

The multinational pediatric precision oncology INFORM registry identified subgroups of patients benefiting with improved progression free survival, refinement of diagnosis, and identification of hereditary cancer predisposition syndromes.
Abstract
INFORM is a prospective, multinational registry gathering clinical and molecular data of relapsed, progressive, or high-risk pediatric patients with cancer. This report describes long-term follow-up of 519 patients in whom molecular alterations were evaluated according to a predefined seven-scale target prioritization algorithm. Mean turnaround time from sample receipt to report was 25.4 days. The highest target priority level was observed in 42 patients (8.1%). Of these, 20 patients received matched targeted treatment with a median progression-free survival of 204 days [95% confidence interval (CI), 99–not applicable], compared with 117 days (95% CI, 106–143; P = 0.011) in all other patients. The respective molecular targets were shown to be predictive for matched treatment response and not prognostic surrogates for improved outcome. Hereditary cancer predisposition syndromes were identified in 7.5% of patients, half of which were newly identified through the study. Integrated molecular analyses resulted in a change or refinement of diagnoses in 8.2% of cases.
Significance:
The pediatric precision oncology INFORM registry prospectively tested a target prioritization algorithm in a real-world, multinational setting and identified subgroups of patients benefiting from matched targeted treatment with improved progression-free survival, refinement of diagnosis, and identification of hereditary cancer predisposition syndromes.
See related commentary by Eggermont et al., p. 2677 .
This article is highlighted in the In This Issue feature, p. 2659
INTRODUCTION
Children and adolescents with relapsed, progressive, or refractory high-risk malignant disease have a particularly poor prognosis. Survival rates of less than 20% following recurrence (1–12) suggest an urgent need for innovative treatment strategies. Based on the potential therapeutic options discovered in several large-scale pediatric sequencing projects (13, 14), a number of comprehensive pediatric precision oncology programs such as pediatric MATCH (15), MOSCATO-01 (16), the ZERO Childhood Cancer Program (17), and the INFORM (INdividualized Therapy FOr Relapsed Malignancies in Childhood) pilot study (18) have been established over recent years, as reviewed by Forrest and colleagues (19) and Mody and colleagues (20). These programs have identified a spectrum of molecularly actionable variants ranging from overexpression of targets to copy-number alterations, gene fusions, point mutations, etc. However, the significance and long-term clinical benefit of such comprehensive and tumor type–agnostic precision oncology studies in a real-world clinical setting are largely unknown for pediatric oncology.
The INFORM registry applies comprehensive molecular profiling in order to provide information on actionable gene variants, which may be used for subsequent clinical trial enrollment or experimental treatment approaches (compassionate or off-label use). One aim of the INFORM registry, and the focus of this report, is to prospectively investigate a predefined (18) seven-scale molecular target prioritization algorithm ranging from “very high” to “very low,” based on the type of alteration and its disease-specific relevance, for its predictive power in a tumor type–agnostic approach. Importantly, the algorithm relies on functional molecular biological relevance of the target rather than evidence for clinical activity of a particular compound. The very high-priority targets are directly actionable genetic alterations with a proven link to tumorigenesis in the specific cancer type. High- and moderate-priority targets are those with a genetic alteration in a known cancer driver or activating an oncogenic pathway, with evidence either in the malignancy of interest or from other tumor types. Intermediate targets include genetic hits that sensitize to a given drug (e.g., activate a pathway) with evidence from tumors other than that being analyzed or highly overexpressed oncogenes that are known to drive the specific malignant disease. Borderline and low-priority targets are those that involve expression changes in oncogenic pathways or otherwise lead to drug sensitivity, with evidence either in the cancer type of interest or from other tumor types. Very low priority covers those with only circumstantial evidence of links to actionable drug targets. The registry routinely documents clinical follow-up, allowing evaluation of clinical benefit for this pediatric patient group with an urgent medical need.
At the time of the data cutoff, 1,051 patients had been registered in INFORM, of whom 519 patients had completed their clinical follow-up and were included in this analysis. This report describes prospective testing of a target prioritization algorithm and long-term clinical follow-up data of this large multinational and comprehensively molecularly profiled pediatric cohort in a real-world clinical setting.
RESULTS
Patients and Baseline Characteristics
Between January 21, 2015, and September 30, 2019, 1,051 patients were registered. Of these, the inclusion criteria were not fulfilled (most frequent reason: no or incomplete samples received, n = 27) for 65 patients, samples were not suitable for molecular analysis for 31 patients, and the molecular analysis was unsuccessful in 29 patients. Patient disposition is shown in Fig. 1. Of the remaining 926 patients, whole-exome sequencing (WES), low-coverage whole-genome sequencing (lcWGS), and DNA methylation analysis were successfully performed in almost all patients, with RNA sequencing successful for 84.3% of patients. No differences in profiling success rates were observed between national (German) or international sites (Supplementary Table S1). In total, 519 patients had completed their clinical follow-up of either at least 2 years or shorter in case of death or loss to follow-up and were subsequently included in this analysis. Patients with ongoing clinical follow-up shorter than 2 years at the time of the data cutoff (September 30, 2019; n = 407) were not included in this analysis. Patients were enrolled in a total of 72 centers in the following eight countries: Austria (n = 5), Finland (n = 5), Germany (n = 396), Greece (n = 3), Poland (n = 2), Sweden (n = 36), Switzerland (n = 13), and the Netherlands (n = 59). Baseline characteristics are provided in Table 1. A total of 14 different pediatric malignant diseases were included in the study, including a group of rare diseases (“other”), resulting in a distribution of 64.5% non–central nervous system (CNS) solid tumors, 26.8% CNS tumors, and 8.7% hematologic malignancies (for details, see Table 1 and Supplementary Table S2). To allow young adults who relapsed after a primary diagnosis during the pediatric age range (e.g., as frequently encountered with sarcomas), patients could be enrolled up to an age of 40 years, provided that they had had their primary pediatric-type diagnosis below the age of 21 years. One hundred and two patients (19.7%) were age ≥18 years at the time of inclusion. Fifty patients (9.6%) with newly diagnosed very high-risk malignancies for which no curative treatment exists [e.g., high-grade glioma (HGG)] were enrolled, whereas the others were enrolled upon refractory disease or relapse. Most patients with solid/CNS tumors (66.5%) were metastatic at enrollment.
Figure 1.
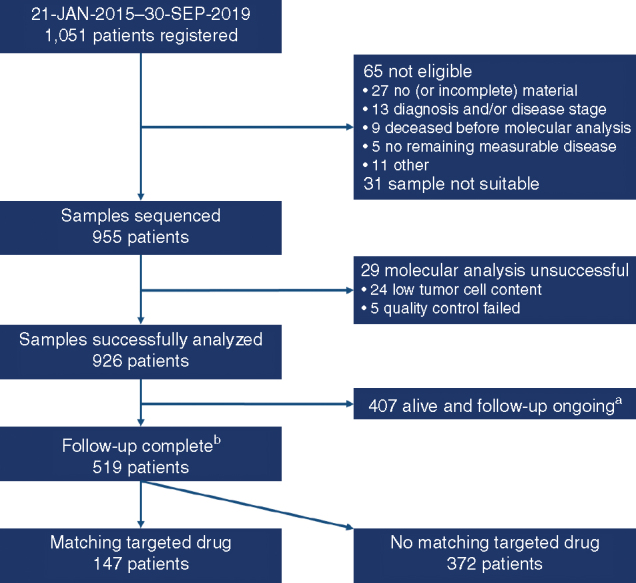
Patient disposition. aRegistered after October 1, 2017, and still alive and with ongoing follow-up (because regular follow-up of 2 years not complete) at the data cutoff. bAt least 2 years of regular follow-up completed, lost to follow-up, or deceased. This includes patients registered after October 1, 2017, who were lost to follow-up or deceased.
Table 1.
