

Abstract
The nucleosome remodeling and deacetylase (NuRD) complex is a chromatin‐modifying assembly that regulates gene expression and DNA damage repair. Despite its importance, limited structural information describing the complete NuRD complex is available and a detailed understanding of its mechanism is therefore lacking. Drawing on information from SEC‐MALLS, DIA‐MS, XLMS, negative‐stain EM, X‐ray crystallography, NMR spectroscopy, secondary structure predictions, and homology models, we applied Bayesian integrative structure determination to investigate the molecular architecture of three NuRD sub‐complexes: MTA1‐HDAC1‐RBBP4, MTA1N‐HDAC1‐MBD3GATAD2CC, and MTA1‐HDAC1‐RBBP4‐MBD3‐GATAD2A [nucleosome deacetylase (NuDe)]. The integrative structures were corroborated by examining independent crosslinks, cryo‐EM maps, biochemical assays, known cancer‐associated mutations, and structure predictions from AlphaFold. The robustness of the models was assessed by jack‐knifing. Localization of the full‐length MBD3, which connects the deacetylase and chromatin remodeling modules in NuRD, has not previously been possible; our models indicate two different locations for MBD3, suggesting a mechanism by which MBD3 in the presence of GATAD2A asymmetrically bridges the two modules in NuRD. Further, our models uncovered three previously unrecognized subunit interfaces in NuDe: HDAC1C‐MTA1BAH, MTA1BAH‐MBD3MBD, and HDAC160–100‐MBD3MBD. Our approach also allowed us to localize regions of unknown structure, such as HDAC1C and MBD3IDR, thereby resulting in the most complete and robustly cross‐validated structural characterization of these NuRD sub‐complexes so far.
Keywords: Bayesian integrative structure determination, chromatin remodeling complexes, cryo‐EM, histone modification, integrative modeling, nucleosome remodeling and deacetylase complex, XLMS
1. INTRODUCTION
The nucleosome remodeling and deacetylase (NuRD) complex is a multi‐protein chromatin‐modifying assembly that is expressed in most metazoan tissues and conserved across animals. 1 , 2 It regulates gene expression and DNA damage repair by modulating nucleosome accessibility in enhancers and promoters for transcription factors and RNA polymerases. 3 , 4 , 5 , 6 , 7 Subunits of NuRD are implicated in human cancers and various congenital defects. 8 , 9 Considerable diversity is observed in subunit isoforms and NuRD‐associated factors across tissues. 10 , 11
NuRD comprises two catalytic modules—a histone deacetylase module and ATP‐dependent chromatin‐remodeling module. 2 , 12 The deacetylase module contains metastasis‐associated proteins (MTA1/2/3) that form a dimeric scaffold for the histone deacetylases (HDAC1/2). It also contains the proteins RBBP4/7, which mediate interactions of NuRD with histone tails and transcription factors. 13 , 14 The chromatin‐remodeling module contains methyl‐CpG DNA‐binding proteins (MBD2/3) that recruit NuRD to methylated and/or hemi‐methylated DNA, GATA‐type zinc‐finger proteins (GATAD2A/B), and an ATP‐dependent DNA translocase (CHD3/4/5). 10 , 12 Note that each of these subunits exists as two or more paralogues that can be incorporated into NuRD; for simplicity, we generally refer to only one paralogue of each subunit herein.
Some structural information is available for the complex. The stoichiometry of the complex, based on a consensus of several recent structural, biochemical and quantitative mass spectrometry studies appears to be 2:2:4:1:1:1 (MTA:HDAC:RBBP:MBD:GATAD2:CHD). 12 , 15 , 16 These data also suggest that NuRD can be thought of as two “modules” with distinct enzymatic activities: a deacetylase sub‐complex comprising MTA1, HDAC2, and RBBP4 (MHR) and a chromatin remodeling sub‐complex comprising CHD4, GATAD2A and MBD3 (MGC). It is notable that MHR also appears to exist in the cell as a distinct assembly, as shown in work that demonstrates an interaction between this sub‐complex and the chromatin regulator PWWP2A. 17 , 18
Experimental structures of parts of the NuRD complex, including the 2:2 MTA1ELM2/SANT‐HDAC1 dimer, RBBP4 bound to the C‐terminal half of MTA1, the MBD domain of MBD3, the coiled‐coil dimer of MBD2 and GATAD2A, and CHD4 bound to a nucleosome have been determined by X‐ray crystallography, cryo‐electron microscopy and NMR spectroscopy. 16 , 19 , 20 , 21 , 22 , 23 Structures of several other NuRD sub‐complexes have also been characterized at various resolutions by negative‐stain electron microscopy, including MHR, as well as two simplified assemblies that are not known to have independent biological functions: (a) a 2:2:2 MTA1N‐HDAC1‐MBD3GATAD2CC (MHM) complex and (b) a 2:2:4:1:1 MTA1‐HDAC1‐RBBP4‐MBD3‐GATAD2A [nucleosome deacetylase (NuDe) complex]. 12 , 23 , 24
Pairwise interactions between domains and subunits within the MHR, MHM, NuDe, and the endogenous NuRD complexes have also been characterized by chemical crosslinking and mass spectrometry (XLMS). 12 , 23 , 25 , 26 , 27 , 28 , 29 A model of the MHM sub‐complex, based on crosslink‐driven rigid‐body docking of known atomic structures together with manual placement of a pair of MTA1‐(RBBP4)2 structures, has also been reported. 12 While this represents the most complete model of NuRD architecture, it was created manually and accounts for only 30% of residues in the NuRD complex. In fact, only 50% of residues in NuRD have known or readily modeled atomic structures, and the structures of proteins such as MBD3, CHD4, and GATAD2A are largely uncharacterized. While more recent artificial intelligence‐based methods such as AlphaFold hold more promise for modeling these uncharacterized regions, significant limitations remain for the modeling of multi‐protein complexes, including size limitations and issues with the modeling of disordered or irregularly structured regions in protein assemblies. 30 These issues, combined with variability in the paralogue composition and significant structural dynamics, suggest that the full structure of the NuRD complex is likely to remain a challenge in the near future.
A striking feature of the NuRD complex is its asymmetric stoichiometry. The MHR complex displays pseudo two‐fold symmetry and yet engages with a 1:1:1 MGC complex. It is known that MBD3 binds to the MHR complex and that the 2:2:4:1:1 MTA1‐HDAC1‐RBBP4‐MBD3‐GATAD2A (NuDe complex) is also asymmetric. 12 In contrast, the MHM sub‐complex, containing only HDAC1, MBD3 and MTA1, has a 2:2:2 stoichiometry, suggesting that it is GATAD2A that introduces the asymmetry. However, the mechanism by which this asymmetry is introduced in NuDe/NuRD is not known. Also, the structure of full‐length MBD3, which contains a significant intrinsically disordered region (IDR; MBD371–213) critical (in the case of MBD2) for recruiting the deacetylase core, is unknown. 31 The localization of full‐length MBD3 in NuDe/NuRD is also not known.
Here, we investigated the molecular architecture of the MHR, MHM, and NuDe sub‐complexes, using Bayesian integrative modeling with the Integrative Modeling Platform (IMP). 32 , 33 , 34 This approach allowed us to combine data from multiple sources, including SEC‐MALLS, DIA‐MS, XLMS, negative‐stain EM, X‐ray crystallography, NMR spectroscopy experiments, secondary structure and homology predictions, and stereochemistry considerations 12 , 16 , 19 , 20 , 22 , 23 , 35 to obtain integrative structures with precisions of 27 Å (MHR), 28 Å (MHM), and 34 Å (NuDe). These structures were corroborated by independent crosslinks, cryo‐EM maps, biochemical assays, known cancer‐associated mutations, and structure predictions from AlphaFold. 24 , 31 , 36 , 37 , 38 The integrative approach facilitated the modeling of proteins with significant regions of unknown structure, such as full‐length MBD3. Our models indicate that MBD3 can potentially localize to two different sites in NuRD, suggesting a mechanism by which MBD3, in the presence of GATAD2A, asymmetrically bridges the deacetylase and chromatin‐remodeling modules. Finally, our models enable us to compare the structure of the MHR complex in the presence and absence of MBD3 and GATAD2A. We show that, while the MHR complex alone is relatively dynamic, the presence of MBD3 and GATAD2A makes it less dynamic.
2. RESULTS
2.1. Integrative modeling workflow
The modeled NuRD subunits, their domains, their representations, and the number of copies in the modeled complexes are shown in Figure 1. The representative paralogues used for all calculations were MTA1, HDAC1, RBBP4, MBD3, and GATAD2A (Figure 1a). The stoichiometry of the modeled proteins was informed by previous DIA‐MS and SEC‐MALLS experiments (Figure 1b). 12 The integrative modeling of the MHR, MHM, and NuDe complexes proceeded in four stages (Figure 2, Section 4). 32 , 33 , 34
FIGURE 1.
