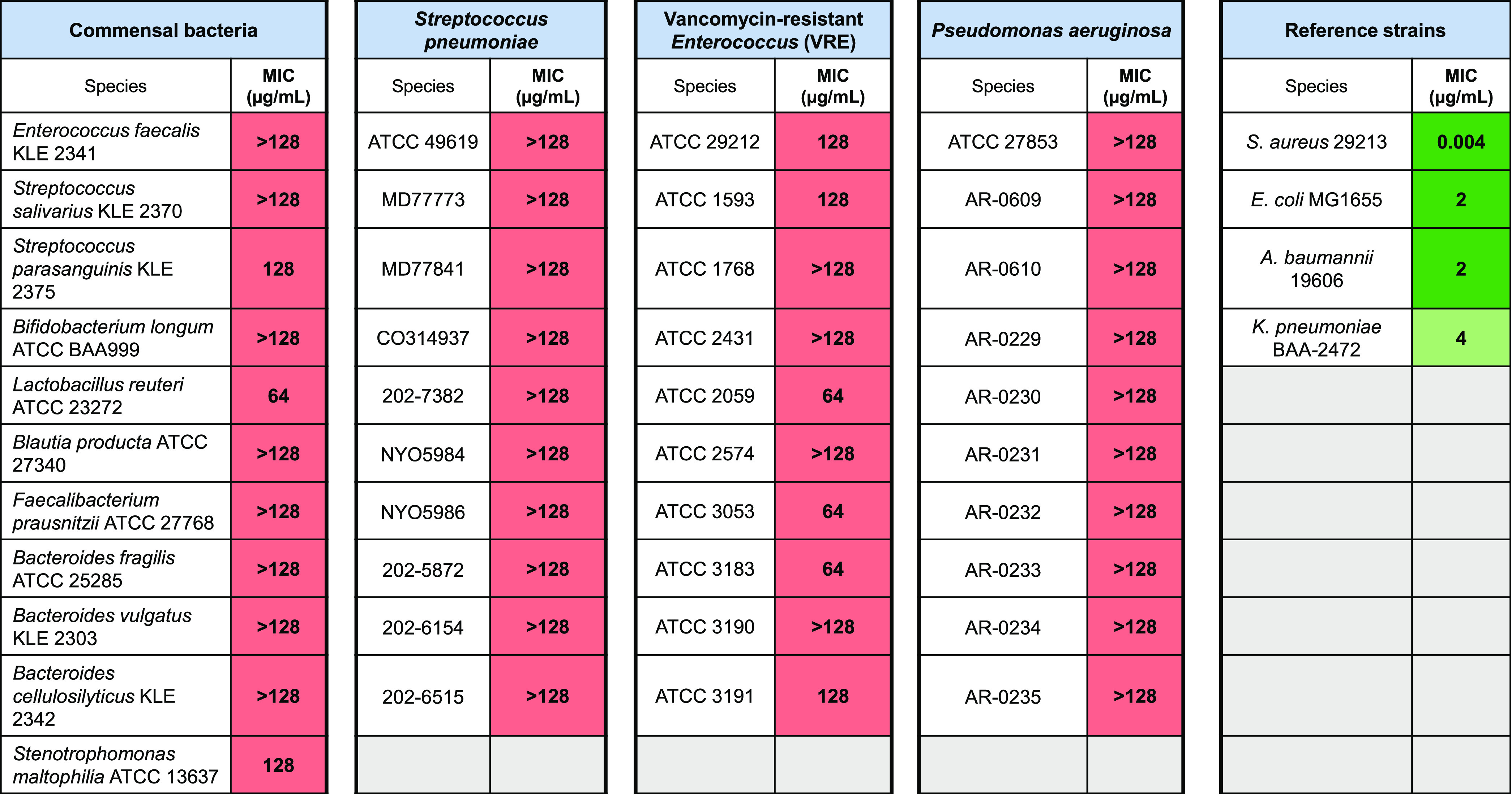
An interesting aspect of FabI as an antibacterial target is the possibility for a relatively narrow spectrum of activity when compared to that observed with most antibiotics (e.g., inhibitors of protein synthesis, DNA replication, and/or cell wall biosynthesis). For example, it has been noted that ESKAPE pathogen P. aeruginosa possesses the FabV isoform, meaning inhibition of FabI will not be lethal in this pathogen.28 Certain Gram-positive pathogens (Streptococcus sp. and Enterococcus sp.) and commensal bacteria are also not reliant on FabI and thus are expected to be insensitive to FabI inhibition, suggesting that FabI inhibitors could be microbiome-sparing.29−31 To explore this possibility, fabimycin was assessed against a panel of pathogens possessing alternate enoyl-acyl carrier protein reductases as well as anaerobic human commensal bacterial species.32−34 In all cases, low/no antibacterial activity was observed against the strains assessed (n = 41), demonstrating the specificity of this FabI inhibitor and its potential for minimal perturbation of the microbiome (Table 1).
Table 1. Spectrum of Fabimycin Activitya.
Given its promising antibacterial activity, fabimycin was advanced through a battery of mechanistic and translational experiments. While separation of (±)-7 on a preparative chiral column was suitable to obtain the quantities of fabimycin required for studies such as MIC assays, it was necessary to optimize the synthetic route to enable scale-up and obtain quantities of the fabimycin required for detailed in vivo tolerability, pharmacokinetic analysis, and efficacy experiments. To this end, the synthetic route shown in Figure 3 was developed and employed to generate gram-scale quantities of enantiopure fabimycin.
Figure 3.