Patient baseline demographic and disease characteristics
| Characteristics (at enrollment) | Received matching targeted drug | Did not receive matching targeted drug | |
|---|---|---|---|
| N | 147 | 372 | |
| Median age (range), y | 13 (0–36) | 12 (1–40) | |
| Sex, n (%) | |||
| Female | 58 (39.5) | 164 (44.1) | |
| Male | 89 (60.5) | 208 (55.9) | |
| Lansky/Karnovsky performance status, n (%) | |||
| 50 | 4 (2.7) | 15 (4.0) | |
| 60 | 2 (1.4) | 18 (4.8) | |
| 70 | 19 (12.9) | 44 (11.8) | |
| 80 | 26 (17.7) | 89 (23.9) | |
| 90 | 46 (31.3) | 92 (24.7) | |
| 100 | 34 (23.1) | 61 (16.4) | |
| Missing | 16 (10.9) | 53 (14.2) | |
| Disease status, n (%) | |||
| Primary diagnosis | 11 (7.5) | 39 (10.5) | |
| Refractory to first-line therapy | 8 (5.4) | 18 (4.8) | |
| Relapse 1 | 71 (48.3) | 198 (53.2) | |
| Relapse 2 | 33 (22.4) | 69 (18.5) | |
| Relapse ≥3 | 23 (15.7) | 44 (11.8) | |
| Unknown | 1 (0.7) | 4 (1.1) | |
| Diagnosis, n (%) | |||
| Hematologic malignancy | 8 (5.4) | 37 (9.9) | |
| Acute lymphoblastic leukemia | 3 (2.0) | 20 (5.4) | |
| Acute myeloid leukemia | 3 (2.0) | 12 (3.2) | |
| Non-Hodgkin lymphoma | 1 (0.7) | 5 (1.3) | |
| Other | 1 (0.7) | 0 | |
| Solid tumor | 101 (68.7) | 234 (62.9) | |
| Desmoplastic small round cell tumor | 2 (1.4) | 6 (1.6) | |
| Ewing sarcoma | 14 (9.5) | 51 (13.7) | |
| Malignant rhabdoid tumor | 1 (0.7) | 9 (2.4) | |
| Neuroblastoma | 21 (14.3) | 51 (13.7) | |
| Osteosarcoma | 17 (11.6) | 23 (6.2) | |
| Rhabdomyosarcoma | 15 (10.2) | 49 (13.2) | |
| Other soft tissue sarcoma | 18 (12.2) | 20 (5.4) | |
| Other | 13 (8.8) | 25 (6.7) | |
| CNS tumor | 38 (25.9) | 101 (27.2) | |
| Atypical teratoid rhabdoid tumor | 3 (2.0) | 5 (1.3) | |
| Ependymoma | 7 (4.8) | 20 (5.4) | |
| High-grade glioma (including DIPG) | 20 (13.6) | 46 (12.4) | |
| Medulloblastoma | 5 (3.4) | 18 (4.8) | |
| Other | 3 (2.0) | 12 (3.2) | |
| Metastatic disease (% of nonhematologic malignancies) | 99 (71.2) | 216 (64.5) | |
| Subjected to molecular analysis at multiple time points (e.g., sequential relapses), n (%) | 17 (11.6) | 28 (7.5) |
Abbreviation: DIPG, diffuse intrinsic pontine glioma.
Target Identification and Distribution
After enrollment and sample receipt, the mean turnaround time to target reporting within the context of a molecular tumor board was 25.4 days. Identified targets were classified by an interdisciplinary review according to a seven-scale prioritization algorithm ranging from “very high” to “very low,” as reported previously (18). In 446 of the 519 patients (85.9%), at least one actionable target was identified, of which 225 (43%) were genetically altered targets (priority levels very high, high, and moderate; Table 2). The distribution of the highest priority level target per patient (multiple actionable alterations could be reported per patient) was: very high, 8.1%; high, 15.0%; moderate, 20.2%; intermediate, 23.9%; borderline, 14.6%; low, 2.3%; very low, 1.0%; and no actionable target available, 14.1% (actionable target not matching one of the priority levels, 0.8%). An overview of all reported actionable targets is provided in Supplementary Table S3. The most frequent very high priority level targets identified were ALK mutations, BRAF mutations, and NTRK fusions (Supplementary Table S4). The distribution of the highest priority level target per patient over the different diagnoses is depicted in Fig. 2. Considering disease groups with relevant case numbers, it was observed that the highest priority level (very high) occurred mostly in neuroblastoma and HGG. Priority levels high, moderate, and intermediate were more dominant in rhabdomyosarcoma and osteosarcoma. Ependymoma, Ewing sarcoma, other soft-tissue sarcomas, and other rare solid tumors tended to have a high proportion of targets of the lowest priority levels only (borderline, low, very low, and no target).
Table 2.
Priority-level distribution (highest priority level per patient)
| Priority level | Highest priority level for which patients received matched targeted drug, n (%) | Highest priority level per patient (all patients), n (%) |
|---|---|---|
| Very high | 20 (13.6) | 42 (8.1) |
| High | 31 (21.1) | 78 (15.0) |
| Moderate | 24 (16.3) | 105 (20.2) |
| Intermediate | 33 (22.4) | 124 (23.9) |
| Borderline | 26 (17.7) | 76 (14.6) |
| Low | 6 (4.1) | 12 (2.3) |
| Very low | 2 (1.4) | 5 (1.0) |
| Not applicable | 5 (3.4) | 4 (0.8) |
| No target | 0 | 73 (14.1) |
| Total | 147 (100) | 519 (100) |
Figure 2.

Distribution of the highest priority level target per patient over diagnoses. For each patient, only the highest priority level target is included. ALL, acute lymphoblastic leukemia; AML, acute myeloid leukemia; ATRT, atypical teratoid rhabdoid tumor; DSRCT, desmoplastic round cell tumor; EPN, ependymoma; EWS, Ewing sarcoma; HGG, high-grade glioma (including diffuse-intrinsic pontine glioma); MB, medulloblastoma; MRT, malignant rhabdoid tumor; NB, neuroblastoma; NHL, non-Hodgkin lymphoma; OS, osteosarcoma; other heme, other hematologic malignancies; RMS, rhabdomyosarcoma; STS, soft-tissue sarcoma. *, Including four patients with a target not matching one of the priority levels.
Treatment and Clinical Outcome
There was a trend to apply matching targeted drugs more frequently in patients with higher priority level targets than in patients with lower priority level targets (Table 2). Treatments matching one of the identified potential targets were applied in 147 patients (33.0% of the 446 patients with a potentially actionable target). The targeted agents applied were mostly small-molecule drugs, as well as immunotherapy (targeted antibodies including immune checkpoint inhibitors) in some instances (Supplementary Table S5a). The median duration of targeted treatment was 92 days. Seventeen patients were enrolled into clinical trials (3.8% of 446 patients with a potential actionable target and 11.6% of the 147 patients who received matching targeted drugs, with one patient enrolled in two trials). The following matching targeted drugs were applied in a trial setting: ceritinib (n = 3), crizotinib (n = 1), dabrafenib (n = 1), larotrectinib (n = 5), olaparib (n = 2), pazopanib (n = 1), ribociclib (n = 3), and tazemetostat (n = 2). All other patients received matching targeted drugs off-label or through compassionate use programs. Further details on drug treatments matching identified targets are provided in Supplementary Table S5a. During the study period, potent selective NTRK inhibitors became available (21–23). Outcome of patients with an NTRK fusion who received an NTRK inhibitor (e.g., larotrectinib) and patients with an NTRK fusion who received an alternative treatment (e.g., because NTRK inhibitors were not available then) is summarized in Supplementary Table S5b.
In the time from registration to completed sample receipt, 6 patients died and 5 had disease progression and were excluded from the progression-free survival (PFS) analyses (resulting in 508 evaluable patients). For overall survival (OS) analysis, the 6 patients who died before completion of sample receipt were excluded (resulting in 513 evaluable patients). Median PFS and OS for the whole cohort were 118 [95% confidence interval (CI), 106–145] and 290 (95% CI, 257–343) days, respectively (survival data for the individual patients can be found in Supplementary Table S2 and data on the disease level are provided in Supplementary Table S6 and Supplementary Fig. S1A–S1F). PFS and OS of all patients who received a matching targeted drug compared with all patients who did not receive matching targeted drugs did not show any significant differences (Fig. 3A and B). Of the 42 patients in whom a very high priority level target was identified, 20 received treatment with a matching targeted drug [all had at least evaluable disease for response evaluation (almost all metastatic)]. This resulted in a median PFS of 204 [95% CI, 99–not applicable (NA)] and OS of 354 days (95% CI, 165–NA), compared with a PFS of 117 (95% CI, 106–143; P = 0.011) and OS of 290 days (95% CI, 256–343; P = 0.32) in all other patients (Fig. 3C and D). At baseline, of the 20 patients with a very high priority level target who received matching targeted treatment, 85% had a Lansky/Karnovsky performance status between 80 and 100, 10% were enrolled at primary diagnosis, 5% were enrolled refractory to first-line therapy, and 75% were enrolled at first or second relapse, whereas this was the case in 66.3%, 9.6%, 5%, and 71.3%, respectively, in the other 499 patients. Five of 20 patients (25%) with a very high priority level target who received matching targeted treatment also received other concomitant treatments, whereas 18 of 22 patients (81.8%) with a very high priority level target who did not receive matching targeted treatment received other conventional treatments (Supplementary Table S4). Comparison of patients with a very high priority level target who received matching targeted drugs and those who did not showed that patients who did not receive matching targeted drugs had comparable PFS to all other patients (Fig. 3E and F). Patients who received targeted drugs matching to other priority level targets did not show a significant PFS or OS improvement (Fig. 3G and H). Of the 372 patients who did not receive matched targeted treatment, 298 patients (80.1%) received other conventional medical systemic treatments, mostly chemotherapy. For the patients with an actionable target who were not treated with a matched targeted drug (n = 299), the following reasons were reported: disease stabilized by other conventional therapy (n = 86), poor performance status due to progression of underlying malignant disease (n = 71), patient/guardians refused (n = 25), patient deceased before finalization of the molecular analysis and molecular tumor board (n = 22), poor performance status due to toxicity from previous treatment or treatments (n = 13), drug not available (n = 13), formulation not available (n = 11), insurance company declined cost coverage (n = 6), and various other reasons (n = 133; n = 20 patients with no information available; multiple reasons could be indicated). A comparable distribution was observed for patients with very high priority level targets who did not receive matched targeted treatment.
