

Abstract
Introduction
Neocortical 3R4R (3‐repeat/4‐repeat) tau aggregates are rarely observed in the absence of amyloid beta (Aβ). 18F‐MK6240 binds specifically to the 3R4R form of tau that is characteristic of Alzheimer's disease (AD). We report four cases with negative Aβ, but positive tau positron emission tomography (PET) findings.
Methods
All Australian Imaging, Biomarkers and Lifestyle study of aging (AIBL) study participants with Aβ (18F‐NAV4694) and tau (18F‐MK6240) PET scans were included. Centiloid <25 defined negative Aβ PET (Aβ–). The presence of neocortical tau was defined quantitatively and visually.
Results
Aβ– PET was observed in 276 participants. Four of these participants (one cognitively unimpaired [CU], two mild cognitive impairment [MCI], one AD) had tau tracer retention in a pattern consistent with Braak tau stages V to VI. Fluid biomarkers supported a diagnosis of AD. In silico analysis of APP, PSEN1, PSEN2, and MAPT genes did not identify relevant functional mutations.
Discussion
Discordant cases were infrequent (1.4% of all Aβ– participants). In these cases, the Aβ PET ligand may not be detecting the Aβ that is present.
Keywords: Alzheimer's disease, amyloid beta (Aβ), discordant, positron emission tomography, tau
1. BACKGROUND
Amyloid beta (Aβ) plaques and tau neurofibrillary tangles (NFTs) are neuropathological hallmarks of Alzheimer's disease (AD). The amyloid‐centric hypothesis posits that Aβ deposition precedes tau deposition and neurodegeneration. 1 , 2 The National Institute on Aging–Alzheimer's Association (NIA‐AA) research framework for AD incorporates biomarkers of Aβ, tau, and neurodegeneration into the AT(N) classification. 3 Individuals across the AD continuum have positive Aβ biomarkers, with or without abnormal tau biomarkers (Aβ+T– or Aβ+T+). 3 Combined Aβ and tau positron emission tomography (PET) biomarker studies show that high neocortical tau is typically observed in association with abnormal Aβ. 4 , 5 , 6 , 7
In cognitively unimpaired (CU) individuals, tau aggregates restricted to the mesial temporal lobe, brainstem, basal forebrain, and olfactory areas with few or absent Aβ plaques has been given the neuropathological designation of primary age‐related tauopathy (PART), 8 whereas the clinical entity of tangle‐predominant dementia (TPD) is observed in very elderly cognitively impaired cohorts. 9 It is common for Aβ– cases with high tau in the mesial temporal lobe to be considered consistent with PART or TPD. On neuropathology, tau deposition in these conditions is typically limited to Braak stages lower than IV, rarely extending to involve the neocortex beyond temporal regions. 8 , 9 Aβ–T+ has also been proposed to represent individuals with microtubule‐associated protein tau (MAPT) mutations. 7 , 10 , 11 Carriers of MAPT mutations such as R406W and V337M produce AD‐like 3‐repeat/4‐repeat (3R4R) cortical tau aggregates that are visually detectable using the tau PET ligand 18F‐AV1451 (flortaucipir) at levels comparable to those observed in AD. 10 Yet the pattern is distinct, with predominant tau PET tracer retention in the anterior temporal pole and frontal cortex, 10 unlike the posterior temporoparietal pattern observed in AD. These entities do not account for discordant low Aβ PET retention and high tau tracer retention, extending into Braak stages V to VI, in a pattern typical for AD.
The alternative consideration in these cases is that the Aβ PET ligand is not detecting the Aβ that is present. PET studies using 11C‐PiB have identified a handful of cases where a low Aβ PET result has been observed, despite neuropathology or alternative biomarker evidence of AD. 12 , 13 , 14 , 15 , 16 , 17 , 18
This study estimates the frequency of cases with discordant low Aβ PET retention and high neocortical tau tracer retention in an observational cohort, characterizes these participants, and hypothesizes that, in these instances, the Aβ PET ligand may not be detecting the Aβ present.
2. METHODS
2.1. Participants
Participants from the Australian Imaging Biomarkers and Lifestyle (AIBL) Flagship study of ageing and the Australian Dementia Network (ADNeT) who completed both an Aβ (18F‐NAV4694) and a tau (18F‐MK6240) PET scan before April 2020 were included in this study. Participants complete cognitive assessments every 12 to 18 months. Based on available clinical and neuropsychology assessments, a multi‐disciplinary clinical review panel determines whether an individual is CU or meets criteria for mild cognitive impairment (MCI) or AD dementia. An individual is deemed to be CU if they perform within 1.5 standard deviation (SD) of published norms for their age group, whereas diagnoses of MCI and AD dementia are determined in accordance with internationally agreed criteria. 19 The full methodology for cohort recruitment and evaluation has been described previously. 20 All relevant institutional review boards have approved this study, and written informed consent was obtained from all participants.