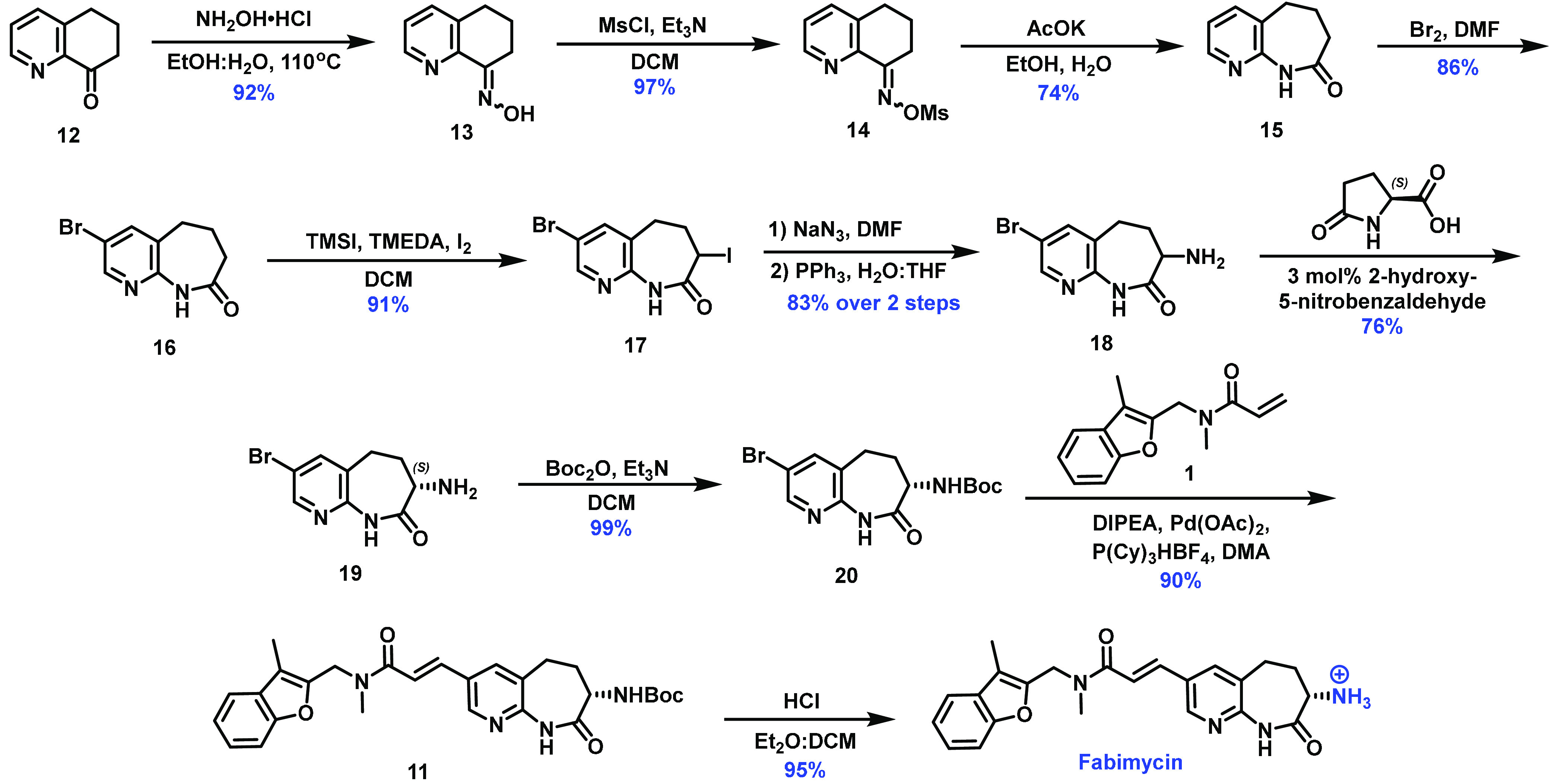
Optimized synthesis of fabimycin. The synthetic route used to access gram-scale quantities of fabimycin, utilizing dynamic kinetic resolution (DKR) to install the critical stereogenic center.
The route proceeds through condensation of quinolone 12 with hydroxylamine, followed by activation of the resulting oxime (13) via treatment with mesyl chloride to generate compound 14 in good yield. After forming azepanone 15 through a Beckmann rearrangement, molecular bromine was used to produce aryl bromide 16. Alpha-iodination of 16 provided dihalogenated 17 in good yield, and compound 17 was subjected to azidation and subsequent Staudinger reduction to afford amine 18. This amine was the key intermediate for enantioenrichment via a dynamic kinetic resolution (DKR), proceeding through epimerization of the amine via imine formation with 2-hydroxy-5-nitrobenzaldehyde followed by selective crystallization with l-pyroglutamic acid.36 Optimization of this critical step involved significant screening (Extended Data, Figure S5). After enantioenrichment (up to 99.5% ee), amine 19 was protected to afford compound 20 (ee maintained) and used in a Heck coupling with acrylic amide 1 to produce 11, which, when subjected to acidic conditions, liberated fabimycin.
Fabimycin was assessed for its antibacterial activity against a panel of multidrug-resistant A. baumannii, E. coli, and K. pneumoniae clinical isolates (54 strains in total). As shown in Figure 4A, fabimycin is markedly more potent than Debio-1452 and Debio-1452-NH3 against all the Gram-negative clinical isolates. All three compounds also maintain high potency versus panels of S. aureus clinical isolates (Figure 4A and Extended Data, Figure S6).
Figure 4.

Antimicrobial activity of fabimycin against clinical isolates. (A) The susceptibility of clinical isolates (Gram-negative species and S. aureus) to fabimycin, Debio-1452-NH3, and Debio-1452. MICs performed in biological triplicate. (B) Further exploration of the breadth of fabimycin’s antibacterial activity against diverse clinical isolate panels of K. pneumoniae and A. baumannii, as compared to levofloxacin. MICs performed in biological duplicate.
As the greatest improvement in activity of fabimycin (relative to Debio-1452-NH3) was against K. pneumoniae and A. baumannii clinical isolates, fabimycin was further assessed against more diverse and expansive clinical isolate panels of these two pathogens to determine the MIC50 and MIC90 values. Excitingly, when assessed against a panel of 100 K. pneumoniae clinical isolates, fabimycin inhibited 90% of the strains at 4 μg/mL, relative to 32 μg/mL for levofloxacin (Figure 4B). Also encouraging was the narrow MIC range for fabimycin, suggesting that intrinsic resistance to this compound is not prevalent in existing bacterial populations. The analogous data in 100 A. baumannii clinical isolates, a panel specifically curated to represent the genomic diversity of the species37 including 27 multidrug-resistant strains, 35 extensively drug-resistant strains, and 1 pan-resistant strain (fabimycin MIC = 1 μg/mL), are also promising, with fabimycin exhibiting a MIC90 value of 8 μg/mL (relative to 32 μg/mL for levofloxacin) and a narrow distribution of MIC values (Figure 4B).
Mode of Action
Spontaneous resistant mutants to fabimycin were generated in E. coli MG1655, A. baumannii 19606, and S. aureus ATCC 29213 at 8×, 16×, and 32× the respective MICs with low frequencies of resistance observed at 8×–16× the MIC for all pathogens (Figure 5A). Importantly, sequencing of the fabI gene in resistant colonies revealed mutations encoding single amino acid changes within the active site of FabI (Figure 5B). While several FabI mutations were observed in fabimycin-resistant A. baumannii, the MIC of the bacteria harboring mutant FabI was often near fabimycin concentrations attained in vivo (as shown later). In E. coli, the most frequently observed FabI mutation to arise in fabimycin-resistant colonies was at G148 (Figure 5B); E. coli with mutations at this position in FabI have been previously shown to have attenuated fitness.19 Encouragingly, the mutant prevention concentration (MPC, the concentration where no resistant mutants are observed) of fabimycin versus E. coli was found to be 64 μg/mL and, impressively, in S. aureus, the MPC was 0.125 μg/mL. A serial passaging experiment of E. coli MG1655 in the presence of a subinhibitory concentration of fabimycin over the course of 21 days led to an 8-fold increase in the MIC for fabimycin, relative to a 128-fold increase in the MIC of ciprofloxacin in the same experiment (Extended Data, Figure 7A). To investigate the rate of killing, a time–kill growth curve was generated that revealed fabimycin slowly kills E. coli over the course of 8 h (Extended Data, Figure 7B); however, fabimycin was not profoundly bactericidal in this experiment, mimicking its progenitor’s (Debio-1452) behavior in S. aureus.21
Figure 5.

Fabimycin mode of action studies. (A) Spontaneous resistance frequencies of S. aureus, E. coli, and A. baumannii versus fabimycin. Data represent three replicates for each pathogen with error bars representing the SEM. (B) Point mutations in FabI observed in fabimycin-resistant colonies, and the corresponding MIC values of fabimycin versus the mutants. All MICs were performed in biological triplicate.