Figure 3.

Survival analyses. A, PFS of patients separated by application of matching targeted drug versus all other patients (P = 0.967). B, OS of patients separated by application of matching targeted drug versus all other patients (P = 0.144). C, PFS of patients separated by application of matching targeted drug in very high priority level patients versus all other patients (P = 0.011). D, OS of patients separated by application of matching targeted drug in very high priority level patients versus all other patients (P = 0.32). E, PFS of patients separated by application of matching targeted drug for very high priority level patients versus very high priority level patients who did not receive matching target drugs versus all other patients (P = 0.034). F, OS of patients separated by application of matching targeted drug for very high priority level patients versus very high priority level patients who did not receive matching target drugs versus all other patients (P = 0.518). G, PFS of patients separated by application of matching targeted drug for very high priority level patients versus high, moderate, and intermediate level patients versus borderline, low, and very low level patients versus all other patients who did not receive matching target drugs. H, OS of patients separated by application of matching targeted drug for very high priority level patients versus high, moderate, and intermediate level patients versus borderline, low, and very low level patients versus all other patients who did not receive matching target drugs.
During the study period, new evidence for certain treatments or relevance of specific alterations emerged that changed the priority levels for some molecular alterations (the algorithm itself did not change). A retrospective analysis using priority levels according to current scientific and medical knowledge did show a change of priority level for 21.6% of the reported alterations that did not show significantly different outcomes per prioritization level. Also, new potential biomarkers were reported even if they could not be confidently ranked by the algorithm (yet). For example, a somatic BRCAness signature was reported as a potential biomarker for PARP inhibition on the basis of preclinical data (24), whereas according to current knowledge a BRCAness signature in the absence of true BRCA family mutation does not represent a biomarker for PARP inhibition and accordingly is not reported anymore.
Diagnosis Refinement
DNA methylation analysis and RNA sequencing for gene fusion detection allowed for reevaluation of tumor diagnosis in specific diseases. For CNS tumor and sarcoma samples, previously described methylation-based classifier scores were applied (25, 26), and furthermore, cancer type–defining fusions (e.g., PAX3/7–FOXO1 in rhabdomyosarcoma or EWSR1 fusions in Ewing sarcomas) were used for comparison between diagnosis at enrollment (mostly based on histology) and the molecularly informed diagnosis. For a total of 257 patients, a statement on molecular confirmation of diagnosis could be made. Of these, diagnosis was confirmed for 240 patients (93.4%). Molecular results suggested a change in diagnosis in 17 patients [6.6%; Ewing sarcoma (n = 7), medulloblastoma (n = 4), primitive neuroectodermal tumor (n = 2), desmoplastic small round cell tumor (n = 1), osteosarcoma (n = 1), ependymoma (n = 1), and HGG (n = 1); Table 3]. For the Ewing sarcoma cases, no typical EWSR1 fusion could be identified based on RNA-sequencing analysis, and the DNA methylation analysis classifier score did not reveal a high score for Ewing sarcoma (26). Thus, these tumors were not considered classic Ewing sarcoma and were classified as sarcoma according to the respective methylation classifier score and/or detected alternative alteration (e.g., in BCOR or CIC). Notably, four cases registered as recurrent medulloblastoma were indicated as HGG (n = 3) or CNS sarcoma (n = 1) by DNA methylation (Table 3). In addition, for four patients with CNS tumors, diagnosis could be further refined (e.g., assignment to specific subgroups within a tumor type).
Table 3.
Molecular diagnosis change or refinement
| ID | Histologic diagnosis at registration | Diagnosis molecularly confirmed | Molecular classification (subgroup) | Conclusive method(s) |
|---|---|---|---|---|
| INF_R_081 | Desmoplastic small round cell tumor | No | Sarcoma (not further classifiable) | RNA-seq/DNA methylation |
| INF_R_037 | Ewing sarcoma | No | Sarcoma (not further classifiable) | RNA-seq/DNA methylation |
| INF_R_075 | Ewing sarcoma | No | Sarcoma (not further classifiable) | RNA-seq/DNA methylation |
| INF_R_101 | Ewing sarcoma | No | Sarcoma (not further classifiable) | RNA-seq/DNA methylation |
| INF_R_262 | Ewing sarcoma | No | Melanoma | RNA-seq/DNA methylation |
| INF_R_119 | Ewing sarcoma | No | Sarcoma with BCOR alteration | RNA-seq/DNA methylation |
| INF_R_228 | Ewing sarcoma | No | Sarcoma with CIC alteration | RNA-seq/DNA methylation |
| INF_R_557 | Ewing sarcoma | No | Sarcoma with CIC alteration | RNA-seq/DNA methylation |
| INF_R_463 | Osteosarcoma | No | Malignant peripheral nerve sheath tumor | DNA methylation |
| INF_R_263 | Ependymoma | No | CNS high-grade neuroepithelial tumor with BCOR alteration | DNA methylation |
| INF_R_003 | High-grade glioma | No | CNS high-grade neuroepithelial tumor with BCOR alteration | DNA methylation |
| INF_R_131 | Medulloblastoma | No | Sarcoma (not further classifiable) | Histology/DNA methylation |
| INF_R_013 | Medulloblastoma | No | High-grade glioma (not further classifiable) | DNA methylation |
| INF_R_223 | Medulloblastoma | No | High-grade glioma (not further classifiable) | DNA methylation |
| INF_R_118 | Medulloblastoma | No | High-grade glioma of receptor tyrosine kinase subgroup | DNA methylation |
| INF_R_178 | Primitive neuroectodermal tumor | No | Pineoblastoma | DNA methylation |
| INF_R_496 | Primitive neuroectodermal tumor | No | Pineoblastoma | DNA methylation |
| INF_R_057 | High-grade glioma | Yes (refinement) | Infantile hemispheric glioma | DNA methylation |
| INF_R_292 | High-grade glioma | Yes (refinement) | Infantile hemispheric glioma | DNA methylation |
| INF_R_707 | High-grade glioma | Yes (refinement) | Pleomorphic xanthoastrocytoma | DNA methylation |
| INF_R_709 | Oligoastrocytoma WHO grade III | Yes (refinement) | K27M-mutated high-grade glioma | DNA methylation |
Note: Patients for whom molecular results suggested a change or refinement in diagnosis.
Abbreviations: RNA-seq, RNA sequencing; WHO, World Health Organization.
Cancer Predisposition
Knowledge about a hereditary cancer predisposition syndrome (CPS) may be critical for the affected patient as it may influence treatment decisions and inform surveillance strategies for the patient and potentially affected relatives. During the informed consent process, patients and/or legal guardians were informed by the treating physician about the potential benefits and adverse effects that knowledge about a genetic cancer predisposition may entail. Patients and/or legal guardians consented to receive any relevant results in 94.2% of cases. We identified pathogenic or likely pathogenic (P/LP) variants in 39 patients (7.5%; Supplementary Table S7a). In 20 of 39 patients diagnosed with a CPS (51.3%), the cancer predisposition was first identified through our systematic screening of constitutional DNA and had not previously been clinically identified (Supplementary Table S7a). In our cohort, constitutional P/LP variants were most frequently identified in TP53 (Li-Fraumeni syndrome; 7/39; 17.9%) in patients with osteosarcoma (n = 4), HGG (n = 1), undifferentiated sarcoma (n = 1), and medulloblastoma (n = 1). Four patients were diagnosed with constitutional mismatch repair deficiency caused by biallelic P/LP variants in the mismatch repair genes PMS2 (HGG, n = 2) or MSH6 (HGG, n = 1; adenocarcinoma, n = 1). Constitutional P/LP variants in CHEK2 (Ewing sarcoma, osteosarcoma, and yolk sac tumor) and in SMARCB1 (ATRT) were found in three patients each. Two patients each carried constitutional P/LP variants in ALK, SDHB, and NF1. Biallelic constitutional P/LP variants in BRCA2 (Fanconi anemia) were also found in two patients (medulloblastoma with homozygous and HGG with compound heterozygous variants). Six patients carried heterozygous P/LP variants in DNA repair genes typically associated with adult-onset cancer (ATM, BRCA1, BRCA2, PALB2, SLX4), for which a driving role in the assessed pediatric tumors was not clear. Alterations in other genes were found in single patients only (Supplementary Table S7a).