2.2. Image acquisition
Radiotracers were manufactured in‐house (Austin Health, Melbourne, Australia). PET scans were acquired on either the Philips TF64 PET/CT or Siemens Biograph mCT. A low‐dose CT was obtained for attenuation correction.
RESEARCH IN CONTEXT
Systematic Review: The authors reviewed the literature using PubMed, conference abstracts, and related presentations. Cases with low amyloid beta (Aβ) positron emission tomography (PET) signal, despite neuropathology or alternative biomarker evidence of Aβ, have been rarely reported.
Interpretation: Four participants from the Australian Imaging Biomarkers and Lifestyle study of aging (AIBL) cohort had a negative Aβ (18F‐NAV4694) PET, but quantitively and visually high neocortical tau (18F‐MK6240) tracer retention, in a pattern consistent with Braak stages V to VI. These discordant cases were rare (1.4% of all Aβ– participants). Fluid biomarkers supported a diagnosis of AD in these cases. Whole exome sequencing did not identify relevant APP, PSEN1, PSEN2, or MAPT mutations. These findings suggest that, similar to prior reports, the Aβ ligand may not be detecting the Aβ that is present in these cases.
Future Directions: Post‐mortem assessments of similar cases would be beneficial to better elucidate the reasons for the low Aβ PET signal.
Aβ PET imaging involved intravenous (IV) administration of 200 MBq (±10%) 18F‐NAV4694 with a 20‐minute acquisition time, commencing 50‐minutes post‐injection. Tau PET imaging involved IV administration of 185 MBq (±10%) 18F‐MK6240, with a 20‐minute acquisition time, commencing 90‐minutes post‐injection.
2.3. Image analysis
Centiloid values were computed from Aβ images using CapAIBL (https://milxcloud.csiro.au/tools/capaibl). 21 Centiloid <25 was used to designate a low (negative) Aβ result.
Tau 18F‐MK6240 PET scans were spatially normalized using the CapAIBL Principal Component Analysis (PCA)‐based approach 22 and scaled using the cerebellar cortex as the reference region. A gray matter inclusion mask and a meninges exclusion mask were applied. Standardized uptake value ratios (SUVR) were estimated in three composite regions‐of‐interest (ROIs): mesial temporal (Me) (comprising entorhinal cortex, hippocampus, parahippocampus, and amygdala); temporoparietal (Te) (comprising inferior and middle temporal gyrus, fusiform, supramarginal and angular gyri, posterior cingulate/precuneus, superior and inferior parietal, and lateral occipital cortex); and the rest of neocortex (R) (comprising the dorsolateral and ventrolateral prefrontal, orbitofrontal, gyrus rectus, superior temporal cortex, and anterior cingulate). 23 The 95th percentile (95%ile) of Aβ– CU participants was used to discriminate between high (+) and low (–) tau burden in each ROI (SUVR thresholds: 1.18 Me, 1.24 Te, and 1.08 R). A peri‐threshold region, comprising between the 90%ile and 99%ile, was also established, resulting in an upper bound of the peri‐threshold range of 1.29 for Me, 1.33 for Te, and 1.17 for R.
For scans where tau tracer retention was above the upper bound of the peri‐threshold range in Te or R, one expert reader (CCR), blind to participant characteristics and quantitative results, visually read the scans using MedImage Software with MedView (version 12) displayed in a white‐black scale. Scans were visually rated for the presence of tau in mesial temporal, temporal, parietal, occipital, posterior cingulate, precuneus, and frontal cortices.
Cases of high neocortical tau were those that had both quantitatively high neocortical tau (tracer retention above the upper bound of the peri‐threshold range in Te and/or R) and were visualized to have tau in neocortical regions.
2.4. Apolipoprotein E (APOE) genotyping
Apolipoprotein E (APOE) genotype was determined from whole blood–extracted DNA as per methodology previously described. 24
2.5. Whole exome sequencing
Whole exome sequencing was performed for participants for whom a blood sample was available (participants 1, 3, and 4). Samples were sequenced on an Illumina NovaSeq platform (150 bp paired‐end reads) using the IDT xGen Exome Research Panel v2 and the Illumina bcl2fastq 2.20.0.422 pipeline to generate the sequence data. Sequence alignment was via the Burrows‐Wheeler Aligner Tool (BWA mem) and variant calls were made with HaplotypeCaller by GATK version 4.0.4.0. Targeted analysis was then undertaken to identify the presence of potential deleterious and functionally relevant genetic variation within the amyloid precursor protein (APP), presenilin 1 (PSEN1), presenilin 2 (PSEN2), and MAPT genes. Sorting Intolerant From Tolerant (SIFT) 25 and Polymorphism Phenotyping version 2 (PolyPhen‐2) 26 programs were used as predictive tools to determine whether any identified amino acid substitutions in a protein‐coding region were likely to affect its function.