Figure 7.

Computational and biophysical evaluation of fabimycin and its enantiomer. (A) Molecular dynamic simulations of fabimycin and its enantiomer using the co-crystal structures in E. coli FabI, demonstrating the enhanced flexibility (decreased stability) of (R)-7. (B) The determined enthalpy changes upon binding to FabI as assessed by isothermal titration calorimetry (ITC), as well as observed stabilization in a differential scanning fluorimetry assay of FabI (from E. coli and A. baumannii) upon compound binding relative to the holoenzyme. Tm values are means of technical triplicates with error shown as the standard deviation.
Crystallography, Molecular Dynamics, and Biophysics
An interesting feature of fabimycin is its considerably enhanced antibacterial activity relative to its enantiomer (R)-7. Assessment in E. coli reveals that each enantiomer accumulates intracellularly to a similar extent (Extended Data, Figure S4). In contrast, evaluation of the two enantiomers in the FabI activity assay shows a significant differential between the two enantiomers (Extended Data, Figure S8), with fabimycin being at least five-fold more potent against A. baumannii and E. coli FabI. Of note, in this enzyme assay IC50 values cannot be accurately determined below ∼10 nM, so the value for fabimycin likely under-represents its biochemical potency. Taken together, the data suggest that the diminished antibacterial activity of (R)-7 relative to fabimycin is due to reduced target engagement and not differential intracellular accumulation.
To further investigate the molecular basis for the observed differential activity between enantiomers, X-ray crystal structures of fabimycin and its enantiomer (R)-7 bound to both E. coli and A. baumannii FabI with NADH cofactor were solved with resolutions ranging from 1.5 to 2.7 Å. The general binding mode of the compounds is similar to what has been previously reported for this class of compounds and Debio-1452 in particular.38 In complex with E. coli FabI, the pyridoazepanone ring forms two hydrogen bonds with the backbone carbonyl and amide nitrogen of conserved residue Ala95 (Figure 6A,B). A water-mediated interaction is also observed between the carbonyl of the lactam and the backbone amide of Gly97. Additionally, a hydrogen bond is formed between the acrylamide linker carbonyl and the conserved residue Tyr156. The benzopyran ring is nestled in a hydrophobic pocket formed by conserved, hydrophobic residues Tyr146, Pro191, Ile153, Met206, and Phe203.
Figure 6.

Co-crystal structures of fabimycin and its enantiomer with FabI. (A) Co-crystal structure of fabimycin with E. coli FabI with NADH cofactor (PDB 7UMW). (B) Co-crystal structure of (R)-7 in E. coli FabI with NADH cofactor (7UM8). (C) Water network surrounding fabimycin in the E. coli FabI active site. (D) Water network surrounding (R)-7 in the E. coli FabI active site.
Similar to E. coli, the hydrogen-bonding network of the pyridoazepanone ring, interactions of the acrylamide carbonyl, and the position of the flanking benzofuran ring are maintained with the analogous amino acid residues in A. baumannii FabI (Extended Data, Figure S9A,B). While no significant changes of the interacting residues can be seen between fabimycin and (R)-7 in A. baumannii FabI, the enhanced resolution of the E. coli crystal structures captures a more nuanced and detailed binding mode. In the E. coli structure, a large water network is observed between the ligand, NADH cofactor, and FabI enzyme. While this network exists in the binding of both enantiomers, it is larger and more tightly structured in the fabimycin co-crystal (see Figure 6C,D).
Intrigued by the relative similarity of the crystal structures, experiments were conducted to evaluate the stability and dynamics of the inhibitor-enzyme complex as well as the strain energy of each enantiomer. The established E. coli co-crystal structure was utilized for computational studies which determined that the less active (R)-enantiomer is more strained relative to fabimycin (∼8.30 kcal/mol higher) in the active site. This is reflected in molecular dynamic simulations that reveal the (R)-enantiomer to be much more flexible in the binding pocket of FabI leading to overall less-productive hydrogen-bond interactions with critical surrounding residues such as Ala95 and Tyr156 (Figure 7A). Isothermal titration calorimetry (ITC) was used to confirm the nanomolar potency of each compound and showed a doubling in enthalpy for fabimycin relative to the less active enantiomer (Figure 7B). Finally, differential scanning fluorimetry (DSF) experiments show that fabimycin enhances the stability of the enzyme–inhibitor complex significantly more than the less active enantiomer in both E. coli and A. baumannii versions of FabI (Figure 7B).
In Vivo Experiments
Given the low resistance frequency and promising data with >200 Gram-negative clinical isolates, experiments were conducted to probe the suitability of fabimycin for in vivo infection models. As a prelude to these studies, fabimycin was evaluated against three human cell lines (HFF-1, A549, HepG2) and for its ability to lyse human red blood cells. These studies revealed fabimycin to be less cytotoxic relative to Debio-1452-NH3 and nonhemolytic even at high concentrations (200 μM, Extended Data, Table S1). Fabimycin also has lower activity against the hERG ion channel relative to both Debio-1452 and Debio-1452-NH3, and lower plasma protein binding relative to Debio-1452 (Extended Data, Table S1). Formulation and MTD studies were performed, and in all cases fabimycin was found to be better tolerated in mice with an MTD of >200 mg/kg (IP injection) relative to 50 mg/kg for Debio-1452-NH3 (Extended Data, Table S2).
An interesting aspect of this compound class is the metabolic instability in mice leading to suboptimal pharmacokinetics;24 indeed, assessment of fabimycin in mouse, rat, and human plasma showed considerable instability in mouse plasma contrasted with excellent stability in rat and human plasma (Figure 8A). While this data suggests the possibility that antibacterial activity could improve as fabimycin moves toward humans, it does complicate the evaluation of this compound class in murine infection models. Thus, as a prelude to efficacy experiments in mouse infection models, a pharmacokinetic study was conducted with fabimycin using neutropenic female BALB/c mice infected with drug-resistant A. baumannii (fabimycin MIC = 2 μg/mL) where single doses of the compound (20, 50, 75, 100 mg/kg) were administered intravenously to the infected mice and blood taken over the course of 8 h. Encouragingly, when dosed at 75 mg/kg, fabimycin concentrations stayed above the MIC for the infectious strain for over 6 h in the thigh tissue with the Cmax nearing the MPC for wild-type E. coli when dosed at 100 mg/kg (Extended Data, Figure S10).
Figure 8.