DISCUSSION
Data from our INFORM registry demonstrate that comprehensive molecular profiling in a real-world multinational setting is feasible and beneficial. A turnaround time of less than 4 weeks is a clinically relevant window for these high-risk patients and particularly notable given the tasks carried out within this period: sample logistics, quality control, WES, lcWGS, RNA sequencing, DNA methylation analysis, bioinformatics processing, curation of target priorities, and conduction of the molecular tumor board. The weekly online interdisciplinary molecular tumor board in which targets and potential matching available approved treatments and matching open trials were discussed with the treating pediatric oncologist has developed into a well-recognized forum where expertise is shared and education is offered to its participants. Importantly, no differences in sequencing success rate were observed between national and international submissions. With a broad variety of pediatric-type hematologic malignancies, solid tumors, and CNS tumors represented, INFORM is truly agnostic to disease type. Notably, hematologic malignancies were somewhat underrepresented because of the availability of effective second- and third-line treatments and studies. The collection of long-term clinical follow-up data allowed for the initial identification of subgroups that may benefit from matched targeted treatment: patients with a very high priority level target who received matching targeted treatment had a significantly longer PFS compared with all other patients. In addition, molecular analyses added important diagnostic specifications and identified previously unknown hereditary cancer predisposition in a considerable number of patients.
In 43% of patients, a genetically altered target was identified (priority levels very high, high, and moderate), which is lower compared with other pediatric molecular profiling platforms [e.g., MOSCATO-01 (60.9%) and ZERO (71.4%); refs. 16, 17]. This discrepancy can be explained by a different definition of actionable altered genes (e.g., somatic TP53 mutations were not considered actionable in INFORM). Importantly, “actionable” refers to a detected molecular alteration or affected pathway that theoretically would be targetable by an approved drug or an investigational agent and does not take into account the availability of a respective drug or trial, particularly in regard to the pediatric population. This may explain the discrepancy between the number of actionable targets identified and the number of patients treated accordingly in INFORM. Furthermore, the methods used for ranking of actionable alterations (e.g., differentiation between genetic and nongenetic alterations) differ between the platforms. The ZERO program applies five levels (tiers; ref. 27) of supporting evidence based on the level of clinical or preclinical evidence in the respective disease (17), whereas the INFORM algorithm applies seven levels of functional molecular biological relevance (18). These differences in prioritization should be taken into account when comparing the clinical outcomes of both platforms. It will be very interesting to compare PFS and OS of both platforms according to both ranking systems at the time the ZERO program publishes long-term follow-up, as well (currently only response data are available; ref. 17). When comparing classification systems used by adult oncology platforms such as MASTER, which applies a similar molecular diagnostic platform as INFORM, one notices that most systems rely on clinical evidence (28). For example, the highest level used in MASTER (NCT/DKTK level m1A) is based on data from a prospective study or a meta-analysis in the same tumor type (29). In MASTER, 17.6% of patients received treatment recommendations of NCT/DKTK levels m1A to m1C (clinical data collected in the same histologic entity), which at this moment is an unobtainable standard in pediatrics because, unfortunately, such clinical evidence is only rarely available in this population (justifying the more functional biological approach used by INFORM).
The reported survival data of the population under study are in line with previous reports of pediatric patients in a phase I/II setting (3, 30), which underlines the enormous medical need in this population. We observed a doubling of PFS for patients with a very high priority level target who received matching targeted treatment compared with all other patients. The baseline performance status of the patients with a very high priority level target who received matching targeted treatment was slightly better compared with all other patients, and in this group, neuroblastoma and HGG were overrepresented. The disease status at enrollment was comparable in both groups, with 80.1% of all patients who were not treated with a matched drug receiving conventional oncologic treatments (mostly chemotherapy), which illustrates that most patients were at least in a sufficient clinical state to be able to receive further therapy. By design, an immortal time bias is observed for patients receiving matching targeted treatment because progression or death in this group could not occur before start of targeted therapy. Poor performance status due to progression of underlying malignant disease was one of the most frequent reasons for not applying matched targeted agents. Importantly, comparison of patients with a very high priority level target who received matching targeted drugs and those who did not showed that patients who did not receive matching targeted drugs had comparable PFS to all other patients and thus confirmed that the respective molecular aberrations are likely predictive for response to a targeted drug and not prognostic for improved outcome. At the start of the registry, a follow-up period of only 2 years was planned (because it was not expected that a relevant number of patients would be alive after 2 years). This could be the reason for not detecting a significant OS difference between target priority levels, in addition to sample size and nonstandardized administration of other local and systemic therapies.
Inherent to its design as a registry with its noninterventional regulatory status, there are a number of obvious limitations to this study. A registry does not define the treatment of choice (e.g., the use of different ALK inhibitors for the same alteration in the same disease) or exclude other treatments in addition to matching targeted treatment. However, because other treatments are applied in all patient categories, this is likely not of relevance for the observed difference in PFS. Furthermore, the fraction of patients with a very high priority level target who received matching targeted treatment as well as other concomitant treatments was low, and it seems unlikely that the improved PFS can be explained by concomitant treatment. It could, however, dilute weaker signals of lower priority level targets, for example. Furthermore, eligibility criteria are less strict than in clinical trials. In addition, response evaluations are not defined with regard to timing or method (in contrast to clinical trials) and therefore not considered an appropriate outcome measure. Still, due to the high a priori chance of relapse in this high-risk population, “progressive disease” does not depend much on the response evaluation method applied. Therefore, its use for PFS determination is considered feasible. Data quality (consistency, accuracy, and completeness) of registries such as INFORM is hampered by the lack of on-site monitoring, audits, and inspections (31), which are especially important for items such as medication information and safety data. Therefore, extensive central monitoring was applied in INFORM to reduce errors and incompleteness as much as possible. Finally, with regard to time-dependent endpoints, a registry such as INFORM has a different baseline definition (completed sample submission and full registration) independent of treatment (or several treatments) compared with a clinical trial. However, despite the limitations of a registry, INFORM has the major advantage that it collects clinical follow-up of all patients regardless of molecular alterations and treatments under real-world clinical conditions, in contrast to clinical trials, which are mostly confined to a defined population with a malignancy harboring a certain biomarker and a respective drug combination.
Unfortunately, compared with adult oncology with numerous new innovative trials, biomarkers, and drugs, pediatric oncology is still lagging behind in access to new compounds (32), which is clearly exemplified by the low number of patients in the INFORM registry enrolled in clinical trials. Optimally, molecular diagnostics should be offered to patients at an earlier stage during their disease course and not only at a stage when precision oncology is essentially “the last hope.” Notably, in INFORM, 32.6% of patients received molecular diagnostics only at second or later relapse. A protocol amendment allowing for enrollment in an earlier disease stage is currently under discussion within the steering committee. Furthermore, despite the availability of a genetically altered target (priority levels very high, high, and moderate), 62% of patients did not receive a targeted agent at all. Surprisingly, 52% patients with a very high priority level target did not receive targeted treatment. Rational combination treatment regimens are important because pathway redundancy, tumor evolution, and molecular cross-talk make it unlikely that targeted monotherapies will result in durable responses, especially in advanced late-stage cancers (20). The clinical outcome data support the requirement of biomarker-driven, cross-entity phase I/II combination trials, as articulated by the INFORM pilot study and others (18, 32). As such, the INFORM consortium has initiated the INFORM2 series of multinational biomarker-driven seamless phase I/II combination trials (33). In addition, other groups have also started large pediatric trial initiatives, such as the European AcSé-ESMART study (clinicaltrials.gov identifier: NCT02813135) and pediatric MATCH in the United States (15). The first INFORM2 trial (INFORM2 NivEnt, clinicaltrials.gov identifier: NCT03838042) is currently recruiting, and further INFORM2 trials are in preparation. In parallel, the INFORM registry will create a continuously growing database for future personalized clinical trials.
In addition to the identification of actionable alterations, the application of the previously described DNA methylation–based classifier for CNS tumors and sarcomas (25, 26) as well as the detection of disease-specific fusions by RNA sequencing contributed to the reevaluation of tumor diagnoses. In a considerable number of cases, changes of the registration diagnosis or refinements were suggested based on molecular results. This is very important for further treatment decisions independent of particular targets.