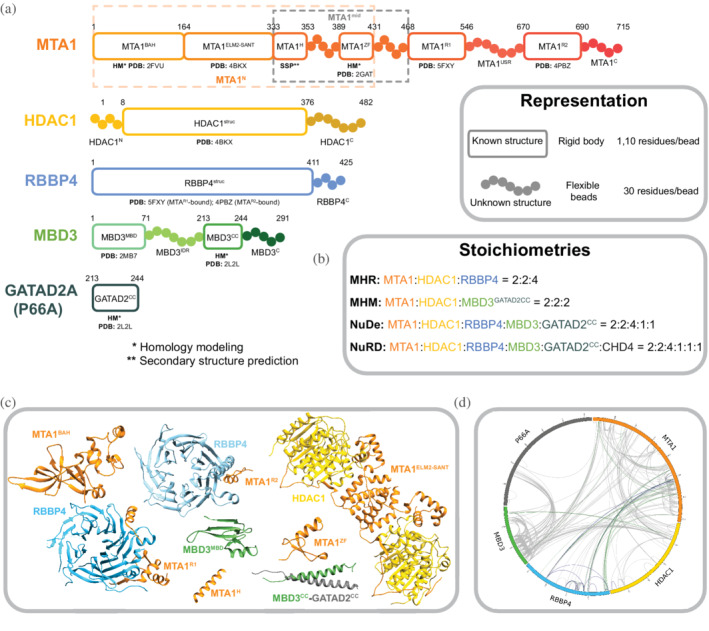
Subunits in nucleosome remodeling and deacetylase (NuRD) sub‐complexes. (a) Sequences and isoforms of NuRD subunits modeled in this study are shown and domains are labeled. Only one paralogue of each subunit is shown. Domains are shown in progressively dark shades along the sequence for MTA1, HDAC1, and MBD3. Regions for which atomic structures exist, or can be predicted, are represented by rectangles whereas regions without known structure are represented by beads. PDB IDs are shown for existing subunit structures and templates of homology models. Orange and gray dashed rectangles represent MTA1N and MTA1mid respectively. Numbering is for the human proteins. (b) Stoichiometries of modeled sub‐complexes and the endogenous NuRD complex. (c) Previously published atomic structures that were used for modeling. PDB codes are given in (a). (d) Crosslinks used in this study, represented as a CIRCOS (http://cx‐circos.net/) plot. The gray, blue, and green lines represent all the BS3/DSS, ADH, and DMTMM crosslinks respectively.
FIGURE 2.
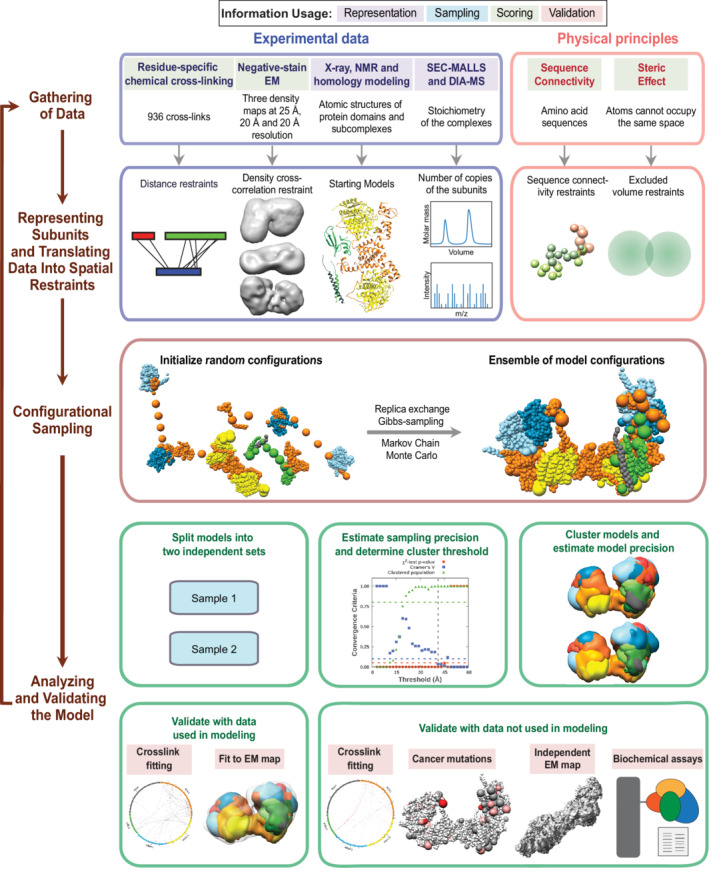
Integrative structure determination of nucleosome remodeling and deacetylase (NuRD) sub‐complexes. Schematic describing the workflow for integrative structure determination of NuRD sub‐complexes. The first row describes the input information. The second‐row details how data are used to encode spatial restraints. The third row describes the sampling method, and the last two rows illustrate the analysis and validation protocol. The background colors of the input information show the stage of modeling in which the information is used, as shown in the legend at the top.
We first represented each protein as a series of beads of sizes that depend on the degree of knowledge of the structure (which can vary throughout the sequence). Protein domains with known atomic structures (such as the MTA1‐HDAC1 dimer) were represented at 1 and 10 residues per bead and modeled as rigid bodies, whereas domains without known structure (such as the MBD3IDR) were coarse‐grained at 30 residues per bead and modeled as flexible strings of beads (Figure 1a). Data from chemical crosslinking combined with mass spectrometry (XL‐MS) were used to restrain the distance between crosslinked residues (Figure 1d). Negative‐stain EM maps were used to restrain the shape of the complexes (Figure S1). 12
The simulations started with randomized configurations for the rigid bodies and flexible beads. Over 40 million models per complex were sampled using a Monte Carlo approach (Replica Exchange Gibbs Sampling MCMC; described in Section 4). The models were scored based on agreement with XL‐MS and EM data, together with additional stereochemical restraints such as connectivity and excluded volume. For each complex, about 20,000 models that sufficiently satisfied the input information were selected for further analysis. 39 , 40
These models were clustered based on structural similarity and the precision of the clusters was estimated (Figures S2–S4). 39 , 40 , 41 The quality of the models was assessed by the fit to input data (Figures S5–S7), as well as data not used in modeling, such as an independent, published crosslinking dataset, 39 cryo‐EM maps, 24 published biochemical data, 24 , 31 , 37 , 38 human cancer‐associated mutations [Catalogue of Somatic Mutations in Cancer (COSMIC)] (Table S1), 36 and predictions from AlphaFold. 30 The robustness of the models was also assessed by jack‐knifing. 1.3% (MHR), 0% (MHM), and 2% (NuDe) crosslinks in the validation set were violated by these models; they are largely similar to the corresponding models computed with the entire dataset (Figure S8, Section 4). The resulting integrative models were visualized in two ways—a representative bead model and a localization probability density map—and represented in UCSF Chimera and ChimeraX. 42 , 43 The bead model represents the centroid of the major cluster, whereas the localization probability density map represents all models in the major cluster, by specifying the probability of a voxel (3D volume unit) being occupied by a bead in the set of superposed cluster models.
2.2. MTA1‐HDAC1‐RBBP4 (MHR)
First, to support the integrative modeling of the MHR complex, an ab initio 3D EM map for the MHR complex was produced by further analysis of the MHR 2D class averages reported in a previous study (EMD‐27557) (Figure S1). 12 Integrative modeling of the 2:2:4 MHR complex produced effectively a single cluster of models (85% of a total of 15,200 models) with a model precision of 27 Å; model precision is the average RMSD between the cluster centroid and models in the cluster (Figure S2). The models fit very well to the input data as measured by the EM and crosslink scores. 98% of the input crosslinks were satisfied within their uncertainty (Figure S5). An adipic acid dihydrazide (ADH)/bis(sulfosuccinimidyl) suberate or disuccinimidyl suberate (BS3/DSS)/dimethoxy triazinyl methyl‐morpholinium chloride (DMTMM) crosslink is violated if the corresponding crosslinked beads are greater than 35/35/25 Å apart in all models in the cluster. The cross‐correlation between the localization probability density map for the models in the major cluster and the input EM map was 0.74, indicating the fit to EM is reasonable but not too high. This could partly be due to unoccupied density in the lobes of the experimental EM map (Figure 3a).
FIGURE 3.
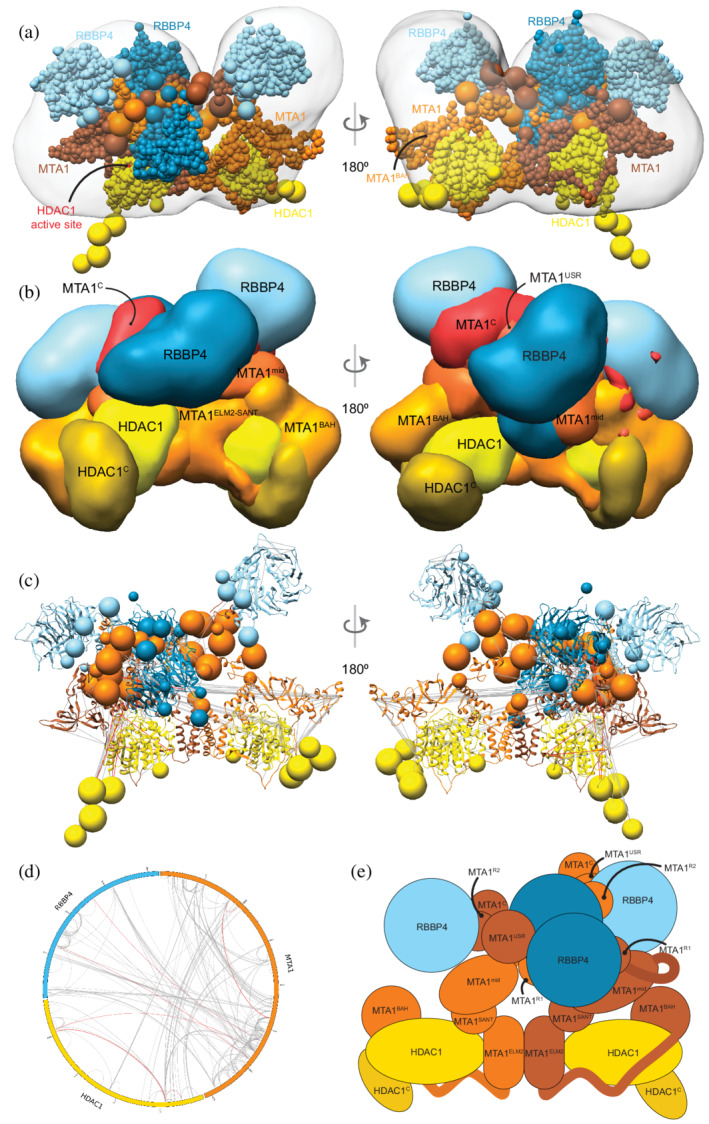
Integrative model of the MTA1‐HDAC1‐RBBP4 (MHR) complex. (a) Representative bead model from the most populated cluster of integrative models for the MHR complex, shown with the MHR EM map. The model is colored by subunit. For MTA1, the two copies are shown in different colors (brown and orange) in panels (a) and (c), to illustrate the crossover. The HDAC1 active site is shown in red. (b) Localization probability density maps showing the position of different domains/subunits in the cluster. The map specifies the probability of any volume element being occupied by a domain in the ensemble of superposed models from the cluster. The domain densities are colored according to Figure 1. These maps are contoured at ~10% of their respective maximum voxel values. (c) Representative bead model from panel (a) with regions of known structure shown in ribbon representation. (d) CX‐CIRCOS (http://cx‐circos.net/) plot for crosslink satisfaction on the ensemble of MHR models from the major cluster. Gray (red) lines indicate satisfied (violated) crosslinks in panels (c) and (d). (e) Schematic representation of the integrative model of the MHR complex. See also Figure 1 and Figures S2 and S5
Surprisingly, the representative bead model from the dominant cluster (cluster centroid model) shows the C‐terminal half of the two MTA subunits (MTA1432–715) crossing over (brown and orange MTAs, Figure 3a). Integrative models of the MHR complex created in the absence of the EM map also showed the MTAs crossing over (Figure S9).