3. RESULTS
Of the 452 participants who had both an Aβ and tau PET scan, 276 had a low Aβ PET result (Aβ−). Of those with a low Aβ PET result, 23 (8% of all Aβ−) were above the tau Te cut‐off (with 9 above the upper bound of the peri‐threshold) and 22 (8% of all Aβ−) were above the tau R cutoff (with 7 above the upper bound). Thus 12 participants (4.3% of all Aβ−) were above the tau upper bound cutoff in Te (n = 5) or R (n = 3), or both Te and R (4). Of these, four participants (1.4% of all Aβ−) were observed to have substantial and unequivocal neocortical tau on visual inspection (Figure 1). Table 1 outlines the demographics and characteristics of these four participants, whereas Table 2 provides the fluid biomarker results for these participants.
FIGURE 1.
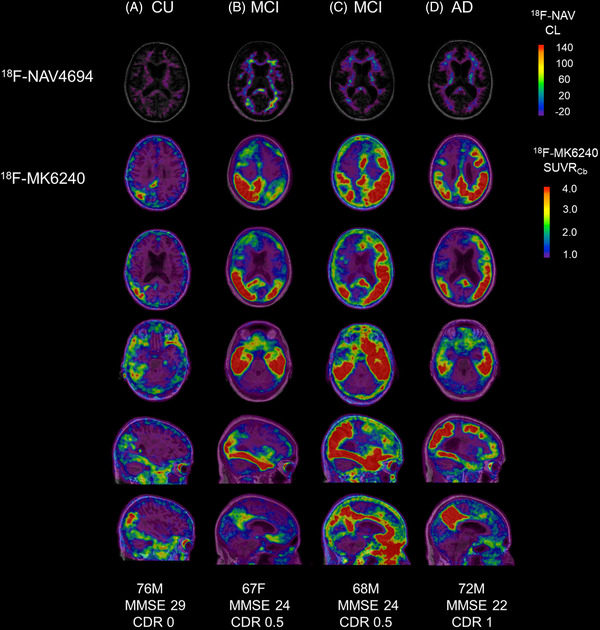
Amyloid beta (Aβ) and tau positron emission tomography (PET) scans of participants 1‐4. Aβ (18F‐NAV4694) (top row) and tau (18F‐MK6240) PET scans (bottom five rows) for participants 1‐4, co‐registered onto a T1‐weighted magnetic resonance imaging (MRI) template. CU, cognitively unimpaired; MCI, mild cognitive impairment; AD, Alzheimer's disease; M, male; F, female; MMSE, Mini‐Mental State Examination; CDR, Clinical Dementia Rating scale; CL, Centiloid; SUVR, standardized uptake value ratio
TABLE 1.
Characteristics of participants with low Aβ PET and high neocortical tau PET tracer retention
| Participant 1 | Participant 2 | Participant 3 | Participant 4 | |
|---|---|---|---|---|
| Age | 76 | 67 | 68 | 72 |
| Sex | M | F | M | M |
| Diagnosis | CU | MCI | MCI | AD |
| Centiloid | −2.50 | 1.43 | 1.82 | 18.40 |
| APOE | E3/E3 | – | E3/E3 | E4/E3 |
| Me SUVR | 1.14 | 2.73 a | 3.27 a | 1.27 |
| Te SUVR | 1.37 a | 3.59 a | 4.01 a | 3.04 a |
| R SUVR | 0.96 | 1.89 a | 2.01 a | 1.68 a |
| MTL atrophy b | 0 | 0 | 2 (1 right, 3 left) | 2.5 (2 right, 3 left) |
| PVWMH c | 0 | 2 | 10 | 2 |
| DWMH c | 1 | 2 | 0 | 1 |
| MMSE | 29 | 24 | 24 | 22 |
| CDR | 0 | 0.5 | 0.5 | 1 |
Abbreviations: AD, Alzheimer's disease; APOE, apolipoprotein E; CDR, Clinical Dementia Rating scale; CU, cognitively unimpaired; DMWHs, deep white matter hyperintensities; MCI, mild cognitive impairment; MMSE, Mini‐Mental State Examination; MTL, mesial temporal lobe; PVMWHs, peri‐ventricular white matter hyperintensities; SUVR, standardized uptake value ratio; Me, mesial temporal; Te, temporo‐parietal; R, rest of brain.
= above the upper bound of the peri‐threshold range for the specified region of interest (ROI).