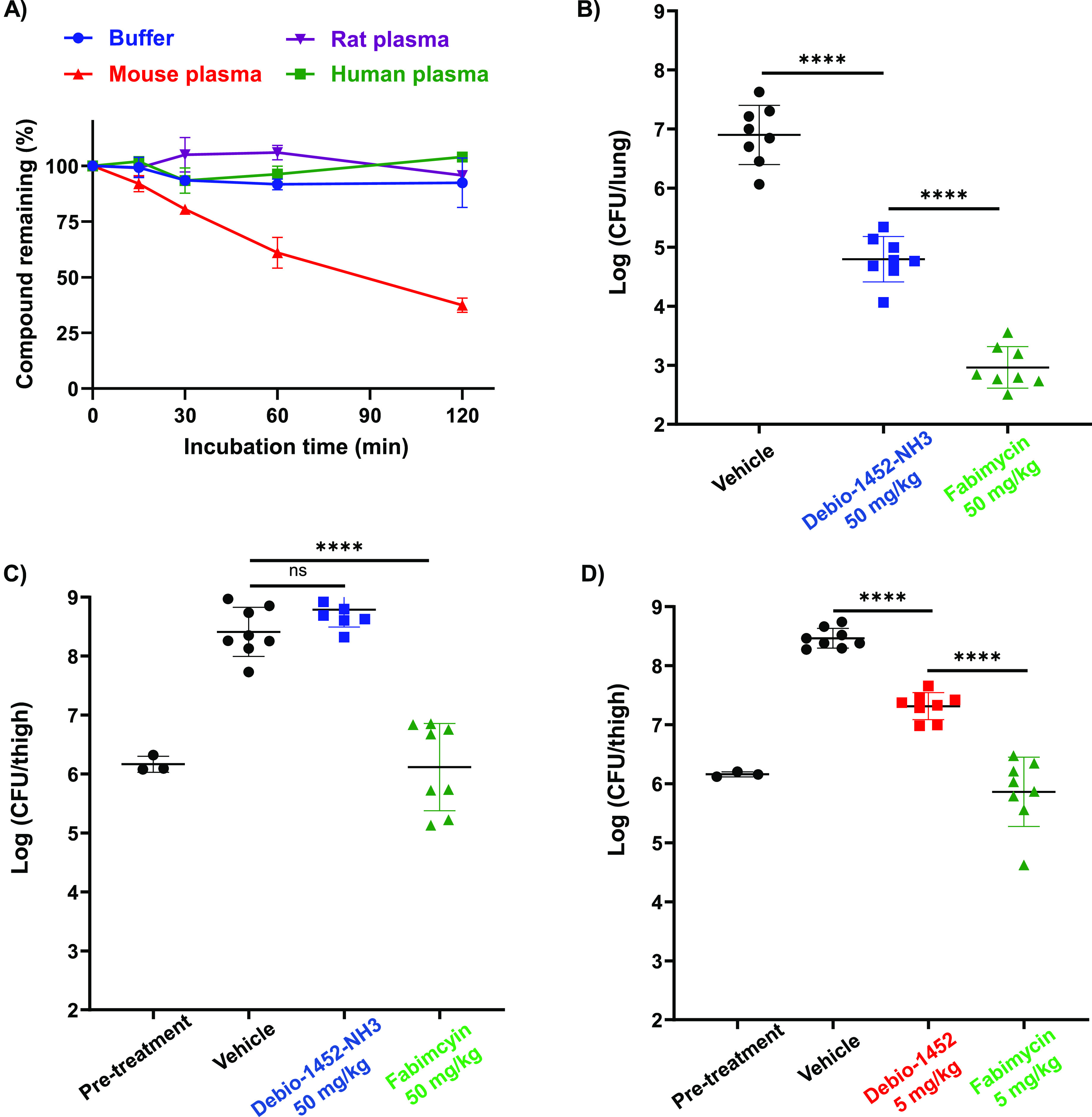
Plasma stability and in vivo efficacy of fabimycin. (A) Assessment of fabimycin stability in plasma. Data shown as the mean and standard deviation from two experiments. (B) Acute pneumonia infections initiated in CD-1 mice with A. baumannii AR-0299 (1.6 × 108 CFUs per mouse intranasally). Mice were treated with vehicle (8 mice) or FabI inhibitor (8 mice per group) 4, 23, and 41 h postinfection (50 mg/kg intramuscular) and the bacterial burden evaluated at 48h postinfection. (C) Neutropenic mouse thigh infection initiated in CD-1 mice with A. baumannii AR-0299 (1.22 × 106 CFUs per mouse intramuscular in thigh) were treated with vehicle (8 mice) or FabI inhibitor (8 mice per group) 2, 6, and 11 h postinfection (50 mg/kg intramuscular), and the bacterial burden was evaluated 26h postinfection. (D) Neutropenic mouse thigh infection initiated in CD-1 mice with S. aureus USA300 LAC (2.3 × 106 CFU per mouse intramuscular in thigh) were treated with vehicle (eight mice) or FabI inhibitor (eight mice per group) 2 and 7 h postinfection (5 mg/kg retro-orbital IV), and the bacterial burden was evaluated 24 h postinfection. Debio-1452-tosylate used. FabI inhibitors formulated with 20% SBE-β-CD in H2O. In B, C, and D statistical significance was determined by one-way ANOVA with Tukey’s multiple comparisons. NS, not significant. ****P < 0.0001. Error bars represent standard deviation.
With formulation, MTD, and pharmacokinetic data in hand, the efficacy of fabimycin was evaluated in murine infection models. To start, a comparative assessment was made of fabimycin and Debio-1452-NH3 in two murine infection models using a dosing regimen (50 mg/kg, intramuscular) near the MTD for Debio-1452-NH3 but well below the MTD for fabimycin. Using an extensively drug-resistant A. baumannii clinical isolate, fabimycin outperformed Debio-1452-NH3 in both lung and neutropenic thigh infection models and achieved a >3-fold decrease in log(CFU/lung) and >2-fold decrease log(CFU/thigh) relative to the vehicle (Figure 8B,C). In a S. aureus (MRSA clinical isolate) neutropenic thigh infection model fabimycin showed significantly greater reduction of bacterial burden relative to Debio-1452 when both were administered at the low dose of 5 mg/kg (Figure 8D). As the goal of these initial models was simply to assess efficacy compared to progenitor compounds, fabimycin dosing was not maximized or optimized. To address this, fabimycin was evaluated in an experiment where neutropenic, A. baumannii-infected mice were treated four times a day (via IV injection) with doses spanning 1.25–75 mg/kg. In this experiment, significant dose-dependent responses were observed beginning at 30 mg/kg (Extended Data, Figure S11A). When using the most efficacious dose (75 mg/kg, four times a day) in a murine neutropenic thigh infection experiment with an extensively resistant NDM-1 containing strain of A. baumannii, fabimycin was able to reduce bacterial burden by nearly 2 log(CFU/thigh) (Extended Data, Figure S11B).
With an effective dose established, we aimed to evaluate fabimycin in a murine model of an infection with high translational value. Urinary tract infections (UTIs) represent one of the biggest risks for healthy individuals in terms of exposure to antibiotic-resistant bacteria with many individuals contracting one in their lifetime (roughly 1 in 2 women and 1 in 10 men);39 UTIs caused by Gram-negative pathogens, particularly those that are drug-resistant, are becoming more frequent and remain a major clinical challenge.39−41 As E. coli is the causative agent in the vast majority of UTIs,42 fabimycin was evaluated in a murine UTI model with a challenging, extensively drug-resistant strain of carbapenem-resistant E. coli (fabimycin MIC = 2 μg/mL). When intravenously dosed at 33.3 mg/kg, three times a day, fabimycin was able to achieve 3.0, 2.8, 2.9, and 1.9 log10 reductions in bacterial load relative to the vehicle in the spleen, bladder, liver, and kidney tissues, respectively (Figure 9). Dose fractionation studies in infected mice also revealed other dosing regiments to be effective in reducing the bacterial load to a significant extent in mouse tissue (Figure 9 and Extended Data, Figure S12).