The MTA1BAH domain (MTA11–164) is positioned distal to the MTA1 dimerization interface (MTA1200–290, MTA1dimer), consistent with its position in an independent EM map (Figure 3b–e). 24 It is proximal to the HDAC1 active site and therefore might regulate HDAC1 activity (Figure 3a). This conclusion is consistent with histone deacetylation assays in which MTA1 was shown to modulate HDAC1 deacetylase activity in NuRD. 38 Further, for one of the MTAs, the MTA1BAH is located near an RBBP4 (Figure 3a,b); MTA1BAH proximity to RBBP4 was also indicated in an independent cryo‐EM map. 24 Finally, MTA1BAH is also proximal to the MTA1mid region (MTA1334–431) containing the predicted helix (H) and zinc finger regions (ZF) (Figure 3b,c).
The MTA1mid region is juxtaposed between MTA1dimer and the MTA1BAH domain (Figure 3b). In contrast, in a previous crosslink‐based MHR model, 12 MTA1mid was proximal to the MTA1BAH domain and distal from the MTA1dimer. The MTA1 C‐terminus (MTA1C; i.e., MTA1692–715) shows considerable conformational heterogeneity and is co‐located with MTA1USR (MTA1547–669), the MTA1 disordered region between the R1 and R2 RBBP4 binding regions (Figure 3b). Overall, many MTA1 domains in the MHR model, such as MTA1BAH domain, MTA1mid, and MTA1C, are exposed and could interact with nucleosomal DNA and/or other proteins.
The HDAC1 C‐terminus (HDAC1C; i.e., HDAC1377–482) interacts with the MTA1BAH domain (Figure 3b,e). Although it has been shown that the MTA1‐HDAC1 dimer can form in the absence of MTA1BAH, 16 this additional interaction between MTA1 and HDAC1 could be functionally important. Consistent with this possibility, mutations in HDAC1C (Δ391–482, S421A, S423A, E426A) have been shown to disrupt binding to NuRD subunits. 37 There are also post‐translational modifications in the HDAC1 tail that might modulate its interaction with MTA1. 37 , 44
Both the MTA1R1‐RBBP4 units are located between the two lobes in the EM map, with one complex in the front and the other at the back (dark blue beads and densities, Figure 3a–c,e). On the other hand, the MTA1R2‐RBBP4 complexes are located in separate lobes (light blue beads and densities, Figure 3a–c,e). The densities of RBBP4 are spread out, indicating that the localization of these subunits in MHR is imprecise (Figure 3b). This is consistent with the structural heterogeneity observed in 2D class averages of the MHR EM data. 12 This flexibility is likely necessary to facilitate RBBP4 interactions with transcription factors and histones.
2.3. MTA1N‐HDAC1‐MBD3GATAD2CC (MHM)
Integrative modeling of the 2:2:2 MHM complex resulted in a major cluster containing 99% of the final 28,836 models. The model precision was 28 Å and 99% of the input crosslinks were satisfied (Figures S3 and S6). The cross‐correlation between the localization probability density map for the models in the major cluster and the input EM map was 0.9.
Our 2:2:2 MHM model shows two binding sites of MBD3 on the MTA1‐HDAC1 dimer (Figure 4a–c). Both copies of MBD3MBD localize close to the MTA1BAH domain, which is consistent with the location observed for MBD2MBD in an independent cryo‐EM map of a 2:2:1 MTA1:HDAC1:MBD2 complex 24 and an independent set of crosslinks 29 (Figure 4a–e). Although there are two MBD3s in our models, a single MBD3IDR localizes to the MTA1 dimerization interface, MTA1dimer (green MBD3, Figure 4b,e). This localization of MBD3IDR is consistent with its previously predicted localization from a crosslink‐based model 12 and the putative localization of MBD2IDR based on cryo‐electron microscopy. 24 It is also supported by two separate mutagenesis and co‐immunoprecipitation studies, one of which showed that MBD2IDR was essential for binding to the MTA1‐HDAC1 dimer, 31 whereas the other showed that MTA1dimer was essential for the interaction with MBD2. 24 Finally, the MBD3CC‐GATAD2CC coiled‐coil domain is exposed.
FIGURE 4.
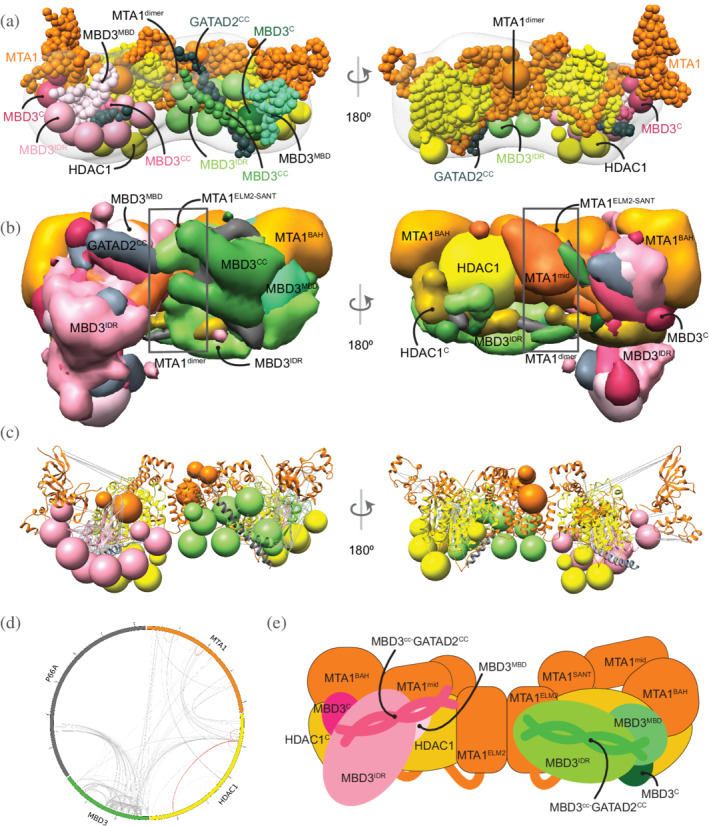
Integrative model of the MTA1N‐HDAC1‐MBD3GATAD2CC (MHM) complex. (a) Representative bead model from the major cluster of analyzed integrative models for the MHM complex, with the corresponding EM map (EMD‐21382), 12 colored by subunit. The domains of the two MBD3s are shown in shades of pink and green, respectively. (b) Localization probability density maps showing the position of different domains in the ensemble of models from the cluster. The domain densities are colored according to Figure 1. (c) The same density maps as (b) (front view), showing the two MBDs in pink and green, respectively, and illustrating that they localize differently on the MTA1‐HDAC1 dimer. The density maps of MTA1mid and GATAD2cc were omitted for clarity. (d) The density maps of the two MBD3IDR domains on the MTA1‐HDAC1 dimer. Most of the maps are contoured at around 20% of their respective maximum voxel values (except MTA1165–333 at 10% and GATAD2cc at 27%). (c) Representative bead model from panel (a) with regions of known structure shown in ribbon representation. (d) CX‐CIRCOS (http://cx‐circos.net/) plot for crosslinks satisfaction on the ensemble of MHM models from the major cluster. Gray (red) lines indicate satisfied (violated) crosslinks in panels (c) and (d). (e) Schematic representation of the integrative model of the MHM complex. Note that MTA1mid in this model corresponds to MTA1334–431. See also Figure 1 and Figures S3 and S6
2.4. MTA1‐HDAC1‐RBBP4‐MBD3‐GATAD2 (NuDe)
In modeling the NuDe complex, we incorporated only the region of GATAD2A that forms a coiled‐coil with MBD3, because of the lack of structural information on GATAD2A and the very small number of XLs involving this subunit. Integrative modeling of the NuDe complex resulted in effectively a single cluster (92% of 19,754 models). The model precision was 34 Å and 99% of the input crosslinks were satisfied (Figures S4 and S7). The cross‐correlation between the localization probability density map for the models in the major cluster and the input EM map was 0.9. Further, we validated these models by an independent set of crosslinks on the endogenous NuRD complex and interacting proteins 29 ; all 89 crosslinks used in this analysis were satisfied.
