

Abstract
Objective
To describe our current understanding of hereditary α-tryptasemia (HαT), how HαT fits into the evolutionary context of tryptases and contemporary framework of mast cellassociated disorders, and to discuss the future clinical and therapeutic landscape for symptomatic individuals with HαT.
Data Sources
Primary peer-reviewed literature Study Selections Basic, clinical, and translational studies describing tryptase gene composition, generation, secretion, and elevation, and the associated clinical impacts of HαT and treatment of such individuals were reviewed.
Results
HαT is a common autosomal dominant genetic trait caused by increased TPSAB1 copy number encoding α-tryptase. Approximately 1 in 20 Caucasians have HαT, making it by far the most common cause for elevated BST levels. While many individuals with HαT may not manifest associated symptoms, the prevalence of HαT is increased in patients with clonal and non-clonal mast cell-associated disorders where it is linked to more prevalent and/or severe anaphylaxis and increased mast cell mediator-associated symptoms. Increased generation of mature α/β-tryptase heterotetramers, and their unique physiochemical properties, may be responsible for some of these clinical findings.
Conclusion
HαT is a common modifier of mast cell-associated disorders and reactions. However, whether HαT may be an independent cause of clinical phenotypes with which it has been associated remains unproven. Correct identification of HαT is critical to accurate interpretation of serum tryptase levels in the clinical evaluation of patients. Beyond HαT, we foresee tryptase genotyping as an important parameter in the standard workup of patients with mast cell-associated disorders and development of therapeutic modalities targeting these patients and associated clinical phenotypes.
Keywords: anaphylaxis, mast cell activation, mastocytosis, venom allergy, HαT, BST
INTRODUCTION
Mast cells are myeloid lineage cells with ancient origins that arise in bone marrow as precursors prior to depositing in peripheral tissues – predominantly skin and mucosae – where they mature into mature allergic effector cells 1,2. Whereas expression of activating receptors such as Mas-related G-protein coupled receptor member X2 (MRGPRX2) and effector molecules such as chymase may be variable 3–5, reflecting the diversity of these cells in different tissues, a general commonality in non-neoplastic mast cells is robust expression of the serine protease tryptase, that accounts for up to 25% of the total protein mass 6,7. Despite evidence that mast cells and tryptase-like secretory proteins are ancient 8,9, human α- and β-tryptases evolved recently in primates 10. Moreover, the multi-gene human tryptase locus is variable, with increased α-tryptase-encoding TPSAB1 copy number resulting in elevated basal serum tryptase (BST) and defining the common genetic trait hereditary α-tryptasemia (HαT) 11,12.
Approximately 5–7% of Western populations have HαT making it the most common cause for elevated BST 13–15. This prevalence is significantly increased among individuals with severe anaphylaxis due to stinging insects or from unknown causes (idiopathic) 16 as well as among individuals with the clonal mast cell disease systemic mastocytosis (SM) where it is associated with an increased prevalence of anaphylaxis and mast cell mediator-associated symptoms 17. Many individuals with HαT do not present with symptoms. However, the modifying effect(s) of this trait on these clonal and non-clonal mast cell-associated disorders and reactions is striking. Furthermore, accumulating evidence suggests HαT may contribute to additional clinical phenotypes, including gastrointestinal symptoms previously believed to be functional 18. Recent discovery of α/β-tryptase heterotetramers and their unique activities have provided functional insights into potential mechanisms underlying some of these clinical associations 19. Herein we describe our current understanding of HαT, how this genetic trait relates to and modifies other disorders, the current state-of-the-art regarding diagnosis and management of symptoms associated with HαT providing current best evidence-based recommendations, and highlight how we envision the future medical landscape may incorporate tryptase genetics into basic and translational studies.
EVOLUTION OF HUMAN TRYPTASES
Tryptases are proteolytic enzymes made predominantly by mast cells in humans. Three soluble tryptases (α, β, and δ) and a membrane-anchored tryptase (γ) have been described 20. However, uncertainty remains about the contribution of these enzymes to homeostasis and disease, and orthologs of these enzymes in other mammals are only modestly conserved. Thus, efforts have been made to identify ancestors with known functions to help answer lingering questions and illuminate roles of mast cells in development of human immunity. Mast cells likely evolved from primitive host defense cells to serve adaptive as well as innate immune functions 21,22. Secreted tryptase-like enzymes appear in histamine-containing granules of mast cell-like cells in a primitive invertebrate suggesting an ancient beginning to these proteins 23. However, as yet there is no evolutionary thread connecting these enzymes with mammalian tryptases, which may be separated by more than half a billion years of evolution. Indeed, it is challenging to trace origins of soluble human tryptases to the comparatively recent time when mammals cleaved off from other vertebrates despite massive deposits of DNA sequence from a wide range of genomes now residing in searchable databases.
Available physical and phylogenetic evidence suggest that soluble human α-, β- and δ-tryptases evolved from ancestral type-1 membrane-anchored proteases similar to γ-tryptase 10. When this occurred is a mystery but given the apparent absence of γ-tryptase and soluble tryptases in non-mammalian vertebrates, the transformation may have occurred early in mammalian evolution. γ-Tryptases, although expressed in humans and rodents, may be vestiges like ginkgos among trees or coelacanths among fishes: “living fossils” retaining ancestral features lost in lineages leading to soluble tryptases. Supporting this hypothesis is the conspicuous absence of γ-tryptase in several major mammalian lineages – including carnivores, bats, and even-toed ungulates (e.g., cattle). Similarities between γ-tryptase and other trypsin-like type I membrane-anchored serine proteases not expressed in mast cells but with demonstrably deeper roots in evolution of non-mammalian vertebrates 24,25 suggest that γ-tryptase itself evolved from ancestral type I serine proteases related to latter-day prostasin, marapsin, testisin and prosemin. Many changes in α- and β-tryptase-encoding genes—including serial duplications, gene conversion events, chimera formation and functionally significant missense and nonsense mutations – have occurred comparatively recently, late in evolution of primates, making these isoforms essentially unique to humans 10.
GENETICS AND DIAGNOSIS OF HEREDITARY ALPHA-TRYPTASEMIA
The human tryptase locus is comprised of four paralogous genes – TPSG1, TPSB2, TPSAB1, and TPSD1 – that form a tight cluster in an unstable sub-telomeric region (p13.3) of chromosome 16, where their number and makeup has been subject to recent modifications featuring duplications, full and partial gene conversions, interstitial inversions, pseudogenization, and variations in copy number attributable in part to non-allelic homologous recombination in a hotspot for such phenomena, as has been recognized for over two decades 26,27. It is important to recognize that a approximately one-third of individuals lack α-tryptase altogether, though racial and ethnic differences in genotype prevalence appear to exist 12. This is not due to deletion of TPSAB1 but rather because this locus may encode either α- or β-tryptase (Fig. 1). While some people lack α-tryptase, and variability in β-tryptase copy number may occur, no individual has been reported to be β-tryptase deficient 12,28. The four common TPSAB1 genotypes are α/α, α/β, and β/β (i.e., no α), with two more β-tryptases residing at TPSB2 (β/β) (Fig. 1). Approximately 5% of individuals inherit extra copies of α-tryptase encoding sequences on a single allele 13–15, and this defines HαT 11,29. Interestingly, it is only these α-tryptase-encoding replications that are associated with elevated BST – likely due to an as yet unidentified modifier of gene expression. Thus, inheritance of α-tryptases can range from none to many.
Figure 1. Reported tryptase haplotypes and genotypes in healthy individuals and in those with HαT.