Figure 9.

In vivo efficacy of fabimycin in a murine UTI model. After inducing diuresis, infection initiated in C3H/HeJ mice (8 per arm, 1.38 * 109 CFU/mouse transurethral) with E. coli AR-0055 and treated with fabimycin (IV) at varying concentrations three times daily with bacterial enumeration at 168h postinfection. Fabimycin formulated in 17% Cremophor EL, 3% SBE-β-CD in H2O which was the formulation used in the vehicle arm (administered intravenously on the same schedule as fabimycin). Colistin was formulated in H2O with 0.9% NaCl and administered subcutaneously. Percentage in red indicates the percentage of animals with bacterial counts below the limit of detection (LOD, indicated by the dotted horizontal line). In A—D statistical significance was determined by one-way ANOVA with Tukey’s multiple comparisons. NS, not significant. *P = 0.0243, ***P < 0.001, ****P < 0.0001. Data represented as the mean with s.e.m.
Discussion
There is a rich history in antibacterial drug discovery of identifying and advancing chemical matter arising from whole-cell phenotypic screens. In contrast, while biochemical screens have uncovered a wealth of scaffolds with promising activity, there has typically been less interest in such compounds due to the documented inability to imbue them with whole-cell antibacterial activity.12,13 In this context, Debio-1452 is an interesting case study as it was developed from a progenitor compound that had only biochemical FabI inhibitory activity—it was first converted into a S. aureus-specific drug (Debio-1452), then to Debio-1452-NH3, and now into fabimycin, which possesses promising activity against important Gram-negative pathogens.
Relative to Debio-1452-NH3, fabimycin has significantly improved activity against A. baumannii, even surpassing the potency increase observed versus E. coli, consistent with the notion that certain FabI inhibitors can be tuned for specific bacterial homologues of FabI if the creation of pathogen-specific antibiotics is of interest.14 The generated X-ray co-crystal structures of fabimycin bound to A. baumannii and E. coli FabI reveal that key hydrogen-bonding interactions are conserved between the two forms of the enzyme, but no significant protein conformational differences in the active site. Given the limitations of the FabI enzymatic assay when evaluating compounds possessing low nanomolar potency, use of multiple biochemical techniques will be critical to the understanding of current and future FabI inhibitors. Overall, a combination of biochemical, structural, biophysical, and computational approaches was used to better understand the differences in whole-cell activity of fabimycin and (R)-7. Biochemical assessment of the inhibitory activities of fabimycin and (R)-7 showed modest differences in potency against purified E. coli FabI (4.75-fold), whereas the differences in MIC activity between the two enantiomers in Gram-negative clinical isolates is much more significant (64–128 fold). Comparison of the co-crystal structures of fabimycin and (R)-7 reveals a more expansive and tightly structured water network in the fabimcyin structure. This result is consistent with ITC data showing fabimycin to have a more favorable binding entropy, DSF results showing a larger change in melting temperature upon compound binding to FabI relative to the less-active (R)-7, and with molecular dynamic simulations showing fabimycin to be much less strained (i.e., more stable) in the E. coli FabI active site. Taken together, these results suggest that the differences in whole-cell activity between the two enantiomers is primarily due to a more favorable water interaction network and higher degree of protein and ligand stability for fabimycin.
Beyond Gram-negative activity, the ability of fabimycin to retain approximately equivalent activity as Debio-1452 against S. aureus is impressive, as Debio-1452 was optimized to treat S. aureus infections and has thus set a high bar for potency versus this pathogen, both in vitro and in vivo.17,43,44 The very low frequency of resistance (1.8 × 10–10 to 5.2 × 10–10, likely due at least in part to critical interactions with the amide backbone of FabI),14,45 outstanding performance in the serial passage experiment, and the extremely low mutation prevention concentration (0.125 μg/mL) suggest great promise for fabimycin against S. aureus, as the MPC is dwarfed by the concentration of fabimycin achieved in mouse plasma (Cmax = 47 μg/mL, t1/2 = 1.4 h).
Inhibition of FabI is attractive due to its orthogonality (relative to the mammalian fatty acid biosynthetic analogue) and its essential nature, for certain bacteria, in the maintenance of cellular membranes.46,47 The nature of the FabI target is such that its inhibition will not be lethal to all types of bacteria, as many bacteria have redundant enzymes or can compensate by exogenous fatty acid uptake;29 indeed, fabimycin has virtually no whole-cell activity versus a sampling of such species. While this includes some pathogenic bacteria, such as P. aeruginosa, it also includes many commensal bacteria, suggesting that a suitable FabI inhibitor could be less damaging to the gut microbiome than the typical broad-spectrum antibiotic.31 In this vein, the activity of fabimycin against UTIs is very promising, given how antibiotic treatment of UTIs is known to trigger Clostridioides difficile infection48 and the recently described “gut–bladder axis” in recurrent UTIs.49,50
Similar to fusidic acid and some other antibiotics,51,52 the advancement of this class of FabI inhibitors has been complicated by the uniquely poor stability of these compounds in mouse plasma. Given the promising activity of fabimycin in mouse infection models and encouraging data that fabimycin is dramatically more stable in rat and human plasma, it is reasonable to believe that fabimycin efficacy may improve as it is used to treat infections in higher organisms. The potency of fabimycin, combined with the very low resistance frequency and apparent lack of pre-existing resistance, bodes well for its translation.
Acknowledgments
This work was supported by the University of Illinois and the NIH (AI136773 to P.J.H. and G.W.L.). K.A.M. and M.P.M. are members of the NIH Chemistry–Biology Interface Training Grant (T32-GM136629). R.J.U. is supported by an NIH Ruth Kirschstein Award (F31AI161953) and was an NSF predoctoral fellow. This work utilized NIAID’s suite of preclinical services for in vitro and in vivo assessment (Contract Numbers HHSN272201700020I_75N93021F00001, HHSN272201700020I_75N93021F00002, and 75N93019D00022_75N93020F00001). The Collaborative Hub for Early Antibiotic Discovery (CHEAD) is grateful for financial support from the Combating Antibiotic-Resistant Bacteria Biopharmaceutical Accelerator (CARB-X). This work was supported by a gift from Anita and Josh Bekenstein and by funding provided under Cooperative Agreement #IDSEP160030-01-00 from Biomedical Advanced Research and Development Authority (BARDA). Its contents are solely the responsibility of the authors and do not necessarily represent the official views of The Assistant Secretary for Preparedness and Response. This project has been funded in part by federal funds from the Military Infectious Disease Research Program Proposals MI210200 and MI220020. We thank L. Li (Metabolomics Center, Roy J. Carver Biotechnology Center, UIUC) for LC-MS/MS analysis. We thank Dean Olson, Lingyang Zhu, and N. Duay at the School of Chemical Sciences NMR Laboratory at UIUC for NMR services. The Bruker 500 MHz NMR spectrometer was obtained with the financial support of the Roy J. Carver Charitable Trust, Muscatine, Iowa.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscentsci.2c00598.
Supporting figures and tables, materials and methods, experimental procedures, and characterization of chemical products including LCMS traces and NMR (1H, 13C) spectra (PDF)
Author Contributions
# E.N.P. and B.N.C. contributed equally.
The authors declare the following competing financial interest(s): The University of Illinois and the Broad Institute have filed patents on some compounds described herein.
