

Abstract
Outcomes for patients with melanoma have improved over the past decade as a result of the development and FDA approval of immunotherapies targeting cytotoxic T lymphocyte antigen-4 (CTLA-4), programmed death-1 (PD-1), and programmed death ligand 1 (PD-L1). However, these therapies do not benefit all patients, and an area of intensive research investigation is identifying biomarkers that can predict which patients are most likely to benefit from them. Here, we report exploratory analyses of the associations of tumor mutational burden (TMB), a 4-gene inflammatory gene expression signature, and BRAF mutation status with tumor response, progression-free survival, and overall survival in patients with advanced melanoma treated as part of the CheckMate 066 and 067 phase III clinical trials evaluating immuno-oncology therapies. In patients enrolled in CheckMate 067 receiving the anti–PD-1 inhibitor nivolumab (NIVO) alone or in combination with the anti–CTLA-4 inhibitor ipilimumab (IPI) or IPI alone, longer survival appeared to associate with high (>median) versus low (≤median) TMB and with high versus low inflammatory signature scores. For NIVO-treated patients, the results regarding TMB association were confirmed in CheckMate 066. In addition, improved survival was observed with high TMB and absence of BRAF mutation. Weak correlations were observed between PD-L1, TMB, and the inflammatory signature. Combined assessment of TMB, inflammatory gene expression signature, and BRAF mutation status may be predictive for response to immune checkpoint blockade in advanced melanoma.
Introduction
Treatment for patients with melanoma has been transformed in the past 10 years by the FDA approval of immuno-oncology (I-O) therapies targeting cytotoxic T lymphocyte antigen-4 (CTLA-4) and programmed death-1 (PD-1; refs. 1, 2). In the phase III CheckMate 066 clinical trial, patients with previously untreated, unresectable, metastatic, BRAF wild-type (BRAFWT) melanoma randomized to receive the PD-1–specific monoclonal antibody nivolumab (NIVO) had significant improvements in overall survival (OS) and progression-free survival (PFS) compared with those randomized to receive dacarbazine, and these improvements were sustained over 5 years of follow-up (3–5). In the phase III CheckMate 067 clinical trial, patients with BRAFWT and V600 mutation–positive (BRAFV600) unresectable or metastatic melanoma assigned NIVO or NIVO combined with the CTLA-4–specific monoclonal antibody ipilimumab (IPI) showed significantly longer PFS and OS than those assigned IPI alone (6, 7). Sustained improvements in OS, PFS, and objective response rate (ORR) were observed in patients receiving NIVO or NIVO+IPI versus IPI with a minimum of 5 years' follow-up (7).
Not all patients respond to immune checkpoint blockade, however, and there is a need to identify biomarkers that can predict which patients are most likely to benefit from these treatments (2, 8). Biomarkers that may predict response to I-O therapy include molecular signatures related to tumor immunogenicity, such as tumor mutational burden (TMB; ref. 9) and microsatellite instability (10). Other molecular signatures that may act as biomarkers of response include mutations in individual genes (11), evidence of adaptive immune resistance [e.g., increase in expression of programmed death ligand 1 (PD-L1; ref. 12) and lymphocyte-activation gene 3 (LAG-3; ref. 13)], and signatures related to tumor inflammation, such as inflammatory gene expression signatures (14). TMB is associated with response to I-O therapy across various tumor types (9, 15, 16), including untreated non–small cell lung cancer (NSCLC; refs. 17, 18). In advanced melanoma, high TMB is associated with response in patients receiving IPI (1, 19), NIVO followed by IPI (20), and NIVO in IPI-naive patients (21).
Mutations in STK11 (22), genes encoding JAK/STAT pathway components (23), and genes encoding antigen presentation pathway components (24) underlie resistance to I-O therapy. Mutations in PTEN (22, 25), NRAS (11), BRAF (11), PBRM1, and other components of the polybromo-associated BRG-/BRM-associated factor (PBAF) complex (26, 27) affect antitumor responses. Mutations affecting IFNγ signaling, which is activated in T-cell inflamed tumor microenvironments (TME), may also influence resistance to I-O therapy (28). In addition, activation of the oncogenic WNT/β-catenin pathway is associated with reduced T-cell infiltration in the TME (28, 29). Therefore, stabilizing mutations in components of the WNT/β-catenin pathway such as the CTNNB1 gene could influence response to I-O therapy (30).
In addition to genomic analyses, gene expression profiling (GEP) studies, including patients with melanoma, show that inflammatory signatures can have predictive value for patients receiving anti–PD-1 therapy (14, 31, 32). Exploratory analyses of patients with metastatic gastroesophageal cancer treated with NIVO+IPI, and patients with advanced hepatocellular carcinoma treated with NIVO, show a correlation between response and an inflammatory gene expression signature consisting of four genes [CD274 (PD-L1), CD8A, LAG3, and STAT1; refs. 33–35], which was developed by investigating gene expression patterns relevant to antitumor immune response and immune suppression within the TME (36–39).
As part of retrospective, exploratory analyses, we evaluated TMB in patients from CheckMate 066 and 067. BRAF V600 mutation status, mutations in genes reported to affect response to I-O therapy or tumor inflammation, and inflammatory signature scores were also assessed in patients from CheckMate 067 (inflammatory signature scores were not assessed in CheckMate 066 due to limited baseline tumor tissue availability). These biomarkers, which represent a comprehensive list of genomic and transcriptomic biomarkers currently being evaluated for I-O, were investigated alone or in combination in terms of their association with clinical response, PFS, and OS in a large multicenter study of patients with advanced melanoma treated with first-line I-O therapy.
Materials and Methods
Patients
CheckMate 066 (NCT01721772) and 067 (NCT01844505) were randomized, double-blind, phase III trials that enrolled adults with previously untreated, histologically confirmed stage III (unresectable) or stage IV melanoma. Eligibility criteria (previously reported; refs. 4, 6) included known BRAF V600 mutation status, Eastern Cooperative Oncology Group performance status score of 0 or 1, and the availability of tissue for biomarker analysis.
Study designs and treatment
The study designs for CheckMate 066 and CheckMate 067 have been previously described (4, 6). In CheckMate 067, randomization to NIVO, NIVO+IPI, or IPI was stratified by tumor PD-L1 expression (≥5% vs. <5%/indeterminate; ref. 6), BRAF V600 mutation status, and metastasis stage (M0/M1A/M1B vs. M1C), whereas in CheckMate 066, patients were confirmed BRAFWT and randomization to either NIVO or dacarbazine was stratified according to tumor PD-L1 expression and metastasis stage (4). In CheckMate 066, OS was the primary endpoint and PFS and ORR were secondary endpoints (4); in CheckMate 067, PFS and OS were primary endpoints and ORR was a secondary endpoint. The trial design for CheckMate 067 was not powered to compare the NIVO and NIVO+IPI trial arms (6, 8).
Exploratory endpoints in both studies included analyses in patients with biomarker-evaluable tumor samples from unresectable or metastatic sites (Supplementary Figs. S1 and S2). In the present analysis, a data cutoff with a minimum of 4 years of follow-up was used for both studies.
The trial protocols for CheckMate 066 and 067 were approved by the institutional review board at each participating study site. Both trials were conducted in accordance with the provisions of the Declaration of Helsinki and with Good Clinical Practice guidelines as defined by the International Conference on Harmonization. All the patients provided written informed consent before enrollment. Data and safety monitoring committees provided oversight of safety and efficacy considerations (4, 6).
Clinical assessments
Between randomization and progression or initiation of subsequent therapy, depending on which occurred first, tumor response was investigator assessed per RECIST v1.1 (40) by computed tomography or magnetic resonance imaging performed initially at 9 weeks then every 6 weeks up to a year (CheckMate 066), and initially at 12 weeks then every 6 weeks for 49 weeks (CheckMate 067), then every 12 weeks until progression or discontinuation due to unacceptable toxicity, whichever occurred first (both studies). Responders had complete (CR) or partial response (PR) as their best overall response (BOR). Nonresponders included patients with stable (SD) or progressive disease (PD) and patients whose BOR could not be determined. For each patient subgroup, ORR was calculated as the fraction of responders of the treated, biomarker-evaluable population. Confirmation of response was not required. PFS and OS were measured from the date of randomization until the date of first radiographic progression or death (PFS), or death only (OS).
Biomarker assessments
The biomarker-evaluable population included patients who had provided informed consent for inclusion in the study and whose samples were adequate for biomarker assessments. Pretreatment tumor tissue, whole blood, and serum samples were collected at baseline as part of this analysis. In both studies, PD-L1 expression was prospectively determined on tumor cells using the Dako PD-L1 IHC 28–8 pharmDx assay (4, 6). CD8 immunohistochemistry (IHC) was performed by pathology-assisted digital scoring at Mosaic Laboratories (Lake Forest, CA) using a monoclonal CD8-specific antibody (clone C8/144B, cat #M710301–2, Agilent Technologies). CD8 IHC scores were expressed as the percentage of CD8+ immune cells of total cells.