Our data demonstrate that a significant fraction of pediatric malignancies develop on the background of a CPS, which is often not clinically evident, although some predispositions (e.g., Li-Fraumeni syndrome) may require adaptation of treatment (e.g., for medulloblastoma; clinicaltrials.gov identifier: NCT02066220). Because INFORM primarily enrolls patients with high-risk tumors, certain CPSs are underrepresented (e.g., NF1 associated with low-grade glioma or RB1 associated with retinoblastoma). In INFORM, we also identified a number of heterozygous alterations in genes involved in DNA double-strand repair and more prominently known to predispose to adult-onset cancers, such as hereditary breast and ovarian cancer (ATM, BRCA1, BRCA2, PALB2, or SLX4). In two of these cases, analysis revealed somatic loss of heterozygosity in the tumor, directly supporting a potential contribution of the event in constitutional DNA to tumor initiation or evolution. Still, some of these constitutional variants potentially are not directly related to the tumor and may represent incidental findings, although this requires a more detailed prospective investigation. Comparably stringent criteria and varying composition of predefined lists containing known cancer predisposition genes may partly explain different rates of reported constitutional variant findings between our study and, for example, the ZERO program (17). To be reported, P/LP variants affecting autosomal recessive genes required a clear association with the respective malignancy or a second hit. In addition, we did not report on findings in clear adult-onset cancer predisposition genes (e.g., HOXB13 or ATR).
In conclusion, the INFORM registry has demonstrated that comprehensive and entity-agnostic pediatric precision oncology in a real-world, multinational setting is feasible. The prioritization algorithm identifies subgroups of pediatric patients with relapsed, progressive, or high-risk cancer benefiting from matched targeted treatment with improved PFS. For this population with an enormous medical need and particularly poor prognosis, this can be considered a hopeful start. Furthermore, molecular characterization of the tumors allows for refinement of diagnosis in some patients and identification of underlying CPS. This achievement has been acknowledged by the German health insurance companies, and negotiations to include INFORM diagnostics in their insurance coverage are ongoing. Despite the steadily decreasing costs of next-generation sequencing, this would be an important sustainability strategy with regard to the still-increasing number of samples submitted. To support such financing models in Germany and other participating countries in the future, a health economic impact analysis is planned as an objective in the still-ongoing registry. In most patients, only lower priority level molecular alterations could be identified, and these could not be shown to be associated with improved outcome when treated accordingly. Next to the methodologic limitations of a registry, an important reason for that can be that the relation between target and targeted drug is not always sufficiently tested. A systematic preclinical in vivo evaluation of targeted compounds in pediatric disease models is warranted [e.g., through ITCC-P4 (https://www.itccp4.eu)]. By doing that, we will learn more about predictive biomarkers, and the priority algorithm can be adapted accordingly. In addition, we propose that next-level precision oncology programs should encompass further layers of molecular and functional data (e.g., gene signatures, liquid biopsy methodologies, single-cell sequencing technologies, proteomics, drug sensitivity profiling), include patients at an earlier time point, and involve combination therapy strategies to hopefully increase the proportion of patients who may experience a clinical benefit.
METHODS
Study Design, Eligibility, and Patients
The INFORM registry is a prospective, noninterventional, multicenter, multinational, and feasibility registry collecting clinical, functional, and molecular data. In a weekly online tumor board, potential matching drugs and trials were discussed by an expert panel and the treating pediatric oncologist. The treating oncologist was involved in a direct discussion with the expert panel allowing for education and practical support, including a discussion on matching open clinical trials and available approved drugs. The use of target information for clinical decision-making was the responsibility of the treating pediatric oncologist. After a pilot phase (18), the registry opened January 21, 2015. A clinical follow-up of 2 years for all patients was planned (longer follow-up allowed). All patients who had finished their clinical follow-up, either at least 2 years or shorter in case of earlier study participation termination (e.g., if the patient died or was lost to follow-up), on September 30, 2019, were included in this report. Patients registered after October 1, 2017, and still alive and with ongoing follow-up less than 2 years were not included in this report. For patients with samples not suitable for molecular analysis and/or unsuccessful molecular analysis, no further clinical follow-up was performed. The registry is still ongoing.
Eligible patients had clinically suspected refractory/relapsed/progressive malignant disease and received a biopsy (solid and CNS tumors) or bone marrow aspiration (e.g., leukemia) as part of their standard-of-care treatment at their local pediatric oncology center. Eligible pediatric diagnoses included high-risk acute lymphoblastic leukemia, acute lymphoblastic leukemia after stem cell transplantation, acute myeloid leukemia, non-Hodgkin lymphoma, desmoplastic small round cell tumor, Ewing sarcoma, malignant rhabdoid tumor, neuroblastoma, osteosarcoma, rhabdomyosarcoma, other soft-tissue sarcoma, ependymoma, HGG (including diffuse-intrinsic pontine glioma), medulloblastoma, and other pediatric cancers, including rare tumors such as nephroblastoma, hepatoblastoma, retinoblastoma, malignant endocrine tumors, germ cell tumors, and others. Patients with a primary diagnosis of HGG (including diffuse-intrinsic pontine glioma), soft-tissue sarcoma, embryonal tumor with multilayered rosettes, rare tumors, and exceptional other cases, for which no curative treatment is established, could already be enrolled at primary diagnosis. To allow young adults who relapsed after having had their primary diagnosis at a pediatric age (e.g., sarcoma), patients could be enrolled up to an age of 40 years, provided that they had their primary pediatric-type diagnosis before the age of 21 years. Patients should have measurable disease, and no established curative treatment options should be available. A life expectancy >3 months and sufficient general condition (Lansky ≥50 or Karnofsky ≥50) were requested.
The study was conducted in accordance with Good Clinical Practice guidelines and the Declaration of Helsinki. All patients or their legally acceptable representative, or both (if possible), provided written informed consent. Approvals for the study protocol (and any modifications thereof) were obtained from independent ethics committees and the institutional review board at each participating center. The study was registered with the German Clinical Trial Register, number DRKS00007623.
Outcomes
The primary objectives of the INFORM registry were (a) to establish logistics for personalized treatment [on an (inter)national scale], (b) to initiate a comprehensive database providing information about individual druggable targets with parallel documentation of clinical follow-up, and (c) to investigate a predefined molecular target prioritization algorithm, based on the type of alteration and its disease-specific relevance, as described previously (18), for its prognostic and predictive power in a tumor-type agnostic approach. Secondary objectives included further evaluation of clinical benefit (PFS and OS), potential diagnostic refinements by molecular methods such as DNA methylation analysis, and identification of possible hereditary predisposition syndromes.
Procedures
Patients were recruited in Austria, Finland, Germany, Greece, Poland, Sweden, Switzerland, and The Netherlands. Before enrollment, the Society for Pediatric Oncology and Hematology in Germany (GPOH) study group of the respective cancer type or the respective national coordinator for other countries was consulted by the treating center to confirm eligibility. Written informed consent was obtained by the local treating physician, and the patient was registered in a globally accessible web portal (MARVIN, XClinical) in a pseudonymized fashion. Fresh-frozen tumor material of the current refractory/relapsed/progressive disease as well as matching nonmalignant material (EDTA blood, saliva) was submitted. After centralized sequencing/molecular analysis, raw data were subjected to bioinformatics processing and biological filtering, resulting in a list of prioritized actionable targets. The definition of “actionable” refers to a detected molecular alteration or affected pathway that theoretically would be targetable by an approved drug or an investigational agent in any phase of clinical development, either directly or indirectly in the affected pathway (16). Alterations included copy-number alterations, single-nucleotide variants, indels, gene fusions, outlier expression of individual genes, and expression of fusion transcripts. In a weekly online molecular tumor board, an expert panel consisting of molecular biologists, pediatric oncologists including the GPOH coordinator of the respective disease (or the respective national coordinator), and pharmacologists discussed, reviewed, and prioritized the identified targets together with the treating pediatric oncologist. Potential matching drugs and trials were also discussed together, but the registry did not give treatment recommendations (giving treatment recommendations would be an intervention and would be an interventional clinical trial according to the German Medicinal Products Act). The use of target information for clinical decision-making on an individual basis remained exclusively the responsibility of the treating pediatric oncologist. Thereafter, the targets were deposited in the globally accessible web portal as the final target report. The treating physician had password-secured access to their patients' data deposited in the web portal. They could use the molecular target information for clinical decision-making on an individual basis. Reporting and discussion of the molecular target information and potential matched treatments and trials with patients and their families was performed by the treating physician, too. Disease evaluations were performed according to routine standard of care in line with (inter)national disease-specific guidelines. Clinical and molecular baseline and follow-up data, including (concomitant) treatments, of all patients were prospectively documented in the web portal by the treating center. Clinical outcome data were centrally monitored in the web portal for plausibility and completeness by INFORM research physicians supported by data management. If necessary, queries for the local sites were generated in the web portal. At baseline, medical history and treatment were checked for plausibility and completeness by research physicians in preparation for the online molecular tumor board. During follow-up, data management monitored for completeness at regular intervals. After follow-up was finished and documentation was completed by the local sites, research physicians checked treatment and response data for plausibility. No site monitoring was performed.