= average (right and left) mesial temporal lobe atrophy scores, as previously described. 27
= Fazekas’ score for the visual assessment of white matter hyperintensities, as previously described. 28
TABLE 2.
Additional biomarker results for participants with low Aβ PET and high neocortical tau PET tracer retention
| Biomarker | Participant 1 | Participant 2 | Participant 3 | Participant 4 | |
|---|---|---|---|---|---|
| CSF | CSF Aβ1‐42 | – | 386pg/mL (low) a | – | – |
| CSF p‐tau | – |
89pg/mL (high) a |
– | – | |
| CSF t‐tau | – | 553pg/mL (high) a | – | – | |
| Plasma | Plasma Aβ b | 1.69 (abnormal) | – | – | – |
| Plasma | Plasma p217+tau c | High | – | High | High |
= reference range for CSF Aβ1‐42 >656 pg/mL, p‐tau <59 pg/mL, t‐tau <304 pg/mL, where the age‐adjusted reference ranges were generated based on an AIBL healthy control cohort (>60 years). 29 Sample collected was tinged pink, with a red blood cell count of 1600/μL.
= AIBL composite value from a combined immunoprecipitation and mass spectrometry method previously described, 30 a measure validated in the AIBL cohort.
The referring diagnosis for participant 2 was MCI due to AD. An 18F‐FDG (fluorodeoxyglucose) ‐PET scan performed for clinical diagnostic purposes showed hypometabolism in the posterior cingulate, precuneus, and bilateral temporoparietal cortices, with relative sparing of the sensorimotor cortices consistent with AD (Figure 2).
FIGURE 2.
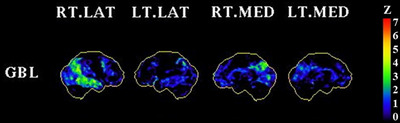
18F‐FDG (fluorodeoxyglucose) PET (positron emission tomography) scan for participant 2. This figure shows a Z‐score map (displayed in NeuroStat 3D‐SSP) of glucose metabolism generated by comparing the 18F‐FDG PET scan of participant 2 against 20 Aβ negative healthy controls with a mean age of 72. The results were normalized to a global (whole brain) reference region
3.1. Whole exome sequencing results
For the three participants for whom whole exome sequencing was performed (participants 1, 3, and 4), no allelic variants identified in APP, PSEN1, and PSEN2 were predicted to be deleterious or damaging using SIFT and PolyPhen‐2. Participant 4 had two missense variants in the MAPT gene (chromosome 17), with a C→T substitution at both rs17651549 and rs63750222. Both variants are predicted to be “deleterious” using SIFT and “probably damaging” using PolyPhen‐2. However, both of these alternative alleles are observed in 24% of the European population. 33 No MAPT mutations were identified for participants 1 and 3.
4. DISCUSSION
In this cohort of 452 participants, 4 participants (0.88%) (or 4/276, 1.4% of all Aβ− participants) had low Aβ PET and high neocortical tau PET tracer retention, quantitatively and visually. This aligns with prior reports suggesting that high neocortical tau tracer retention is rare in the absence of an abnormal Aβ PET. 4 , 5 , 6 , 7
The four participants with low Aβ PET and high neocortical tau PET tracer retention had a tau distribution pattern consistent with that typically observed in Aβ+ individuals with AD. Participant 1, who was CU, had mesial temporal and asymmetric, right temporoparietal cortical 18F‐MK6240 tracer retention, whereas the other three participants (two classified as having amnestic MCI and one with AD dementia) had high 18F‐MK6240 tracer retention in mesial temporal, posterior cingulate, precuneus, lateral temporal, lateral parietal, lateral occipital, and prefrontal cortices, characteristic of AD Braak stages V to VI. 29 This pattern is consistent with the regions of high 18F‐MK6240 binding reported previously for Aβ+ individuals across the AD continuum. 30 The distribution of 18F‐MK6240 binding has been shown to match the post‐mortem regional distribution of tau NFT, as observed for two individuals with AD dementia in a prior study. 31 Autoradiography has also shown that 18F‐MK6240 has a very high affinity for mixed 3R4R tau isoforms in AD over 3R or 4R isoforms present in non‐AD tauopathies. 32 Although whole exome sequencing identified two missense mutations in the MAPT gene in participant 4, these alternative alleles are observed not infrequently in European populations. The absence of these missense mutations in the other participants and their moderately high minor allele frequency in the general population suggest that they are unlikely to be the driver of the observed discordance. However, it cannot be ruled out that these missense variants may contribute to the observed discordance, in combination with variants in other genes that were not investigated. Of note, whole exome sequencing did not identify known mutations in the MAPT gene (such as R406W), which have been associated with the formation of mixed 3R4R tau aggregates.