In contrast to the MHM model, MBD3 is localized more precisely in NuDe. It is juxtaposed next to the MTA1BAH and MTA1mid domains (Figure 5a–e). An independent cryo‐EM map of MTA11–546‐HDAC1‐MBD2‐RBBP4 24 and an independent set of crosslinks 29 also showed that MBD3 was proximal to MTA1BAH and MTA1dimer. Similar to the MHM model, MBD3IDR extends toward MTA1dimer.
FIGURE 5.
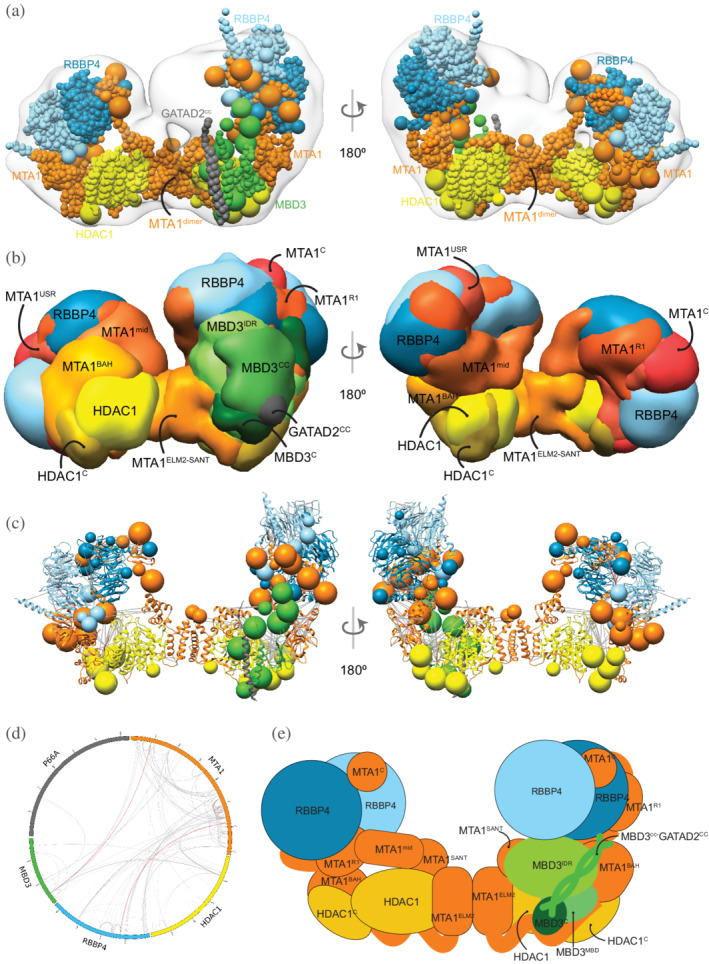
Integrative model of the nucleosome deacetylase (NuDe) complex. (a) Representative bead model from the dominant cluster of integrative models for the NuDe complex, with the corresponding EM map (EMD‐22904), 12 colored by subunit. (b) Localization probability density maps showing the position of different domains in the ensemble of models from the cluster. The domain densities are colored according to Figure 1. Maps are contoured at ~10% of their respective maximum voxel values (except GATAD2CC at 20%). (c) Representative bead model from panel (a) with regions of known structure shown in ribbon representation. (d) CX‐CIRCOS (http://cx‐circos.net/) plot for crosslinks satisfaction on the ensemble of NuDe models from the major cluster. Gray (red) lines indicate satisfied (violated) crosslinks in panels (c) and (d). (e) Schematic representation of the integrative model of the NuDe complex. See also Figure 1 and Figures S4, S7, and S10
From protein–protein distance maps of the cluster, HDAC160–100 and MTA1BAH are most proximal to MBD3 (Figures S10a and S10b). The MBD3CC‐GATAD2CC coiled‐coil is exposed. The MBD3MBD domain is buried, consistent with the failure of MBD3 to bind DNA in NuRD noted in immunoprecipitation experiments (Figure 5a–e). 38 Interestingly, several nucleosome‐interacting domains such as MTA1BAH and MTA1ZF are co‐localized in the NuDe model (Figure 5a–e).
Similar to the MHR models, the HDAC1C domain is proximal to MTA1BAH (Figure S10c). In contrast to the MHR models, which showed crossover of MTAs, the two MTAs are well‐separated in NuDe (Figure 5a–e). The localization of RBBPs is also more precise in NuDe than in MHR (Figures 3b and 5b).
2.4.1. Mapping COSMIC mutations
We next consulted the COSMIC database for somatic, confirmed pathogenic, point mutations of the NuRD subunits, MTA1, HDAC1, RBBP4, and MBD3. 36 In total, 356‐point mutations were identified and mapped onto the cluster of NuDe integrative models (Methods, 5.1 COSMIC data analysis). Analysis of these mutations revealed that 24% of residues with three or more reported COSMIC mutations mapped to exposed regions that are known to bind to nucleosomes and transcription factors, such as the HDAC1 active site and RBBP4 H3 interaction site (Figure 6, Figure S11, Table S1). This number is significantly higher than the number expected based on random chance (9%) (Section 4). However, 44% of the residues with three or more reported COSMIC mutations, in comparison to 41% in the random set, mapped to subunit interfaces in NuDe. This suggests that protein–protein interfaces within NuRD are not over‐represented in the COSMIC database, whereas mutations at exposed surfaces are. This perhaps indicates that interactions of NuRD subunits with other macromolecules, such as nucleosomes and/or transcription factors, are crucial for the function of the complex. Therefore, mutations that impair binding of NuRD to its binding partners could contribute to the pathogenesis of the disease.
FIGURE 6.
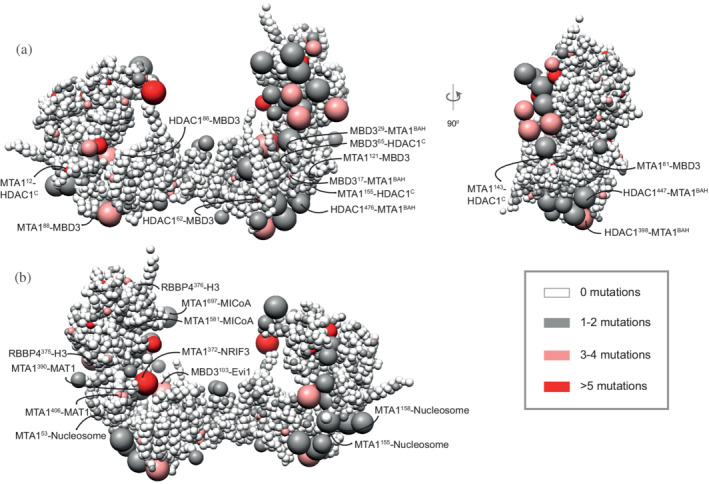
COSMIC mutations mapped onto the NuDe integrative model. Somatic pathogenic point mutations from the COSMIC database 36 mapped onto the representative bead model of the NuDe complex (Figure 5a). (a) Mutations of residues that map to previously undescribed protein–protein interfaces within our model. (b) Mutations on residues that map to exposed binding sites between modeled proteins and known binding partners. A bead is colored according to the maximum number of mutations on any residue in the bead, according to the legend. Representative mutations are labeled in both (a) and (b). See also Table S1 and Figure S12
Moreover, of the 24% of mutations that map to exposed regions, half map to regions of unknown structure (regions for which no experimental structure or reliable model is available), such as MTA1USR and MBD3IDR (Figure 6, Table S1). The functional significance of these mutations is therefore difficult to predict but could indicate that these regions of unknown structure also have important roles in protein stability, regulating interactions with binding partners of NuRD, or interactions between NuRD subunits. An important additional consideration for all these disease‐causing mutations is that many of the NuRD subunits function in cellular contexts independent of other NuRD subunits, and so in some cases, these mutations may be rationalized in the context of other functional roles.
2.4.2. Docking the nucleosome
We next attempted to dock the CHD4‐nucleosome structure 21 into the cleft in the NuDe structure between the MTA1 C‐terminal arms (Figure 7). Although there are limitations to this docking, this positioning of the nucleosome indicates its size complementarity to the integrative model, further corroborating the latter. This placement allows for the histone tails to be located toward the HDAC1 active site. It also accommodates the known interactions between the RBBPs and the histone H3 (Figure 7). The partial CHD4 structure is exposed. MTA1mid, which contains the zinc finger, can also potentially interact with the nucleosome in this position. Finally, MBD3 does not interact with the nucleosome, since MBD3MBD is buried in our model of NuDe (Figure 5b), consistent with MBD3 in NuRD failing to bind DNA in immunoprecipitation experiments. 38
FIGURE 7.
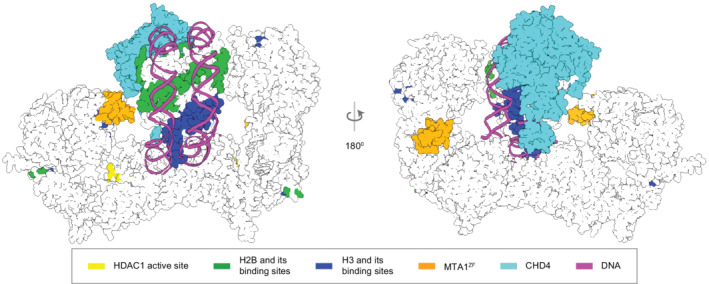
Integrative model of NuDe complex with the nucleosome. The CHD4‐nucleosome structure 21 is placed in the cleft of the NuDe integrative model. The regions with known atomic structure are shown in the NuDe integrative model from Figure 5a. CHD4, histones, DNA and the corresponding NuDe subunit residues they are proposed to bind to, are depicted in the same color, as given by the legend.