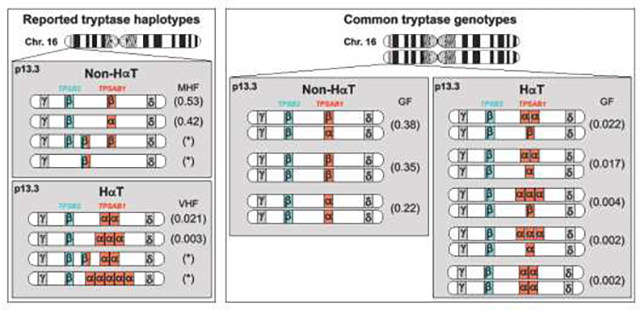
(Left) Reported tryptase haplotypes for secreted alpha- (α) and beta-tryptases (β) arising from TPSAB1 and TPSB2 on chromosome 16 (Chr. 16) p13.3, with associated minor haplotype frequencies (MHF) and variant haplotype frequencies (VHF) in parentheses. (Right) Common tryptase genotypes with reported genotype frequencies (GF).
Because of a high degree of sequence homology between α- and β-tryptases, conventional clinical next-generation sequencing including whole exome or genome sequencing (WES or WGS, respectively) or microarray-based comparative genomic hybridization (aCGH) are not able to delineate tryptase genotype or copy number. However, a droplet digital polymerase chain reaction (ddPCR) assay has been developed for clinical use that can accurately calculate α- and β-tryptase copy number arising from TPSAB1 and TPSB2 11,30. Rarely, β-tryptase copy number variation may occur 15,17,28,31,32. While this is not associated with increased BST, it can confound interpretation of a tryptase genotype and associated BST level. In these rare cases, as has been reported 33, examining inheritance patterns of BST and tryptase genotype can be exceedingly useful, as tryptase haplotypes (Fig. 1) – sequences at TPSAB1 and TPSB2 – do not independently segregate and are co-inherited in near complete linkage disequilibrium such that they can be followed through family pedigrees.
BIOLOGICAL FUNCTIONS OF TRYPTASES AND HETEROTETRAMERS
Enzymatically active secreted tetrameric α- and β-tryptases are generated from pro-tryptase zymogens via step-wise proteolytic processing. Whereas pro-tryptases are constitutively secreted by mast cells and comprise BST levels, mature tryptases are stored in secretory granules and following mast cell activation are released with other pre-formed mediators such as histamine, ostensibly contributing to symptoms of immediate hypersensitivity 28,34. Given the potentially primordial origins of mast cells and tryptases, a number of studies have sought to identify biologically relevant substrates for tryptases that impact health and disease. Some of these properties include effects on tissue remodeling including the degradation of extracellular matrix proteins and promotion of angiogenesis and fibrosis, stimulation of nerves, contribution to smooth muscle contractility and proliferation directly, as well as indirectly via cleavage of certain peptides (e.g., vasoactive intestinal peptide) and promotion of acute vascular permeability (Figure 2) 28. However, because α-tryptase tetramers lack enzymatic activity, these studies are largely limited to evaluation of β-tryptase activities.
Figure 2. Putative effects of mature tryptases in HαT.
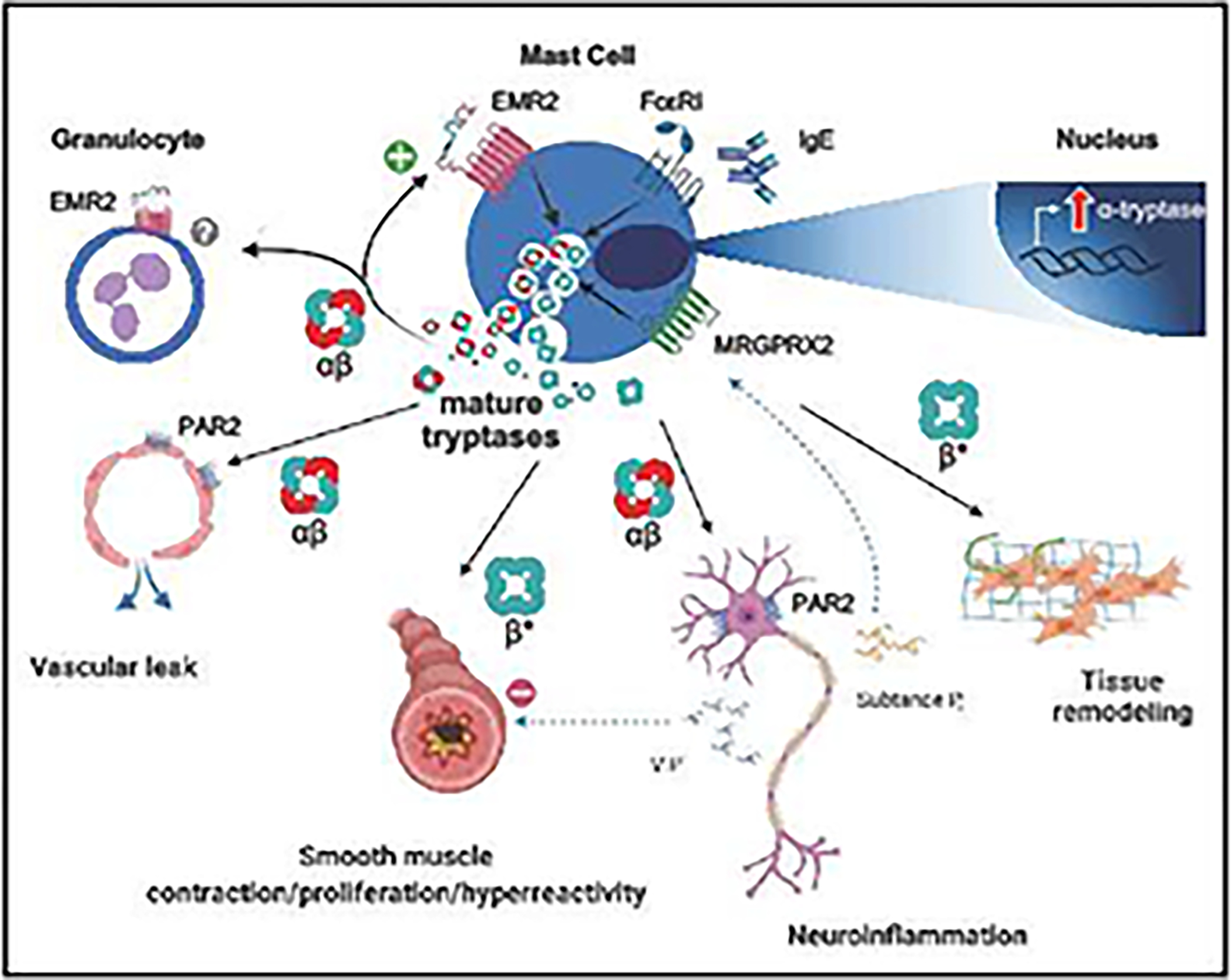
Increased TPSAB1 copy number encoding α-tryptase is associated with generation of mature αβ-tryptase heterotetramers. Following mast cell activation and release of secretory granule contents, mature αβ-tryptases can contribute to mast cell degranulation in an autocrine manner through cleavage of the mechanoreceptor EMR2. αβ-Tryptases also selectively cleave and activate PAR2, potentially leading to increased acute vascular permeability and neuroinflammation. Mature β-tryptases have been shown to promote proliferation of connective tissue fibroblasts and airway smooth muscle cells which is associated with extracellular matrix remodeling and fibrosis. In the airways, mature β-tryptases have also been shown to promote smooth muscle tone, potentially in part by cleaving the bronchodilator VIP. *It is currently unknown whether αβ-tryptases have differential effects on connective tissue and airway phenotypes. EMR2, EGF-Like Module-Containing Mucin-Like Hormone Receptor-Like 2; PAR2, protease-activated receptor 2; VIP, vasoactive intestinal peptide; MRGPRX2, Mas-related G protein-coupled receptor-X2; FcεRI, high affinity IgE receptor.