In this study, participants with low Aβ PET and high neocortical tau PET were examined specifically to explore potential explanations for this combination of biomarker results. In primary age related tauopathy and tangle predominant dementia (PART/TPD), the distribution of tau involves the mesial temporal cortex, and can extend into the temporal lobes. We, along with other investigators, have documented elevated mesial temporal tau in the absence of Aβ, which may reflect PART. 34 , 35 , 36 , 37 However, this study is restricted to participants with extensive cortical tau deposition, well beyond that typical of PART, and more consistent with AD. The extent of neocortical tracer retention and relatively younger age of these participants suggest that these cases do not simply reflect PART/TPD.
Aβ tracer 18F‐NAV4694 has a binding profile comparable to that of 11C‐PiB. 38 Aβ burden in this study was measured in Centiloids, and participants 1 to 3 had Centiloid values ranging from −2.5 to 1.8. A Centiloid value of zero represents the mean Aβ burden derived from scans of young healthy individuals, 39 and Centiloid values less than 10 have been observed to reflect the absence of any neuritic plaques at post‐mortem. 40 Although the Centiloid value approached the pre‐specified threshold for participant 4, the low Aβ burden was in relative discordance with the extensive neocortical tau PET retention observed. We have observed recently that widespread tau deposition in the neocortex on PET occurs primarily above an Aβ burden of 40 Centiloid. 6
The four participants with discordant Aβ and tau PET results had alternative biomarker findings that were supportive of a diagnosis of AD, despite the low Aβ PET results. Participant 1 had a plasma Aβ result available, measured using a combined immunoprecipitation and mass spectrometry method, which has been evaluated in the AIBL cohort. 41 An AIBL composite result (described previously) 41 was consistent with this individual being Aβ positive. Participants 1, 3, and 4 had high plasma p217+tau. Plasma p217+tau is a recently developed biomarker that detects tau phosphorylation at epitopes 217 and 212, 42 and evaluation in the AIBL cohort has shown that a high level of p217+tau has good concordance with both abnormal Aβ and tau PET. 43 Participant 2 had both an 18F‐FDG PET scan and cerebrospinal fluid (CSF) analysis. The observed 18F‐FDG PET pattern of glucose hypometabolism in the posterior cingulate, precuneus, and bilateral temporoparietal cortices with relative sparing of the sensorimotor cortices is consistent with the pattern of hypometabolism observed in AD. This participant's low CSF Aβ1‐42 and elevated CSF phosphorylated tau (p‐tau) and total tau (t‐tau) is also characteristic of AD. However, it should be noted that the CSF sample was tinged pink and had a red cell count of 1600/μL. Blood contamination has been observed to decrease CSF Aβ1‐42 and Aβ40 levels, proportional to the amount of blood present. 44 Of note, blood contamination <5000 red blood cells/μL, as in our case, has been observed to have a negligible impact on Aβ1‐42 levels. 45 The impact on p‐tau and t‐tau is less clear. With these caveats, CSF results for participant 2 may also suggest a diagnosis of AD.
The patterns of tau tracer retention, relative discordance between the Aβ and tau PET findings, and alternative biomarker evidence suggesting a diagnosis of AD in these four participants raises the possibility that these participants have Aβ deposits undetected by the PET ligand. To our knowledge, there are no reports of this occurring with 18F‐NAV4694; however, a handful of cases have been reported using 11C‐PiB, 12 , 13 , 14 , 15 , 16 , 17 , 18 and it has been reported in a multi‐center study in which participants were scanned using 11C‐PiB, as well as Aβ ligands 18F‐Florbetapir and 18F‐Florbetaben. 46 Similar to the current study, participants with low Aβ and high tau in a meta‐temporal composite ROI in this latter multi‐center study were young (mean age of 66.2), four of seven were APOE ε4 negative, and had a tau (18F‐Flortaucipir) PET pattern consistent with a pattern typically observed in AD in the majority of cases reported, but in contrast to this study, four of seven participants had an atypical clinical phenotype. 46
There may be different Aβ conformations that are undetected by the ligand, and this has been observed in non‐human primates that have the same amino acid sequence as human Aβ, but the aggregates have a different conformation. 47 , 48 In one report, a patient was found to lack high‐affinity binding sites for 3H‐PiB, despite detection of extensive Aβ deposition. 15 A few prior reports using 11C‐PiB identified APP mutations associated with low 11C‐PiB signal. 13 , 18 APP mutation E693Delta, for example, was observed to result in alternative Aβ processing, leading to a propensity toward formation of oligomers over fibrillar Aβ. 13 In this study, for the three participants for whom a blood sample was available for processing, whole exome sequencing did not identify any known APP, PSEN1, or PSEN2 mutations. An undetected novel mutation in these individuals cannot be excluded. Post‐mortem assessments of these cases would be beneficial to better elucidate the reasons for the low Aβ PET signal.