2.4.3. Comparison to structure predictions by AlphaFold‐Multimer
We compared our models to models predicted by AlphaFold‐Multimer for a 2:2:1 MTA11–350‐HDAC11–376‐MBD31–291‐GATAD2A213–244 complex (Figure S12). 45 In the top AlphaFold prediction, the MTA1‐HDAC1 dimer closely resembles the corresponding crystal structure, 16 as expected, given that AlphaFold is trained on the PDB. The localization of MTA1BAH, MTA1H, and MBD3 are broadly consistent with our integrative model and the cryo‐EM map from an independent study (Millard, 2021). MBD3MBD is proximal to MTA1BAH, as predicted in our model. MBD3IDR is near the MTA1 dimerization interface. Specifically, the N‐terminal part of the MBD3IDR (MBD3125–175) winds an irregular path toward the MTA1dimer, whereas the C‐terminal part of the MBD3IDR (MBD3125–175), which is predicted to be ordered, forms a compact structure at the MTA1dimer (Figure S12). The MBD3CC‐GATAD2A coiled‐coil sits diagonally across the MTA1 dimerization interface. This prediction, however, has several clashes (a phenomenon that we and others have observed in other predictions made using AlphaFold multimer, unpublished) and violates some of the input crosslinks. Thus, 94%, 87%, 94%, 87%, 61%, and 61% of the HDAC1‐HDAC1, MTA1‐MTA1, HDAC1‐MTA1, MBD3‐MTA1, MBD3‐MBD3, and HDAC1‐MBD3 crosslinks, respectively, were satisfied by the top AlphaFold model.
3. DISCUSSION
Here, we obtained structural models of the MTA1‐HDAC1‐RBBP4 (MHR), MTA1N‐HDAC1‐MBD3 (MHM), and MTA1‐HDAC1‐RBBP4‐MBD3‐GATAD2A (NuDe) complexes using Bayesian integrative modeling with IMP. Our approach has the several advantages over other modeling strategies. First, models generated by IMP can incorporate full‐length protein sequences, including regions that are predicted to be intrinsically disordered and/or might only form structure once assembled in a complex. IMP allows for a multi‐scale representation, with regions of known atomic structure and unknown structure represented at higher and lower resolutions, respectively. This feature facilitated the modeling of NuRD proteins with significant regions of unknown structure, such as full‐length MBD3, for the first time. Second, the Bayesian inference framework in IMP allows us to combine data from several experiments at multiple resolutions, for example, negative‐stain EM and XLMS, by considering the data uncertainty, and without using arbitrary weights, making the sampling more objective and accurate. 32 , 33 , 46 , 47 Finally, the output of IMP is an ensemble of models consistent with the data, instead of a single model. This allows us to obtain precise uncertainty bounds on the structure as a measure of model precision. 39 , 40 , 41 , 48
3.1. Comparison between IMP and AlphaFold
Integrative models are computed by satisfying spatial restraints based on information specific to a given system, whereas AI‐based methods such as AlphaFold predict structures relying heavily on features learnt from general databases of protein structures. The advantages of the latter methods are that they are fast, easy to use, and generate structures at atomic resolution. However, the predicted structures may not fit experimental data for a given system. For example, for our complex, the top prediction from AlphaFold violated a significant number of crosslinks. In contrast, integrative structure determination by IMP produces an ensemble of models consistent with the input information, instead of a single structure. In addition, rigorous validation of the integrative models based on fit to input information, jack‐knifing, and consistency with information not used in modeling, is an essential part of the integrative approach.
3.2. NuDe complex is more ordered than MHR
A comparison of MTA1 and RBBP4 in the MHR and NuDe models suggests that these subunits are more conformationally heterogenous in MHR, as shown by the broader localization probability densities for the C‐terminal half of MTA1 and RBBPs in MHR (volume enclosed by the corresponding maps = 1,400 nm3) compared to NuDe (volume enclosed = 1,300 nm3). Also, the cross‐correlation of the MHR localization probability density to the corresponding EM map is lower than that of NuDe, indicating higher heterogeneity for the former. This result suggests that dynamics in the MHR sub‐complex might be damped in the presence of MBD3‐GATAD2A.
3.3. MBD3IDR —MTA1dimer interaction
In our MHM models, one MBD3IDR is near the MTA1dimer, consistent with the previously predicted localization of MBD3IDR based on chemical crosslinks 12 and MBD2IDR based on a cryo‐electron density map (Figure 4c–e). 24 Two separate mutagenesis and co‐immunoprecipitation studies have shown that the MBDIDR and the MTA1 dimerization interface are each essential for MBD2 interaction with the MTA1‐HDAC1 dimer. 24 , 31 Despite the corresponding region of MBD2 being disordered in solution when the protein is in isolation, 31 MBD3125–175 is predicted to be ordered based on PONDR® analysis (Figure S13) (http://www.pondr.com). 49 , 50 Because this region is well conserved across species, 51 it is likely that it becomes ordered upon binding, similar to the region of MTA1 that winds irregularly across the surface of HDAC1 (MTA1165–226). Further, the crosslinks between MBD3IDR and MTA1 involve a loop (MTA1229–236) of the MTA1dimer that is not visible in the MTA1‐HDAC1 crystal structure. It is possible that this region of MTA1 may also become ordered upon binding MBD3. It is also worth noting that, unlike the NuDe and MHR complexes, MHM has not been observed in cells or in material purified from cells. MHM was created as part of a reductionist strategy toward understanding NuRD assembly.
3.4. Model of MBD3 binding in NuRD
The stoichiometry of MBD3 in NuRD is intriguing. The MHM complex has two copies of MBD3, though it is likely that this is not a physiologically relevant complex, whereas a single MBD3 is seen in the NuDe and NuRD complexes. 12 Based on our integrative models, we propose a two‐state mechanism to explain the asymmetric binding of MBD3 in NuRD (Figure 8).
FIGURE 8.
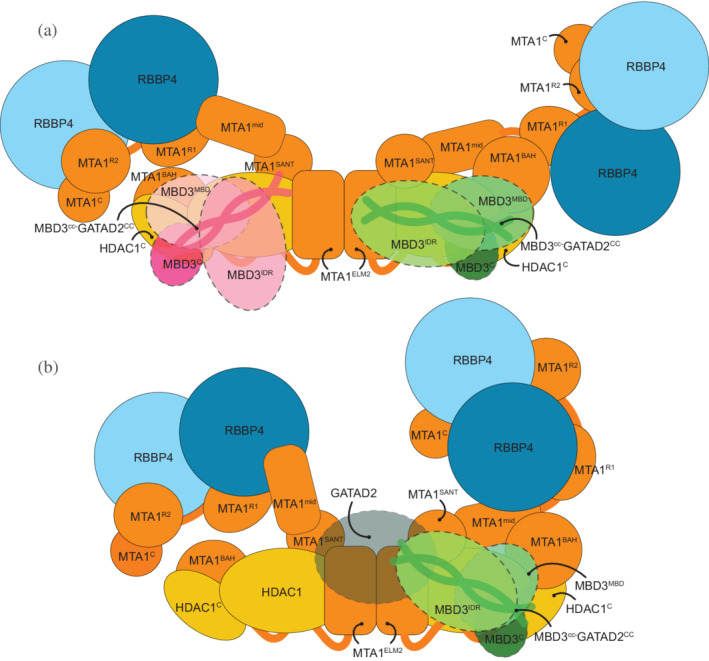
Model of MBD3 binding in nucleosome remodeling and deacetylase (NuRD) The figure shows two states of MBD3 in NuRD. (a) In the first state, the MTA1 dimerization interface is accessible for MBD3IDR to bind. (b) In the second state, upon binding, MBD3 recruits GATAD2A and the chromatin remodeling module and shifts to one end of the MTA1‐HDAC1 dimer. GATAD2A localizes near MTA1dimer, precluding a second MBD3 from binding to it.
In the first state (Figure 8a), the C‐terminal arms of MTA1 in MHR are heterogenous and adopt a range of configurations including an extended, open configuration 24 and crossed‐over configuration (Figure 3, MHR models). Functionally, this flexibility could play a role in facilitating interactions with histones as well as the transcriptional regulators such as PWWP2A that can recruit MHR to target sites. 17 In the open state, the MTA1 dimerization interface is accessible for MBD3IDR to bind. This MBD3IDR‐MTA1dimer interaction is critical for MBD recruitment to the deacetylase module (Figure 4, MHM models). 24 , 31 Although there are two MBD3 binding sites on the MTA1‐HDAC1 dimer, only one interacts with the MTA1 dimer. This is probably the MBD3 that is present in the physiological NuDe/NuRD complex.
In the second state (Figure 8b), upon binding to MTA1dimer, MBD3 recruits GATAD2A and the chromatin remodeling module, and shifts to one end of the MTA1‐HDAC1 dimer (Figure 5, NuDe models). In this state, GATAD2A localizes near MTA1dimer, precluding a second MBD3 from binding to it. Although we did not model full GATAD2A in NuDe due to unavailability of structures and crosslinks involving the protein, the proximity of CHD4, and hence GATAD2A, to the MTA1dimer in our coarse nucleosome docking supports this idea (Figure 7). This possibly explains how GATAD2A introduces asymmetry of MBD3 binding in NuRD. Moreover, upon binding the chromatin remodeling module, the C‐terminal arms of MTA1 with the RBBPs are less heterogenous and adopt a closed configuration (Figures 3b and 5b). This reduced flexibility of MTA1C‐RBBP in NuDe may reflect a functional distinction between the MHR and NuDe sub‐complexes, the nature of which is not currently clear.