Exploratory genomic and transcriptomic analyses were performed by whole-exome sequencing (WES) and RNA sequencing (RNA-seq), respectively, using formalin-fixed, paraffin-embedded (FFPE) pretreatment tumor samples. Tumor specimens were collected before randomization. Tissue blocks were stored at an ambient temperature and slides were sectioned as needed for IHC and genomic analyses. Slides were stored at refrigeration temperature and serum samples were stored at temperatures of −20°C at sites and −70°C at a central storage facility. Whole blood for peripheral blood mononuclear cell (PBMC) isolation was collected before dosing and shipped at ambient temperature to the processing laboratory, where PBMCs were isolated using the aliquot method and stored in liquid nitrogen. Dual DNA and RNA extraction from FFPE tumor tissue was carried out using the AllPrep DNA/RNA FFPE Kit (cat #80234, Qiagen). RNA concentration and integrity were evaluated using Qubit RNA HS Assay Kit (cat #Q32852, Thermo Fisher Scientific) and Agilent RNA 6000 Nano Kit (cat #5067–1511) reagents, respectively. DNA concentration was assessed using Qubit BR DNA Assay Kit (cat #Q32853, Thermo Fisher Scientific). Whole blood was collected in ethylenediaminetetraacetic acid tubes and stored at −80°C until ready for DNA extraction using QIAmp Blood DNA Extraction Kit (cat #51106, Qiagen); concentration was assessed using Qubit BR DNA Assay Kit (cat #Q32853).
WES and TMB
Libraries derived from WES for both tumor and blood DNA samples were prepared using the Agilent SureSelectXT v5 Kit following validated standard operating procedures (SOP) for both DNA extracted from FFPE tissue and whole blood at Expression Analysis (Raleigh, NC). Briefly, extracted DNA was fragmented using a sonication instrument (Covaris). WES libraries were then prepared using 65–100 ng DNA as input template, and subsequently entered into a hybridization capture step targeting exonic regions of the genome. Enrichment of libraries and addition of a sample barcode index was achieved via a post-capture polymerase chain reaction (PCR) step. Library concentration was quantified using the KAPA Library Quantification Kit (Roche, cat# 07960140001) and library fragment size analyzed using Agilent D1000 ScreenTape Assay on the TapeStation system, respectively. Library quantification involves qPCR amplification of six prediluted DNA Standards (provided with KAPA Library Quantification Kit) and diluted library samples using primers targeting the Illumina P5 and P7 flow cell oligo sequences. Average Cq for each DNA standard is plotted against log10 to generate a standard curve. Using absolute quantification, concentrations of diluted libraries were then estimated against the standard curve. Finally, equimolar amounts of libraries were pooled and sequenced using Illumina HiSeq2500 generating 2 × 100 paired-end (PE) reads and targeting 100× depth of coverage. Sequence alignment, variant calling, and TMB calculation were performed as described in Chang and colleagues (41). TMB was calculated as total observed missense mutations in pretreatment tumor samples using matched normal (blood) samples from each patient for reference (41). For each trial, TMB was either log10 transformed (when used as a continuous variable) or the median pretreatment TMB score across all evaluable patients was used as a cutoff to group patients by high TMB (>median) or low TMB (≤ median) status. Biomarker analyses included BRAF mutation status, which was reported as part of patients' clinical characteristics. Somatic mutations in select genes previously shown to affect response to I-O treatment or tumor inflammation (including BRAF) were also evaluated by WES.
GEP and inflammatory gene expression signature scoring
RNA-seq libraries were prepared using the TruSeq RNA Access Library Kit (cat #20020189, Illumina) following validated SOP at Expression Analysis. Briefly, total RNA samples were fragmented and converted into indexed cDNA libraries using 100–200 ng RNA as input template. Indexed libraries were then subsequently enriched for coding RNA using hybrid capture probes specific for coding RNA. Libraries were quantified and qualified as described in the previous section. Equimolar amounts of libraries were pooled and sequenced using the Illumina HiSeq2500 to a depth of 50 million 2 × 50 PE reads. Reads were aligned to the Ensembl GRCh37 v75 Human reference genome using STAR (http://star.mit.edu/cluster/) and gene-level expression estimates were calculated using RNA-Seq by Expectation Maximization as previously described (42). Quality control metrics were calculated using Picard (http://broadinstitute.github.io/picard). Samples with fewer than 30 million reads, and an estimated library size less than 10 million were excluded from analysis. CohortMatcher (https://github.com/golharam/cohort-matcher) was used to confirm sample identity by comparison with matched tumor and normal (blood) WES results. Scores for the 4-gene inflammatory signature and other inflammatory gene expression signatures were calculated by z-scoring (a measurement describing a value's distance from the mean) the log2 counts per million values for each component gene in the signature, then calculating the median of the z-scored values for all component genes (CD274, CD8A, LAG3, and STAT1) within a sample as the signature score (33, 35). All four genes were weighted equally in calculating the signature score of each sample. The median signature score across all evaluable patients was used to group individual patient scores as high (>median) or low (≤median) for inflammatory gene expression. RNA-seq was not performed on patient samples from CheckMate 066 due to limited baseline tumor tissue availability.
Statistical analyses
Statistical analyses were performed using R versions 3.4.4 or higher. Survival analyses used version 2.41–3 or higher of the “survival” package and visualized using version 0.4.2 or higher of the “survminer” package. Patient characteristics and clinical outcomes were compared between the biomarker-evaluable population and the intent-to-treat (ITT) population using frequency statistics and descriptive statistics. Hazard ratios (HR) and their 95% confidence intervals (CI) for PFS or OS in the TMB and inflammatory signature patient subgroups were obtained by univariate Cox proportional hazards, stratified by PD-L1 expression (≥5% vs. <5%/indeterminate), BRAF V600 mutation status, and disease stage, and were illustrated with Kaplan–Meier plots. The association between different biomarkers was explored using linear regression analysis, Pearson's correlation coefficient, and scatter plots. The association between response with biomarkers was assessed by descriptive statistics, box plots, logistic regression models, and receiver operating characteristic (ROC) plots by trial arm. ROC analyses were completed using version 1.10.0 or higher of the “pROC” package and visualized using version 2.2.0 or higher of the “plotROC” package. All other plots were generated using version 2.2.1 or higher of the “ggplot” package.
Considering that these exploratory analyses were performed retrospectively on a subset of the ITT population, HRs and areas under the curve (AUC) are reported with 95% CIs instead of P values (43). The studies were not powered for formal statistical comparison between treatment arms or within biomarker subgroups.
Data deposition
Raw sequencing data cannot be made publicly available for all patients owing to restrictions on patient consent preventing sharing of potentially identifiable genetic data. Informed consent for data sharing was obtained from 16 TMB-evaluable patients in the CheckMate 066 trial as well as from 61 TMB-evaluable patients and 38 GEP-evaluable patients in the CheckMate 067 trial. Sequence data, TMB, and 4-gene inflammatory signature scores from these patients support the conclusions of this article and have been deposited in the European Genome-phenome Archive under accession numbers EGAS00001004564, EGAS00001004555, and EGAS00001004567. TMB and 4-gene inflammatory signature scores are in Supplementary Data Files S1–S3. More information on Bristol Myers Squibb's data sharing policy can be found here: https://www.bms.com/researchers-and-partners/clinical-trials-and-research/disclosure-commitment.html.
Results
Patients
In CheckMate 067, 176 (56.2%) and 97 (31.0%) of the 313 patients treated with NIVO, 184 (58.8%) and 85 (27.2%) of the 313 patients treated with NIVO+IPI, and 178 (57.2%) and 87 (28.0%) of the 311 patients treated with IPI had pretreatment samples evaluable for TMB and GEP, respectively (Supplementary Fig. S1). In CheckMate 066, 418 patients were randomized, of whom 206 received treatment with NIVO and 205 received dacarbazine; 119 of 411 treated patients (29.0%) had TMB-evaluable pretreatment samples, 52 in the NIVO arm and 67 in the dacarbazine arm (Supplementary Fig. S2).
Demographics and baseline patient characteristics were generally comparable between the biomarker-evaluable and the ITT populations in both CheckMate 066 (4) and 067 (Supplementary Tables S1–S3; ref. 6). TMB distribution (CheckMate 067 and 066; Supplementary Figs. S3 and S4) and inflammatory signature scores (CheckMate 067; Supplementary Fig. S5) were similar across treatment arms. Median missense mutations for all TMB-evaluable patients in CheckMate 066 and 067 were 203.5 and 157, respectively (Supplementary Figs. S3 and S4). The fractions of patients by M stage, BRAF V600 mutation rate, PD-L1 expression, and sex were similar between TMB-evaluable patients (CheckMate 067 and CheckMate 066), GEP-evaluable patients (CheckMate 067), and ITT patients (Supplementary Tables S1–S3). Within each treatment arm, ORR (Supplementary Tables S4–S6), PFS, and OS were similar between as-treated patients and biomarker-evaluable patients (Supplementary Figs. S6–S11).