Supplementary Material
References
- Konychev A.; Heep M.; Moritz R. K.; Kreuter A.; Shulutko A.; Fierlbeck G.; Bouylout K.; Pathan R.; Trostmann U.; Chaves R. L. Safety and efficacy of daptomycin as first-line treatment for complicated skin and soft tissue infections in elderly patients: an open-label, multicentre, randomized phase IIIb trial. Drugs Aging 2013, 30 (10), 829–836. 10.1007/s40266-013-0114-8. [DOI] [PubMed] [Google Scholar]
- Moran G. J.; Fang E.; Corey G. R.; Das A. F.; De Anda C.; Prokocimer P. Tedizolid for 6 days versus linezolid for 10 days for acute bacterial skin and skin-structure infections (ESTABLISH-2): a randomised, double-blind, phase 3, non-inferiority trial. Lancet Infect Dis 2014, 14 (8), 696–705. 10.1016/S1473-3099(14)70737-6. [DOI] [PubMed] [Google Scholar]
- File T. M.; Goldberg L.; Das A.; Sweeney C.; Saviski J.; Gelone S. P.; Seltzer E.; Paukner S.; Wicha W. W.; Talbot G. H.; Gasink L. B. Efficacy and Safety of Intravenous-to-oral Lefamulin, a Pleuromutilin Antibiotic, for the Treatment of Community-acquired Bacterial Pneumonia: The Phase III Lefamulin Evaluation Against Pneumonia (LEAP 1) Trial. Clin Infect Dis 2019, 69 (11), 1856–1867. 10.1093/cid/ciz090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO Antibacterial products in clinical development for priority pathogens. https://www.who.int/observatories/global-observatory-on-health-research-and-development/monitoring/antibacterial-products-in-clinical-development-for-priority-pathogens (accessed June 13, 2022).
- Rice L. B. Federal funding for the study of antimicrobial resistance in nosocomial pathogens: no ESKAPE. J. Infect Dis 2008, 197 (8), 1079–81. 10.1086/533452. [DOI] [PubMed] [Google Scholar]
- Boucher H. W.; Talbot G. H.; Bradley J. S.; Edwards J. E.; Gilbert D.; Rice L. B.; Scheld M.; Spellberg B.; Bartlett J. Bad bugs, no drugs: no ESKAPE! An update from the Infectious Diseases Society of America. Clin Infect Dis 2009, 48 (1), 1–12. 10.1086/595011. [DOI] [PubMed] [Google Scholar]
- Cassini A.; Hogberg L. D.; Plachouras D.; Quattrocchi A.; Hoxha A.; Simonsen G. S.; Colomb-Cotinat M.; Kretzschmar M. E.; Devleesschauwer B.; Cecchini M.; Ouakrim D. A.; Oliveira T. C.; Struelens M. J.; Suetens C.; Monnet D. L.; Attributable deaths and disability-adjusted life-years caused by infections with antibiotic-resistant bacteria in the EU and the European Economic Area in 2015: a population-level modelling analysis. Lancet Infect. Dis. 2019, 19 (1), 56–66. 10.1016/S1473-3099(18)30605-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Global burden of bacterial antimicrobial resistance in 2019: a systematic analysis. Lancet 2022, 399 (10325), 629–655. 10.1016/S0140-6736(21)02724-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter M. F.; Drown B. S.; Riley A. P.; Garcia A.; Shirai T.; Svec R. L.; Hergenrother P. J. Predictive compound accumulation rules yield a broad-spectrum antibiotic. Nature 2017, 545 (7654), 299–304. 10.1038/nature22308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao S.; Adamiak J. W.; Bonifay V.; Mehla J.; Zgurskaya H. I.; Tan D. S. Defining new chemical space for drug penetration into Gram-negative bacteria. Nat. Chem. Biol. 2020, 16 (12), 1293–1302. 10.1038/s41589-020-00674-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz K. A.; Hergenrother P. J. Facilitating Compound Entry as a Means to Discover Antibiotics for Gram-Negative Bacteria. Acc. Chem. Res. 2021, 54 (6), 1322–1333. 10.1021/acs.accounts.0c00895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne D. J.; Gwynn M. N.; Holmes D. J.; Pompliano D. L. Drugs for bad bugs: confronting the challenges of antibacterial discovery. Nat. Rev. Drug Discov 2007, 6 (1), 29–40. 10.1038/nrd2201. [DOI] [PubMed] [Google Scholar]
- Tommasi R.; Brown D. G.; Walkup G. K.; Manchester J. I.; Miller A. A. ESKAPEing the labyrinth of antibacterial discovery. Nat. Rev. Drug Discov 2015, 14 (8), 529–42. 10.1038/nrd4572. [DOI] [PubMed] [Google Scholar]
- Yao J.; Rock C. O. Resistance Mechanisms and the Future of Bacterial Enoyl-Acyl Carrier Protein Reductase (FabI) Antibiotics. Cold Spring Harb Perspect Med. 2016, 6 (3), a027045. 10.1101/cshperspect.a027045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seefeld M. A.; Miller W. H.; Newlander K. A.; Burgess W. J.; Payne D. J.; Rittenhouse S. F.; Moore T. D.; DeWolf W. E. Jr.; Keller P. M.; Qiu X.; Janson C. A.; Vaidya K.; Fosberry A. P.; Smyth M. G.; Jaworski D. D.; Slater-Radosti C.; Huffman W. F. Inhibitors of bacterial enoyl acyl carrier protein reductase (FabI): 2,9-disubstituted 1,2,3,4-tetrahydropyrido[3,4-b]indoles as potential antibacterial agents. Bioorg. Med. Chem. Lett. 2001, 11 (17), 2241–4. 10.1016/S0960-894X(01)00405-X. [DOI] [PubMed] [Google Scholar]
- Payne D. J.; Miller W. H.; Berry V.; Brosky J.; Burgess W. J.; Chen E.; DeWolf Jr W. E. Jr.; Fosberry A. P.; Greenwood R.; Head M. S.; Heerding D. A.; Janson C. A.; Jaworski D. D.; Keller P. M.; Manley P. J.; Moore T. D.; Newlander K. A.; Pearson S.; Polizzi B. J.; Qiu X.; Rittenhouse S. F.; Slater-Radosti C.; Salyers K. L.; Seefeld M. A.; Smyth M. G.; Takata D. T.; Uzinskas I. N.; Vaidya K.; Wallis N. G.; Winram S. B.; Yuan C. C.; Huffman W. F. Discovery of a novel and potent class of FabI-directed antibacterial agents. Antimicrob. Agents Chemother. 2002, 46 (10), 3118–24. 10.1128/AAC.46.10.3118-3124.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittke F.; Vincent C.; Chen J.; Heller B.; Kabler H.; Overcash J. S.; Leylavergne F.; Dieppois G. Afabicin, a First-in-Class Antistaphylococcal Antibiotic, in the Treatment of Acute Bacterial Skin and Skin Structure Infections: Clinical Noninferiority to Vancomycin/Linezolid. Antimicrob. Agents Chemother. 2020, 64 (10), e00250-20. 