In the second state, the MBD3MBD is buried and fails to bind DNA, also noted in previous experiments. 38 MBD3 is less effective at distinguishing methylated and unmethylated DNA compared to other MBD paralogs. 52 Considering these facts, it is possible that the major role of MBD3 here is to connect its two enzymatic modules rather than to recruit NuRD to DNA, methylated or otherwise. Experiments in mouse ES cells showed that MBD3 deletion resulted in loss of integrity of NuRD in these cells, supporting its role in NuRD assembly. 53
The novel NuRD protein interfaces predicted by our model need to be confirmed by future experiments. High‐resolution structures of regions such as MBD3IDR will delineate their roles in NuRD, although they will continue to be a challenge to characterize by empirical methods. Ultimately, a complete atomic characterization of the NuRD complex will aid in understanding NuRD‐mediated regulation of gene expression.
4. MATERIALS AND METHODS
4.1. Integrative modeling
The integrative structure determination of the NuRD sub‐complexes proceeded through four stages (Figure 2). 32 , 33 , 34 The modeling protocol (i.e., Stages 2, 3, and 4) was scripted using the Python Modeling Interface package, a library for modeling macromolecular complexes based on open‐source IMP package, version 2.13.0 (https://integrativemodeling.org). 34 The current procedure is an updated version of previously described protocols. 39 , 41 , 48 , 54 , 55 , 56 , 57 Files containing the input data, scripts, and output results are publicly available at https://github.com/isblab/nurd. Integrative structures of MHR, MHM, and NuDe complexes are deposited to the wwPDB.
4.1.1. Stage 1: Gathering data
The stoichiometry and isoforms of subunits were based on DIA‐MS and SEC‐MALLS experiments (Figure 1). 12 Known atomic structures were used for the MTA1‐HDAC1 dimer, MTA1R1 and MTA1R2 domains in complex with RBBP4, and MBD domain of MBD3 (Figure 1). 16 , 19 , 20 , 23 The MTA1BAH domain, MTA1H, MTA1ZF, and MBD3CC‐GATAD2ACC structures were homology‐modeled based on the structures of related templates (Figure 1a). 22 , 35 , 58
The shapes of the complexes were based on 3D negative‐stain EM maps; MHR:EMD‐27557 (25 Å), MHM: EMD‐21382 (20 Å), and NuDe: EMD‐22904 (20 Å). 12 The negative‐stained EM map for the MHR complex was produced by further analysis of data reported in a previous study (Figure S1). 12 Twenty‐five thousand one hundred fifty‐five particle images were subjected to multiple rounds of 2D classification in CryoSparc, 59 following which an ab initio 3D reconstruction was obtained and refined by homogenous 3D refinement. The final map was produced from 13,299 particles and had an estimated resolution of ~25 Å according to the FSC0.143 criterion.
Chemical crosslinks informed the relative localization of the NuRD subunits. For modeling the MHR complex, 387 BS3/DSS (bis(sulfosuccinimidyl)suberate and disuccinimidyl suberate), 34 DMTMM (dimethoxy triazinyl methyl‐morpholinium chloride), and 19 ADH (adipic acid dihydrazide) crosslinks were used. 12 For MHM (NuDe), 539 (312) BS3/DSS, 0 (40) DMTMM, and 0 (19) ADH crosslinks were used for modeling.
The models were validated by independent EM maps, 24 biochemical assays, 31 , 37 , 38 and human cancer‐associated mutations on NuRD proteins. 36
4.1.2. Stage 2: Representing the system and translating data into spatial restraints
The stoichiometry and representation of subunits is shown (Figure 1). The domains with known atomic structures were represented in a multi‐scale manner with 1 and 10 residues per bead to maximize computational efficiency. These domains were modeled as rigid bodies where the relative distances between beads is constrained during sampling. In contrast, domains without known structure were coarse‐grained at 30 residues per bead and modeled as flexible strings of beads.
We next encoded the spatial restraints into a scoring function based on the information gathered in Stage 1, as follows:
Crosslink restraints: The Bayesian crosslinks restraint 46 was used to restrain the distances spanned by the crosslinked residues. 60 The restraint accounts for ambiguity (multiple copies of a subunit) via a compound likelihood term that considers multiple residue pairs assigned to an individual crosslink. 60 Intra‐subunit crosslinks are also considered ambiguous when there are multiple copies of a subunit.
EM restraints: The Bayesian EM density restraint was used to restrain the shape of the modeled complexes and was based on the cross‐correlation between the Gaussian Mixture Model (GMM) representations of the NuRD subunits and the GMM representation of the corresponding negative‐stain EM density maps. 61
Excluded volume restraints: The excluded volume restraints were applied to each bead, using the statistical relationship between the volume and the number of residues that it covered. 32
Sequence connectivity restraints: We applied the sequence connectivity restraints, using a harmonic upper distance bound on the distance between consecutive beads in a subunit, with a threshold distance equal to twice the sum of the radii of the two connected beads. The bead radius was calculated from the excluded volume of the corresponding bead, assuming standard protein density. 60
4.1.3. Stage 3: Structural sampling to produce an ensemble of structures that satisfies the restraints
We aimed to maximize the precision at which the sampling of good‐scoring solutions was exhaustive (Stage 4). The sampling runs relied on Gibbs sampling, based on the Replica Exchange Monte Carlo algorithm. 39 , 41 The positions of the rigid bodies (domains with known structure) and flexible beads (domains with unknown structure) were sampled.
The initial positions of the flexible beads and rigid bodies in all complexes were randomized, with one exception. For MHR, we were able to unambiguously dock the structure of the MTA1‐HDAC1 core in the EM map, with the help of the previous EM map (EMD‐3399). 23 Hence, the position of the corresponding rigid body was fixed throughout.
The Monte Carlo moves included random translations of individual beads in the flexible segments and rigid bodies (around 3.7 and 1.3 Å respectively). A model was saved every 10 Gibbs sampling steps, each consisting of a cycle of Monte Carlo steps that moved every bead and rigid body once.
The sampling produced a total of 40 million MHR, 48 million MHM, and 80 million NuDe integrative models.
4.1.4. Stage 4: Analyzing and validating the ensemble of structures and data
The sampled models were analyzed to assess sampling exhaustiveness and estimate the precision of the structure, its consistency with input data and consistency with data not used in modeling. The structure was further validated by experiments based on the predictions from the models. We used the analysis and validation protocol published earlier. 33 , 39 , 40 , 41 Assessment began with a test of the thoroughness of structural sampling, including structural clustering of the models, estimating model precision, and visualizing the variability in the ensemble of structures using localization probability density maps. 40 The precision of a domain refers to its positional variation in an ensemble of superposed models. It can also be visualized by the localization probability density map for the domain. A localization probability density map specifies the probability of a voxel (3D volume unit) being occupied by a bead in a set of superposed models. The models and densities were visualized with UCSF Chimera and ChimeraX. 42 , 43
Determining good‐scoring models
Starting from the millions of sampled models, first, we selected models obtained after score equilibration and clustered them based on the restraint scores. 39 For further analysis, we considered 15,200 MHR, 28,836 MHM, and 19,754 NuDe good‐scoring models that satisfy the data restraints sufficiently well.
Clustering and structure precision
We next assessed the sampling exhaustiveness and performed structural clustering. 39 , 40 , 41 Integrative structure determination resulted in effectively a single cluster for all complexes, at a precision of 27 Å (MHR), 28 Å (MHM), and 34 Å (NuDe). The cluster precision is the bead RMSD from the cluster centroid model averaged over all models in the cluster. 40
Fit to input information
The dominant clusters from each modeled NuRD sub‐complex satisfied over 95% of all the BS3/DSS, ADH, and DMTMM crosslinks used; a crosslink is satisfied by a cluster of models if the corresponding Cα‐Cα distance in any model in the cluster is less than 35, 35, 25 Å for BS3/DSS, ADH, and DMTMM crosslinks respectively. The agreement between the models and the corresponding EM maps was computed by calculating the cross‐correlation of the combined localization probability densities of all subunits for the major cluster with the experimental EM map using the fitmap tool in UCSF Chimera (Figures 3, 4, 5). 42 The remainder of the restraints are harmonic, with a specified standard deviation. The cluster generally satisfied the excluded volume and sequence connectivity restraints. A restraint is satisfied by a cluster of models if the restrained distance in any model in the cluster (considering restraint ambiguity) is violated by less than 3 standard deviations, specified for the restraint. Most of the violations are small, and can be rationalized by local structural fluctuations, coarse‐grained representation of the model, and/or finite structural sampling.
Jack‐knifing
The robustness of the models was assessed by jack‐knifing, that is, generating models using a subset of the input crosslinks. For each modeled sub‐complex, we generated models based on a randomly selected subset consisting of 80% of the BS3/DSS crosslinks and the corresponding EM maps, and used the remaining 20% of the crosslinks for validation. Our analysis showed that 1/79 (MHR), 0/111 (MHM), and 1/64 (NuDe) validation crosslinks were violated in these models. The resultant models largely resemble the original results, although they are lower in precision (Figure S8).