Recently α/β-tryptase heterotetramers have been shown to form naturally, exhibiting enhanced stability and unique activities 19. At ostensibly physiologic concentrations, α/β-tryptase heterotetramers are uniquely able to cleave and activate the mechanosensing adhesion GPCR EGF-like module-containing mucin-like hormone receptor-like 2 (EMR2) encoded by Adhesion G protein-coupled receptor E2 (ADGRE2) in vitro (Figure 2). Interestingly, a gain-of-function missense variant in ADGRE2 has been associated with a severe familial form of vibratory urticaria 35. In affected individuals forearm vibration using a laboratory vortex, led to significant localized symptoms of erythema, edema, and pruritic hives. Because individuals with HαT have frequently been reported to present with similar skin manifestations – often triggered by vibratory stimuli such as running a lawnmower or hand mixer – the association between tryptase genotypes and response to vibratory challenge was examined 19. In this study increasing ratios of α- to β-tryptase were associated with more severe vibratory symptom scores among both healthy volunteers and individuals with HαT.
At the same concentrations used in studies of EMR2 activation, α/β-tryptase heterotetramers were also shown to selectively cleave and activate Protease activated receptor-2 (PAR2) on multiple cellular targets (Figure 2) 13,19. Importantly, α/β-tryptase heterotetramers were shown to be uniquely capable of inducing acute vascular endothelial cell permeability in a PAR2-dependent manner 13. The in vivo consequences of selective PAR2 activation by α/β-tryptase heterotetramers are unproven. However, selective activation by α/β-tryptase heterotetramers may contribute to certain clinical phenotypes associated with HαT, including modification of anaphylaxis severity and pain sensation.
HαT-ASSOCIATED PHENOTYPES: WHEN TO CONSIDER GENETIC TESTING?
In the first series of publications describing HαT 30,36,37 a variety of systemic signs and symptoms were reported which included systemic immediate hypersensitivity reactions, cutaneous flushing and pruritus, seemingly functional upper and lower gastrointestinal (GI) symptoms, connective tissue abnormalities and associated pain, as well as symptoms suggestive of autonomic dysfunction. As more individuals with HαT were identified, and the realization was made that this was a common genetic trait, it has likewise become clear that many individuals with HαT – perhaps the majority – may be asymptomatic 15,30. Despite this, some symptoms including severe anaphylaxis in a number of clinical contexts 13,14,30 discussed later in this review, as well as certain skin and GI complaints have been validated as significantly associated with HαT in well-controlled or unselected populations 30 (Table 1).
Table 1.
Clinical features associated with hereditary alpha-tryptasemia (HαT).
| Manifestation | Reported Prevalence | Association Supported in Independent Cohorts |
|---|---|---|
|
| ||
| Basal serum tryptase >8ng/mL * | 100% | Yes |
| Chronic gastroesophageal reflux symptoms | 56–77% | No |
| Arthralgia | 44–45% | No |
| Body pain/Headache | 33–47% | No |
| Flushing/Pruritus | 32–55% | Yes |
| Sleep disruption | 22–39% | No |
| Systemic immediate hypersensitivity reaction | 21–28% | Yes † |
| Retained primary dentition | 20–33% | Yes |
| Lower GI symptoms | 14–90% | Yes |
| Systemic venom reaction | 14–22% | Yes |
| Congenital skeletal abnormality | 11–26% | No |
| GI food sensitivity | 0–39% | No |
| Joint Hypermobility | 0–28% | No |
Rarely BST among individuals with 2α3β genotypes have been reported with BST as low as 6.5 ng/mL, however this genotype may result from increased β-tryptase encoding copy number, which is not associated with inherited elevations in BST and has not been observed in Mendelian family studies. GI – gastrointestinal.
Associated with idiopathic anaphylaxis and prevalence of anaphylaxis in patients with systemic mastocytosis.
One emerging story, which exemplifies the complexity of understanding HαT-associated symptoms is that of lower GI manifestations. In initial reports, HαT was linked to irritable bowel syndrome (IBS), as symptomatic individuals frequently presented with seemingly functional lower GI complaints frequently meeting Rome III criteria. A more recent examination of a large cohort of well-phenotyped patients with classical IBS found no association with HαT 18; this finding has been recapitulated in a subsequent smaller study 32. However, two independent studies have demonstrated that symptomatic individuals with HαT have increased mast cells with atypical features in gut mucosae 18,38 which were associated with increased intestinal epithelial cell pyroptosis in one of the studies – suggestive of occult inflammation despite no gross evidence of classical inflammatory disease. These changes were associated with changes in lymphocyte populations in the gut tissue and peripheral blood and associated with increased antibodies directed towards GI-associated proteins. Mechanisms underlying these phenomena remain an area of investigation, however these studies demonstrate GI complaints in the context of HαT may have histopathologic and clinical phenotypes which overlap with, but are distinct from, IBS.
Taking a thorough patient history is the most important first step in evaluating the need of genetic testing for HαT. Tryptase genotyping by ddPCR should be considered in patients who present with symptoms of mast cell activation, have a history of anaphylaxis, GI symptoms that are associated with nocturnal awakening, or gut inflammation that is not responding to therapy or is atypical and have a BST level greater than 8 ng/mL; in rare cases genotyping may be considered in patients with BST >6 ng/mL particularly if there is a family history of elevated BST >8 ng/mL. Establishing a diagnosis of HαT may reduce the need for further invasive and cumbersome work-up including tissue and/or bone marrow biopsies in individuals who do not otherwise display “red flags” suggestive of clonal mast cell or other myeloid dyscrasias (Table 2) 34. Moreover, identifying elevated BST in the absence of HαT or advanced kidney disease, should prompt a clinical evaluation for clonal disease within the myeloid compartment 39.
Table 2.
“Red Flags” - signs and symptoms suggestive of clonal myeloid or mast cell disease
| • Lymphadenopathy |
| • Hepatosplenomegaly |
| • CBC abnormalities |
| – thrombocytopenia |
| – anemia |
| – pancytopenia |
| – polycythemia |
| – neutrophilia |
| – hypereosinophilia (AEC >1500 cells/μL) |
| • Eosinophilic tissue infiltration and/or inflammation |
| • Anaphylaxis |
| – idiopathic |
| – venom |
| – severe reactions with syncope and/or hemodynamic instability |
| • Urticaria pigmentosa / Darier’s sign |
| • Premature osteopenia/osteoporosis or pathological fracture |
| • BST discordant with TPSAB1 copy number* |
Each additional TPSAB1 copy number encoding α-tryptase increases BST by approximately 9–10 ng/mL, but definitely cut-offs for discordance have not yet been established; AEC – absolute eosinophil count; BST – basal serum tryptase.
IMPACT OF HαT ON THE PRESENTATION AND MANAGEMENT OF CLONAL MAST CELL DISEASE
Increased germline copies of alpha-tryptase–encoding sequences at TPSAB1 are more prevalent among individuals with systemic mastocytosis and are associated with an increased relative risk for anaphylaxis among these patients 13,14. In the first study, HαT was found in 5.3% of healthy individuals, 5.6 % of controls with non-atopic disease and 12.2 % of patients diagnosed with systemic mastocytosis 13. Those with mastocytosis and concomitant HαT were also at increased risk for systemic anaphylaxis when compared to the general population (relative risk = 9.5 %; P = 0.007). Similarly, a follow-up study reported HαT in 17.2% of mastocytosis patients and 4.4% of the control population 14. In the latter study, the highest prevalence was found in those with indolent systemic mastocytosis. More severe mediator-related symptoms were also observed in those with concomitant HαT and mastocytosis in the large multicenter European study, when examined using a validated symptom scoring tool. More recently, a smaller single-center study confirmed the association between HαT and clonal mast cell disease, with 18% having HαT 32. However, this study failed to identify differences in phenotypes based upon the concomitant presence of HαT in mastocytosis patients, likely due to an insufficient sample-size to detect meaningful differences. Thus, identifying individuals with increased TPSAB1 germline copy number appears to be an important biomarker to be included in risk assessment models relating to mastocytosis. It remains unknown at present whether patients with both HαT and mastocytosis will require more intense or different medical management.