The limitations of this study are the lack of availability of a more comprehensive panel of validated non‐Aβ/non‐tau PET AD biomarkers, the lack of an available blood sample for participant 2, and the absence of post‐mortem correlation. Although blood‐based biomarkers for AD (plasma Aβ and p‐tau) are very promising, these biomarkers are not yet as established as CSF AD biomarkers.
5. CONCLUSION
In this cohort, high neocortical tau PET retention in combination with low Aβ PET was rare (1.4% of all Aβ− individuals). The four participants with unequivocal discordant PET findings had alternative biomarker findings that supported a diagnosis AD, suggesting that similar to prior reports, the Aβ PET ligand may not be detecting the Aβ present in these cases.
CONFLICT OF INTERESTS
This work was supported by the National Health and Medical Research Council (NHMRC) (grant numbers APP1140853, APP1132604). Natasha Krishnadas was supported by a cofunded PhD scholarship from Australian Rotary Health and the Estate of Bartolina Peluso. Simon M. Laws has received institutionally administered grants as follows: NHMRC APP1151854, APP1161706, APP1191535, APP2001320, APP2007656; Department of Health (Government of Western Australia) ‐ Multiple Sclerosis Western Australia ‐ Australian Alzheimer's Research Foundation ‐ Edith Cowan University, Strategic Research Centre Support ‐ Florey Institute of Neuroscience and Mental Health. Simon M. Laws has participated on the Cytox Group Ltd ‐ Scientific Advisory Board (pro bono). Victor L Villemagne has received consulting fees from IXICO, Eli Lilly, Life molecular imaging, and Hospicom, and has received payment/honoraria from ACE Barcelona. Christopher C. Rowe has received grants from Cerveau Technologies (institution), Eisai (institution), and Biogen (institution). Christopher C. Rowe has received consulting fees from Nutricia (speaker fee), Prothera, and Biogen (for preparation of educational material). Christopher C. Rowe has participated on a data safety board/advisory board for Cerveau Technologies (unpaid). Vincent Doré, Tenielle Porter, Fiona Lamb, and Svetlana Bozinovski do not report any disclosures.
ACKNOWLEDGMENTS
The data used in the preparation of this article were obtained from the Australian Imaging Biomarkers and Lifestyle (AIBL) flagship study of ageing, funded by the Commonwealth Scientific and Industrial Research Organization (CSIRO), National Health and Medical Research Council of Australia (NHMRC), and participating institutions. AIBL researchers are listed at www.aibl.csiro.au. The authors acknowledge Prof. Steve Collins and Dr. Qiao‐Xin Li for their assistance with interpreting the cerebrospinal fluid (CSF) data, and Dr. James Doecke for assistance with interpreting the plasma amyloid beta (Aβ) results. The authors thank all participants who took part in the study, as well as their families. This study was supported financially by the National Health and Medical Research Council (NHMRC) (grant numbers APP1140853, APP1132604). Christopher C. Rowe was the recipient of a research grant from Cerveau who supplied the MK6240 tau tracer precursor for research use. Natasha Krishnadas was supported by a co‐funded PhD scholarship from Australian Rotary Health/Bartolina Peluso.
Krishnadas N, Doré V, Laws SM, et al. Exploring discordant low amyloid beta and high neocortical tau positron emission tomography cases. Alzheimer's Dement. 2022;14:e12326. 10.1002/dad2.12326
REFERENCES
- 1. Hardy J, Allsop D. Amyloid deposition as the central event in the aetiology of Alzheimer's disease. Trends Pharmacol Sci. 1991;12:383‐388. [DOI] [PubMed] [Google Scholar]
- 2. Masters CL, Beyreuther K. Alzheimer's centennial legacy: prospects for rational therapeutic intervention targeting the Abeta amyloid pathway. Brain. 2006;129:2823‐2839. [DOI] [PubMed] [Google Scholar]
- 3. Jack CR, Bennett DA, Blennow K, et al. NIA‐AA Research Framework: toward a biological definition of Alzheimer's disease. Alzheimers Dement. 2018;14:535‐562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jack CR, Wiste HJ, Botha H, et al. The bivariate distribution of amyloid‐beta and tau: relationship with established neurocognitive clinical syndromes. Brain. 2019;142:3230‐3242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pontecorvo MJ, Devous MD, Sr. , Navitsky M, et al. Relationships between flortaucipir PET tau binding and amyloid burden, clinical diagnosis, age and cognition. Brain. 2017;140:748‐763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Doré V, Krishnadas N, Bourgeat P, et al. Relationship between amyloid and tau levels and its impact on tau spreading. Eur J Nucl Med Mol Imaging. 