Clinical outcomes
Association of TMB with tumor response and survival
Median TMB values were numerically higher in responders to NIVO (both trials), NIVO+IPI, or IPI (CheckMate 067) compared with nonresponders (Fig. 1A and Table 1 for CheckMate 067 data; Supplementary Fig. S12 and Supplementary Table S7 for CheckMate 066 data). ORR was also numerically higher in patients with high versus low TMB irrespective of PD-L1 expression with NIVO (both trials) or NIVO+IPI (CheckMate 067) and appeared highest in patients with high TMB and PD-L1 expression treated with NIVO or NIVO+IPI (Supplementary Tables S8 and S9). For both studies, ROC curves also highlighted the potential predictive value of TMB in identifying responders to NIVO+IPI, as well as to IPI (CheckMate 067) and to NIVO (CheckMate 066, 067), but not to dacarbazine (CheckMate 66; Supplementary Figs. S13 and S14). In CheckMate 067, the AUC was 67.2 (95% CI, 59.1–75.3) for NIVO, 60.5 (95% CI, 52.3–68.7) for NIVO+IPI, and 64.7 (95% CI, 55.1–74.4) for IPI (Supplementary Fig. S13). In CheckMate 066, TMB yielded an AUC (95% CI) of 66.4 (51.1–81.8) with NIVO but had lower predictive value for response to dacarbazine (AUC, 39.8; 95% CI, 24.3–55.3; Supplementary Fig. S14). The association of TMB with response was observed regardless of PD-L1 expression levels (Supplementary Figs. S15 and S16).
Figure 1.
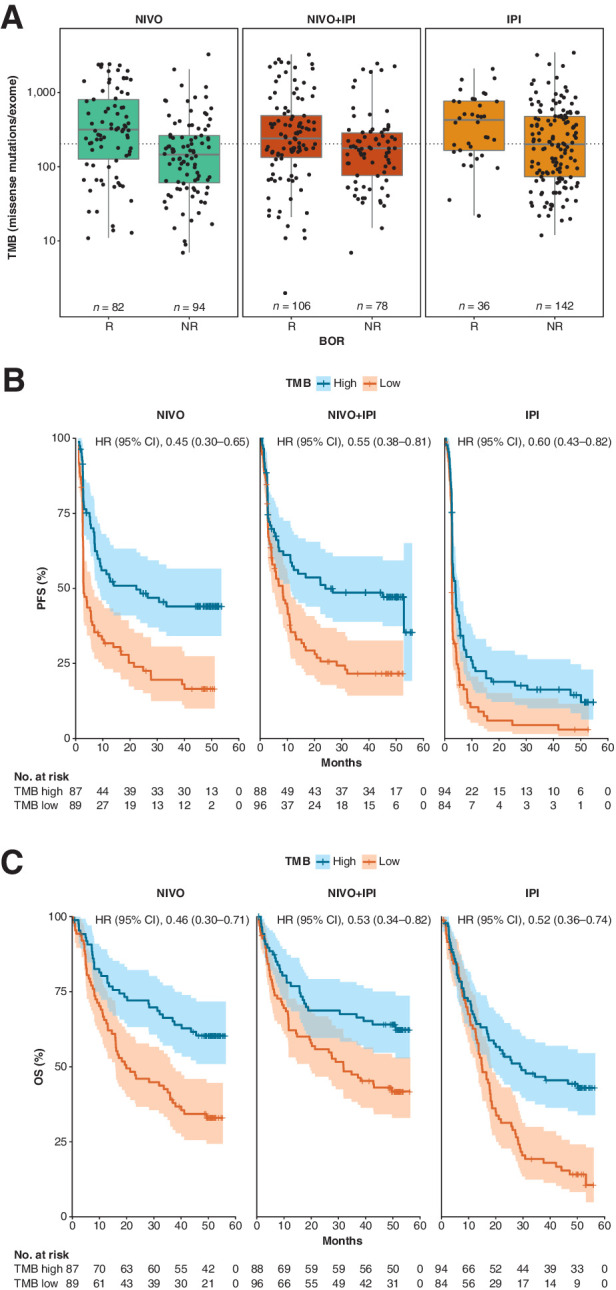
Clinical response and survival by TMB status in CheckMate 067. A, Distribution of TMB in responders and nonresponders with NIVO, NIVO+IPI, or IPI. Boxes extend from the first to third quartiles, the middle line shows the median, and the whiskers extend to the most extreme data point that is no more than 1.5 times the IQR from the box. Dotted line represents the median TMB across all treatment arms. B, Kaplan–Meier curve for PFS by TMB status for TMB-evaluable patients treated with NIVO, NIVO+IPI, or IPI. C, Kaplan–Meier curve for OS by TMB status for TMB-evaluable patients treated with NIVO, NIVO+IPI, or IPI. HRs (95% CI) for high versus low TMB were obtained with univariate Cox proportional hazards models. NR, nonresponders; R, responders.
Table 1.
ORR by TMB status in CheckMate 067.
| Treatment | TMB class, N | Nonresponders, na | Responders, nb | ORR (%) |
|---|---|---|---|---|
| NIVO | Low, 89 | 61 | 28 | 31.5 |
| High, 87 | 33 | 54 | 62.1 | |
| As-treated population, 313 | 172 | 141 | 45.0 | |
| NIVO+IPI | Low, 96 | 47 | 49 | 51.0 |
| High, 88 | 31 | 57 | 64.8 | |
| As-treated population, 313 | 130 | 183 | 58.5 | |
| IPI | Low, 84 | 72 | 12 | 14.3 |
| High, 94 | 70 | 24 | 25.5 | |
| As-treated population, 311 | 251 | 60 | 19.3 |
aNonresponders include patients experiencing SD or PD or those whose BOR could not be determined.
bResponders include patients who achieved CR or PR.
In CheckMate 067, PFS appeared to be longer in patients with high TMB (>203.5 total missense mutations) in all treatment arms, with HRs for high versus low TMB of 0.45 (95% CI, 0.30–0.65) in the NIVO arm, 0.55 (95% CI, 0.38–0.81) in the NIVO+IPI arm, and 0.60 (95% CI, 0.43–0.82) in the IPI arm (Fig. 1B). In addition, OS appeared to be longer in patients with high TMB in all treatment arms, with HRs for high versus low TMB of 0.46 (95% CI, 0.30–0.71) in the NIVO arm, 0.53 (95% CI, 0.34–0.82) in the NIVO+IPI arm, and 0.52 (95% CI, 0.36–0.74) in the IPI arm (Fig. 1C). Similar results were observed in patients with BRAFWT tumors receiving NIVO compared with dacarbazine in CheckMate 066. In the NIVO arm, 23 of 52 patients (44.2%) had high TMB, whereas in the dacarbazine arm, 36 of 67 patients (53.7%) had high TMB (Supplementary Fig. S17). PFS and OS in the NIVO arm appeared longer in patients with high TMB status compared with those with low TMB (HR for PFS, 0.34; 95% CI, 0.16–0.72 and OS, 0.45; 95% CI, 0.21–0.95), whereas no difference was observed in the dacarbazine arm by TMB status (HR for PFS, 0.68; 95% CI, 0.40–1.20, and OS, 0.69; 95% CI, 0.41–1.20; Supplementary Fig. S17). TMB did not correlate with tumor PD-L1 expression and the distribution of TMB was not correlated with PD-L1 expression levels in either of the trials (Supplementary Figs. S18–S21). The separation of PFS and OS by high versus low TMB was also similar across metastatic stages and PD-L1 expression levels in CheckMate 067 (Supplementary Figs. S22–S25; Supplementary Table S10).
Association of TMB and BRAF mutation status with survival
Within the TMB-evaluable population in CheckMate 067, BRAFV600 tumors had lower median TMB than BRAFWT (176 missense mutations in BRAFV600 vs. 240 in BRAFWT) across all treated patients (Supplementary Fig. S26). In patients with BRAFV600 tumors, median TMB was similar for responders and nonresponders, whereas median TMB in BRAFWT was numerically higher for responders (Fig. 2A). The suggested increase in PFS in patients with high versus low TMB appeared more pronounced in patients with BRAFWT versus patients with BRAFV600 across all treatment arms (Fig. 2B). The same effect was observed in the NIVO and IPI arms for OS (Fig. 2C). A trend toward longer OS in patients with high versus low TMB was also observed with BRAFV600 tumors with NIVO+IPI (Fig. 2C). Survival appeared longer in patients with BRAFWT tumors treated with NIVO but not with dacarbazine in CheckMate 066 (Supplementary Fig. S17). When PD-L1 expression was considered concurrently with BRAF mutation status, a trend toward higher PFS, OS, and ORR was observed in patients with ≥5% PD-L1 and BRAFWT or BRAFV600 treated with NIVO, patients with ≥5% PD-L1 and BRAFV600 treated with NIVO+IPI, and patients with ≥5% PD-L1 and BRAFWT treated with IPI. This trend was not observed in patients with BRAFWT treated with NIVO+IPI or in patients with BRAFV600 treated with IPI (Supplementary Figs. S27 and S28; Supplementary Table S11).