A predisposition to cancer (CPS) may be critical for the affected patient as it may influence treatment decisions but could also affect carriers within the family. Prior to any analyses of constitutional DNA, patients and/or legal guardians received genetic counseling. To allow for a more precise interpretation of somatic variants detected in tumors, constitutional DNA isolated from white blood cells, alternatively from saliva, was sequenced in parallel. Variants identified in tumor tissues were reviewed in the constitutional genetic code from normal blood cells to infer true somatic mutations. If the patient and/or legal guardians had consented to investigation for a genetic cancer predisposition during the informed consent process, constitutional DNA of each patient was screened for damaging alterations in a predefined list of 157 known cancer predisposition genes (Supplementary Table S7b). Potential constitutional alterations were assessed by human geneticists according to American College of Medical Genetics and Genomics (ACMG) criteria (34), and only likely pathogenic (ACMG class 4) or pathogenic (ACMG class 5) variants with a probable relation to the patient's cancer diagnosis were reported to the treating physician, and genetic counseling of the patient and the family was recommended. Constitutional variant findings were incorporated in the target prioritization only if they represented an actionable target.
Molecular Profiling and Prioritization Algorithm
Within the INFORM pilot phase between October 2013 and January 2015, logistic and analytic pipelines necessary for rapid and comprehensive molecular profiling in a clinical setting have been established and described (18). In brief, fresh-frozen tumor material and nonmalignant DNA (e.g., from blood sample) was subjected to WES, lcWGS, RNA sequencing, RNA-based gene expression array, and DNA-methylation. RNA-sequencing data were used for identification of actionable fusion genes and for assessment of outlier gene expression of actionable genes in a given sample compared with a within- and across-malignant disease type reference series. The platform has an overall validation rate of 99.3% (range, 98.2%–100%) across the three test systems (WES, RNA sequencing, WGS) used. Only those variants receiving a “high-confidence” score based on an in-house quality assessment were considered. This metric takes into account features such as mappability of the region, coverage at the position, strand bias of reads, and number of variant reads in the control sample. No strict cutoffs in terms of variant allele fraction are applied, but in practice the quality score gives a lower variant allele fraction limit of approximately 0.05 for reported variants.
As previously described (18), actionable and tumor biologically relevant findings were prioritized in a standardized way based on a seven-scale score ranging from “very high” to “very low,” depending on the alteration type and its disease-specific relevance. As new evidence for certain treatments or relevance of specific alterations emerged during the study period, the target priority level could have changed over time. For the present analysis, the priority levels reported back to the treating physician after the respective interdisciplinary review were used.
Statistical Analysis
Survival time was defined as time since all necessary samples were received for molecular analysis. Patients who died or had a disease progression in the time between registration and sample receipt completion were excluded from the PFS analyses. Patients who died in the time between registration and sample receipt completion were excluded from OS analyses. OS is defined as time since all necessary samples were received to death from any cause. PFS is defined as time since all necessary samples were received until progression or death. PFS and OS comparisons were performed by log-rank tests.
Data Availability
WES, lcWGS, RNA-sequencing, and methylation data generated by this study are available from the European Genome Archive, accession number EGAS00001005112.
Authors' Disclosures
C.M. van Tilburg reports grants from German Cancer Aid (DKH 111234), German Childhood Oncology Foundation (DKS 2014.12), German Cancer Consortium (DKTK, Heidelberg, Germany) via the German Cancer Research Center (DKFZ) for a molecular diagnostics group and support to the DKFZ Genomics and Proteomics Core Facility, German Federal Ministry of Education and Research (01KX2025), Bild e.V. “Ein Herz für Kinder” (PÃ.-24151), Scheu Family, and German Federal Ministry of Health (ZMVI1-2520IGW004) during the conduct of the study; personal fees from Novartis and personal fees from Bayer outside the submitted work. E. Pfaff reports grants from German Cancer Aid (DKH 111234), German Childhood Oncology Foundation (DKS 2014.12), German Cancer Consortium (DKTK, Heidelberg, Germany) via the German Cancer Research Center (DKFZ) for a molecular diagnostics group and support to the DKFZ Genomics and Proteomics Core Facility, German Federal Ministry of Education and Research (01KX2025), Bild e.V. “Ein Herz für Kinder” (PÄ-24151), Scheu Family, and German Federal Ministry of Health (ZMVI1–2520IGW004) during the conduct of the study. K.W. Pajtler reports German Cancer Aid (DKH 111234), German Childhood Oncology Foundation (DKS 2014.12), German Cancer Consortium (DKTK, Heidelberg, Germany) via the German Cancer Research Center (DKFZ) for a molecular diagnostics group and support to the DKFZ Genomics and Proteomics Core Facility, German Federal Ministry of Education and Research (01KX2025), Bild e.V. “Ein Herz für Kinder” (PÄ-24151), Scheu Family, German Federal Ministry of Health (ZMVI1–2520IGW004), Structure- and Innovation Fund, Baden-Württemberg. B.C. Jones reports grants from German Cancer Aid, German Childhood Oncology Foundation, German Cancer Consortium (DKTK, Heidelberg, Germany) via the German Cancer Research Center (DKFZ) for a molecular diagnostics group and support to the DKFZ Genomics and Proteomics Core Facility, German Federal Ministry of Education and Research, Bild e.V. “Ein Herz für Kinder,” Scheu Family, and German Federal Ministry of Health during the conduct of the study. A. von Stackelberg reports personal fees from Amgen, Novartis, and personal fees from Jazz outside the submitted work. R. Meisel reports grants from DKTK (German Cancer Consortium) and DKH (German Cancer Aid) during the conduct of the study; personal fees from Novartis Pharma, Vertex Pharmaceuticals, Bluebird Bio, and personal fees from Bellicum Pharma outside the submitted work. D. Reinhardt reports grants from Novartis, Bluebirdbio, personal fees from BMS, personal fees from Hexal, grants and personal fees from Jazz, and personal fees from Janssen & Janssen outside the submitted work. J.H. Klusmann reports grants from European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program (grant agreement #714226) and St. Baldrick's Robert J. Arceci Innovation Award during the conduct of the study; personal fees from Roche, personal fees from Bluebird Bio, personal fees from Novartis, personal fees from Jazz Pharmaceuticals, grants from German Research Foundation (DFG; KL2374/5–1 and KL2374/3–1), BMBF (01GM1911D myPred), and EU Horizon 2020 (#116064) outside the submitted work. G. Fleischhack reports other support from Novartis Oncology during the conduct of the study; grants from German Children Cancer Foundation outside the submitted work. C.M. Kramm reports grants from DKFZ, Heidelberg, Germany, and other support from Novartis, Berlin, Germany, during the conduct of the study; grants and other support from Deutsche Kinderkrebsstiftung, Bonn, Germany, outside the submitted work. A.O. von Bueren reports personal fees from Consulting or Advisory Role and other support from Travel, Accommodations, Expenses outside the submitted work. M. Fischer reports grants from Bild hilft e.V. during the conduct of the study; personal fees from Novartis, personal fees from Bayer, personal fees from Roche, personal fees from Janssen, and personal fees from Bristol-Myers Squibb outside the submitted work. B. Burkhardt reports grants from German Childhood Cancer Foundation during the conduct of the study; German Cancer Aid, Roche, Miltenyi, and grants from Novartis outside the submitted work. W. Wößmann reports grants from German Childhood Cancer Foundation during the conduct of the study; other support from Takeda outside the submitted work. S. Hecker-Nolting reports grants from Förderkreis krebskranke Kinder Stuttgart e.V. during the conduct of the study; grants from EISAI outside the submitted work. I.B. Brecht reports grants from Rare tumours outside the submitted work. M. Schwab reports grants from Robert Bosch Stiftung and grants from Deutsche Forschungsgemeinschaft (DFG) im Rahmen der Exzellenzstrategie des Bundes und der Länder (EXC 2180—390900677) during the conduct of the study; grants from Green Cross WellBeing Co. Ltd, grants from Gilead Sciences Inc., other support from Agena Bioscience GmbH, and grants from Robert Bosch GmbH outside the submitted work; Pharmacogenetics and Genomics, Editor in Chief Drug Research, Editor in Chief Genome Medicine, Section Editor Honoraria for oral presentations at academically organized congresses and meetings. R. Tremmel reports grants from Robert Bosch Stiftung during the conduct of the study. T. Milde reports grants from BioMed Valley Discoveries outside the submitted work. D. Reuss reports a patent for NF1 antibody licensed to Merckmillipore. F. Sahm reports other support from Illumina, other support from AbbVie, and other support from Bayer outside the submitted work. A. von Deimling reports a patent for methylation-based classification pending. U. Dirksen reports grants from German Cancer Aid, grants from German Cancer Aid, and grants from German Cancer Aid outside the submitted work. D.T.W. Jones reports grants from German Cancer Aid, grants from German Childhood Oncology Foundation, grants from German Federal Ministry of Health, grants from German Federal Ministry of Education and Research (01KX2025), and grants from Bild e.V. “Ein Herz für Kinder” during the conduct of the study. J.J. Molenaar reports grants from ZonMW, grants from ERC START, and grants from NWO Vidi during the conduct of the study. D. Capper reports a patent for DNA methylation-based tumor classification pending. S.M. Pfister reports grants from ITCC-P4 Consortium (IMI-2) funded, involving Eli Lilly, Roche, Bayer, Pfizer, AstraZeneca, PharmaMar, and Johnson & Johnson, grants from Structure- and Innovation Fund, Baden-Württemberg, grants from German Cancer Consortium (DKTK, Heidelberg, Germany) via the German Cancer Research Center (DKFZ) for a molecular diagnostics group and support to the DKFZ Genomics and Proteomics Core Facility, grants from German Cancer Aid (DKH 111234), grants from German Childhood Oncology Foundation (DKS 2014.12), grants from German Federal Ministry of Education and Research (01KX2025), grants from Bild e.V. “Ein Herz für Kinder” (PÄ-24151), and grants from Scheu Family during the conduct of the study; in addition, S.M. Pfister has a patent for DNA-methylation based method for classifying tumor species issued. O. Witt reports grants from German Cancer Aid (DKH 111234), grants from German Childhood Oncology Foundation (DKS 2014.12), grants from German Cancer Consortium (DKTK, Heidelberg, Germany) via the German Cancer Research Center (DKFZ) for a molecular diagnostics group and support to the DKFZ Genomics and Proteomics Core Facility, grants from German Federal Ministry of Education and Research (01KX2025), grants from Bild e.V. “Ein Herz für Kinder” (PÄ-24151), grants from Scheu Family, and grants from German Federal Ministry of Health (ZMVI1–2520IGW004) during the conduct of the study; other support from Day One Biopharmaceuticals, grants from BioMed Valley Discoveries, other support from Novartis, other support from BMS, other support from Janssen, other support from Roche, other support from Bayer, and other support from AstraZeneca outside the submitted work. No disclosures were reported by the other authors.