2021;48:2225‐2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Leuzy A, Smith R, Ossenkoppele R, et al. Diagnostic performance of RO948 F 18 tau positron emission tomography in the differentiation of Alzheimer disease from other neurodegenerative disorders. JAMA Neurol. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Crary JF, Trojanowski JQ, Schneider JA, et al. Primary age‐related tauopathy (PART): a common pathology associated with human aging. Acta Neuropathol. 2014;128:755‐766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jellinger KA, Attems J. Neurofibrillary tangle‐predominant dementia: comparison with classical Alzheimer disease. Acta Neuropathol. 2007;113:107‐117. [DOI] [PubMed] [Google Scholar]
- 10. Jones DT, Knopman DS, Graff‐Radford J, et al. In vivo (18)F‐AV‐1451 tau PET signal in MAPT mutation carriers varies by expected tau isoforms. Neurology. 2018;90:e947‐e54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Smith R, Puschmann A, Schöll M, et al. 18F‐AV‐1451 tau PET imaging correlates strongly with tau neuropathology in MAPT mutation carriers. Brain. 2016;139:2372‐2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Leinonen V, Alafuzoff I, Aalto S, et al. Assessment of beta‐amyloid in a frontal cortical brain biopsy specimen and by positron emission tomography with carbon 11‐labeled Pittsburgh Compound B. Arch Neurol. 2008;65:1304‐1309. [DOI] [PubMed] [Google Scholar]
- 13. Tomiyama T, Nagata T, Shimada H, et al. A new amyloid beta variant favoring oligomerization in Alzheimer's‐type dementia. Ann Neurol. 2008;63:377‐387. [DOI] [PubMed] [Google Scholar]
- 14. Cairns NJ, Ikonomovic MD, Benzinger T, et al. Absence of Pittsburgh compound B detection of cerebral amyloid beta in a patient with clinical, cognitive, and cerebrospinal fluid markers of Alzheimer disease: a case report. Arch Neurol. 2009;66:1557‐1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rosen RF, Ciliax BJ, Wingo TS, et al. Deficient high‐affinity binding of Pittsburgh compound B in a case of Alzheimer's disease. Acta Neuropathol. 2010;119:221‐233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sojkova J, Driscoll I, Iacono D, et al. In vivo fibrillar beta‐amyloid detected using [11C]PiB positron emission tomography and neuropathologic assessment in older adults. Arch Neurol. 2011;68:232‐240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ikonomovic MD, Abrahamson EE, Price JC, et al. Early AD pathology in a [C‐11]PiB‐negative case: a PiB‐amyloid imaging, biochemical, and immunohistochemical study. Acta Neuropathol. 2012;123:433‐447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Schöll M, Wall A, Thordardottir S, et al. Low PiB PET retention in presence of pathologic CSF biomarkers in Arctic APP mutation carriers. Neurology. 2012;79:229‐236. [DOI] [PubMed] [Google Scholar]
- 19. Fowler C, Rainey‐Smith SR, Bird S, et al. Fifteen Years of the Australian Imaging, Biomarkers and Lifestyle (AIBL) study: progress and observations from 2,359 older adults spanning the spectrum from cognitive normality to Alzheimer's disease. J Alzheimers Dis Rep. 2021;5:443‐468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ellis KA, Bush AI, Darby D, et al. The Australian Imaging, Biomarkers and Lifestyle (AIBL) study of aging: methodology and baseline characteristics of 1112 individuals recruited for a longitudinal study of Alzheimer's disease. Int Psychogeriatr. 2009;21:672‐687. [DOI] [PubMed] [Google Scholar]
- 21. Bourgeat P, Dore V, Fripp J, et al. Implementing the centiloid transformation for (11)C‐PiB and beta‐amyloid (18)F‐PET tracers using CapAIBL. Neuroimage. 2018;183:387‐393. [DOI] [PubMed] [Google Scholar]
- 22. Dore V, Bourgeat P, Burnham SC, et al. Automated reporting of tau PET quantification on the brain surface. Alzheimer's & Dementia. 2019;15:P1269‐P. [Google Scholar]
- 23. Villemagne V, Dore V, Bourgeat P. The Tau MeTeR composites for the generation of continuous and categorical measures of tau deposits in the brain. J Mol Med Ther 2017; 1 (1):25‐32. [Google Scholar]
- 24. Porter T, Burnham SC, Milicic L, et al. Utility of an Alzheimer's disease risk‐weighted polygenic risk score for predicting rates of cognitive decline in preclinical Alzheimer's disease: A prospective longitudinal study. J Alzheimers Dis. 2018;66:1193‐1211. [DOI] [PubMed] [Google Scholar]