10.1128/AAC.00250-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlowsky J. A.; Kaplan N.; Hafkin B.; Hoban D. J.; Zhanel G. G. AFN-1252, a FabI inhibitor, demonstrates a Staphylococcus-specific spectrum of activity. Antimicrob. Agents Chemother. 2009, 53 (8), 3544–8. 10.1128/AAC.00400-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker E. N.; Drown B. S.; Geddes E. J.; Lee H. Y.; Ismail N.; Lau G. W.; Hergenrother P. J. Implementation of permeation rules leads to a FabI inhibitor with activity against Gram-negative pathogens. Nat. Microbiol 2020, 5 (1), 67–75. 10.1038/s41564-019-0604-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter M. F.; Hergenrother P. J. The challenge of converting Gram-positive-only compounds into broad-spectrum antibiotics. Ann. N.Y. Acad. Sci. 2019, 1435 (1), 18–38. 10.1111/nyas.13598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan N.; Albert M.; Awrey D.; Bardouniotis E.; Berman J.; Clarke T.; Dorsey M.; Hafkin B.; Ramnauth J.; Romanov V.; Schmid M. B.; Thalakada R.; Yethon J.; Pauls H. W. Mode of action, in vitro activity, and in vivo efficacy of AFN-1252, a selective antistaphylococcal FabI inhibitor. Antimicrob. Agents Chemother. 2012, 56 (11), 5865–5874. 10.1128/AAC.01411-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seefeld M. A.; Miller W. H.; Newlander K. A.; Burgess W. J.; DeWolf W. E. Jr.; Elkins P. A.; Head M. S.; Jakas D. R.; Janson C. A.; Keller P. M.; Manley P. J.; Moore T. D.; Payne D. J.; Pearson S.; Polizzi B. J.; Qiu X.; Rittenhouse S. F.; Uzinskas I. N.; Wallis N. G.; Huffman W. F. Indole naphthyridinones as inhibitors of bacterial enoyl-ACP reductases FabI and FabK. J. Med. Chem. 2003, 46 (9), 1627–35. 10.1021/jm0204035. [DOI] [PubMed] [Google Scholar]
- Ramnauth J.; Surman M. D.; Sampson P. B.; Forrest B.; Wilson J.; Freeman E.; Manning D. D.; Martin F.; Toro A.; Domagala M.; Awrey D. E.; Bardouniotis E.; Kaplan N.; Berman J.; Pauls H. W. 2,3,4,5-Tetrahydro-1H-pyrido[2,3-b and e][1,4]diazepines as inhibitors of the bacterial enoyl ACP reductase, FabI. Bioorg. Med. Chem. Lett. 2009, 19 (18), 5359–5362. 10.1016/j.bmcl.2009.07.094. [DOI] [PubMed] [Google Scholar]
- Takhi M.; Sreenivas K.; Reddy C. K.; Munikumar M.; Praveena K.; Sudheer P.; Rao B. N.; Ramakanth G.; Sivaranjani J.; Mulik S.; Reddy Y. R.; Narasimha Rao K.; Pallavi R.; Lakshminarasimhan A.; Panigrahi S. K.; Antony T.; Abdullah I.; Lee Y. K.; Ramachandra M.; Yusof R.; Rahman N. A.; Subramanya H. Discovery of azetidine based ene-amides as potent bacterial enoyl ACP reductase (FabI) inhibitors. Eur. J. Med. Chem. 2014, 84, 382–394. 10.1016/j.ejmech.2014.07.036. [DOI] [PubMed] [Google Scholar]
- Hergenrother P. J.; Geddes E. J.; Drown B. S.; Motika S. E.; Parker S. E.. Antibiotics Effective for Gram-Negative Pathogens. WO2019/177975A1, 2019.
- Gerusz V. B.; Bravo J.; Pauls H.; Berman J.; Finn T.. Novel Compounds and their Use. WO2021/123372A1, 2021.
- Hergenrother P. J.; Parker E. N.; Hung D.; Serrano-Wu M.; Lee K. K.. FabI inhibitors for Gram-negative pathogens. US Provisional Patent Application No. 63/156,145. 2021.
- Geddes E. J.; Li Z.; Hergenrother P. J. An LC-MS/MS assay and complementary web-based tool to quantify and predict compound accumulation in E. coli. Nat. Protoc 2021, 16 (10), 4833–4854. 10.1038/s41596-021-00598-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu L.; Lin J.; Ma J.; Cronan J. E.; Wang H. Triclosan resistance of Pseudomonas aeruginosa PAO1 is due to FabV, a triclosan-resistant enoyl-acyl carrier protein reductase. Antimicrob. Agents Chemother. 2010, 54 (2), 689–698. 10.1128/AAC.01152-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons J. B.; Frank M. W.; Subramanian C.; Saenkham P.; Rock C. O. Metabolic basis for the differential susceptibility of Gram-positive pathogens to fatty acid synthesis inhibitors. Proc. Natl. Acad. Sci. U. S. A. 2011, 108 (37), 15378–15383. 10.1073/pnas.1109208108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu L.; Bi H.; Ma J.; Hu Z.; Zhang W.; Cronan J. E.; Wang H. The two functional enoyl-acyl carrier protein reductases of Enterococcus faecalis do not mediate triclosan resistance. mBio 2013, 4 (5), e00613–13. 10.1128/mBio.00613-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao J.; Carter R. A.; Vuagniaux G.; Barbier M.; Rosch J. W.; Rock C. O. A Pathogen-Selective Antibiotic Minimizes Disturbance to the Microbiome. Antimicrob. Agents Chemother. 2016, 60 (7), 4264–73. 10.1128/AAC.00535-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai Y.; Meyer K. J.; Iinishi A.; Favre-Godal Q.; Green R.; Manuse S.; Caboni M.; Mori M.; Niles S.; Ghiglieri M.; Honrao C.; Ma X.; Guo J. J.; Makriyannis A.; Linares-Otoya L.; Bohringer N.; Wuisan Z. G.; Kaur H.; Wu R.; Mateus A.; Typas A.; Savitski M. M.; Espinoza J. L.; O’Rourke A.; Nelson K. E.; Hiller S.; Noinaj N.; Schaberle T. F.; D’Onofrio A.; Lewis K. A new antibiotic selectively kills Gram-negative pathogens. Nature 2019, 576 (7787), 459–464. 10.1038/s41586-019-1791-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leimer N.; Wu X.; Imai Y.; Morrissette M.; Pitt N.; Favre-Godal Q.; Iinishi A.; Jain S.; Caboni M.; Leus I. V.; Bonifay V.; Niles S.; Bargabos R.; Ghiglieri M.; Corsetti R.; Krumpoch M.; Fox G.; Son S.; Klepacki D.; Polikanov Y. S.; Freliech C. A.; McCarthy J. E.; Edmondson D. G.; Norris S. J.; D’Onofrio A.; Hu L. T.; Zgurskaya H. I.; Lewis K. A selective antibiotic for Lyme disease. Cell 2021, 184 (21), 5405–5418. 10.1016/j.cell.2021.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poyet M.; Groussin M.; Gibbons S. M.; Avila-Pacheco J.; Jiang X.; Kearney S. M.; Perrotta A. R.; Berdy B.; Zhao S.; Lieberman T. D.; Swanson P. K.; Smith M.; Roesemann S.; Alexander J. E.; Rich S. A.; Livny J.; Vlamakis H.; Clish C.; Bullock K.; Deik A.; Scott J.; Pierce K. A.; Xavier R. J.; Alm E. J. A library of human gut bacterial isolates paired with longitudinal multiomics data enables mechanistic microbiome research. Nat. Med. 2019, 25 (9), 1442–1452. 10.1038/s41591-019-0559-3. [DOI] [PubMed] [Google Scholar]