Another consideration is whether HαT as a genetic trait might influence homeostasis within the marrow microenvironment and predispose to the development of myeloid disorders including mastocytosis. In support of this possibility, individuals with HαT and mast cell activation symptoms have increased bone marrow mast cells, which are atypical 36,40, hypogranular and abnormally localized. These findings led investigators to suggest such HαT-associated mast cell abnormalities might contribute to or predispose for the development of mastocytosis. Moreover, the increased prevalence observed in systemic mastocytosis may also support this conclusion. However, it is also likely that concomitant HαT increases the likelihood that an individual with mastocytosis comes to medical attention due to the presence of both increased symptom severity as well as elevated BST levels.
The presence of HαT in association with clonal mast cell disease also has implications for diagnosis. This is because a serum total tryptase persistently > 20 ng/ml is a minor criterion for the diagnosis of systemic mastocytosis, in the absence of an associated myeloid neoplasm. These serum tryptase levels are also followed to monitor disease progression and response to therapeutic intervention. Adjustments to the baseline tryptase measurement thus must be considered if BST in those with HαT is to be used as a minor diagnostic criterion.
At the time of the writing of this manuscript, a validated method to adjust such values based on α-tryptase copy number is not yet available. One suggestion is that the basal tryptase level in mastocytosis patients with HαT be corrected by dividing the tryptase level by one plus the number of extra α-tryptase copies 41. However, in populations where the average BST for individuals with one extra TPSAB1 copy is approximately 15 ng/mL this formula may overcorrect, particularly in individuals with higher order copy number (Table 3). An alternative approach is to use prediction intervals based upon these population data for individuals with HαT in the absence of mastocytosis 42.
Table 3.
Tryptase genotypes and associated basal serum tryptase levels dictated by additional TPSAB1 copy encoding α-tryptase.
| Additional TPSAB1 copy number | 0 | 1 | 2 | 3 | 4 |
|---|---|---|---|---|---|
|
| |||||
|
Tryptase Genotypes* (TPSAB1, TPSB2) |
β,β/β,β; α,β/β,β; α,β/α,β; α,β/β,β,β; β,β/β,β,β; β/α,β; β/β,β |
αα,β/β,β; αα,β/α,β; αα,ββ/β,β; αα,ββ/α,β |
αα,β/αα,β; ααα,β/β,β; ααα,β/α,β |
ααα,β/αα,β | ααααα,β/β,β; ααααα,β/α,β |
|
| |||||
| BST (ng/mL), median (range) | 4.1 (0–10.4) | 13.6 (6.5–33.9) | 22.5 (10.5–39.5) | 27.3 (23.4–40) | 37 (25.5–62.7) |
Tryptase genotypes identified to date for this number of TPSAB1 replications. In all cases TPSAB1 replications associated with elevated BST identified encode α-tryptase.
HαT AND ANAPHYLAXIS SEVERITY IN VENOM ALLERGIC PATIENTS
In unselected healthy volunteers, those found to have HαT are two- to threefold more likely (16%) to report Hymenoptera venom-triggered anaphylaxis (HVA) compared to those who do not have HαT 16. However, recent larger studies showed an increased prevalence of HαT only among individuals with severe HVA (principally Mueller grade IV), with a prevalence of roughly doubled relative to the expected prevalence in the general population or in overall cohorts of patients with venom allergy (from 5.5% to ~10%) 13,43. Thus, HαT does not appear to contribute to the development of sensitization to Hymenoptera venom but rather modifies the clinical outcome, potentially by unique activities of α/β-tryptase heterotetramers that may increase vascular permeability and anaphylaxis severity 13,19. Given the high frequency (81% and 82%) of detecting HαT among those with HVA 13,43, elevated BST and no evidence of clonal MC disease, and recent observations that HαT is a most common cause (65%) of elevated BST levels in patients with HVA 43, we recommend that tryptase genotyping be considered in the clinical workup of all individuals with HVA and BST levels >8 ng/mL as well as in individuals with a history of, or at risk for, severe venom anaphylaxis and a normal BST level of 6–8 ng/mL. Currently, no individual has been reported or observed with HαT and BST< 6 ng/mL, including individuals with HVA 13,15,30,31,36,37,43. The association between this genetic trait and severe venom anaphylaxis appears to be independent of concomitant clonal MC disease; nevertheless, the relatively high and unexpected prevalence of concomitant HαT and clonal MC disease of (8.6–15.8%) in different HVA cohorts suggests that the relative risk for severe HVA imparted by HαT and SM are additive 13,14,16,43–45.
The ability to risk-stratify individuals with venom allergy and identify those at risk of severe HVA is important not only for anticipatory counseling of patients but also for guiding therapy, as lifelong VIT is currently recommended for individuals with clonal MC disease and HVA 44,45. Because at the time of writing this manuscript, longitudinal data are lacking the authors feel it is prudent to continue lifelong VIT in patients with severe HVA and HαT, until such time that the long-term data become available.
IMPLICATIONS OF HαT FOR NON-CLONAL MAST CELL DISORDERS
In the current classification of mast cell disorders, idiopathic mast cell disorders are a subcategory further divided to include idiopathic anaphylaxis (IA), chronic spontaneous urticaria (CSU), idiopathic histaminergic angioedema (IHA) and idiopathic mast cell activation syndrome (iMCAS) 46. In one study, patients with IA were three times as likely (17% versus 5%) to be diagnosed with HαT and accounted for all individuals with a baseline serum tryptase ≥11.4 ng/ml with IA in whom systemic mastocytosis had been ruled out with bone marrow biopsy 13. These patients did not have increased markers of mast cell activation in bone marrow mast cells when compared to patients with clonal mast cell disease 47. Moreover, cultured mast cells from individuals with HαT have not been shown to be hyperactive in vitro 30, suggesting that increased mast cell reactivity is unlikely to be the cause for this clinical observation.
Like IA, symptomatic individuals with HaT are more often female 48,49, so one can speculate there may be an influence from hormones as seen in asthma and other atopic diseases 50, as well as in anaphylaxis in mice 51. Although HαT is present from birth – de novo increases in TPSAB1 have yet to be observed or reported – many individuals report symptom onset later in life, frequently after puberty. In one study of over 900 patients in an Austrian allergy practice with an elevated serum tryptase – in whom tryptase genotyping was not performed –children ages 1–10 years had the lowest proportion of elevated BST values and cutaneous symptoms were the most prevalent symptom with anaphylaxis being rare 52.
The increased prevalence of HαT in IA, the associated increased frequency of anaphylaxis and severity of mast cell mediator symptoms among SM patients with HαT, as well as the increased severity of anaphylaxis among venom allergic patients HαT, suggests that this genetic trait may modify mast cell-mediated symptoms and reactions more generally. However, other than aiding in risk stratification of SM and venom allergic patients, and proposed extensions to the duration of VIT, diagnosing HαT is not associated with clinical experience which would support a modification of management relating to other associated allergic diseases. This is in part because no specific or targeted therapies for HαT are currently available. Additional studies in patients with IgE-mediated food and drug allergy as well as non-IgE mast cell-mediated reactions are also needed to better understand whether HαT may modify these other clinical phenotypes. One recent study failed to identify any specific symptoms associated with HαT 32. Unfortunately, the study design was not powered to detect differences in symptoms were they found to be present at the expected prevalence 30,33. However, this study reported that over 40% of individuals referred for symptoms of mast cell activation who did not have evidence of clonal mast cell disease were found to have HαT, suggesting a possible association with developing such symptoms.