Figure 2.
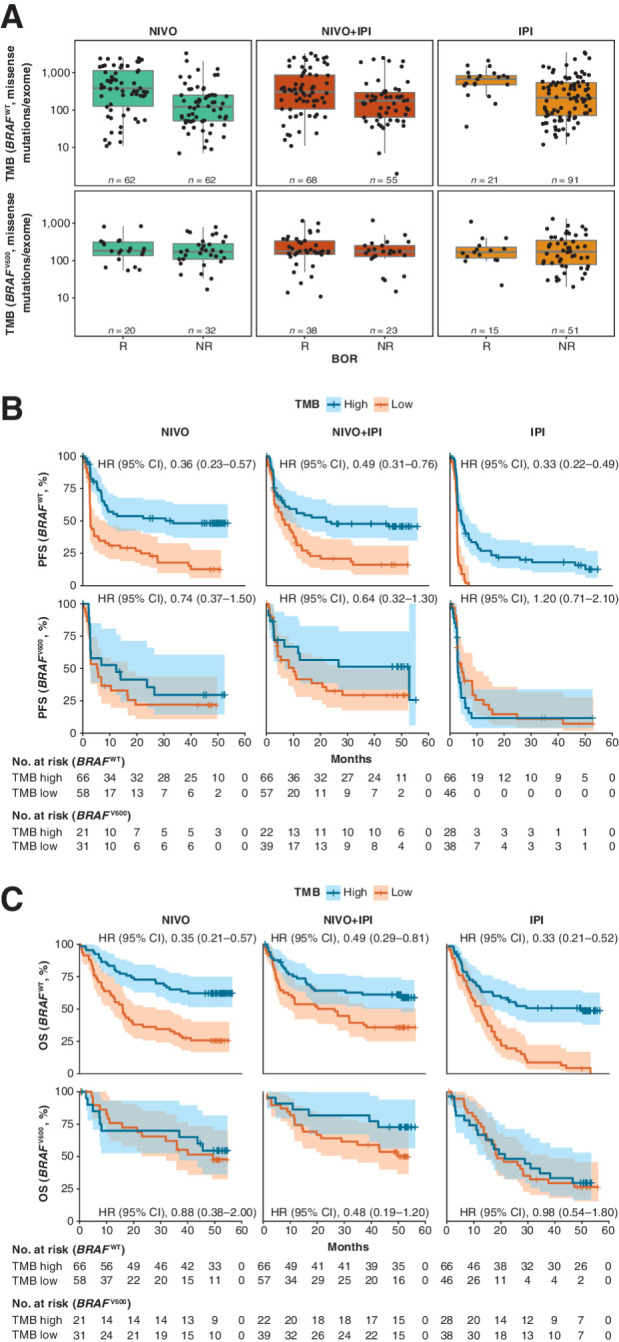
Clinical response and survival by TMB status and BRAF mutation status in CheckMate 067. A, Distribution of TMB by BRAF mutation status in responders and nonresponders treated with NIVO, NIVO+IPI, or IPI. Number of responders and nonresponders by treatment arm and BRAF mutation status is indicated on the figure. Boxes extend from the first to third quartiles, the middle line shows the median, and the whiskers extend to the most extreme data point that is no more than 1.5 times the IQR from the box. B, Kaplan–Meier curve for PFS comparing TMB-high and TMB-low patient subgroups by BRAF mutation status with NIVO, NIVO+IPI, or IPI. C, Kaplan–Meier curve for OS comparing TMB-high and TMB-low patient subgroups by BRAF mutation status with NIVO, NIVO+IPI, or IPI. HRs (95% CI) for high versus low TMB were obtained with univariate Cox proportional hazards models. NR, nonresponders; R, responders.
Association of an inflammatory signature with tumor response and survival
In CheckMate 067, the median inflammatory signature score was numerically higher in patients who responded to NIVO, NIVO+IPI, or IPI compared with nonresponders (Fig. 3A). Across treatment arms, ORR was suggested to be higher in patients with high signature scores compared with low signature scores (Supplementary Table S12). ROC curves supported the potential predictive value of the inflammatory signature score in identifying responders across treatment arms with AUC of 68.9 (95% CI, 58.3–79.5) for NIVO, 62.3 (95% CI, 50.1–74.6) for NIVO+IPI, and 59.2 (95% CI, 46.3–72.1) for IPI (Supplementary Fig. S29).
Figure 3.
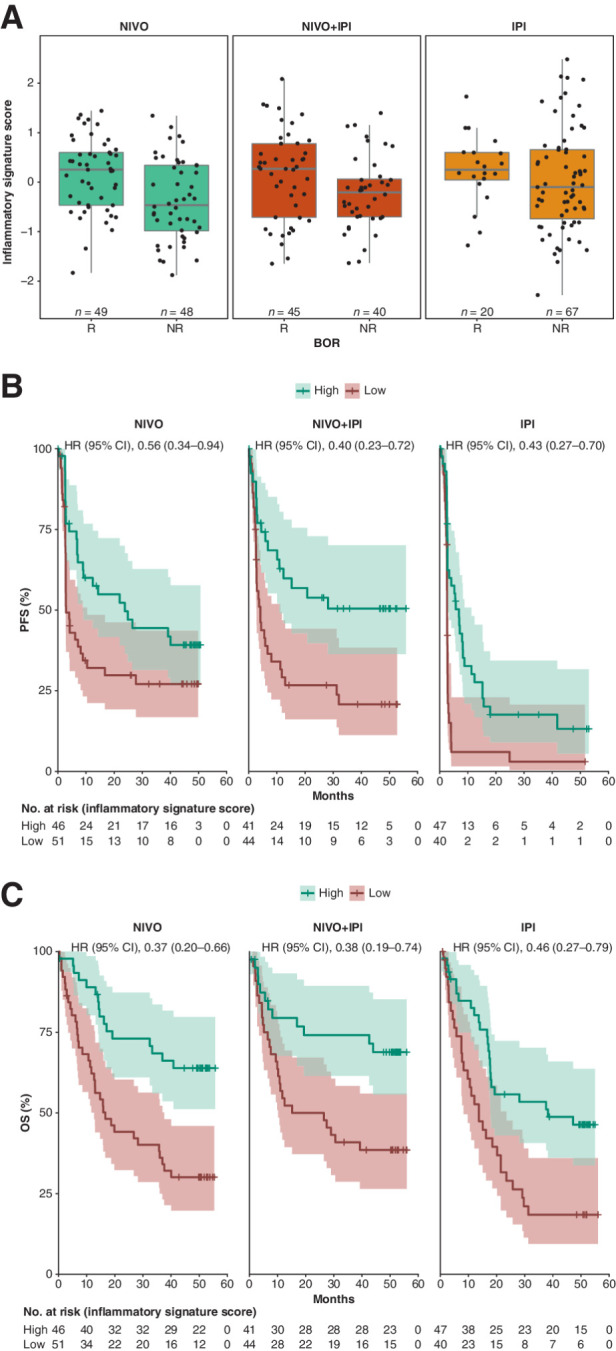
Clinical response and survival by inflammatory signature score in CheckMate 067. A, Distribution of inflammatory signature scores in responders and nonresponders treated with NIVO, NIVO+IPI, or IPI. Number of responders and nonresponders is indicated on the figure. Boxes extend from the first to third quartiles, the middle line shows the median, and the whiskers extend to the most extreme data point that is no more than 1.5 times the IQR from the box. B, Kaplan–Meier curve for PFS comparing patient subgroups with high or low inflammatory signature scores for NIVO, NIVO+IPI, or IPI. C, Kaplan–Meier curve for OS comparing patient subgroups with high or low inflammatory signature score status for NIVO, NIVO+IPI, or IPI. HRs (95% CI) for high versus low TMB were obtained with univariate Cox proportional hazards models. NR, nonresponders; R, responders.
PFS appeared generally longer in patients with signature scores above the dataset median (> −0.04) for all treatment arms, with HRs of 0.56 (95% CI, 0.34–0.94), 0.40 (95% CI, 0.23–0.72), and 0.43 (95% CI, 0.27–0.70) comparing scores above and below the median signature score in the NIVO, NIVO+IPI, and IPI arms, respectively. OS also appeared longer in patients with high signature scores for all treatment arms, with HRs of 0.37 (95% CI, 0.20–0.66), 0.38 (95% CI, 0.19–0.74), and 0.46 (95% CI, 0.27–0.79), respectively (Fig. 3B and C).