Fit to data not used in modeling
The MHR integrative models were supported by histone deacetylation assays, mutagenesis, and co‐immunoprecipitation, showing that MTA1 and the HDAC1C regulate HDAC1 deacetylase activity and NuRD assembly. 37 , 38 The localization of domains such as MTA1BAH and RBBP4 were validated by their consistency with independently determined cryo‐EM maps. 24
The MHM integrative models were supported by independent cryo‐EM maps of the complex showing similar localizations for MBD2MBD and MTA1BAH. 24 The MBD3IDR‐MTA1dimer interaction was also supported by two separate mutagenesis and co‐immunoprecipitation studies. 24 , 31
The NuDe integrative models were corroborated by immunoprecipitation experiments showing that the MBD domain of MBD3 is buried in NuRD. 38 They were also supported by independent cryo‐EM maps showing that MBD3 is proximal to MTA1BAH, and biochemical assays showing the importance of HDAC1C interactions in NuRD. 24 , 37 The mapping of cancer mutations to protein–protein interfaces in the NuDe model also supported them (Figure 6, Figure S11, Table S1). 36 We also validated the NuDe integrative models by an independent set of crosslinks on the endogenous NuRD complex and interacting proteins. 29 We mapped these crosslinks onto paralogs of the subunits represented in our models. Of the 155 crosslinks in this set, 89 were within and between subunits represented in our models. All crosslinks were satisfied by the dominant cluster of the NuDe sub‐complex, at a threshold of 35 Å. Finally, we also compared our model to predictions from Alphafold‐Multimer. 45
Mapping COSMIC mutations
We obtained a total of 356 somatic, confirmed pathogenic, point mutations for the modeled NuRD subunits (MTA1, HDAC1, RBBP4, MBD3) from the COSMIC database. 36 For each subunit, point mutations were selected from search results based on the presence of census genes and correct documentation of current structures. To ensure the mutations studied significantly affect the function, folding, and protein–protein interaction of the protein, the “confirmed pathogenic” and “somatic” filters were applied in all cases. To test the significance of the mapping, we considered residues with three or more reported mutations. 62 We generated an equal number of random mutations for each subunit and mapped them onto the NuDe integrative model. For this analysis, a mutation was considered to be at an interface if the average distance of the corresponding residue to a residue in an interacting protein is less than 5 Å.
AUTHOR CONTRIBUTIONS
Shreyas Arvindekar: Conceptualization (lead); investigation (equal); methodology (equal); software (lead); validation (equal); visualization (lead); writing – original draft (equal); writing – review and editing (equal). Matthew J. Jackman: Data curation (supporting); validation (supporting); visualization (supporting); writing – original draft (supporting); writing – review and editing (supporting). Jason K. K. Low: Data curation (supporting). Shruthi Viswanath: Conceptualization (lead); funding acquisition (lead); investigation (lead); methodology (equal); resources (lead); supervision (lead); validation (equal); visualization (equal); writing – original draft (equal); writing – review and editing (equal). Joel P. Mackay: Conceptualization (equal); data curation (equal); funding acquisition (equal); investigation (equal); methodology (equal); supervision (supporting); validation (supporting); writing – original draft (supporting); writing – review and editing (supporting). Michael J. Landsberg: Conceptualization (equal); data curation (equal); funding acquisition (equal); investigation (equal); methodology (equal); supervision (supporting); validation (supporting); writing – original draft (supporting); writing – review and editing (supporting).
FUNDING INFORMATION
This work has been supported by the following grants: Department of Atomic Energy (DAE) TIFR grant RTI 4006 and Department of Science and Technology (DST) SERB grant SPG/2020/000475 from the Government of India to Shruthi Viswanath, National Health and Medical Research Council of Australia project grants: APP1012161, APP1063301, APP1126357 to Michael J. Landsberg and Joel P. Mackay and a fellowship (APP1058916) from the same organization to Joel P. Mackay.
CONFLICT OF INTEREST
None declared.
Supporting information
Appendix S1 Supporting Information
ACKNOWLEDGMENTS
We thank Vinothkumar Kutti Ragunath and lab members Satwik Pasani, Praveen Roy DS, and Varun Ullanat for useful comments on the manuscript. Molecular graphics images were produced using the UCSF Chimera and UCSF ChimeraX packages from the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by NIH P41 RR001081, NIH R01‐GM129325, and National Institute of Allergy and Infectious Diseases).
Arvindekar S, Jackman MJ, Low JKK, Landsberg MJ, Mackay JP, Viswanath S. Molecular architecture of nucleosome remodeling and deacetylase sub‐complexes by integrative structure determination. Protein Science. 2022;31(9):e4387. 10.1002/pro.4387
Review Editor: Nir Ben‐Tal
Funding information Department of Atomic Energy, Government of India, Grant/Award Number: RTI 4006; Department of Science and Technology India, Grant/Award Number: SPG/2020/000475; National Health and Medical Research Council of Australia, Grant/Award Numbers: APP1012161, APP1058916, APP1063301, APP1126357
Contributor Information
Michael J. Landsberg, Email: m.landsberg@uq.edu.au.
Joel P. Mackay, Email: joel.mackay@sydney.edu.au.
Shruthi Viswanath, Email: shruthiv@ncbs.res.in.
DATA AVAILABILITY STATEMENT
Files containing the input data, scripts, and output results are publicly available at https://github.com/isblab/nurd. EM maps of MHR are deposited in the EMDB (EMD 27557). MHR, MHM, NuDe Integrative structures are deposited in the wwPDB.
REFERENCES
- 1. Basta J, Rauchman M. Chapter 3 ‐ the nucleosome remodeling and deacetylase complex in development and disease. In: Laurence J, Beusekom MV, editors. Translating epigenetics to the clinic. Boston: Academic Press, 2017; p. 37–72. 10.1016/B978-0-12-800802-7.00003-4. [DOI] [Google Scholar]
- 2. Denslow SA, Wade PA. The human Mi‐2/NuRD complex and gene regulation. Oncogene. 2007;26:5433–5438. 10.1038/sj.onc.1210611. [DOI] [PubMed] [Google Scholar]
- 3. Bornelöv S, Reynolds N, Xenophontos M, et al. The nucleosome remodeling and deacetylation complex modulates chromatin structure at sites of active transcription to fine‐tune gene expression. Mol Cell. 2018;71:56–72.e4. 10.1016/j.molcel.2018.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Li D‐Q, Kumar R. Mi‐2/NuRD complex making inroads into DNA‐damage response pathway. Cell Cycle Georget Tex. 2010;9:2071–2079. 10.4161/cc.9.11.11735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Reynolds N, O'Shaughnessy A, Hendrich B. Transcriptional repressors: Multifaceted regulators of gene expression. Development. 2013;140:505–512. 10.1242/dev.083105. [DOI] [PubMed] [Google Scholar]
- 6. Smeenk G, Wiegant WW, Vrolijk H, Solari AP, Pastink A, van Attikum H. The NuRD chromatin–remodeling complex regulates signaling and repair of DNA damage. J Cell Biol. 2010;190:741–749. 10.1083/jcb.201001048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yoshida T, Hazan I, Zhang J, et al. The role of the chromatin remodeler Mi‐2β in hematopoietic stem cell self‐renewal and multilineage differentiation. Genes Dev. 2008;22:1174–1189. 10.1101/gad.1642808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Basta J, Rauchman M. The nucleosome remodeling and deacetylase complex in development and disease. Transl Res J Lab Clin Med. 2015;165:36–47. 10.1016/j.trsl.2014.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Toh Y, Nicolson GL. The role of the MTA family and their encoded proteins in human cancers: Molecular functions and clinical implications. Clin Exp Metastasis. 2009;26:215–227. 10.1007/s10585-008-9233-8. [DOI] [PubMed] [Google Scholar]
- 10. Burgold T, Barber M, Kloet S, et al. The nucleosome remodelling and deacetylation complex suppresses transcriptional noise during lineage commitment. EMBO J. 2019;38:e100788. 10.15252/embj.2018100788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hoffmann A, Spengler D. Chromatin remodeling complex NuRD in neurodevelopment and neurodevelopmental disorders. Front Genet. 2019;10:682. 10.3389/fgene.2019.00682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Low JKK, Silva APG, Sharifi Tabar M, et al. The nucleosome remodeling and deacetylase complex has an asymmetric, dynamic, and modular architecture. Cell Rep. 2020;33:108450. 10.1016/j.celrep.2020.108450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hong W, Nakazawa M, Chen Y‐Y, et al. FOG‐1 recruits the NuRD repressor complex to mediate transcriptional repression by GATA‐1. EMBO J. 2005;24:2367–2378. 10.1038/sj.emboj.7600703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lejon S, Thong SY, Murthy A, et al. Insights into association of the NuRD complex with FOG‐1 from the crystal structure of an RbAp48·FOG‐1 complex. J Biol Chem. 2011;286:1196–1203. 10.1074/jbc.M110.195842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhang W, Aubert A, Gomez de Segura JM , et al. The nucleosome remodeling and deacetylase complex NuRD Is built from preformed catalytically active sub‐modules. J Mol Biol. 2016;428:2931–2942. 10.1016/j.jmb.2016.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Millard CJ, Watson PJ, Celardo I, et al. Class I HDACs share a common mechanism of regulation by inositol phosphates. Mol Cell. 2013;51:57–67. 10.1016/j.molcel.2013.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Link S, Spitzer RMM, Sana M, et al. PWWP2A binds distinct chromatin moieties and interacts with an MTA1‐specific core NuRD complex. Nat Commun. 2018;9:4300. 10.1038/s41467-018-06665-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pünzeler S, Link S, Wagner G, et al. Multivalent binding of PWWP2A to H2A.Z regulates mitosis and neural crest differentiation. EMBO J. 2017;36:2263–2279. 10.15252/embj.201695757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Alqarni SSM, Murthy A, Zhang W, et al. Insight into the architecture of the NuRD complex. J Biol Chem. 2014;289:21844–21855. 10.1074/jbc.M114.558940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cramer JM, Scarsdale JN, Walavalkar NM, Buchwald WA, Ginder GD, Williams DC. Probing the dynamic distribution of bound states for methylcytosine‐binding domains on DNA. J Biol Chem. 2014;289:1294–1302. 10.1074/jbc.M113.512236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Farnung L, Ochmann M, Cramer P. Nucleosome‐CHD4 chromatin remodeler structure maps human disease mutations. eLife. 2020;9:e56178. 10.7554/eLife.56178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gnanapragasam MN, Scarsdale JN, Amaya ML, et al. p66α–MBD2 coiled‐coil interaction and recruitment of Mi‐2 are critical for globin gene silencing by the MBD2–NuRD complex. Proc Natl Acad Sci. 2011;108:7487–7492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Millard CJ, Varma N, Saleh A, et al. The structure of the core NuRD repression complex provides insights into its interaction with chromatin. Elife. 2016;2016:e13941. 10.7554/eLife.13941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Millard CJ, Fairall L, Ragan TJ, Savva CG, Schwabe JWR. The topology of chromatin‐binding domains in the NuRD deacetylase complex. Nucleic Acids Res. 2020;48:12972–12982. 10.1093/nar/gkaa1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kloet SL, Baymaz HI, Makowski M, et al. Towards elucidating the stability, dynamics and architecture of the nucleosome remodeling and deacetylase complex by using quantitative interaction proteomics. FEBS J. 2015;282:1774–1785. 10.1111/febs.12972. [DOI] [PubMed] [Google Scholar]