HαT VIEWED IN THE CONTEXT OF PRIMARY ATOPIC DISORDERS
While the initial descriptions of HαT were consistent with a monogenic cause of allergic disease, or a Primary Atopic Disorder (PAD) 30,36,37, our understanding of HαT has since evolved considerably. PADs refer to congenital disorders, usually monogenic, which present with symptoms related to allergic reactions or inflammation 53. PADs can range from classically syndromic – such as the facial and skeletal dysmorphism, developmental delay autoimmunity, infection, and severe atopic disease seen in Phosphoglucomutase-3 (PGM3) deficiency – to the focally reactive – such as severe familial vibratory urticaria resulting from a damaging ADGRE2 variant. As such, symptoms of atopy in PADs can vary based on the system impacted by the underlying genetic variants, from marked IgE elevation to chronic allergic inflammation such as eczema and eosinophilic GI disease, to mast cell activation and anaphylaxis. The host of symptoms reported in association with HαT not clearly related to mast cell function, therefore could be considered to fall into the category of a syndromic PAD. However, the realization that HαT is relatively common has raised the probability that some of the disease associations reported thus far have been due to ascertainment, observational, and/or referral biases.
Many of the symptoms seen in the initial cohorts still require validation in unbiased cohorts. In the meantime, the important question raised is whether HαT should be considered a PAD. Evidence points to HαT as a risk factor for mast cell-mediated reactions including anaphylaxis 13,17,30, and clearly demonstrates HαT is the overwhelming cause for elevated BST in the general population. It is noteworthy that many monogenic diseases display variable penetrance and expressivity; even among disorders known to cause atopy, allergic disease is not always present 53. Likewise, it is estimated that as many as two-thirds of individuals HαT may not manifest symptoms. Among PADs, common variant-derived genetic risk for allergy impact the penetrance and expressivity of symptoms in these patients 54–57, and it is likely that HαT can modify certain PADs to promote the expression of allergic phenotypes as seen in acquired disorders such as venom allergy and SM as well. It is therefore perhaps better to contextualize single gene variants with respect to their overall impact. In this context, the impact of HαT on various clinical outcomes remains to be fully determined. However, based upon the preponderance of available clinical data, HαT appears to lie somewhere on the spectrum between a PAD and a common genetic risk factor for, or modifier of, certain allergic phenotypes, and is best thought of currently as a genetic trait.
MANAGEMENT STRATEGIES IN SYMPOMATIC INDIVIDUALS WITH HαT
As described above, the clinical phenotypes associated with HαT can be varied and affect multiple organ including those in the skin and GI tract suggestive of mast cell mediator release. Treatment strategies to date have largely been adopted from those published for the management of clonal and non-clonal mast cell-associated disorders focused on improving symptoms with anti-histamines and other mast cell-directed therapies including mast cell stabilizers, leukotriene modifiers, aspirin, and in certain specific situations corticosteroids 58. As elsewhere in medical management, it is similarly important that medications are trialed step-wise fashion and unhelpful modalities discontinued in order to prevent the real risk of polypharmacy. Caution is similarly followed when using supratherapeutic doses and combinations of sedating and non-sedating antihistamines as overdose can lead to drowsiness, blurred vision, nausea, vomiting, increased heart rate, confusion, loss of balance/dizziness, drowsiness, headache, and agitation and in rare instances frank hallucinations or pseudo-seizures 59–62. These untoward side effects can particularly HαT since these symptoms are perceived to overlap with those frequently associated with HαT. Caution is further warranted when prescribing courses of systemic corticosteroids, and an inventory should be made of the frequency and cumulative doses received from various providers since many patients receive regular courses of steroids following anaphylactoid or anaphylactic reactions – particularly from emergency departments – and the harm profile of steroids is well-established even with short term use (<30 days in a 12-month period) 63,64.
Since HαT is associated with an increased risk for anaphylaxis in multiple contexts, individuals with recurrent anaphylaxis should have their treatment targeted at mitigating risk of recurrence. If first-line oral agents are unsuccessful, two retrospective studies in symptomatic individuals with HαT suggest omalizumab may be beneficial for certain symptoms, in particular skin and respiratory symptoms related to mast cell activation, as well as anaphylaxis 31,65. A recent randomized placebo controlled clinical trial of omalizumab in idiopathic anaphylaxis (IA) demonstrated modestly favorable, although not statistically significant treatment benefit 66. At least two of the trial participants had HαT; one was in the placebo arm, and one was in the treatment arm. The HαT participant in the treatment cohort experienced a significant reduction in episodes of anaphylaxis during the treatment period.
Unfortunately – and somewhat surprisingly – omalizumab has not been reported to provide significant benefit for gastrointestinal manifestations among symptomatic individuals with HαT 65. Perhaps less unexpectedly, pain, connective tissue abnormalities, and symptoms suggestive of autonomic dysfunction also have not been reported to improve with omalizumab – and were also reported to be largely refractory to mast cell-targeted therapies. Strategies for treatment of these kinds of symptoms frequently require a multi-disciplinary approach and should be based on a differential diagnosis directed by history and physical, imaging, and targeted laboratory testing. Of paramount importance is the requirement to ensure additional diagnoses are correctly made; since HαT is common, additional diagnoses should be anticipated in highly symptomatic patients as the rule, not the exception.
While data are incomplete at the time of this manuscript, the authors feel it is important that individuals with HαT be considered at risk and undergo evaluation for premature bone loss. This recommendation is based upon 1) clinical observations linking elevated BST of unknown causes with premature osteopenia and osteoporosis 67, 2) increased and distinct populations of bone marrow mast cells in symptomatic individuals 36,40, and 3) frequent receipt of systemic corticosteroids 68.
An increased prevalence of autoinflammatory or autoimmune disease has not been reported among individuals with HαT and population-based studies have not identified elevations in BST levels to be associated with these conditions. Nevertheless, because HαT affects 1 in 20 individuals in the U.S., roughly the same number with these conditions would be expected to have HαT. Interestingly, some of the same kinds of treatments used in autoimmune diseases – including certain Janus and tyrosine kinase inhibitors, calcineurin inhibitors, and IL-6 blockade – have been used or are in clinical trials (NCT03770273) for patients with systemic mastocytosis 69–71. Whether these therapies might have specific benefit to patients with autoimmune or inflammatory disorders and concomitant HαT remains an area of clinical interest.
FUTURE THERAPEUTIC AND RESEARCH DIRECTIONS
To date, only two relatively small cohorts of ostensibly healthy adults has been screened prospectively for HαT in order to validate certain associated phenotypes (Table 1) 30,32. While many of these phenotypes - including clonal mast cell disease and idiopathic anaphylaxis - are likely too rare to evaluate in this manner, more common symptoms may be amenable to this kind of approach. In the first study, 100 individuals from the ClinSeq® cohort - comprised of ostensibly healthy adult volunteers - were prospectively screened with 91 available for follow-up evaluation. Nine individuals with HαT were compared to 82 controls; systemic venom reactions, cutaneous flushing and pruritus, IBS-like symptoms, retained primary dentition, and autonomic symptom scores were significantly associated with HαT. Using a similar approach, the two authors of a more recent study screened a biorepository of patients with various medical conditions for HαT, 100 of which were randomly chosen for clinical evaluation. From these, 10 individuals with HαT were compared to 24 without, after 65 controls were excluded. This latter smaller study did not confirm the associations found in the first study. However, given that only 3/9 from the initial report were found to be symptomatic, the follow-up study design was not powered to detect any clinical phenotype at this prevalence, even if it was completely absent from the control group. These conflicting results highlight the difficulties in study design when considering common genetic conditions. Moreover, the prevalence of this trait, and the potential for significant referral, ascertainment, and observational biases in case series can further cloud our understanding in future studies. Additional large-scale validation studies in well-defined patient populations not selected for based upon serum tryptase, HαT, or other potentially confounding findings – such as those undertaken among individuals with HVA and SM 13,14 – are of paramount importance for further assessment of clinical phenotypes associated with HαT.