- Marrakchi H.; Dewolf W. E. Jr.; Quinn C.; West J.; Polizzi B. J.; So C. Y.; Holmes D. J.; Reed S. L.; Heath R. J.; Payne D. J.; Rock C. O.; Wallis N. G. Characterization of Streptococcus pneumoniae enoyl-(acyl-carrier protein) reductase (FabK). Biochem. J. 2003, 370 (3), 1055–1062. 10.1042/bj20021699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong J. D.; Eng K. K.; Keller J. L.; Purick R. M.; Hartner F. W.; Choi W.-B.; Askin D.; Volante R.P. An Efficient Asymmetric Synthesis of (R)-3-Amino-2,3,4,5-tetrahydro-1H-[1]benzazepin-2-one. Tetrahedron Lett. 1994, 35 (20), 3239–3242. 10.1016/S0040-4039(00)76874-2. [DOI] [Google Scholar]
- Galac M. R.; Snesrud E.; Lebreton F.; Stam J.; Julius M.; Ong A. C.; Maybank R.; Jones A. R.; Kwak Y. I.; Hinkle K.; Waterman P. E.; Lesho E. P.; Bennett J. W.; Mc Gann P. A Diverse Panel of Clinical Acinetobacter baumannii for Research and Development. Antimicrob. Agents Chemother. 2020, 64 (10), e00840-20. 10.1128/AAC.00840-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao N. K.; Nataraj V.; Ravi M.; Panchariya L.; Palai K.; Talapati S. R.; Lakshminarasimhan A.; Ramachandra M.; Antony T. Ternary complex formation of AFN-1252 with Acinetobacter baumannii FabI and NADH: Crystallographic and biochemical studies. Chem. Biol. Drug Des 2020, 96 (2), 704–713. 10.1111/cbdd.13686. [DOI] [PubMed] [Google Scholar]
- Sihra N.; Goodman A.; Zakri R.; Sahai A.; Malde S. Nonantibiotic prevention and management of recurrent urinary tract infection. Nat. Rev. Urol 2018, 15 (12), 750–776. 10.1038/s41585-018-0106-x. [DOI] [PubMed] [Google Scholar]
- Flores-Mireles A. L.; Walker J. N.; Caparon M.; Hultgren S. J. Urinary tract infections: epidemiology, mechanisms of infection and treatment options. Nat. Rev. Microbiol 2015, 13 (5), 269–284. 10.1038/nrmicro3432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goebel M. C.; Trautner B. W.; Grigoryan L. The Five Ds of Outpatient Antibiotic Stewardship for Urinary Tract Infections. Clin Microbiol Rev. 2021, 34 (4), e0000320. 10.1128/CMR.00003-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielubowicz G. R.; Mobley H. L. Host-pathogen interactions in urinary tract infection. Nat. Rev. Urol 2010, 7 (8), 430–441. 10.1038/nrurol.2010.101. [DOI] [PubMed] [Google Scholar]
- Flamm R. K.; Rhomberg P. R.; Kaplan N.; Jones R. N.; Farrell D. J. Activity of Debio1452, a FabI inhibitor with potent activity against Staphylococcus aureus and coagulase-negative Staphylococcus spp., including multidrug-resistant strains. Antimicrob. Agents Chemother. 2015, 59 (5), 2583–2587. 10.1128/AAC.05119-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafkin B.; Kaplan N.; Murphy B. Efficacy and Safety of AFN-1252, the First Staphylococcus-Specific Antibacterial Agent, in the Treatment of Acute Bacterial Skin and Skin Structure Infections, Including Those in Patients with Significant Comorbidities. Antimicrob. Agents Chemother. 2016, 60 (3), 1695–701. 10.1128/AAC.01741-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radka C. D.; Rock C. O.. Mining Fatty Acid Biosynthesis for New Antimicrobials. Annu. Rev. Microbiol. 2022, 76, 10.1146/annurev-micro-041320-110408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne D. J.; Warren P. V.; Holmes D. J.; Ji Y.; Lonsdale J. T. Bacterial fatty-acid biosynthesis: a genomics-driven target for antibacterial drug discovery. Drug Discov Today 2001, 6 (10), 537–544. 10.1016/S1359-6446(01)01774-3. [DOI] [PubMed] [Google Scholar]
- Yao J.; Rock C. O. Bacterial fatty acid metabolism in modern antibiotic discovery. Biochim Biophys Acta Mol. Cell Biol. Lipids 2017, 1862 (11), 1300–1309. 10.1016/j.bbalip.2016.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staley C.; Vaughn B. P.; Graiziger C. T.; Sadowsky M. J.; Khoruts A. Gut-sparing treatment of urinary tract infection in patients at high risk of Clostridium difficile infection. J. Antimicrob. Chemother. 2017, 72 (2), 522–528. 10.1093/jac/dkw499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worby C. J.; Schreiber H. L. t.; Straub T. J.; van Dijk L. R.; Bronson R. A.; Olson B. S.; Pinkner J. S.; Obernuefemann C. L. P.; Munoz V. L.; Paharik A. E.; Azimzadeh P. N.; Walker B. J.; Desjardins C. A.; Chou W. C.; Bergeron K.; Chapman S. B.; Klim A.; Manson A. L.; Hannan T. J.; Hooton T. M.; Kau A. L.; Lai H. H.; Dodson K. W.; Hultgren S. J.; Earl A. M. Longitudinal multi-omics analyses link gut microbiome dysbiosis with recurrent urinary tract infections in women. Nat. Microbiol 2022, 7 (5), 630–639. 10.1038/s41564-022-01107-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schembri M. A.; Nhu N. T. K.; Phan M. D. Gut-bladder axis in recurrent UTI. Nat. Microbiol 2022, 7 (5), 601–602. 10.1038/s41564-022-01113-z. [DOI] [PubMed] [Google Scholar]
- Njoroge M.; Kaur G.; Espinoza-Moraga M.; Wasuna A.; Dziwornu G. A.; Seldon R.; Taylor D.; Okombo J.; Warner D. F.; Chibale K. Semisynthetic Antimycobacterial C-3 Silicate and C-3/C-21 Ester Derivatives of Fusidic Acid: Pharmacological Evaluation and Stability Studies in Liver Microsomes, Rat Plasma, and Mycobacterium tuberculosis culture. ACS Infect Dis 2019, 5 (9), 1634–1644. 10.1021/acsinfecdis.9b00208. [DOI] [PubMed] [Google Scholar]
- Smith P. W.; Zuccotto F.; Bates R. H.; Martinez-Martinez M. S.; Read K. D.; Peet C.; Epemolu O. Pharmacokinetics of beta-Lactam Antibiotics: Clues from the Past To Help Discover Long-Acting Oral Drugs in the Future. ACS Infect Dis 2018, 4 (10), 1439–1447. 10.1021/acsinfecdis.8b00160. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.