Supplementary Material
Acknowledgments
This work was supported by the German Cancer Aid (111234), the German Childhood Oncology Foundation (DKS 2014.12, DKS 2018.18), the German Federal Ministry of Health (ZMVI1-2520IGW004), the German Federal Ministry of Education and Research (01KX2025) and Structure- and Innovation Fund, Baden-Württemberg. Funding support was provided by the German Cancer Consortium (DKTK, Heidelberg, Germany) via the German Cancer Research Center (DKFZ) for a molecular diagnostics group and support to the DKFZ Genomics and Proteomics Core Facility. Bild e.V. “Ein Herz für Kinder” generously has supported molecular analyses for non-German patients (PÄ-24151). In addition, we are very grateful for a donation by the Scheu Family. The authors acknowledge the support of the German Cancer Research Center (DKFZ) and Heidelberg University Hospital, Heidelberg, Germany. The Dutch iTHER program is acknowledged for their contribution, in particular Monique den Boer, Judith Boer, and Esther Hulleman. The staff at the Swedish Childhood Tumor Biobank, Karolinska Institute are acknowledged for their crucial contribution concerning the Swedish patients. Kerstin Ottawa of the NCT Trial Center, Heidelberg, Germany, has supported data management and played a major role in central monitoring. We would like to convey our heartfelt thanks to Carsten Maus (Genomics and Proteomics Core Facility, DKFZ), Christopher Previti, Lena Weiser (Omics IT and Data Management Core Facility, DKFZ), and Rolf Kabbe (Division of Pediatric Neurooncology, DKFZ) for their dedicated contribution in the bioinformatics analyses. We thank all patients and parents for their participation. We are grateful for the valuable support by all local sites, GPOH study centers, and national coordinating institutions in all participating countries for their participation, especially for documentation and the provision of high-quality tumor material by pathology and neuropathology departments and respective logistic personnel.
The publication costs of this article were defrayed in part by the payment of publication fees. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Footnotes
Note: Supplementary data for this article are available at Cancer Discovery Online (http://cancerdiscovery.aacrjournals.org/).
Authors' Contributions
C.M. van Tilburg: Data curation, formal analysis, validation, investigation, visualization, methodology, writing–original draft, writing–review and editing. E. Pfaff: Data curation, formal analysis, validation, investigation, writing–original draft, writing–review and editing. K.W. Pajtler: Resources, data curation, formal analysis, validation, investigation, writing–original draft, writing–review and editing. K.P.S. Langenberg: Data curation, investigation, writing–review and editing. P. Fiesel: Investigation, writing–review and editing. B.C. Jones: Data curation, formal analysis, validation, investigation, writing–review and editing. G.P. Balasubramanian: Investigation, writing–review and editing. S. Stark: Investigation, writing–review and editing. P.D. Johann: Investigation, writing–review and editing. M. Blattner-Johnson: Investigation, writing–review and editing. K. Schramm: Investigation, writing–review and editing. N. Dikow: Investigation, writing–review and editing. S. Hirsch: Investigation, writing–review and editing. C. Sutter: Investigation, writing–review and editing. K. Grund: Investigation, writing–review and editing. A. von Stackelberg: Writing–review and editing. A.E. Kulozik: Writing–review and editing. A. Lissat: Writing–review and editing. A. Borkhardt: Writing–review and editing. R. Meisel: Writing–review and editing. D. Reinhardt: Writing–review and editing. J.H. Klusmann: Writing–review and editing. G. Fleischhack: Writing–review and editing. S. Tippelt: Writing–review and editing. D. von Schweinitz: Writing–review and editing. I. Schmid: Writing–review and editing. C.M. Kramm: Writing–review and editing. A.O. von Bueren: Writing–review and editing. G. Calaminus: Writing–review and editing. P. Vorwerk: Writing–review and editing. N. Graf: Writing–review and editing. F. Westermann: Investigation, writing–review and editing. M. Fischer: Writing–review and editing. A. Eggert: Writing–review and editing. B. Burkhardt: Writing–review and editing. W. Wößmann: Writing–review and editing. M. Nathrath: Writing–review and editing. S. Hecker-Nolting: Writing–review and editing. M.C. Frühwald: Writing–review and editing. D.T. Schneider: Writing–review and editing. I.B. Brecht: Writing–review and editing. P. Ketteler: Writing–review and editing. S. Fulda: Writing–review and editing. E. Koscielniak: Writing–review and editing. M.T. Meister: Writing–review and editing. M. Scheer: Writing–review and editing. S. Hettmer: Writing–review and editing. M. Schwab: Writing–review and editing. R. Tremmel: Writing–review and editing. I. Øra: Writing–review and editing. C. Hutter: Writing–review and editing. N.U. Gerber: Writing–review and editing. O. Lohi: Writing–review and editing. B. Kazanowska: Writing–review and editing. A. Kattamis: Writing–review and editing. M. Filippidou: Investigation, writing–review and editing. B. Goemans: Writing–review and editing. C.M. Zwaan: Writing–review and editing. T. Milde: Investigation, writing–review and editing. N. Jäger: Investigation, writing–review and editing. S. Wolf: Investigation, writing–review and editing. D. Reuss: Investigation, writing–review and editing. F. Sahm: Investigation, writing–review and editing. A. von Deimling: Investigation, writing–review and editing. U. Dirksen: Writing–review and editing. A. Freitag: Data curation, formal analysis, visualization, writing–review and editing. R. Witt: Investigation, project administration, writing–review and editing. P. Lichter: Investigation, writing–review and editing. A. Kopp-Schneider: Formal analysis, validation, investigation, visualization, methodology, writing–review and editing. D.T.W. Jones: Conceptualization, resources, supervision, funding acquisition, investigation, methodology, writing–review and editing. J.J. Molenaar: Conceptualization, supervision, funding acquisition, investigation, writing–review and editing. D. Capper: Conceptualization, supervision, investigation, writing–review and editing. S.M. Pfister: Conceptualization, resources, supervision, funding acquisition, investigation, methodology, writing–review and editing. O. Witt: Conceptualization, resources, supervision, funding acquisition, investigation, methodology, writing–review and editing.