The 4-gene inflammatory signature correlated with a 10-gene IFNγ signature derived from 19 patients with melanoma and associated with response to pembrolizumab (r = 0.93; ref. 14), an 18-gene tumor inflammation signature (TIS) derived using baseline clinical samples from multiple tumor types and found to enrich for response to pembrolizumab (r = 0.96; ref. 44), and a 13-gene inflammatory signature, the absence of which was found to mediate cancer immune evasion in melanoma samples (r = 0.89; Supplementary Fig. S30; ref. 30). Notably, the Pearson correlation between the 4-gene inflammatory signature and tumor PD-L1 expression was weaker (r = 0.39) compared with the correlation between the 4-gene inflammatory signature and other inflammatory signatures (Supplementary Figs. S30 and S31). In contrast with the observation of BRAFV600 tumors appearing to have lower TMB than BRAFWT tumors (176 missense mutations in BRAFV600 vs. 240 missense mutations in BRAFWT; Supplementary Fig. S26), BRAFV600 tumors had numerically higher median 4-gene inflammatory signature scores than BRAFWT (0.29 in BRAFV600 vs. −0.10 in BRAFWT; Supplementary Fig. S32).
Composite biomarker analysis
The correlation between log10TMB and the 4-gene inflammatory signature was weak [r = 0.283 (95% CI, 0.157–0.400), all arms combined; Fig. 4A]. Consistent with this result, as CD8A is one of the genes in the 4-gene inflammatory signature, there was no correlation between TMB and expression of CD8, as measured by IHC (Supplementary Fig. S33). Patients who had both high 4-gene inflammatory signature scores and high TMB values appeared to have higher ORR with NIVO and NIVO+IPI (75.0% and 66.7%, respectively) versus IPI (27.6%). The worst ORRs were observed for patients whose tumors were low for both TMB and the 4-gene inflammatory signature score (22.2%, 25.0%, and 9.5% for NIVO, NIVO+IPI, and IPI, respectively; Fig. 4A; Supplementary Table S13). The same trends were observed when subgroup analyses combining TMB and the 4-gene inflammatory signature were conducted for PFS and OS (Fig. 4B and C). Similarly, greater ORRs were observed in patients with high TMB and high CD8 expression treated with NIVO and NIVO+IPI (72.2% and 69.6%) compared with IPI (23.3%; Supplementary Table S14). However, the small patient subgroups should be considered when interpreting these analyses (Fig. 4; Supplementary Tables S13 and S14).
Figure 4.

Clinical response and survival by combined analysis of TMB and the inflammatory signature in CheckMate 067. A, Scatter plot of the distribution of inflammatory signature scores and TMB categorized in responders and nonresponders treated with NIVO, NIVO+IPI, or IPI in CheckMate 067. ORR (%) is provided for each quadrant of inflammatory signature score and TMB by treatment arm. B, Kaplan–Meier curve for PFS comparing patient subgroups based on combined analysis of TMB and the inflammatory signature in CheckMate 067. C, Kaplan–Meier curve for OS comparing patient subgroups based on combined analysis of TMB and the inflammatory signature in CheckMate 067. HRs (95% CI) for each comparison were obtained with univariate Cox proportional hazards models. NR, nonresponders; R, responders.
Relationship between candidate gene mutations and response
The association of somatic mutations in genes affecting either response to I-O therapy or tumor inflammation with response was assessed in CheckMate 067. For most genes examined, mutations were mainly observed in tumors with high TMB (Fig. 5A–C; Supplementary Fig. S34). For most genes and pathways assessed, there were more wild-type tumors than tumors harboring mutations (Supplementary Table S15). When the antigen presentation complex, IFNγ signaling pathway, NRF–KEAP complex, PBAF complex, PTEN–STK11 pathway, RAS/RAF pathway, and WNT/β-catenin pathways were examined, there was no strong evidence of an association between mutational status of the tumor and response to treatment across arms of CheckMate 067 (Supplementary Table S16).
Figure 5.

Heatmaps representing the relationship of candidate gene mutations to BOR, TMB, and inflammatory score in CheckMate 067. Patients in each arm are ordered by increasing TMB; inflammatory score is shown for patients with evaluable GEP results. ORR is provided as a fraction of responders over total mutated tumors for each gene assessed. A, Patients in the NIVO arm (n = 176). B, Patients in the NIVO+IPI arm (n = 184). C, Patients in the IPI arm (n = 178). FS, frameshift; NR, nonresponders; R, responders.
In the case of β-catenin, mutations in amino acids 31–45 disrupt an N-terminal degradation box, leading to WNT-independent expression of downstream genes (45). Despite the association of the WNT/β-catenin pathway with lack of immune infiltration reported elsewhere (30), response rates and survival did not appear reduced by stabilizing mutations in CTNNB1 or by mutations in individual genes of the WNT/β-catenin pathway in any treatment arm (Supplementary Fig. S35; Supplementary Tables S17 and S18).
Discussion
Ongoing research on the role of immunogenicity and inflammation in the context of tumor response to I-O therapy has highlighted the association of TMB and gene expression signatures of inflammation with response to PD-1 blockade across different tumor types (14, 15, 32, 33, 35). In this robust multicenter study evaluating several biomarkers in a large cohort of patients with advanced melanoma, we find that baseline high TMB may be associated with improved treatment efficacy in patients who receive first-line NIVO, NIVO+IPI, or IPI, consistent with previous studies in patients with melanoma receiving I-O therapy (1, 15, 19–21). TMB is also associated with response to anti–PD-1 therapy in other tumor types, including advanced and metastatic NSCLC (17, 18), previously treated urothelial carcinoma (9), and small-cell lung cancer (SCLC; ref. 16). Consistent with the current study, in studies of NIVO+IPI, high TMB is associated with improved PFS in advanced and metastatic NSCLC (46–48), improved efficacy in SCLC (16), and response in castration-resistant prostate cancer (49).
The results presented herein from CheckMate 066 suggest a potential association between high TMB and improved efficacy in patients with BRAFWT tumors who receive NIVO compared with dacarbazine. Considering the weak evidence for a potential association between high TMB and efficacy with dacarbazine, the results of this study are more consistent with TMB potentially predicting response to I-O therapy.
In the combined analysis of TMB and BRAF mutation status, we observed that median TMB was numerically lower in BRAFV600 tumors than in BRAFWT tumors. This is consistent with previous studies in other tumor types suggesting that driver mutations, including BRAF, are associated with lower TMB (50, 51). Interestingly, each BRAF mutation type may differentially associate with TMB, with BRAF V600K associating with higher TMB than BRAF V600E (52). In our study, stratifying by TMB and BRAF (albeit with subgroups of limited size) showed that in patients treated with NIVO or IPI, PFS and OS appeared to be the longest in patients with high TMB and BRAFWT. TMB did not associate with PFS and OS in BRAFV600 tumors, although a trend toward longer OS was observed in patients with high TMB and BRAFV600 tumors treated with NIVO+IPI. This is consistent with the generally lower TMB observed in BRAFV600 tumors than in BRAFWT tumors. Meanwhile, a numerically higher median inflammatory score was observed in BRAFV600 tumors than in BRAFWT tumors. The interpretation of this observation should be treated with caution considering the small size of the patient subgroups involved in these analyses (Supplementary Tables S1–S3). Possible explanations contributing to the observed differential distribution of TMB and inflammatory signature scores in BRAFV600 tumors include the fact that BRAFV600 melanoma arises early in life with less opportunity for cumulative ultraviolet (UV) damage (53), whereas high TMB is associated with increased age and UV exposure (54). Alternatively, as demonstrated in an earlier study, constitutive activation of the mitogen-activated protein kinase (MAPK) pathway in BRAFV600 melanoma regulates cytokine production (55), which may ultimately influence immune regulation within the TME. Further investigation into such possibilities is warranted.
Efficacy in CheckMate 067 did not appear affected by the presence of mutations in select genes and pathways, including stabilizing mutations in CTNNB1 that are proposed to activate the WNT/β-catenin pathway and thereby lead to tumor immune evasion (30). However, the small size of the patient subgroups in our study should be considered when interpreting our results.
PD-L1 expression is associated with immune infiltration and response to PD-1 blockade across multiple tumor types (56). Interestingly, survival analyses in our current study suggested that high TMB may be associated with improved PFS and OS irrespective of PD-L1 expression levels (Supplementary Figs. S24 and S25). Meanwhile, Madore and colleagues (57) demonstrated that lack of PD-L1 expression was associated with lower TMB and reduced survival in patients with melanoma. Although low TMB and PD-L1 expression levels have previously been associated with worse clinical outcomes following I-O therapy (12), results from the current study suggest that patients with low TMB and tumor-cell PD-L1 expression below 5% may still benefit from NIVO or NIVO+IPI (Supplementary Table S8). Our analyses also showed a low correlation between tumor PD-L1 expression and TMB, supporting the suggestion that these are independent biomarkers for I-O therapy (58). Furthermore, in contrast with previous reports showing an association between PD-L1 expression and the TIS in patients with advanced melanoma treated with pembrolizumab (44, 59, 60), and an association between PD-L1 expression and a gene expression signature including high CD8A expression in archival melanoma samples (57), we found that the 4-gene inflammatory signature only weakly correlated with PD-L1 expression. This observation might be explained by mechanisms regulating the mRNA stability of the CD274 transcript and/or post-translational mechanisms.