The relatively recent discovery that HαT is the common cause for elevated BST has and will likely continue to impact the diagnostic use of tryptases in mast cell disorders and reactions. Indeed, the incorporation of BST as a minor criterion for systemic mastocytosis is likely to require revision, as stated above. In addition, while baseline variability of BST had previously been reported, the impact of this variability has now been more fully appreciated and shown to significantly affect the specificity of the previously published algorithm 20% + 2ng/mL (20+2) for confirmation of anaphylaxis among individuals with elevated BST due to both HαT and SM 72. A new threshold ratio has been proposed – where an increase in tryptase over baseline of ~68% would constitute a significant rise in BST consistent with anaphylaxis. This new ratio rule is identical to 20+2 at the median BST for healthy individuals (in whom SM and HαT has been excluded) but demonstrated improved specificity among those with elevated BST, without impacting sensitivity. However, this new approach requires validation.
While α/β-tryptase heterotetramers are a compelling explanation for some symptoms associated with HαT, this mechanism seems to be a dissatisfying hypothetical basis for other symptoms. Potentially consistent with this supposition, are reports that many symptoms associated with HαT failed to respond to mast cell directed therapies including omalizumab. Additional studies are ongoing to address these questions, but one intriguing observation was the finding of increased intestinal epithelial cell pyroptosis and class-switched memory B cells in GI mucosa of symptomatic individuals with HαT 18 – where symptoms were largely refractory to mast cell-directed therapies. Whether HαT is modifying another disease phenotype, or this provides a clue to an additional mechanism by which α-tryptase over-expression may impact epithelial cell inflammation and/or homeostasis, remains an area of ongoing research.
Looking forward, we envision the impact of tryptase genotyping on current and future clinical trials to extend well beyond ascertainment of the presence or absence of HαT alone. A recent poignant example of this was a study of asthmatic patients treated with omalizumab 73. In a post-hoc analysis of phase III clinical trial data, it was shown that lacking α-tryptase – and conversely having more active β-tryptase-containing alleles – was associated with Th2 low asthma that was poorly responsive to omalizumab therapy and higher mature tryptase levels in the respiratory tract. This has led to a phase II trial of an anti-tryptase neutralizing antibody (NCT04092582), the results from which are not public at the time of writing this manuscript. We anticipate tryptase genotyping – as used in this study – will be an important risk stratifying component of future clinical trials in patients with mast cell-associated disorders and reactions.
In addition to tryptase neutralization, which would seem likely to have specific benefit in symptomatic individuals with HαT, other targeted therapies in various stages of clinical trials are also intriguing candidates. Monoclonal antibodies targeting IL-33 signaling and sialic acid–binding immunoglobulin-type lectins-8 (SIGLEC-8) also show mechanistic promise 74.
An additional class of drugs which may have a possible future role are kinase inhibitors such as BTK inhibitors that have recently been shown to reduce mast cell reactivity in humans 75. While preliminary data from phase II clinical trials in chronic spontaneous urticaria (NCT03137069) failed to reach statistical significance in this disorder, these and similar inhibitors may have benefit in mast cell-associated disorders provided these potentially cytotoxic treatments can be used safely administered without significant suppression of the immune or hematopoietic systems. All such interventions will require further clinical study in prospective randomized clinical trials, though the variability expressivity, moderate penetrance on clinical phenotypes, and relatively common nature of HαT have thus far frustrated efforts to design prospective studies. At the time of this manuscript, to the authors knowledge, there are no active clinical trials for symptomatic individuals with HαT.
CONCLUSION
Since it was first revealed in 2014 that elevated BST levels could be inherited in an autosomal dominant Mendelian manner, much has been learned about HαT. We now know that HαT is common and associated generally with modest increases in mast cells within the bone marrow and GI mucosa as well as with systemic mastocytosis where having both leads to more prevalent anaphylaxis and mast cell-mediator symptoms. HαT is also associated with more severe anaphylaxis among venom allergic patients where it is the most common cause for elevated BST in those individuals. Moreover, HαT is nearly three times more common among individuals with idiopathic anaphylaxis – ostensibly due to modification of reaction severity in these patients in a manner similar to that seen with SM. Since α-tryptase lacks enzymatic activity, the mechanism(s) underlying these clinical associations were initially cryptic. However, the identification of α/β-tryptase heterotetramers and their unique properties may help explain at least some of these findings. Despite these advances, much remains to be learned how HαT may contribute to and/or modify these and other associated phenotypes and/or diseases. However, given how broadly mast cells have been implicated in health and disease, HαT provides a unique lens in which to study the mast cell compartment in these contexts. Future studies will undoubtedly both refine and codify our understanding of this common genetic trait as well as the effects more broadly of variable tryptase gene composition in humans.
Funding
This research was supported by the Division of Intramural Research of the National Institute of Allergy and Infectious Diseases, NIH. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. This work was also supported in part by the Gatorade Trust through funds distributed by the University of Florida, Department of Medicine, and extramural funding via 1R21TR002639–01A1 (SCG).
Abbreviations used:
- aCGH
microarray-based comparative genomic hybridization
- BST
basal serum tryptase
- ddPCR
droplet digital PCR
- ADGRE2
Adhesion G Protein-Coupled Receptor E2
- GPCR
G-protein coupled receptor
- HαT
hereditary alpha-tryptasemia
- HUVEC
human vascular endothelial cells
- ISM
indolent systemic mastocytosis
- MDS
myelodysplastic syndrome
- MRGPRX2
Mas-related G-protein coupled receptor member X2
- PAR2
Protease activated receptor-2
- TPSAB1
Tryptase alpha/beta 1
- TPSB2
Tryptase beta 2
- TPSG1
Tryptase gamma 1
- WES
whole exome sequencing
- WGS
whole genome sequencing
- PGM3
Phosphoglucomutase-3
Footnotes
Declaratrion of competing interest
VCU receives royalties from Thermo Fisher for their tryptase test that are shared with LBS as its inventor. None of the remaining authors have relevant conflicts of interest to report.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Metcalfe DD. Mast cells and mastocytosis. Blood. 2008;112(4):946–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Olivera A, Beaven MA, Metcalfe DD. Mast cells signal their importance in health and disease. J Allergy Clin Immunol. 2018;142(2):381–393. [DOI] [PubMed] [Google Scholar]