References
- 1. Antony R, Wong KE, Patel M, Olch AJ, McComb G, Krieger Met al. A retrospective analysis of recurrent intracranial ependymoma. Pediatr Blood Cancer 2014;61:1195–201. [DOI] [PubMed] [Google Scholar]
- 2. Athale UH, Duckworth J, Odame I, Barr R. Childhood atypical teratoid rhabdoid tumor of the central nervous system: a meta-analysis of observational studies. J Pediatr Hematol Oncol 2009;31:651–63. [DOI] [PubMed] [Google Scholar]
- 3. Bautista F, Di Giannatale A, Dias-Gastellier N, Fahd M, Valteau-Couanet D, Couanet Det al. Patients in pediatric phase I and early phase II clinical oncology trials at Gustave Roussy: a 13-year center experience. J Pediatr Hematol Oncol 2015;37:e102–10. [DOI] [PubMed] [Google Scholar]
- 4. Creutzig U, Zimmermann M, Dworzak MN, Gibson B, Tamminga R, Abrahamsson Jet al. The prognostic significance of early treatment response in pediatric relapsed acute myeloid leukemia: results of the international study Relapsed AML 2001/01. Haematologica 2014;99:1472–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kempf-Bielack B, Bielack SS, Jurgens H, Branscheid D, Berdel WE, Exner GUet al. Osteosarcoma relapse after combined modality therapy: an analysis of unselected patients in the Cooperative Osteosarcoma Study Group (COSS). J Clin Oncol 2005;23:559–68. [DOI] [PubMed] [Google Scholar]
- 6. Kobrinsky NL, Sposto R, Shah NR, Anderson JR, DeLaat C, Morse Met al. Outcomes of treatment of children and adolescents with recurrent non-Hodgkin's lymphoma and Hodgkin's disease with dexamethasone, etoposide, cisplatin, cytarabine, and l-asparaginase, maintenance chemotherapy, and transplantation: Children's Cancer Group Study CCG-5912. J Clin Oncol 2001;19:2390–6. [DOI] [PubMed] [Google Scholar]
- 7. London WB, Castel V, Monclair T, Ambros PF, Pearson AD, Cohn SLet al. Clinical and biologic features predictive of survival after relapse of neuroblastoma: a report from the International Neuroblastoma Risk Group project. J Clin Oncol 2011;29:3286–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. MacDonald TJ, Aguilera D, Kramm CM. Treatment of high-grade glioma in children and adolescents. Neuro Oncol 2011;13:1049–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Oberlin O, Rey A, Lyden E, Bisogno G, Stevens MC, Meyer WHet al. Prognostic factors in metastatic rhabdomyosarcomas: results of a pooled analysis from United States and European cooperative groups. J Clin Oncol 2008;26:2384–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ramaswamy V, Remke M, Bouffet E, Faria CC, Perreault S, Cho YJet al. Recurrence patterns across medulloblastoma subgroups: an integrated clinical and molecular analysis. Lancet Oncol 2013;14:1200–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Stahl M, Ranft A, Paulussen M, Bolling T, Vieth V, Bielack Set al. Risk of recurrence and survival after relapse in patients with Ewing sarcoma. Pediatr Blood Cancer 2011;57:549–53. [DOI] [PubMed] [Google Scholar]
- 12. von Stackelberg A, Volzke E, Kuhl JS, Seeger K, Schrauder A, Escherich Get al. Outcome of children and adolescents with relapsed acute lymphoblastic leukaemia and non-response to salvage protocol therapy: a retrospective analysis of the ALL-REZ BFM Study Group. Eur J Cancer 2011;47:90–7. [DOI] [PubMed] [Google Scholar]
- 13. Grobner SN, Worst BC, Weischenfeldt J, Buchhalter I, Kleinheinz K, Rudneva VAet al. The landscape of genomic alterations across childhood cancers. Nature 2018;555:321–7. [DOI] [PubMed] [Google Scholar]
- 14. Ma X, Liu Y, Liu Y, Alexandrov LB, Edmonson MN, Gawad Cet al. Pan-cancer genome and transcriptome analyses of 1,699 paediatric leukaemias and solid tumours. Nature 2018;555:371–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Allen CE, Laetsch TW, Mody R, Irwin MS, Lim MS, Adamson PCet al. Target and agent prioritization for the Children's Oncology Group–National Cancer Institute Pediatric MATCH Trial. J Natl Cancer Inst 2017;109:djw274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Harttrampf AC, Lacroix L, Deloger M, Deschamps F, Puget S, Auger Net al. Molecular screening for cancer treatment optimization (MOSCATO-01) in pediatric patients: a single-institutional prospective molecular stratification trial. Clin Cancer Res 2017;23:6101–12. [DOI] [PubMed] [Google Scholar]
- 17. Wong M, Mayoh C, Lau LMS, Khuong-Quang DA, Pinese M, Kumar Aet al. Whole genome, transcriptome and methylome profiling enhances actionable target discovery in high-risk pediatric cancer. Nat Med 2020;26:1742–53. [DOI] [PubMed] [Google Scholar]
- 18. Worst BC, van Tilburg CM, Balasubramanian GP, Fiesel P, Witt R, Freitag Aet al. Next-generation personalised medicine for high-risk paediatric cancer patients—the INFORM pilot study. Eur J Cancer 2016;65:91–101. [DOI] [PubMed] [Google Scholar]
- 19. Forrest SJ, Geoerger B, Janeway KA. Precision medicine in pediatric oncology. Curr Opin Pediatr 2018;30:17–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mody RJ, Prensner JR, Everett J, Parsons DW, Chinnaiyan AM. Precision medicine in pediatric oncology: lessons learned and next steps. Pediatr Blood Cancer 2017;64:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Drilon A, Laetsch TW, Kummar S, DuBois SG, Lassen UN, Demetri GDet al. Efficacy of larotrectinib in TRK fusion-positive cancers in adults and children. N Engl J Med 2018;378:731–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hong DS, DuBois SG, Kummar S, Farago AF, Albert CM, Rohrberg KSet al. Larotrectinib in patients with TRK fusion-positive solid tumours: a pooled analysis of three phase 1/2 clinical trials. Lancet Oncol 2020;21:531–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Laetsch TW, DuBois SG, Mascarenhas L, Turpin B, Federman N, Albert CMet al. Larotrectinib for paediatric solid tumours harbouring NTRK gene fusions: phase 1 results from a multicentre, open-label, phase 1/2 study. Lancet Oncol 2018;19:705–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Engert F, Kovac M, Baumhoer D, Nathrath M, Fulda S. Osteosarcoma cells with genetic signatures of BRCAness are susceptible to the PARP inhibitor talazoparib alone or in combination with chemotherapeutics. Oncotarget 2017;8:48794–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Capper D, Jones DTW, Sill M, Hovestadt V, Schrimpf D, Sturm Det al. DNA methylation-based classification of central nervous system tumours. Nature 2018;555:469–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Koelsche C, Schrimpf D, Stichel D, Sill M, Sahm F, Reuss DEet al. Sarcoma classification by DNA methylation profiling. Nat Commun 2021;12:498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Harris MH, DuBois SG, Glade Bender JL, Kim A, Crompton BD, Parker Eet al. Multicenter feasibility study of tumor molecular profiling to inform therapeutic decisions in advanced pediatric solid tumors: the Individualized Cancer Therapy (iCat) Study. JAMA Oncol 2016;2:608–15. [DOI] [PubMed] [Google Scholar]
- 28. Horak P, Heining C, Kreutzfeldt S, Hutter B, Mock A, Hullein Jet al. Comprehensive genomic and transcriptomic analysis for guiding therapeutic decisions in patients with rare cancers. Cancer Discov 2021 Jun 11 [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 29. Leichsenring J, Horak P, Kreutzfeldt S, Heining C, Christopoulos P, Volckmar ALet al. Variant classification in precision oncology. Int J Cancer 2019;145:2996–3010. [DOI] [PubMed] [Google Scholar]
- 30. Carceller F, Bautista FJ, Jimenez I, Hladun-Alvaro R, Giraud C, Bergamaschi Let al. Prognostic factors of overall survival in children and adolescents enrolled in dose-finding trials in Europe: an innovative therapies for children with cancer study. Eur J Cancer 2016;67:130–40. [DOI] [PubMed] [Google Scholar]
- 31. McGettigan P, Alonso Olmo C, Plueschke K, Castillon M, Nogueras Zondag D, Bahri Pet al. Patient registries: an underused resource for medicines evaluation: operational proposals for increasing the use of patient registries in regulatory assessments. Drug Saf 2019;42:1343–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Laetsch TW, DuBois SG, Bender JG, Macy ME, Moreno L. Opportunities and challenges in drug development for pediatric cancers. Cancer Discov 2021;11:545–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. van Tilburg CM, Witt R, Heiss M, Pajtler KW, Plass C, Poschke Iet al. INFORM2 NivEnt: the first trial of the INFORM2 biomarker driven phase I/II trial series: the combination of nivolumab and entinostat in children and adolescents with refractory high-risk malignancies. BMC Cancer 2020;20:523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster Jet al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17:405–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
WES, lcWGS, RNA-sequencing, and methylation data generated by this study are available from the European Genome Archive, accession number EGAS00001005112.