Gene expression signatures of inflammation and T-cell populations are associated with response to I-O therapy in patients with melanoma (32, 61, 62). We showed that the 4-gene inflammatory signature correlated with published gene expression signatures predictive for response to pembrolizumab in patients with metastatic melanoma (14, 30). Consistent with exploratory analyses in metastatic gastroesophageal cancer and advanced hepatocellular carcinoma (33, 35), our results suggest that high inflammatory signature scores may associate with improved efficacy in patients across treatment arms of CheckMate 067. These observations suggest that inflammation assessed by GEP may be a valid marker of tumor inflammation and may be assessed as a predictive biomarker in clinical trials evaluating novel therapy combinations (e.g., NCT04133948) for patients less likely to respond to standard therapy.
In our study, correlation analyses and concurrent assessment of TMB and the inflammatory signature within the same patient subgroups suggested that they are independent markers of response in all treatment arms. This supports data from previous studies showing that TMB and gene expression underlying inflammation appear to be independent biomarkers in patients with melanoma and other malignancies (32, 63). These results highlight that neoantigens and inflammation could serve distinct roles as biomarkers. Combined evaluation of TMB and gene expression signatures also suggests that efficacy of I-O therapy is improved in patients with both high TMB and signature scores compared with patients with either high TMB or high signature scores (32). On the basis of these encouraging results, the development of a clinical assay combining PD-L1, TMB, and the inflammatory gene expression signature could be considered.
This study was limited by the exploratory and retrospective nature of the biomarker analyses as well as by baseline sample availability, lack of consent for select biomarker analyses, and sample degradation. In addition, there are many other variables that can ultimately influence response. Neither study was statistically powered to compare treatment arms in patient subgroups; therefore, P values are not reported. The 95% CIs were unadjusted for multiple comparisons, and their interpretation should be limited to the exploratory context.
In conclusion, our analyses highlight the potential predictive value of TMB and suggest that a combined approach to evaluating TMB and the inflammatory signature may differentiate response to I-O therapy in patients with advanced melanoma. These hypotheses need to be validated in prospective studies using predefined cutoffs for TMB and inflammatory signature score assessment to better understand the predictive value of these biomarkers in the context of I-O therapy.
Authors' Disclosures
F.S. Hodi reports other support from Bristol Myers Squibb during the conduct of the study; grants and personal fees from Bristol Myers Squibb and Novartis, and personal fees from Merck, Genentech, EMD Serono, Sanofi, Pionyr, Surface Oncology, Apricity, Bicara, Checkpoint, Compass, Torque, Aduro, Psioxus, Eisai, Idera, Takeda, Biocenter, Gossamer, Iovance, and Trillium outside the submitted work; and a patent for MICA-related disorders pending, licensed, and with royalties paid, a patent for tumor antigens and uses thereof issued, a patent for angiopoiten-2 biomarkers predictive of anti-immune checkpoint response pending, a patent for compositions and methods for identification, assessment, prevention, and treatment of melanoma using PD-L1 isoforms pending, a patent for therapeutic peptides issued, a patent for methods of using pembrolizumab and trebananib pending, a patent for vaccine compositions and methods for restoring NKG2D pathway function against cancers pending, licensed, and with royalties paid, a patent for antibodies that bind to MHC class I polypeptide-related sequence A pending, licensed, and with royalties paid, and a patent for anti-galectin antibody biomarkers predictive of anti-immune checkpoint and anti-angiogenesis responses pending. J.D. Wolchok reports grants and personal fees from Bristol Myers Squibb during the conduct of the study; personal fees from Boehringer Ingelheim, Chugai, Daiichi Sankyo, Dragonfly, Eli Lilly, F-Star, Idera, Kyowa Hakko Kirin, Maverick Therapeutics, Merck, Polynoma, Psioxus, Recepta, Takara Bio, Trieza, Trishula, Sellas, Surface Oncology, Syndax, Syntalogic, and Werewolf Therapeutics and personal fees and other support from Georgiamune, Tizona, Imvaq, Beigene, Linneaus, Apricity, and Arsenal IO; and a patent for xenogeneic DNA vaccines licensed and with royalties paid from Merial, a patent for myeloid-derived suppressor cell (MDSC) assay licensed and with royalties paid from Serametrix, a patent for anti-PD1 antibody licensed to Agenus, a patent for anti-CTLA4 antibodies licensed to Agenus, a patent for anti-GITR antibodies and methods of use thereof licensed to Agenus/Incyte, a patent for Alphavirus replicon particles expressing TRP2 issued, a patent for Newcastle disease viruses for cancer therapy issued, a patent for engineered vaccinia viruses for cancer immunotherapy pending, a patent for anti-CD40 agonist mAb fused to monophosphoryl lipid A (MPL) for cancer therapy pending, a patent for CAR+ T cells targeting differentiation antigens as means to treat cancer pending, a patent for identifying and treating subjects at risk for checkpoint blockade therapy–associated colitis pending, a patent for immunosuppressive follicular helper-like T cells modulated by immune checkpoint blockade pending, and a patent for phosphatidylserine-targeting agents and uses thereof for adoptive T-cell therapies pending. D. Schadendorf reports grants from Bristol Myers Squibb, Amgen, Novartis, MSD, and Roche and personal fees from Bristol Myers Squibb, Novartis, MSD, Roche, Philogen, Pierre Fabre, Array, Pfizer, Sanofi, Regeneron, Sun Pharma, Merck-Serono, and Merck outside the submitted work. J. Larkin reports grants and personal fees from Roche, Novartis, Bristol Myers Squibb, Pfizer, Merck, Immunocore, Achilles, Nektar, and Aveo and personal fees from iOnctura, Incyte, Dynavax, GlaxoSmithKline, Eisai, Iovance, Boston Biomedical, Pierre Fabre, AstraZeneca, EUSA Pharma, touchIME, touchExperts, YKT Global, Apple Tree, MSD, Ervaxx, GlaxoSmithKline, Ipsen, Aptitude, Calithera, Ultimovacs, Seagen, and CRUK outside the submitted work. G.V. Long reports personal fees from Aduro Biotech Inc., Amgen Inc., Array Biopharma Inc., Boehringer Ingelheim International GmbH, Bristol Myers Squibb, Hexel AG, Highlight Therapeutics S.L., Merck Sharpe and Dohme, Novartis Pharma AG, OncoSec, Pierre Fabre, QBiotics Group Limited, Regeneron Pharmaceuticals, SkylineDX B.V., and Specialized Therapeutics Australia Pty Ltd. outside the submitted work. X. Qian reports employment at Bristol Myers Squibb during the time when the data for this publication were generated and analyzed, as well as a patent for PCT patent publication WO2020/198676, filing date March 27, 2020, to Bristol Myers Squibb. A. Saci has a patent for PCT patent publication WO2020/198676, filing date March 27, 2020, to Bristol Myers Squibb. T.C. Young reports other support from Bristol Myers Squibb during the conduct of the study; other support from Bristol Myers Squibb outside the submitted work; a patent for PCT patent publication WO2020/198676 A1 licensed, filing date March 27, 2020, to Bristol Myers Squibb; and is an employee of and owns stock in Bristol Myers Squibb. S. Srinivasan has a patent for PCT patent publication WO2020/198676, filing date March 27, 2020, to Bristol Myers Squibb. H. Chang reports other support from Bristol Myers Squibb outside the submitted work, as well as a patent for PCT patent publications WO2020/198672 and WO2020/198676, filing date March 27, 2020, to Bristol Myers Squibb. H. Tang reports personal fees from other support from Bristol Myers Squibb during the conduct of the study. M. Wind-Rotolo reports other support from Bristol Myers Squibb during the conduct of the study; other support from Bristol Myers Squibb outside the submitted work; and a patent for PCT patent publication WO2020/198676, filing date March 27, 2020, to Bristol Myers Squibb. J.I. Rizzo reports a patent for PCT patent publication WO2020/198676, filing date March 27, 2020. D.G. Jackson reports other support from Bristol Myers Squibb during the conduct of the study; other support from Sanofi outside the submitted work; and a patent for WO2020/198676, filing date March 27, 2020, to Bristol Myers Squibb. P.A. Ascierto reports grants and personal fees from Bristol Myers Squibb, Roche-Genentech, and Sanofi; personal fees and other support from MSD; and personal fees from Novartis, Array, Merck Serono, Pierre Fabre, Incyte, MedImmune, AstraZeneca, Syndax, Sun Pharma, Idera, Ultimovax, Sandoz, Immunocore, 4SC, Alkermes, Italfarmaco, Nektar, Boehringer Ingelheim, Eisai, Regeneron, Daiichi Sankyo, Pfizer, OncoSec, Nouscom, Takis, Lunaphore, and Seagen outside the submitted work. No disclosures were reported by the other authors.