- 3.Irani AA, Schechter NM, Craig SS, DeBlois G, Schwartz LB. Two types of human mast cells that have distinct neutral protease compositions. Proc Natl Acad Sci U S A. 1986;83(12):4464–4468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Oskeritzian CA, Zhao W, Min HK, et al. Surface CD88 functionally distinguishes the MCTC from the MCT type of human lung mast cell. J Allergy Clin Immunol. 2005;115(6):1162–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Subramanian H, Gupta K, Ali H. Roles of Mas-related G protein-coupled receptor X2 on mast cell-mediated host defense, pseudoallergic drug reactions, and chronic inflammatory diseases. J Allergy Clin Immunol. 2016;138(3):700–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hallgren J, Pejler G. Biology of mast cell tryptase. An inflammatory mediator. FEBS J. 2006;273(9):1871–1895. [DOI] [PubMed] [Google Scholar]
- 7.Schwartz LB, Lewis RA, Austen KF. Tryptase from human pulmonary mast cells. Purification and characterization. J Biol Chem. 1981;256(22):11939–11943. [PubMed] [Google Scholar]
- 8.Cavalcante MC, de Andrade LR, Du Bocage Santos-Pinto C, et al. Colocalization of heparin and histamine in the intracellular granules of test cells from the invertebrate Styela plicata (Chordata-Tunicata). J Struct Biol. 2002;137(3):313–321. [DOI] [PubMed] [Google Scholar]
- 9.Crivellato E, Travan L, Ribatti D. The phylogenetic profile of mast cells. Methods Mol Biol. 2015;1220:11–27. [DOI] [PubMed] [Google Scholar]
- 10.Trivedi NN, Tong Q, Raman K, Bhagwandin VJ, Caughey GH. Mast cell alpha and beta tryptases changed rapidly during primate speciation and evolved from gamma-like transmembrane peptidases in ancestral vertebrates. J Immunol. 2007;179(9):6072–6079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lyons JJ. Hereditary Alpha Tryptasemia: Genotyping and Associated Clinical Features. Immunol Allergy Clin North Am. 2018;38(3):483–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Trivedi NN, Tamraz B, Chu C, Kwok PY, Caughey GH. Human subjects are protected from mast cell tryptase deficiency despite frequent inheritance of loss-of-function mutations. J Allergy Clin Immunol. 2009;124(5):1099–1105 e1091–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lyons JJ, Chovanec J, O’Connell MP, et al. Heritable risk for severe anaphylaxis associated with increased alpha-tryptase-encoding germline copy number at TPSAB1. J Allergy Clin Immunol. 2020. [DOI] [PubMed] [Google Scholar]
- 14.Greiner G, Sprinzl B, Gorska A, et al. Hereditary alpha tryptasemia is a valid genetic biomarker for severe mediator-related symptoms in mastocytosis. Blood. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Robey RC, Wilcock A, Bonin H, et al. Hereditary Alpha-Tryptasemia: UK Prevalence and Variability in Disease Expression. J Allergy Clin Immunol Pract. 2020. [DOI] [PubMed] [Google Scholar]
- 16.O’Connell MP, Lyons JJ. Hymenoptera venom-induced anaphylaxis and hereditary alpha-tryptasemia. Curr Opin Allergy Clin Immunol. 2020;20(5):431–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu R, Lyons JJ. Hereditary Alpha-Tryptasemia: a Commonly Inherited Modifier of Anaphylaxis. Curr Allergy Asthma Rep. 2021;21(5):33. [DOI] [PubMed] [Google Scholar]
- 18.Konnikova L, Robinson TO, Owings AH, et al. Small intestinal immunopathology and GI-associated antibody formation in hereditary alpha-tryptasemia. J Allergy Clin Immunol. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Le QT, Lyons JJ, Naranjo AN, et al. Impact of naturally forming human alpha/beta-tryptase heterotetramers in the pathogenesis of hereditary alpha-tryptasemia. J Exp Med. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Caughey GH. Tryptase genetics and anaphylaxis. J Allergy Clin Immunol. 2006;117(6):1411–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hellman LT, Akula S, Thorpe M, Fu Z. Tracing the Origins of IgE, Mast Cells, and Allergies by Studies of Wild Animals. Front Immunol. 2017;8:1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stevens RL, Adachi R. Protease-proteoglycan complexes of mouse and human mast cells and importance of their beta-tryptase-heparin complexes in inflammation and innate immunity. Immunol Rev. 2007;217:155–167. [DOI] [PubMed] [Google Scholar]
- 23.Wong GW, Zhuo L, Kimata K, Lam BK, Satoh N, Stevens RL. Ancient origin of mast cells. Biochem Biophys Res Commun. 2014;451(2):314–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Raymond WW, Sommerhoff CP, Caughey GH. Mastin is a gelatinolytic mast cell peptidase resembling a mini-proteasome. Arch Biochem Biophys. 2005;435(2):311–322. [DOI] [PubMed] [Google Scholar]
- 25.Yuan J, Beltman J, Gjerstad E, et al. Expression and characterization of recombinant gamma-tryptase. Protein Expr Purif. 2006;49(1):47–54. [DOI] [PubMed] [Google Scholar]
- 26.Badge RM, Yardley J, Jeffreys AJ, Armour JA. Crossover breakpoint mapping identifies a subtelomeric hotspot for male meiotic recombination. Hum Mol Genet. 2000;9(8):1239–1244. [DOI] [PubMed] [Google Scholar]
- 27.Martin J, Han C, Gordon LA, et al. The sequence and analysis of duplication-rich human chromosome 16. Nature. 2004;432(7020):988–994. [DOI] [PubMed] [Google Scholar]
- 28.Lyons JJ, Yi T. Mast cell tryptases in allergic inflammation and immediate hypersensitivity. Curr Opin Immunol. 2021;72:94–106. [DOI] [PubMed] [Google Scholar]
- 29.Luskin KT, White AA, Lyons JJ. The Genetic Basis and Clinical Impact of Hereditary Alpha-Tryptasemia. J Allergy Clin Immunol Pract. 2021. [DOI] [PubMed] [Google Scholar]
- 30.Lyons JJ, Yu X, Hughes JD, et al. Elevated basal serum tryptase identifies a multisystem disorder associated with increased TPSAB1 copy number. Nat Genet. 2016;48(12):1564–1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Giannetti MP, Weller E, Bormans C, Novak P, Hamilton MJ, Castells M. Hereditary alpha-tryptasemia in 101 patients with mast cell activation-related symptomatology including anaphylaxis. Ann Allergy Asthma Immunol. 2021. [DOI] [PubMed] [Google Scholar]
- 32.Chollet MB, Akin C. Hereditary alpha tryptasemia is not associated with specific clinical phenotypes. J Allergy Clin Immunol. 2021. [DOI] [PubMed] [Google Scholar]
- 33.Lyons JJ. On the complexities of tryptase genetics and impacts on clinical phenotypes. J Allergy Clin Immunol. 2021;In submission. [DOI] [PubMed] [Google Scholar]
- 34.Lyons JJ, Schwartz LB. Clinical Approach to a Patient with Elevated Serum Tryptase: Implications of Acute Versus Basally Elevated Levels. 1 ed: Springer, Cham; 2020. [Google Scholar]
- 35.Boyden SE, Desai A, Cruse G, et al. Vibratory Urticaria Associated with a Missense Variant in ADGRE2. N Engl J Med. 2016;374(7):656–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lyons JJ, Sun G, Stone KD, et al. Mendelian inheritance of elevated serum tryptase associated with atopy and connective tissue abnormalities. J Allergy Clin Immunol. 2014;133(5):1471–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sabato V, Chovanec J, Faber M, Milner JD, Ebo D, Lyons JJ. First Identification of an Inherited TPSAB1 Quintuplication in a Patient with Clonal Mast Cell Disease. J Clin Immunol. 2018;38(4):457–459. [DOI] [PubMed] [Google Scholar]
- 38.Hamilton MJ, Zhao M, Giannetti MP, et al. Distinct Small Intestine Mast Cell Histologic Changes in Patients With Hereditary Alpha-tryptasemia and Mast Cell Activation Syndrome. Am J Surg Pathol. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lyons JJ. Inherited and acquired determinants of serum tryptase levels in humans. Ann Allergy Asthma Immunol. 2021;In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Giannetti MP, Akin C, Hufdhi R, et al. Patients with mast cell activation symptoms and elevated baseline serum tryptase level have unique bone marrow morphology. J Allergy Clin Immunol. 2021;147(4):1497–1501 e1491. [DOI] [PubMed] [Google Scholar]