- 25. Ng PC, Henikoff S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31:3812‐3814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Adzhubei I, Jordan DM, Sunyaev SR. Predicting functional effect of human missense mutations using PolyPhen‐2. Curr Protoc Hum Genet. 2013;Chapter 7:Unit7.20‐Unit7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Scheltens P, Leys D, Barkhof F, et al. Atrophy of medial temporal lobes on MRI in “probable” Alzheimer's disease and normal ageing: diagnostic value and neuropsychological correlates. J Neurol Neurosurg Psychiatry. 1992;55:967‐972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Fazekas F, Chawluk JB, Alavi A, Hurtig HI, Zimmerman RA. MR signal abnormalities at 1.5 T in Alzheimer's dementia and normal aging. AJR Am J Roentgenol. 1987;149:351‐356. [DOI] [PubMed] [Google Scholar]
- 29. Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del Tredici K. Staging of Alzheimer disease‐associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 2006;112:389‐404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kreisl WC, Lao PJ, Johnson A, et al. Patterns of tau pathology identified with (18) F‐MK‐6240 PET imaging. Alzheimers Dement. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pascoal TA, Benedet AL, Tudorascu DL, et al. Longitudinal 18F‐MK‐6240 tau tangles accumulation follows Braak stages. Brain. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Aguero C, Dhaynaut M, Normandin MD, et al. Autoradiography validation of novel tau PET tracer [F‐18]‐MK‐6240 on human postmortem brain tissue. Acta Neuropathol Commun. 2019;7:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Genomes Project C , Auton A, Brooks LD, Durbin RM, et al. A global reference for human genetic variation. Nature. 2015;526:68‐74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Krishnadas N, Doré V, Groot C, et al. Mesial temporal tau in amyloid‐β‐negative cognitively normal older persons. Alzheimers Res Ther. 2022;14:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Groot C, Doré V, Robertson J, et al. Mesial temporal tau is related to worse cognitive performance and greater neocortical tau load in amyloid‐β‐negative cognitively normal individuals. Neurobiol Aging. 2021;97:41‐48. [DOI] [PubMed] [Google Scholar]
- 36. Maass A, Lockhart SN, Harrison TM, et al. Entorhinal tau pathology, episodic memory decline, and neurodegeneration in aging. J Neurosci. 2018;38:530‐543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lowe VJ, Bruinsma TJ, Wiste HJ, et al. Cross‐sectional associations of tau‐PET signal with cognition in cognitively unimpaired adults. Neurology. 2019;93:e29‐e39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rowe CC, Pejoska S, Mulligan RS, et al. Head‐to‐head comparison of 11C‐PiB and 18F‐AZD4694 (NAV4694) for beta‐amyloid imaging in aging and dementia. J Nucl Med. 2013;54:880‐886. [DOI] [PubMed] [Google Scholar]
- 39. Klunk WE, Koeppe RA, Price JC, et al. The Centiloid Project: standardizing quantitative amyloid plaque estimation by PET. Alzheimers Dement. 2015;11:1‐15.e1‐4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Amadoru S, Doré V, McLean CA, et al. Comparison of amyloid PET measured in Centiloid units with neuropathological findings in Alzheimer's disease. Alzheimers Res Ther. 2020;12:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nakamura A, Kaneko N, Villemagne VL, et al. High performance plasma amyloid‐beta biomarkers for Alzheimer's disease. Nature. 2018;554:249‐254. [DOI] [PubMed] [Google Scholar]
- 42. Triana‐Baltzer G, Moughadam S, Slemmon R, et al. Development and validation of a high‐sensitivity assay for measuring p217+tau in plasma. Alzheimers Dement (Amst). 2021;13:e12204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Doré V, Doecke JD, Saad ZS, et al. Plasma p217+tau versus NAV4694 amyloid and MK6240 tau PET across the Alzheimer's continuum. Alzheimers Dement (Amst). 2022;14:e12307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Janelidze S, Stomrud E, Brix B, Hansson O. Towards a unified protocol for handling of CSF before β‐amyloid measurements. Alzheimer's research & therapy. 2019;11:63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bjerke M, Portelius E, Minthon L, et al. Confounding factors influencing amyloid Beta concentration in cerebrospinal fluid. International journal of Alzheimer's disease. 2010;2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Windon C IL, Mundada N, Apostolova L, et al. Clinical and PET imaging characteristics of amyloid negative, tau positive cases. Tau2022 Global Conference abstract. 2022.
- 47. Rosen RF, Walker LC, LeVine III H. PIB binding in aged primate brain: enrichment of high‐affinity sites in humans with Alzheimer's disease. Neurobiology of aging. 2011;32:223‐234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Levine H, 3rd , Walker LC. Molecular polymorphism of Abeta in Alzheimer's disease. Neurobiol Aging. 2010;31:542‐548. [DOI] [PMC free article] [PubMed] [Google Scholar]