Supplementary Material
Acknowledgments
The authors acknowledge the contributions of Peter Szabo and Nathan Siemers for development of the inflammatory gene expression signature consisting of four genes. Medical writing and editorial support were provided by Katerina Pipili, PhD, and Jay Rathi, MA, of Spark Medica Inc., and Kathryn Woods, PhD, of Complete HealthVizion for an earlier draft of this article, funded by Bristol Myers Squibb. All authors met the ICMJE authorship criteria and ensure the integrity of the work. Neither honoraria nor payments were made for authorship. In addition to interpretation of the data, all authors participated in drafting, review, and final approval of the article. This study was supported by Bristol Myers Squibb.
The publication costs of this article were defrayed in part by the payment of publication fees. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Footnotes
Note: Supplementary data for this article are available at Cancer Immunology Research Online (http://cancerimmunolres.aacrjournals.org/).
Authors' Contributions
F.S. Hodi: Data curation, formal analysis, supervision, investigation, writing–original draft, writing–review and editing. J.D. Wolchok: Conceptualization, investigation, writing–review and editing. D. Schadendorf: Visualization, writing–original draft, writing–review and editing, providing patients and samples. J. Larkin: Writing–original draft, writing–review and editing. G.V. Long: Resources, supervision, writing–original draft, project administration, writing–review and editing, contribution of patients and tissue. X. Qian: Conceptualization, resources, data curation, formal analysis, supervision, validation, investigation, project administration, writing–review and editing. A. Saci: Formal analysis, investigation. T.C. Young: Conceptualization, data curation, formal analysis, validation, visualization, methodology, writing–original draft, writing–review and editing, data interpretation. S. Srinivasan: Validation, methodology, development of pipeline and methods for variant calling and validation. H. Chang: Resources, supervision, validation, methodology, writing–review and editing. H. Tang: Conceptualization, data curation, formal analysis, validation, methodology, writing–review and editing. M. Wind-Rotolo: Conceptualization, formal analysis, writing–original draft, writing–review and editing, data interpretation. J.I. Rizzo: Data curation, formal analysis, supervision, methodology, writing–original draft, writing–review and editing. D.G. Jackson: Data curation, software, formal analysis, supervision, methodology, writing–original draft, writing–review and editing. P.A. Ascierto: Conceptualization, data curation, supervision, writing–review and editing.
References
- 1. Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med 2014;371:2189–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Havel JJ, Chowell D, Chan TA. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat Rev Cancer 2019;19:133–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ascierto PA, Long GV, Robert C, Brady B, Dutriaux C, Di Giacomo AM, et al. Survival outcomes in patients with previously untreated BRAF wild-type advanced melanoma treated with nivolumab therapy. Three-year follow-up of a randomized phase 3 trial. JAMA Oncol 2019;5:187–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Robert C, Long GV, Brady B, Dutriaux C, Maio M, Mortier L, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med 2015;372:320–30. [DOI] [PubMed] [Google Scholar]
- 5. Robert C, Long GV, Brady B, Dutriaux C, Di Giacomo AM, Mortier L, et al. Five-year outcomes with nivolumab in patients with wild-type BRAF advanced melanoma. J Clin Oncol 2020;38:3937–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, et al. Combined nivolumab and ipilimumab or monotherapy in previously untreated melanoma. N Engl J Med 2015;373:23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Rutkowski P, Lao CD, et al. Five-year survival with combined nivolumab and ipilimumab in advanced melanoma. N Engl J Med 2019;381:1535–46. [DOI] [PubMed] [Google Scholar]
- 8. Hodi FS, Chiarion-Sileni V, Gonzalez R, Grob JJ, Rutkowski P, Cowey CL, et al. Nivolumab plus ipilimumab or nivolumab alone versus ipilimumab alone in advanced melanoma (CheckMate 067): 4-year outcomes of a multicentre, randomised, phase 3 trial. Lancet Oncol 2018;19:1480–92. [DOI] [PubMed] [Google Scholar]
- 9. Galsky MD, Saci A, Szabo PM, Azrilevich A, Horak C, Lambert A, et al. Impact of zumor (sic) mutation burden on nivolumab efficacy in second-line urothelial carcinoma patients: exploratory analysis of the phase II CheckMate 275 study. Ann Oncol 2017;28:V296–7. [Google Scholar]
- 10. Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med 2015;372:2509–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Johnson DB, Lovly CM, Flavin M, Panageas KS, Ayers GD, Zhao Z, et al. Impact of NRAS mutations for patients with advanced melanoma treated with immune therapies. Cancer Immunol Res 2015;3:288–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Morrison C, Pabla S, Conroy JM, Nesline MK, Glenn ST, Dressman D, et al. Predicting response to checkpoint inhibitors in melanoma beyond PD-L1 and mutational burden. J Immunother Cancer 2018;6:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ascierto PA, Melero I, Bhatia S, Bono P, Sanborn RE, Lipson EJ, et al. Initial efficacy of anti-lymphocyte activation gene-3 (anti–LAG-3; BMS-986016) in combination with nivolumab (nivo) in pts with melanoma (MEL) previously treated with anti–PD-1/PD-L1 therapy. J Clin Oncol 35, 2017 (suppl; abstr 9520). [Google Scholar]
- 14. Ayers M, Lunceford J, Nebozhyn M, Murphy E, Loboda A, Kaufman DR, et al. IFN-γ–related mRNA profile predicts clinical response to PD-1 blockade. J Clin Invest 2017;127:2930–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Samstein RM, Lee CH, Shoushtari AN, Hellmann MD, Shen R, Janjigian YY, et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat Genet 2019;51:202–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hellmann MD, Callahan MK, Awad MM, Calvo E, Ascierto PA, Atmaca A, et al. Tumor mutational burden and efficacy of nivolumab monotherapy and in combination with ipilimumab in small-cell lung cancer. Cancer Cell 2018;33:853–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Carbone DP, Reck M, Paz-Ares L, Creelan B, Horn L, Steins M, et al. First-line nivolumab in stage IV or recurrent non–small cell lung cancer. N Engl J Med 2017;376:2415–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Mutational landscape determines sensitivity to PD-1 blockade in non–small cell lung cancer. Science 2015;348:124–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Van Allen EM, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 2015;350:207–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yusko E, Vignali M, Wilson RK, Mardis ER, Hodi FS, Horak C, et al. Association of tumor microenvironment T-cell repertoire and mutational load with clinical outcome after sequential checkpoint blockade in melanoma. Cancer Immunol Res 2019;7:458–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Riaz N, Havel JJ, Makarov V, Desrichard A, Urba WJ, Sims JS, et al. Tumor and microenvironment evolution during immunotherapy with nivolumab. Cell 2017;171:934–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Skoulidis F, Goldberg ME, Greenawalt DM, Hellmann MD, Awad MM, Gainor JF, et al. STK11/LKB1 mutations and PD-1 inhibitor resistance in KRAS-mutant lung adenocarcinoma. Cancer Discov 2018;8:822–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N Engl J Med 2016;375:819–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Garrido F, Aptsiauri N, Doorduijn EM, Garcia Lora AM, van Hall T. The urgent need to recover MHC class I in cancers for effective immunotherapy. Curr Opin Immunol 2016;39:44–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhao J, Chen AX, Gartrell RD, Silverman AM, Aparicio L, Chu T, et al. Immune and genomic correlates of response to anti–PD-1 immunotherapy in glioblastoma. Nat Med 2019;25:462–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Miao D, Margolis CA, Gao W, Voss MH, Li W, Martini DJ, et al. Genomic correlates of response to immune checkpoint therapies in clear cell renal cell carcinoma. Science 2018;359:801–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pan D, Kobayashi A, Jiang P, Ferrari de Andrade L, Tay RE, Luoma AM, et al. A major chromatin regulator determines resistance of tumor cells to T-cell–mediated killing. Science 2018;359:770–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Trujillo JA, Sweis RF, Bao R, Luke JJ. T-cell–inflamed versus non–T-cell–inflamed tumors: a conceptual framework for cancer immunotherapy drug development and combination therapy selection. Cancer Immunol Res 2018;6:990–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Li X, Xiang Y, Li F, Yin C, Li B, Ke X. WNT/β-catenin signaling pathway regulating T-cell inflammation in the tumor microenvironment. Front Immunol 2019;10:2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature 2015;523:231–5. [DOI] [PubMed] [Google Scholar]