- 41.Valent P, Akin C, Hartmann K, et al. Revised Diagnostic Criteria and Classification of Mast Cell Disorders: A Consensus Proposal. Blood. 2021;In submission. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Park YH, Liu Y, Chovanec J, et al. Incorporating tryptase genotyping in the evaluation of clonal myeloid disorders. Blood. 2021;In submission. [Google Scholar]
- 43.Selb J, Rijavec M, Erzen R, et al. Routine KIT p.D816V screening identifies clonal mast cell disease in Hymenoptera allergic patients regularly missed using baseline tryptase levels alone. J Allergy Clin Immunol. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bonadonna P, Perbellini O, Passalacqua G, et al. Clonal mast cell disorders in patients with systemic reactions to Hymenoptera stings and increased serum tryptase levels. J Allergy Clin Immunol. 2009;123(3):680–686. [DOI] [PubMed] [Google Scholar]
- 45.Zanotti R, Lombardo C, Passalacqua G, et al. Clonal mast cell disorders in patients with severe Hymenoptera venom allergy and normal serum tryptase levels. J Allergy Clin Immunol. 2015;136(1):135–139. [DOI] [PubMed] [Google Scholar]
- 46.Valent P, Akin C, Bonadonna P, et al. Proposed Diagnostic Algorithm for Patients with Suspected Mast Cell Activation Syndrome. J Allergy Clin Immunol Pract. 2019;7(4):1125–1133 e1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Carter MC, Desai A, Komarow HD, et al. A distinct biomolecular profile identifies monoclonal mast cell disorders in patients with idiopathic anaphylaxis. J Allergy Clin Immunol. 2018;141(1):180–188 e183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Moneret-Vautrin DA, Gay G. [The so-called “idiopathic” anaphylaxis: allergic and pseudo-allergic reactions]. Allerg Immunol (Paris). 1991;23(3):89–93. [PubMed] [Google Scholar]
- 49.Webb LM, Lieberman P. Anaphylaxis: a review of 601 cases. Ann Allergy Asthma Immunol. 2006;97(1):39–43. [DOI] [PubMed] [Google Scholar]
- 50.Osman M Therapeutic implications of sex differences in asthma and atopy. Arch Dis Child. 2003;88(7):587–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hox V, Desai A, Bandara G, Gilfillan AM, Metcalfe DD, Olivera A. Estrogen increases the severity of anaphylaxis in female mice through enhanced endothelial nitric oxide synthase expression and nitric oxide production. J Allergy Clin Immunol. 2015;135(3):729–736 e725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fellinger C, Hemmer W, Wohrl S, Sesztak-Greinecker G, Jarisch R, Wantke F. Clinical characteristics and risk profile of patients with elevated baseline serum tryptase. Allergol Immunopathol (Madr). 2014;42(6):544–552. [DOI] [PubMed] [Google Scholar]
- 53.Lyons JJ, Milner JD. Primary atopic disorders. J Exp Med. 2018;215(4):1009–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Clark H, Granell R, Curtin JA, et al. Differential associations of allergic disease genetic variants with developmental profiles of eczema, wheeze and rhinitis. Clin Exp Allergy. 2019;49(11):1475–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cotterchio M, Lowcock E, Bider-Canfield Z, et al. Association between Variants in Atopy-Related Immunologic Candidate Genes and Pancreatic Cancer Risk. PLoS One. 2015;10(5):e0125273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jansen PR, Petrus NCM, Venema A, et al. Higher Polygenetic Predisposition for Asthma in Cow’s Milk Allergic Children. Nutrients. 2018;10(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Winters A, Bahnson HT, Ruczinski I, et al. The MALT1 locus and peanut avoidance in the risk for peanut allergy. J Allergy Clin Immunol. 2019;143(6):2326–2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Castells M, Butterfield J. Mast Cell Activation Syndrome and Mastocytosis: Initial Treatment Options and Long-Term Management. J Allergy Clin Immunol Pract. 2019;7(4):1097–1106. [DOI] [PubMed] [Google Scholar]
- 59.Bahji A, Kasurak E, Sterling M, Good L. Misuse and dependence of dimenhydrinate: A mixed studies systematic review. J Psychiatr Res. 2021;136:581–588. [DOI] [PubMed] [Google Scholar]
- 60.Simons FE. H1-receptor antagonists. Comparative tolerability and safety. Drug Saf. 1994;10(5):350–380. [DOI] [PubMed] [Google Scholar]
- 61.Thomas A, Nallur DG, Jones N, Deslandes PN. Diphenhydramine abuse and detoxification: a brief review and case report. J Psychopharmacol. 2009;23(1):101–105. [DOI] [PubMed] [Google Scholar]
- 62.Kim H, Kim SH, Kim JB. Antihistamines as a common cause of new-onset seizures: a single-center observational study. Neurol Sci. 2021;42(6):2505–2508. [DOI] [PubMed] [Google Scholar]
- 63.Waljee AK, Rogers MA, Lin P, et al. Short term use of oral corticosteroids and related harms among adults in the United States: population based cohort study. BMJ. 2017;357:j1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Stanbury RM, Graham EM. Systemic corticosteroid therapy--side effects and their management. Br J Ophthalmol. 1998;82(6):704–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mendoza Alvarez LB, Barker R, Nelson C, et al. Clinical response to omalizumab in patients with hereditary alpha-tryptasemia. Ann Allergy Asthma Immunol. 2020;124(1):99–100 e101. [DOI] [PubMed] [Google Scholar]
- 66.Carter MC, Maric I, Brittain EH, et al. A randomized double-blind, placebo-controlled study of omalizumab for idiopathic anaphylaxis. J Allergy Clin Immunol. 2021;147(3):1004–1010 e1002. [DOI] [PubMed] [Google Scholar]
- 67.Carosi G, Guabello G, Longhi M, Grifoni F, Passeri E, Corbetta S. Hypertryptasemia and Mast Cell-Related Disorders in Severe Osteoporotic Patients. Mediators Inflamm. 2020;2020:5785378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.van Staa TP, Leufkens HG, Abenhaim L, Zhang B, Cooper C. Oral corticosteroids and fracture risk: relationship to daily and cumulative doses. Rheumatology (Oxford). 2000;39(12):1383–1389. [DOI] [PubMed] [Google Scholar]
- 69.Correia O, Duarte AF, Quirino P, Azevedo R, Delgado L. Cutaneous mastocytosis: Two pediatric cases treated with topical pimecrolimus. Dermatol Online J. 2010;16(5):8. [PubMed] [Google Scholar]
- 70.Ustun C, DeRemer DL, Akin C. Tyrosine kinase inhibitors in the treatment of systemic mastocytosis. Leuk Res. 2011;35(9):1143–1152. [DOI] [PubMed] [Google Scholar]
- 71.Dowse R, Ibrahim M, McLornan DP, Moonim MT, Harrison CN, Radia DH. Beneficial effects of JAK inhibitor therapy in Systemic Mastocytosis. Br J Haematol. 2017;176(2):324–327. [DOI] [PubMed] [Google Scholar]
- 72.Mateja A, Wang Q, Chovanec J, et al. Defining baseline variability of serum tryptase levels improves accuracy in identifying anaphylaxis. J Allergy Clin Immunol. 2021;In revision. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Maun HR, Jackman JK, Choy DF, et al. An Allosteric Anti-tryptase Antibody for the Treatment of Mast Cell-Mediated Severe Asthma. Cell. 2019;179(2):417–431 e419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lyons JJ, Metcalfe DD. Targeting Mast Cells with Biologics. Immunol Allergy Clin North Am. 2020;40(4):667–685. [DOI] [PubMed] [Google Scholar]
- 75.Dispenza MC, Krier-Burris RA, Chhiba KD, Undem BJ, Robida PA, Bochner BS. Bruton’s tyrosine kinase inhibition effectively protects against human IgE-mediated anaphylaxis. J Clin Invest. 2020;130(9):4759–4770. [DOI] [PMC free article] [PubMed] [Google Scholar]