- 26. Le Guezennec X, Vermeulen M, Brinkman AB, et al. MBD2/NuRD and MBD3/NuRD, two distinct complexes with different biochemical and functional properties. Mol Cell Biol. 2006;26:843–851. 10.1128/MCB.26.3.843-851.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Smits AH, Jansen PWTC, Poser I, Hyman AA, Vermeulen M. Stoichiometry of chromatin‐associated protein complexes revealed by label‐free quantitative mass spectrometry‐based proteomics. Nucleic Acids Res. 2013;41:e28. 10.1093/nar/gks941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Spruijt CG, Bartels SJJ, Brinkman AB, et al. CDK2AP1/DOC‐1 is a bona fide subunit of the Mi‐2/NuRD complex. Mol Biosyst. 2010;6:1700–1706. 10.1039/C004108D. [DOI] [PubMed] [Google Scholar]
- 29. Spruijt CG, Gräwe C, Kleinendorst SC, Baltissen MPA, Vermeulen M. Cross‐linking mass spectrometry reveals the structural topology of peripheral NuRD subunits relative to the core complex. FEBS J. 2021;288:3231–3245. 10.1111/febs.15650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jumper J, Evans R, Pritzel A, et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596:583–589. 10.1038/s41586-021-03819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Desai MA, Webb HD, Sinanan LM, et al. An intrinsically disordered region of methyl‐CpG binding domain protein 2 (MBD2) recruits the histone deacetylase core of the NuRD complex. Nucleic Acids Res. 2015;43:3100–3113. 10.1093/nar/gkv168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Alber F, Dokudovskaya S, Veenhoff LM, et al. Determining the architectures of macromolecular assemblies. Nature. 2007;450:683–694. 10.1038/nature06404. [DOI] [PubMed] [Google Scholar]
- 33. Rout MP, Sali A. Principles for integrative structural biology studies. Cell. 2019;177:1384–1403. 10.1016/j.cell.2019.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Russel D, Lasker K, Webb B, et al. Putting the pieces together: Integrative modeling platform software for structure determination of macromolecular assemblies. PLoS Biol. 2012;10:e1001244. 10.1371/journal.pbio.1001244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Connelly JJ, Yuan P, Hsu H‐C, Li Z, Xu R‐M, Sternglanz R. Structure and function of the Saccharomyces cerevisiae Sir3 BAH domain. Mol Cell Biol. 2006;26:3256–3265. 10.1128/MCB.26.8.3256-3265.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Forbes S, Clements J, Dawson E, et al. Cosmic 2005. Br J Cancer. 2006;94:318–322. 10.1038/sj.bjc.6602928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pflum MKH, Tong JK, Lane WS, Schreiber SL. Histone deacetylase 1 phosphorylation promotes enzymatic activity and complex formation. J Biol Chem. 2001;276:47733–47741. 10.1074/jbc.M105590200. [DOI] [PubMed] [Google Scholar]
- 38. Zhang Y, Ng H‐H, Erdjument‐Bromage H, Tempst P, Bird A, Reinberg D. Analysis of the NuRD subunits reveals a histone deacetylase core complex and a connection with DNA methylation. Genes Dev. 1999;13:1924–1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Saltzberg DJ, Viswanath S, Echeverria I, Chemmama IE, Webb B, Sali A. Using integrative modeling platform to compute, validate, and archive a model of a protein complex structure. Protein Sci. 2021;30:250–261. 10.1002/pro.3995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Viswanath S, Chemmama IE, Cimermancic P, Sali A. Assessing exhaustiveness of stochastic sampling for integrative modeling of macromolecular structures. Biophys J. 2017;113:2344–2353. 10.1016/j.bpj.2017.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Saltzberg D, Greenberg CH, Viswanath S, et al. Modeling biological complexes using integrative modeling platform. In: Bonomi M, Camilloni C, editors. Biomolecular simulations: Methods and protocols, methods in molecular biology. New York, NY: Springer, 2019; p. 353–377. 10.1007/978-1-4939-9608-7_15. [DOI] [PubMed] [Google Scholar]
- 42. Pettersen EF, Goddard TD, Huang CC, et al. UCSF Chimera—A visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 43. Pettersen EF, Goddard TD, Huang CC, et al. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 2021;30:70–82. 10.1002/pro.3943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rathert P, Dhayalan A, Murakami M, et al. Protein lysine methyltransferase G9a acts on non‐histone targets. Nat Chem Biol. 2008;4:344–346. 10.1038/nchembio.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Evans R, O'Neill M, Pritzel A, et al. Protein complex prediction with AlphaFold‐Multimer. bioRxiv. 2022. 10.1101/2021.10.04.463034. [DOI]
- 46. Rieping W, Habeck M, Nilges M. Inferential structure determination. Science. 2005;309:303–306. 10.1126/science.1110428. [DOI] [PubMed] [Google Scholar]
- 47. Schneidman‐Duhovny D, Pellarin R, Sali A. Uncertainty in integrative structural modeling. Curr Opin Struct Biol. 2014;28:96–104. 10.1016/j.sbi.2014.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Webb B, Viswanath S, Bonomi M, et al. Integrative structure modeling with the integrative modeling platform. Protein Sci. 2018;27:245–258. 10.1002/pro.3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Romero P, Obradovic Z, Kissinger C, Villafranca JE, Dunker AK. Identifying disordered regions in proteins from amino acid sequence. In: Proceedings of international conference on neural networks (ICNN'97). Presented at the proceedings of international conference on neural networks (ICNN'97). 1997;vol. 1:pp. 90–95. 10.1109/ICNN.1997.611643. [DOI] [Google Scholar]
- 50. Romero P, Obradovic Z, Li X, Garner EC, Brown CJ, Dunker AK. Sequence complexity of disordered protein. Proteins. 2001;42:38–48. . [DOI] [PubMed] [Google Scholar]
- 51. Cramer JM, Pohlmann D, Gomez F, et al. Methylation specific targeting of a chromatin remodeling complex from sponges to humans. Sci Rep. 2017;7:40674. 10.1038/srep40674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Liu K, Lei M, Wu Z, et al. Structural analyses reveal that MBD3 is a methylated CG binder. FEBS J. 2019;286:3240–3254. 10.1111/febs.14850. [DOI] [PubMed] [Google Scholar]
- 53. Kaji K, Caballero IM, MacLeod R, Nichols J, Wilson VA, Hendrich B. The NuRD component Mbd3 is required for pluripotency of embryonic stem cells. Nat Cell Biol. 2006;8:285–292. 10.1038/ncb1372. [DOI] [PubMed] [Google Scholar]
- 54. Ganesan SJ, Feyder MJ, Chemmama IE, et al. Integrative structure and function of the yeast exocyst complex. Protein Sci. 2020;29:1486–1501. 10.1002/pro.3863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Gutierrez C, Chemmama IE, Mao H, et al. Structural dynamics of the human COP9 signalosome revealed by cross‐linking mass spectrometry and integrative modeling. Proc Natl Acad Sci. 2020;117:4088–4098. 10.1073/pnas.1915542117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kim SJ, Fernandez‐Martinez J, Nudelman I, et al. Integrative structure and functional anatomy of a nuclear pore complex. Nature. 2018;555:475–482. 10.1038/nature26003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Viswanath S, Bonomi M, Kim SJ, et al. The molecular architecture of the yeast spindle pole body core determined by Bayesian integrative modeling. Mol Biol Cell. 2017;28:3298–3314. 10.1091/mbc.E17-06-0397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Tjandra N, Omichinski JG, Gronenborn AM, Clore GM, Bax A. Use of dipolar 1H‐15N and 1H‐13C couplings in the structure determination of magnetically oriented macromolecules in solution. Nat Struct Biol. 1997;4:732–738. 10.1038/nsb0997-732. [DOI] [PubMed] [Google Scholar]
- 59. Punjani A, Rubinstein JL, Fleet DJ, Brubaker MA. cryoSPARC: Algorithms for rapid unsupervised cryo‐EM structure determination. Nat Methods. 2017;14:290–296. 10.1038/nmeth.4169. [DOI] [PubMed] [Google Scholar]
- 60. Shi Y, Fernandez‐Martinez J, Tjioe E, et al. Structural characterization by cross‐linking reveals the detailed architecture of a coatomer‐related heptameric module from the nuclear pore complex. Mol Cell Proteomics. 2014;13:2927–2943. 10.1074/mcp.M114.041673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Bonomi M, Hanot S, Greenberg CH, et al. Bayesian weighing of electron cryo‐microscopy data for integrative structural modeling. Structure. 2019;27:175–188.e6. 10.1016/j.str.2018.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Alsulami AF, Torres PHM, Moghul I, et al. COSMIC cancer gene census 3D database: Understanding the impacts of mutations on cancer targets. Brief Bioinform. 2021;22:bbab220. 10.1093/bib/bbab220. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1 Supporting Information
Data Availability Statement
Files containing the input data, scripts, and output results are publicly available at https://github.com/isblab/nurd. EM maps of MHR are deposited in the EMDB (EMD 27557). MHR, MHM, NuDe Integrative structures are deposited in the wwPDB.