- 31. Weber J, Del Vecchio M, Mandala M, Gogas H, Arance AM, Dalle S, et al. Adjuvant nivolumab (NIVO) versus ipilimumab (IPI) in resected stage III/IV melanoma: 3-year efficacy and biomarker results from the phase 3 CheckMate 238 trial. Ann Oncol 2019;30:v533–v63. [Google Scholar]
- 32. Cristescu R, Mogg R, Ayers M, Albright A, Murphy E, Yearley J, et al. Pan-tumor genomic biomarkers for PD-1 checkpoint blockade-based immunotherapy. Science 2018;362:eaar3593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sangro B, Melero I, Wadhawan S, Finn RS, Abou-Alfa GK, Cheng AL, et al. Association of inflammatory biomarkers with clinical outcomes in nivolumab-treated patients with advanced hepatocellular carcinoma. J Hepatol 2020;73:1460–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hodi FS, Wolchok JD, Schadendorf D, Larkin J, Qian M, Saci A, et al. Genomic analyses and immunotherapy in advanced melanoma [abstract]. In:Proceedings of the American Association for Cancer Research Annual Meeting 2019; 2019 Mar 29–Apr 3; Atlanta, GA. Philadelphia (PA): AACR; Cancer Res 2019;79(13 Suppl):Abstract nr CT037. [Google Scholar]
- 35. Lei M, Siemers NO, Pandya D, Chang H, Sanchez T, Harbison C, et al. Analyses of PD-L1 and inflammatory gene expression association with efficacy of nivolumab ± ipilimumab in gastric cancer/gastroesophageal junction cancer. Clin Cancer Res 2021;27:3926–35. [DOI] [PubMed] [Google Scholar]
- 36. Hadrup S, Donia M, Thor Straten P. Effector CD4 and CD8 T cells and their role in the tumor microenvironment. Cancer Microenviron 2013;6:123–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ascierto PA, McArthur GA. Checkpoint inhibitors in melanoma and early phase development in solid tumors: what's the future? J Transl Med 2017;15:173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol 2008;26:677–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Meissl K, Macho-Maschler S, Muller M, Strobl B. The good and the bad faces of STAT1 in solid tumours. Cytokine 2017;89:12–20. [DOI] [PubMed] [Google Scholar]
- 40. Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 2009;45:228–47. [DOI] [PubMed] [Google Scholar]
- 41. Chang H, Sasson A, Srinivasan S, Golhar R, Greenawalt DM, Geese WJ, et al. Bioinformatic methods and bridging of assay results for reliable tumor mutational burden assessment in non–small cell lung cancer. Mol Diagn Ther 2019;23:507–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-seq data with or without a reference genome. BMC Bioinformatics 2011;12:323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wasserstein RL, Lazar NA. The ASA statement on p-values: context, process, and purpose. Am Stat 2016;70:129–33. [Google Scholar]
- 44. Danaher P, Warren S, Lu R, Samayoa J, Sullivan A, Pekker I, et al. Pan-cancer adaptive immune resistance as defined by the tumor inflammation signature (TIS): results from The Cancer Genome Atlas (TCGA). J Immunother Cancer 2018;6:63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Morin PJ, Sparks AB, Korinek V, Barker N, Clevers H, Vogelstein B, et al. Activation of β-catenin-Tcf signaling in colon cancer by mutations in β-catenin or APC. Science 1997;275:1787–90. [DOI] [PubMed] [Google Scholar]
- 46. Hellmann MD, Ciuleanu TE, Pluzanski A, Lee JS, Otterson GA, Audigier-Valette C, et al. Nivolumab plus ipilimumab in lung cancer with a high tumor mutational burden. N Engl J Med 2018;378:2093–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ready N, Hellmann MD, Awad MM, Otterson GA, Gutierrez M, Gainor JF, et al. First-line nivolumab plus ipilimumab in advanced non-small-cell lung cancer (CheckMate 568): outcomes by programmed death ligand 1 and tumor mutational burden as biomarkers. J Clin Oncol 2019;37:922–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hellmann MD, Nathanson T, Rizvi H, Creelan BC, Sanchez-Vega F, Ahuja A, et al. Genomic features of response to combination immunotherapy in patients with advanced non–small cell lung cancer. Cancer Cell 2018;33:843–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sharma P, Pachynski RK, Narayan V, Flechon A, Gravis G, Galsky MD, et al. Initial results from a phase II study of nivolumab (NIVO) plus ipilimumab (IPI) for the treatment of metastatic castration-resistant prostate cancer (mCRPC; CheckMate 650). J Clin Oncol 37, 2019 (suppl 7S; abstr 142). [Google Scholar]
- 50. Patel R, Halligan K, Ramkissoon S, Ross J, Weintraub L. TBIO-08. Tumor mutational burden and driver mutations: further insight into the genomic landscape of pediatric brain tumors. Neuro Oncol 2018;20:i181–i2. [Google Scholar]
- 51. Dudnik E, Peled N, Nechushtan H, Wollner M, Onn A, Agbarya A, et al. BRAF mutant lung cancer: programmed death ligand 1 expression, tumor mutational burden, microsatellite instability status, and response to immune check-point inhibitors. J Thorac Oncol 2018;13:1128–37. [DOI] [PubMed] [Google Scholar]
- 52. Pires da Silva I, Wang KYX, Wilmott JS, Holst J, Carlino MS, Park JJ, et al. Distinct molecular profiles and immunotherapy treatment outcomes of V600E and V600K BRAF-mutant melanoma. Clin Cancer Res 2019;25:1272–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bauer J, Büttner P, Murali R, Okamoto I, Kolaitis NA, Landi MT, et al. BRAF mutations in cutaneous melanoma are independently associated with age, anatomic site of the primary tumor, and the degree of solar elastosis at the primary tumor site. Pigment Cell Melanoma Res 2011;24:345–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Cirenajwis H, Lauss M, Ekedahl H, Törngren T, Kvist A, Saal LH, et al. NF1-mutated melanoma tumors harbor distinct clinical and biological characteristics. Mol Oncol 2017;11:438–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Sumimoto H, Imabayashi F, Iwata T, Kawakami Y. The BRAF-MAPK signaling pathway is essential for cancer-immune evasion in human melanoma cells. J Exp Med 2006;203:1651–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Herbst RS, Soria JC, Kowanetz M, Fine GD, Hamid O, Gordon MS, et al. Predictive correlates of response to the anti–PD-L1 antibody MPDL3280A in cancer patients. Nature 2014;515:563–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Madore J, Strbenac D, Vilain R, Menzies AM, Yang JY, Thompson JF, et al. PD-L1 negative status is associated with lower mutation burden, differential expression of immune-related genes, and worse survival in stage III melanoma. Clin Cancer Res 2016;22:3915–23. [DOI] [PubMed] [Google Scholar]
- 58. Yarchoan M, Albacker LA, Hopkins AC, Montesion M, Murugesan K, Vithayathil TT, et al. PD-L1 expression and tumor mutational burden are independent biomarkers in most cancers. JCI Insight 2019;4:e126908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ribas A, Robert C, Schachter J, Long GV, Arance A, Carlino MS, et al. Tumor mutational burden (TMB), T cell-inflamed gene expression profile (GEP) and PD-L1 are independently associated with response to pembrolizumab (Pembro) in patients with advanced melanoma in the KEYNOTE (KN)-006 study [abstract]. In:Proceedings of the American Association for Cancer Research Annual Meeting 2019; 2019 Mar 29–Apr 3; Atlanta, GA. Philadelphia (PA): AACR; Cancer Res 2019;79(13 Suppl):Abstract nr 4217. [Google Scholar]
- 60. Ott PA, Bang YJ, Piha-Paul SA, Razak ARA, Bennouna J, Soria JC, et al. T-cell–inflamed gene-expression profile, programmed death ligand 1 expression, and tumor mutational burden predict efficacy in patients treated with pembrolizumab across 20 cancers: KEYNOTE-028. J Clin Oncol 2019;37:318–27. [DOI] [PubMed] [Google Scholar]
- 61. Edwards J, Wilmott JS, Madore J, Gide TN, Quek C, Tasker A, et al. CD103+ tumor-resident CD8+ T cells are associated with improved survival in immunotherapy-naive melanoma patients and expand significantly during anti–PD-1 treatment. Clin Cancer Res 2018;24:3036–45. [DOI] [PubMed] [Google Scholar]
- 62. Gide TN, Quek C, Menzies AM, Tasker AT, Shang P, Holst J, et al. Distinct immune cell populations define response to anti–PD-1 monotherapy and anti–PD-1/anti–CTLA-4 combined therapy. Cancer Cell 2019;35:238–55. [DOI] [PubMed] [Google Scholar]
- 63. Dummer R, Brase JC, Garrett J, Campbell CD, Gasal E, Squires M, et al. Adjuvant dabrafenib plus trametinib versus placebo in patients with resected, BRAFV600-mutant, stage III melanoma (COMBI-AD): exploratory biomarker analyses from a randomised, phase 3 trial. Lancet Oncol 2020;21:358–72. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.